

Abstract
The identity of the potassium channel underlying the slow, apamin-insensitive component of the afterhyperpolarization current (sIAHP) remains unknown. We studied sIAHP in CA1 pyramidal neurons using simultaneous whole-cell recording, calcium fluorescence imaging, and flash photolysis of caged compounds. Intracellular calcium concentration ([Ca2+]i) peaked earlier and decayed more rapidly than sIAHP. Loading cells with low concentrations of the calcium chelator EGTA slowed the activation and decay of sIAHP. In the presence of EGTA, intracellular calcium decayed with two time constants. When [Ca2+]i was increased rapidly after photolysis of DM-Nitrophen, both apamin-sensitive and apamin-insensitive outward currents were activated. The apamin-sensitive current activated rapidly (<20 msec), whereas the apamin-insensitive current activated more slowly (180 msec). The apamin-insensitive current was reduced by application of serotonin and carbachol, confirming that it was caused by sIAHP channels. When [Ca2+]i was decreased rapidly via photolysis of diazo-2, the decay of sIAHPwas similar to control (1.7 sec). All results could be reproduced by a model potassium channel gated by calcium, suggesting that the channels underlying sIAHP have intrinsically slow kinetics because of their high affinity for calcium.
Keywords: afterhyperpolarization, intracellular calcium concentration, potassium channel, hippocampus, pyramidal neurons, current
In many neurons throughout the nervous system, a train of action potentials is followed by a prolonged afterhyperpolarization (AHP). The currents generating the AHP are thought to be calcium-activated potassium currents, because they require calcium influx for activation and are blocked when intracellular calcium is strongly buffered (Sah, 1996). The currents underlying the AHP can be separated into two distinct types. One of these (IAHP), activates rapidly (<5 msec) after calcium influx and decays with a time constant of several hundred milliseconds. This current is voltage-independent and is blocked by the peptide apamin (Pennefather et al., 1985; Sah and McLachlan, 1991). The channels underlying IAHPare SK-type calcium-activated potassium channels (Marty, 1989; Sah, 1996). A second type of AHP current [slow IAHP(sIAHP)] has much slower kinetics. After calcium influx, it activates with a time constant of >100 msec and decays with a time constant of ∼1.5 sec. This current is also voltage-independent but is unaffected by apamin (Sah, 1996). In hippocampal pyramidal cells, sIAHP is thought to be restricted to the apical dendrites (Sah and Bekkers, 1996) and is modulated by second messenger systems (Nicoll, 1988; Knopfel et al., 1990; Pedarzani and Storm, 1993; Sah and Isaacson, 1995). It is therefore a key determinant of the repetitive firing properties of CA1 pyramidal neurons.
Molecular cloning studies have identified two families of genes coding for calcium-activated potassium channels (Vegara et al., 1998). One family codes for large conductance, voltage-sensitive channels, which are blocked by tetraethylammonium (TEA) ions and charybdotoxin. These channels are identified with BK-type calcium-activated potassium channels (Vegara et al., 1998). In neurons, the macroscopic current associated with activation of these channels is designatedIC. IC contributes to action potential repolarization in some neurons but is not active during the slow afterhyperpolarization (Sah, 1996). A second gene family codes for channels with low conductance, which are not blocked by charybdotoxin and are less sensitive to block by TEA. Three genes have been identified: SK1, SK2, and SK3 (Kohler et al., 1996). When SK2 and SK3 are expressed in oocytes they form channels that are blocked by apamin and thus have been identified withIAHP (Hirschberg et al., 1998). The identity of the channel underlying sIAHP is not known. SK1 forms channels that are apamin-insensitive; however, its kinetic properties, which are similar to those of SK2 and SK3 (Hirschberg et al., 1998), make it an unlikely candidate to mediate sIAHP (see Discussion).
Several different mechanisms have been proposed to underlie the slow kinetics of sIAHP (Sah, 1996). Lancaster and Zucker (1994) have suggested that the channels underlying sIAHP have rapid kinetics, and other factors such as calcium diffusion and clearance determine the sIAHP time course. In this study we have reexamined the relationship between intracellular calcium transients and activation of sIAHP using a fast calcium-imaging system and manipulation of intracellular calcium by photolysis of caged compounds. We find that the time course of sIAHP is best explained by a model in which the underlying channels have intrinsically slow deactivation kinetics because of their high affinity for calcium.
MATERIALS AND METHODS
All procedures were in accordance with Institutional Animal Care and Ethics Committee guidelines. Experiments were performed on brain slices prepared from rats aged 16–21 d. Rats were anesthetized with sodium pentobarbital (50 mg/kg) and decapitated, and the brain was rapidly immersed in ice cold Ringer’s solution. Transverse hippocampal slices (300–350 μm thick) were prepared on a vibratome (Camden), and allowed to recover for 1 hr at 30°C. They were subsequently stored at room temperature. Slices were superfused with Ringer’s solution containing (in mm): 119 NaCl, 2.5 KCl, 1.3 Mg2Cl2, 2.5 CaCl2, 1.0 Na2PO4, 26.2 NaHCO3, and 11 glucose, which was equilibrated with 5% CO2 and 95% O2. Patch electrodes were filled with an internal solution containing (in mm): 135 KMeSO4, 8 NaCl, 10 HEPES, 2 Mg2ATP, and 0.3 Na3-GTP, pH 7.3 with KOH (osmolarity, 290 mOsm). Calcium fluorophores and caged compounds were added to this solution at the indicated concentrations. Whole-cell patch-clamp recordings were obtained from the somata of CA1 pyramidal neurons using infrared differential interference contrast techniques on an Olympus Optical (Tokyo, Japan) BX-50 microscope equipped with a 60× water immersion objective (numerical aperture, 0.9; Olympus). Currents were recorded using an Axopatch-1D amplifier (Axon Instruments, Foster City, CA) and digitized at 0.5 kHz using custom software running on a Macintosh 7500 computer (Apple Computer, Cupertino, CA) under Igor Pro (WaveMetrics, Lake Oswego, OR). sIAHPs were evoked by brief (50–100 msec) depolarizing voltage steps to 0 mV from a holding potential of −50 mV.
In some experiments, tetraethylammonium (1–5 mm) was included in the perfusing Ringer’s solution to block currents mediated by large-conductance calcium-activated potassium channels. No differences were noted in outward currents in the presence of TEA.
Fluorescence measurements. Fluorescence measurements were made using a monochromator-based illumination system (Polychrome II; T.I.L.L. Photonics, Planegg, Germany). In most experiments, the calcium indicator used was Oregon Green BAPTA-1 (Kd, 170 nm in the absence of Mg; Molecular Probes, Eugene, OR) added to the internal solution at a concentration of 50 μm. In some cases, fluo3 (Molecular Probes) was used instead. No differences were noted in the results, and the data from the two dyes have been lumped together. Cells were allowed to load with the calcium indicators for at least 5 min after breaking in to allow the cells to equilibrate with dye. Care was taken to select cells in which the apical dendrite was visible in the plane of section of the slice for up to 150 μm from the soma. Cells were illuminated at 488 nm with an exposure of 10 msec. Images were acquired with an in-line transfer, cooled CCD camera (T.I.L.L. Photonics) in which the scan lines were binned by two in both horizontal and vertical directions, giving a spatial resolution of 0.33 μm/pixel. A region of interest selected from the full frame was captured at 20 Hz. Subsequently, the data were analyzed off-line using Vision (T.I.L.L. Photonics). In each frame, regions of interest (ROIs) were selected, and the background from a nearby, similar-size ROI was subtracted. Distances from the soma were calculated from the point of emergence of the apical dendrite from the soma. ROIs were rectangular regions, usually 10 pixels long. Traces were corrected for bleaching by fitting a line to data points collected during the baseline and after the calcium transient had returned to resting levels and subtracting the fitted line from the data. All data are presented either as percent ΔF/F or ΔF.
Caged compounds used were DM-Nitrophen, diazo-2, and diazo-3 (all from Molecular Probes). Diazo-2 and diazo-3 were dissolved directly in the internal solution at the required concentration. DM-Nitrophen was made up in internal solution in which MgATP had been replaced with NaATP to avoid loading the compound with Mg2+. It was loaded with calcium to a concentration of 60–70%. The lamp used for photolysis was a pulsed xenon arc lamp (T.I.L.L. Photonics), which illuminated the entire field of view and discharged ∼80 J in 2 msec.
Apamin was purchased from Sigma (St. Louis, MO) and made up as a 20 μm stock solution and kept frozen until required at −20°C. All data are presented as mean ± SEM.
Kinetic model of Ca-activated K channels. A Markov model of a Ca-activated K channel was used to predict the sIAHP time course. The model assumed that the binding of four Ca ions is required for channel activation, and that the binding sites are identical and independent. The assumption of three or more Ca binding sites is supported by analogy with other Ca-binding proteins, including SK channels (Hirschberg et al., 1998).
The reaction rates were set to rb = 10 μm/sec, ru = 0.5/sec,ro = 600/sec, and rc = 400/sec. The rates were chosen based on the following three assumptions concerning the Ca-activated K channels: (1) the steady-state dose–response curve has a steep activation above the resting [Ca2+]i of 50 nm and has an EC50 of 150 nm, so that it is efficiently activated by small increases in [Ca2+]i; (2) when [Ca2+]i falls rapidly, the decay of sIAHP is limited by the channel closing and Ca dissociation rates to give a time constant of ∼1.5 sec; and (3) the peak open probability of the channel is ∼0.6, and its mean open time is 2.5 msec based on estimates from noise analysis of sIAHP (Sah and Isaacson, 1995). The technique for calculating the current time course for a given reaction scheme and agonist concentration transient has been described previously (Benveniste et al., 1990). The software implementing the kinetic model was written as a set of custom programs within AxoGraph (Axon Instruments).
The kinetic model was driven with [Ca2+]i transients calculated from the dendritic ΔF/F fluorescence measurements in the apical dendrite over the region 50–100 μm from the soma. Previous studies suggest that the channels underlying sIAHP are concentrated in this region (Sah and Bekkers, 1996). The fluorescence transients were converted to absolute [Ca2+]i based on the measured Ca affinity of Oregon Green. The Kd is ∼170 nm, and the fluorescence at saturating levels of [Ca2+]i is 14 times fluorescence in zero [Ca2+]i (Haugland, 1996). The fluorescence-to-concentration conversion also requires the resting [Ca2+]i level, which has been estimated at 50 nm (Schiller et al., 1995). Thus,
RESULTS
Whole-cell patch clamp recordings were obtained from the somata of CA1 pyramidal neurons. The fluorescence change in a given region, relative to the resting fluorescence in that region (ΔF/F), provides an index of [Ca2+]i and permits comparison between different intracellular regions. The sIAHP, evoked in response to a 50 msec voltage step from −50 to +10 mV, and ΔF/Ftransients simultaneously recorded from the soma, 66, 100, and 165 μm from the soma in one cell, are shown in Figure1. The calcium transients indicate that [Ca2+]i peaks at a higher level and decays more rapidly in the dendrites than in the soma, consistent with previous reports (Jaffe et al., 1992; Markram et al., 1995). After the voltage step, sIAHP rises slowly, peaks at several hundred milliseconds, and then decays over several seconds. When the sIAHP and associated calcium transients are normalized and superimposed (Fig. 1B), [Ca2+]i peaks before sIAHP in all regions. At the soma, the time constant of decay for ΔF/F is similar to that for sIAHP, but in regions distant from the soma, ΔF/F decays more rapidly than sIAHP. To quantify the kinetics of sIAHP, we fit the current with the difference of two exponentials (Fig. 1A). Under control conditions, the rising phase had a time constant of 233 ± 35 msec, whereas the decay had a time constant of 1.46 ± 0.16 sec (n = 10). In comparison, the decay of the ΔF/F transient in non-nuclear somatic regions had a time constant of 1.5 ± 0.11 sec (n = 11). In regions 10–50 μm from the soma, the time constant of decay was 614 ± 51 msec (n = 16); 50–100 μm from the soma it was 384 ± 35 msec (n = 16); 100–150 μm from the soma it was 283 ± 32 msec; and 150–200 μm from the soma it was ∼167 msec (n = 2).
Fig. 1.
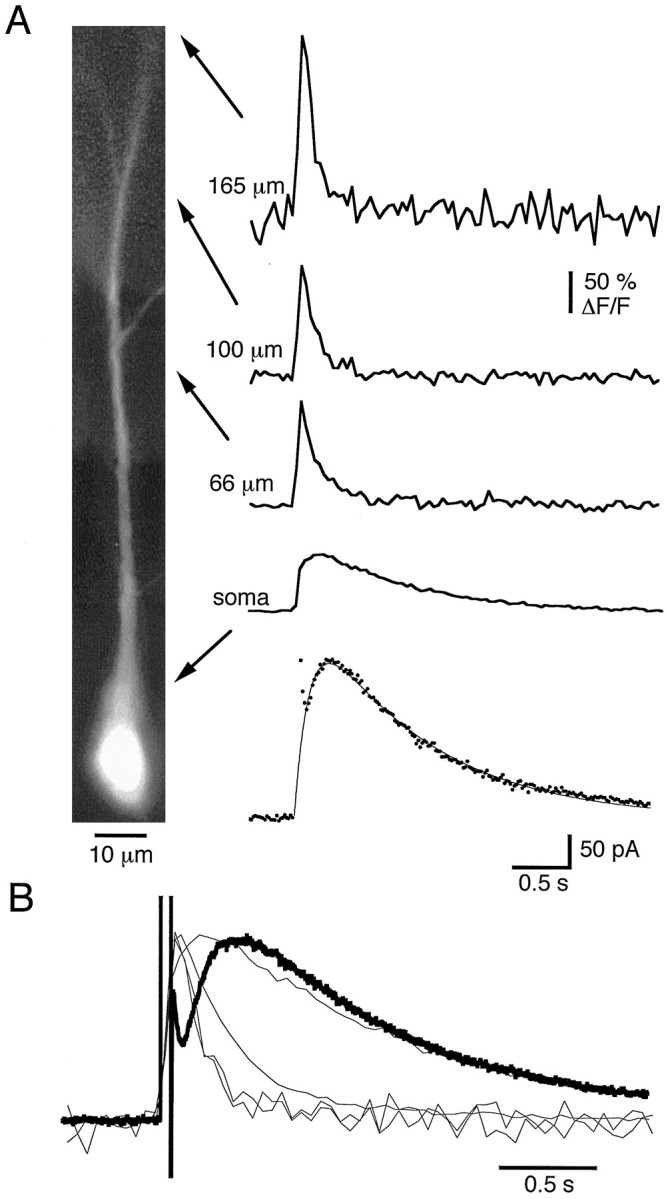
Calcium transients associated with the slow afterhyperpolarization in a pyramidal neuron. A, Photomontage of a CA1 pyramidal cell filled with 50 μmOregon Green. The bottom trace shows the membrane current (sIAHP) in response to a 50 msec voltage step from −50 to +10 mV. The smooth lineis the best fit to a difference of two exponentials (a*exp(−t/τ1) −a*exp(−t/τ2)) with τ1 = 1.2 sec and τ2 = 180 msec. Simultaneously recorded calcium transients measured at the indicated locations are shown as ΔF/F.B, The sIAHP and the associated calcium transients have been normalized and superimposed.
Both sIAHP and the associated calcium transients were markedly reduced by addition of cadmium (100 μm;n = 3) to the external solution (Fig.2A), showing that both are attributable to calcium influx via voltage-gated calcium channels. An identifying characteristic of sIAHP is its negative modulation by a range of transmitter systems (Sah, 1996). This modulation is thought to be downstream of calcium influx and may be attributable to phosphorylation of the underlying channels (Nicoll, 1988; Knopfel et al., 1990). Consistent with this, addition of the β-adrenergic agonist isoprenaline (20 μm;n = 3) or the muscarinic agonist carbachol (10–20 μm; n = 5) inhibited sIAHP without reducing the associated calcium transients (Fig. 2B).
Fig. 2.

Cadmium blocks both the calcium transient and sIAHP, but isoprenaline only reduces sIAHP. A, The sIAHP (top traces) and the associated calcium transients (bottom traces) in response to a 150 msec voltage step from −50 to +10 mV are shown in control Ringer’s solution and after addition of 100 μmcadmium. Records obtained in cadmium are indicated with anasterisk. B, The sIAHP (top traces) and the associated calcium transients (bottom traces) in response to a 100 msec voltage step from −50 to +10 mV are shown in control Ringer’s solution and after addition of 10 μmisoprenaline. Records obtained in the presence of isoprenaline are indicated with an asterisk.
Potassium currents activated by uncaging calcium
What is the reason for the slow rising phase of sIAHP? One suggestion is that it may be attributable to diffusion of calcium to the K channels from its site of influx (Lancaster and Zucker, 1994). This hypothesis requires that the K channels are well separated from the site of calcium influx. Furthermore, it implies that a fast increase in [Ca2+]i delivered close to the K channels will activate them more rapidly. To test this idea we used photorelease of calcium from DM-Nitrophen to raise [Ca2+]i rapidly throughout the cell. DM-Nitrophen binds Ca2+ with an affinity of 5 nm in its unphotolysed state. After photolysis, itsKd for Ca2+ increases to 3 mm, thereby releasing bound calcium. DM-Nitrophen was 60–70% loaded with calcium in the absence of Mg2+and introduced into the cell at a concentration of 2 mm. To confirm that uncaging DM-Nitrophen did raise intracellular calcium rapidly, we simultaneously measured free calcium levels by including Oregon Green in the recording pipette. Apamin (100 nm) was added to the bath to block SK channels. The addition of DM-Nitrophen did not markedly affect intracellular calcium dynamics. A voltage step delivered to the soma activated sIAHP, and the associated calcium transients were similar to those recorded under control conditions (Fig.3A,B, compare with Fig.1A). After uncaging DM-Nitrophen with a UV flash (see Materials and Methods), [Ca2+]iincreased rapidly throughout the cell (Fig. 3D). Despite this, an outward current with slow activation kinetics was observed (Fig. 3C; n = 9). The outward current was fit with the difference of two exponentials and had an activation time constant of 183 ± 13 msec (n = 5), similar to the time constant of activation for sIAHP after a voltage step (233 msec). Thus, even when free calcium is increased rapidly, the channels underlying sIAHP activate slowly.
Fig. 3.

sIAHP activates slowly after photorelease of calcium from DM-Nitrophen. A, sIAHP recorded after a depolarizing voltage step from a holding potential of −50 mV. B, Simultaneous recording of calcium transients in the same neuron.C, Membrane current recorded after photolysis of DM-Nitrophen. D, Simultaneous recording of calcium transients. Apamin (100 nm) was present throughout.
When apamin was not present, DM-Nitrophen photolysis produced a rapidly activating outward current, which rose with a time constant of 15 ± 6 msec (n = 9; Fig.4A,B). This result indicates that apamin-sensitive, calcium-activated potassium channels are expressed in hippocampal pyramidal neurons and can be rapidly activated after photolysis of caged calcium (Gurney et al., 1987). The amplitude of the apamin-sensitive current activated by photolysis of DM-Nitrophen was generally larger than that of the apamin-insensitive current (Fig. 4A, compare with C). In nine cells, at a holding potential of −50 mV, the apamin-sensitive current had an amplitude of 128 ± 22 pA, whereas the amplitude of the apamin-insensitive current was 55 ± 8 pA. It is generally thought that apamin-sensitive potassium channels do not contribute to the afterhyperpolarization in hippocampal pyramidal neurons (Lancaster and Nicoll, 1987; Storm, 1989). However, activation of these channels by calcium entry via voltage-activated calcium channels could also be demonstrated after long (150–200 msec) depolarizing voltage steps to +10 mV (Fig. 4C) (Oh et al., 1997; Norris et al., 1998). The amplitude of the apamin-sensitive current activated by a voltage step was variable from cell to cell, ranging from 30 to 200 pA. With shorter voltage steps (50–150 msec), the apamin-sensitive component was small, and the outward current was dominated by the apamin-insensitive sIAHP. The relatively large amplitude of the apamin-sensitive current evoked by photolysis of DM-Nitrophen may be attributable to the higher peak [Ca2+]i levels that can be attained after uncaging of DM-Nitrophen (see Discussion).
Fig. 4.

Apamin-sensitive outward current in CA1 pyramidal neurons. A, Membrane currents after photolysis of DM-Nitrophen before and after (asterisk) addition of 100 nm apamin. B, Same traces as inA, but the traces have been normalized and superimposed.C, Slow outward currents generated in response to the 200 msec voltage step from a holding potential of −50 mV before and after (asterisk) addition of apamin.
If the slow, apamin-insensitive component of the outward current after a UV flash is attributable to activation of sIAHP channels, it should be inhibited by activation of second messenger pathways activated by adrenergic, serotonergic, and muscarinic receptors (Nicoll, 1988; Pedarzani and Storm, 1993). To test this, we loaded cells with DM-Nitrophen and ensured that sIAHP activated by a voltage step could be blocked by application of serotonin (10 μm) (Fig. 5A), which activates protein kinase A (Pedarzani and Storm, 1993). Photolysis of DM-Nitrophen in the presence of serotonin only activated a fast outward current (Fig. 5B), which could be entirely blocked by apamin (data not shown), indicating that this current is not blocked by activation of these second messenger systems. Serotonin was then washed out, and recovery of sIAHP was confirmed (Fig.5A). Subsequent photolysis of DM-Nitrophen activated a slow outward apamin-insensitive current (Fig. 5B;n = 2). Similar results were obtained with isoprenaline (10 μm; n = 3), which activates β-adrenergic receptors, and carbachol (10–20 μm;n = 3), which activates muscarinic acetylcholine receptors. In summary, the slow apamin-insensitive current generated in response to DM-Nitrophen photolysis was inhibited by activating second messenger systems, which are known to block sIAHP. Furthermore, these results are consistent with the suggestion that modulation of sIAHP is downstream from the rise in free calcium (Knopfel et al., 1990; Muller and Connor, 1991).
Fig. 5.

The slow outward current activated with flash photolysis is reduced by application of 5-HT. A, Membrane currents in response to a 100 msec voltage step from −50 to +10 mV are shown in control Ringer’s solution (control), in the presence of 10 μm5-HT (asterisk), and after washout of 5-HT (wash). B, The membrane current in response to photorelease of calcium is shown during application of 5-HT (asterisk) and after its washout (wash).
Effect of calcium buffers
Loading cells with low concentrations of calcium chelators slowed the kinetics of sIAHP, as previously reported (Schwindt et al., 1992b; Zhang et al., 1996). In cells loaded with 2 mm EGTA, the rising phase of sIAHP was slowed to 633 ± 130 msec, whereas the decay was 3.5 ± 0.7 sec (n = 4) (Fig.6A). Similar effects were seen when cells were loaded with either BAPTA (1 mm) or EDTA (2 mm). In cells loaded with calcium chelators, the ΔF/F transients decayed with two time constants. At the soma, the fast component was very small, and the slow component had a time constant of 2.9 ± 0.3 sec. In regions farther from the soma the two components were more easily separable (Fig. 6B). In the region 10–50 μm from the soma, [Ca2+]i decayed with time constants of 284 ± 119 msec and 1.8 ± 0.5 sec, and in the region 50–100 μm from the soma, the two time constants were 89 ± 26 and 804 ± 180 msec (n = 4). The discrepancy between the peak of the calcium transient and that of the sIAHP was accentuated in the presence of calcium buffers, and it was clear that [Ca2+]ipeaked well before sIAHP (Jahromi et al., 1999).
Fig. 6.

Effect of intracellular EGTA on sIAHP and calcium transients.A, sIAHP recorded from a cell loaded with 2 mm EGTA in response to a depolarizing voltage step from a holding potential of −50 mV. B, Simultaneous recording of calcium transients at the indicated locations. C, The sIAHP and calcium transients fromA and B have been normalized and superimposed. D, The calcium transient recorded at 70 μm from the soma is shown in greater detail, together with exponential fits to the fast and slow components.
Effects of caged BAPTA, diazo-2
To examine the effect of rapidly chelating calcium on sIAHP we loaded cells with diazo-2. Diazo-2 binds Ca2+ with a Kd of 2.2 μm in its unphotolyzed state. After photolysis,Kd for Ca2+ increases to 70 nm. Thus, diazo-2 acts as a poor calcium buffer in its unphotolyzed state, and it becomes a strong calcium buffer after photolysis. As diazo-2 diffused into pyramidal neurons a small outward current was gradually activated, there was a reduction in peak amplitude of sIAHP, and the time course of sIAHP was slowed (Fig.7A). In cells loaded with 2 mm diazo-2 for 5 min, the time constant for the activation of sIAHP was 554 ± 97 msec, and the decay time constant was prolonged to 3.43 ± 0.53 sec (n= 11). These effects are likely attributable to the calcium-buffering properties of diazo-2. Uncaging of diazo-2 during the decay phase of the sIAHP rapidly (<50 msec) clamped [Ca2+]i to baseline or below baseline levels (Fig. 7D) and accelerated the decay of sIAHP (Fig. 7C). The time constant of decay of sIAHP after photolysis of diazo-2 was 1.65 ± 0.04 sec (n = 16). This is very similar to the decay time constant for sIAHP under control conditions (1.46 ± 0.16 sec). In no instance was a rapid deactivation of sIAHP seen after photolysis of diazo-2. However, in some cells a sudden increase in the amplitude of the outward current was observed. Because diazo-2 does not discriminate very well between calcium and magnesium, this increase may reflect a transient reduction in intracellular [Mg2+]i (Lancaster et al., 1991). Loading cells with the control compound diazo-3 (2 mm) had no effects on sIAHP either during loading or after a UV flash (n = 3; data not shown).
Fig. 7.

Rapid chelation of [Ca2+]i does not shut off sIAHP. A, sIAHP recorded from a cell loaded with 2 mm diazo-2. B, Simultaneous recording of calcium transients from the indicated locations. C, When diazo-2 was photolyzed at the peak of sIAHP, its decay was accelerated.D, Simultaneous recording of calcium transients demonstrates photolysis of diazo-2 clamped [Ca2+]i to baseline or below baseline levels in all neuronal compartments.
DISCUSSION
In this study we have examined the relationship between intracellular calcium and the slow apamin-insensitive, calcium-activated potassium current in hippocampal pyramidal neurons. Under physiological conditions, this current is activated by calcium entering the neuron via voltage-gated calcium channels and is the primary cause of the pronounced spike frequency adaptation seen in these cells (Madison and Nicoll, 1984). The definitive features of this current are (1) its slow activation and decay and (2) its characteristic modulation by a range of second messenger-coupled neurotransmitter systems (Sah, 1996). In CA1 pyramidal cells, it has been argued that this current may be restricted to the apical dendritic tree (Sah and Bekkers, 1996).
After calcium influx, intracellular free calcium rises to a peak within 20–50 msec (see also (Jaffe et al., 1992; Markram et al., 1995). In contrast, sIAHP activates with a time constant of 233 msec. Calcium transients at the soma decay with kinetics similar to the decay of sIAHP. However, in the apical dendritic tree, where sIAHP channels are thought to be located (Andreason and Lambert, 1995; Sah and Bekkers, 1996), free calcium decays at a rate severalfold faster than sIAHP. Similar observations have also recently been reported by Jahromi et al. (1999), and the mismatch between the kinetics of calcium transients and sIAHP has also been noted in vagal neurons (Lasser-Ross et al., 1997; Moore et al., 1998). In contrast to the apamin-insensitive current, activation of the apamin-sensitive potassium current is fast and appears to follow changes in [Ca2+]i (Fig. 4) (Gurney et al., 1987).
Rapid release of calcium by photolysis of DM-Nitrophen activated an outward current with kinetics and pharmacology similar to sIAHP. Thus, sIAHPchannels activate slowly, even when [Ca2+]i increases rapidly. Furthermore, rapid chelation of intracellular calcium did not immediately shut off sIAHP. Instead, sIAHP decayed as slowly as in control conditions. We can be confident that the photolysis of caged compounds altered [Ca2+]i in the expected ways, because we simultaneously measured it with fluorescence imaging. Our results are not consistent with those of Lancaster and Zucker (1994), who suggested that the sIAHP current could be rapidly activated after release of caged calcium and rapidly deactivated after calcium chelation. Our use of apamin to exclude contributions from apamin-sensitive potassium currents may explain the discrepancies between our results and those of Lancaster and Zucker (1994).
Several hypothesis have been considered to explain the slow time course of sIAHP. (1) Activation of sIAHPmay be slow because of the diffusion of calcium to a site distant from its point of influx (Lancaster and Zucker, 1994; Zhang et al., 1996). This seems unlikely given that, during depolarization, calcium rises quickly in all parts of the cell. Furthermore, the rising phase of sIAHP has a high temperature sensitivity, making it unlikely that it reflects diffusion of calcium to a distant site. The temperature sensitivity may arise from the binding of calcium to an endogenous buffer; however, the existence of buffers with temperature-sensitive properties has not been demonstrated in CA1 pyramidal neurons. Finally, in the presence of an exogenous mobile calcium buffer such as EGTA, we would expect the delivery of calcium to be accelerated (Irving et al., 1990). In fact, the rising phase is slower in the presence of EGTA. (2) The slow time course of sIAHP may be attributable to calcium-induced calcium release (CICR) (Sah and McLachlan, 1991). This mechanism appears to hold for sensory vagal neurons (Moore et al., 1998). Although CICR has been suggested to play a role in activating sIAHP in CA3 pyramidal neurons in slice cultures (Tanabe et al., 1998), there is no evidence for CICR in CA1 pyramidal neurons in situ (Zhang et al., 1996). (3) The channels underlying sIAHP may not be gated by calcium directly but may require some second messenger for its activation (Schwindt et al., 1992a; Lasser-Ross et al., 1997; Moore et al., 1998). This also seems unlikely, because there is no delay between the uncaging of calcium and the foot of the potassium current. Second messenger-mediated responses typically exhibit latencies of >20 msec to the foot of the response (Sodickson and Bean, 1996). (4) The slow activation of sIAHP may be attributable to delayed facilitation of the calcium channels, which supply the calcium to activate sIAHP (Cloues et al., 1997; Marrion and Tavalin, 1998). This hypothesis requires that the K channels underlying sIAHP respond rapidly to changes in [Ca2+]i. However, our results clearly demonstrate that this is not the case. (5) The channels underlying sIAHP may have intrinsically slow activation and inactivation kinetics (Hocherman et al., 1992; Schwindt et al., 1992a;Sah, 1993). We have explored this final possibility by constructing a kinetic model of a potassium channel that is gated by the binding of intracellular calcium to a high-affinity site (see Materials and Methods). Model channels were exposed to the [Ca2+]i transients recorded in the apical dendrite under several different experimental conditions. The predicted current transients reproduced all of our main findings, indicating that such channels could underlie the sIAHP.
The average ΔF/F time course recorded in the apical dendrite 50–100 μm from the soma was converted to a [Ca2+]i transient as described in Materials and Methods. The [Ca2+]itransient peaked at ∼850 nm and then decayed back to the resting level of 50 nm. This [Ca2+]i transient was applied to the model channels and produced a theoretical sIAHPwith kinetics similar to the recorded current (Fig.8A,B). The activation time constant was 250 msec, and the decay time constant was 1.6 sec (cf. 230 msec and 1.5 sec for the recorded current).
Fig. 8.

The experimental data are reproduced well by a simple kinetic scheme. The sIAHP was modeled using a simple Markov scheme as described in Materials and Methods. A, Observed calcium transient in the proximal dendritic tree (light trace) and the theoretical potassium current in response to the calcium transient.B, sIAHP recorded in response to a voltage step, for comparison with A. The activation time constant of the model sIAHP was 250 msec, and the decay time constant was 1.6 sec. C, In the presence of EGTA the model predicts that both the rise and decay of the theoretical potassium current will be slowed relative to control. The model current had an activation time constant of 690 msec and decay time constant of 2.2 sec. D, sIAHPrecorded in the presence of EGTA for comparison with C.E, Rapidly clamping calcium to baseline levels at the peak of the potassium current accelerates the decay of the theoretical potassium current. F, Representative records after photolysis of diazo-2. G, Reducing the probability of channel opening reduces the amplitude of the theoretical potassium current and accelerates its decay. H, sIAHPs recorded in control Ringer’s solution and in the presence of isoprenaline. The decay time constants were 1.13 sec in control and 0.835 sec in isoprenaline.
The experimental sIAHP activated more rapidly after flash photolysis of DM-Nitrophen (180 msec) than after a voltage step (230 msec). This may be a result of a higher level of [Ca2+]i produced by DM-Nitrophen photolysis. It has been estimated that [Ca2+]i peaks briefly at tens of micromolar concentrations immediately after photolysis (Zucker, 1993). The [Ca2+]i decay after DM-Nitrophen photolysis was modeled by increasing the peak of the ΔF/F transient from 190 to 240%, which produced a [Ca2+]i transient that peaked briefly at 3.3 μm. The resulting theoretical current was fit with the difference of two exponentials. The activation time constant was 180 msec, and the decay time constant was 1.5 sec (cf. 180 msec and 1.7 sec for the recorded current after DM-Nitrophen photolysis).
When EGTA (2 mm) was included in the patch pipette, it reduced the peak amplitude of the dendritic [Ca2+]i transient to 600 nm and slowed the decay. The time constant of the slow component could not be measured accurately because of bleaching. However, a double-exponential fit to the first few seconds of the observed transient suggested a lower limit of ∼1.2 sec for the slow component. This value is consistent with the measured time constant of 1.15 sec for Ca unbinding from EGTA in an in vitro system (Neher, 1986). The actual Ca clearance rate from a dendrite will be slower than this because of rebinding of calcium to EGTA. The model K channels were driven with a [Ca2+]itransient calculated from a double-exponential fluorescence transient with time constants of 90 msec and 1.5 sec. This [Ca2+]i transient produced a theoretical current with slower activation and inactivation kinetics (Fig. 8C,D). The model current had an activation time constant of 690 msec and a decay time constant of 2.2 sec, similar to the values of 633 msec and 3.5 sec for the experimental current recorded with added EGTA.
It was assumed that the photolyzed diazo-2 clamped [Ca2+]i to 30 nm, and the resulting transient was applied to the model K channel. After photolysis, the simulated current decayed to below baseline with a time constant of 1.6 sec, compared with 1.7 sec for the recorded current transients (Fig. 8E,F).
Isoprenaline has been shown to reduce the open probability of the K current underlying sIAHP (Sah and Isaacson, 1995). This was simulated by reducing the opening rate in the kinetic model to ro = 100/sec, which reducedpo from 0.6 to 0.2. When the control [Ca2+]i transient was applied to this modified kinetic model, it produced a smaller, faster K current with a decay time constant of 996 msec. Thus, our kinetic model predicts that the sIAHP current in the presence of low concentrations of modulators should decay with a faster time constant. Indeed, when the amplitude of the sIAHP was reduced by application of isoprenaline, the decay constant of the sIAHP was 742 ± 174 msec (n = 4; Fig. 8G,H). In contrast, reducing the amplitude of sIAHP to a similar extent with Cd2+ did not alter the kinetics of the current.
Recently, small-conductance calcium-activated potassium channels have been cloned, and three genes, SK1, SK2, and SK3, have been identified (Kohler et al., 1996). When expressed in Xenopus oocytes, SK2 and SK3 form channels that are blocked by apamin, whereas SK1 forms channels that are insensitive to apamin. It has therefore been suggested that SK2 and SK3 may code for channels underlying apamin-sensitive AHP (IAHP), whereas SK1 may code for the channels underlying sIAHP. A Markov model has been proposed for the SK channels, based on single-channel recording and analysis of SK2 channels (Hirschberg et al., 1998). When the control [Ca2+]itransient was applied to a model SK channel, it produced a theoretical K current with fast activation and inactivation kinetics, similar to the apamin-sensitive current we have recorded. Furthermore, when [Ca2+]i transients evoked by voltage step or by DM-Nitrophen photolysis were applied to model SK channels in the low open probability mode, the simulated K current after DM-Nitrophen photolysis was more than eight times larger than the current after a voltage step, consistent with our experimental results.
Taken together, the above results demonstrate that a K channel that is directly gated by [Ca2+]i could underlie the sIAHP. However, the kinetic properties of this channel need to be slower than those previously reported for SK channels (Hirschberg et al., 1998). Specifically, the sIAHP channel must have slower calcium binding and unbinding rates. Our experiments cannot rule out the hypothesis that activation of sIAHP may be attributable to the generation of a second messenger after calcium influx. In this model, the time course of sIAHP would be caused by the slow generation, binding, and degradation of the putative second messenger. However, this model has difficulty explaining the faster sIAHP decay observed in the presence of modulators such as noradrenaline and 5-HT. Also, the absence of any delay between DM-Nitrophen photolysis and the foot of the K current is inconsistent with a second messenger-dependent response. Both of these results are elegantly explained by the model we have presented, suggesting that the sIAHP channels are directly gated by calcium. It has recently been reported that calmodulin is an integral component of SK channels, and gating of SK channels is mediated by binding of calcium to calmodulin (Xia et al., 1998). It is therefore possible that the slow kinetics of sIAHP may be attributable to the presence of a different calcium-binding protein interacting with SK1 channels, which confers on them slow kinetics. Alternatively, the channels underlying sIAHP may belong to a novel class of channel.
Footnotes
P.S. is a Sylvia and Charles Viertel senior medical research fellow. J.C. is supported by a Queen Elizabeth II fellowship from the Australian Research Council.
Correspondence should be addressed to Pankaj Sah, Division of Neuroscience, John Curtin School of Medical Research, G.P.O. Box 334, Canberra ACT 2601, Australia.
REFERENCES
- 1.Andreason M, Lambert JDC. The excitability of CA1 pyramidal cell dendrites is modulated by a local Ca2+-dependent K+-conductance. Brain Res. 1995;698:193–203. doi: 10.1016/0006-8993(95)00910-i. [DOI] [PubMed] [Google Scholar]
- 2.Benveniste M, Clements J, Vyklicky L, Jr, Mayer ML. A kinetic analysis of the modulation of N-methyl-d-aspartic acid receptors by glycine in mouse cultured hippocampal neurones. J Physiol (Lond) 1990;428:333–357. doi: 10.1113/jphysiol.1990.sp018215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cloues RK, Tavalin SJ, Marrion NV. B-adrenergic stimulation selectively inhibits long-lasting L-type calcium channel facilitation in hippocampal pyramidal neurons. J Neurosci. 1997;7:6493–6503. doi: 10.1523/JNEUROSCI.17-17-06493.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gurney AL, Tsien RY, Lester HA. Activation of a potassium current by rapid photochemically generated step increases of intracellular calcium in rat sympathetic neurons. Proc Natl Acad Sci USA. 1987;84:3496–3500. doi: 10.1073/pnas.84.10.3496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Haugland RP. Handbook of fluorescent probes and research chemicals. Molecular Probes; Eugene, OR: 1996. [Google Scholar]
- 6.Hirschberg B, Maylie J, Adelman JP, Marrion NV. Gating of recombinant small-conductance Ca-activated K+ channels by calcium. J Gen Physiol. 1998;111:565–581. doi: 10.1085/jgp.111.4.565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hocherman SD, Werman R, Yarom Y. An analysis of the long-lasting after-hyperpolarization of guinea-pig vagal motoneurones. J Physiol (Lond) 1992;456:325–349. doi: 10.1113/jphysiol.1992.sp019339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Irving M, Maylie J, Sizto NL, Chandler WK. Intracellular diffusion in the presence of mobile buffers. Application to proton movement in muscle. Biophys J. 1990;57:717–721. doi: 10.1016/S0006-3495(90)82592-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jaffe DB, Johnston D, Lasser-Ross N, Lisman JE, Miyakawa H, Ross WN. The spread of Na+ spikes determines the pattern of dendritic Ca2+ entry into hippocampal neurons. Nature. 1992;357:244–246. doi: 10.1038/357244a0. [DOI] [PubMed] [Google Scholar]
- 10.Jahromi BS, Zhang L, Carlen PL, Pennefather P. Differential time-course of slow afterhyperpolarizations and associated Ca2+ transients in rat CA1 pyramidal neurons: further dissociation by Ca2+ buffer. Neuroscience. 1999;88:719–726. doi: 10.1016/s0306-4522(98)00203-6. [DOI] [PubMed] [Google Scholar]
- 11.Knopfel T, Vranesic I, Gahwiler BH, Brown DA. Muscarinic and β-adrenergic depression of the slow Ca2+ activated potassium conductance in hippocampal CA3 pyramidal cells is not mediated by a reduction of the depolarization-induced cytosolic Ca2+ transients. Proc Natl Acad Sci USA. 1990;87:4083–4087. doi: 10.1073/pnas.87.11.4083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kohler M, Hischberg B, Bond CT, Kinzie JM, Marrion NV, Maylie J, Adelman JP. Small-conductance, calcium-activated potassium channels from mammalian brain. Science. 1996;273:1709–1714. doi: 10.1126/science.273.5282.1709. [DOI] [PubMed] [Google Scholar]
- 13.Lancaster B, Nicoll RA. Properties of two calcium-activated hyperpolarizations in rat hippocampal neurones. J Physiol (Lond) 1987;389:187–204. doi: 10.1113/jphysiol.1987.sp016653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lancaster B, Zucker RS. Photolytic manipulation of Ca2+ and the time course of slow, Ca2+-activated potassium current in rat hippocampal neurones. J Physiol (Lond) 1994;475:229–239. doi: 10.1113/jphysiol.1994.sp020064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lancaster B, Nicoll RA, Perkel DJ. Calcium activates two types of potassium channels in rat hippocampal neurons in culture. J Neurosci. 1991;11:23–32. doi: 10.1523/JNEUROSCI.11-01-00023.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lasser-Ross N, Ross W, Yarom Y. Activity-dependent [Ca2+]i changes in guinea pig vagal motoneurons: relationship to the slow afterhyperpolarization. J Neurophysiol. 1997;78:825–834. doi: 10.1152/jn.1997.78.2.825. [DOI] [PubMed] [Google Scholar]
- 17.Madison DV, Nicoll RA. Control of repetitive discharge of rat CA1 pyramidal neurones in vitro. J Physiol (Lond) 1984;354:319–331. doi: 10.1113/jphysiol.1984.sp015378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Markram H, Helm PJ, Sakmann B. Dendritic calcium transients evoked by single back-propagating action potentials in rat neocortical pyramidal neurons. J Physiol (Lond) 1995;485:1–20. doi: 10.1113/jphysiol.1995.sp020708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Marrion NV, Tavalin SJ. Selective activation of Ca2+-activated K+ channels by co-localised Ca2+ channels in hippocampal neurons. Nature. 1998;395:900–905. doi: 10.1038/27674. [DOI] [PubMed] [Google Scholar]
- 20.Marty A. The physiological role of calcium-dependent channels. Trends Neurosci. 1989;12:420–425. doi: 10.1016/0166-2236(89)90090-8. [DOI] [PubMed] [Google Scholar]
- 21.Moore KA, Cohen AS, Kao JPY, Weinreich D. Ca2+-induced Ca2+ release mediates a slow post-spike hyperpolarization in rabbit vagal afferent neurons. J Neurophysiol. 1998;79:688–694. doi: 10.1152/jn.1998.79.2.688. [DOI] [PubMed] [Google Scholar]
- 22.Muller W, Connor JA. Cholinergic input uncouples Ca2+ changes from K+ conductance activation and amplifies intradendritic Ca2+ changes om hippocampal neurons. Neuron. 1991;6:901–905. doi: 10.1016/0896-6273(91)90230-w. [DOI] [PubMed] [Google Scholar]
- 23.Neher E. Concentration profiles of intracellular calcium in the presence of a diffusible chelator. In: Heinemann U, Klee U, Neher E, Singer W, editors. Calcium electrogenesis and neuronal functioning. Berlin; Springer: 1986. pp. 80–96. [Google Scholar]
- 24.Nicoll RA. The coupling of neurotransmitter receptors to ion channels in the brain. Science. 1988;241:545–551. doi: 10.1126/science.2456612. [DOI] [PubMed] [Google Scholar]
- 25.Norris CM, Halpain S, Foster TC. Reversal of age-related alterations in synaptic plasticity by blockade of L-type Ca2+ channels. J Neurosci. 1998;18:3171–3179. doi: 10.1523/JNEUROSCI.18-09-03171.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Oh MM, Power JM, Disterhoft JF. Apamin decreases the afterhyperpolarization in rabbit CA1 neurons. Soc Neurosci Abstr. 1997;17:1745. [Google Scholar]
- 27.Pedarzani P, Storm JF. PKA mediates the effects of monoamine transmitters on the K+ current underlying the slow spike frequency adaptation in hippocampal neurons. Neuron. 1993;11:1023–1035. doi: 10.1016/0896-6273(93)90216-e. [DOI] [PubMed] [Google Scholar]
- 28.Pennefather P, Lancaster B, Adams PR, Nicoll RA. Two distinct Ca-dependent K currents in bullfrog sympathetic ganglionic cells. Proc Natl Acad Sci USA. 1985;82:3040–3044. doi: 10.1073/pnas.82.9.3040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sah P. Kinetic properties of a slow apamin insensitive Ca2+ activated K+ current in guinea pig vagal neurones. J Neurophysiol. 1993;69:361–366. doi: 10.1152/jn.1993.69.2.361. [DOI] [PubMed] [Google Scholar]
- 30.Sah P. Calcium2+-activated K+ currents in neurones: types, physiological roles and modulation. Trends Neurosci. 1996;19:150–154. doi: 10.1016/s0166-2236(96)80026-9. [DOI] [PubMed] [Google Scholar]
- 31.Sah P, Bekkers JM. Apical dendritic location of slow-afterhyperpolarization current in hippocampal pyramidal neurons: implications for the integration of LTP. J Neurosci. 1996;16:4537–4542. doi: 10.1523/JNEUROSCI.16-15-04537.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sah P, Isaacson JS. Channels underlying the slow afterhyperpolarization in hippocampal pyramidal neurons: neurotransmitters modulate the open probability. Neuron. 1995;15:435–441. doi: 10.1016/0896-6273(95)90047-0. [DOI] [PubMed] [Google Scholar]
- 33.Sah P, McLachlan EM. Ca2+ activated K+ currents underlying the afterhyperpolarization in guinea pig vagal neurons: a role for Ca2+ activated Ca2+ release. Neuron. 1991;7:257–264. doi: 10.1016/0896-6273(91)90264-z. [DOI] [PubMed] [Google Scholar]
- 34.Schiller J, Helmchen F, Sakmann B. Spatial profile of dendritic calcium transients evoked by action potentials in rat neocortical pyramidal neurones. J Physiol (Lond) 1995;487:583–600. doi: 10.1113/jphysiol.1995.sp020902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Schwindt PC, Spain WJ, Crill WE. Calcium-dependent potassium currents in neurones from cat sensorimotor cortex. J Neurophysiol. 1992a;67:216–226. doi: 10.1152/jn.1992.67.1.216. [DOI] [PubMed] [Google Scholar]
- 36.Schwindt PC, Spain WJ, Crill WE. Effects of intracellular calcium chelation on voltage-dependent and calcium-dependent currents in cat neocortcal neurons. Neuroscience. 1992b;47:571–578. doi: 10.1016/0306-4522(92)90166-y. [DOI] [PubMed] [Google Scholar]
- 37.Sodickson DL, Bean BP. GABAB receptor-activated inwardly rectifying potassium current in dissociated hippocampal CA3 neurons. J Neurosci. 1996;16:6374–6385. doi: 10.1523/JNEUROSCI.16-20-06374.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Storm JF. An after-hyperpolarization of medium duration in rat hippocampal pyramidal cells. J Physiol (Lond) 1989;409:171–190. doi: 10.1113/jphysiol.1989.sp017491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Tanabe M, Gahwiler BH, Gerber U. L-type Ca2+ channels mediate the slow Ca2+-dependent afterhyperpolarization current in rat CA3 pyramidal cells in vitro. J Neurophysiol. 1998;80:2268–2273. doi: 10.1152/jn.1998.80.5.2268. [DOI] [PubMed] [Google Scholar]
- 40.Vegara C, Latorre R, Marrion NV, Adelman JP. Calcium-activated potassium channels. Curr Opin Neurobiol. 1998;8:321–329. doi: 10.1016/s0959-4388(98)80056-1. [DOI] [PubMed] [Google Scholar]
- 41.Xia X-M, Falker B, Rivard A, Wayman G, Johnson-Pais T, Keen JE, Ishii T, Hirschberg B, Bond CT, Lutsenko S, Maylie J, Adelman JP. Mechanism of calcium gating in small-conductance calcium-activated potassium channels. Nature. 1998;395:503–507. doi: 10.1038/26758. [DOI] [PubMed] [Google Scholar]
- 42.Zhang L, Pennefather P, Velumian A, Tymianski M, Charlton M, Carlen PL. Potentiation of a slow Ca2+-dependent K+ current by intracellular Ca2+ chelators in hippocampal CA1 neurons of rat brain slices. J Neurophysiol. 1996;74:2225–2241. doi: 10.1152/jn.1995.74.6.2225. [DOI] [PubMed] [Google Scholar]
- 43.Zucker RS. The calcium concentration clamp: spikes and reversible pulses using the photolabile chelator DM-nitrophen. Cell Calcium. 1993;14:87–100. doi: 10.1016/0143-4160(93)90079-l. [DOI] [PubMed] [Google Scholar]