

Abstract
Electrical stimulation of the cerebellar fastigial nucleus (FN) elevates regional cerebral blood flow (rCBF) and arterial pressure (AP) and provides long-lasting protection against focal and global ischemic infarctions. We investigated which neuronal element in FN, perikarya or axons, mediates this central neurogenic neuroprotection and whether it also protects against excitotoxicity. In anesthetized rats, the FN was stimulated for 1 hr, and ibotenic acid (IBO) was microinjected unilaterally into the striatum. In unstimulated controls, the excitotoxic lesions averaged ∼40 mm3. Stimulation of FN, but not dentate nucleus (DN), significantly reduced lesion volumes up to 80% when IBO was injected 15 min, 72 hr, or 10 d, but not 30 d, thereafter. In other rats, intrinsic neurons of FN or DN were destroyed by pretreatment with IBO. Five days later, the FN was stimulated, and 72 hr later, IBO was microinjected into the striatum. Lesions of FN, but not DN, abolished neuroprotection but not the elevations of rCBF and AP elicited from FN stimulation. Excitotoxic lesions of FN, but not DN, also abolished the 37% reduction in focal ischemic infarctions produced by middle cerebral artery occlusion. Excitation of intrinsic FN neurons provides long-lasting, substantial, and reversible protection of central neurons from excitotoxicity, as well as focal ischemia, whereas axons in the nucleus, probably collaterals of ramified brainstem neurons, mediate the elevations in rCBF, which do not contribute to neuroprotection. Long-lived protection against a range of injuries is an unrecognized function of FN neurons transmitted over pathways distinct from those regulating rCBF.
Keywords: fastigial nucleus, excitotoxicity, focal cerebral ischemia, neuroprotection, cerebral blood flow, cerebellum
In rat, electrical stimulation of the cerebellar fastigial nucleus (FN) for 1 hr reduces, by ∼50%, the volume of a focal ischemic infarction produced by occlusion of the middle cerebral artery (MCA) (Reis et al., 1991, 1998; Zhang and Iadecola, 1993; Golanov et al., 1996; Galea et al., 1998a). Stimulation also salvages over 50% of the hippocampal pyramidal neurons of the CA1 region, which undergo delayed degeneration after global cerebral ischemia (Golanov et al., 1998). The effects of FN stimulation are long lasting and reversible (Reis et al., 1998). The mechanism mediating this central neurogenic neuroprotection (Reis et al., 1997) is unknown but cannot be attributed to elevations of regional cerebral blood flow (rCBF) associated with FN stimulation (Yamamoto et al., 1993; Reis et al., 1998). Proposed mechanisms include a reduction in immune reactivity of cerebral microvessels (Galea et al., 1998a,b) and reduced neuronal excitability (Golanov and Reis, 1999).
It is not known which neuronal elements within the FN are responsible for mediating the neuroprotection nor the projection pathways involved. Although electrical stimulation of the FN excites intrinsic neurons, it also activates axons projecting to or through the FN, including efferents from Purkinje cells of the overlying cerebellar vermis (De Camilli et al., 1984) and axons arising from a system of brainstem neurons with collaterals that project into the cerebellum (Deitrichs and Haines, 1985). The fact that intrinsic neurons and axons in FN may subserve different autonomic functions has been shown. Thus, whereas electrical stimulation of the FN elevates arterial pressure (AP) and rCBF but does not alter regional cerebral glucose utilization (rCGU) (Nakai et al., 1983; Golanov et al., 1996), selective stimulation of intrinsic FN neurons with excitatory amino acids lowers AP, rCBF, and rCGU (Chida et al., 1986, 1989). Moreover, although destruction of FN neurons by excitotoxins eliminates the depressor responses to chemical stimulation, it does not affect the elevations of AP and rCBF elicited electrically. Thus, intrinsic FN neurons promote sympathoinhibition, reduced cerebral metabolism, and rCBF, whereas axons innervating the region initiate sympathoexcitation and cerebrovascular vasodilation.
In the present study, we sought to determine which neuronal element in FN is responsible for neuroprotection. Because focal ischemic infarctions are large and may have functional effects which extend beyond the lesion (e.g., by “diachisis”) (Bidmon et al., 1997), including remote effects on cerebellar metabolism (Baron et al., 1984;Nagasawa et al., 1994), we first investigated whether FN stimulation would protect against excitotoxic lesions of striatum and, if so, whether such neuroprotection would persist after intrinsic neurons of the FN were destroyed. We report the following: (1) electrical stimulation of the FN results in a substantial, long-lasting, and reversible reduction in excitotoxicity; (2) chronic and selective destruction of intrinsic FN neurons abolishes the neuroprotection but preserves stimulation-elicited elevations of rCBF and AP; and (3) intrinsic FN neurons are also responsible for protection against focal ischemia. The neuronal system represented in neurons of the rostral FN and its projections mediate protection of the brain against several modes of neuronal injury.
MATERIALS AND METHODS
General procedures. All studies, except those of focal ischemia, were performed on 98 adult male Sprague Dawley rats weighing 250–450 gm. Studies of focal ischemia were performed on 34 adult rats (300–450 gm) of the spontaneously hypertensive strain. Rats were fed lab chow ad libitum and maintained in a thermally controlled (27°C), 12 hr light/dark cycle (lights on, 7.00 A.M.; lights off, 7:00 P.M.) environment. All surgical procedures were performed aseptically.
Rats were anesthetized with 1.8–2.5% halothane blown over the nose. A thin-walled polyethylene catheter was placed in the femoral artery for continuous recording of AP and to sample arterial blood gases. Pulsatile AP was monitored with a strain gauge transducer, and the signal was amplified and recorded on channels of a chart recorder. Mean arterial pressure (MAP) measurements represent the average AP recorded through the duration of the surgical procedure.
In some experiments, rCBF was measured during brain stimulation (Golanov and Reis, 1996) by laser Doppler flowmetry (LDF) using a PeriFlux PF3 (Perimed, Piscataway, NJ) flowmeter equipped with a 2 mW helium–neon laser with a wavelength of 632.8 nm. Flow values were expressed in arbitrary perfusion units (PU). The probe (tip diameter of 1.0 mm; model PF303, Perimed) was mounted on a micromanipulator (David Kopf, Tujunga, CA) and placed on the thin layer of bone remaining after shaving a 2 × 2 mm square over the parietal cortex. A drop of mineral oil was then applied to fill in the space between dura and probe. The analog output was fed into an analog-to-digital converter and chart recorder. Before probe placement, the underlying cortex was examined under magnification to define a recording site devoid of arterioles or venules, because recordings from these larger vessels may give erroneous results. Placements yielding recording perfusion measurements between 50 and 100 PU were considered suitable for recording, and the probe was then fixed in place. Cerebrovascular resistance (CVR) was calculated by dividing arterial pressure (in millimeters mercury) by flow (in perfusion units). Data are expressed as percentage of change of baseline CVR evoked by stimulation recorded at the peak rise in rCBF.
Core temperature was continuously maintained at 37 ± 0.5°C by use of heating pad electronically controlled by a rectal probe. Brain temperature was continuously measured during each experiment via a flexible thermocouple inserted into the temporalis muscle. The temperature of the temporalis muscle accurately parallels brain temperature and, therefore, has been demonstrated to be a reliable indirect estimate of brain temperature (Busto et al., 1987; Miyazawa and Hossmann, 1992). After instrumentation, rats were mounted in a stereotaxic apparatus (David Kopf) with the bite bar adjusted to −11 mm below the interaural line. Paco2, Pao2, and pH were measured by a blood-gas analyzer in 0.1 ml of arterial blood sampled at the beginning of the experiment and at the end of stimulation. Hematocrit (Hct) and plasma glucose were measured at the beginning and end of all procedures.
Electrical stimulation. The posterior cerebellar vermis and lower medulla were exposed by a small occipital craniotomy to reveal the calamus scriptorius, which was used as stereotaxic zero. A monopolar electrode, fabricated from Teflon-insulated stainless steel wire (outer diameter of 150 μm), carried in stainless steel tubing and exposed at the tip for 100 μm, was lowered into the cerebellum with a posterior inclination of 10 degrees. The area of the FN from which stimulation produces neuroprotection coincides with the area from which stimulation elevates AP and also rCBF (Reis et al., 1991; Golanov et al., 1996). To localize the most active site, the electrode was placed 5 mm rostral to, 0.8 mm lateral to, and 2 mm above stereotaxic zero. The electrode was then lowered in steps of 0.2 mm, stimulating at each site with a 5 sec train (pulses of 0.5 msec, 50 Hz; stimulus current of 10–20 μA). Once the most active site in the FN was identified (i.e., that site from which the largest rise in AP was elicited along a track), the electrode was fixed in place. The threshold current was determined by increasing simulation currents for each train in steps of 5 μA, starting from 5 μA until the increase in blood pressure reached 10 mmHg. The average threshold current required to elevate AP was 31.8 ± 1.18 μA (n = 71).
To elicit neuroprotection, the FN was stimulated with intermittent trains of pulses (1 sec on, 1 sec off; 0.5 msec pulse durations) at 50 Hz, with current intensities increasing gradually to five times threshold current. In some animals, stimulation elevated AP ∼150 mmHg. When necessary, AP was maintained below 150 mmHg, within the autoregulated range for rCBF in rat (Dirnagl and Pulsinelli, 1990), by slowly withdrawing 2–4 ml of blood into a syringe (Nakai et al., 1983) to counteract the rise in AP. Stimulation was continued for 1 hr. At the termination of stimulation, blood was reinfused. Such “controlled hemorrhage” does not modify the size of focal ischemic infarctions nor their salvage by FN stimulation. Stimulation sites were verified histologically postmortem.
To stimulate the dentate nucleus (DN), electrodes were positioned at a site 5 mm rostral, 2.5 mm lateral, and 2 mm above the stereotaxic zero and stimulated with intermittent trains of pulses (1 sec on, 1 sec off; 0.5 msec) at 50 Hz, 100 μA for 1 hr. Stimulation of DN does not alter AP and rCBF, nor does it evoke neuroprotection (Reis et al., 1991,1998). In sham-stimulated controls, the electrode was inserted 5 mm rostral to, 0.8 mm lateral to, and 1.3 mm above the calamus and left in place for 1 hr. After completion of the procedures, wounds were closed and covered with topical anesthetic, catheters were capped, anesthesia was discontinued, and animals were returned to their cages.
Microinjection of ibotenic acid into the striatum. Rats subjected to intrastriatal microinjections were, with the exception of the group treated at the time of stimulation (see below), reanesthetized with 1.8–2.5% halothane 72 hr, 10 d, or 30 d after stimulation of FN or DN. Two small burr holes were drilled in the calvarium over the head of the striatum at sites 2.7 mm lateral to and 0.7 mm rostral to bregma. The dental drill was continuously irrigated with saline at room temperature to prevent overheating of the underlying cortex. Ibotenic acid (IBO) or its vehicle [phosphate buffer (PB) 0.1 m, pH 7.3) was microinjected into the striatum through capillary glass pipettes (∼55 μm tip outer diameter). The micropipette was lowered 4.5 mm below the cortical surface. PB (360 nl) was injected into the left and IBO (23 nmol dissolved in 360 nl of PB) into the right striatum. Solutions were administered slowly by hand over 3 min, and the pipette was then left in place for 5 min to limit diffusion of IBO up the pipette track. In rats in which injections were made just after a stimulation epoch, the calvarium was prepared in advance so that the microinjection pipette could be rapidly positioned and the striata injected. After injections, wounds were closed and covered with topical anesthetic, anesthesia was discontinued, and animals were returned to their cages. Twenty-four hours later, they were deeply anesthetized with halothane and killed by decapitation.
Brains were removed, immediately frozen in liquid freon, and stored at −20°C until analysis. They were serially sectioned coronally at 20 μm thickness in a cryostat at −20°C with sections sampled every 200 μm and stained with thionin. The boundary of the lesion was defined by the sharp delineation of loss of Nissl-stained cells. The borders of the lesions were confirmed microscopically. The cross-sectional area was digitized and computed by tracing the lesion on MCID software (Imaging Research, St. Catharines, Ontario, Canada). Salvage was expressed as the percentage of decrease of lesion volume in FN-stimulated compared with the lesion volume in control (sham-stimulated) animals.
Ibotenic acid lesions of deep cerebellar nuclei. Intrinsic neurons of the FN or DN were selectively destroyed by IBO. In an initial experiment, rats were anesthetized with halothane (1.8–2.5%) and instrumented to record AP from one femoral artery. Using stereotaxic coordinates, capillary micropipettes filled with IBO or vehicle were inserted into FN or DN through burr holes placed in the calvarium, as described above. Lesions of the FN were made at six sites, three on each side. To target the fastigial nucleus, the injection pipette was first inserted (with reference to the calamus scriptorius as stereotaxic zero) at anterior 5 mm, lateral 0.8 mm, and dorsal 1.6 mm. IBO or vehicle was injected over 3 min by hand. After injections, the pipette was removed and reinserted. Additional lesions were placed ipsilaterally in FN at sites 4.8 and 4.6 mm anteriorly. The contralateral FN was then treated similarly. The total dose–injection per hemisphere was 3.89 nmol in 30 nl, for a total of 23 nmol (dissolved in 360 nl of PB).
In other animals, the DN was comparably injected bilaterally at six sites with the same volume and concentration of IBO. Microinjections into DN were made with the pipette positioned 5 mm anteriorly, 2.5 mm laterally, and 1.5 mm above calamus scriptorius, with lesions also placed at 4.8 and 4.6 mm anteriorly. In sham-lesioned controls, equal volumes of PB were microinjected into six sites in FN to mirror injections made into FN with IBO. At the completion of all procedures, wounds were closed, the femoral arterial cannula was ligated, halothane was discontinued, and animals were returned to their cages.
Five days later, rats were reanesthetized with halothane and instrumented to record AP from the remaining femoral artery, and rCBF by LDF over the parietal cortex. Stimulating electrodes were inserted into an active site of FN as described above, threshold currents were measured, and stimulation continued for 1 hr (50 Hz at five times threshold current), while recording AP and rCBF. After stimulation, wounds were closed, anesthesia was discontinued, and rats were returned to their cages.
Seventy two hours later, the rats were reanesthetized and prepared for microinjections of IBO and PB into striatum as described above. In all animals, IBO was microinjected into one striatum and saline into the other. Wounds were closed, and animals were allowed to recover. Twenty four hours later, they were deeply anesthetized with halothane and killed by decapitation, brains were removed, and the distribution and magnitude of the striatal and cerebellar lesions were determined as described.
Occlusion of the MCA. MCA occlusions were performed in spontaneously hypertensive male rats, which were used because the intersubject variability of ischemic lesion are small (Duverger and MacKenzie, 1988; Reis et al., 1991, 1998). However, the percentage of salvage elicited by FN stimulation is comparable with that of rats of the Sprague Dawley strain (Reis et al., 1991).
Six groups of 4–12 rats were studied. In an initial procedure, three of the groups received cerebellar microinjections of IBO or PBS into FN or IBO into DN, and animals were allowed to recover. Five days later, rats were reanesthetized with halothane (1.8–2.5% in 100% O2) and placed in a stereotaxic frame, and AP was recorded while maintaining body temperature and blood gases. Three additional groups were run in parallel. These rats received real or sham stimulation of FN or DN. In all groups, the MCA was exposed before stimulating the cerebellum and, immediately after stimulation, the MCA was cauterized distal to the lenticulostriatal branches by a modified method of Tamura et al. (1981). The arterial cannula was recapped, wounds were closed, and animals were returned to their cages. Twenty-four hours later, they were anesthetized and decapitated, and brains were removed, sectioned, and stained with thionin. The distribution of cerebellar lesions and distribution and volumes of the ischemic lesions were mapped and measured using MCID software (Imaging Research). Infarction volumes were corrected for edema by calculating a coefficient of the ratio of the noninfarcted versus infarcted hemispheres. Salvage was expressed as the percentage of decrease of infarction volume in FN-stimulated compared with the infarction volume in control (sham-stimulated) animals.
Statistical methods. Data are expressed as mean ± SEM. Multiple comparisons were analyzed using ANOVA and the Student’s–Newman–Keuls (SNK) tests. Differences were considered significant at p < 0.05.
RESULTS
Effects of IBO in sham-stimulated rats before or after blockade of NMDA receptors
IBO (23 nmol in 360 nl of 0.1 m PBS) was microinjected unilaterally into the striatum of sham-stimulated rats. The dose and volume were selected, on the basis of pilot experiments, to produce lesions almost entirely confined to the body of the nucleus.
As expected (e.g., Volpe et al., 1998), at the level of the injection, IBO destroyed virtually all striatal neurons (Figs.1A,2B). Also damaged was a thin strip of overlying cerebral cortex surrounding the cannula track (Figs. 1A, arrow, 2B). In some cases, the lesion extended ventrally to destroy some olfactory tubercle and a small portion of orbitofrontal cortex (Fig.2B, Sham Stimulated). The lesion covered an area of ∼2.5 mm. The maximum area was ∼20 mm2(Fig. 2A) at the site of injection, 9.7 mm rostral to the interaural line. The lesion became progressively smaller at sites ∼1.5 mm ahead of or behind the injection site (Fig. 2), consistent with diffusion of IBO. The average lesion volume in sham-stimulated rats was ∼40 mm3 (Table1). In the contralateral striatum, vehicle alone produced small lesions that averaged 1.1 ± 0.3 mm3 (n = 40; all groups), with most damage localized along the pipette track in the cortex (data not shown).
Fig. 1.
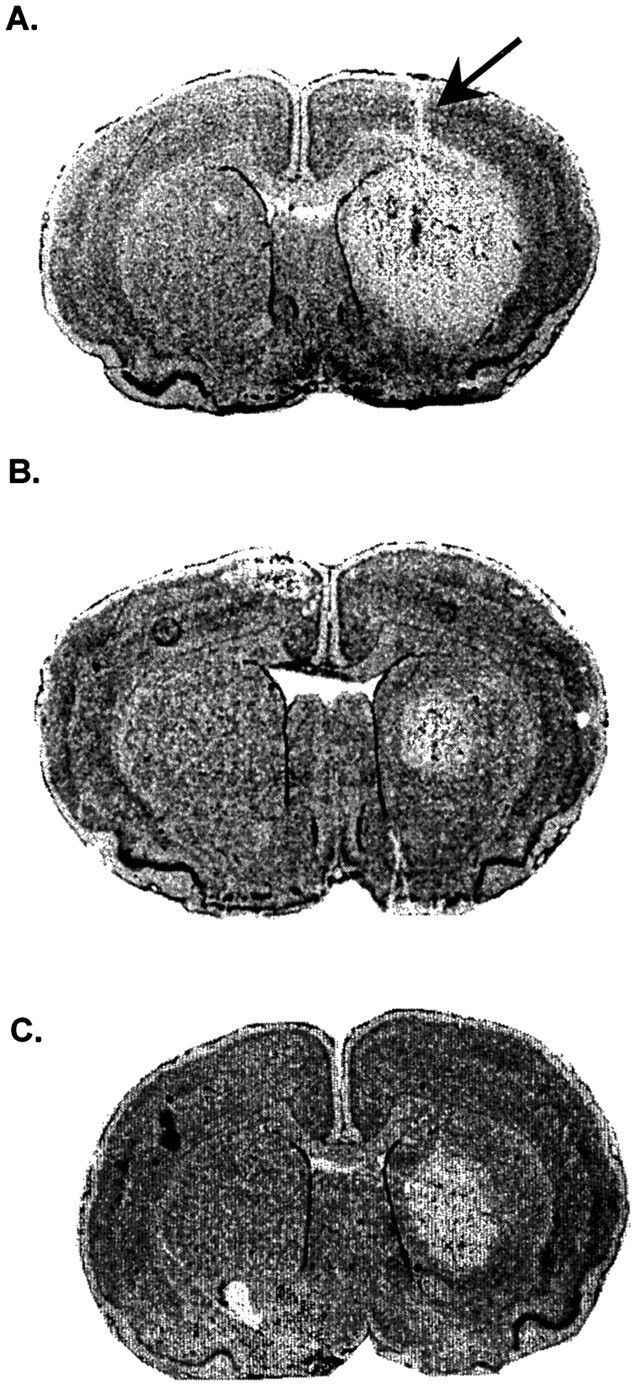
Effect of FN stimulation on excitotoxic lesions of rat striatum. A, Distribution of excitotoxic lesion in the right striatum 24 hr after microinjection of IBO (23 nmol in 360 nl of PB). Arrow indicates cannula track. Left striatum was injected with equal volume of PB. B, Reduction in excitotoxic lesion produced by 1 hr of electrical stimulation of FN 3 d before injecting IBO. C, Reduction in excitotoxic lesion produced by intraperitoneal pretreatment with MK801. Nissl-stained sections.
Fig. 2.

Effect of FN stimulation on area and distribution of unilateral excitotoxic striatal lesions in rat striatum. FN was simulated for 1 hr 3 d before injection of IBO. Lesion volumes were estimated 24 hr thereafter. A, Cross-sectional areas, expressed as mean ± SEM, at different levels of brain along the rostrocaudal axis at sites rostral to the interaural line. Each group represents five sham-stimulated (filled triangles), DN-stimulated (open circles), or FN-stimulated (open squares) rats. *p < 0.05. B, Distribution of lesions at different rostrocaudal levels in representative cases from each group described in A. Stippledepicts the extent of the lesion. Numbers represent the distance from interaural line. Note that only FN stimulation reduces lesion size.
Table 1.
Volume of excitotoxic lesions (average ± SEM) in rats resulting from striatal microinjection of IBO
| Interval between FN stimulation and IBO | Lesion volume (mm3) | ||
|---|---|---|---|
| Sham-FN stim | FN stim | DN stim | |
| <15 min | 37.2 ± 6.9 (n = 11) | 20.2 ± 2.4 (n = 12)* | 42.4 ± 2.0 (n = 4) |
| 72 hr | 41.4 ± 7.4 (n = 5) | 7.8 ± 1.6 (n = 5)* | 43.0 ± 5.3 (n = 5) |
| 10 d | 45.1 ± 2.6 (n = 5) | 24.4 ± 7.9 (n = 5)* | 44.8 ± 6.2 (n = 5) |
| 30 d | 46.7 ± 11.4 (n = 3) | 36.8 ± 4.2 (n = 3) | 33.4 ± 5.5 (n = 3) |
IBO was injected immediately after FN stimulation or at longer time intervals after stimulation compared with DN-stimulated or sham-FN-stimulated controls. The neuroprotective effect is present immediately after stimulation, conditionally protects for at least 10 d, and is reversed by 30 d after stimulation.
*p < 0.05 compared with matching control; ANOVA and SNK test.
To determine whether IBO destroyed striatal neurons by stimulating NMDA receptors, we pretreated rats with the competitive NMDA receptor antagonist MK801 [(+)-5-methyl-10,11-dihydro-5H-dibenzo [a,d] cyclohepten-5,10-imine maleate] (Wong et al., 1986). MK801 (4 mg/kg in isotonic saline) was administered intraperitoneally 30 min before intrastriatal microinjection of IBO (23 nm in 360 nl). Such treatment reduces NMDA-mediated excitotoxicity to other excitotoxins (Beal et al., 1988; Foster et al., 1988).
Treatment with MK801 reduced the volume of IBO-induced lesions by 80% (8 ± 1.5 mm3; n = 5; untreated controls, 41 ± 1.3 mm3; n = 4;p < 0.001). The region within the striatum protected by MK801 was primarily in the periphery, although neuronal death was still observed at the site of injection (Fig. 1C). Thus, in this model, IBO neurotoxicity can primarily be attributed to activation of the NMDA receptor, particularly in the periphery of the striatal lesion.
Effects of FN stimulation on excitotoxic lesions of striatum
IBO (23 nmol in 360 nl of 0.1 m PBS) was microinjected into the striatum at 15 min, 72 hr, 10 d, or 30 d after stimulating the FN for 1 hr (50 Hz; 1 sec on, 1 sec off; stimulus currents five times threshold). Rats were killed 24 hr later. The location of electrode sites in the FN in 15 representative animals is shown in Figure 3. For each group, sham-stimulated controls were prepared and processed in parallel. Lesion volumes in the sham-stimulated animals did not differ between groups (ANOVA; p > 0.5) (Table 1).
Fig. 3.

Location of stimulation sites in the FN (filled circles) and DN (open circles) in 15 representative experiments each.CST, Cortical spinal tract; DN, dentate nucleus; FN, fastigial nucleus; STT, spinothalamic tract; VII, facial nerve nucleus;IV, ventricle IV.
Stimulation of the FN substantially and significantly reduced the volumes of excitotoxic lesions in the striatum (Figs. 1, 2,4; Table1). Moreover, the effects of stimulation were long-lasting. Thus, lesion volumes were reduced by 46% when IBO was injected immediately after stimulation and 82% when excitotoxin was injected 72 hr later (Figs. 1, 2, 4; Table 1). Thereafter, the protective effects began to wane (Fig. 4, Table 1). When lesions were placed 10 d after stimulation, salvage was reduced to 46% of control, although still significant. By 30 d, neuroprotection disappeared (Fig. 4, Table 1). Thus, the effect of FN stimulation is reversible.
Fig. 4.

Persistence of the neuroprotective actions of FN stimulation. Data are expressed as percentage of lesion volume in matched sham-stimulated rats (open bar). Eachsolid bar represents volume of lesions placed at various times between 1 hr of FN stimulation and microinjection of IBO.Hatched bar represents normalized data from all groups in which the DN was stimulated before IBO treatment. (n= 5–12; *p < 0.05). Mean volume for all sham stimulated rats was 40.9 ± 2.4 mm3(n = 24).
In general, the area of salvage resulted from a significant and concentric shrinkage of the lesion, as exemplified in cases injected 72 hr after stimulation (Figs. 1, 2). The distribution of salvage was primarily restricted to the periphery of the lesion which, interestingly, is the region primarily protected by pretreatment with MK801 (Fig. 1C).
Effects of DN stimulation on excitotoxic lesions of striatum
To control for nonspecific effects of electrical stimulation, the DN was stimulated for 1 hr (50 Hz; 1 sec on, 1 sec off; 100 μA), and IBO was injected, as above, at 15 min, 72 hr, 10 d, or 30 d, and animals were killed 24 hr later. The location of electrode sites in 15 representative animals is shown in Figure 3. The volume and distribution of the lesions produced in rats after stimulation of DN did not differ between groups (ANOVA) (Table 1) and, when averaged overall (41.7 ± 2.47 mm3; n = 17), did not differ from sham-stimulated controls (p > 0.5) (Figs. 2, 4; Table 1).
Effects of MAP, blood gases, glucose, hematocrit, hemorrhage, and brain temperature
Physiological variables
MAP, blood gases (Paco2, PaO2, and pH), glucose, and Hct were measured in all rats (Table 2) during real or sham stimulation of cerebellar nuclei in all groups. All animals were spontaneously breathing 100% O2. The values, in general, did not differ from those of anesthetized rats reported previously (Caggiano and Kraig, 1998). Compared with normal blood-gas values in unanesthetized animals (Loeb and Quimby, 1989), rats were significantly but variably hyperoxic, with lower (yet still hyperoxic) values obtained in some subgroups (e.g., Table2, <15 minutes, DN-Stim), most probably reflecting a variable degree of atelectasis commonly seen in anesthetized rat (Nathan and Reis, 1975). The anesthetized rats were also modestly hypercarbic and acidotic, probably reflecting a modest hypoventilation.
Table 2.
MAP, Hct, blood glucose, and blood gases
| <15 min | 72 hr | 10 d | 30 d | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Sham-FN | DN-Stim | FN-Stim | Sham-FN | DN-Stim | FN-Stim | Sham-FN | DN-Stim | FN-Stim | Sham-FN | DN-Stim | FN-Stim | |
| MAP | 102.8 ± 3.3 | 96.1 ± 5.6 | 108.8 ± 4.0 | 116.7 ± 6.4 | 104.0 ± 6.7 | 122.8 ± 1.2 | 92.5 ± 7.9 | 96.8 ± 8.6 | 103.5 ± 9.5 | 105.3 ± 4.3 | 106.8 ± 2.2 | 111.5 ± 6.5 |
| Hct (%) | 42.1 ± 0.5 | 43.1 ± 0.5 | 43.7 ± 0.5 | 45.6 ± 0.6 | 42.3 ± 0.8 | 44.9 ± 0.6 | 42.7 ± 1.2 | 42.8 ± 0.6 | 44.6 ± 1.1 | 43.5 ± 0.5 | 42.8 ± 0.5 | 47.9 ± 1.2 |
| Glu (mg/dl) | 119.5 ± 11 | 115.0 ± 18 | 134.7 ± 9.9 | 154.4 ± 16.7 | 125.6 ± 10.7 | 146.9 ± 9.5 | 100.5 ± 9.5 | 87.5 ± 16.5 | 179.5 ± 38.3 | 130.5 ± 83.5 | 152.8 ± 20.7 | 109.8 ± 21.2 |
| pH | 7.26 ± 0.02 | 7.27 ± 0.0 | 7.29 ± 0.02 | 7.31 ± 0.03 | 7.24 ± 0.06 | 7.29 ± 0.02 | 7.26 ± 0.03 | 7.28 ± 0.02 | 7.34 ± 0.02 | 7.22 ± 0.02 | 7.24 ± 0.03 | 7.27 ± 0.04 |
| pCO2 | 60.2 ± 3.1 | 65.7 ± 2.9 | 54.0 ± 2.0 | 53.5 ± 3.9 | 59.8 ± 5.7 | 58.7 ± 1.8 | 49.0 ± 4.1 | 47.6 ± 2.5 | 44.9 ± 2.8 | 51.0 ± 4.2 | 57.3 ± 5.7 | 44.7 ± 5.5 |
| pO2 | 300.3 ± 31 | 161.7 ± 30 | 311.0 ± 30.0 | 244.5 ± 50.3 | 250.2 ± 54.3 | 276.2 ± 38.4 | 235.6 ± 46.1 | 182.6 ± 25.4 | 307.4 ± 48.4 | 192.3 ± 56.8 | 239.6 ± 63.1 | 247.7 ± 63.5 |
| n | 11 | 4 | 12 | 5 | 5 | 5 | 5 | 5 | 5 | 3 | 3 | 3 |
MAP, Hct, blood glucose (Glu), and blood gases in Sprague Dawley rats averaged from samples taken before and immediately after sham or real stimulation of FN or DN. Physiological variables are grouped with reference to the time interval after stimulation and before the placement of excitotoxic lesions in striatum. Values are expressed as mean ± SEM; n, number of rats. There were no significant differences between groups (p > 0.05; ANOVA and SNK test).
Comparisons between groups, although not significant (p > 0.05; ANOVA and SNK test), indicate sporadic changes in some variables, which were neither systematic nor correlated with neuroprotection. Thus, the Hct varied by no more than 10% between groups at any time and differed slightly only in DN-stimulated rats at 72 hr and FN-stimulated rats at 30 d, both groups without neuroprotection. MAP was modestly elevated in the FN-stimulated group at 72 hr, but not at 10 d, despite protection in both, whereas blood glucose was elevated in the FN-stimulated group at 10 d, but not at 72 hr, a time of maximal neuroprotection. The random nature of the changes and the facts that hyperoxia (Roos et al., 1998) is not neuroprotective, whereas hyperglycemia and hypercarbia with associated acidosis are not only nonprotective but may even exacerbate lesion size (Browning et al., 1997), indicate that changes in blood gases, Hct, and/or blood sugars do not relate to evoked neuroprotection.
Hemorrhage
To control for effects of withdrawal and replacement of blood on lesion volume, the FN was stimulated while gradually increasing the stimulus current to five times threshold so that AP was only elevated to 122 ± 9.4 mmHg from baseline of 103.4 ± 6.7 mmHg, well within the range of cerebrovascular autoregulation in rat (Dirnagl and Pulsinelli, 1990). Stimulation was continued for 1 hr, and blood was not withdrawn. IBO was injected intrastriatally 72 hr later. The average lesion volume of the group was 14.1 ± 4.4 mm3 (n = 4), which did not differ significantly from FN-stimulated rats in which blood was withdrawn and replaced (7.8 ± 3.6 mm3; p> 0.5) (Table 1). Thus, withdrawal of blood does not affect lesion size.
Temperature
To determine whether FN stimulation reduced brain temperature, a possible mechanism promoting neuroprotection (Maier et al., 1998), we estimated intracortical temperature with a thermistor probe inserted into temporal muscle during 1 hr of FN stimulation. Baseline temporal muscle temperature in anesthetized rats was comparable between groups (FN-stimulated, 36.8 ± 0.2; DN-stimulated, 37.0 ± 0.3°C). Brain temperature was increased slightly and not significantly by FN stimulation compared with control (FN-stimulated, 5.2 ± 1.0; DN-stimulataed, 2.2 ± 1.3% increase from baseline temperature;n = 4 per group; p > 0.05). Hyperthermia has been shown to exacerbate ischemic injury (Dietrichs et al., 1990) and, therefore, this slight increase in brain temperature cannot account for evoked neuroprotection. Thus, salvage elicited from FN cannot be attributed to nonspecific effects of stimulation, to a reduction in brain temperature, or to withdrawal of blood.
Effects of excitotoxic lesions of FN on neurogenic protection against striatal injury
We investigated which neuronal elements in FN, when stimulated, initiated neuroprotection. Two principal experiments, each with three subgroups, were performed.
FN stimulation alone
In the first experiment (Table 3, Intact Cerebellum + Stimulation), groups of rats were instrumented to record AP and rCBF and to compute CVR. The FN (Table 3, FN Stimulation) or DN (DN Stimulation) were stimulated for 1 hr (50 Hz; 1 sec on, 1 sec off; stimulus currents five times threshold for FN or 100 μA for DN). For sham-stimulation, an electrode was inserted in FN for 1 hr but not was stimulated (Table 3, Sham Stimulation) (see Materials and Methods for details). AP and rCBF were continuously measured. After termination of stimulation, animals’ wounds were closed, and animals were returned to their cages. Seventy two hours later, they were reanesthetized, and IBO (23 nmol in 360 nl of 0.1 m PBS) was microinjected into one striatum and PBS into the other. Twenty four hours later, rats were killed, and brains were examined.
Table 3.
Volumes of striatal excitotoxic lesions (average ± SEM) in rats in association with cerebellar lesion and FN stimulation
| Group (n) | Lesion volume (mm3) | Percent of sham striatal lesion |
|---|---|---|
| Intact cerebellum + stimulation | ||
| Sham stimulation (4) | 41.3 ± 1.3 | 100.0 |
| FN stimulation (5) | 15.5 ± 4.5 | 37.5* |
| DN stimulation (4) | 40.4 ± 4.9 | 97.8 |
| Cerebellar lesions + FN stimulation | ||
| FN-lesion (6) | 29.1 ± 7.5 | 70.5 |
| DN-lesion (4) | 13.0 ± 3.7 | 31.5* |
| FN-sham-lesion (5) | 23.2 ± 6.1 | 56.2* |
The FN was stimulated 5 d after bilateral FN lesion, DN lesion, or after injection of vehicle alone into the FN (FN-sham-lesion). Three days later, IBO was microinjected into the striatum, and 24 hr later, volumes were compared with matched nonlesioned (Intact) FN-stimulated rats and control DN- or sham-FN-stimulated rats. Lesion of the intrinsic FN neurons abolishes the neuroprotective effects of FN stimulation.
*p < 0.05 compared with matching control; ANOVA and SNK test.
As expected, electrical stimulation of the FN in intact controls reduced the striatal lesion by ∼64% (15.5 ± 4.5 vs 41.3 ± 1.3 mm3; p < 0.05) (Table 3). The volumes of the striatal lesions in sham-stimulated and DN-stimulated rats (Table 3) did not differ from each other or from those obtained in similar groups in the previous study (Table 1). Electrical stimulation of FN, but not DN, significantly elevated AP and rCBF and reduced CVR, as expected (Table4).
Table 4.
Values (±SEM) of AP, percentage of increases in rCBF, and change in percentage of baseline CVR
| Group (n) | Peak AP (mmHg) | Peak rCBF (PU as % increase from baseline) | Δ Percentage of baseline CVR |
|---|---|---|---|
| Control stimulation | |||
| Sham stimulated (3) | 90.7 ± 4.8 | 4.3 ± 4.7 | 96.7 ± 7.1 |
| DN stimulated (3) | 96.8 ± 3.2 | 3.0 ± 5.8 | 111.1 ± 9.6 |
| FN stimulation | |||
| Intact (4) | 110.9 ± 6.2 | 34.0 ± 6.0 | 79.0 ± 1.4*§ |
| FN-lesion (4) | 114.0 ± 3.8 | 50.0 ± 7.9 | 75.8 ± 2.7*§ |
| FN-sham-lesion (4) | 110.4 ± 4.5 | 53.0 ± 10.7 | 76.5 ± 7.3*§ |
| DN-lesion (4) | 122.0 ± 7.4 | 93.6 ± 27.1 | 77.8 ± 8.6*§ |
Values (±SEM) of AP, percentage of increases in rCBF, and change in percentage of baseline CVR (absolute millimeters mercury per absolute perfusion units) at peak elevation of rCBF in Sprague Dawley rats evoked by electrical stimulation of the FN in cerebellar-lesioned animals (FN-lesion, DN-lesion), in nonlesioned controls (Intact, FN-sham-lesion), or in nonlesioned animals after control stimulation (DN stimulated, Sham stimulated; data taken at 10 min after the onset of stimulation, a time point that parallels average time after onset of FN stimulation when peak rise in rCBF is elicited). Note that FN stimulation significantly reduces CVR relative to control stimulated groups and that this effect persists after lesion of FN intrinsic neurons.
*p < 0.05 compared with sham- and DN-stimulated controls; ANOVA and SNK test.
§p < 0.05 compared with baseline CVR within group; ANOVA and SNK test.
FN stimulation after cerebellar lesions
In the second experiment, we investigated the effects of destroying FN neurons on the FN-mediated protection against striatal neurotoxicity (Table 3, Cerebellar Lesions + FN Stimulation). In this study, rats were anesthetized and instrumented to record AP. IBO (23 nmol in 360 nl of PBS; see Materials Methods for details of injection) was either injected into the FN (Table 3, FN-Lesion) or into the DN (DN-Lesion). As control, PBS was injected into the FN (FN-Sham-Lesion). IBO injected into FN transiently (∼5 min) reduced AP (−20.0 ± 3.3 mmHg; n = 32), confirming previous studies (Chida et al., 1986, 1989, 1990). Injections into DN were without effect. PBS injected briefly (∼20 sec) elevated AP (18.3 ± 4.9 mmHg; n = 10). Rats were allowed to recover. The treatments did not produce ataxia or significant weight loss.
Five days later, the rats were reanesthetized and instrumented to record AP, rCBF, and CVR. A stimulating electrode was placed in FN and stimulated for 1 hr as above. Seventy two hours later, under anesthesia, IBO was injected into the striatum, and 24 hr later, the rats were killed and brains were removed to measure striatal lesions and analyze the extent of neuronal damage in the cerebellum.
FN stimulation, in all three groups, significantly elevated AP and rCBF and reduced CVR. These reductions in CVR were significant when compared with baseline values and with that in control stimulated animals. Responses to FN stimulation did not differ in rats with cerebellar lesion compared with those without lesion (Fig.5, Table 4). Stimulation of DN, as expected, was without effect (Tables 3, 4). Stimulation of the FN in rats with excitotoxic FN lesions no longer significantly reduced striatal lesion volume (Table 3) and distribution (Fig.6A). In contrast, excitotoxic lesions of the striata were significantly reduced in size by FN stimulation in rats in which saline was injected into the FN or in which the DN was destroyed by IBO (Table 3).
Fig. 5.
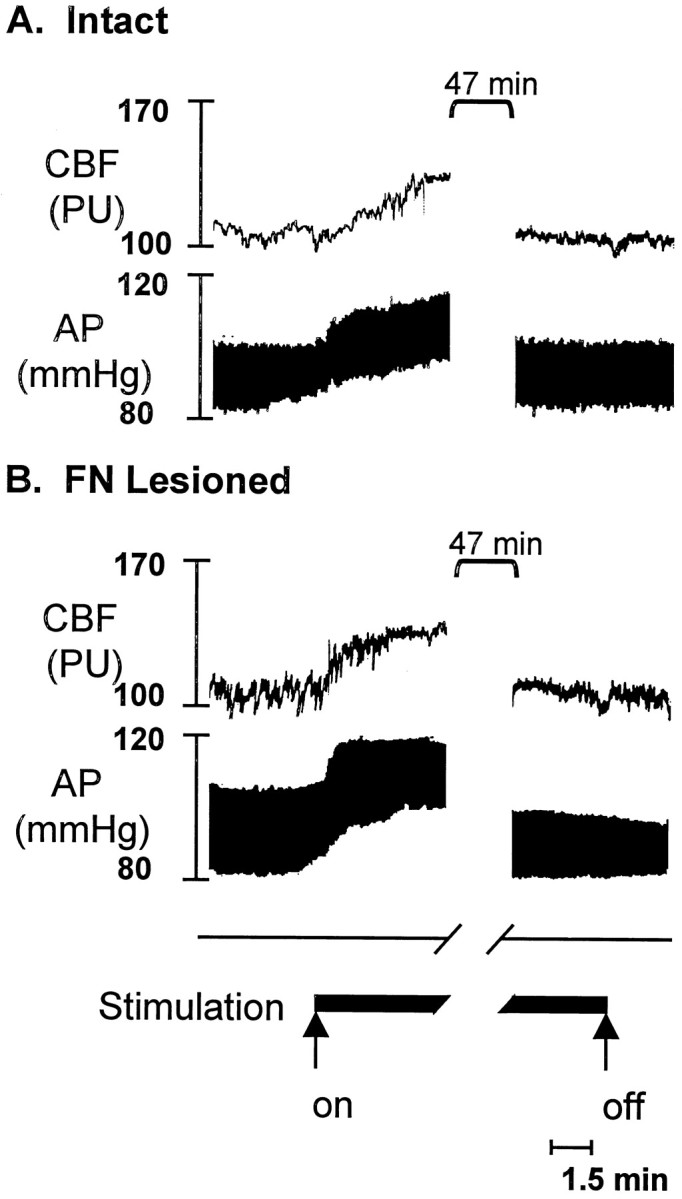
Changes in AP and rCBF in anesthetized rats elicited by electrical stimulation of FN with or without excitotoxic lesions of the nucleus. rCBF was measured by laser-Doppler flowmetry; IBO or vehicle were microinjected 5 d earlier. A, Intact rat during and at end of electrical stimulation of FN (50 Hz; 1 sec on, 1 sec off stimulus current five times threshold). Note rapid rise in rCBF and AP, which recovers to baseline after 1 hr, characteristics of the fastigial pressor response. B, FN-lesioned rat. Note preservation of stimulation-evoked elevations in rCBF and AP, despite destruction of intrinsic neurons of FN as verified histologically.
Fig. 6.
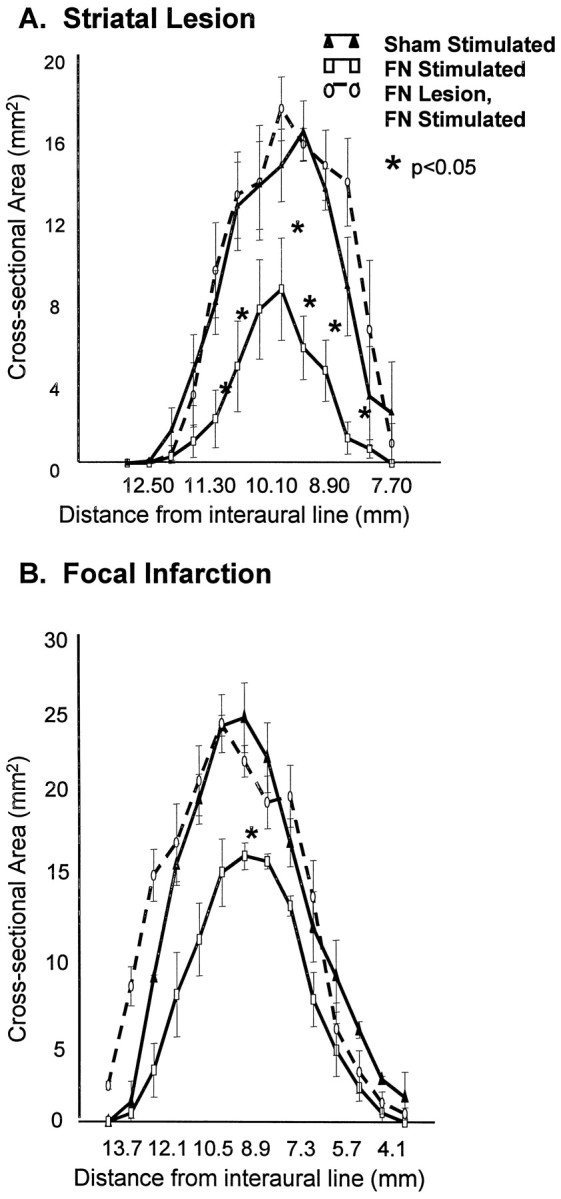
Excitotoxic lesions of FN reduces the salvage produced by FN stimulation of both excitotoxic lesions and focal ischemic infarctions. Intrinsic FN neurons were destroyed by IBO 5 d before FN stimulation or sham stimulation. Average cross-sectional area (n = 5–6 per group; mean ± SEM; *p < 0.05, FN-stimulated compared with sham-stimulated) are plotted relative to distance from interaural line as in Figure 2. Groups of rats comprising sham-stimulated (filled triangles), FN-stimulated without excitotoxic lesions (open circles), and FN-stimulated (open squares) with excitotoxic lesions of FN were compared. A, Effects on excitotoxic lesions. IBO was microinjected into striatum 3 d after stimulating the FN, and rats were killed 24 hr thereafter. B, Volume of focal ischemic infarctions. Note that destruction of intrinsic lesions of FN abolish neuroprotection with either lesion.
Histological examination of the cerebella of rats in which IBO was microinjected into FN demonstrated that the majority of large intrinsic neurons of the anterior third of the nucleus had disappeared and were replaced by small non-neuronal cells representing glia, macrophages, and leukocytes (Fig. 7A–D). The DN was not damaged. Rats in which IBO was injected into DN had cytologically comparable changes, but with neuronal loss confined to the lateral nucleus and without damage evident in FN. In rats in which the FN was injected with saline, the large Nissl-stained intrinsic neurons of FN were basically undamaged, although some small cell infiltration was seen (Fig.7E,F), probably reflecting mechanical damage.
Fig. 7.
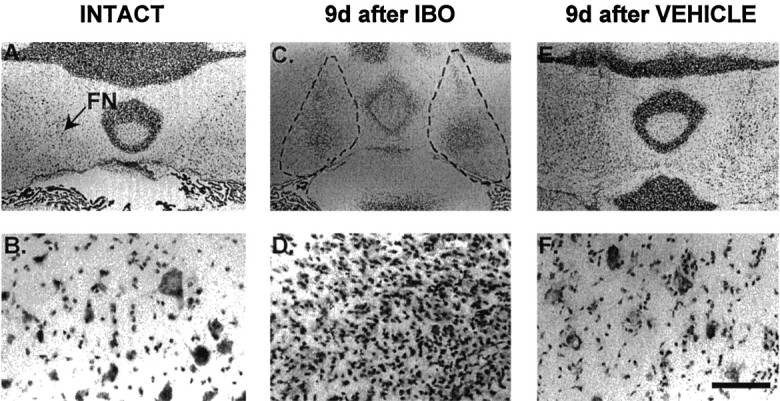
Representative lesions of FN produced by injection of IBO into FN 9 d earlier visualized at low (scale bar, 1.5 mm) and higher (scale bar, 0.125 mm) magnifications. A,B, Naive control. Arrow indicates left FN. Note large Nissl-stained FN neurons at higher power.C, D, IBO treatment. The broken lines outline the area of complete destruction of local neurons with reactive gliosis 9 d after microinjection of IBO. Note loss of large neurons. E, F, The FN 9 d after injection of vehicle. Note accumulation of small stained cells about some neurons.
Physiological variables
MAP, blood gases, glucose, and hematocrit did not differ between groups (Table 5), nor did they differ from values reported in the previous experiment (Table 2).
Table 5.
MAP, Hct, blood glucose, and blood gases
| Control stimulation | FN stimulation | |||||
|---|---|---|---|---|---|---|
| Sham-FN | DN-stim | Intact | FN-lesion | DN-lesion | FN-sham-lesion | |
| MAP (mmHg) | 90.5 ± 2.2 | 95.1 ± 1.9 | 90.0 ± 3.5 | 108.8 ± 2.7 | 102.0 ± 5.6 | 104.2 ± 2.4 |
| Hct (%) | 42.5 ± 2.9 | 43.7 ± 1.6 | 45.1 ± 0.9 | 42.0 ± 1.3 | 38.7 ± 2.9 | 40.4 ± 4.6 |
| Glu (mg/dl) | 107.0 ± 0.0 | 131.0 ± 15.0 | 127.0 ± 0.0 | 138.0 ± 49.0 | 127.5 ± 0.0 | 118.5 ± 18.9 |
| pH | 7.31 ± 0.12 | 7.26 ± 0.16 | 7.32 ± 0.03 | 7.31 ± 0.04 | 7.35 ± 0.07 | 7.34 ± 0.04 |
| pCO2 | 62.3 ± 8.9 | 68.2 ± 4.4 | 64.3 ± 1.4 | 66.4 ± 6.2 | 57.6 ± 9.7 | 54.9 ± 4.9 |
| pO2 | 339.1 ± 109 | 316.9 ± 125 | 320.2 ± 123.6 | 329.5 ± 64.8 | 303.5 ± 73.4 | 223.2 ± 52.3 |
| n | 4 | 4 | 5 | 6 | 4 | 5 |
MAP, Hct, blood glucose (Glu), and blood gases in Sprague Dawley rats evoked by electrical stimulation of the FN in cerebellar-lesioned animals (FN-lesion, DN-lesion), in nonlesioned controls (Intact, FN-sham-lesion), or in nonlesioned animals after control stimulation (DN-stim, Sham-stim). Values are averaged from samples taken before and immediately after stimulation and are expressed as mean ± SEM; n, number of rats. There were no significant differences between groups (p > 0.05; ANOVA and SNK test).
This study, therefore, indicates that neuroprotection elicited from FN is abolished by selective destruction of intrinsic neurons while preserving the stimulus-locked elevations in rCBF and AP (Fig. 5, Table4). These findings indicate that neuroprotection is elicited from FN neurons, whereas, in confirmation of earlier studies (Chida et al., 1986, 1989, 1990), the effects on rCBF and AP result from stimulation of axons projecting into or through the nucleus.
Effects of cerebellar lesions on focal ischemic infarctions
We investigated whether intrinsic neurons of FN also mediate the reductions in focal ischemic infarctions elicited by occlusion of the MCA (Reis et al., 1991, 1998; Yamamoto et al., 1993). This study, like the previous one, consisted of two experiments, each comprised of three subgroups (Table 6).
Table 6.
Volumes of MCA infarction (average ± SEM), corrected for edema, in rats in association with cerebellar lesion and FN stimulation
| Group (n) | Infarction volume (mm3) | Percent of sham MCA Occlusion |
|---|---|---|
| Intact cerebellum + stimulation | ||
| Sham stimulation (3) | 119.0 ± 6.0 | 100.0 |
| FN stimulation (3) | 75.2 ± 7.6 | 63.0* |
| DN stimulation (4) | 132.5 ± 9.7 | 90.1 |
| Cerebellar lesion + FN stimulation | ||
| FN-lesion (12) | 126.3 ± 8.3 | 105.8 |
| DN-lesion (4) | 87.3 ± 5.1 | 73.1* |
| FN-sham-lesion (6) | 95.9 ± 3.1 | 80.4 |
The FN was stimulated 5 d after bilateral FN lesion, after DN lesion, or after injecting vehicle alone in the FN (FN-sham-lesion). Immediately after FN stimulation, the MCA was occluded, and 24 hr later, volumes were compared with matched nonlesioned (Intact) FN-stimulated rats and control DN- or FN-stimulated rats. Bilateral excitotoxic lesions of the intrinsic FN neurons abolish the reductions in infarction volume elicited by FN stimulation.
*p < 0.05 compared with matching control; ANOVA and SNK test.
Neuroprotection with FN stimulation
In the first experiment (Table 6, Intact Cerebellum + Stimulation), the FN (FN Stimulation) or DN (DN Stimulation) was stimulated or an electrode was inserted into FN but not stimulated for 1 hr, as described above and in Materials and Methods. At the end of stimulation, the MCA was exposed and ligated, wounds were closed, and animals were returned to their cages. Twenty-four hours later, they were killed, and the distribution and size of lesions was measured.
Occlusion of the MCA in sham-stimulated rats produced ischemic infarctions whose distribution (Fig. 6B) and volumes (Table 6) were comparable with those reported previously (Reis et al., 1991, 1998). The average lesion volume was 119.0 ± 6.0 mm3 (n = 3). At the level of maximum damage, the lesion extended through all layers of the cerebral cortex, dorsally into the primary motor area, ventrally to the pyriform cortex, and medially to the lateral edge of the caudate-putamen (Fig.8B). In the rostrocaudal axis, the lesion involved large portions of the parietal, insular, temporal, and occipital cortices.
Fig. 8.
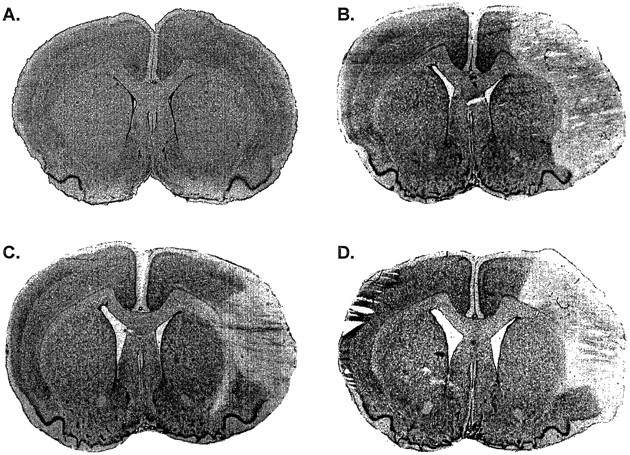
Effect of FN stimulation in spontaneously hypertensive rats with or without excitotoxic lesions of FN on focal ischemic infarctions produced by occlusion of MCA. A, Naive rat. B, Twenty-four hours after MCA occlusion.C, MCA occlusion immediately after 1 hr of FN stimulation. The rat was killed 24 hr later. Note reduction of lesion area and distribution. D, MCA occlusion immediately after 1 hr of FN stimulation in a rat in which the FN was destroyed by IBO 5 d earlier. Note that the stimulation no longer salvaged the lesion compared with C. Nissl staining.
FN stimulation reduced infarct volume by 37% (75.2 ± 7.6 mm3; n = 3; p < 0.01) (Figs. 6B, 8C; Table 6). Stimulation of the DN was without effect and averaged 132.5 ± 9.7 mm3 (n = 4), not significantly different from infarction volumes in control animals (Table 6). In a parallel experiment, the stimulation current was increased rapidly to five times threshold and also maintained for 1 hr, but blood was not withdrawn. The maximum AP in this group was 148 mmHg. Lesion volume for the group was 65.4 ± 5.9 mm3, which also did not differ from the blood-withdrawal group (75.2 ± 7.6 mm3; p > 0.05). Thus, not only does withdrawal of blood not affect lesion size, but the elevations of AP associated with FN stimulation also do not modify lesion volume.
FN stimulation after cerebellar lesions
In the second experiment (Table 6, Cerebellar Lesions + FN Stimulation), rats were anesthetized and IBO was injected into FN (FN-Lesion) or DN (DN-Lesion), or PBS was injected into the FN (FN-Sham-Lesion). After 5 d of recovery, these rats were reanesthetized and instrumented to record AP and rCBF, the FN was stimulated for 1 hr, and the MCA was occluded. Twenty-four hours later, all rats were killed, brains were removed, and the distribution and volumes of the focal ischemic infarctions and the extent of cerebellar lesions were determined. IBO injected into the FN or DN destroyed intrinsic neurons in the respective areas as in the previous study (data not shown), whereas saline was without effect. FN stimulation in rats treated with saline or after DN stimulation reduced infarction volumes to the same extent as in rats without cerebellar injections (Table 6). However, the neuroprotective effect of FN stimulation was completely abolished by excitotoxic lesions of the FN (Fig.8D, Table 6).
Physiological variables
MAP, blood gases, glucose, and hematocrit did not differ between groups (Table 7). In all animals in this experiment, blood glucose levels were substantially higher than those obtained in Sprague Dawley rats (Tables 2, 5).
Table 7.
Physiological variables in spontaneously hypertensive rats in groups comparable with those of Table 5
| Control stimulation | FN stimulation | |||||
|---|---|---|---|---|---|---|
| Sham-FN | DN-Stim | Intact | FN-lesion | DN-lesion | FN-sham-lesion | |
| MAP (mmHg) | 100.4 ± 7.9 | 112.5 ± 8.8 | 98.6 ± 5.0 | 110.5 ± 2.4 | 123.7 ± 2.0 | 116.2 ± 2.4 |
| Hct (%) | 46.1 ± 0.5 | 47.6 ± 0.3 | 45.8 ± 0.1 | 40.8 ± 1.3 | 44.1 ± 0.4 | 45.2 ± 1.2 |
| Glu (mg/dl) | 202.0 ± 22.0 | 184.1 ± 13.5 | 176.5 ± 8.5 | 405.1 ± 64.0 | 200.4 ± 46.1 | 180.6 ± 69.6 |
| pH | 7.36 ± 0.03 | 7.33 ± 0.04 | 7.41 ± 0.06 | 7.33 ± 0.03 | 7.29 ± 0.03 | 7.34 ± 0.02 |
| pCO2 | 54.9 ± 2.3 | 60.9 ± 7.2 | 54.7 ± 0.8 | 54.0 ± 2.6 | 51.1 ± 6.4 | 54.6 ± 5.4 |
| pO2 | 323.8 ± 78.6 | 306.1 ± 23.2 | 245.3 ± 174.9 | 173.1 ± 17.5 | 154.1 ± 28.1 | 142.9 ± 0.7 |
| n | 3 | 4 | 3 | 12 | 4 | 6 |
Values are expressed as mean ± SEM; n, number of rats. There were no significant differences between groups (p > 0.05; ANOVA and SNK test).
This study indicates that histologically verified excitotoxic lesions of FN, which destroy local neurons but preserve axons of the region mediating the FPR, will abolish the salvage of focal ischemic infarctions elicited by FN stimulation. Thus, as with salvage of excitotoxic lesions, intrinsic neurons of the FN, not fibers of passage, initiate neuroprotection.
DISCUSSION
Effects of stimulation of FN on excitotoxicity
In the first experiment, we investigated whether electrical stimulation of the FN would protect against excitotoxic, as well as ischemic, infarctions (Golanov et al., 1998; Reis et al., 1998). Excitotoxic lesions were produced by microinjection of IBO into the striatum. IBO, a structurally rigid glutamate analog that binds with high affinity to the NMDA and, to a lesser degree, to AMPA receptors (MacLennan and Lodge, 1979), destroys neuronal perikarya, sparing axons, and non-neuronal cells (Coyle et al., 1978). The area of neuronal damage was maximal at the site of injection and diminished radially over a distance 1.5 mm from the core, a pattern compatible with diffusion (Guiliano et al., 1989). Systemic administration of MK801, a noncompetitive inhibitor of the NMDA channel (Wong et al., 1986), blocked neuronal loss in the periphery of the lesion but not around the cannula tip (core). Thus, damage in the periphery resulted from activation of NMDA receptors. The failure of treatment to salvage neurons in the core most likely represents a disproportionate amount of agonist compared with antagonist or a differential activation of the AMPA receptor at increased IBO concentration (Martin et al., 1998).
As demonstrated here and in previous studies (Reis et al., 1991, 1998;Golanov et al., 1998), salvage cannot be attributed to variations in blood gases, anesthesia, AP, brain temperature, or to controlled hemorrhage. All rats were hyperoxemic as a consequence of breathing 100% O2, and some groups had modest respiratory acidosis, a common effect in studies in which animals are not paralyzed and ventilated. Moreover, FN-evoked neuroprotection was also present in ventilated, normocarbic animals (Golanov and Reis, 1999). Although halothane has been reported by some (Warner et al., 1995), but not all (Browning et al., 1997), to be neuroprotective, possibly by blocking NMDA receptors (Beirne et al., 1998), all rats were subject to the same anesthetic regimen, making it highly unlikely that concentrations differed systematically between groups. Salvage cannot be attributed to hypertension, which some claim may reduce ischemia-induced infarction (Ogilvy et al., 1996) because FN stimulation elevated AP to the same extent in rats in which FN neurons were destroyed but protection was abolished. Brain temperature, a variable influencing lesion volume (Maier et al., 1998) did not change during stimulation and, hence, cannot contribute to salvage. Finally, controlled hemorrhage, commonly used to equalize AP during FN stimulation (Nakai et al., 1983), did influence responses for lesion volumes were comparable between groups in which blood was or was not withdrawn. Thus, modest variations in physiological variables occurred in random groups and did not correlate with neuroprotection.
As with focal ischemia (Reis et al., 1991, 1998), the neuroprotection evoked from FN was substantial, long-lasting, and reversible. The distribution of salvage was also similar: it was confined to a rim of tissue surrounding an irretrievable core, a pattern corresponding to the distribution of neurons in the ischemic penumbra, which are the ones protected not only by FN stimulation but by pharmacological interventions (Wahlestedt et al., 1993). Interestingly, in excitotoxic and ischemic injury, the area of salvage represents the area in which the toxic stimulus, IBO or hypoxia, respectively, are submaximal. The pattern of salvage therefore can be interpreted to represent a shift to the right of the dose–response curve for the excitotoxicity of IBO because neurons at the rim, exposed to the lowest dosage, are salvaged. Salvage cannot be attributed to alterations in the binding sites for IBO, because the maximal binding of MK801 is unaltered by stimulation (Glickstein et al., 1997), nor is it likely to result from interference in movement of agent through the brain, because diffusion of dyes from the injection site are not affected by FN stimulation (Glickstein et al., 1997). Irrespective of mechanism, the experiment indicates that the central neurogenic neuroprotection elicited from FN is not specific to ischemic injuries, nor is it topographically restricted to cerebral cortex.
Cerebellar substrate for neuroprotection
The area of the FN eliciting neuroprotection, the rostral ventromedial quadrant, is the region of the nucleus from which electrical stimulation potently elevates AP (Miura and Reis, 1970;Takahashi et al., 1995), elevates rCBF globally without modifying rCGU (Nakai et al., 1983), and releases catecholamines from adrenal medulla (Del Bo et al., 1983a), arginine vasopressin from pituitary (Del Bo et al., 1983b), and renin from the kidney (Manning et al., 1985), a response called the fastigial pressor response (FPR) (Miura and Reis, 1970. In unanesthetized animals, such stimulation also elicits a range of consummatory behaviors (Reis et al., 1973). However, electrical stimulation of FN excites not only intrinsic neurons but also axons projecting to and/or through the nucleus. These arise from Purkinje cells of midline cerebellar cortex (De Camilli et al., 1984) and also from axons of collateralized brainstem neurons innervating, via collaterals, not only cerebellum but also regions of the autonomic centers of brainstem, including lateral hypothalamus, periaqueductal gray, nucleus solitarii, parabrachial nucleus, and dorsal tegmental nucleus of pons (Deitrichs, 1985; Deitrichs and Haines, 1985; Deitrichs et al., 1994; Ruggiero et al., 1997). That neurons and axons within FN have different autonomic functions has been demonstrated previously (Chida et al., 1986). Thus, stimulation of fastigial perikarya with excitatory amino acids reduces AP, rCBF, and rCGU, the fastigial depressor response (Chida et al., 1986), whereas, as confirmed here, after FN neurons are destroyed, the FPR persists. Because the FPR is also preserved after removal of the midline cerebellar cortex (Chida et al., 1989) but disappears after bilateral lesions of the rostral ventrolateral medullary reticular nucleus (RVL) (Chida et al., 1990), we have proposed that the FPR is initiated from collaterals of brainstem neurons, themselves innervating RVL (Chida et al., 1989), whereas the FDR is initiated from FN neurons themselves (Fig.9).
Fig. 9.

Possible neuronal circuits mediating neuroprotection and changes in rCBF, rCGU, and AP elicited by stimulation of the FN. A, Neuroprotection results from excitation of intrinsic neurons of FN, which relay through unidentified pathways to protect cortex and striatum. The reduction in AP, rCBF, and rCGU evoked by chemically stimulating FN depends on RVL because bilateral lesions block these responses (Chida et al., 1990).B, Elevations in rCBF and AP elicited by electrical stimulation of FN result from antidromic excitation of brainstem neurons projecting to FN and, as proposed (Chida et al., 1990), collaterally to RVL. It is excitation of reticulospinal neurons of RVL which initiate the elevations of AP and rCBF (for review, see Reis et al., 1994). SG, Sympathetic ganglion.
To determine whether neuroprotection resulted from excitation of intrinsic neurons or axons, neurons of FN were destroyed by an excitotoxin and, 5 d thereafter, the FN was stimulated. Three days later, IBO was injected into the striatum and, 24 hr later, the volumes and distribution of the lesions were assessed. Lesions of FN, but not adjacent DN, abolished neuroprotection. However, as seen previously (Chida et al., 1990), such lesions did not impair the stimulation-evoked elevations of rCBF and AP, indicating as expected the excitotoxin did not damage axons (Iadecola et al., 1987). We also confirmed that stimulation of FN neurons mediates protection against focal ischemia.
These findings have several implications. First, they indicate that neuroprotection is a function of neurons and not axons, and as such it is linked to the network mediating the fastigial depressor response. Second, the findings further supports the view (Reis et al., 1998) that neuroprotection cannot be attributed to the associated elevations of rCBF. The conclusion was argued previously on grounds that neuroprotection lasts for weeks whereas elevations in rCBF are stimulus-locked and that comparable elevations in rCBF elicited from RVL are not neuroprotective (Yamamoto et al., 1993). This study adds the fact that whereas excitotoxic lesions of FN abolish neuroprotection, elevations in rCBF are preserved. Third, the study indicates that the cerebellar elements and, hence, pathways mediating protection and elevations of rCBF differ (Fig. 9). This fact helps to explain why stimulation of RVL, the nucleus essential for the primary elevations of rCBF elicited from RVL (Underwood et al., 1992), is not neuroprotective (Yamamoto et al., 1993). Fourth, the fact that FN lesions abolished stimulation-induced protection against excitotoxic and focal ischemic lesions suggests that the neuronal pathways subserving protection are the same. Presumably, a similar pathway may protect against global ischemia, as well (Golanov et al., 1998). Finally, it indicates a heretofore unrecognized function of the FN: neuroprotection.
The pathways from the rostral FN that might mediate neuroprotection and the cellular mechanisms mediating it are unknown. Projections from rostral and caudal FN in large part differ (Ito, 1984), with the rostral FN projecting to all vestibular nuclei, the dorsal and paramecia medullary reticular formation, several potentially relevant nuclei of pons including locus ceruleus and parabrachial nuclei, subareas of the periaqueductal gray, the centromedian–parafasicular complex (Batton et al., 1977; Haroian, 1982), and even substantia nigra (Snider et al., 1976) and amygdala (Heath and Harper, 1974). It is most likely that the neuroprotective projection is indirect and involves multiple synapses. The cellular mechanism(s) by which stimulation of FN protects the brain against ischemia and excitotoxicity is also unknown. Conditional neuroprotective stimulation of FN stimulation has other effects that predict neuroprotection, including long-lasting suppression of inflammatory responses in cerebral microvessels (Galea et al., 1998a,b) and reduction in neuronal excitability (Golanov and Reis, 1999). The neuroanatomical and cellular substrates of this neuroprotection are presently under investigation.
Footnotes
This research was supported by National Heart, Lung, and Blood Institute Grant P01 HL18947, National Eye Institute Grant T32 EY07138, National Institute of Neurological Diseases and Stroke Grant R01NS36154, American Heart Association, and Irving Harris Foundation.
Correspondence should be addressed to Dr. Donald J. Reis, Division of Neurobiology, Cornell University Medical College, 411 East 69th Street, New York, NY 10021.
REFERENCES
- 1.Baron JC, Rougemont D, Soussaline F, Bustany P, Crouzel C, Bousser MG, Comar D. Local interrelationships of cerebral oxygen consumption and glucose utilization in normal subjects and in ischemic stroke patients: a positron tomography study. J Cereb Blood Flow Metab. 1984;4:140–149. doi: 10.1038/jcbfm.1984.22. [DOI] [PubMed] [Google Scholar]
- 2.Batton RR, III, Jayaraman A, Ruggiero DA, Carpenter MB. Fastigial efferent projections in the monkey: an autoradiographic study. J Comp Neurol. 1977;174:281–306. doi: 10.1002/cne.901740206. [DOI] [PubMed] [Google Scholar]
- 3.Beal MF, Kowall NW, Swartz KJ, Ferrante RJ, Martin JB. Systemic approaches to modifying quinolinic acid striatal lesions in rats. J Neurosci. 1988;8:3901–3908. doi: 10.1523/JNEUROSCI.08-10-03901.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Beirne JP, Pearlstein RD, Massey GW, Warner DS. Effect of halothane in cortical cell cultures exposed to N-methyl-d-aspartate. Neurochem Res. 1998;23:17–23. doi: 10.1023/a:1022489017731. [DOI] [PubMed] [Google Scholar]
- 5.Bidman H-J, Oermann E, Schleicher A, Kato K, Kinscherf R, Buchkremer-Ratzmann I, Witte OW, Zilles K. Copper-zinc superoxide dismutase and isolectin B4 binding are markers for associative and transhemispheric diachisis induced by focal ischemia in rat cortex. Neurosci Lett. 1997;228:163–166. doi: 10.1016/s0304-3940(97)00389-3. [DOI] [PubMed] [Google Scholar]
- 6.Browning JL, Heizer ML, Widmayer MA, Baskin DS. Effects of halothane, α-chloralose, and pCO2 on injury volume and CSF β-endorphin levels in focal cerebral ischemia. Mol Chem Neuropathol. 1997;31:29–42. doi: 10.1007/BF02815158. [DOI] [PubMed] [Google Scholar]
- 7.Busto R, Dietrich WD, Globus MY-T, Valdes I, Scheinberg P, Ginsberg MD. Small differences in intraischemia brain temperature critically determine the extent of ischemic neuronal injury. J Cereb Blood Flow Metab. 1987;7:729–738. doi: 10.1038/jcbfm.1987.127. [DOI] [PubMed] [Google Scholar]
- 8.Caggiano AO, Kraig RP. Neuronal nitric oxide synthase expression is induced in neocortical astrocytes after spreading depression. J Cereb Blood Flow Metab. 1998;18:75–87. doi: 10.1097/00004647-199801000-00008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chida K, Iadecola C, Underwood M, Reis DJ. A novel vasodepressor response elicited from the rat cerebellar fastigial nucleus: the fastigial depressor response. Brain Res. 1986;370:378–382. doi: 10.1016/0006-8993(86)90498-1. [DOI] [PubMed] [Google Scholar]
- 10.Chida K, Iadecola C, Reis DJ. Global reduction in cerebral blood flow and metabolism elicited from intrinsic neurons of fastigial nucleus. Brain Res. 1989;500:177–192. doi: 10.1016/0006-8993(89)90312-0. [DOI] [PubMed] [Google Scholar]
- 11.Chida K, Iadecola C, Reis DJ. Lesions of rostral ventrolateral medulla abolish some cardio- and cerebrovascular components of the cerebellar fastigial pressor and depressor responses. Brain Res. 1990;508:93–104. doi: 10.1016/0006-8993(90)91122-w. [DOI] [PubMed] [Google Scholar]
- 12.Coyle JT, Molliver ME, Kuhar MJ. In situ injection of kainic acid: a new method for selectively lesioning neural cell bodies while sparing axons of passage. J Comp Neurol. 1978;180:301–323. doi: 10.1002/cne.901800208. [DOI] [PubMed] [Google Scholar]
- 13.De Camilli P, Miller PE, Levitt P, Walter U, Greengard P. Anatomy of cerebellar Purkinje cells in the rat determined by a specific immunohistochemical marker. Neuroscience. 1984;11:761–817. doi: 10.1016/0306-4522(84)90193-3. [DOI] [PubMed] [Google Scholar]
- 14.Deitrichs E. Divergent axon collaterals to cerebellum and amygdala from neurons in the parabrachial nucleus locus coeruleus and some adjacent nuclei: a fluorescent double labeling study using rhodamine labeled latex microspheres and fast blue as retrograde tracers. Anat Embryol. 1985;172:75–82. doi: 10.1007/BF00318986. [DOI] [PubMed] [Google Scholar]
- 15.Deitrichs E, Haines DE. Observations on the cerebello-hypothalamic projection, with comments on non-somatic cerebellar circuits. Arch Ital Biol. 1985;123:133–139. [PubMed] [Google Scholar]
- 16.Deitrichs E, Haines DE, Roste GK, Rose LS. Hypothalamocerebellar and cerebellohypothalamic projections: circuits for regulating nonsomatic cerebellar activity? Histol Histopathol. 1994;9:603–614. [PubMed] [Google Scholar]
- 17.Del Bo A, Ross CA, Pardal JF, Saavedra JM, Reis DJ. Fastigial stimulation in rats releases adrenomedullary catecholamines. Am J Physiol. 1983a;244:R801–R809. doi: 10.1152/ajpregu.1983.244.6.R801. [DOI] [PubMed] [Google Scholar]
- 18.Del Bo A, Sved AF, Reis DJ. Fastigial stimulation releases vasopressin in amounts that elevate arterial pressure. Am J Physiol. 1983b;244:H687–H694. doi: 10.1152/ajpheart.1983.244.5.H687. [DOI] [PubMed] [Google Scholar]
- 19.Dietrichs WD, Busto R, Valdes I, Loor Y. Effects of normothermic versus mild hyperthermic forebrain ischemia in rats. Stroke. 1990;21:1318–1325. doi: 10.1161/01.str.21.9.1318. [DOI] [PubMed] [Google Scholar]
- 20.Dirnagl U, Pulsinelli WA. Autoregulation of cerebral blood flow in experimental focal brain ischemia. J Cereb Blood Flow Metab. 1990;10:327–336. doi: 10.1038/jcbfm.1990.61. [DOI] [PubMed] [Google Scholar]
- 21.Duverger D, MacKenzie ET. The quantification of cerebral infarction following focal ischemia in the rat: influence of strain, arterial pressure, blood glucose concentration, and age. J Cereb Blood Flow Metab. 1988;8:449–461. doi: 10.1038/jcbfm.1988.86. [DOI] [PubMed] [Google Scholar]
- 22.Foster AC, Gill R, Woodruff GN. Neuroprotective effects of MK-801 in vivo: selectivity and evidence for delayed degeneration mediated by NMDA receptor activation. J Neurosci. 1988;8:4745–4754. doi: 10.1523/JNEUROSCI.08-12-04745.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Galea E, Golanov EV, Feinstein DL, Kobylarz KA, Glickstein SB, Reis DJ. Cerebellar stimulation reduces inducible nitric oxide synthase expression and protects brain from ischemia. Am J Physiol. 1998a;274:H2035–H2045. doi: 10.1152/ajpheart.1998.274.6.H2035. [DOI] [PubMed] [Google Scholar]
- 24.Galea E, Glickstein SB, Feinstein DL, Golanov EV, Reis DJ. Stimulation of cerebellar fastigial nucleus inhibits interleukin-1β-induced cerebrovascular inflammation. Am J Physiol. 1998b;275:H2053–H2063. doi: 10.1152/ajpheart.1998.275.6.H2053. [DOI] [PubMed] [Google Scholar]
- 25.Glickstein SB, Turner BA, Golanov EV, Regunathan S, Reis DJ. Electrical stimulation of cerebellar fastigial nucleus protects against excitotoxic degeneration of striatal neurons. J Cereb Blood Flow Metab. 1997;17:S127. [Google Scholar]
- 26.Golanov EV, Reis DJ. Contribution of oxygen-sensitive neurons of the rostral ventrolateral medulla to hypoxic cerebral vasodilation in the rat. J Physiol (Lond) 1996;495:201–216. doi: 10.1113/jphysiol.1996.sp021585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Golanov EV, Reis DJ. Neuroprotective electrical stimulation of cerebellar fastigial nucleus attenuates expression of periinfarction depolarizing waves (PIDS) and inhibits cortical spreading depression. Brain Res. 1999;818:304–315. doi: 10.1016/s0006-8993(98)01169-x. [DOI] [PubMed] [Google Scholar]
- 28.Golanov EV, Yamamoto S, Reis DJ. Electrical stimulation of cerebellar fastigial nucleus fails to rematch blood flow and metabolism in focal ischemic infarctions. Neurosci Lett. 1996;210:181–184. doi: 10.1016/0304-3940(96)12682-3. [DOI] [PubMed] [Google Scholar]
- 29.Golanov EV, Liu F, Reis DJ. Stimulation of cerebellum protects hippocampal neurons from global ischemia. NeuroReport. 1998;9:819–824. doi: 10.1097/00001756-199803300-00010. [DOI] [PubMed] [Google Scholar]
- 30.Guiliano R, Ruggiero DA, Morrison S, Ernsberger P, Reis DJ. Cholinergic regulation of arterial pressure by the CA area of the rostral ventrolateral medulla. J Neurosci. 1989;9:923–942. doi: 10.1523/JNEUROSCI.09-03-00923.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Haroian AJ. Cerebello-olivary projections in the rat: an autoradiographic study. Brain Res. 1982;235:125–130. doi: 10.1016/0006-8993(82)90201-3. [DOI] [PubMed] [Google Scholar]
- 32.Heath RG, Harper JW. Ascending projections of the cerebellar fastigial nucleus to the hippocampus, amygdala, and other temporal lobe sites: evoked potential and histological studies in monkeys and cats. Exp Neurol. 1974;45:268–287. doi: 10.1016/0014-4886(74)90118-6. [DOI] [PubMed] [Google Scholar]
- 33.Iadecola C, Arneric SP, Baker HD, Tucker LW, Reis DJ. Role of local neurons in the cerebrocortical vasodilation elicited from cerebellum. Am J Physiol. 1987;252:R1082–R1091. doi: 10.1152/ajpregu.1987.252.6.R1082. [DOI] [PubMed] [Google Scholar]
- 34.Ito M. The cerebellum and neural control. Raven; New York: 1984. [Google Scholar]
- 35.Loeb WF, Quimby FW. The clinical chemistry of laboratory animals, p 519. Pergamon; New York: 1989. [Google Scholar]
- 36.MacLennon H, Lodge D. The antagonism of amino acid-induced excitation of spinal neurones in the cat. Brain Res. 1979;169:83–90. doi: 10.1016/0006-8993(79)90375-5. [DOI] [PubMed] [Google Scholar]
- 37.Maier CM, Ashern KV, Cheng ML, Lee JE, Yenari MA, Steinberg GK, Kirsch JR. Optimal depth and duration of mild hypothermia in a focal model of transient cerebral ischemia: effects on neurologic outcome, infarct size, apoptosis, and inflammation. Stroke. 1998;29:2171–2180. doi: 10.1161/01.str.29.10.2171. [DOI] [PubMed] [Google Scholar]
- 38.Manning JW, Hartle DK, Ammons WS, Koyama S. The median preoptic area in cardiovascular reflex activity. J Auton Nerv Syst. 1985;12:239–249. doi: 10.1016/0165-1838(85)90064-5. [DOI] [PubMed] [Google Scholar]
- 39.Martin LJ, Al-Abdulla NA, Brambrink AM, Kirsch JR, Sieber FE, Portera-Cailliau C. Neurodegeneration in excitotoxicity, global cerebral ischemia, and target deprivation: a perspective on the contributions of apoptosis and necrosis. Brain Res Bull. 1998;46:281–309. doi: 10.1016/s0361-9230(98)00024-0. [DOI] [PubMed] [Google Scholar]
- 40.Miura M, Reis DJ. A blood pressure response from fastigial nucleus and its relay pathway in brainstem. Am J Physiol. 1970;219:1330–1336. doi: 10.1152/ajplegacy.1970.219.5.1330. [DOI] [PubMed] [Google Scholar]
- 41.Miyazawa T, Hossmann K-A. Methodological requirements for accurate measurements of brain and body temperature during global forebrain ischemia of rat. J Cereb Blood Flow Metab. 1992;12:817–822. doi: 10.1038/jcbfm.1992.113. [DOI] [PubMed] [Google Scholar]
- 42.Nagasawa H, Kogure K, Fujiwara T, Itoh M, Ido T. Metabolic disturbances in exo-focal brain areas after cortical stroke studied by positron emission tomography. J Neurol Sci. 1994;123:147–153. doi: 10.1016/0022-510x(94)90217-8. [DOI] [PubMed] [Google Scholar]
- 43.Nakai M, Iadecola C, Ruggiero DA, Tucker LW, Reis DJ. Electrical stimulation of cerebellar fastigial nucleus increases cerebral cortical blood flow without change in local metabolism: evidence for an intrinsic system in brain for primary vasodilation. Brain Res. 1983;260:35–49. doi: 10.1016/0006-8993(83)90762-x. [DOI] [PubMed] [Google Scholar]
- 44.Nathan MA, Reis DJ. Hypoxemia, atelectasis, and the elevation of arterial pressure and heart rate in paralyzed artificially ventilated rat. Life Sci. 1975;16:1103–1120. doi: 10.1016/0024-3205(75)90195-2. [DOI] [PubMed] [Google Scholar]
- 45.Ogilvy CS, Chu D, Kaplan S. Mild hypothermia, hypertension, and mannitol are protective against infarction during experimental intracranial temporary vessel occlusion. Neurosurgery. 1996;38:1202–1209. doi: 10.1097/00006123-199606000-00030. [DOI] [PubMed] [Google Scholar]
- 46.Reis DJ, Doba N, Nathan MA. Predatory attack, grooming and consummatory behavior evoked by electrical stimulation of cerebellar nuclei in cat. Science. 1973;182:845–847. doi: 10.1126/science.182.4114.845. [DOI] [PubMed] [Google Scholar]
- 47.Reis DJ, Berger SB, Underwood MD, Khayata M. Electrical stimulation of cerebellar fastigial nucleus reduces ischemic infarction elicited by middle cerebral artery occlusion in rat. J Cereb Blood Flow Metab. 1991;11:810–818. doi: 10.1038/jcbfm.1991.139. [DOI] [PubMed] [Google Scholar]
- 48.Reis DJ, Golanov EV, Ruggiero DA, Sun M-K. Sympatho-excitatory neurons of the rostral ventrolateral medulla are oxygen sensors and essential elements in the tonic and reflex control of the systemic and cerebral circulations. J Hypertens. 1994;12:S159–S180. [PubMed] [Google Scholar]
- 49.Reis DJ, Golanov EV, Galea E, Feinstein DL. Central neurogenic neuroprotection: central neural systems that protect the brain from hypoxia and ischemia. Ann NY Acad Sci. 1997;835:168–186. doi: 10.1111/j.1749-6632.1997.tb48628.x. [DOI] [PubMed] [Google Scholar]
- 50.Reis DJ, Golanov EV, Kobylarz KA, Yamamoto S. Brief electrical stimulation of cerebellar fastigial nucleus conditions long-lasting salvage from focal cerebral ischemia: conditioned central neurogenic neuroprotection. Brain Res. 1998;780:161–165. [PubMed] [Google Scholar]
- 51.Roos JA, Jackson-Friedman C, Lyden P. Effects of hyperbaric oxygen on neurologic outcome for cerebral ischemia in rats. Acad Emerg Med. 1998;5:18–24. doi: 10.1111/j.1553-2712.1998.tb02569.x. [DOI] [PubMed] [Google Scholar]
- 52.Ruggiero DA, Anwar M, Golanov EV, Reis DJ. The pedunculopontine tegmental nucleus issues collaterals to the fastigial nucleus and rostral ventrolateral reticular nucleus in the rat. Brain Res. 1997;760:272–276. doi: 10.1016/s0006-8993(97)00397-1. [DOI] [PubMed] [Google Scholar]
- 53.Snider RS, Maiti A, Snider SR. Cerebellar pathways to ventral midbrain and nigra. Exp Neurol. 1976;53:714–728. doi: 10.1016/0014-4886(76)90150-3. [DOI] [PubMed] [Google Scholar]
- 54.Takahashi S, Crane AM, Jehle J, Cook M, Kennedy C, Sokoloff L. Role of the cerebellar fastigial nucleus in the physiological regulation of cerebral blood flow. J Cereb Blood Flow Metab. 1995;15:128–142. doi: 10.1038/jcbfm.1995.15. [DOI] [PubMed] [Google Scholar]
- 55.Tamura A, Graham DI, McCulloch J, Teasdale GM. Focal cerebral ischaemia in the rat. I. Description of a technique and early neuropathological consequences following middle cerebral artery occlusion. J Cereb Blood Flow Metab. 1981;1:53–60. doi: 10.1038/jcbfm.1981.6. [DOI] [PubMed] [Google Scholar]
- 56.Underwood MD, Iadecola C, Sved A, Reis DJ. Stimulation of C1 area neurons globally increases regional cerebral blood flow but not metabolism. J Cereb Blood Flow Metab. 1992;12:844–855. doi: 10.1038/jcbfm.1992.116. [DOI] [PubMed] [Google Scholar]
- 57.Volpe BT, Wildmann J, Altar CA. Brain-derived neurotrophic factor prevents the loss of nigral neurons induced by excitotoxic striatal-pallidal lesions. Neuroscience. 1998;83:741–748. doi: 10.1016/s0306-4522(97)00424-7. [DOI] [PubMed] [Google Scholar]
- 58.Wahlestedt C, Golanov E, Yamamoto S, Yee F, Ericson H, Yoo H, Inturrisi CE, Reis DJ. Antisense oligodeoxynucleotides to the NMDA-R1 receptor channel protect cortical neurons from excitotoxicity and reduce focal ischaemic infarctions. Nature. 1993;363:260–263. doi: 10.1038/363260a0. [DOI] [PubMed] [Google Scholar]
- 59.Warner DS, Ludwig PS, Pearlstein R, Brinkhous AD. Halothane reduces focal ischemic injury in the rat when brain temperature is controlled. Anesthesiology. 1995;82:1237–1245. doi: 10.1097/00000542-199505000-00019. [DOI] [PubMed] [Google Scholar]
- 60.Wong EH, Kemp JA, Priestley T, Knight AR, Woodruff GN, Iversen LL. The anticonvulsant MK-801 is a potent N-methyl-d-aspartate antagonist. Proc Natl Acad Sci USA. 1986;83:7104–7108. doi: 10.1073/pnas.83.18.7104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Yamamoto S, Golanov EV, Reis DJ. Reductions in focal ischemic infarctions elicited from cerebellar fastigial nucleus do not result from elevations in cerebral blood flow. J Cereb Blood Flow Metab. 1993;13:1020–1024. doi: 10.1038/jcbfm.1993.128. [DOI] [PubMed] [Google Scholar]
- 62.Zhang FY, Iadecola C. Fastigial stimulation increases ischemic blood flow and reduces brain damage after focal ischemia. J Cereb Blood Flow Metab. 1993;13:1013–1019. doi: 10.1038/jcbfm.1993.127. [DOI] [PubMed] [Google Scholar]