

Abstract
Nuclear factor κB (NF-κB) appears to participate in the excitotoxin-induced apoptosis of striatal medium spiny neurons. To elucidate molecular mechanisms by which this transcription factor contributes to NMDA receptor-triggered apoptotic cascades in vivo, rats were given the NMDA receptor agonist quinolinic acid (QA) by intrastriatal infusion, and the role of NF-κB in the induction of apoptosis-related genes and gene products was evaluated. QA administration induced time-dependent NF-κB nuclear translocation. The nuclear NF-κB protein after QA treatment was comprised mainly of p65 and c-Rel subunits as detected by gel supershift assay. Levels of c-Myc and p53 mRNA and protein were markedly increased at the time of QA-induced NF-κB nuclear translocation. Immunohistochemical analysis showed that c-Myc and p53 induction occurred in the excitotoxin-sensitive medium-sized striatal neurons. NF-κB nuclear translocation was blocked in a dose-dependent manner by the cell-permeable recombinant peptide NF-κB SN50, but not by the NF-κB SN50 control peptide. NF-κB SN50 significantly inhibited the QA-induced elevation in levels of c-Myc and p53 mRNA and protein. Pretreatment or posttreatment with NF-κB SN50, but not the control peptide, also substantially reduced the intensity of QA-induced internucleosomal DNA fragmentation. The results suggest that NF-κB may promote an apoptotic response in striatal medium-sized neurons to excitotoxic insult through upregulation of c-Myc and p53. This study also provides evidence indicating an unique signaling pathway from the cytoplasm to the nucleus, which regulates p53 and c-Myc levels in these neurons during apoptosis.
Keywords: transcription factor, NF-κB, quinolinic acid, tumor suppressor gene, apoptosis, Huntington’s disease
Excitotoxicity has long been implicated in the pathogenesis of degenerative disorders, such as Huntington’s disease (HD), Parkinson’s disease (PD), and Alzheimer’s disease (AD) (Choi, 1992; Coyle and Puttfarcken, 1993). The selective neuronal degeneration occurring in many of these conditions appears to involve an apoptotic process (Portera-Cailliau et al., 1995; Cotman and Su, 1996; Anglade et al., 1997; Mochizuki et al., 1997). Recent preclinical observations suggest that glutamate agonist-induced neuronal destruction, at least in part, also occurs by an apoptotic mechanism (Ankarcrona et al., 1993; Bonfoco et al., 1995; Qin et al., 1996; Simonian et al., 1996).
The time course of transcription factor expression in tissues undergoing apoptosis, as well as the effect of transcription and translation inhibitors on neuronal injury, suggest that some transcription factors may participate in the regulation of the destructive process (Pittman et al., 1994; Hughes et al., 1997). One such transcription factor is nuclear factor κB (NF-κB). NF-κB is expressed in neurons and glia throughout the mammalian CNS (Kaltschmidt et al., 1993, 1994) and can be activated by glutamate receptor agonists (Guerrini et al., 1995; Kaltschmidt et al., 1995). Increased NF-κB levels have recently been observed in areas of neuronal degeneration in animal models of ischemia, as well as in patients with AD and PD (Terai et al., 1996; Clemens et al., 1997;Hunot et al., 1997; Kaltschmidt et al., 1997). Although these NF-κB changes could reflect epiphenomena or the activation of defense mechanisms (Barger et al., 1995; Mattson et al., 1997), a contribution to pathogenesis would seem more likely because the changes appear limited to brain regions undergoing neurodegeneration.
NF-κB participates in the control of a broad range of physiological and pathological processes (Baeuerle and Henkel, 1994; O’Neil and Kaltschmidt, 1997; Siebenlist, 1997). Its role in the regulation of apoptosis, however, has been rather controversial (Baeuerle and Baltimore, 1996; Baichwal and Baeuerle, 1997; Lipton, 1997). Evidence suggesting that NF-κB protects dividing cells against apoptosis derives mainly from studies of tumor necrosis factor-induced cell death (Antwerp et al., 1996; Beg and Baltimore, 1996) but has been observed in certain other situations, including an NF-κB p65 subunit knock-out animal model (Beg et al., 1995; Wu et al., 1996;Ozaki et al., 1997; Taglialatela et al., 1997). In contrast, other studies have suggested that NF-κB promotes apoptotic cell death in a variety of cell injury models (Lin et al., 1995a; Grilli et al., 1996;Grimm et al., 1996; Marinovich et al., 1996; Clemens et al., 1997, 1998). Previously, we have reported that quinolinic acid (QA)-induced striatal cell death in rats displays many hallmarks of apoptosis (Qin et al., 1996) and that the cell-permeable peptide NF-κB SN50, which blocks NF-κB nuclear translocation, diminishes QA-induced apoptosis (Qin et al., 1998). These observations suggest that NF-κB positively regulates excitotoxin-induced apoptosis in postmitotic striatal neurons in vivo.
Many factors could contribute to the antipodal consequences of NF-κB activation for cell survival. Because NF-κB interacts with a broad array of genes, it is hardly surprising that different genetic programs may be stimulated in different cell types, exposed to different apoptotic triggers, under various experimental or naturally occurring conditions. It is important, therefore, to study the regulation of those genes involved in excitotoxin-induced apoptosis by NF-κB in the neurons of interest. To elucidate biochemical events possibly linking NF-κB activation to the excitotoxin-induced apoptosis of striatal medium spiny neurons, we have studied the role of NF-κB activation on QA-induced c-Myc and p53 regulation and on the induction of neuronal apoptosis in the excitotoxic HD model.
MATERIALS AND METHODS
Animals and drug administration. Male Sprague Dawley rats weighing 300–350 gm were purchased from Taconic Farms (Germantown, NY). All procedures were performed in accordance withNational Institutes of Health Guidelines for the Care and Use of Laboratory Animals.
Intrastriatal stereotaxic drug administration was performed as described previously (Qin et al., 1996). To study the time course of QA-induced NF-κB activation, rats were infused intrastriatally with QA (60 nmol) or saline (1 μl) and killed 3, 6, 12, or 24 hr later. Striatal tissues were dissected for electrophoresis mobility shift assay (EMSA). To study the effect of QA on c-Myc and p53, rats were treated and killed as described above, and striatal tissues were used for Western blot analysis of c-Myc and p53 protein levels or for Northern blot analysis of c-Myc and p53 mRNA levels. Some animals were perfused transcardially with a solution containing 4% paraformaldehyde, 0.2% picric acid, and 0.05% glutaraldehyde, pH 7.4, and their brains were assessed immunohistochemically. To study the effect of NF-κB inhibitory peptide on the QA-induced nuclear translocation of NF-κB, rats were infused intrastriatally with NF-κB SN50 (Lin et al., 1995b) or NF-κB SN50 control peptide (5–20 μg; Biomol, Plymouth Meeting, PA) 15 min before QA and killed 12 hr later. Striatal tissues were used for nuclear protein extraction and EMSA. To study the effect of NF-κB SN50 on QA-induced inhibitor κB (IκB) degradation, rats received this recombinant peptide (20 μg) 15 min before QA and were killed 12 hr later. Striatal proteins were extracted and used for Western blot analysis. To study the effect of NF-κB SN50 on the QA-induced increase in c-Myc and p53, rats received 20 μg of this recombinant peptide 15 min before QA and were killed 24 hr later. Striatal tissues were used for Northern and Western blot analysis. To study the effect of NF-κB SN50 on QA-induced internucleosomal DNA fragmentation, three experiments were performed. In the first, rats were pretreated with NF-κB SN50 or NF-κB SN50 control peptide (5–20 μg) 15 min before QA and were killed 24 hr later. In the second, rats were pretreated with NF-κB SN50 (20 μg) 15 min before QA and were killed 12, 24, or 48 hr later. In the third experiment, one group of rats received NF-κB SN50 (20 μg) 15 min before QA, whereas other groups were first infused intrastriatally with QA (60 nmol) and 2, 4, and 6 hr later were given NF-κB SN50 (20 μg) intrastriatally. All animals were killed 24 hr after QA administration, and their striatal tissues were used for DNA extraction.
Western blot analysis. Western blot analysis was performed as described previously (Wosikowski et al., 1995) with modifications. Striatal tissues were homogenized and then sonicated in a lysing buffer containing 10 mm Tris-HCl, pH 7.4, 150 mm NaCl, 1% Triton X-100, 1% sodium deoxycholate, 0.1% SDS, 5 mm EDTA, 1 mm PMSF, 0.28 U/ml aprotinin, 50 μg/ml leupeptin, 1 mm benzamidine, and 7 μg/ml pepstatin A. Protein concentrations were determined using a BCA protein assay kit (Pierce, Rockford, IL). Samples were mixed with loading buffer and boiled for 5 min. An aliquot of 30 μg of protein from each sample was separated on 12% SDS-PAGE gel using constant current. Proteins were subsequently transferred to Immobilon-P membranes (Millipore, Bedford, MA) with a semidry blotting system. After blocking for 1 hr in PBS with 0.1% Tween 20 (PBST) and 5% nonfat dry milk, membranes were incubated for 3 hr with primary antibodies in PBST containing 3% nonfat dry milk. Membranes were then washed and incubated with a horseradish peroxidase-conjugated secondary antibody in PBST containing 3% nonfat dry milk for 1 hr. Immunoreactivity was detected by enhanced chemiluminescence autoradiography (ECL kit; Amersham, Arlington Heights, IL) in accordance with the manufacturer’s instructions. The following primary antibodies were used: antibody against IκB-α was rabbit polyclonal antibody IκB-α (FL) (Santa Cruz Biotechnology, Santa Cruz, CA); antibodies against c-Myc and p53 were mouse monoclonal antibodies c-Myc (C-33) (Santa Cruz), p53 (Ab-1) and p53 (Ab-3) (Calbiochem, Cambridge, MA), and p53 (Pab 240) (Santa Cruz).
Northern blot analysis. Total mRNA was extracted from injected striatum and isolated by cesium chloride gradient centrifugation. After electrophoresis on 1% agarose gel containing formaldehyde, mRNA was transferred to Duralose membranes (Stratagene, La Jolla, CA). cDNA probes for p53 and c-Myc mRNA were labeled with [32P]dCTP by the random priming method. Hybridization was performed at 42°C for 16 hr. Membranes were then washed twice at room temperature with 2× SSC containing 0.1% SDS and twice at 50°C in 0.1× SSC containing 0.1% SDS. The results were quantitatively analyzed using a Betascope Model 603 Blot Analyzer (Betagen, Waltham, MA).
EMSA. Striatal nuclear proteins were prepared as described previously (Qin et al., 1998). Protein concentrations were determined with a BCA kit (Pierce). Double strand DNA oligonucleotides containing consensus sequences for NF-κB and activator protein 1 (AP-1) (Promega, Madison, WI) were labeled with [32P]ATP by T4 polynucleotide kinase (Promega). Nuclear proteins (8–12 μg) were incubated with radiolabeled DNA probes (40,000 cpm) for 15 min at room temperature in the binding buffer (Promega). For supershift assay, nuclear proteins were incubated with antibodies (4 μg) to p65, p50, p52, Rel B or c-Rel [NF-κB P65 (A), NF-κB p50 (C-19), NF-κB p52 (K-27), Rel B (19), c-Rel (N) (all from Santa Cruz)] for 1 hr at room temperature before adding labeled DNA probes. The reaction mixture was then electrophoresed on 4.5% nondenaturing polyacrylamide gel with 0.5× Tris borate-EDTA buffer. Autoradiograms were developed by exposing the vacuum-dried gels to x-ray film at −80°C with intensifying screens for 24–48 hr. Results were quantitatively evaluated with an image analyzer (NIH Image 1.60).
Immunohistochemistry. Perfused brains were post-fixed overnight in the same perfusate as described above and then immersed in 30% sucrose. Coronal sections of 25 μm thickness were cut with a cryostat and washed in 0.1 m Tris-buffered saline (TBS) three times 10 min each and incubated in TBS with 0.25% Triton X-100 for 30 min. Free-floating sections were then blocked with 1% normal goat serum for 1 hr at room temperature and incubated with primary antibodies in the above solution at 4°C for 48 hr. Sections were subsequently washed and incubated with secondary antibodies using a Vectastain Elite kit (Vector Laboratories, Burlingame, CA) according to the manufacturer’s protocol. Finally, sections were counterstained with thionin. Two antibodies, mouse monoclonal antibody (C-33; Santa Cruz) and rabbit polyclonal antibody (C-19; Santa Cruz), were used to detect c-Myc immunoreactivity; mouse monoclonal antibody (Ab-3; Calbiochem) and mouse monoclonal antibody (Pab 246, Santa Cruz) were used to detect p53 immunoreactivity. Antibody specificity was evaluated by preabsorption with specific peptides or omission of primary antibodies.
Genomic DNA preparation and DNA electrophoresis. Striatal genomic DNA was prepared as described previously (Qin et al., 1996). Briefly, striatal tissues were homogenized in a buffer containing 100 mm NaCl, 25 mm EDTA, 10 mmTris-HCl, pH 8.0, 0.5% SDS, and 0.5 μg/ml RNase. Homogenates were incubated at 55°C and then extracted with phenol/chloroform/isoamyl alcohol (25:24:1). DNA pellets were washed once with precooled 80% alcohol, vacuum-dried, and resuspended in Tris-EDTA buffer. DNA fragments were separated on 2% agarose gel (3:1; NuSieve, Rockland, ME) and detected with an UV transilluminator after staining with ethidium bromide.
Statistical analysis. For comparison of multiple means, ANOVA followed by Dunnett t test was performed using the original data. For comparison of two means, the Student’s ttest was used. After statistical comparison, some data were converted to percent of control for presentation in bar figures.
RESULTS
QA effects on NF-κB activation
QA induced a marked time-dependent increase in NF-κB binding activity in striatal nuclear extracts, with the peak occurring 12 hr after QA administration (Fig.1A). Nuclear NF-κB binding activity was not significantly affected in vehicle-treated control animals (Fig. 1B). The specificity of NF-κB binding was confirmed with unlabeled probes and mutant NF-κB probes. NF-κB binding activity was almost completely eliminated by adding a 60-fold excess of unlabeled NF-κB probes but not by adding unlabeled AP-1 probes (Fig. 2A). A single base mutation in NF-κB DNA binding motif eliminated upper band binding but only slightly reduced lower band activity. In other studies, only upper bands, which were markedly altered by QA treatment, were quantitatively analyzed. Supershift assays showed that preincubation with anti-p65 or anti-c-Rel antibodies reduced NF-κB binding activity in nuclear extracts. The addition of anti-p65 antibody to the assay also produced a supershifted band, but the addition of anti-c-Rel antibody failed to produce an appreciable supershifted band. This may indicate loss of DNA binding ability of antibody–c-Rel complex. The results suggest that NF-κB subunits translocated to the nucleus after QA treatment were mainly comprised of p65 and c-Rel (Fig.2B). In vehicle-treated animals, no appreciable supershifted signals were detectable (data not shown).
Fig. 1.
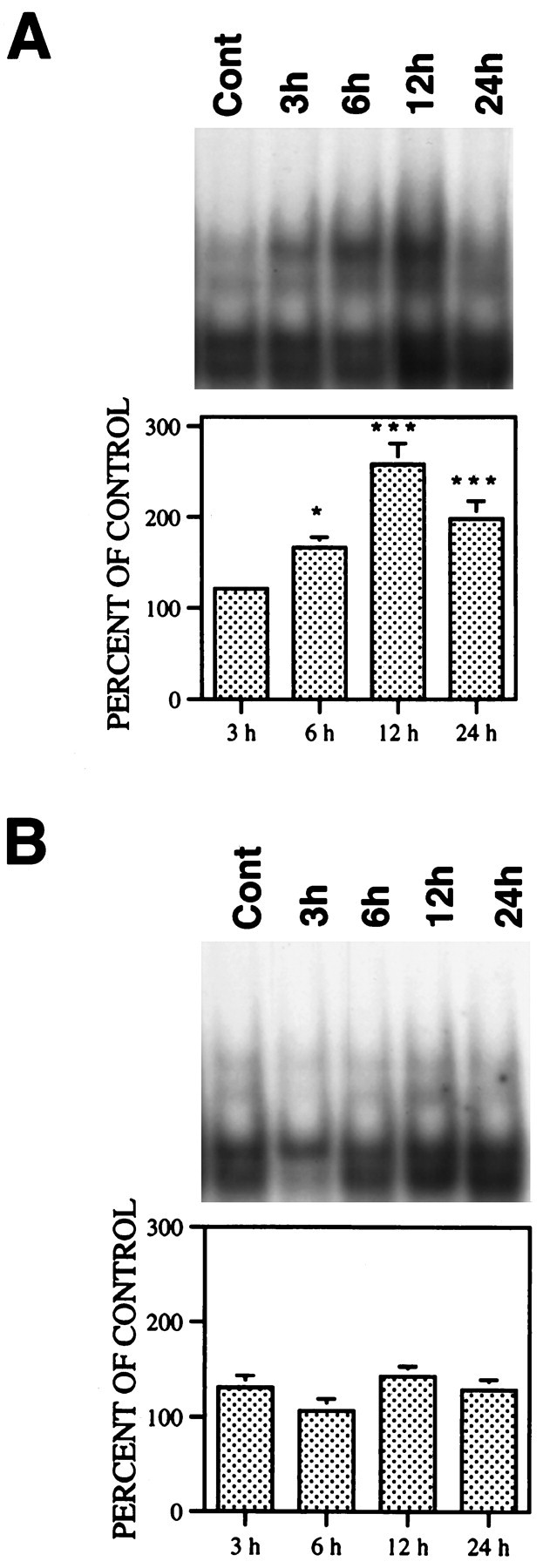
Effect of QA on NF-κB binding activity in nuclear extracts. Rats received QA (60 nmol in 1 μl) or saline intrastriatally and were killed 3, 6, 12, and 24 hr later. Striatal nuclear proteins were extracted and analyzed by EMSA. A, QA treatment. B, Vehicle treatment. Results from four rats in each group were quantitatively analyzed with an image analyzer and expressed as percent of control (nontreated animals; mean ± SEM). Statistical comparisons between treated and control groups were performed by ANOVA followed by a Dunnett t test. *p < 0.05; ***p < 0.001.
Fig. 2.

Specificity of QA-induced NF-κB binding activity and NF-κB subunits detected in nuclear extracts. Rats were treated as described in legend for Figure 1 and killed 12 hr after QA treatment. Striatal nuclear proteins were extracted and analyzed by EMSA.A, Specificity of NF-κB binding. In lane 3 from the left, unlabeled competitors were added to the assay mixtures 15 min before radioactively labeled NF-κB probes. B, Supershift assay. Antibodies specific for individual NF-κB subunit were added to assay 1 hr before radioactively labeled NF-κB probes. The arrowindicates supershifted band.
QA effects on c-Myc and p53
Levels of p53 mRNA were significantly increased starting 6 hr and peaking 12 hr after QA treatment, as determined by Northern blot analysis (Fig. 3A). Meanwhile, levels of p53 immunoreactivity were also substantially elevated 12–24 hr after QA but were not increased by vehicle treatment (Fig.3B,C) as revealed by Western blot analysis. The increase in p53 immunoreactivity was detectable with several p53 antibodies [p53, Ab-1, p53 (Pab 240), and p53 (Ab-3)], but these antibodies react with both wild-type and mutant p53 under denaturing conditions. Similarly, QA treatment also markedly increased levels of c-Myc mRNA and c-Myc proteins (Fig.4A,B). No change in c-Myc protein levels occurred in vehicle-treated animals (Fig. 4C).
Fig. 3.

Effect of QA on p53 mRNA and protein levels. Rats were treated with QA or saline and then killed as described in legend for Figure 1. Striatal mRNA or proteins were extracted and used for Northern or Western blot analysis. Results from three to four rats in each group were quantitatively analyzed with an image analyzer and expressed as percent of control (nontreated animals; mean ± SEM). Statistical comparisons of treated groups with control groups were performed by ANOVA followed by the Dunnett t test. **p < 0.01. ***p < 0.001.A, QA treatment, p53 mRNA. B, QA treatment, p53 protein. C, Vehicle treatment, p53 protein.
Fig. 4.

Effect of QA on c-Myc mRNA and protein levels. Rats were treated with QA or saline and then killed as described in legend for Figure 1. Striatal mRNA and proteins were extracted and used for Northern and Western blot analysis. Results from three to four rats in each group were quantitatively analyzed with an image analyzer and expressed as percent of control (nontreated animals; mean ± SEM). Statistical comparisons of treated groups with control groups were performed by ANOVA followed by the Dunnett t test. *p < 0.05; ***p < 0.001.A, QA treatment. B, Vehicle treatment. The arrow indicates the loading of total RNA in eachlane was approximately equal.
In agreement with these results obtained by Western blot analysis, large increases in both c-Myc and p53 immunoreactivity were also detected by immunohistochemistry 12–48 hr after QA administration. The rise in p53 immunoreactivity could be measured equally well with two p53 antibodies [p53 (Ab-3) and p53 (Pab 246)]. P53 (Pab 246) reacts to wild-type but not mutant p53 under nondenaturing conditions. Microscopic analysis of counterstained brain sections showed that the increased levels of c-Myc and p53 immunoreactivity occurred mainly in medium-sized neurons and only occasionally in glia (Fig.5C,D). No apparent increase in either c-Myc or p53 immunoreactivity occurred after vehicle treatment (Fig. 5A,B).
Fig. 5.

Effect of QA on c-Myc and p53 expression: immunohistochemical studies. Rats were treated and then killed as described in legend for Figure 1. Fixed brain sections were immunostained with c-Myc or p53 monoclonal antibodies. Representative results from animals 24 hr after QA or saline administration are presented showing the neuronal localization of c-Myc [stained with c-Myc (c-33); p53, stained with p53 (Pab 246)] immunoreactivity.A, B, Saline treatment. C,D, QA treatment. Arrows indicate typical striatal neurons expressing high levels of c-Myc and p53 (C, D). Scale bar, 25 μm.
NF-κB SN50 effects on QA-induced IκB-α degradation, NF-κB nuclear translocation, and AP-1 activation
The QA-induced increase in nuclear NF-κB binding activity was inhibited by pretreatment with NF-κB SN50 (5–20 μg) in a dose-dependent manner (Fig.6A). However, intrastriatally administered NF-κB SN50 did not have an appreciable effect on the QA-induced rise in AP-1 binding activity (Fig.6B). Moreover, the same doses of NF-κB SN50 control peptide failed to alter the QA-induced NF-κB nuclear translocation or increase in AP-1 binding (Fig. 6C,D). As expected, NF-κB SN50 (20 μg) alone did not modify IκB-α levels in untreated controls and, when used in combination with QA, had no significant effect on QA-induced degradation of IκB-α (Fig.7).
Fig. 6.

Effect of NF-κB SN50 on QA-induced alterations in NF-κB and AP-1 binding activities. Rats received intrastriatally administered NF-κB SN50 (5–20 μg) or control peptide (5–20 μg) 15 min before QA (60 nmol) and were killed 12 hr later. Striatal nuclear proteins were extracted and NF-κB and AP-1 binding activities measured by EMSA. Results from six rats in each group were quantitatively analyzed with an image analyzer and expressed as percent of control (saline treated; mean ± SEM) animals. Statistical comparisons were performed by ANOVA followed by the Dunnettt test. ***p < 0.001 (comparison of QA plus NS, QA plus 5 μg of SN50, QA plus 10 μg of SN50, or QA plus 20 μg of SN50 with control). #p < 0.05 (comparison of QA plus 5 μg of SN50, QA plus 10 μg of SN50, or QA plus 20 μg of SN50 with QA plus NS). A, NF-κB SN50 treatment, NF-κB binding. B, NF-κB SN50 treatment, AP-1 binding. C, NF-κB SN50 control peptide treatment, NF-κB binding. D, NF-κB SN50 control peptide treatment, AP-1 binding.
Fig. 7.

Effects of NF-κB SN50 on QA-induced degradation of IκB-α. Rats received intrastriatally administered NF-κB SN50 (20 μg) 15 min before intrastriatal injection of QA (60 nmol) and were killed 12 hr later. Striatal proteins were extracted for Western blot analysis. Results from six rats in each group were quantitatively analyzed with an image analyzer and expressed as percent of control (saline-treated animals; mean ± SEM). Statistical comparisons were performed by ANOVA followed by the Dunnett t test. *p < 0.05; **p < 0.01 (comparison of QA with control or QA plus 20 μg of SN50 with control).
NF-κB nuclear translocation blockade effects on QA-induced c-Myc and p53 expression
The QA-induced increases in striatal p53 mRNA and protein were diminished by NF-κB SN50. Quantitative analysis of the data indicated that NF-κB SN50 (20 μg) significantly reduced the QA-induced rise in levels of c-Myc mRNA from 1311 ± 169 to 700 ± 42% of control and protein from 735 ± 123 to 431 ± 49% of control (p < 0.001) (Fig.8A,B). Similarly, NF-κB SN50 (20 μg) also attenuated the QA-induced increase in levels of p53 mRNA from 230 ± 7 to 169 ± 9% of control (p < 0.001) and protein from 260 ± 47 to 131 ± 37% of control (p < 0.01) (Fig. 8C,D).
Fig. 8.

Effect of NF-κB SN50 on QA-induced increases in p53 and c-Myc mRNA and protein levels. Rats received intrastriatally administered NF-κB SN50 (20 μg) 15 min before QA (60 nmol) and were killed 12 or 24 hr later. Levels of p53 and c-Myc mRNA or proteins were measured by Northern or Western blot analysis. Results from four rats in each group were quantitatively analyzed with an image analyzer and expressed as percent of control (saline treated animals; mean ± SEM). Statistical comparisons were performed by ANOVA followed by the Dunnett t test or by Student’s t test. ***p < 0.001 (comparison of QA plus NS with control or QA plus 20 μg of SN50 with control). ##p < 0.01; ###p < 0.001 (comparison of QA plus NS with QA plus 20 μg of SN50).A, p53 mRNA. B, p53 protein.C, c-Myc mRNA. D, c-Myc protein. Thearrow indicates the loading of total RNA in eachlane was approximately equal.
NF-κB nuclear translocation blockade effects on QA-induced internucleosomal DNA fragmentation
Pretreatment with NF-κB SN50 (5–20 μg) produced a dose-dependent inhibition of QA-induced internucleosomal DNA fragmentation in animals assayed 24 hr after excitotoxin administration (Fig. 9A). In contrast, pretreatment with NF-κB SN50 control peptide (5–20 μg) had no effect on QA-induced internucleosomal DNA fragmentation (Fig.9B). To determine whether NF-κB SN50 merely delays onset of DNA fragmentation rather than actually blocking it, rats were pretreated with a single dose of NF-κB SN50 (20 μg), and striatal DNA fragmentation was assayed at various times after QA administration. The results showed that NF-κB SN50 inhibited QA-induced internucleosomal DNA fragmentation at all times examined (12, 24, and 48 hr) (Fig. 9C). To determine whether NF-κB SN50 inhibits DNA fragmentation when given shortly after QA treatment, animals received a single dose of NF-κB SN50 either 15 min before or 2, 4, or 6 hr after QA infusion. Biochemical evaluation 24 hr after QA administration indicated that pretreatment with NF-κB SN50 was most effective in reducing QA-induced internucleosomal DNA fragmentation. Some attenuation of QA-induced DNA fragmentation occurred when NF-κB SN50 was administered 2 or 4 hr after QA but not when given 6 hr later (Fig. 9D).
Fig. 9.

Effect of NF-κB SN50 on QA-induced internucleosomal DNA fragmentation. A, B, Rats received intrastriatally administered NF-κB SN50 (5–20 μg,A) or control peptide (5–20 μg, B) 15 min before QA (60 nmol) and were killed 24 hr later. Genomic DNA was extracted and electrophoresed on 2% agarose gel. Eachlane shows results from three animals. C, Rats received intrastriatally administered NF-κB SN50 (20 μg) 15 min before QA (60 nmol) and were killed 12, 24, and 48 hr later. Genomic DNA was extracted and electrophoresed on 2% agarose gel. Eachlane shows results from three animals. D, Rats received intrastriatally administered NF-κB SN50 (20 μg) 15 min before QA (60 nmol) or were given intrastriatal QA (60 nmol) and then NF-κB SN50 (20 μg) 2, 4, or 6 hr later. Animals were killed 24 hr after QA, and genomic DNA was extracted and electrophoresed on 2% agarose gel. Each lane shows results from three animals.
DISCUSSION
The present results provide additional support for the possibility that NF-κB participates in a signaling cascade culminating in the apoptotic demise of rat striatal neurons exposed to the excitotoxin QA (Qin et al., 1996, 1998). It now appears that QA stimulation of glutamate receptors of the NMDA subtype leads to the selective degradation of IκB-α, a cytosolic protein that binds NF-κB. Our data further suggest that the resultant release and nuclear translocation of NF-κB augments expression of the proapoptotic genes p53 and c-Myc, because blockade of NF-κB translocation with NF-κB SN50 inhibits the QA-induced rise in mRNA and protein levels of both p53 and c-Myc, as well as the severity of internucleosomal DNA fragmentation. These in vivo findings could have important implications for the treatment of CNS degenerative disorders, as well as other conditions in which excitotoxicity and NF-κB activation may contribute to neuronal death.
As a transcription factor, NF-κB is known to influence the expression of a broad array of genes. In the present study, NF-κB activation by QA preceded the appearance of maximum internucleosomal DNA cleavage (Qin et al., 1998). It is thus conceivable that NF-κB may be able to induce gene products that stimulate apoptosis in striatal neurons. Among potential candidates for this role, c-Myc and p53 are of particular interest. Previous studies have shown that NF-κB binding sites are present in the promoter regions of both genes (Duyao et al., 1990; Kessler et al., 1992; Wu and Lozano, 1994; Lee et al., 1995), but whether these binding sites contribute to the regulation of p53 and c-Myc expression in neurons has yet to be established. Here, we find that QA increases neuronal levels of p53 and c-Myc mRNA and protein in striatal neurons and that the induction of these genes and gene products occurs in close temporal relation to NF-κB activation by IκB degradation and the subsequent appearance of the biochemical and morphological stigmata of apoptosis. The increase in p53 protein levels by QA observed in the present studies is consistent with finding reported by Hughes et al. (1996). These observations raise the question as to whether the temporal association in this instance could signify a causal relationship.
The possibility that NF-κB activation triggers c-Myc and p53 induction and the apoptotic death of rat striatal neurons was addressed by studies with NF-κB SN50. This recombinant peptide contains two functional regions: the hydrophobic domain of the signal peptide from Kaposi fibroblast growth factor, which confers membrane permeability, and a nuclear localization sequence (residues 360–369), which selectively blocks the nuclear translocation of NF-κB in a saturable, competitive manner (Lin et al., 1995b). In agreement with our previous results (Qin et al., 1998), we found that NF-κB SN50, but not its control peptide, attenuated the QA-induced increase in nuclear NF-κB activity in a dose-dependent manner. We also found that the inhibitory peptide had no effect on IκB-α degradation but did diminish the QA-induced rise in the expression of both p53 and c-Myc mRNA and protein. These studies suggest that NF-κB nuclear translocation upregulates the expression of both p53 and c-Myc in striatal neurons. The observation that NF-κB SN50 reduced p53 and c-Myc induction by only 50–80% could indicate that its inhibitory action was incomplete or that other signaling pathways besides NF-κB contribute to c-Myc and p53 responses to NMDA receptor stimulation.
Both c-Myc and p53 serve as critical regulators of the cell cycle and of apoptotic mechanisms in normal and malignant cells (Kastan et al., 1995; Packham and Cleveland, 1995; Selivanova and Wiman, 1995). The important contribution of p53 to apoptotic processes in postmitotic neurons is now especially well documented (Slack et al., 1996; Hughes et al., 1997; Jordan et al., 1997). Moreover, several studies have linked p53 expression to the neuronal damage in the CNS after exposure to ischemia or excitotoxins (Chopp et al., 1992; Li et al., 1993; Sakhi et al., 1994). Conversely, p53 null mice have been found to resist ischemic or excitotoxic insult (Crumrine et al., 1994; Hermeking and Eick, 1994; Morrison et al., 1996; Xiang et al., 1996). Similar evidence suggests the participation of c-Myc as a proapoptotic regulator in cells of various types and under variety of conditions (Packham and Cleveland, 1995; Nakagomi et al., 1996; Packham et al., 1996). Indeed, c-Myc and p53 have been reported to act synergistically in the induction of apoptosis (Saito and Ogawa, 1995). In this study, we found that increased levels of both c-Myc and p53 occurred in the medium-sized efferent neurons but not in the large-sized interneurons, which are relatively resistant to QA. The increased p53 immunoreactivity in striatal neurons could also be detected with an antibody specific for the wild form of p53, indicating that the wild type of p53 is induced (for review, see Hughes et al., 1997). Interestingly, kainic acid-induced elevations in c-Myc and p53 immunoreactivities occurred in both medium- and large-sized striatal neurons, and these c-Myc and p53-positive large neurons evidenced rapid degeneration (data not shown). The upregulation of c-Myc and p53 in vulnerable medium-sized efferent neurons by NF-κB observed in this study is consistent with the view that NF-κB plays a proapoptotic role in striatal neurons exposed to excitotoxic insult. More direct support for this possibility derives from our findings with NF-κB SN50; this inhibitor of NF-κB activation produced a dose-dependent reduction in the intensity of the QA-induced internucleosomal DNA fragmentation, a hallmark of apoptosis. Similar treatment with NF-κB SN50 control peptide had no effect on QA-induced internucleosomal DNA fragmentation. NF-κB SN50 even evidenced some protective activity when given a few hours after QA treatment. On the other hand, NF-κB SN50 had no effect when administered 6 hr after QA, because by then NF-κB activation had already taken place.
Apoptotic processes in mature neurons are tightly regulated, and underlying mechanisms are necessarily complex. Previous studies have identified a complicated pattern of QA-induced alterations in transcription factors in addition to the NF-κB changes studied here. These included time-dependent increases in AP-1 and decreases in octamer 1 and SP-1 binding activities occurring in association with the apoptotic destruction of rat striatal neurons (Qin et al., 1998). Concomitant changes in signaling molecules other than NF-κB could exert a critical influence on apoptotic mechanisms involving NF-κB. Moreover, different combinations of NF-κB subunits may activate different sets of target genes (Perkins et al., 1992). In view of previous observations suggesting a proapoptotic role for c-Rel (Abbadie et al., 1993), the fact that we were able to detect c-Rel in the nucleus after QA treatment could be of considerable importance. Clearly, NF-κB function depends on many factors, few of which are now understood. Apparent discrepancies between studies of this transcription factor are thus hardly surprising (Barger et al., 1995;Mattson et al., 1997).
Results of the present investigation not only support earlier observations suggesting that NF-κB plays a proapoptotic role in certain models of cell death (Abbadie et al., 1993; Lin et al., 1995a;Grimm et al., 1996; Marinovich et al., 1996) but also detail some of the molecular events linking NF-κB activation to an apoptotic program in an animal model. In addition, our findings may shed light on recent reports of increased NF-κB levels in certain neurodegenerative disorders (Terai et al., 1996; Hunot et al., 1997; Kaltschmidt et al., 1997; Clemens et al., 1998). Current evidence suggests that NF-κB elevations in these CNS disorders may be injurious to vulnerable neurons by either or both of two mechanisms, one involving cytokine-mediated inflammatory reactions, the other involving the direct activation of apoptotic processes. In either case, NF-κB could be an important target for new drug development.
Footnotes
Correspondence should be addressed to Dr. Thomas N. Chase, Chief, Experimental Therapeutics Branch, National Institute of Neurological Diseases and Stroke, National Institutes of Health, Building 10, Room 5C103, 10 Center Drive, MSC 1406, Bethesda, MD 20892.
REFERENCES
- 1.Abbadie C, Kabrun N, Bouali F, Smardova J, Stehelin D, Vandenbunder B, Enrietto PJ. High levels of c-Rel expression are associated with programmed cell death in the developing avian embryo and in bone marrow cells in vitro. Cell. 1993;75:899–912. doi: 10.1016/0092-8674(93)90534-w. [DOI] [PubMed] [Google Scholar]
- 2.Anglade P, Vyas S, Javoy-Agid F, Herrero MT, Michel PP, Marquez J, Mouatt-Prigent A, Ruberg M, Hirsch EC, Agid Y. Apoptosis and autophagy in nigral neurons of patients with Parkinson’s disease. Histol Histopathol. 1997;12:25–31. [PubMed] [Google Scholar]
- 3.Ankarcrona M, Dypbukt JM, Bonfoco E, Zhivotovsky B, Orrenius S, Lipton SA, Nicotera P. Glutamate-induced neuronal death: a succession of necrosis or apoptosis dependent on mitochondria function. Neuron. 1993;15:961–973. doi: 10.1016/0896-6273(95)90186-8. [DOI] [PubMed] [Google Scholar]
- 4.Antwerp DJV, Martin SJ, Kafri T, Green DR, Verma IM. Suppression of TNF-α-induced apoptosis by NF-κB. Science. 1996;274:787–789. doi: 10.1126/science.274.5288.787. [DOI] [PubMed] [Google Scholar]
- 5.Baeuerle PA, Baltimore D. NF-κB: ten years after. Cell. 1996;87:13–20. doi: 10.1016/s0092-8674(00)81318-5. [DOI] [PubMed] [Google Scholar]
- 6.Baeuerle PA, Henkel T. Function and activation of NF-κB in the immune system. Annu Rev Immunol. 1994;12:141–179. doi: 10.1146/annurev.iy.12.040194.001041. [DOI] [PubMed] [Google Scholar]
- 7.Baichwal V, Baeuerle PA. Apoptosis: activate NF-κB or die? Curr Biol. 1997;7:R94–R96. doi: 10.1016/s0960-9822(06)00046-7. [DOI] [PubMed] [Google Scholar]
- 8.Barger SW, Horster D, Furukawa K, Goodman Y, Krieglstein J, Mattson MP. Tumor necrosis factors α and β protect neurons against amyloid β-peptide toxicity: evidence for involvement of a κB-binding factor and attenuation of peroxide and Ca2+ accumulation. Proc Natl Acad Sci USA. 1995;92:9328–9332. doi: 10.1073/pnas.92.20.9328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Beg AA, Baltimore D. An essential role for NF-κB in preventing TNF-α-induced cell death. Science. 1996;274:782–784. doi: 10.1126/science.274.5288.782. [DOI] [PubMed] [Google Scholar]
- 10.Beg AA, Sha WC, Bronson RT, Ghosh S, Baltimore D. Embryonic lethality and liver degeneration in mice lacking the Rel A component of NF-κB. Nature. 1995;376:167–170. doi: 10.1038/376167a0. [DOI] [PubMed] [Google Scholar]
- 11.Bonfoco E, Krainc D, Ankarcrona M, Nicotera P, Lipton SA. Apoptosis and necrosis: two distinct events induced, respectively, by mild and intense insults with N-methyl-d-aspartate or nitric oxide/superoxide in cortical cell cultures. Proc Natl Acad Sci USA. 1995;92:7162–7166. doi: 10.1073/pnas.92.16.7162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Choi DW. Excitotoxic cell death. J Neurobiol. 1992;23:1261–1276. doi: 10.1002/neu.480230915. [DOI] [PubMed] [Google Scholar]
- 13.Chopp M, Li Y, Zhang ZG, Freytag SO. p53 expression in brain after middle cerebral artery occlusion in the rat. Biochem Biophys Res Commun. 1992;182:1201–1207. doi: 10.1016/0006-291x(92)91859-o. [DOI] [PubMed] [Google Scholar]
- 14.Clemens JA, Stephenson DT, Smalstig EB, Dixon EP, Little SP. Global ischemia activates nuclear factor-κB in forebrain neurons of rats. Stroke. 1997;28:1073–1081. doi: 10.1161/01.str.28.5.1073. [DOI] [PubMed] [Google Scholar]
- 15.Clemens JA, Stephenson DT, Yin T, Smalstig EB, Panetta JA, Little SP. Drug-induced neuroprotection from global ischemia is associated with prevention of persistent but not transient activation of nuclear factor-κB in rats. Stroke. 1998;29:177–182. doi: 10.1161/01.str.29.3.677. [DOI] [PubMed] [Google Scholar]
- 16.Cotman CW, Su JH. Mechanisms of neuronal death in Alzheimer’s disease. Brain Pathol. 1996;6:493–506. doi: 10.1111/j.1750-3639.1996.tb00878.x. [DOI] [PubMed] [Google Scholar]
- 17.Coyle JT, Puttfarcken P. Oxidative stress, glutamate, and neurodegenerative disorders. Science. 1993;262:689–695. doi: 10.1126/science.7901908. [DOI] [PubMed] [Google Scholar]
- 18.Crumrine RC, Thomas AL, Morgan PF. Attenuation of p53 expression protects against focal ischemic damage in transgenic mice. J Cereb Blood Flow Metab. 1994;14:887–891. doi: 10.1038/jcbfm.1994.119. [DOI] [PubMed] [Google Scholar]
- 19.Duyao MP, Buckler AJ, Sonenshein GE. Interaction of an NF-κB-like site upstream of the c-Myc promoter. Proc Natl Acad Sci USA. 1990;87:4727–4731. doi: 10.1073/pnas.87.12.4727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Grilli M, Pizzi M, Memo M, Spano P. Neuroprotection by aspirin and sodium salicylate through blockade of NF-κB activation. Science. 1996;274:1383–1385. doi: 10.1126/science.274.5291.1383. [DOI] [PubMed] [Google Scholar]
- 21.Grimm S, Bauer MKA, Baeuerle PA, Schulze-Osthoff K. Bcl-2 down-regulations the activity of transcription factor NF-κB induced upon apoptosis. J Cell Biol. 1996;134:13–23. doi: 10.1083/jcb.134.1.13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Guerrini L, Blasi F, Denis-Donini S. Synaptic activation of NF-κB by glutamate in cerebellar granule neurons in vitro. Proc Natl Acad Sci USA. 1995;92:9077–9081. doi: 10.1073/pnas.92.20.9077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hermeking H, Eick D. Mediation of c-Myc-induced apoptosis by p53. Science. 1994;265:2091–2093. doi: 10.1126/science.8091232. [DOI] [PubMed] [Google Scholar]
- 24.Hughes PE, Alexi T, Yoshida T, Schida SS, Knusel B. Excitotoxic lesion of rat brain with quinolinic acid induces expression of p53 messenger RNA and protein and p53-inducible genes bax and Gadd-45 in brain areas showing DNA fragmentation. Neuroscience. 1996;74:1143–1160. doi: 10.1016/0306-4522(96)00174-1. [DOI] [PubMed] [Google Scholar]
- 25.Hughes P, Alexi T, Schreiber SS. A role for the tumor suppressor gene p53 regulating neuronal apoptosis. NeuroReport. 1997;8:5–12. [PubMed] [Google Scholar]
- 26.Hunot S, Brugg B, Ricard D, Michel PP, Muriel M-P, Ruberg M, Faucheux BA, Agid Y, Hirsch EC. Nuclear translocation of NF-κB is increased in dopaminergic neurons of patients with Parkinson’s disease. Proc Natl Acad Sci USA. 1997;94:7531–7536. doi: 10.1073/pnas.94.14.7531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Jordan J, Galino MF, Prehn JHM, Weichselbaum RR, Beckett M, Ghadge GD, Roos RP, Leiden JM, Miller RJ. p53 expression induces apoptosis in hippocampal pyramidal neuron cultures. J Neurosci. 1997;17:1397–1405. doi: 10.1523/JNEUROSCI.17-04-01397.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kaltschmidt C, Kaltschmidt B, Baeuerle PA. Brain synapses contain inducible forms of the transcription factor NF-κB. Mech Dev. 1993;43:135–147. doi: 10.1016/0925-4773(93)90031-r. [DOI] [PubMed] [Google Scholar]
- 29.Kaltschmidt C, Kaltschmidt B, Neumann H, Wekerle H, Baeuerle PA. Constitutive NF-κB activity in neurons. Mol Cell Biol. 1994;14:3981–3992. doi: 10.1128/mcb.14.6.3981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kaltschmidt C, Kaltschmidt B, Baeuerle PA. Stimulation of ionotropic glutamate receptor activates transcription factor NF-κB in primary neurons. Proc Natl Acad Sci USA. 1995;92:9618–9622. doi: 10.1073/pnas.92.21.9618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kaltschmidt C, Uherek M, Volk B, Baeuerle PA, Kaltschmidt C. Transcription factor NF- κB is activated in primary neurons by β amyloid peptides and in neurons surrounding early plaques from patients with Alzheimer’s disease. Proc Natl Acad Sci USA. 1997;94:2642–2647. doi: 10.1073/pnas.94.6.2642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kastan MB, Canman CE, Leonard CJ. p53, cell cycle control and apoptosis: implications for cancer. Cancer Metastasis Rev. 1995;14:3–15. doi: 10.1007/BF00690207. [DOI] [PubMed] [Google Scholar]
- 33.Kessler DJ, Duyao MP, Spicer DB, Sonenshein GE. NF-κB-like factors mediate interleukin 1 induction of c-Myc gene transcription in fibroblasts. J Exp Med. 1992;176:787–792. doi: 10.1084/jem.176.3.787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lee H, Arsura M, Wu M, Duyao M, Buckler AJ, Sonenshein GE. Role of Rel-related factors in control of c-Myc gene transcription in receptor-mediated apoptosis of murine B cell WEHI 231 line. J Exp Med. 1995;181:1169–1177. doi: 10.1084/jem.181.3.1169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Li Y, Chopp M, Zhang ZG, Zaloga C, Niewenhuis L, Gautam S. p53-immunoreactive protein and p53 mRNA expression after transient middle cerebral artery occlusion in rats. Stroke. 1993;25:849–856. doi: 10.1161/01.str.25.4.849. [DOI] [PubMed] [Google Scholar]
- 36.Lin K-I, Lee S-W, Narayanan R, Baraban JM, Hardwick JM. Thiol agents and Bcl-2 identify an α virus-induced apoptosis pathway that requires activation of the transcription factor NF-κB. J Cell Biol. 1995a;131:1149–1161. doi: 10.1083/jcb.131.5.1149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lin Y-Z, Yao S, Veach RA, Torgerson TR, Hawiger J. Inhibition of nuclear translocation of transcription factor NF-κB by a synthetic peptide containing a cell membrane-permeable motif and nuclear localization sequence. J Biol Chem. 1995b;270:14255–14258. doi: 10.1074/jbc.270.24.14255. [DOI] [PubMed] [Google Scholar]
- 38.Lipton SA. Janus faces of NF-κB: neurodestruction versus neuroprotection. Nat Med. 1997;3:20–22. doi: 10.1038/nm0197-20. [DOI] [PubMed] [Google Scholar]
- 39.Marinovich M, Viviani B, Corsini E, Ghilardi F, Galli CL. NF-κB activation by triphenyltin triggers apoptosis in HL-60 cells. Exp Cell Res. 1996;226:98–104. doi: 10.1006/excr.1996.0207. [DOI] [PubMed] [Google Scholar]
- 40.Mattson MP, Goodman Y, Luo H, Fu W, Furukawa K. Activation of NF-κB protects hippocampal neurons against oxidative stress-induced apoptosis: evidence for induction of manganese superoxide dismutase and suppression of peroxynitrite production and protein tyrosine nitration. J Neurosci Res. 1997;49:681–697. doi: 10.1002/(SICI)1097-4547(19970915)49:6<681::AID-JNR3>3.0.CO;2-3. [DOI] [PubMed] [Google Scholar]
- 41.Mochizuki H, Mori H, Mizuno Y. Apoptosis in neurodegenerative disorders. J Neural Transm Suppl. 1997;50:125–140. doi: 10.1007/978-3-7091-6842-4_13. [DOI] [PubMed] [Google Scholar]
- 42.Morrison RS, Wenzel HJ, Kinoshita Y, Robbins CA, Donehower LA, Schwartzkroin PA. Loss of the p53 tumor suppressor gene protects neurons from kainate-induced cell death. J Neurosci. 1996;16:1337–1345. doi: 10.1523/JNEUROSCI.16-04-01337.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Nakagomi T, Asai A, Kanemitsu H, Narita K, Kushiyuki Y, Tamura A, Kirino T. Up- regulation of c-Myc gene expression following focal ischemia in the rat brain. Neurol Res. 1996;18:559–563. doi: 10.1080/01616412.1996.11740470. [DOI] [PubMed] [Google Scholar]
- 44.O’Neil LAJ, Kaltschmidt C. NF-κB: a crucial transcription factor for glial and neuronal cell function. Trends Neurosci. 1997;20:252–258. doi: 10.1016/s0166-2236(96)01035-1. [DOI] [PubMed] [Google Scholar]
- 45.Ozaki N, Takeda H, Iwahashi H, Kitano S, Hanazawa S. NF-κB inhibitors stimulate apoptosis of rabbit mature osteoclasts and inhibit bone reabsorption by these cells. FEBS Lett. 1997;410:297–300. doi: 10.1016/s0014-5793(97)00653-4. [DOI] [PubMed] [Google Scholar]
- 46.Packham G, Cleveland JL. c-Myc and apoptosis. Biochem Biophys Acta. 1995;1242:11–28. doi: 10.1016/0304-419x(94)00015-t. [DOI] [PubMed] [Google Scholar]
- 47.Packham G, Porter CW, Cleveland JL. c-Myc induces apoptosis and cell cycle progression by separable, yet overlapping, pathways. Oncogene. 1996;13:461–469. [PubMed] [Google Scholar]
- 48.Perkins ND, Schmid RM, Duckett CS, Leung K, Rice NR, Nabel GJ. Distinct combinations of NF-κB subunits determine the specificity of transcriptional activation. Proc Natl Acad Sci USA. 1992;89:1529–1533. doi: 10.1073/pnas.89.5.1529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Pittman RN, Mills JC, DiBenedetto AJ, Hynicka WP, Wang S. Neuronal cell death: searching for the smoking gun. Curr Opin Neurobiol. 1994;4:87–94. doi: 10.1016/0959-4388(94)90036-1. [DOI] [PubMed] [Google Scholar]
- 50.Portera-Cailliau C, Hedreen JC, Price DL, Koliatsos VE. Evidence for apoptotic cell death in Huntington’s disease and excitotoxic animal models. J Neurosci. 1995;15:3775–3787. doi: 10.1523/JNEUROSCI.15-05-03775.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Qin Z-H, Wang Y, Chase TN. Stimulation of N-methyl-d-aspartate receptors induces apoptosis in rat brain. Brain Res. 1996;725:166–177. doi: 10.1016/0006-8993(96)00200-4. [DOI] [PubMed] [Google Scholar]
- 52.Qin Z-H, Wang Y, Nakai M, Chase TN. Transcription factor NF-κB contributes to excitotoxin-induced apoptosis in rat striatum. Mol Pharmacol. 1998;53:33–42. doi: 10.1124/mol.53.1.33. [DOI] [PubMed] [Google Scholar]
- 53.Saito Y, Ogawa K. Wild type p53 and c-Myc co-operation in generating apoptosis of a rat hepatocellular carcinoma cell line FAA-HTC1. Oncogene. 1995;11:1013–1018. [PubMed] [Google Scholar]
- 54.Sakhi S, Bruce A, Sun N, Tocco G, Baudry M. p53 induction is associated with neuronal damage in the central nervous system. Proc Natl Acad Sci USA. 1994;91:7525–7529. doi: 10.1073/pnas.91.16.7525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Selivanova G, Wiman KG. p53: a cell cycle regulator activated by DNA damage. Adv Cancer Res. 1995;66:143–180. doi: 10.1016/s0065-230x(08)60253-5. [DOI] [PubMed] [Google Scholar]
- 56.Siebenlist U. NFκB/IκB proteins. Their role in cell growth, differentiation and development. Biochim Biophys Acta. 1997;1332:R7–R13. doi: 10.1016/s0304-419x(96)00038-8. [DOI] [PubMed] [Google Scholar]
- 57.Simonian NA, Getz RL, Leveque JC, Konradi C, Coyle JT. Kainate induces apoptosis in neurons. Neuroscience. 1996;74:675–683. doi: 10.1016/0306-4522(96)00141-8. [DOI] [PubMed] [Google Scholar]
- 58.Slack RS, Belliveau DJ, Rosenberg M, Atwal J, Lochmuller H, Aloyz R, Haghighi A, Lach B, Seth P, Cooper E, Miller FD. Adenovirus-mediated gene transfer of the tumor suppressor, p53, induces apoptosis in postmitotic neurons. J Cell Biol. 1996;135:1085–1096. doi: 10.1083/jcb.135.4.1085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Taglialatela G, Robinson R, Perez-Polo R. Inhibition of Nuclear factor Kappa B (NF-κB) activity induces nerve growth factor-resistant apoptosis in PC12 cells. J Neurosci Res. 1997;47:155–162. [PubMed] [Google Scholar]
- 60.Terai K, Matsuo A, Mcgeer PL. Enhancement of immunoreactivity for NF-κB in the hippocampal formation and cerebral cortex of Alzheimer’s disease. Brain Res. 1996;735:159–168. doi: 10.1016/0006-8993(96)00310-1. [DOI] [PubMed] [Google Scholar]
- 61.Wosikowski K, Regis JT, Robey RW, Alvarez M, Buters JT, Gudas JM, Bates SE. Normal p53 status and function despite the development of drug resistance in human breast cancer cells. Cell Growth Differ. 1995;6:1395–1403. [PubMed] [Google Scholar]
- 62.Wu H, Lozano G. NF-κB activation of p53. J Biol Chem. 1994;269:20067–20074. [PubMed] [Google Scholar]
- 63.Wu M, Lee H, Bellas RE, Schauer SL, Arsura M, Katz D, FitzGerald MJ, Rothstein TL, Scherr DH, Sonenshein GE. Inhibition of NF-κB/Rel induces apoptosis of murine B cells. EMBO J. 1996;15:4682–4690. [PMC free article] [PubMed] [Google Scholar]
- 64.Xiang H, Hochman DW, Saya H, Fujiwara T, Schwartzkroin PA, Morrison RS. Evidence for p53-mediated modulation of neuronal viability. J Neurosci. 1996;16:6753–6765. doi: 10.1523/JNEUROSCI.16-21-06753.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]