

Abstract
Nuclear factor-κB (NF-κB) is activated in brain cells after various insults, including cerebral ischemia and epileptic seizures. Although cell culture studies have suggested that the activation of NF-κB can prevent neuronal apoptosis, the role of this transcription factor in neuronal injury in vivo is unclear, and the specific κB subunits involved are unknown. We now report that mice lacking the p50 subunit of NF-κB exhibit increased damage to hippocampal pyramidal neurons after administration of the excitotoxin kainate. Gel-shift analyses showed that p50 is required for the majority of κB DNA-binding activity in hippocampus. Intraventricular administration of κB decoy DNA before kainate administration in wild-type mice resulted in an enhancement of damage to hippocampal pyramidal neurons, indicating that reduced NF-κB activity was sufficient to account for the enhanced excitotoxic neuronal injury in p50−/− mice. Cultured hippocampal neurons from p50−/− mice exhibited enhanced elevations of intracellular calcium levels and increased levels of oxidative stress after exposure to glutamate and were more vulnerable to excitotoxicity than were neurons from p50+/+ and p50+/− mice. Collectively, our data demonstrate an important role for the p50 subunit of NF-κB in protecting neurons against excitotoxic cell death.
Keywords: calcium, epileptic seizures, hippocampus, kainic acid, stroke, superoxide dismutase, transcription, tumor necrosis factor
Nuclear factor-κB (NF-κB) exists in the cytosol as an inducible three subunit complex consisting of two (prototypical) subunits of 50 kDa (p50) and 65 kDa (p65; RelA) and an inhibitory subunit called I-κB. NF-κB activation occurs when I-κB is induced to dissociate from the complex. The p50–p65 dimer then translocates to the nucleus and binds to 5′ regulatory elements of genes responsive to NF-κB (for review, see Baeuerle and Baltimore, 1996; Mattson, 1998). NF-κB is expressed in the nervous system and shows a low level of constitutive activity in neurons (Kaltschmidt et al., 1994). NF-κB activity is increased greatly in neurons after seizure activity (Prasad et al., 1994; Grilli et al., 1996; Rong and Baudry, 1996; Matsuoka et al., 1999) and ischemia (Salminen et al., 1995; Clemens et al., 1997, 1998; Carroll et al., 1998; Zhang et al., 1998). Injury-related signals that can activate NF-κB in neurons include tumor necrosis factor (Barger et al., 1995), glutamate (Kaltschmidt et al., 1995), nerve growth factor (Carter et al., 1996;Maggirwar et al., 1998), and reactive oxygen species (Schreck and Baeuerle, 1994; Guo et al., 1998). Because the activation of NF-κB is associated with cell injury and death in many different pathological settings, it has been proposed that the activation of NF-κB contributes to the cell death process (Grilli et al., 1996; Clemens et al., 1997). However, cell culture studies suggest that the activation of NF-κB represents a cytoprotective response that can, in fact, prevent neuronal apoptosis (Cheng et al., 1994; Barger et al., 1995;Mattson et al., 1997; Taglialatela et al., 1997). Gene targets that may mediate the anti-apoptotic action of NF-κB in neurons include those encoding manganese superoxide dismutase (Mn-SOD; Mattson et al., 1997) and the calcium-binding protein calbindin D28k (Cheng et al., 1994).
Although the activation of NF-κB in neurons may be beneficial, the activation of NF-κB in microglia may stimulate potentially neurotoxic cascades involving the production of nitric oxide and excitotoxins (Barger and Harmon, 1997; Kim and Ko, 1998). It is therefore unclear whether the net result of the activation of NF-κB in neurons and glial cells after brain injury in vivo is beneficial or detrimental for neurons. Knock-out of the p65 subunit of NF-κB by targeted gene disruption results in embryonic lethality (Beg et al., 1995). In contrast, mice develop normally in the absence of the p50 subunit of NF-κB, although such p50-deficient mice exhibit altered lymphocyte responses when challenged with lipopolysaccharide and infectious agents (Sha et al., 1995; Snapper et al., 1996). It is not known whether p50 plays roles in cellular responses to brain injury. In the present study we used p50 knock-out mice and a method for suppression of NF-κB activation, using κB decoy DNA (Mattson et al., 1997), to determine the role of NF-κB directly in the death of hippocampal neurons after the administration of the seizure-inducing excitotoxin kainate. The data suggest that p50 is required for kainate-induced κB DNA-binding activity in hippocampus and that the activation of NF-κB protects hippocampal pyramidal neurons against excitotoxic injury.
MATERIALS AND METHODS
Mice and procedures for administration of kainate and κB decoy DNA. The gene-targeting strategy used to generate lines of mice lacking p50 has been described previously (Sha et al., 1995;Snapper et al., 1996). Homozygous p50 knock-out (p50−/−) mice exhibit no overt phenotype but do exhibit abnormalities in responses of B lymphocytes to bacterial lipopolysaccharide and reduced resistance to certain infectious agents. Mice were maintained on a random C57BL/6 × 129 background. Experiments were performed in 3-month-old male mice (25–30 gm body weight); the mice were fed ad libitum and maintained on a 12 hr light/dark cycle. Kainate was administered via stereotaxic injection into the dorsal hippocampus by methods detailed in our previous studies (Bruce et al., 1996; Guo et al., 1999). Briefly, kainate (0.1 or 0.2 μg in a volume of 0.5 μl) was injected unilaterally into dorsal hippocampus (dorsoventral, −2.0; mediolateral, +2.4; anteroposterior, −0.8 from bregma) of anesthetized mice. All mice that were administered kainate exhibited seizures within the first hour after injection. The mice were killed at designated time points after kainate administration, and brain tissue was prepared for histological analyses or for gel-shift assays as described previously (Bruce et al., 1996; Guo et al., 1998). κB decoy DNA was prepared by annealing single-stranded oligonucleotides of the following sequences: 5′-GAGGGGACTTTCCCT-3′ and 5′-AGGGAAAGTCCCCTC-3′. Control DNA with a scrambled sequence was prepared by annealing oligonucleotides of the following sequences: 5′-GATGCGTCTGTCGCA-3′ and 5′-TGCGACAGACGCATC-3′ Stocks of double-stranded DNA were prepared at a concentration of 2 mm in saline. Decoy DNA was administered via stereotaxic injection into the lateral ventricles as described previously (Smith-Swintosky et al., 1994).
Histological procedures. Coronal brain sections (30 μm) were cut on a freezing microtome and were stained with cresyl violet. Nissl-positive undamaged neurons were counted in hippocampal regions CA1, CA3, and hilus (three 45× fields per region per section and three sections per hippocampus); counts were performed without knowledge of the genotype or treatment history of the mice. Immunohistochemistry was performed in free-floating sections, using the methods described previously (Bruce et al., 1996). Briefly, the sections were incubated for 2 hr at room temperature in a solution containing 0.2% Triton X-100 and 1% normal horse serum in PBS. Then the sections were incubated overnight at 4°C in primary antibody (a mouse monoclonal antibody against microtubule-associated protein-2 (MAP-2); 1:250 dilution; Sigma, St. Louis, MO) in PBS. Sections then were washed with PBS and incubated for 2 hr at room temperature in the presence of biotinylated secondary antibody, followed by a washing in PBS. Next the sections were incubated for 1 hr in ABC reagent (Vector Laboratories, Burlingame, CA), washed in PBS, and incubated for 5 min in nickel-enhanced DAB solution (Vector Laboratories). The sections were air-dried, mounted, observed, and photographed under bright-field optics.
Gel-shift and supershift assays. Nuclear protein extracts were prepared according to the methods reported previously, with some modifications (Guo et al., 1998). Briefly, after being removed from storage at −80°C, brain tissues were immersed immediately in 1 ml of ice-cold lysis buffer containing (in mm) 10 KCl, 1.5 MgCl2, 0.5 dithiothreitol, 0.5 phenylmethylsulfonyl fluoride, and 10 mm HEPES, pH 7.9, plus 2 μg/ml pepstatin A, 2 μg/ml leupeptin, and 2 μg/mll-leucinethiol. Samples were homogenized immediately on ice with a Dounce homogenizer, using pestles A (10 strokes) and B (5 strokes). Samples were kept on ice for 15 min, and then 25 μl of 10% Nonidet P-40 was added in the homogenate. After a brief vortexing, they were incubated on ice for another 20 min and were centrifuged at 12,500 rpm for 30 sec. The pelleted nuclei were resuspended in 50–100 μl of extraction buffer consisting of (in mm) 420 NaCl, 1.5 MgCl2, 0.2 EDTA, 0.5 dithiothreitol, 0.5 phenylmethylsulfonyl fluoride, and 20 HEPES, pH 7.9, plus 2 μg/ml pepstatin A, 2 μg/ml leupeptin, and 2 μg/mll-leucinethiol and were incubated on ice for 30 min. The nuclear suspension was centrifuged at 12,500 rpm for 15 min at 4°C, and the supernatant containing the nuclear protein extracts was saved. Protein concentration of the nuclear extract was determined by the Bio-Rad protein assay reagent (Richmond, CA). Aliquots of the nuclear extracts were stored at −80°C for the gel-shift/supershift assay. The gel-shift assay was performed with a commercial DNA-binding protein detection system (Life Technologies, Gaithersburg, MD), as described by the manufacturer. Briefly, a 5 μg aliquot of extracted nuclear protein was preincubated in a reaction buffer containing (in mm) 500 NaCl, 5 EDTA, 5 dithiothreitol, and 50 Tris, pH 7.5, plus 20% glycerol and 0.4 mg/ml salmon sperm DNA. After 15 min of incubation on ice, approximately 1 × 105 cpm of32P end-labeled double-stranded oligonucleotide containing the κB consensus sequence 5-TCAGAGGGGACTTTCCGAGAGG-3was added to the reaction, and the mixture was incubated for 20 min at room temperature. Then 2 ml of 0.1% bromphenol blue dye was added to each sample, and a 25 μl aliquot of the sample was electrophoresed through a 6% nondenaturing polyacrylamide gel for 105 min at 150 V. The gel was dried and exposed to x-ray film, using intensifier screens at −70°C. The specificity of the identified κB-binding proteins in the nuclear extracts was determined by adding a 100-fold excess of unlabeled competitor DNA to the reaction. For gel supershift analysis, extracted nuclear proteins (5 μg) were incubated with 1 μg of polyclonal antibodies against the p65, p50, and/or c-Rel proteins (Santa Cruz Biotechnology, Santa Cruz, CA) for 45 min before the addition of32P-labeled double-stranded NF-κB oligonucleotide; then gel-shift analysis was performed as described above.
Primary hippocampal cell cultures and measurements of neuronal survival, intracellular calcium levels, and levels of reactive oxygen species. The method for establishing primary cultures of hippocampal neurons from postnatal day 1 mice was described in our previous study (Guo et al., 1999). Cells were maintained in Neurobasal medium with B27 supplements (Life Technologies), and experiments were performed in 8-d-old cultures. Immediately before experimental treatment the medium was replaced with Locke’s buffer [containing (in mm) 154 NaCl, 5.6 KCl, 2.3 CaCl2, 1.0 MgCl2, 3.6 NaHCO3, 5 glucose, and 5 HEPES, pH 7.2]. Glutamate was prepared as a 200× stock in Locke’s buffer. Neuron survival was quantified as described previously (Cheng et al., 1994). Neurons in premarked microscope fields were counted before, and at indicated time points after, exposure to glutamate. Neurons with intact neurites and a cell body that was smooth and round to oval-shaped were considered viable, whereas neurons with beaded or fragmented neurites and a cell body that was shrunken and rough in appearance were considered nonviable. Analyses were performed without knowledge of the treatment history of the cultures. Intracellular free calcium levels ([Ca2+]i) were quantified by fluorescence ratio imaging of the calcium indicator dye fura-2, using methods described previously (Cheng et al., 1994). Briefly, the cells were loaded with the acetoxymethylester form of fura-2 (30 min incubation in the presence of 10 μmfura-2) and imaged with a Zeiss AttoFluor system with a 40× oil objective. The average [Ca2+]i in individual neuronal cell bodies was determined from the ratio of the fluorescence emissions obtained by using two different excitation wavelengths (334 and 380 nm). The system was calibrated with solutions containing either no Ca2+ or a saturating level of Ca2+ (1 mm) by the formula: [Ca2+]i =KD[(R −Rmin)/(Rmax−R)](Fo/Fs). Peroxide levels were measured by confocal microscope analysis of cellular dichlorofluorescein (DCF) fluorescence, as described previously (Mattson et al., 1995).
RESULTS
NF-κB DNA-binding activity is decreased and neuronal damage is increased after kainate administration in hippocampus of mice lacking p50
Gel-shift analysis of NF-κB DNA-binding proteins in hippocampal nuclear extracts from control (saline-injected) wild-type mice revealed two prominent bands (Fig.1A). The intensity of the upper band was increased markedly after kainate administration, whereas the intensity of the lower band was not changed (Fig.1A). The upper shifted band was completely absent in p50−/− mice, both under basal conditions and after kainate administration, whereas the lower shifted band was present in both p50+/+ and p50−/− mice (Fig. 1A). Supershift analysis showed that the upper band was shifted by an antibody against p50 and by an antibody against p65, but not by an antibody against c-Rel (Fig. 1B). In contrast, the lower band was not shifted by any of the three antibodies. These results indicated that the upper shifted band consists of p50/p65 heterodimers and is the major kainate-inducible κB-binding complex present in hippocampus. Excess unlabeled κB DNA eliminated the NF-κB binding activity of the upper shifted band, demonstrating the specificity of binding of those proteins to the DNA (Fig.1C). The lower shifted band, which was partially competed by cold competitor DNA, appears to correspond to the novel neuronal κB-binding protein recently described by Moerman et al. (1999) for which physiological roles remain to be determined.
Fig. 1.

κB DNA-binding activity is greatly reduced in hippocampal tissue from p50 knock-out mice. A, Gel-shift analysis of nuclear extracts from hippocampi of p50−/− mice and p50+/+ mice that had been killed 4 hr after intrahippocampal administration of either saline or kainate (KA). Similar results were obtained in two additional experiments. LS, Lower shifted band. B, Supershift analysis of κB-binding proteins present in nuclear extracts of hippocampus from a wild-type mouse (8 hr after kainate). Samples of nuclear extracts (5 μg) were preincubated for 45 min (before the addition of radiolabeled κB DNA) in the absence of additions (lane 1) or in the presence of a 200-fold excess of unlabeled cold probe or antibody against p65, p50, and/or c-Rel, as indicated. LS, Lower shifted band.C, Samples of nuclear extracts (5 μg) were preincubated for 45 min in the absence of additions (lane 1) or in the presence of a 100- or 50-fold excess of unlabeled cold probe (lanes 2, 3).
We next injected saline or kainate into the dorsal hippocampus unilaterally in wild-type and p50−/−mice and then killed the mice either 6 or 24 hr later. Analyses of cresyl violet-stained coronal sections of hippocampus from wild-type mice showed that ∼30 and 70% of CA1 and CA3 neurons were damaged (in the kainate-injected hippocampus) at the 6 and 24 hr time points, respectively (Figs. 2,3A,B). There was a significant enhancement of neuronal degeneration in region CA1 at the 6 hr time point and in regions CA3 and CA1 at the 24 hr time point in the p50−/− mice. Kainate also caused more damage to hilar neurons at the 24 hr time point (Fig.3B). At the dose that was used, kainate caused little or no damage to neurons in the uninjected hippocampus of p50+/+ mice. However, in p50−/− mice there was extensive damage to CA1 and CA3 neurons in the contralateral (uninjected) hippocampus (Fig. 3C). Collectively, these data demonstrate increased vulnerability to excitotoxicity of hippocampal pyramidal neurons in mice lacking p50.
Fig. 2.
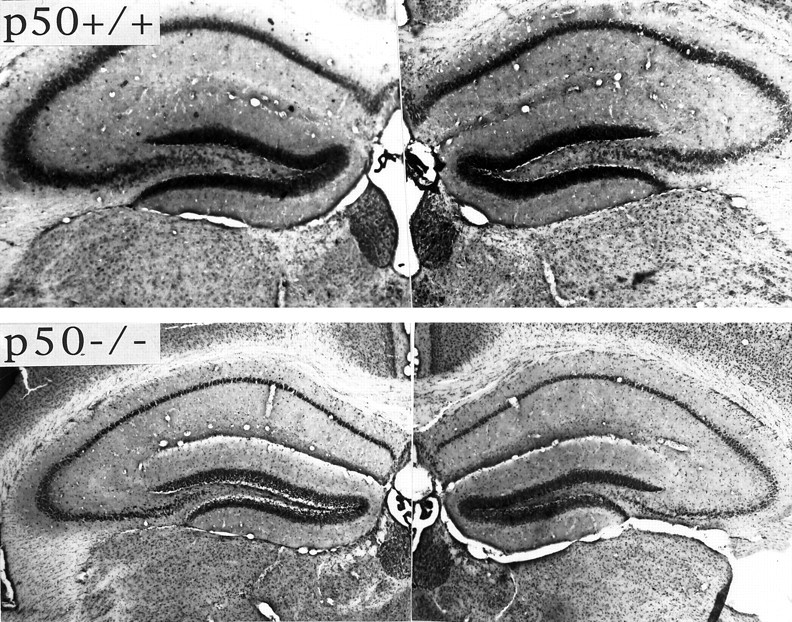
Cresyl violet-stained sections of hippocampi from p50+/+ (top) and p50−/− (bottom) mice 24 hr after the administration of kainate into the right hippocampus. Note that the extent of neuronal damage in regions CA3 and CA1 of right hippocampus is greater in the p50−/− mouse and that there is also damage to CA1 neurons in the contralateral hippocampus of the p50−/− mouse, but not in the p50+/+ mouse.
Fig. 3.

Vulnerability of hippocampal neurons to kainate-induced damage is increased in mice lacking p50.A,B, p50+/+ and p50−/− mice were administered either saline or 0.2 μg of kainate into the dorsal hippocampus. Mice were killed either 6 hr (A) or 24 hr (B) later, and the numbers of undamaged neurons in regions CA1, CA3, and hilus of the injected hippocampus were quantified (see Materials and Methods). Values are the mean and SD (n = 8 mice per group). *p < 0.05 and **p < 0.01 as compared with the corresponding Control value. ##p < 0.01 as compared with the corresponding value for group p50+/+, KA; ANOVA with Scheffé’s post hoctests. C, p50+/+ and p50−/− mice were administered either saline or 0.2 μg of kainate into the dorsal hippocampus. The mice were killed 24 hr later, and the numbers of undamaged neurons in regions CA1, CA3, and hilus of the contralateral (uninjected) hippocampus were quantified. Values are the mean and SD (n = 8 mice per group). *p < 0.05 as compared with p50+/+, saline value. ###p < 0.001 as compared with the corresponding value for group p50+/+, KA; ANOVA with Scheffé’s post hoc tests.
κB decoy DNA increases the vulnerability of hippocampal neurons to excitotoxicity
We (Mattson et al., 1997) and others (Taglialatela et al., 1997) have shown that administration of κB decoy DNA can suppress NF-κB activity and increase vulnerability of cultured neurons to apoptosis induced by several different insults. In the present study we administered double-stranded κB decoy DNA or control double-stranded DNA with a scrambled sequence (see Materials and Methods) into the lateral ventricles of adult wild-type mice (two injections at 24 and 2 hr before kainate administration) and then killed the mice either 8 hr after kainate administration for gel-shift analysis of NF-κB activity or 24 hr after kainate for quantification of damage to hippocampal neurons. For these studies the dose of kainate was reduced to 0.1 μg to reduce the extent of neuronal damage in control mice so that any exacerbation of damage by κB decoy DNA could be detected more readily. In a preliminary experiment we administered fluorescein-tagged κB decoy DNA (60 μg) into the lateral ventricles, killed the mice 2 hr later, and prepared coronal brain sections for examination with a microscope with epifluorescence illumination. Many neurons throughout the hippocampus exhibited intense fluorescence, including cells corresponding to Nissl-positive pyramidal neurons in region CA1 (Fig.4A), indicating that the DNA was taken up by the neurons. Weaker fluorescence was present in cells in neuropil, suggesting that the DNA also was taken up by glia. The cell-associated fluorescence was attributable to the uptake of intact labeled DNA, because no cell-associated fluorescence was observed after intraventricular administration of fluorescein-labeled decoy DNA that had been preincubated with DNase-I (data not shown). These results are consistent with previous studies that demonstrated the penetration of oligonucleotides of similar size as our κB decoy DNA into both neurons and glia in vivo (Grzanna et al., 1998) and showed their specific efficacy in modulating target cell function (Meiri et al., 1997; Engelmann et al., 1998).
Fig. 4.

Intraventricular administration of κB decoy DNA results in increased vulnerability of hippocampal neurons to kainate-induced damage. A, Micrographs showing κB decoy DNA-associated fluorescence (top panel) and Nissl staining (bottom panel) in region CA1 of a hippocampal section from a mouse that was killed 2 hr after the intraventricular administration of 60 μg of fluorescein-labeled κB decoy DNA. Note that many neurons exhibit intense fluorescence.B, Gel-shift analysis showing NF-κB DNA-binding activity in hippocampal nuclear extracts from wild-type mice that had received intraventricular injections of vehicle, 60 μg of κB decoy DNA (Decoy), or 60 μg of scrambled control DNA (ScDNA) at 24 and 2 hr before the administration of either saline or kainate (KA; 0.1 μg injection into the dorsal hippocampus). The mice were killed 8 hr after kainate administration, and nuclear extracts of hippocampal tissue were prepared and subjected to gel-shift analysis (3 μg of nuclear extract per lane). Similar results were obtained in two additional experiments.C, Mice received intraventricular injections of 60 μg of κB decoy DNA (Decoy) or scrambled control DNA (ScDNA) at 24 and 2 hr before the administration of either saline or kainate (0.1 μg injection into the dorsal hippocampus). The mice were killed 24 hr later, and the numbers of undamaged neurons in regions CA1, CA3, and hilus of the ipsilateral hippocampus were quantified. Values are the mean and SD (n = 8 mice per group). *p < 0.05 as compared with the corresponding Control value; #p < 0.01 as compared with each of the other values for that region of hippocampus; ANOVA with Scheffé’spost hoc tests.
We next performed gel-shift analyses to determine whether levels of NF-κB activity were altered in hippocampal cells after intraventricular administration of κB decoy DNA. The kainate-induced increase in NF-κB activity was suppressed completely in hippocampus of mice that were administered κB decoy DNA but was unaffected in mice that were administered control scrambled DNA (Fig.4B). Quantification of neuronal damage in hippocampus 24 hr after kainate administration revealed a marked enhancement of the degeneration of CA1 pyramidal neurons and hilar neurons in the mice that were administered κB decoy DNA as compared with mice that were administered saline or scrambled control DNA (Fig.4C). When taken together with the data obtained in studies of p50−/− mice, the data suggest that NF-κB plays a key neuroprotective role in the kainate model of excitotoxic neuronal injury.
Increased vulnerability of hippocampal neurons lacking p50 to excitotoxicity is correlated with increased intracellular calcium levels
To determine whether the increased vulnerability of hippocampal neurons in p50−/− mice in vivo was the result of a lack of p50 specifically in neurons and to begin to examine the underlying mechanisms, we studied primary hippocampal neurons in cultures established from p50+/+, p50+/−, and p50−/− mice. The vulnerability of hippocampal neurons to increasing concentrations of glutamate was determined in cultures from mice of each genotype. Neurons from p50−/− mice were significantly more vulnerable to glutamate toxicity than were neurons from p50+/+ mice, and the vulnerability of neurons from p50+/− mice was intermediate to that of neurons from p50+/+ and p50−/− mice (Fig.5A). Glutamate neurotoxicity is mediated by calcium influx through NMDA receptor channels and voltage-dependent calcium channels (Mattson et al., 1993; Choi, 1994). We recently provided evidence that the activation of NF-κB in cultured rat hippocampal neurons results in the stabilization of cellular calcium homeostasis such that calcium responses to glutamate are decreased (Furukawa and Mattson, 1998). To determine whether a lack of p50 affects neuronal calcium homeostasis after exposure to glutamate, we measured calcium responses to glutamate, using the calcium indicator dye fura-2 in hippocampal neurons from p50+/+, p50+/−, and p50−/− mice. Basal levels of [Ca2+]i were ∼70–75 nm in neurons of each genotype (Fig.5B,C). Exposure of neurons from p50+/+ mice to 10 μm glutamate resulted in a rapid increase of [Ca2+]i to a peak level of ∼210 nm within 30 sec and a subsequent reduction to a plateau level of ∼175 nm by 13 min after treatment. Both the peak (380 nm) and plateau (300 nm) [Ca2+]i were significantly greater in neurons from p50−/− mice as compared with neurons from p50+/+ mice (Fig. 5B,C). The [Ca2+]iresponse to glutamate in neurons from p50+/− mice was intermediate to that of p50+/+ and p50−/− mice. MAP-2 is a dendritic microtubule-associated protein that is sensitive to calcium-mediated proteolysis; levels of MAP-2 immunoreactivity decrease in molecular layers of CA1 and CA3 after kainate administration (Smith-Swintosky et al., 1996; Arias et al., 1997). Consistent with a role for increased levels of [Ca2+]iin the increased vulnerability of hippocampal neurons to excitotoxicityin vivo, we found that the extent of decrease in MAP-2 immunoreactivity in molecular layers of region CA1 was greater in p50−/− mice than in p50+/+ mice (Fig.6).
Fig. 5.

Cultured hippocampal neurons from p50−/− mice exhibit increased neuronal death and enhanced elevations of intracellular calcium levels after exposure to glutamate. A, Hippocampal cultures from p50+/+, p50+/−, and p50−/− mice were exposed for 24 hr to the indicated concentrations of glutamate, and the neuronal survival was quantified. Values are the mean and SD of determinations made in four to six cultures. *p < 0.05 and *p < 0.01 as compared with the corresponding value for p50+/+ mice; ANOVA with Scheffé’spost hoc tests. B, The [Ca2+]i was measured in neurons from p50+/+, p50+/−, and p50−/− mice at the indicated time points before and after exposure to 5 μm glutamate. Each data point represents the mean of 10–15 neurons. C, The basal (immediately before exposure to glutamate), peak (1 min after glutamate), and plateau (13 min after glutamate) levels of [Ca2+]i were quantified in neurons from p50+/+, p50+/−, and p50−/− mice. Values are the mean and SD (n = 4 cultures, with measurements made in 10–15 neurons per culture). *p < 0.05 and *p < 0.01 as compared with the value for cultures from p50+/+ mice; ANOVA with Scheffé’spost hoc tests.
Fig. 6.

Kainate-induced loss of MAP-2 immunoreactivity in hippocampus is exacerbated in mice lacking p50. Micrographs of MAP-2 immunoreactivity in hippocampi of p50+/+ and p50−/− mice, 24 hr after intrahippocampal administration of 0.2 μg kainate. Note the greater loss of MAP-2 immunoreactivity in molecular layers of region CA1 in the p50−/− mouse as compared with the p50+/+ mouse. Similar results were obtained in analyses of six p50+/+ and six p50−/− mice.
We had shown previously that glutamate-induced elevations of [Ca2+]i lead to increased levels of reactive oxygen species in cultured hippocampal neurons (Mattson et al., 1995). In the present study we found that the magnitude of glutamate-induced increase in levels of reactive oxygen species (measured with the probe DCF; see Materials and Methods) was greater in neurons from the p50−/− mice than in neurons from p50+/+ mice. Basal levels of DCF fluorescence (average pixel intensity per neuron; mean ± SD of determinations made in four cultures with 12–15 neurons analyzed per culture) were 29 ± 8 in neurons from p50+/+ mice and 37 ± 6 in neurons from p50−/− mice. Levels of DCF fluorescence 2 hr after exposure to 5 μm glutamate were 100 ± 14 in neurons from p50+/+ mice and 144 ± 15 in neurons from p50−/− mice (p < 0.05).
DISCUSSION
Although NF-κB activation occurs in neurons after injury to the nervous system, the role of this activation in the injury outcome has been unclear. Many investigators that documented increased NF-κB activity under conditions in which neurons were dying assumed that NF-κB was contributing to the cell death (Grilli et al., 1996). However, subsequent cell culture studies provided evidence that the activation of NF-κB can protect neurons against oxidative and metabolic insults (Barger et al., 1995; Mattson et al., 1997) and death after trophic factor withdrawal (Taglialatela et al., 1997). We found that excitotoxic damage to hippocampal neurons in vivo was increased in mice lacking the p50 subunit of NF-κB and after the administration of κB decoy DNA, two conditions in which injury-induced NF-κB activity was suppressed. On the basis of experiments that used pharmacological agents such as aspirin that inhibit NF-κB activation, it was proposed that NF-κB activity contributes to neuronal death (Grilli et al., 1996; Clemens et al., 1997). Unfortunately, such agents are not specific inhibitors of NF-κB and possess antioxidant and anti-inflammatory actions that may account for their neuroprotective actions (Barneoud and Curet, 1999). In contrast, we studied mice in which NF-κB activity was inhibited selectively by the genetic deletion of p50 or treatment with κB decoy DNA. Our data strongly support a neuroprotective role for p50 and NF-κB activation in vivo, at least in the present model of excitotoxic neuronal degeneration.
Our gel-shift analyses revealed two distinct shifted bands. The upper band appears to consist of p50/p65 heterodimers, because the band was shifted by both p50 and p65 antibodies and because it was absent in p50−/− mice. The lower shifted band appears to correspond to the band recently described by Moerman et al. (1999) in their study that characterizes κB-binding proteins in neural cells. Their data are consistent with a role for this κB-binding protein in the nervous system. However, the lower shifted band was present in p50−/− mice and was not affected by kainate, suggesting that it is unlikely to play a role in the increased vulnerability of hippocampal neurons p50−/− mice and κB decoy DNA-treated mice to excitotoxicity.
Several findings suggest that the excitoprotective action of NF-κB activation in vivo results from its activation in neurons. First, previous cell culture studies have shown that the activation of NF-κB in neurons results in increased resistance to apoptosis and excitotoxicity (Barger et al., 1995; Barger and Mattson, 1996; Goodman and Mattson, 1996). Second, blockade of the NF-κB activity that used decoy DNA or pharmacological agents greatly increases the vulnerability of cultured neurons to several different insults (Mattson et al., 1997; Taglialatela et al., 1997). Third, activation of NF-κB in microglial cells induces the production of inflammatory cytokines and toxins that may be damaging to neurons (Colasanti et al., 1995;Barger and Harmon, 1997; Bonaiuto et al., 1997; Kim and Ko, 1998). Suppression of NF-κB in microglia therefore should reduce neuronal damage in vivo, a result not observed in the present study. In addition, increased levels of NF-κB activity measured within 4–8 hr of kainate administration are not likely to represent activation in microglia, because microglial activation appears to be delayed by at least 24–48 hr in this model (Andersson et al., 1991). However, a contribution of NF-κB activation in glia to neuroprotection in vivo cannot be ruled out, because astrocytes and microglia produce several different neurotrophic factors and cytokines that can protect neurons against excitotoxicity and apoptosis (for review, see Mattson and Furukawa, 1996).
The p65 subunit of NF-κB is necessary for normal embryonic development and survival (Beg et al., 1995), whereas mice lacking p50−/− survive and reproduce relatively normally (Sha et al., 1995). However, when mice lacking p50 are challenged by exposure to infectious agents, they exhibit altered immune responses (Sha et al., 1995; Snapper et al., 1996). Recent studies of p50−/− mice suggest increased vulnerability to apoptosis of cells from several different tissues. Exposure of p50−/− mice to murine encephalomyocarditis virus results in increased apoptosis of fibroblasts (Schwarz et al., 1998), and increased apoptosis may contribute to the reduced eosinophilia in allergic airway inflammation (Yang et al., 1998). Our gel-shift analyses indicate that p50 is required for the majority of κB DNA-binding activity in hippocampus, and our analyses of neuronal vulnerability to excitotoxicity reveal an important role for p50 in protecting hippocampal neurons against excitotoxic injury in vivo and in cell culture. Overactivation of glutamate receptors in hippocampal neurons can induce either apoptosis or necrosis in vivo (Pollard et al., 1994;Yang et al., 1997) and in cell culture (Ankarcrona et al., 1995). We did not determine whether the increased vulnerability of neurons lacking p50 resulted from increased apoptosis and/or increased necrosis. However, previous cell culture studies have shown that the activation of NF-κB is particularly effective in preventing neuronal apoptosis (Barger et al., 1995; Mattson et al., 1997; Taglialatela et al., 1997). An anti-apoptotic function of NF-κB is strongly suggested by data from studies of lymphocyte cell death also (Wang et al., 1996;Wu et al., 1996). Interestingly, both pro-apoptotic signaling cascades (Mattson et al., 1998; Duan et al., 1999) and NF-κB (Kaltschmidt et al., 1993; Meberg et al., 1996) can be activated in synaptic terminals, suggesting important roles for neurodegenerative and neuroprotective signaling at the level of the synapse. Such synaptic signaling mechanisms likely play key roles in the kainate model of excitotoxic injury that was used in the present study.
Recent studies are elucidating the identity of the κB-responsive genes that may mediate increased resistance of neurons to excitotoxic and apoptotic insults. The antioxidant enzyme Mn-SOD is induced in hippocampal neurons by tumor necrosis factor-α (TNFα) in vivo (Bruce et al., 1996) and in cell culture in which treatment with κB decoy DNA prevents Mn-SOD induction (Mattson et al., 1997). Overexpression of Mn-SOD protects cultured neural cells and neuronsin vivo against oxidative and ischemic injury (Keller et al., 1998). We previously reported that Mn-SOD levels increase in hippocampal pyramidal neurons after kainate administration (Bruce et al., 1996). We have found that levels of TNFα and Mn-SOD are increased to a much greater extent after kainate administration in hippocampal pyramidal neurons in p50+/+mice as compared with p50−/− mice (Z. Yu and M. P. Mattson, unpublished data), suggesting that the p50 subunit of NF-κB plays an important role in the induction of TNFα and Mn-SOD in hippocampal neurons after excitotoxic insults. Other κB-responsive genes that have been linked to neuroprotection are those encoding members of the inhibitors of apoptosis (IAP) family (Xu et al., 1997; Stehlik et al., 1998; Simons et al., 1999) and the calcium-binding protein calbindin (Cheng et al., 1994).
It is well known that there is differential vulnerability of subpopulations of neurons in the hippocampus to excitotoxic and metabolic insults. For example, CA3 and CA1 neurons are more vulnerable than dentate granule cells to seizure-induced injury (Nadler et al., 1978), whereas CA1 neurons are selectively vulnerable to transient global forebrain ischemia (Pulsinelli and Brierley, 1979). It is not known whether different subpopulations of hippocampal neurons express different levels of NF-κB subunits nor whether they exhibit differential activation of NF-κB or express different κB-responsive genes after injury. Our data suggest that p50 and NF-κB activation plays a particularly important role in protecting CA1 neurons against excitotoxic injury. A comparison of the data in Figure 3, Aand B, indicates that CA1 neurons in p50−/− mice die much more rapidly as compared with CA1 neurons in wild-type mice and with CA3 neurons in p50−/− mice. In addition, there was marked degeneration of CA1 and CA3 neurons in the hippocampus contralateral to that receiving kainate in p50−/− mice, whereas there was little or no degeneration of these neuronal populations in the contralateral hippocampus of wild-type mice. Although not extensively studied, available data also suggest differential expression of κB-responsive genes among hippocampal neurons under basal conditions and after brain injury. For example, Mn-SOD is expressed at higher levels in CA3 neurons than in CA1 or dentate granule neurons under basal conditions (Akai et al., 1990), and Mn-SOD increases in CA3 and CA1 neurons after kainate administration (Bruce et al., 1996). Calbindin D28k, another putative gene target of NF-κB (Cheng et al., 1994), is induced in hippocampal CA1 and dentate granule neurons by seizures and stress (Krugers et al., 1996; Lee et al., 1997) and is therefore another potential mediator of κB-induced neuroprotection. Collectively, the emerging data suggest important roles for NF-κB in modifying neuronal injury responses and further suggest a potential role for differential NF-κB signaling in selective neuronal vulnerability in various neurodegenerative disorders. The ability of NF-κB to prevent excitotoxic neuronal death suggests that therapeutic interventions aimed at activating NF-κB may prove beneficial in the many different neurodegenerative conditions (e.g., stroke, epileptic seizures, and Alzheimer’s and Parkinson’s diseases) that are believed to involve an excitotoxic component.
Footnotes
This work was supported by grants to M.P.M. from the National Institute on Aging and National Institute of Neurological Diseases and Stroke and to D.Z. from the National Institute of Mental Health. We thank W. Fu, J. Partin, and J. Yu for technical assistance.
Correspondence should be addressed to Dr. Mark P. Mattson, 211 Sanders-Brown Building, University of Kentucky, Lexington, KY 40536.
REFERENCES
- 1.Akai F, Maeda M, Suzuki K, Inagaki S, Takagi H, Taniguchi N. Immunocytochemical localization of manganese superoxide dismutase (Mn-SOD) in the hippocampus of the rat. Neurosci Lett. 1990;115:19–23. doi: 10.1016/0304-3940(90)90510-g. [DOI] [PubMed] [Google Scholar]
- 2.Andersson PB, Perry VH, Gordon S. The kinetics and morphological characteristics of the macrophage–microglial response to kainic acid-induced neuronal degeneration. Neuroscience. 1991;42:201–214. doi: 10.1016/0306-4522(91)90159-l. [DOI] [PubMed] [Google Scholar]
- 3.Ankarcrona M, Dypbuky JM, Bonfoco E, Zhivotovsky B, Orrenius S, Lipton SA, Nicotera P. Glutamate-induced neuronal death: a succession of necrosis or apoptosis depending on mitochondrial function. Neuron. 1995;15:961–973. doi: 10.1016/0896-6273(95)90186-8. [DOI] [PubMed] [Google Scholar]
- 4.Arias C, Arrieta I, Massieu L, Tapia R. Neuronal damage and MAP2 changes induced by the glutamate transport inhibitor dihydrokainate and by kainate in rat hippocampus in vivo. Exp Brain Res. 1997;116:467–476. doi: 10.1007/pl00005774. [DOI] [PubMed] [Google Scholar]
- 5.Baeuerle P, Baltimore D. NF-κB: ten years after. Cell. 1996;87:13–20. doi: 10.1016/s0092-8674(00)81318-5. [DOI] [PubMed] [Google Scholar]
- 6.Barger SW, Harmon AD. Microglial activation by Alzheimer amyloid precursor protein and modulation by apolipoprotein E. Nature. 1997;388:878–881. doi: 10.1038/42257. [DOI] [PubMed] [Google Scholar]
- 7.Barger SW, Mattson MP. Induction of neuroprotective κB-dependent transcription by secreted forms of the Alzheimer’s β-amyloid precursor. Mol Brain Res. 1996;40:116–126. doi: 10.1016/0169-328x(96)00036-8. [DOI] [PubMed] [Google Scholar]
- 8.Barger SW, Horster D, Furukawa K, Goodman Y, Krieglestein J, Mattson MP. Tumor necrosis factors α and β protect neurons against amyloid β-peptide toxicity: evidence for involvement of a κB-binding factor and attenuation of peroxide and Ca2+ accumulation. Proc Natl Acad Sci USA. 1995;92:9328–9332. doi: 10.1073/pnas.92.20.9328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Barneoud P, Curet O. Beneficial effects of lysine acetylsalicylate, a soluble salt of aspirin, on motor performance in a transgenic model of amyotrophic lateral sclerosis. Exp Neurol. 1999;155:243–251. doi: 10.1006/exnr.1998.6984. [DOI] [PubMed] [Google Scholar]
- 10.Beg AA, Sha WC, Bronson RT, Ghosh S, Baltimore D. Embryonic lethality and liver degeneration in mice lacking the RelA component of NF-κB. Nature. 1995;376:167–170. doi: 10.1038/376167a0. [DOI] [PubMed] [Google Scholar]
- 11.Bonaiuto C, McDonald PP, Rossi F, Cassatella MA. Activation of nuclear factor-κB by β-amyloid peptides and interferon-γ in murine microglia. J Neuroimmunol. 1997;77:51–56. doi: 10.1016/s0165-5728(97)00054-4. [DOI] [PubMed] [Google Scholar]
- 12.Bruce AJ, Boling W, Kindy MS, Peschon J, Kraemer PJ, Carpenter MK, Holtsberg FW, Mattson MP. Altered neuronal and microglial responses to brain injury in mice lacking TNF receptors. Nat Med. 1996;2:788–794. doi: 10.1038/nm0796-788. [DOI] [PubMed] [Google Scholar]
- 13.Carroll JE, Howard EF, Hess DC, Wakade CG, Chen Q, Cheng C. Nuclear factor-κB activity during cerebral reperfusion: effect of attenuation with N-acetylcysteine treatment. Mol Brain Res. 1998;56:186–191. doi: 10.1016/s0169-328x(98)00045-x. [DOI] [PubMed] [Google Scholar]
- 14.Carter BD, Kaltschmidt C, Kaltschmidt B, Offenhauser N, Bohm-Matthaei R, Baeuerle PA, Barde YA. Selective activation of NF-κB by nerve growth factor through the neurotrophin receptor p75. Science. 1996;272:542–545. doi: 10.1126/science.272.5261.542. [DOI] [PubMed] [Google Scholar]
- 15.Cheng B, Christakos S, Mattson MP. Tumor necrosis factors protect neurons against excitotoxic/metabolic insults and promote maintenance of calcium homeostasis. Neuron. 1994;12:139–153. doi: 10.1016/0896-6273(94)90159-7. [DOI] [PubMed] [Google Scholar]
- 16.Choi DW. Calcium and excitotoxic neuronal injury. Ann NY Acad Sci. 1994;747:162–171. doi: 10.1111/j.1749-6632.1994.tb44407.x. [DOI] [PubMed] [Google Scholar]
- 17.Clemens JA, Stephenson DT, Smalstig EB, Dixon EP, Little SP. Global ischemia activates nuclear factor-κB in forebrain neurons of rats. Stroke. 1997;28:1073–1080. doi: 10.1161/01.str.28.5.1073. [DOI] [PubMed] [Google Scholar]
- 18.Clemens JA, Stephenson DT, Yin T, Smalstig EB, Panetta JA, Little SP. Drug-induced neuroprotection from global ischemia is associated with prevention of persistent but not transient activation of nuclear factor-κB in rats. Stroke. 1998;29:677–682. doi: 10.1161/01.str.29.3.677. [DOI] [PubMed] [Google Scholar]
- 19.Colasanti M, Persichini T, Menegazzi M, Mariotto S, Giordano E, Caldarera CM, Sogos V, Lauro GM, Suzuki H. Induction of nitric oxide synthase mRNA expression. Suppression by exogenous nitric oxide. J Biol Chem. 1995;270:26731–26733. doi: 10.1074/jbc.270.45.26731. [DOI] [PubMed] [Google Scholar]
- 20.Duan W, Rangnekar V, Mattson MP. Par-4 production in synaptic compartments following apoptotic and excitotoxic insults: evidence for a pivotal role in mitochondrial dysfunction and neuronal degeneration. J Neurochem. 1999;72:2312–2322. doi: 10.1046/j.1471-4159.1999.0722312.x. [DOI] [PubMed] [Google Scholar]
- 21.Engelmann M, Landgraf R, Lorscher P, Conzelmann C, Probst JC, Holsboer F, Reul JM. Downregulation of brain mineralocorticoid and glucocorticoid receptor by antisense oligodeoxynucleotide treatment fails to alter spatial navigation in rats. Eur J Pharmacol. 1998;361:17–26. doi: 10.1016/s0014-2999(98)00702-x. [DOI] [PubMed] [Google Scholar]
- 22.Furukawa K, Mattson MP. The transcription factor NF-κB mediates increases in calcium currents and decreases in NMDA and AMPA/kainate-induced currents in response to TNFα in hippocampal neurons. J Neurochem. 1998;70:1876–1886. doi: 10.1046/j.1471-4159.1998.70051876.x. [DOI] [PubMed] [Google Scholar]
- 23.Goodman Y, Mattson MP. Ceramide protects hippocampal neurons against excitotoxic and oxidative insults and amyloid β-peptide toxicity. J Neurochem. 1996;66:869–872. doi: 10.1046/j.1471-4159.1996.66020869.x. [DOI] [PubMed] [Google Scholar]
- 24.Grilli M, Pizzi M, Memo M, Spano P. Neuroprotection by aspirin and sodium salicylate through blockade of NF-κB activation. Science. 1996;274:1383–1385. doi: 10.1126/science.274.5291.1383. [DOI] [PubMed] [Google Scholar]
- 25.Grzanna R, Dubin JR, Dent GW, Ji Z, Zhang W, Ho SP, Hartig PR. Intrastriatal and intraventricular injections of oligodeoxynucleotides in the rat brain: tissue penetration, intracellular distribution, and c-fos antisense effects. Mol Brain Res. 1998;63:35–52. doi: 10.1016/s0169-328x(98)00238-1. [DOI] [PubMed] [Google Scholar]
- 26.Guo Q, Robinson N, Mattson MP. Secreted APPα counteracts the pro-apoptotic action of mutant presenilin-1 by activation of NF-κB and stabilization of calcium homeostasis. J Biol Chem. 1998;273:12341–12351. doi: 10.1074/jbc.273.20.12341. [DOI] [PubMed] [Google Scholar]
- 27.Guo Q, Fu W, Sopher BL, Miller MW, Ware CB, Martin GM, Mattson MP. Increased vulnerability of hippocampal neurons to excitotoxic necrosis in presenilin-1 mutant knock-in mice. Nat Med. 1999;5:101–106. doi: 10.1038/4789. [DOI] [PubMed] [Google Scholar]
- 28.Kaltschmidt C, Kaltschmidt B, Baeuerle PA. Brain synapses contain inducible forms of the transcription factor NF-κB. Mech Dev. 1993;43:135–147. doi: 10.1016/0925-4773(93)90031-r. [DOI] [PubMed] [Google Scholar]
- 29.Kaltschmidt C, Kaltschmidt B, Neumann H, Wekerle H, Baeuerle PA. Constitutive NF-κB activity in neurons. Mol Cell Biol. 1994;14:3981–3992. doi: 10.1128/mcb.14.6.3981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kaltschmidt C, Kaltschmidt B, Baeuerle PA. Stimulation of ionotropic glutamate receptors activates transcription factor NF-κB in primary neurons. Proc Natl Acad Sci USA. 1995;92:9618–9622. doi: 10.1073/pnas.92.21.9618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Keller JN, Kindy MS, Holtsberg FW, St. Clair D, Yen HC, Germeyer A, Steiner SM, Bruce-Keller AJ, Hutchins JB, Mattson MP. Mitochondrial Mn-SOD prevents neural apoptosis and reduces ischemic brain injury: suppression of peroxynitrite production, lipid peroxidation, and mitochondrial dysfunction. J Neurosci. 1998;18:687–697. doi: 10.1523/JNEUROSCI.18-02-00687.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kim WK, Ko KH. Potentiation of N-methyl-d-aspartate-mediated neurotoxicity by immunostimulated murine microglia. J Neurosci Res. 1998;54:17–26. doi: 10.1002/(SICI)1097-4547(19981001)54:1<17::AID-JNR3>3.0.CO;2-K. [DOI] [PubMed] [Google Scholar]
- 33.Krugers HJ, Koolhaas JM, Medema RM, Korf J. Prolonged subordination stress increases calbindin-D28k immunoreactivity in the rat hippocampal CA1 area. Brain Res. 1996;729:289–293. [PubMed] [Google Scholar]
- 34.Lee S, Williamson J, Lothman EW, Szele FG, Chesselet MF, Von Hagen S, Sapolsky RM, Mattson MP, Christakos S. Early induction of mRNA for calbindin-D28k and BDNF but not NT-3 in rat hippocampus after kainic acid treatment. Mol Brain Res. 1997;47:183–194. doi: 10.1016/s0169-328x(97)00043-0. [DOI] [PubMed] [Google Scholar]
- 35.Maggirwar SB, Sarmiere PD, Dewhurst S, Freeman RS. Nerve growth factor-dependent activation of NF-κB contributes to survival of sympathetic neurons. J Neurosci. 1998;18:10356–10365. doi: 10.1523/JNEUROSCI.18-24-10356.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Matsuoka K, Kitamura Y, Okazaki M, Terai K, Taniguchi T. Kainic acid-induced activation of nuclear factor-κB in rat hippocampus. Exp Brain Res. 1999;124:215–222. doi: 10.1007/s002210050616. [DOI] [PubMed] [Google Scholar]
- 37.Mattson MP. Free radicals, calcium, and the synaptic plasticity– cell death continuum: emerging roles of the transcription factor NF-κB. Int Rev Neurobiol. 1998;42:103–168. doi: 10.1016/s0074-7742(08)60609-1. [DOI] [PubMed] [Google Scholar]
- 38.Mattson MP, Furukawa K. Programmed cell life: anti-apoptotic signaling and therapeutic strategies for neurodegenerative disorders. Restor Neurol Neurosci. 1996;9:191–205. doi: 10.3233/RNN-1996-9401. [DOI] [PubMed] [Google Scholar]
- 39.Mattson MP, Kumar K, Cheng B, Wang H, Michaelis EK. Basic FGF regulates the expression of a functional 71 kDa NMDA receptor protein that mediates calcium influx and neurotoxicity in cultured hippocampal neurons. J Neurosci. 1993;13:4575–4588. doi: 10.1523/JNEUROSCI.13-11-04575.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Mattson MP, Lovell MA, Furukawa K, Markesbery WR. Neurotrophic factors attenuate glutamate-induced accumulation of peroxides, elevation of [Ca2+]i and neurotoxicity, and increase antioxidant enzyme activities in hippocampal neurons. J Neurochem. 1995;65:1740–1751. doi: 10.1046/j.1471-4159.1995.65041740.x. [DOI] [PubMed] [Google Scholar]
- 41.Mattson MP, Goodman Y, Luo H, Fu W, Furukawa K. Activation of NF-κB protects hippocampal neurons against oxidative stress-induced apoptosis: evidence for induction of Mn-SOD and suppression of peroxynitrite production and protein tyrosine nitration. J Neurosci Res. 1997;49:681–697. doi: 10.1002/(SICI)1097-4547(19970915)49:6<681::AID-JNR3>3.0.CO;2-3. [DOI] [PubMed] [Google Scholar]
- 42.Mattson MP, Keller JN, Begley JG. Evidence for synaptic apoptosis. Exp Neurol. 1998;153:35–48. doi: 10.1006/exnr.1998.6863. [DOI] [PubMed] [Google Scholar]
- 43.Meberg PJ, Kinney WR, Valcourt EG, Routtenberg A. Gene expression of the transcription factor NF-κB in hippocampus: regulation by synaptic activity. Mol Brain Res. 1996;38:179–190. doi: 10.1016/0169-328x(95)00229-l. [DOI] [PubMed] [Google Scholar]
- 44.Meiri N, Ghelardini C, Tesco G, Galeotti N, Dahl D, Tomsic D, Cavallaro S, Quattrone A, Capaccioli S, Bartolini A, Alkon DL. Reversible antisense inhibition of Shaker-like Kv1.1 potassium channel expression impairs associative memory in mouse and rat. Proc Natl Acad Sci USA. 1997;94:4430–4434. doi: 10.1073/pnas.94.9.4430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Moerman AM, Mao X, Lucas MM, Barger SW. Characterization of a neuronal κB-binding factor distinct from NF-κB. Mol Brain Res. 1999;67:303–315. doi: 10.1016/s0169-328x(99)00091-1. [DOI] [PubMed] [Google Scholar]
- 46.Nadler J, Perry B, Cotman CW. Intraventricular kainic acid preferentially destroys hippocampal pyramidal cells. Nature. 1978;271:676–677. doi: 10.1038/271676a0. [DOI] [PubMed] [Google Scholar]
- 47.Pollard H, Charriaut-Marlangue C, Cantagrel S, Represa A, Robain O, Moreau J, Ben-Ari Y. Kainate-induced apoptotic cell death in hippocampal neurons. Neuroscience. 1994;63:7–18. doi: 10.1016/0306-4522(94)90003-5. [DOI] [PubMed] [Google Scholar]
- 48.Prasad AV, Pilcher WH, Joseph SA. Nuclear factor-κB in rat brain: enhanced DNA-binding activity following convulsant-induced seizures. Neurosci Lett. 1994;170:145–148. doi: 10.1016/0304-3940(94)90260-7. [DOI] [PubMed] [Google Scholar]
- 49.Pulsinelli WA, Brierley JB. A new model of bilateral hemispheric ischemia in the unanesthetized rat. Stroke. 1979;10:267–272. doi: 10.1161/01.str.10.3.267. [DOI] [PubMed] [Google Scholar]
- 50.Rong Y, Baudry M. Seizure activity results in a rapid induction of nuclear factor-κB in adult but not juvenile rat limbic structures. J Neurochem. 1996;67:662–668. doi: 10.1046/j.1471-4159.1996.67020662.x. [DOI] [PubMed] [Google Scholar]
- 51.Salminen A, Liu PK, Hsu CY. Alteration of transcription factor binding activities in the ischemic rat brain. Biochem Biophys Res Commun. 1995;212:939–944. doi: 10.1006/bbrc.1995.2060. [DOI] [PubMed] [Google Scholar]
- 52.Schreck R, Baeuerle PA. Assessing oxygen radicals as mediators in activation of inducible eukaryotic transcription factor NF-κB. Methods Enzymol. 1994;234:151–163. doi: 10.1016/0076-6879(94)34085-4. [DOI] [PubMed] [Google Scholar]
- 53.Schwarz EM, Badorff C, Hiura TS, Wessely R, Badorff A, Verma IM, Knowlton KU. NF-κB-mediated inhibition of apoptosis is required for encephalomyocarditis virus virulence: a mechanism of resistance in p50 knock-out mice. J Virol. 1998;72:5654–5660. doi: 10.1128/jvi.72.7.5654-5660.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Sha WC, Liou HC, Tuomanen EI, Baltimore D. Targeted disruption of the p50 subunit of NF-κB leads to multifocal defects in immune responses. Cell. 1995;80:321–330. doi: 10.1016/0092-8674(95)90415-8. [DOI] [PubMed] [Google Scholar]
- 55.Simons M, Beinroth S, Gleichmann M, Liston P, Korneluk RG, MacKenzie AE, Bahr M, Klockgether T, Robertson GS, Weller M, Schulz JB. Adenovirus-mediated gene transfer of inhibitors of apoptosis protein delays apoptosis in cerebellar granule neurons. J Neurochem. 1999;72:292–301. doi: 10.1046/j.1471-4159.1999.0720292.x. [DOI] [PubMed] [Google Scholar]
- 56.Smith-Swintosky VL, Pettigrew LC, Craddock SD, Culwell AR, Rydel RE, Mattson MP. Secreted forms of β-amyloid precursor protein protect against ischemic brain injury. J Neurochem. 1994;63:781–784. doi: 10.1046/j.1471-4159.1994.63020781.x. [DOI] [PubMed] [Google Scholar]
- 57.Smith-Swintosky VL, Pettigrew LC, Sapolsky RM, Phares C, Craddock SD, Brooke SM, Mattson MP. Metyrapone, an inhibitor of glucocorticoid production, reduces brain injury induced by focal and global ischemia and seizures. J Cereb Blood Flow Metab. 1996;16:585–598. doi: 10.1097/00004647-199607000-00008. [DOI] [PubMed] [Google Scholar]
- 58.Snapper CM, Zelazowski P, Rosas FR, Kehry MR, Tian M, Baltimore D, Sha WC. B cells from p50/NF-κB knock-out mice have selective defects in proliferation, differentiation, germ-line CH transcription, and Ig class switching. J Immunol. 1996;156:183–191. [PubMed] [Google Scholar]
- 59.Stehlik C, de Martin R, Kumabashiri I, Schmid JA, Binder BR, Lipp J. Nuclear factor (NF)-κB-regulated X-chromosome-linked IAP gene expression protects endothelial cells from tumor necrosis factor α-induced apoptosis. J Exp Med. 1998;188:211–216. doi: 10.1084/jem.188.1.211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Taglialatela G, Robinson R, Perez-Polo JR. Inhibition of nuclear factor-κB (NF-κB) activity induces nerve growth factor-resistant apoptosis in PC12 cells. J Neurosci Res. 1997;47:155–162. [PubMed] [Google Scholar]
- 61.Wang C-Y, Mayo MW, Baldwin AS. TNF- and cancer therapy-induced apoptosis: potentiation by inhibition of NF-κB. Science. 1996;274:784–787. doi: 10.1126/science.274.5288.784. [DOI] [PubMed] [Google Scholar]
- 62.Wu M, Lee H, Bellas RE, Schauer SL, Arsura M, Katz D, Fitzgerald MF, Rothstein TL, Sherr DH, Sonenshein GE. Inhibition of NF-κB/Rel induces apoptosis of murine B cells. EMBO J. 1996;15:4682–4690. [PMC free article] [PubMed] [Google Scholar]
- 63.Xu DG, Crocker SJ, Doucet JP, St-Jean M, Tamai K, Hakim AM, Ikeda JE, Liston P, Thompson CS, Korneluk RG, MacKenzie A, Robertson GS. Elevation of neuronal expression of NAIP reduces ischemic damage in the rat hippocampus. Nat Med. 1997;3:997–1004. doi: 10.1038/nm0997-997. [DOI] [PubMed] [Google Scholar]
- 64.Yang DD, Kuan CY, Whitmarsh AJ, Rincon M, Zheng TS, Davis RJ, Rakic P, Flavell RA. Absence of excitotoxicity-induced apoptosis in the hippocampus of mice lacking the Jnk3 gene. Nature. 1997;389:865–870. doi: 10.1038/39899. [DOI] [PubMed] [Google Scholar]
- 65.Yang L, Cohn L, Zhang DH, Homer R, Ray A, Ray P. Essential role of nuclear factor-κB in the induction of eosinophilia in allergic airway inflammation. J Exp Med. 1998;188:1739–1750. doi: 10.1084/jem.188.9.1739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Zhang S, Tobaru T, Zivin JA, Shackelford DA. Activation of nuclear factor-κB in the rabbit spinal cord following ischemia and reperfusion. Mol Brain Res. 1998;63:121–132. [PubMed] [Google Scholar]