

Abstract
Hexamers of connexins (Cxs) form hemichannels that dock tightly in series via their extracellular domains to give rise to gap junction channels. Here we examined the ability of a variety of C-terminal Cx32 mutations, most of which have been identified in X-linked Charcot–Marie–Tooth disease, to form hemichannels and to complete gap junction channels using the Xenopus oocyte system. First, we show that undocked wild-type Cx32 hemichannels at the plasma membrane can be detected as opening channels activated by depolarization. We have been able to estimate the efficiency of assembly of complete channels by measuring the time-dependent incorporation of preformed hemichannels into gap junction channels after cell-to-cell contact. These data offer strong evidence that hemichannels are the direct precursors of gap junction channels. Of 11 Cx32 mutants tested, a group of 5 mutations prevented the formation of functional hemichannels at the cell surface, whereas 4 mutations were fully able to form precursors but reduced the ability of hemichannels to assemble into complete channels, and 2 mutants formed channels normally. The data revealed that a minimum length of human Cx32 including the residue Arg-215 is required for the expression of hemichannels at the cell surface and that the efficiency of hemichannel incorporation into complete channels decreased gradually with the progressive shortening of the cytoplasmic C-terminal domain.
Keywords: connexin, voltage gating, gap junction channel formation, hereditary motor sensory neuropathy, X-linked Charcot–Marie–Tooth disease, Xenopus oocyte
Gap junctions are clusters of intercellular channels that form adherent regions where the direct passage of ions and other small molecules between neighboring cells occurs (Revel and Karnovsky, 1967; Gilula et al., 1982). Connexins (Cxs) are the structural proteins of these intercellular channels in vertebrates, of which 15 isotypes of this multigene family have been cloned so far in rodents, as well as several orthologs in other species (Bruzzone et al., 1996). In human, mutations of the Cx32 gene were found in X-linked forms of Charcot–Marie–Tooth disease (CMTX) (Bergoffen et al., 1993), a hereditary peripheral neuropathy that is characterized by abnormally low nerve conduction and chronic weakness with progressive muscular atrophy and sensory loss of the distal extremities.
Cx32 is thought to form gap junction channels between the adjacent folds at noncompact myelin (Bergoffen et al., 1993). These reflexive channels may act as an adhesive structure as well as constituting a radial pathway for the rapid passage of ions, small metabolites, and second messenger molecules between adjacent cytoplasmic compartments of the perinuclear and adaxonal regions of Schwann cells (Balice-Gordon et al., 1998). To date, >130 different CMTX mutations have been identified, and they are distributed throughout all topological domains of the Cx32 molecule (Bone et al., 1997).
Attempts to explain the molecular and cellular mechanisms leading to the CMTX phenotype from the location and type of mutation have been hampered by our limited knowledge concerning the molecular structure and contribution of the various Cx32 domains to channel formation. It is currently accepted that the assembly of connexins into channels is a two-step process. First, connexins oligomerize into connexons or hemichannels in the intracellular compartment before their transport to the plasma membrane (Musil and Goodenough, 1993; Kumar et al., 1995), and then the hemichannels come in contact and dock with another hemichannel from a neighboring cell to form the complete channel. Most of the events and molecular mechanisms involved in the successive stages of channel formation are currently not well understood. There is increasing evidence that hemichannels are the immediate precursors of gap junction channels, but direct proof of this is still not forthcoming. It is expected that during the process of channel formation hemichannels are closed until they dock with another hemichannel, inducing the newly formed channel to open (Loewenstein et al., 1981; Chow and Young, 1987). However, the opening of solitary hemichannels in the plasma membrane can be induced by certain pharmacological manipulations (DeVries and Schwartz, 1992; Li et al., 1996) or by membrane potential depolarization (Paul et al., 1991; Gupta et al., 1994; Ebihara et al., 1995; Ebihara, 1996).
Here, we report that human Cx32 is also able to form solitary hemichannels that open on depolarization. Taking advantage of our ability to detect functionally undocked hemichannels at the cell surface, we have characterized the normal process of the incorporation of hemichannels into complete channels and examined the specific stage of channel formation altered by several C-terminal Cx32 mutants associated with CMTX.
MATERIALS AND METHODS
Construction of human Cx32 mutants. The human Cx32 (HCx32) cDNA (kindly provided by N. M. Kumar; Kumar and Gilula, 1986) containing the complete coding region was inserted into the plasmid pBScMXT (Gupta et al., 1994) between the 5′ and 3′ flanking regions of noncoding Xenopus β-globin sequence to boost expression. Eight HCx32 mutants found in patients with the X-linked form of Charcot–Marie–Tooth disease and two non-CMTX-related mutations were introduced into the wild-type cDNA by PCR site-directed mutagenesis. To generate the mutants, the following oligonucleotide primers were used: E208K (sense, 5′-CGCGACATTGAGGATGATGCAGAT-3′; antisense, 5′-AAGGTGGTGTACCTCATCATCCGG-3′), which introduced a newNru restriction site; Y211stop [sense 5′-TAGACCCCAAGGCCTCTCTCTGCC-3′ (stopTAG); antisense, 5′-GACCACCTCGGCCACATTGAGGAT-3′], with a new translationally silentXbaI site; R215W (sense 5′-TGGGCCTGTGCCCGCCGAGCCCAG-3′; antisense, 5′-TATGATGAGGTACACCACCTCGGC-3′), with a new NdeI restriction site; R215Q (sense, 5′-CAGGCCTGTGCCCGCCGAGCC-3′; antisense, 5′-GATGATGAGGTACACCACCTCGG-3′), which created a newStuI site; R215stop [sense, 5′-TGATGCCACATACCAGGCAACCTG-3′ (stopTGA); antisense, 5′-GATGATGAGGTACACCACCTCGG-3′]; C217stop (sense, stopTAG; antisense, 5′-GGCCCGGATGATGAGGTACACCAC-3′); R220stop (sense, stopTGA; antisense, 5′-GCGGGCACAGGCCCGGATGATGAG-3′); R238H(sense, 5′-CCGGAATACAAGCAGAATGAGATC-3′; antisense, 5′-AGACAGGTGGTGGCCGAAGCCCGA-3′), with a new AccIII site;R265stop (sense, stopTAG; antisense, 5′-GCGCAGTATGTCTTTCAGGGAGCCAT-3′); and S281stop (sense, stopTAG; antisense, 5′-GCAGCGGTCGCTCTTTTCAGCCAG-3′). The PCR products were digested by DpnI endonuclease to eliminate the template and with Pfu DNA polymerase (Stratagene, La Jolla, CA) to remove the bases extended onto the 3′ ends before ligation. The whole coding region of the C280G mutation amplified from genomic DNA using primers 1F and 2R (Ionasescu et al., 1994) was directly inserted into theEcoRV site of the pBScMXT vector. Mutants were screened by size or restriction enzyme analysis and confirmed by DNA sequencing of the entire coding region on both strands.
In vitro transcription of cRNAs. Capped cRNAs were transcribed by T3 RNA polymerase from 10 μg ofSalI-linearized plasmid. The cRNA synthesis was performed under standard reaction conditions (protocol and reagents from Promega, Madison, WI) in the presence of the cap analog m7G(5′)ppp(5′)G (Boehringer Mannheim, Mannheim, Germany). After DNase digestion and purification, cRNAs were quantified by absorbance (260 nm), and the proportion of full-length transcripts (>90%) was checked by agarose gel electrophoresis.
Expression in Xenopus oocytes. Adult femaleXenopus laevis frogs were purchased from Nasco (Fort Atkinson, WI), and oocytes were prepared as previously described (Barrio et al., 1997). Oocytes in stages V and VI were co-injected with an antisense oligonucleotide directed against Xenopus Cx38 mRNA to block endogenous expression (10 ng/oocyte; Barrio et al., 1991), and with the in vitro-transcribed cRNA of wild-type and CMTX mutants (0.1–0.5 μg/μl, 50 nl/oocyte).
Translation in oocytes. Seventy-two hours after RNA injection, oocytes were homogenized with lysis buffer (in mm: 5 Tris, 5 EDTA, 5 EGTA, and 20 PMSF and 10 mg/ml leupeptin) and centrifuged to remove the yolk. Membranes were precipitated by centrifugation (100,000 × g, 30 min) after 30 min incubation in alkaline solution (20 mm NaOH). The membrane pellet was resuspended in loading buffer (25 mm Tris, 0.5% SDS, 18% glycerol, and 143 mm2-mercaptoethanol). For each mutant, four oocyte equivalents per lane were loaded in a 4–20% Tris-glycine SDS-PAGE gel (Bio-Rad, Hercules, CA). After separation, proteins were transferred to nitrocellulose membranes and visualized with 0.2% Ponceau S. Immunodetection was performed with the M12.13 antibody (Goodenough et al., 1988; Chemicon, Temecula, CA), which recognizes the cytoplasmic loop of Cx32. Specific bands were detected by enhanced chemiluminescence (ECL kit; Amersham, Buckinghamshire, England) following the instructions of the manufacturer.
Macroscopic hemijunctional and junctional currents. Oocytes injected with the wild-type and mutated Cx32 cRNAs were recorded in ND96 medium (in mm: 96 NaCl, 2 KCl, 1 MgCl2, 1.8 CaCl2, and 5 HEPES, pH 7.40) using microelectrodes of 0.5–1 MΩ filled with 2 mKCl, 10 mm EGTA, and 10 mm HEPES, pH 7.20. To explore the channel-forming ability of mutated Cx32, the vitelline membrane was removed 24 hr after injection, and oocytes were paired to facilitate the formation of cell-to-cell channels. The macroscopic junctional currents (Ij) were measured and analyzed as previously described (Barrio et al., 1997). The transmembrane currents attributable to opening hemichannels (Ihj) were directly recorded under voltage clamp with two electrodes in isolated oocytes, and in the coupled pairs, the currents were calculated asIhj = Itotal −Ij for each cell of the pair.
Patch–clamp recordings. The patch-clamp experiments were performed according to standard techniques. Borosilicate glass pipettes (LE16; Dagan, Minneapolis, MN) of 1.5–4 MΩ resistance were filled with ND96 solution, and the bath solution was standard internal solution (in mm: 100 KCl, 10 HEPES, and 10 EGTA); under these conditions, the expected membrane potential of the oocyte was near zero, as confirmed by the lack of appreciable differences in unitary conductance and the reversal potential of hemichannels between cell-attached and inside–outside configurations. We used an Axopatch 1D amplifier (Axon Instruments, Foster City, CA) connected to a personal computer compatible through a Digi-Data (Jessup, MD) 1200 interface, and data acquisition and analysis were performed using the pClamp 6. The currents filtered at 1 kHz were sampled at 2 and 20 kHz. Patch recordings in cell-attached and inside-out configurations were performed in oocytes for which positive expression was previously measured at the macroscopic level.
RESULTS
We have evaluated the hemichannel- and channel-forming ability of 11 mutants lying between residues 208–283 of the HCx32 molecule. Nine of these mutations have been implicated in CMTX; their location is illustrated in Figure1A. The group of nonsense CMTX mutations leading to truncated proteins are as follows: Y211stop (Tan et al., 1996), C217stop (Ionasescu et al., 1994, 1996), R220stop (Fairweather at al., 1994; Bone et al., 1995; Ionasescu et al., 1996), and S281stop (Nelis et al., 1997). An additional truncation generated by a 29-bp deletion beginning at amino acid 265, which produces a stop codon directly after position 265 has also been tested (R265stop; Ionasescu et al., 1996). Furthermore, the group of missense CMTX mutations analyzed includes E208K (Fairweather et al., 1994), R215W (Fairweather et al., 1994; Ressot et al., 1996; Bort et al., 1997), R238H (Nelis et al., 1997) and C280G, not previously published. The novel C280G mutation was detected in one Hungarian family by direct sequencing of the Cx32 gene (see methods in Silander et al., 1997), a 30-year-old male being diagnosed with CMT based on electrophysiological study and nerve biopsy findings. His subjectively healthy mother was found to carry the mutation and showed decreased nerve conduction. The mutation was not present in three unaffected family members (father and two maternal cousins) and 100 Finnish controls.
Fig. 1.

Hemichannel- and channel-forming ability of the CMTX mutants. A, Diagrams of the topological domains of HCx32 molecule and the location of the nine CMTX mutations and two nonrelated CMTX mutants, R215stop and R215Q, tested.M1, M4, Transmembrane domains; E1,E2, extracellular loops; CL, cytoplasmic loop;NT, N terminal; CT, C terminal. B, Sample records of macroscopic transmembrane currents induced by depolarizations from −40 to +120 mV by increments of 20 mV and of 10 sec duration in isolated oocytes expressing the wild-type HCx32 and mutants. Depolarizations of +40 mV or greater induced slowly activating outward currents, which became larger as higher positive potentials were applied in oocytes expressing HCx32. Currents became inward and declined with a slow time course when the potentials returned to −40 mV. The induction of large currents with these unusual characteristics suggested the presence of Cx32 hemichannels preformed in the plasma membrane, which were normally closed at resting potentials and that open with depolarization (also see single channel recordings in Fig.2). A group of mutants, E208K, Y211stop, R215stop, R215W, and R215Q, did not show detectable levels of hemichannels at the cell surface, whereas the oocytes expressing the C217stop, R220stop, R238H, R265stop, C280G, and S281stop mutants developed large hemichannel currents.C, Percentage of coupling in oocytes induced between nonsense (top) and missense (bottom) mutants relative to wild-type pairs. Coupling values are from the experiments in Figure 4. Nondetectable levels of coupling (NC) were found in pairs expressing the E208K, Y211stop, R215stop, R215W, and R215Q mutants. The C217stop, R220stop, R265stop, and S281stop mutants induced lower levels of coupling, whereas the R238H and C280G showed similar coupling to wild-type pairs.Asterisks indicate t test values that differ statistically (*p < 0.05; **p < 0.001).
Hemichannel-forming ability of C-terminal CTMX mutants
It is currently accepted that the first step leading to channel formation commences with the synthesis of connexins, their correct insertion into the membrane, and their hexameric assembly into connexons that are targeted and incorporate as hemichannels in the plasma membrane. To evaluate the presence of preformed hemichannels at the cell surface, unpaired Xenopus oocytes expressing wild-type HCx32 and C-terminal mutants (Fig. 1B) were voltage-clamped to a holding potential of −40 mV, and positive pulses were applied up to +120 mV depolarizations. In wild-type cells, macroscopic recordings showed typically slow activating outward currents detectable at depolarizations of +40 mV or greater and larger currents at greater positive voltages. Currents became inward and declined with a slow time course when the potentials returned to −40 mV. In contrast, oocytes injected exclusively with the antisense oligonucleotide directed against the endogenous connexin component (Barrio et al., 1991) did not exhibit this type of current. Thus, the induction of large currents with these unusual characteristics suggested the presence of Cx32 hemichannels preformed in the plasma membrane, which were normally closed at resting potentials and that open with depolarization. Cell-attached single-channel patch-clamp recordings from oocytes expressing human Cx32 allowed us to confirm the presence of opening hemichannels (Fig.2A), which displayed properties consistent with those observed at the macroscopic level. With smaller depolarizations, the first hemichannel openings typically occurred after a long latency after the pulse onset, in agreement with the delayed activation of macroscopic currents. With larger pulses, latencies shorted, and the open time duration and number of hemichannels open increased progressively. The unitary currents became inward when returning to the holding potential of −40 mV, and the hemichannels showed a characteristic flickering activity with frequent and recurrent transitions between the open and fully closed states before actually closing. This activity was consistent with the slow kinetics of the macroscopic tail currents. Patches containing one or two hemichannels were selected to study single-channel properties. The HCx32 hemichannels recorded in physiological solutions showed a main open state conductance of ∼17 pS and fast transitions (<1–2 msec) between the main open and fully closed states. In addition, transitions to at least two subconductance states of 12 and 4 pS were often observed (Fig. 2B). Unitary currents associated with the main open state did not rectify into the voltage range explored, from −40 to +120 mV, and reversed near −10 mV (Fig.3A). These properties of HCx32 hemichannels differ markedly from those previously reported for the Cx46 hemichannels (Trexler et al., 1996), indicating that the unitary characteristics of different hemichannels depend on their connexin composition.
Fig. 2.

Cell-attached single-channel recordings of HCx32 hemichannels. A, Depolarization of +40 mV and greater induced opening events whose properties are consistent with those observed at macroscopic level in Figure 1B. Unitary currents became inward returning to a holding potential of −40 mV and exhibited a characteristic bursting activity before complete closure. B, Selected patch records containing two open hemichannels at +60 mV. Hemichannel activity showed fast transitions between closed (C) and main open (O) states of 17 pS and less frequently to, at least, two subconductance states (S1 andS2) of 12 and 4 pS.
Fig. 3.
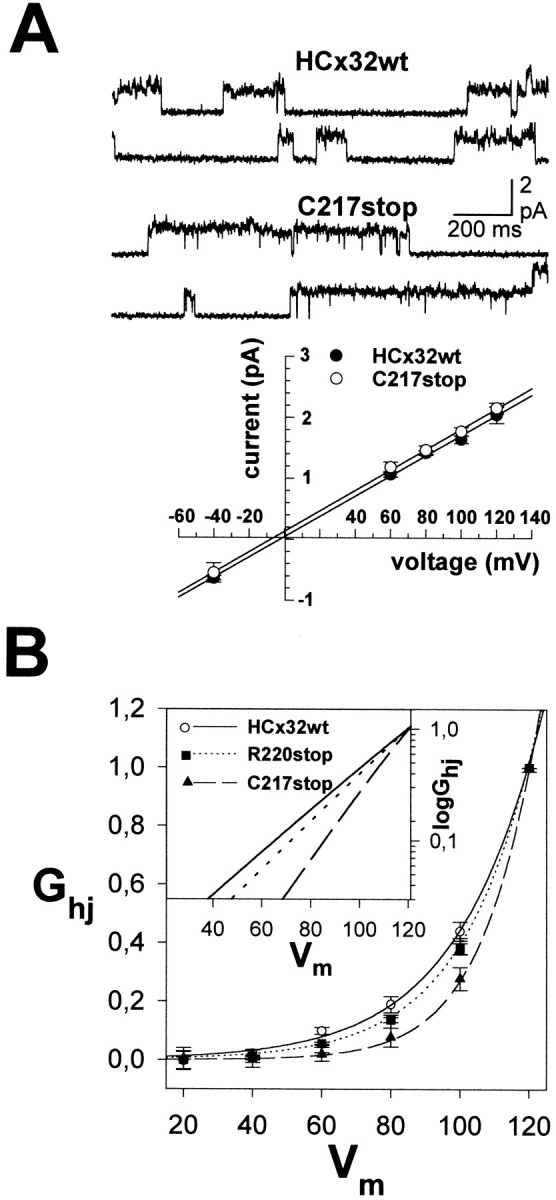
Properties of functional CMTX hemichannels.A, Unitary properties of C217stop and wild-type hemichannels. Selected single-channel recordings in cell-attached configuration of patches containing two open hemichannels at +80 mV show fast current transitions between closed and main open states and less frequently to subconductance states (top). The unitary current–voltage relationships of the main open state for wild-type and C217stop hemichannels were linear from +120 to −40 mV, and the currents reversed near −5 mV (bottom). The unitary conductance values calculated from the slope of the linear fits were of 16.9 ± 0.4 and 17.5 ± 1.0 pS for wild-type and C217stop hemichannels, respectively. Each pointrepresents mean values of 12–16 patches from different oocytes. Error bars indicate SEM. B, Voltage dependence and limiting voltage sensitivity (inset) of hemichannels comprising wild-type HCx32 and the C217stop and R220stop mutants. Hemijunctional conductance (Ghj) measured at the end of pulses was normalized relative to the value for +120 mV pulse. Note the progressive shift of the threshold of activation (inset) and conductance–voltage curves toward higher positive potentials with the shortening of the C-terminal domain. Each point is the mean value of 10 measurements. Error bars indicate SEM.
All HCx32 mutants were efficiently translated in oocytes and gave rise to proteins of the expected sizes, as revealed by Western blot analysis (data not shown). Those oocytes expressing mutants located at more distal regions, C217stop, R220stop, R238H, R265stop, C280G, and S281stop, developed large hemijunctional conductances whose levels were comparable to those found in the wild-type Cx32 oocytes. In contrast, oocytes injected with the E208K and Y211stop mutants did not show detectable levels of opening hemichannels in the voltage range explored (Fig. 1B). We also constructed a nonrelated CMTX mutant with a premature stop codon at position 215, which was incapable of forming functional hemichannels in the plasma membrane. These results indicate that there is a minimum length of the carboxyl tail needed to retain hemichannel-forming ability at the cell surface. In this context, we observed that when Arg-215 was substituted by Trp in the full-length Cx32 (R215W), the ability to form hemichannels was completely impaired. Because the hydrophobic nature could account for R215W effects, we tested the R215Q substitution, which also led to a complete loss of function. Thus, we concluded that the presence of Arg-215 is required for Cx32 to retain the ability to form functional hemichannels, a function that could not be rescued by the other four positively charged amino acids, Arg-219, -220, -223, and -224.
The C-terminal mutations did not change the unitary properties of hemichannels, because even in the case of the shortest mutant with hemichannel function, C217stop, its unitary conductance in the main open state was close to ∼17 pS; moreover, currents reverted near −5 mV and behaved linearly from −40 to +120 mV (Fig. 3A). However, the C217stop, R220stop, and R265stop hemichannels exhibited novel voltage-gating properties at macroscopic levels (Fig.3B). The activation of wild-type hemichannels was detectable at depolarizations of +40 mV or greater, whereas the threshold for the R265stop and R220stop hemichannels increased to +50–60 mV, and that for C217stop hemichannels increased to depolarizations of +80 mV or greater. In parallel, the curves of activation shifted progressively toward higher positive potentials. The voltage-gating properties of R238H and C280G mutants were as those of the wild-type hemichannels.
Gap junction channel-forming ability of C-terminal CTMX mutants
Making hemichannels is only half the process in cell-to-cell channel formation. The final stage of channel formation involves the aligning, recognition and docking of hemichannels in adjacent cells via their extracellular domains (Dahl et al., 1991, 1994; Meyer et al., 1992). The formation of the channel is completed by the final gating of the two closed hemichannels to an open configuration. Channel formation between paired cells was estimated from the development of coupling after cell-to-cell contact by measuring the macroscopic junctional conductance. The mutations located closest to the cytoplasmic–membrane boundary, mutants R215W, R215Q, R215stop, Y211stop, and E208K, produced a complete loss in the ability to form complete channels (Fig.1C). A second group of mutations that included the truncations C217stop, R220stop and R265stop, and S281stop developed very low, moderate, or almost normal levels of junctional conductance, respectively, and a third group formed by the R238H and C280G mutants showed similar levels of coupling to the wild-type HCx32 (Fig.1C). Thus, it appears that only those mutants that induced the expression of functional hemichannels were also able to form complete intercellular channels and that the effects of the mutations depended on the combined action of their location and the type of mutation. The removal of the last three amino acids of the carboxyl tail, S281stop, or the C280G substitution had little or no effect on the channel-forming ability. The additional deletion of the C-terminal tail up to positions 264 and 219 markedly reduced the coupling to nearly half the value of the wild type, whereas the R238H substitution in this region did not alter the channel-forming ability. The C217stop mutant, which has a shorter C-terminal tail, in which only four extra amino acids were deleted with respect to the R220stop mutant, still retained the ability to form channels, but the efficiency to induce coupling was reduced by >95%. These results indicate that although the majority of the C-terminal domain is not a structural requirement to form gap junction channels, this domain plays a critical role determining the efficiency of channel formation.
A discrepancy was observed with the mutants C217stop, R220stop, and R265stop between the large amount of channel precursors and the low levels of coupling induced. To evaluate the channel-forming efficiency of those mutants with respect to the wild-type Cx32, the cRNA concentrations injected were empirically adjusted to those that induced similar levels of macroscopic hemijunctional conductance (Fig.4A, insets). Under these conditions, the temporal pattern of mutated hemichannels at the cell surface of unpaired oocytes expressing wild-type and mutated Cx32 overlapped, indicating that they underwent similar processing as wild-type ones. The time course of hemichannel expression at the cell surface was characterized by a delay of 6–12 hr after cRNA injection until macroscopic currents attributable to hemichannel openings became detectable in the plasma membrane. This delay probably reflects the time required for connexin synthesis, assembly into hemichannels, and transport to plasma membrane. Hemichannels accumulated rapidly, reached peak values between 48 and 72 hr, and then declined slowly and progressively with time. In the same experiments, the set of paired oocytes expressing the S281stop, R265stop, R220stop, and C217stop mutants developed a lower degree of coupling, 89, 44, 37, and 5% of the wild-type pairs, respectively. Thus, the C217stop mutant is an example of a fully functional mutation with respect to the formation of hemichannels but with an almost complete inability to induce functional channels. The lower coupling induced could be accounted for by a reduction in the assembly of precursors into gap junction channels, or alternatively, it may reflect the failure of newly formed channels to achieve the final open conformation. To discern between these two possibilities, we used an experimental paradigm in which the pool size of solitary hemichannels in both cells of each pair was monitored in parallel with channel formation when oocytes were brought into contact manually and compared with the amount of hemichannels expressed in oocytes that remained isolated (Fig. 4). The time course of wild-type channel formation was approximately sigmoidal, with a latency of minutes or a few hours until coupling became detectable (>10 nS). Coupling was preceded by a slow increment of junctional conductance during the first 24 hr after pairing and was followed by a rapidly increasing phase (48–72 hr after pairing) during which the rate of channel formation accelerated progressively. During the last phase of coupling (72–144 hr after pairing), coincident with the decreasing phase of hemichannel expression, the junctional conductance increased slowly (Fig. 4A, top andbottom). Thus, in contrast to the pool of undocked hemichannels whose expression declined with time, the hemichannels assembled into complete gap junction channels seemed to constitute more stable structures, because the pool of newly formed channels tended to increase progressively in nascent gap junctions over the same time scale. These results also reveal that the formation of junctional channels is favored over the persistence of solitary hemichannels, suggesting that once hemichannels are assembled, the undocking process is less probable or does not occur. Our observation that the hemichannel and complete channel undergo different rates of removal at the cell surface of Xenopus oocytes is consistent with the recent observation in which the degree of dye coupling does not decrease significantly when 85% of connexin staining disappeared, with a half life of ∼1 hr, from apposed mammalian cell membranes (Laing et al., 1997).
Fig. 4.

Efficiency of the CMTX hemichannels to assemble into intercellular channels. A, To evaluate the efficiency of assembly, the wild-type and mutated cRNA concentrations were empirically adjusted to those that induced similar levels of hemijunctional conductance (insets). The time course of channel formation of the homotypic pairs expressing the C217stop and R220stop (top) and R265stop (bottom) mutants indicates slower coupling, reaching lower levels than the respective control wild types. The reduction in coupling was small for S281stop (bottom), moderate in the case of R265stop and R220stop, and marked for C217stop. The deficient channel formation induced by C217stop and R265stop alone could be rescued partially in heterotypic combinations by the wild-type Cx32 hemichannels (broken lines). B, Measurement of the hemichannel pools in isolated and paired oocytes injected with the wild-type and C217stop, R220stop, and R265stop mutants. Data are from the experiments in A. In the paired oocytes expressing the mutants there was an excess of solitary hemichannels relative to those found in the wild-type Cx32 pairs, revealing an inefficient assembly of hemichannels into complete channels. Eachpoint in A and Brepresents mean values of 20–25 measurements from four experiments. Error bars indicate SEM. C, Percentage of hemichannels incorporated into complete channels. The amount of incorporation was calculated as the difference of hemijunctional conductances between isolated and paired oocytes, and in case of the mutants their values were normalized relative to the percentage of wild-type incorporation. The reduction was consistent with the lower levels of coupling induced by the C217stop, R220stop, and R265stop mutants. h.a.i., Hours after injection; gj, junctional conductance; ghj, hemijunctional conductance.
According to the assumption that newly formed channels were formed by the progressive docking of preformed hemichannels contributed by each cell of a pair, the size of the hemichannel pools measured in both cells of the pair decreased relative to that found in the unpaired oocytes (Fig. 4B), the reduction being similar in each cell of the pair, and its time course was parallel to the coupling development. This reduction was not complete, because a certain amount of solitary hemichannels remained at the cell surfaces of paired oocytes when longer pairing times were analyzed. The percentage of hemichannels incorporated into complete channels was estimated as the difference between the macroscopic hemijunctional conductance in paired oocytes relative to that found in parent isolated oocytes. It varied between 30 and 60% depending on the batches of oocytes used and, more importantly, on the level of precursor expression, because less incorporation occurred at lower levels of hemichannel expression (data not shown). The percentage of incorporation relative to the total number of hemichannels delivered to the plasma membrane would necessarily be smaller, because approximately half of the hemichannel pool was removed from the membrane before they could be docked.
The pool of channel precursors that remained as undocked hemichannels in paired oocytes was abnormally large for CMTX mutants that induced low levels of coupling (Fig. 4B). The percentage of hemichannels incorporated into complete channels also decreased relative to that of wild-type Cx32 by up to 58 and 41%, respectively, in the case of the R265stop and R220stop but only 3.4% for C217stop (Fig. 4C). This reduced incorporation to channels is consistent with the lower levels of coupling previously measured, indicating that this group of mutants altered the assembly process of two hemichannels into an intercellular channel. Interestingly, the inefficient assembly could be partially rescued by wild-type hemichannels when oocytes that expressed wild-type HCx32 were paired with those expressing the C217stop or R265stop mutants (Fig.4A, broken lines). These heterotypic combinations favored the development of coupling relative to the combinations of mutants alone, although this rescue effect was slight in the case of the hybrid junctions with the C217stop mutant and more pronounced in combination with the R265stop hemichannels. These data indicate that channel assembly is a cooperative process, the efficiency of which results from the combined action of both contributing hemichannels.
DISCUSSION
Assembly of preformed hemichannels into gap junction channels
Our data provide electrophysiological evidence that human Cx32 forms hemichannels at the cell surface and that such hemichannels are the immediate precursors of the gap junction channels. The efficiency of the processes leading to the channel formation is only just beginning to be understood. The efficiency of connexin assembly into hemichannels reported in vivo and in vitro is as low as ∼20% (Musil and Goodenough, 1993; Falk et al., 1997), similar to that for other ionic channels (for review, see Green and Millar, 1995). In the present study, when the efficiency of hemichannel assembly into complete channels was first determined, it appeared to represent a similarly inefficient process, considering jointly the continuous removal of unpaired hemichannels already present at the cell surface and the percentage of hemichannels incorporated into complete channels. Taken together, it may be that only 5% of HCx32 is incorporated into gap junction channels. Additional experimentation will be required to confirm whether the low efficiency of hemichannel assembly is a common feature among different connexins and different cell types.
In this and previous studies of nascent gap junctions using induced cell pairs, the development of coupling follows a characteristic sigmoidal time course (Dahl et al., 1991; Valiunas et al., 1997). Channel formation first proceeds slowly and later increases rapidly until reaching saturating values. This indicates that gap junction formation is an autofacilitated process rather than a simple addition of one channel at a time in an independent manner. Our ability to quantitate the pool of channel precursors allowed us to observe that the initial slow rate of channel formation occurs even when large amounts of preformed hemichannels are already present at the cell surface of both contributing cells. Thus, the formation of the first complete channels of nascent gap junctions may be by itself a limiting factor in coupling development. Accordingly, the CMTX mutants with a limited ability to form complete channels showed a slower rate of channel formation during longer periods (Fig.4A).
Role of the C-terminal domain in the hemichannel and channel formation
In our studies of distinct Cx32 C-terminal mutants, two types of effects were found depending on the location and type of mutation (Table 1). Five mutations (E208K, Y211stop, R215W, R215Q, and R215stop) prevented the formation of functional hemichannels at the cell surface, whereas four mutants (C217stop, R220stop, R265stop, and S281stop) reduced the ability of hemichannels to assemble into complete channels. Conversely, the R238H and C280G mutants were fully capable of forming functional intercellular channels. Taken together these data indicate that a minimum length of the protein, which includes the presence of Glu-208 and Arg-215 residues, is a structural requirement for the formation of functional hemichannels at the cell surface. Accordingly, the cellular localization of the E208K mutant transfected into mammalian cells was entirely cytoplasmatic (Deschênes et al., 1997), and R215W staining at adjacent cell surfaces was greatly reduced (Omari et al., 1996), indicating that trafficking to the plasma membrane is altered. However, the extrapolation of these results to Xenopusoocytes might be limited given the differences in subcellular location of R142W and E186K mutants reported between the two cell types (Bruzzone et al., 1994; Deschênes et al., 1997).
Table 1.
Hemichannel- and channel-forming ability of C-terminal Cx32 mutants associated with CMTX expressed in Xenopus oocytes
| CMTX mutants | Opening hemichannels (threshold of activation) | Gap junction channel formation (%) | Incorporation of hemichannels into gap junction channels (%) |
|---|---|---|---|
| HCx32wt | Yes (≥ + 40 mV) | 100 | 100 |
| E208K | ND | NC | |
| Y211stop | ND | NC | |
| R215W | ND | NC | |
| R215Q1-a | ND | NC | |
| R215stop1-a | ND | NC | |
| C217stop | Yes (≥ + 80 mV) | 5* | 3* |
| R220stop | Yes (≥ + 60 mV) | 37* | 41* |
| R238H | Yes (≥ + 40 mV) | 103 | 98 |
| R265stop | Yes (≥ + 50 mV) | 44* | 58* |
| C280G | Yes (≥ + 40 mV) | 98 | 95 |
| S281stop | Yes (≥ + 40 mV) | 89** | 91** |
ND, Nondetectable levels of hemichannel activation; NC, nondetectable levels of electrical coupling. Percentages of coupling and hemichannel incorporation are relative to the wild-type values.
Non-related CMTX mutations.
*,**t test values that differ statistically: *P < 0.001; **P < 0.05.
Our findings from the truncated C-terminal CMTX mutants are consistent with previous results obtained from the rat Cx43 (Durham et al., 1992) and rat Cx32 (Rabadan-Diehl et al., 1994). In the present study, we could ascribe the complete loss of channel formation to the absence of functional hemichannels at the cell surface, whereas the gradual decrease of coupling associated with the progressive shortening of the carboxyl tail length can be accounted for by a defect in the assembly of mutated hemichannels into complete channels (Table 1). The role that the carboxyl domain plays in facilitating assembly is intriguing. It has been well established that the binding site between hemichannels is complex and involves several segments of the two extracellular domains (Dahl et al., 1994; White et al., 1994; Haubrich et al., 1996). There is evidence that hemichannel binding could involve hydrophobic interactions, hydrogen bonds, and ionic attractions (Goodenough and Gilula, 1974; Manjunath et al., 1984; Ghoshroy et al., 1995), and that multiple weak bonds can contribute to the stability of two hemichannels docking. Based on ultrastructural studies and consistent with the evidence of a disulfide-bonded β-sheet provided by scanning cysteine mutagenesis studies (Foote et al., 1998), the extracellular surface of hemichannels forms six protrusions (Hoh et al., 1991, 1993; Perkins et al., 1997). This has led to the proposal that a “lock and key” mechanism mediates docking (Perkins et al., 1998). Because each extracellular protrusion may also include an extension of the intramembranous α-helical structure (Tibbitts et al., 1990), most likely contributed to by the third and fourth transmembrane segments as the predicted lengths postulated to span the bilayer are clearly excessive, it is possible that the mutations such as C217stop and R220stop located close to the inner membrane–cytoplasmic boundary may induce a conformational change of the binding surface, which may reduce the strength and/or affinity of binding between hemichannels. Such a structural change seems less possible in the case of the C-terminal truncations, because R265stop is located more distally. An alternative explanation is that the C-terminal domain might be involved in a global conformational change of molecule needed to stabilize the assembly between two hemichannels. The assumption that there is an “induced fit” rather than a lock and key mechanism governing hemichannel–hemichannel binding is speculative at this time, but it might explain the striking correlation observed in this study between the gradual reduction of hemichannel incorporation into complete channels (Fig. 4C) and the progressive increments of the voltage needed to induce hemichannel opening (Fig. 3B). We interpret this linkage between both processes as attributable to the existence of a common gate for voltage and chemical binding; even the structure–function determinants of both gatings must not be the same. Based on the novel voltage-gating properties of mutated hemichannels (C217stop, R220stop, and R265stop), it is conceivable that the C-terminal truncations may cause conformational changes in the molecule, which would affect the energy barrier profile of the gate in a manner that increases the activation energy for opening. If this is so, we would also expect a limited ability of hemichannels to open by the chemical mechanism and, consequently, a reduction in the efficiency of hemichannel assembly into gap junction channels.
Cx32 mutations and CMTX disease
As more becomes known about CMTX mutants, it is evident that a greater diversity of pathomechanisms seems to cause the disease. To date, 27 mutants of the 160 mutations identified in CMTX patients have been analyzed using exogenous expression systems, including the 9 mutants described here (Bruzzone et al., 1994; Rabadan-Diehl et al., 1994; Omari et al., 1996; Deschênes et al., 1997; Oh et al., 1997; Ressot et al., 1998). Twelve mutants have been reported as null mutations, whereas 15 mutants still produce functional intercellular channels, although their efficiency in most cases has not been addressed. In our series, at least four of six functional mutants have a limited ability to form complete channels (Table 1). Mutations may also impair channel function, such as has been reported in the case of two mutants that reduce the junctional permeability or open probability (Oh et al., 1997), and for the group of mutants that exhibit altered voltage- and pH-gating properties (Ressot et al., 1998). In this context, we found that five of six functional junctions (C217stop, R220stop, R238H, R265stop, and S281stop) showed novel voltage-gating properties. The effects were not pronounced, but, interestingly, the changes were mutation-specific.
The severity of the CMTX clinical phenotype seems to correlate with the location and type of mutation in the Cx32 gene (Ionasescu et al., 1996;Deschênes et al., 1997), although strict correlations between genotype and clinical phenotype are still to be established. In this context, our results suggest that different mutations could lead to severe phenotypes by altering different mechanisms. The severe phenotype of E208K (Deschênes et al., 1997) and Y211stop (Hahn et al., 1990) mutants may be caused by the complete loss of channel function. The possibility of dominant negative interactions with other myelin proteins has also been proposed based on the defective trafficking of E208K to the cell surface (Deschênes et al., 1997). In the case of the C217stop, R220stop, and R265stop mutants, which form hemichannels whose assembly is inefficient, they may cause a severe clinical phenotype (Ionasescu et al., 1996) by limiting channel formation or owing to an excess of solitary hemichannels, which could adversely affect cell viability if they were to open. The presence of open hemichannels may have a deleterious effect, as has been demonstrated in oocytes overexpressing Cx46, in which they induced swelling and cell lysis because they are normally open at resting membrane potential (Paul et al., 1991). In the case of HCx32 hemichannels, opening on depolarization is unlikely given the high positive voltage required, but the possibility that opening may be induced by other physiological agents is presently under investigation.
Footnotes
This research was supported by Fondo de Investigaciones Sanitarias (FIS) Grant 95/0643, Fundación “la Caixa” Grant 97/123-00, and Ministerio de Educación y Cultura Grant PM97-0021 (to L.C.B.). L.C.B. is member of the European Charcot–Marie–Tooth Consortium (BIOMED2 Grant CT961614). J.M.G.-H. is a postdoctoral fellow of the Comunidad Autónoma de Madrid. C.C. is the recipient of an FIS fellowship. We are grateful to N. M. Kumar for providing the HCx32 cDNA. We acknowledge the cooperation of the Hungarian family in this study and Dr. V. Karcagi for providing data on the patients. We also thank R. Barquero for invaluable technical assistance with the oocyte experiments.
Correspondence should be addressed to Dr. Luis C. Barrio, Departamento de Investigación, Unidad de Neurología Experimental–Consejo Superior de Investigaciones Científicas, Hospital “Ramón y Cajal,” Carretera de Colmenar km 9.1, 28034 Madrid, Spain.
REFERENCES
- 1.Balice-Gordon RJ, Bone LJ, Scherer SS. Functional gap junctions in the Schwann cell myelin sheath. J Cell Biol. 1998;142:1095–1104. doi: 10.1083/jcb.142.4.1095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Barrio LC, Suchyna T, Bargiello TA, Xu LX, Roginski RS, Bennett MVL, Nicholson BJ. Gap junctions formed by connexins 26 and 32 alone and in combination are differently affected by applied voltage. Proc Natl Acad Sci USA. 1991;88:8410–8414. doi: 10.1073/pnas.88.19.8410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Barrio LC, Capel J, Jarillo JA, Castro C, Revilla A. Species-specific voltage-gating properties of connexin-45 junctions expressed in Xenopus oocytes. Biophys J. 1997;73:757–769. doi: 10.1016/S0006-3495(97)78108-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bergoffen J, Scherer SS, Wang S, Oronzi M, Scott L, Bone J, Paul DL, Chen K, Lensch MW, Chance PF, Fischbeck KH. Connexin mutations in X-linked Charcot-Marie-Tooth disease. Science. 1993;262:2039–2042. doi: 10.1126/science.8266101. [DOI] [PubMed] [Google Scholar]
- 5.Bone LJ, Dahl N, Lensch MW, Chance PF, Kelly T, Le Guern E, Magi S, Parry G, Shapiro H, Wang S, Fischbeck KH. New connexin32 mutations associated with X-linked dominant Charcot-Marie-Tooth disease. Neurology. 1995;45:1863–1866. doi: 10.1212/wnl.45.10.1863. [DOI] [PubMed] [Google Scholar]
- 6.Bone LJ, Deschênes SM, Balice-Gordon RJ, Fischbeck KH, Scherer SS. Connexins32 and X-linked Charcot-Marie-Tooth disease. Neurobiol Dis. 1997;4:221–230. doi: 10.1006/nbdi.1997.0152. [DOI] [PubMed] [Google Scholar]
- 7.Bort S, Nelis E, Timmerman V, Sevilla T, Cruz-Martinez A, Martinez F, Millán JM, Arpa J, Vílchez JJ, Prieto J, Van Broeckhoven C, Palau F. Mutational analysis of the MPZ, PMP22 and Cx32 genes in patients of Spanish ancestry with Charcot-Marie-Tooth disease and hereditary neuropathy with liability to pressure palsies. Hum Genet. 1997;99:746–754. doi: 10.1007/s004390050442. [DOI] [PubMed] [Google Scholar]
- 8.Bruzzone R, White TW, Scherer SS, Fischbeck KH, Paul DL. Null mutations of connexin32 in patients with X-linked Charcot-Marie-Tooth disease. Neuron. 1994;13:1253–1260. doi: 10.1016/0896-6273(94)90063-9. [DOI] [PubMed] [Google Scholar]
- 9.Bruzzone R, White TW, Paul DL. Connections with connexins: The molecular basis of direct intercellular signaling. Eur J Biochem. 1996;238:1–27. doi: 10.1111/j.1432-1033.1996.0001q.x. [DOI] [PubMed] [Google Scholar]
- 10.Chow I, Young SH. Opening of single junction channels during the formation of electrical coupling between embryonic muscle cells. Dev Biol. 1987;122:332–337. doi: 10.1016/0012-1606(87)90298-3. [DOI] [PubMed] [Google Scholar]
- 11.Dahl G, Levine E, Rabadan-Diehl C, Werner R. Cell/cell channel formation involved disulfide exchange. Eur J Biochem. 1991;197:141–144. doi: 10.1111/j.1432-1033.1991.tb15892.x. [DOI] [PubMed] [Google Scholar]
- 12.Dahl G, Nonner W, Werner R. Attempts to define functional domains of gap junction proteins with synthetic peptides. Biophys J. 1994;67:1816–1822. doi: 10.1016/S0006-3495(94)80663-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Deschênes SM, Walcott JL, Wexler TL, Scherer SS, Fischbeck KH. Altered trafficking of mutant connexin32. J Neurosci. 1997;17:9077–9084. doi: 10.1523/JNEUROSCI.17-23-09077.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.DeVries SH, Schwartz EA. Hemi-gap-junction channels in solitary horizontal cell of catfish retina. J Physiol (Lond) 1992;445:201–230. doi: 10.1113/jphysiol.1992.sp018920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Durham B, Liu S, Taffet S, Trabda-Janik E, Delmar M, Tetryshyn R, Zheng S, Perzova R, Vallano ML. Immunolocalization and expression of functional and nonfunctional cell-to-cell channels from wild-type and mutant rat heart connexin43 cDNA. Circ Res. 1992;70:1233–1243. doi: 10.1161/01.res.70.6.1233. [DOI] [PubMed] [Google Scholar]
- 16.Ebihara L. Xenopus connexin38 forms hemi-gap-junctional channels in nonjunctional plasma membrane of Xenopus oocytes. Biophys J. 1996;96:742–748. doi: 10.1016/S0006-3495(96)79273-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ebihara L, Berthoud VM, Beyer EC. Distinct behavior of connexin56 and connexin46 gap junctional channels can be predicted from the behavior of their hemi-gap-junctional channels. Biophys J. 1995;68:1796–1803. doi: 10.1016/S0006-3495(95)80356-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Fairweather N, Bell C, Cochrane S, Chelly J, Wang S, Mostacciuolo ML, Monaco AP, Haites NE. Mutations in the connexin 32 gene in X-linked dominant Charcot-Marie-Tooth disease (CMTX1). Hum Mol Genet. 1994;3:29–34. doi: 10.1093/hmg/3.1.29. [DOI] [PubMed] [Google Scholar]
- 19.Falk MM, Buehler LK, Kumar NM, Gilula NB. Cell-free synthesis and assembly of connexins into functional gap junction membrane channels. EMBO J. 1997;16:2703–2716. doi: 10.1093/emboj/16.10.2703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Foote CI, Xhu X, Zhou L, Nicholson BJ. The pattern of disulfide linkages in the extracellular loop regions of Cx32 suggest a model for the docking interfaces of gap junctions. J Cell Biol. 1998;140:1187–1197. doi: 10.1083/jcb.140.5.1187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ghoshroy S, Goodenough DA, Sosinsky GE. Preparation, characterization, and structure of half gap junctional layers split with urea and EGTA. J Membr Biol. 1995;146:15–28. doi: 10.1007/BF00232677. [DOI] [PubMed] [Google Scholar]
- 22.Gilula NB, Reves OR, Steinbach A. Metabolic coupling, ionic coupling and cell contacts. Nature. 1982;235:262–265. doi: 10.1038/235262a0. [DOI] [PubMed] [Google Scholar]
- 23.Green WN, Millar NS. Ion-channel assembly. Trends Neurosci. 1995;18:280–287. [PubMed] [Google Scholar]
- 24.Goodenough DA, Gilula NB. The splitting of hepatocyte gap junctions and zonulae occludentes with hypertonic disaccharides. J Cell Biol. 1974;61:575–590. doi: 10.1083/jcb.61.3.575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Goodenough DA, Paul DL, Jesaitis L. Topological distribution of two connexin32 antigenic sites in intact and split rodent hepatocyte gap junctions. J Cell Biol. 1988;107:1817–1824. doi: 10.1083/jcb.107.5.1817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gupta VK, Berthoud VM, Atal N, Jarillo JA, Barrio LC, Beyer EC. Bovine connexin44, a lens gap junction protein: molecular cloning, immunological characterization and functional expression. Invest Ophthalmol Vis Sci. 1994;35:3747–3758. [PubMed] [Google Scholar]
- 27.Hahn AF, Brown WF, Koopman WJ, Feasby TE. X-linked dominant hereditary motor and sensory neuropathy. Brain. 1990;113:1511–1525. doi: 10.1093/brain/113.5.1511. [DOI] [PubMed] [Google Scholar]
- 28.Haubrich S, Schwarz HJ, Bukauskas F, Lichtenberg-Frate H, Traub O, Weingart R, Willecke K. Incompatibility of connexin 40 and 43 hemichannels in gap junctions between mammalian cells is determined by intracellular domains. Mol Biol Cell. 1996;7:1995–2006. doi: 10.1091/mbc.7.12.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hoh JH, Lal R, John SA, Revel J-P, Arnsdorf MF. Atomic force microscopy and dissection of gap junctions. Science. 1991;235:1405–1408. doi: 10.1126/science.1910206. [DOI] [PubMed] [Google Scholar]
- 30.Hoh JH, Sosinsky GE, Revel J-P, Hansma PK. Structure of the extracellular surface of the gap junction by atomic force microscopy. Biophys J. 1993;65:149–163. doi: 10.1016/S0006-3495(93)81074-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ionasescu VV, Searby C, Ionasescu R. Point mutations of the connexin32 (GJβ1) gene in X-linked dominant Charcot-Marie-Tooth neuropathy. Hum Mol Genet. 1994;3:355–358. doi: 10.1093/hmg/3.2.355. [DOI] [PubMed] [Google Scholar]
- 32.Ionasescu VV, Ionasescu R, Searby C. Correlation between connexin 32 gene mutations and clinical phenotype in X-linked dominant Charcot-Marie-Tooth neuropathy. Am J Med Genet. 1996;63:486–491. doi: 10.1002/(SICI)1096-8628(19960614)63:3<486::AID-AJMG14>3.0.CO;2-I. [DOI] [PubMed] [Google Scholar]
- 33.Kumar NM, Gilula NB. Cloning and characterization of human and rat liver cDNAs coding for a gap junction protein. J Cell Biol. 1986;103:767–776. doi: 10.1083/jcb.103.3.767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kumar NM, Friend DS, Gilula NB. Synthesis and assembly of human β1 gap junctions in BHK cell by DNA transfection with the human β1 DNA. J Cell Sci. 1995;108:3725–3734. doi: 10.1242/jcs.108.12.3725. [DOI] [PubMed] [Google Scholar]
- 35.Laing JG, Tadros PN, Westphale EM, Beyer EC. Degradation of connexin43 gap junctions involves both the proteasome and lysosome. Exp Cell Res. 1997;236:482–492. doi: 10.1006/excr.1997.3747. [DOI] [PubMed] [Google Scholar]
- 36.Li H, Liu T-F, Lazrak A, Peracchia C, Goldberg GS, Lampe GP, Johnson RG. Properties and regulation of gap junctional hemichannels in plasma membranes of cultured cells. J Cell Biol. 1996;134:1019–1030. doi: 10.1083/jcb.134.4.1019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Loewenstein WR, Kanno Y, Scolar SJ. Quantum jumps of conductance during formation of membrane channels at cell-to-cell junction. Nature. 1981;274:133–136. doi: 10.1038/274133a0. [DOI] [PubMed] [Google Scholar]
- 38.Manjunath CK, Goings GE, Page E. Detergent sensitivity and splitting of isolated liver gap junctions. J Membr Biol. 1984;78:147–155. doi: 10.1007/BF01869201. [DOI] [PubMed] [Google Scholar]
- 39.Meyer RA, Laird DW, Revel J-P, Johnson R. Inhibition of gap junction and adherens junction assembly by connexin and N-CAM antibodies. J Cell Biol. 1992;119:179–189. doi: 10.1083/jcb.119.1.179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Musil LS, Goodenough DA. Multisubunits assembly of a integral plasma membrane channel protein, gap junction connexin43, occurs after exit from the ER. Cell. 1993;74:1065–1077. doi: 10.1016/0092-8674(93)90728-9. [DOI] [PubMed] [Google Scholar]
- 41.Nelis E, Simokovic S, Timmerman V, Löfgren A, Backhovens H, De Jonghe P, Martin J, Van Broeckhoven C. Mutation analysis of the connexin 32 (Cx32) gene in Charcot-Marie-Tooth neuropathy type 1: identification of five new mutations. Hum Mutat. 1997;9:47–52. doi: 10.1002/(SICI)1098-1004(1997)9:1<47::AID-HUMU8>3.0.CO;2-M. [DOI] [PubMed] [Google Scholar]
- 42.Oh S, Ri Y, Bennett MVL, Trexler EB, Verselis VK, Bargiello TA. Changes in permeability caused by connexin 32 mutations underlie X-linked Charcot-Marie-Tooth disease. Neuron. 1997;19:927–938. doi: 10.1016/s0896-6273(00)80973-3. [DOI] [PubMed] [Google Scholar]
- 43.Omari Y, Mesnil M, Yamasaki H. Connexin 32 mutations from X-linked Charcot-Marie-Tooth disease patients: functional defects and dominant negative effects. Mol Biol Cell. 1996;7:907–916. doi: 10.1091/mbc.7.6.907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Paul DL, Ebihara L, Takemoto LJ, Swenson KI, Goodenough DA. Connexin46, a novel lens gap junction protein, induces voltage-gated currents in nonjunctional plasma membrane of Xenopus oocytes. J Cell Biol. 1991;115:1077–1089. doi: 10.1083/jcb.115.4.1077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Perkins G, Goodenough DA, Sosinsky GE. Three-dimensional structure of the gap junction connexon. Biophys J. 1997;72:533–544. doi: 10.1016/s0006-3495(97)78693-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Perkins G, Goodenough DA, Sosinsky GE. Structural considerations for connexon docking. In: Werner R, editor. Gap junction. IOS; Amsterdam: 1998. pp. 13–17. [Google Scholar]
- 47.Rabadan-Diehl C, Dahl G, Werner R. A connexin-32 mutation associated with Charcot-Marie-Tooth disease does not affect channel formation in oocytes. FEBS Lett. 1994;351:90–94. doi: 10.1016/0014-5793(94)00819-1. [DOI] [PubMed] [Google Scholar]
- 48.Ressot C, Latour P, Blanquet-Grossard F, Sturtz F, Duthel S, Battin J, Corbillon E, Ollagnon E, Serville F, Vanderberghe A, Dautigny A, Pham-Dinh D. X-linked dominant Charcot-Marie-Tooth neuropathy (CMTX): new mutations in the connexin32 gene. Hum Genet. 1996;98:172–175. doi: 10.1007/s004390050183. [DOI] [PubMed] [Google Scholar]
- 49.Ressot C, Gomés D, Dautigny A, Pham-Dinh D, Bruzzone R. Connexin32 mutations associated with X-linked Charcot-Marie-Tooth disease show two distinct behaviors: loss of function and altered gating properties. J Neurosci. 1998;18:4063–4075. doi: 10.1523/JNEUROSCI.18-11-04063.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Revel J-P, Karnovsky MJ. Hexagonal array of subunits in intercellular junctions of the mouse heart and liver. J Cell Biol. 1967;33:C7–C12. doi: 10.1083/jcb.33.3.c7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Silander K, Meretoja P, Pihko H, Jovonen V, Issakainen J, Aula P, Savontaus M-L. Screening for Cx32 mutations in Charcot-Marie-Tooth disease families with possible X-linked inheritance. Hum Genet. 1997;100:391–397. doi: 10.1007/s004390050522. [DOI] [PubMed] [Google Scholar]
- 52.Tan CC, Ainsworth PJ, Hahn AF, MacLeod P. Novel mutations in the connexin 32 gene associated with X-linked Charcot-Marie-Tooth disease. Hum Mutat. 1996;7:167–171. doi: 10.1002/(SICI)1098-1004(1996)7:2<167::AID-HUMU14>3.0.CO;2-0. [DOI] [PubMed] [Google Scholar]
- 53.Tibbitts TT, Casper LD, Phillips WC, Goodenough DA. Diffraction diagnosis of protein folding in gap junction connexons. Biophys J. 1990;57:1025–1036. doi: 10.1016/S0006-3495(90)82621-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Trexler EB, Bennett MVL, Bargiello TA, Verselis VK. Voltage gating and permeation in a gap junction hemichannel. Proc Natl Acad Sci USA. 1996;93:5836–5841. doi: 10.1073/pnas.93.12.5836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Valiunas V, Bukauskas F, Weingart R. Conductances and selective permeability of connexon-43 gap junction channels examined in neonatal rat heart cells. Circ Res. 1997;80:708–719. doi: 10.1161/01.res.80.5.708. [DOI] [PubMed] [Google Scholar]
- 56.White TW, Bruzzone R, Wolfram S, Paul DL, Goodenough DA. Selective interactions among the multiple connexin proteins expressed in the vertebrate lens: the second extracellular domain is a determinant of compatibility between connexins. J Cell Biol. 1994;125:879–892. doi: 10.1083/jcb.125.4.879. [DOI] [PMC free article] [PubMed] [Google Scholar]