

Abstract
Cocaine is a highly addictive drug that binds to the dopamine transporter (DAT), inhibits the reuptake of dopamine, and initiates multiple actions within midbrain dopaminergic systems. Using the rhesus monkey, we have investigated the consequences of in utero cocaine exposure on the expression of DAT in the fetal brain. By using the selective DAT ligand [125I]RTI-121and tyrosine hydroxylase (TH) immunocytochemistry, we found that DAT binding sites are highly developed by day 70 of gestation and show a distribution pattern similar to TH. The rank order of specific 3β-(4-[125I]iodophenyl)tropane-2β-carboxylic acid isopropyl ester ([125I]RTI-121) binding densities was substantia nigra–ventral tegmental area > putamen > caudate > lateral hypothalamus > accumbens > linear/interfascicular nuclei ≥ globus pallidus > prefrontal cortex. Furthermore, we observed that DAT mRNA was differentially expressed within fetal midbrain dopamine neurons with the highest levels detected in the ventral tier of the substantia nigra pars compacta, and the lowest levels in the ventral tegmental area and the linear/interfascicular nuclei. In utero cocaine exposure between days 22 and 70 significantly increased DAT mRNA expression, and the density of [125I]RTI-121 binding sites within midbrain dopamine neurons in the 70-d-old fetus. This increased DAT expression is accompanied by other presynaptic and postsynaptic neuronal changes, which collectively suggest that midbrain dopamine neurons are hypoactive after prolonged cocaine exposure, a state that may be a contributing factor in the development of attention deficit disorders observed in subjects exposed prenatally to cocaine.
Keywords: cocaine, dopamine transporter, tyrosine hydroxylase, fetus, rhesus monkey, [125I]RTI-121, in situhybridization histochemistry, autoradiography, immunocytochemistry
Dopamine neurotransmission is important for the control of movement, reward-reinforced learning, cognition, and emotion (Wickens, 1990; Glenthoj, 1995; Gerfen and Wilson, 1996). Pathophysiology of this system is implicated in a number of disorders, including schizophrenia, mood disorder, and drug addiction (Koob, 1992; Brown and Gershon, 1993; Self and Nestler, 1995;Knable et al., 1997). Disturbances of dopamine neurotransmission may also play an important role in attention deficit hyperactivity disorder (ADHD) (Hunt et al., 1994; Searight et al., 1995; Sagvolden and Sergeant, 1998). The main symptoms of ADHD, which is a complex developmental disorder, are inattentiveness, impulsivity, and hyperactivity (Stevenson, 1992; Hunt et al., 1994; Cook et al., 1995;Solanto, 1998; Taylor, 1998). Some of the same symptoms are also described frequently in children who were exposed to cocaine and other drugs in utero (Struthers and Hansen, 1992; Mayes et al., 1995,1998; Tronick et al., 1996; Chasnoff et al., 1998).
Cocaine binds to dopamine, norepinephrine, and serotonin transporters and thus affects many physiological functions (Ritz et al., 1987;Fischman, 1988). However, the acute reinforcing properties of cocaine are attributable primarily to its binding to the dopamine transporter (DAT), which affects the midbrain–rostral forebrain dopaminergic circuitry (Ritz et al., 1987; Koob and LeMoal, 1997). Chronic use of cocaine leads to neuronal adaptations, including upregulation of the cAMP pathway, which contributes to addiction with symptoms such as emotional distress, drug craving, and relapse (Koob and LeMoal, 1997;Nestler and Aghajanian, 1997). Fetal cocaine exposure through maternal use of the drug also results in long-term adaptations, as evidenced by alterations in dopamine release, dopamine receptors, receptor–G-protein coupling, and increased sensitivity to stressors in the offspring (Dow-Edwards et al., 1990; Scalzo et al., 1990; Friedman et al., 1996; Keller, et al., 1996; Choi et al., 1998; Spear et al., 1998).
In an effort to characterize cocaine-induced alterations in the developing fetus, we have used a nonhuman primate model, which exhibits disruption of the midbrain–rostral forebrain dopamine circuitry after gestational cocaine exposure (Rønnekleiv et al., 1998). The rhesus monkey is a particularly useful model because its in uterodevelopment is similar to that of the human (Gribnau and Geijsberts, 1981); in particular, the development of the dopamine neurocircuitry exhibits many of the same characteristics (Verney et al., 1991;Rønnekleiv and Naylor, 1995; Aubert et al., 1997; Fang et al., 1997;Unis et al., 1998). Our previous studies demonstrated that gestational cocaine exposure decreases the mRNA expression of tyrosine hydroxylase (TH), the rate-limiting enzyme in dopamine synthesis, in the midbrain by the 60th d of gestation (Rønnekleiv and Naylor, 1995). This treatment also increases the expression of mRNAs encoding several dopamine receptor subtypes as well as dopamine receptor binding in striatal regions of the fetal rhesus monkey (Choi and Rønnekleiv, 1996; Fang et al., 1997). Because cocaine may cause these changes through its action on the DAT, in the present study, we evaluated the expression of DAT mRNA and binding sites in both normal and cocaine-treated day 70 fetal monkeys using quantitative ligand autoradiography and in situ hybridization.
MATERIALS AND METHODS
Animals and cocaine treatment. Animal care and use were in accordance with institutional guidelines based on National Institutes of Health standards. The animal model used in this study was described previously in detail (Rønnekleiv and Naylor, 1995). Briefly, adult female rhesus monkeys (Macaca mulatta) were paired with fertile males for 3 d, and the second day was chosen as the day of conception. When pregnancy was confirmed, at days 21–23 after mating, cocaine (3 mg dissolved in 50 μl of 0.9% saline/kg) was injected intramuscularly four times daily at 8 A.M. and 12, 4, and 8 P.M. until the fetus was delivered. This cocaine treatment gave peak plasma cocaine levels of 800–1000 ng/ml in the mother at 10–15 min after the injection and 150–460 ng/ml in the fetus at ∼45 min after the cocaine injection (Rønnekleiv and Naylor, 1995).
Tissue preparation. On day 70 of gestation, the fetuses were delivered by cesarean section. A total of 20 fetuses were used in this study, and preferably they were matched according to body weight and sex. However, both males and females were used, and each experiment had one set of male–female match, because we have found that the day 60–70 fetal brains respond similarly to cocaine irrespective of sex (Rønnekleiv and Naylor, 1995). Each fetal brain was dissected into three or four coronal blocks, which were soaked in ice-cold 20% sucrose for 2 hr for binding studies (control group, four males and one female; cocaine group, five males) or fixed in 4% paraformaldehyde for 6 hr on ice for immunocytochemistry and in situhybridization studies (control group, three males and two females; cocaine group, four males and one female). The tissue blocks were frozen at −55° C and stored in liquid nitrogen until sectioned coronally on a cryostat at 15 μm.
In vitro binding autoradiography. We used 3β-(4-[125I]iodophenyl)tropane-2β-carboxylic acid isopropyl ester ([125I]RTI-121), which binds specifically to the DAT (Boja et al., 1995; Carroll et al., 1995), to determine DAT distribution in the fetal brain and to characterize cocaine-induced changes. The method for in vitro autoradiography for the DAT was performed as described previously (Boja et al., 1995; Staley et al., 1995) with minor modifications as follows. On the day of an assay, sections were fixed lightly in 0.5% paraformaldehyde in 0.1 mphosphate buffer, pH 7.4, for 5 min and rinsed briefly in 0.1m phosphate buffer. The light fixation was needed to keep the fragile fetal tissue sections on the slides, and preliminary test experiments determined that [125I]RTI-121 binding in fetal monkey striatal sections was not influenced by the fixation described above. To disassociate the binding of injected cocaine to the dopamine transporter, all sections were preincubated in a large volume (400 ml) of 10 mm sodium phosphate buffer containing (in mm): 10 Na2HPO4, 1.8 KH2PO4, 50 NaCl, 2.8 KCl, and 10 NaI (assay buffer, pH 7.4) for 30 min. This procedure is necessary and effective because disassociation of membrane-bound cocaine is rapid, with a disassociation half-life in seconds (Reith et al., 1981). Thereafter, the sections were incubated with 150 pm [125I]RTI-121 (2200 Ci/mmol; New England Nuclear, Boston, MA) in the assay buffer for 60 min at 22–23°C. Nonspecific binding was evaluated by treating a parallel set of slides with the same concentration of iodinated ligand in the presence of 100 μm (−)-cocaine. After the incubation, the tissue sections were rinsed twice for 20 min each in ice-cold assay buffer, followed by a brief rinse in ice-cold deionized water, and immediately dried with a stream of cold, dry air. The sections from controls and cocaine-exposed animals and an autoradiographic 125I microscale used as standard (Amersham, Arlington Heights, IL), were co-exposed to tritium sensitive-film (Hyperfilm-3H;, Amersham) for up to 72 hr at −80°C. The exception was the hypothalamus, which was exposed for up to 2 weeks, and the frontal cortex, which was exposed for up to 3 weeks.
Immunocytochemistry and in situ hybridization histochemistry. Immunocytochemistry for TH was performed according to previously described methods with minor modifications as outlined below (Rønnekleiv and Naylor, 1995). Briefly, tissue sections were reacted overnight at 4°C with a monoclonal TH antibody (DiaSorin, Inc, Stillwater, MN) at a 1:5000 dilution. A 30 min postprimary wash in phosphate buffer was followed by a 60 min room temperature incubation with biotinylated IgG at 1:300 (Vector Laboratories, Burlingame, CA), another 30 min wash, and a 60 min room temperature incubation with either streptavidin-HRP (1.5 μg/ml, NeutraLite; Molecular Probes, Eugine, OR) or streptavidin-Cy3 (1:500; Jackson ImmunoResearch; West Grove, PA). The HRP slides were washed for 30 min in phosphate buffer and then reacted with DAB (Aldrich, Milwaukee, WI) by the glucose oxidase–β-d-glucose method (Itoh et al., 1979). The slides were allowed to air dry, quickly dipped in xylene, and coverslipped with Polymount (Poly Scientific Research and Development Corp., Bay Shore, NY). The Cy3 fluorescent slides were washed for 2–3 hr in phosphate buffer and coverslipped with buffered glycerol, pH 7.4.
A monkey DAT-specific riboprobe was used for the in situhybridization histochemistry studies. A pGEM-3Zf(+) plasmid, containing a 410 bp insert corresponding to the C-terminal segment of rhesus monkey DAT cDNA sequence, was cloned in our laboratory (Choi and Rønnekleiv, 1996). The 35S-rUTP-labeled antisense cRNA probe was transcribed with SP6 RNA polymerase from the DAT plasmid construct, linearized with EcoRI, and purified through a G-50 Nick column (Pharmacia Biotech, Alemeda, CA). In situ hybridization experiments were performed with minor modifications of previously described methods (Fang et al., 1998). Briefly, sections were fixed in 4% paraformaldehyde for 40 min in 0.3m Sorensen’s buffer, pH 7.4, rinsed briefly in Sorensen’s buffer, treated in 0.1 mtriethanolamine (TEA) followed by 0.25% acetic anhydride, and finally treated with proteinase K (1 μg/ml) for 2 min at 37°C. Sections were prehybridized for 60 min at 58°C (60% formamide, 10% dextran sulfate, 1× Denhardt’s solution, 2× SSC, 125 μg/ml yeast transfer RNA, and 100 mm dithiothreitol) and then quickly rinsed in 2× SSC buffer. The 35S-labeled riboprobe was diluted with hybridization buffer and used at a final saturating concentration of 2 × 104dpm/μl. Subsequently, the sections were covered with glass coverslips, sealed, and hybridized for at least 18 hr at 57°C. After hybridization, the slides were rinsed in 2× SSC buffer, reacted with RNase (20 μg/ml) for 30 min at 37°C, and washed in 2.0, 1.0, and 0.5× SSC at 55–60°C to a final stringency of 0.1× SSC at 65°C. The sections were dehydrated in ethanol and together with autoradiographic [14C]microscales (Amersham) were exposed to hyperfilm-βmax x-ray film (New England Nuclear) for 5 d. The slides were then dipped in Eastman Kodak (Rochester, NY) NTB-2 nuclear track emulsion and exposed for up to 16 d at 4°C. In some instances, sequential in situ hybridization for DAT and immunocytochemistry for TH were performed on the same tissue sections. The in situhybridization procedure was performed first, essentially as described above with the exception of eliminating the TEA–acetic anhydride treatment and ethanol dehydration. After the final posthybridization wash in 0.1× SSC, sections for the combined procedure were reacted for TH immunocytochemistry as described above, and streptavidin Cy3 was used as marker. The slides were then dipped in Kodak NTB-2 nuclear track emulsion and exposed for up to 35 d at 4°C.
Quantification and image analysis. Quantification of film images of DAT binding and mRNA was performed using an Apple Power Macintosh computer equipped with the NIH Image 1.61 program. For quantification of [125I]RTI-121 (DAT) binding densities, six sections from each area were matched and analyzed in control (n = 4–5) and cocaine-treated (n = 5) fetuses. Film images of these sections were scanned on a flat-bed scanner (ScanJet IIcx/T; Hewlett–Packard, Palo Alto, CA), and the optical densities of the signals were measured and quantified. The optical density was converted to nanocuries per milligram using the standard autoradiographic125I microscale. The specific binding equals the total binding minus the nonspecific binding. Data were expressed as mean ± SEM and analyzed by a two-tailed Student’st test. Bright-field and pseudocolor autoradiograms were generated from the autoradiographic film using the Adobe (Mountain View, CA) Photoshop and Macromedia (San Francsico, CA) FreeHand software programs. For quantification of DAT mRNA signals, film images of six sections from the rostral, medial, and caudal midbrain of matched control (n = 5) and cocaine-treated (n = 5) fetuses were analyzed, and an average optical density value was obtained, which was used for further analysis. The autoradiographic 14C microscale was used as a standard to calculate the density of DAT mRNA signals, and the optical density was converted to nanocuries per gram. Statistical analysis was performed using a paired two-tailed Student’st test. Sections from control and cocaine-exposed brains were always treated identically.
To illustrate the distribution of TH-IR images, immunostained sections from fetal brains were scanned directly from the slides and digitized using the Power Macintosh computer outfitted with a slide scanner (Polaroid Sprint Scan 35 Plus with Pathscan Enabler) and the appropriate software. TH-IR and DAT mRNA images were also evaluated and photographed under fluorescent and dark-field illumination using aZeiss (Thornwood, NY) microscope. Dark-field and fluorescent views of photomicrographs were illustrated from film negatives or color slides using the slide scanner and the Adobe Photoshop software.
To determine the concentration of TH-containing cells in the ventral midbrain, cells within 250 × 250 μm2 were counted under bright-field illumination with a 16× objective using an eyepiece square grid reticle on a Leitz (Wetzlar, Germany) Laborlux microscope. Five to seven sections at ∼300 μm intervals were counted from the rostral to caudal midbrain of each animal without knowledge of treatment groups. Within each section, cells were counted in the ventral (SNv) and dorsal (SNd) substantia nigra, the ventral tegmental area (VTA), and the linear/interfascicular (Li/IF) area. The mean number of cells from each area was calculated and used for further analysis. Statistical differences between saline- and cocaine-treated fetuses were evaluated using ANOVA. Values were expressed as group mean and SEM.
RESULTS
Fetal growth and development
In this quantitative study, the control fetal groups consisted of seven males and three females, and the cocaine-treated groups consisted of nine males and one female. Maternal cocaine treatment from day 22 to day 70 of gestation did not significantly affect fetal body weight, crown–rump length, or head circumference. Body weight of control and cocaine-treated fetuses was 27.25 ± 1.43 and 28.13 ± 1.28 gm, respectively. The crown–rump lengths of the two fetal groups were 7.78 ± 0.11 and 7.80 ± 0.10 cm, and the head circumference was 8.06 ± 0.12 and 8.18 ± 0.14 cm, respectively.
Distribution of TH-IR in day 70 fetal monkey brain
To assess the distribution of dopamine neurons in the fetal brain, we performed immunocytochemistry with a monoclonal antibody to TH. In the most rostral brain sections, TH-IR fibers were found primarily in the intermediate layer of the developing prefrontal cortex (Pfc) (Figs. 1A,2A). Sections at the level of the striatum revealed dense TH-IR fibers in the caudate (Cd), putamen (Put), and nucleus accumbens (ACB), which exhibited a patchy appearance (Figs.1B,C, 2B). This was in contrast to the more uniform distribution of fibers observed in day 60 fetuses, suggesting that the patch matrix compartments begin to develop between days 60 and 70 in the nonhuman primate (O. K. Rønnekleiv unpublished observations). At this stage in gestation, a relatively large, undifferentiated subventricular zone destined to form additional striatal neurons was observed dorsal and medial to the caudate, which supports findings by others (Goldman-Rakic, 1981). The subventricular zone, however, did not contain immunoreactive TH (Fig.1B,C). In the olfactory bulbs (OB), TH-IR cells and fibers were found in the glomerular and plexiform layers (Fig. 1B). Cells immunoreactive to TH were also present in the olfactory tubercle area, diagonal band, and striatum of the day 70 fetus. These cells were small (6–10 μm in diameter), round or pyramidal shaped, with one to three processes. The latter population of dopamine cells have previously been documented in adult human and nonhuman primates but have not been described in the rodent (Dubach et al., 1987; Betarbet et al., 1997; Ikemoto et al., 1998).
Fig. 1.
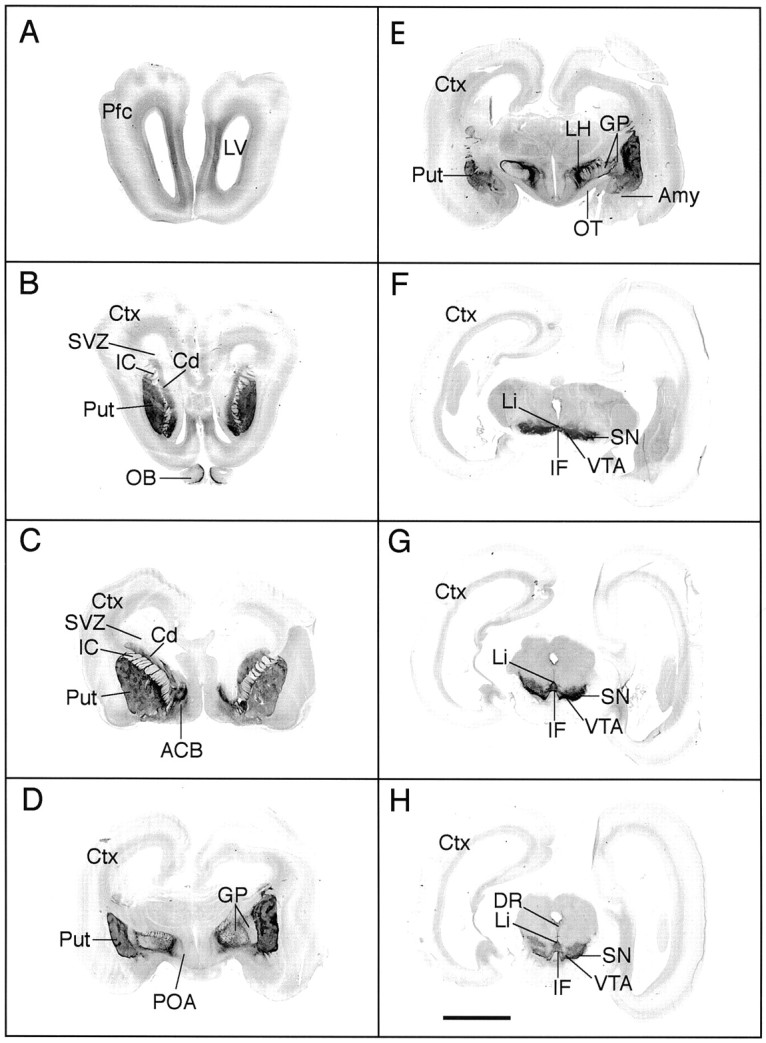
Distribution of immunoreactive TH (IR-TH) in day 70 fetal monkey brain. Images of TH stained coronal sections from the prefrontal cortex (Pfc) to the midbrain were scanned and digitized directly from the slides. The dark stain, indicative of IR-TH, can be seen in the prefrontal cortex (Pfc; A), olfactory bulbs (OB; B), caudate nucleus (Cd; B, C), putamen (Put; B–E), nucleus accumbens (ACB; C), globus pallidus (GP; D, E), lateral hypothalamus (LH; E), substantia nigra (SN; F–H), ventral tegmental area (VTA; F–H), linear nucleus (Li), and interfascicular nucleus (IF;F–H). The striatal TH fiber stain was patchy at this stage in gestation (B–E). IR-TH was not found in the subventricular zone (SVZ), an area destined to form additional striatal neurons. Amy, Amygdala;Ctx, cortex; DR, dorsal raphé;IC, internal capsule; OT, optic tract;POA, preoptic area. Scale bar, 5 mm.
Fig. 2.

Dark-field views of photomicrographs of some of the sections in Figure 1 with a more detailed illustration of IR-TH.A, TH fibers in the prefrontal cortex (Pfc) were primarily concentrated in the intermediate layer. B, C, The TH fiber distribution in the striatum was clearly patchy in appearance, and some of the fibers crossed the internal capsule (IC). C,D, A distinct network of fibers was observed in the internal and external segment of the globus pallidus (GPi, GPe). D, Within the hypothalamus TH cells and fibers were found in the periventricular, arcuate (Arc), and lateral hypothalamic nuclei; a distinct fiber bundle connects the lateral hypothalamus (LH) and the Arc.E, F, At day 70, TH cells and fibers within the ventral midbrain were densely intermingled; however, the various TH neuronal groups described in adult primates were also found at this stage in development (also see Fig. 4A).Cd, Caudate; IF, interfascicular nucleus;LV, lateral ventricle; Li, linear nucleus; OT, optic tract; Put, putamen;SN, substantia nigra; 3V, third ventricle; VTA, ventral tegemental area. Scale bar:A, B, 500 μm; C–F, 1.0 mm.
At the level of the preoptic area, TH-IR cells were located ventrally along the optic chiasm and dorsally in the paraventricular nucleus. Dense, patchy TH fiber stain was present in the putamen, and relatively dense concentrations of TH-IR fiber bundles were found in the external and internal segments of the globus pallidus (Figs.1D, 2C). Dense fiber stain and scattered cells were also located in the lateral preoptic area (Fig.1D).
At the level of the basal hypothalamus, TH-IR fibers exhibited a dense and patchy distribution in the putamen (Fig. 1E). The TH-IR fibers were also dense in the central nucleus of the amygdala, globus pallidus, and lateral hypothalamus (Figs. 1E,2D). A distinct TH-IR fiber tract extended from the dorsal hypothalamus to the rostral arcuate nucleus of the hypothalamus (Figs. 1E, 2D). TH-IR cells were found in the zona incerta of the dorsal hypothalamus, the periventricular area, and the arcuate nucleus of the hypothalamus.
At the level of the midbrain, the different layers of TH-IR neurons that have been described in the adult primate (Haber et al., 1995) were also observed in the day 70 fetus. Thus, TH-IR neurons were found in the dorsal and ventral tiers of the substantia nigra pars compacta and in the cell columns of the ventral tier (Figs. 1F–H,2E,F). TH-IR neurons were also present in the VTA, ventral periaqueductal gray, linear nucleus, and interfascicular nucleus (Figs. 1F–H,2E,F).
Distribution of [125I]RTI-121 binding sites in day 70 fetal monkey brain
To investigate the development of DAT binding sites in the day 70 fetal monkey brain, we used the ligand [125I]RTI-121, which is highly specific for the dopamine transporter. This analysis revealed a defined pattern of distribution of [125I]RTI-121 binding sites (Fig. 3). The nonspecific binding in different regions varied from 0.001 to 0.08 nCi/mg of tissue with a mean value of 0.04 ± 0.01 nCi/mg. The rank order of specific [125I]RTI-121 binding densities was substantia nigra–ventral tegmental area > putamen > caudate > lateral hypothalamus > accumbens > linear/interfascicular nuclei ≥ globus pallidus > prefrontal cortex. There was a paucity of DAT binding sites in the prefrontal cortex, which correlated with the faint TH-IR (Figs.2A, 3A). These binding sites were observed in the intermediate zone of the prefrontal cortex. In comparison, ligand binding in the caudate and putamen from rostral to caudal was quite dense and exhibited a patchy appearance as described above for the TH fiber input (Fig.3B–E). The [125I]RTI-121 binding sites were dense in the patches and significantly less dense in the surrounding matrix area (p < 0.05; Table1). Overall, the ligand binding in the putamen was significantly higher than that found in the caudate and accumbens (ANOVA, p < 0.05 and p < 0.01, respectively). DAT binding sites, although slight, were also observed in the globus pallidus and a relatively high concentration was found in the lateral hypothalamus (Fig. 3, Table 1). DAT binding sites were not detected in the medial preoptic or medial basal hypothalamus, although these regions had substantial amount of TH-positive cells and fibers (Figs. 1-3). The highest density of [125I]RTI-121 binding sites in the fetal brain was measured in the SN/VTA area, which also had high concentration of densely packed dopamine cells and fibers (Figs. 3-5, Table 1). In contrast, faint label was found in the Li/IF, an area of the midbrain which also contained a high concentration of TH-positive dopamine neurons (Figs. 1-3). Overall the distribution of [125I]RTI-121 binding sites coincides well with the distribution of immunoreactive TH (Figs. 1, 3).
Fig. 3.

DAT binding sites are highly expressed in day 70 fetal monkey brain. Bright-field view of autoradiograms illustrating the distribution of DAT binding sites, labeled with [125I]RTI-121, in coronal sections from rostral (A) to caudal (H). The darker the images, the denser are the binding sites.A, The DAT binding sites were faint (0.08 ± 0.03 nCi/mg) in the prefrontal cortex (Pfc) and were illustrated within the intermediate layer after 3 weeks of film exposure. B–E, Within the caudate (Cd) and the putamen (Put) the DAT binding sites were dense and patchy in appearance, similar to the TH fiber stain illustrated in Figure 1. DAT binding sites were also present in the nucleus accumbens (ACB), globus pallidus (GP), and lateral hypothalamus (LH) and exhibited a distribution pattern similar to IR-TH. F–H, The highest density of DAT binding sites was measured in the substantia nigra (SN) and the ventral tegmental area (VTA) of the fetal midbrain. F–H, Light binding densities were found in the linear nucleus (Li) and interfascicular nucleus (IF), whereas binding sites were not detected in the dorsal Raphé (DR). The nonspecific binding is 0.04 ± 0.01 nCi/mg.Ctx, Cortex; IC, internal capsule;OB, olfactory bulb; OT, optic tract;POA, preoptic area. Scale bar, 2 mm.
Table 1.
Effect of gestational cocaine exposure on the density of [125I]RTI-121 binding sites (nanocuries per milligram) in day 70 fetal monkey brain
| Area | Treatment | |
|---|---|---|
| Control | Cocaine | |
| Forebrain | ||
| Prefrontal cortex | 0.08 ± 0.03 | 0.11 ± 0.03 |
| Accumbens | 1.32 ± 0.33 | 1.23 ± 0.20 |
| Caudate | ||
| Patch | 3.46 ± 0.63* | 4.34 ± 0.65** |
| Matrix | 1.61 ± 0.22* | 2.43 ± 0.36** |
| Putamen | ||
| Patch | 4.40 ± 0.86* | 5.20 ± 0.56** |
| Matrix | 2.91 ± 0.53* | 3.20 ± 0.34** |
| Globus pallidus | 0.71 ± 0.24 | 1.24 ± 0.10 |
| Lateral hypothalamic area | 1.99 ± 0.19 | 2.02 ± 0.23 |
| Midbrain | ||
| Substantia nigra/ventral tegmental area | 6.05 ± 0.54 | 8.56 ± 0.67*** |
| Linear nucleus/interfascicular nucleus | 0.75 ± 0.15 | 1.88 ± 0.37*** |
The values represent the mean density ± SEM of DAT binding sites in the different brain regions, measured using the specific ligand [125I]RTI-121. Data were analyzed by two-tailed Student’s t test. The highest density of DAT binding sites was found in the SN/VTA, and DAT binding sites in this area was significantly increased in cocaine-exposed fetuses. Cocaine exposure also significantly increased DAT binding sites in the linear and interfascicular nuclei.
*,**Within the caudate and the putamen [125I]RTI-121 binding site densities were significantly higher in the patch compared with the matrix compartments in both control (*p < 0.05) and cocaine-exposed (**p < 0.01) fetuses.
***p < 0.05 comparing cocaine-treated fetuses (n = 5) versus saline-treated control animals (n = 4–5).
Fig. 4.
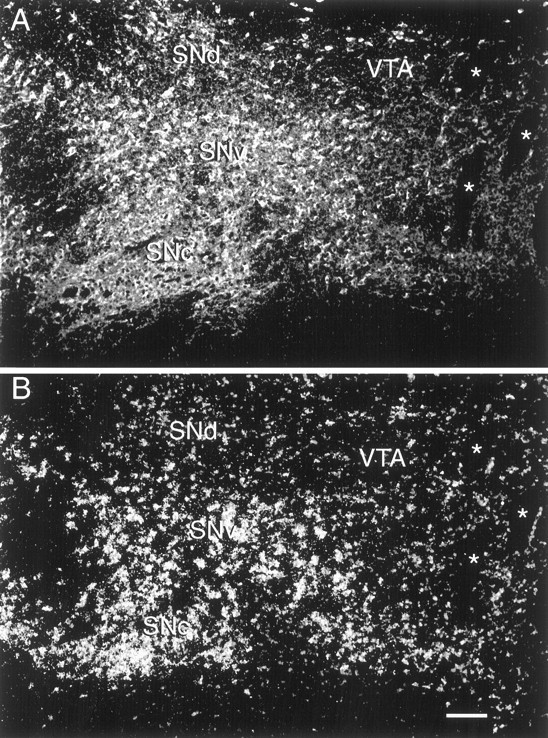
DAT mRNA is expressed in TH-containing neurons of the fetal midbrain. Fluorescent immunocytochemistry (A) coupled with in situhybridization (B) on the same tissue section revealed that TH-containing neurons of the ventral tier (SNv) including the columns of the SN (SNc), contain high concentration of autoradiographic silver grains indicative of a high concentration of DAT mRNA (B). DAT mRNA was more lightly expressed in TH neurons of the dorsal tier of the SN (SNd) and the VTA. The neurons containing clusters of silver grains (B) were hybridized with a35S-labeled DAT riboprobe. Immunocytochemistry was performed after the in situ hybridization procedure with a TH monoclonal antibody visualized with the Cy-3 fluorochrome.★Bundles of myelinated fibers of the oculomotor nerve. Scale bar, 100 μm.
Fig. 5.

High-power view of TH and DAT co-localization in ventral tier neurons of the fetal substantia nigra.A, Fluorescent photomicrograph illustrating IR-TH neurons in the ventral tier of the SN. B, Dark-field photomicrograph of the same section showing silver grains, indicative of DAT mRNA, surrounding cell nuclei (blue nuclei). Scale bar, 50 μm.
Differential localization of DAT mRNA in midbrain dopamine neurons
We used in situ hybridization with a DAT riboprobe to document the distribution of DAT mRNA in the fetal brain and combinedin situ hybridization and fluorescence immunocytochemistry to determine its expression in TH-positive neurons of the midbrain. DAT mRNA was found in the midbrain from rostral to caudal and was co-localized with TH-IR (Fig. 4). An example of a high-power view of DAT mRNA and TH-IR co-localization in ventral tier neurons is illustrated in Figure5. We were, however, unable to detect expression of DAT mRNA in any of the rostral forebrain regions where TH-positive cells were found. Within the midbrain, DAT mRNA was most highly expressed in dopamine neurons of the ventral tier of the SN and was lightly expressed in the dorsal SN, VTA, and Li/IF area (see Figs.4, 8). Quantitative analysis of film images revealed that the density of DAT mRNA was significantly higher in the SNv in comparison with that in other midbrain regions (ANOVA, p < 0.05–0.01; see Fig. 8).
Fig. 8.

Group data of cocaine-induced increase in DAT mRNA expression in the fetal VTA. Distribution and quantitative analysis of DAT mRNA in midbrain tissue sections obtained from control and cocaine-treated fetal monkeys (n = 5 each) are shown. Values represent the mean density of DAT mRNA levels (nanocuries per gram of tissue) in the ventral and dorsal tiers of the substantia nigra (SNv,SNd, respectively), the ventral tegmental area (VTA), and the linear nucleus/interfascicular nucleus (Li/IF) in the fetal midbrain. The expression of DAT mRNA was significantly increased in the VTA of cocaine-treated fetuses compared with saline-treated controls (★p < 0.03, paired two-tailed Student’st test).
Effect of cocaine on the density of [125I]RTI-121 binding sites
Based on the evidence that cocaine exposure in uterohas specific action within the fetal midbrain–rostral forebrain dopamine circuitry (Rønnekleiv et al., 1998), we hypothesize that cocaine initiates these actions through binding to the dopamine transporter. To assess the effects of in utero cocaine exposure on DAT binding in the fetal brain, we compared the distribution and density of [125I]RTI-121 binding sites in brain sections obtained from control and cocaine-treated fetuses. This analysis revealed that the distribution of DAT binding sites was similar in control (n = 4–5) and cocaine-treated fetal brains (n = 5) (Table 1). However, the fetal monkeys exposed to cocaine between days 22 and 70 of gestation exhibited a significant increase in binding density of [125I]RTI-121 in the SN/VTA and the Li/IF (p < 0.05) in comparison with controls (Figs. 6,7; Table 1).
Fig. 6.

In utero cocaine exposure caused increased DAT binding sites in the fetal ventral midbrain. Pseudocolor autoradiograms show DAT binding site densities, labeled with the specific ligand [125I]RTI-121 in matched sections through the rostral and caudal midbrain of a control (a, c) and cocaine-exposed (b,d) fetus. Each color represents a range of DAT binding densities (nanocuries per milligram) calculated from the calibrated 125I standards exposed along with the brain sections. Analogous sections from each brain were determined using standard anatomical landmarks. Scale bar, 2 mm.
Fig. 7.

Group data of cocaine-induced increase in DAT binding sites in the fetal midbrain. Quantitative analysis and distribution of DAT binding site densities labeled with [125I]RTI-121 (nanocuries per milligram of tissue) in brain sections obtained from control and cocaine-exposed fetal monkeys are shown. DAT binding site density was significantly increased in the substantia nigra/ventral tegmental area (SN/VTA) and the linear nucleus/interfascicular nucleus (Li/IF) area in cocaine-treated fetuses compared with saline-treated controls (★p< 0.05, two-tailed Student’s t test).n, Number of animals. Pfc, prefrontal cortex; ACB, nucleus accumbens; Cd, caudate; Put, putamen; GP, globus pallidus; LH, lateral hypothalamus.
Effect of cocaine on the expression of dopamine transporter mRNA
Next we determined the effects of prenatal cocaine exposure on the expression of DAT mRNA using in situ hybridization analysis of tissue sections through the fetal midbrain. Similar to observations in the controls, DAT mRNA was found exclusively in the midbrain of cocaine-exposed fetuses. Densitometric analysis of film images revealed that DAT mRNA in sections from cocaine-exposed fetuses was significantly increased in the VTA (n = 5;p < 0.03) and approaching significance in the Li/IF (p = 0.052) in comparison with findings in control fetuses (Fig. 8). DAT mRNA levels were also elevated in the SN as a result of cocaine exposure, but again, the increase did not reach statistical significance (Fig. 8). To determine whether cocaine exposure had altered the development of dopamine cells, we counted the number of TH-IR cells within specific midbrain regions from control and cocaine-exposed fetuses. There were significant regional differences in the distribution of TH-IR cells, in that the concentration of cells in the ventral tier of the SN was significantly higher than that of the dorsal tier of the SN and the VTA (p < 0.05) in both control and cocaine-treated animals. There was, however, no significant change in TH cell concentration in any midbrain regions in cocaine-treated fetuses in comparison with controls (Table 2).
Table 2.
Number of TH-immunoreactive neurons per unit area in day 70 fetal monkey midbrain
| Treatment | Area | |||
|---|---|---|---|---|
| SNv | SNd | VTA | Li/IF | |
| Control | 81.1 ± 1.6* | 57.4 ± 7.3 | 58.0 ± 5.5 | 69.2 ± 3.4 |
| Cocaine | 83.2 ± 4.4* | 48.6 ± 2.9 | 62.2 ± 5.1 | 64.0 ± 3.5 |
Values represent estimates of mean number of cells ± SEM counted in 250 × 250 μm2 in the ventral substantia nigra (SNv), dorsal SN (SNd), ventral tegmental area (VTA), and linear/interfascicular (Li/IF) nuclei from control and cocaine-treated fetuses. Data were analyzed by ANOVA and revealed that cocaine treatment did not affect the number of TH-IR neurons (p> 0.05). In both control and cocaine-exposed fetuses the number of TH-IR cells in the SNv was significantly higher than that in the other midbrain regions (*p < 0.05; n = 5).
DISCUSSION
In this study we have shown that the DAT is highly developed before midgestation in the primate and that cocaine exposure from day 22 to day 70 of gestation significantly increases DAT mRNA expression and binding site density in the midbrain of the day 70 fetus. These findings suggest that during early development, the primate midbrain dopamine neurons are particularly vulnerable to gestational environmental insults.
Differential expression of DAT in fetal midbrain dopamine neurons
Previously, we demonstrated that DAT mRNA can be detected in RNA extracted from the fetal monkey midbrain by day 45 of gestation and that midbrain DAT mRNA expression is greatly increased by day 60 (Choi and Rønnekleiv, 1996). In the present study we found that DAT mRNA is expressed in midbrain dopa-mine neurons and is differentially located within the fetal midbrain by day 70. The dopamine neurons in the SN of primates have a distinct organization that is different from that found in rodents. In particular these neurons are organized into dorsal and ventral tiers and columns within the substantia nigra pars compacta (Haber et al., 1995). Our results indicate that fetal dopamine neurons of the ventral tier and columns in the SN densely express DAT mRNA as evidenced by the high concentration of grains. In contrast, TH-containing neurons of the dorsal tier of the SN, the VTA, and the raphé nuclei display low levels of DAT mRNA. A similar DAT mRNA distribution pattern has also been described in the adult monkey (Haber et al., 1995), suggesting that the organization of the primate fetal dopamine neurons is similar to that of adults, although these neurons continue to develop throughout gestation (Levitt and Rakic, 1982;Aubert et al., 1997). It is well known that dopamine neurons are distributed in various regions of the rostral forebrain (Hökfelt et al., 1984). In addition, in contrast to rodents, primates have a population of dopamine cells within the olfactory tubercle and striatum (Dubach et al., 1987; Betarbet et al., 1997; Ikemoto et al., 1998). These rostral, primate-specific dopamine cells were detected as early as day 70 in the fetus. Using in situ hybridization we explored DAT mRNA expression in diencephalic and more rostral brain regions, but none of these neurons contained detectable levels of DAT mRNA. Given the findings reported for the adult rat (Cerruti et al., 1994), we suggest that DAT mRNA may be present in these rostral dopamine neurons of the monkey but are below the level of detectability by the methods we used.
DAT binding sites and immunoreactive TH have similar distribution pattern
In this investigation the DAT-specific radioligand [125I]RTI-121 was used to measure transporter density in the fetal brain (Boja et al., 1995; Carroll et al., 1995). In most regions examined we found a close correlation between [125I]RTI-121 binding sites and immunoreactive TH. For example, in the striatum TH was expressed in a patch–matrix distribution pattern with the densest staining in patches. Similarly, [125I]RTI-121 binding sites were found as dense patches with less dense areas in between. In contrast, dopamine-β-hydroxylase (DBH) immunoreactivity, which is used to differentiate dopamine from norepinephrine, was weak in most forebrain regions, including the striatum, suggesting a sparse noradrenergic innervation of the fetal rostral forebrain at this stage in development (O. K. Rønnekleiv, unpublished observations).
One of the important findings made in the course of this investigation is that [125I]RTI-121 binding sites are highly developed in multiple brain regions by day 70 of gestation in the primate. In this respect, the DAT binding sites were most dense in the SN/VTA area of the fetal midbrain and less dense in terminal field areas of the rostral forebrain. This is in contrast to observations made in adult brains, in which DAT binding sites were found to be highest in the striatum area in comparison with midbrain regions (Kaufman et al., 1991; Reader et al., 1998). A different developmental profile of the DAT has been reported in the rat, in which DAT binding sites in the fetal midbrain and striatum are extremely low, even close to birth (Shearman et al., 1996). The significance of these developmental differences is currently unclear but may imply that dopamine neurons in primates develop earlier and are more sensitive to prenatal modification at an earlier time point compared with the developing rat.
Gestational cocaine exposure increases the expression of DAT in the fetal midbrain
A significant finding made in the course of the present study is that cocaine exposure in utero increased the expression of DAT in the fetal midbrain. The increased density of DAT mRNA and binding sites were not due to an increased concentration of dopamine neurons because this was not found in cocaine-treated animals. In contrast to the midbrain, [125I]RTI-121 binding sites were not significantly different from controls in the striatum and surrounding areas of the rostral forebrain. This would suggest that in utero cocaine exposure primarily affects dopamine transport mechanisms of the midbrain at this time in gestation. To our knowledge, a comparable study has not been reported in other species. However, several studies have treated rats and mice prenatally with cocaine and then explored the consequences at various time points postnatally. One such study found that striatal DAT binding sites were increased during postnatal days 1–5 and decreased at postnatal days 14 and 35 in offspring from rats treated with cocaine during gestation until birth (Leslie et al., 1994). Other investigators have also documented a decrease in DAT in the striatum in infants, juvenile and young adult animals after cocaine withdrawal at term (Pumford et al., 1993; Leslie et al., 1994; Stadlin et al., 1994;Collins and Meyer, 1996). In adult subjects, prolonged cocaine intake increases DAT binding sites in the striatal area and/or the ventral midbrain when assessed shortly after termination of treatment (Alburges et al., 1993; Wilson et al., 1994; Staley et al., 1994; Aloyo et al., 1995; Tella et al., 1996; Little et al., 1999). Interestingly, animals that receive prolonged cocaine treatment display increased DAT ligand binding immediately after the last dose of cocaine, but withdrawal for 3 weeks decreases DAT binding sites in the striatal area and the ventral midbrain (Wilson et al., 1994). The decrease in DAT ligand binding sites in rostral forebrain regions after a period of cocaine withdrawal is observed in both rats and monkeys and in the rat is accompanied by a decrease in DAT mRNA in midbrain nuclei (Sharpe et al., 1991; Farfel et al., 1992; Cerruti et al., 1994; Pilotte et al., 1994). Therefore, chronic exposure to cocaine may result in an immediate increase in the population of dopamine transporters, whereas, abstinence from cocaine after a period of exposure reduces its concentration (Sharpe et al., 1991; Farfel et al., 1992; Alburges et al., 1993; Byrnes et al., 1993; Cerruti et al., 1994; Leslie et al., 1994; Pilotte et al., 1994; Stadlin et al., 1994; Wilson et al., 1994;Collins and Meyer, 1996; Tella et al., 1996).
It is evident that in mature animals prolonged cocaine exposure acts at the level of the dopamine cell bodies and also in the terminal field regions to alter DAT activity (Sharpe et al., 1991; Farfel et al., 1992; Cerruti et al., 1994; Pilotte et al., 1994; Wilson et al., 1994;Tella et al., 1996). By way of comparison, cocaine exerts its action at the level of the midbrain dopamine neurons to increase DAT activity in the primate fetus. The mechanism by which cocaine exerts these effects is not obvious. One could speculate that cocaine binding to transporters in the fetal midbrain can prevent reuptake and consequently increase dopamine in the extracellular space. This might cause induction of new transporters in an effort to clear dopamine from synapses within the brain. One argument against a dopamine-stimulated upregulation of DAT is that significant alteration in dopamine levels with l-DOPA or α-methyltyrosine administration do not alter DAT ligand binding sites (Moody et al., 1996). Another explanation for DAT up-regulation is that it could be related to the pharmacodynamics of cocaine binding to a specific site on the transporter. Such a possibility was suggested by Tella et al. (1996), who found that prolonged cocaine administration but not administration of the dopamine uptake blocker GBR-12909 increased [125I]RTI-121 binding in the ventral midbrain as well as the striatum. Cocaine and GBR compounds bind to overlapping but nonidentical binding domains on the transporter (Saadouni et al., 1994). It is also possible that the upregulation of DAT expression is coupled to cocaine-induced alterations of the feedback mechanisms involving dopamine receptors and intracellular transduction pathways (Simon et al., 1997; Vaughan et al., 1997;Batchelor and Shenk, 1998). In this respect, dopamine D2 receptor antagonists inhibit upregulation of dopamine uptake induced by repeated cocaine intake (Parsons et al., 1993). Because D2 receptor binding sites are not yet expressed in the fetal monkey SN/VTA, but D1 sites are present (Fang et al., 1997), we hypothesize that increased extracellular dopamine may activate D1 receptors on striatal–substantia nigra GABA terminals and thereby increase GABA release, which in turn may inhibit the dopamine neurons (Cameron and Williams, 1993). In fact, we have found in the cocaine-exposed 70-d-old fetus that D1 receptor binding sites are increased on presumed striatal–SN projection neurons (Fang et al., 1997). This hypothesis is supported by our previous findings that TH mRNA is reduced in the fetal midbrain, and dopamine D1 and D2 receptor subtypes are up-regulated in the striatal area of cocaine exposed animals (Rønnekleiv and Naylor, 1995; Fang et al., 1997). The mechanism by which these cocaine-induced alterations within the dopamine circuitry would cause, or be the result of, increased expression of DAT is unknown and needs further investigation.
The midbrain dopaminergic system is important for the control of movement, reward-based learning, cognition, and emotion (Wickens, 1990;Nestler, 1994; Glenthoj, 1995; Gerfen and Wilson, 1996). Interestingly, genetic analysis has revealed a significant association between ADHD and the human DAT gene (Cook et al., 1995; Rowe et al., 1998; Waldman et al., 1998). Among other transmitter candidates, the specific role of dopamine in this disorder is unknown, but based on clinical observations and the results obtained from animal models, it has been proposed that ADHD may result from reduced activity of dopamine neurons that ultimately causes behavioral problems (Sagvolden and Sergeant, 1998). We have currently found in the fetal primate that gestational cocaine exposure increased the expression of DAT in the midbrain, suggesting a specific action of cocaine on midbrain dopamine neurons. Based on our previous findings, increased DAT activity is accompanied by other presynaptic and postsynaptic neuronal alterations, which collectively suggest that the dopamine neurons have decreased activity (Rønnekleiv et al., 1998). These conclusions are further supported by findings of reduced spontaneous activity of midbrain dopamine neurons in juvenile and adult animals prenatally exposed to cocaine (Minabe et al., 1992; Wang and Pitts, 1994). Such changes within the midbrain dopamine circuitry may be a contributing factor to the development of attention deficit disorders observed in subjects exposed prenatally to cocaine and other drugs of abuse.
Footnotes
This work was supported by National Institutes of Health Grant DA 07165, Population P30 Program Project Grant HD 18185, and Animal Resources Branch Grant RR 00163 for operation of the Oregon Regional Primate Research Center. We thank Barry R. Naylor and Martha A. Bosch for excellent technical help during the study and Drs. David K. Grandy and Martin J. Kelly for critical comments during the preparation of this manuscript.
Correspondence should be addressed to Dr. Oline K. Rønnekleiv, Department of Physiology and Pharmacology, Oregon Health Sciences University, 3181 Southwest Sam Jackson Park Road, Portland, OR 97201-3098.
REFERENCES
- 1.Alburges ME, Narang N, Wamsley JK. Alterations in the dopaminergic receptor system after chronic administration of cocaine. Synapse. 1993;14:314–323. doi: 10.1002/syn.890140409. [DOI] [PubMed] [Google Scholar]
- 2.Aloyo VJ, Pazdalski PS, Kirifides AL, Harvey JA. Behavioral sensitization, behavioral tolerance and increased [3H]WIN 35,428 binding in rabbit caudate nucleus after repeated injections of cocaine. Pharmacol Biochem Behav. 1995;52:335–340. doi: 10.1016/0091-3057(95)00101-2. [DOI] [PubMed] [Google Scholar]
- 3.Aubert I, Brana C, Pellevoisin C, Giros B, Caille I, Carles D, Vital C, Bloch B. Molecular anatomy of the development of the human substantia nigra. J Comp Neurol. 1997;379:72–87. doi: 10.1002/(sici)1096-9861(19970303)379:1<72::aid-cne5>3.0.co;2-f. [DOI] [PubMed] [Google Scholar]
- 4.Batchelor M, Shenk JO. Protein kinase A activity may kinetically upregulate the striatal transporter for dopamine. J Neurosci. 1998;18:10304–10309. doi: 10.1523/JNEUROSCI.18-24-10304.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Betarbet R, Turner R, Chockkan V, DeLong MR, Allers KA, Walters J, Levey AI, Greenamyre JT. Dopaminergic neurons intrinsic to the primate striatum. J Neurosci. 1997;17:6761–6768. doi: 10.1523/JNEUROSCI.17-17-06761.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Boja JW, Cadet JL, Kopajtic TA, Lever J, Seltzman HH, Wyrick CD, Lewin AH, Abraham P, Carroll FI. Selective labeling of the dopamine transporter by the high affinity ligand 3β-(4-[125I]iodophenyl)tropane-2β-carboxylic acid isopropyl ester. Mol Pharmacol. 1995;47:779–786. [PubMed] [Google Scholar]
- 7.Brown AS, Gershon S. Dopamine and depression. J Neural Transm. 1993;91:75–109. doi: 10.1007/BF01245227. [DOI] [PubMed] [Google Scholar]
- 8.Byrnes JJ, Pritchard GA, Koff JM, Miller LG. Prenatal cocaine exposure: decreased sensitization to cocaine and decreased striatal dopamine transporter binding in offspring. Neuropharmacology. 1993;32:721–723. doi: 10.1016/0028-3908(93)90087-j. [DOI] [PubMed] [Google Scholar]
- 9.Cameron DL, Williams JT. Dopamine D1 receptors facilitate transmitter release. Nature. 1993;366:344–347. doi: 10.1038/366344a0. [DOI] [PubMed] [Google Scholar]
- 10.Carroll FI, Kotian P, Dehghani A, Gray JL, Kuzemko MA, Parham KA, Abraham P, Lewin AH, Boja JW, Kuhar MJ. Cocaine and 3β-(4′-substituted phenyl)tropane-2β-carboxylic acid ester and amide analogues. New high-affinity and selective compounds for the dopamine transporter. J Med Chem. 1995;38:379–388. doi: 10.1021/jm00002a020. [DOI] [PubMed] [Google Scholar]
- 11.Cerruti C, Pilotte NS, Uhl G, Kuhar MJ. Reduction in dopamine transporter mRNA after cessation of repeated cocaine administration. Mol Brain Res. 1994;22:132–138. doi: 10.1016/0169-328x(94)90040-x. [DOI] [PubMed] [Google Scholar]
- 12.Chasnoff IJ, Anson A, Hatcher R, Stenson H, Iaukea K, Randolph LA. Prenatal exposure to cocaine and other drugs: outcome at four to six years. Ann NY Acad Sci. 1998;386:314–328. [PubMed] [Google Scholar]
- 13.Choi SJ, Mazzio E, Kolta MG, Soliman KFA. Prenatal cocaine exposure affects postnatal dopaminergic systems in various regions of the rat brain. Ann NY Acad Sci. 1998;844:293–302. [PubMed] [Google Scholar]
- 14.Choi WS, Rønnekleiv OK. Effects of in utero cocaine exposure on the expession of mRNAs encoding the dopamine transporter and the D1, D2 and D5 dopamine receptor subtypes in fetal rhesus monkey. Dev Brain Res. 1996;96:249–260. doi: 10.1016/0165-3806(96)00123-x. [DOI] [PubMed] [Google Scholar]
- 15.Collins LM, Meyer JS. Prenatal cocaine alters dopamine transporter binding in postnatal day 10 rat striatum. Synapse. 1996;23:335–343. doi: 10.1002/(SICI)1098-2396(199608)23:4<335::AID-SYN12>3.0.CO;2-W. [DOI] [PubMed] [Google Scholar]
- 16.Cook EH, Jr, Stein MA, Krasowski MD, Cox NJ, Olkon DM, Kiefer JE, Leventhal BL. Association of attention-deficit dissorder and the dopamine transporter gene. Am J Hum Genet. 1995;56:993–998. [PMC free article] [PubMed] [Google Scholar]
- 17.Dow-Edwards DL, Freed LA, Fico TA. Structural and functional effects of prenatal cocaine exposure in adult rat brain. Dev Brain Res. 1990;57:263–268. doi: 10.1016/0165-3806(90)90052-z. [DOI] [PubMed] [Google Scholar]
- 18.Dubach M, Schmidt R, Kunkel D, Bowden DM, Martin R, German DC. Primate neostriatal neurons containing tyrosine hydroxylase: immunohistochemical evidence. Neurosci Lett. 1987;75:205–210. doi: 10.1016/0304-3940(87)90298-9. [DOI] [PubMed] [Google Scholar]
- 19.Fang Y, Janowsky A, Rönnekleiv OK. Cocaine exposure in fetal rhesus monkey: consequences for dopamine D1- and D2-like receptor binding densities. Dev Brain Res. 1997;104:163–174. doi: 10.1016/s0165-3806(97)00151-x. [DOI] [PubMed] [Google Scholar]
- 20.Fang Y, Kelly MJ, Rønnekleiv OK. Proopiomelanocotin (POMC) mRNA expression: distribution and region-specific down-regulation by chronic morphine in female guinea pig hypothalamus. Mol Brain Res. 1998;55:1–8. doi: 10.1016/s0169-328x(97)00348-3. [DOI] [PubMed] [Google Scholar]
- 21.Farfel GM, Kleven MS, Woolverton WL, Seiden LS, Perry BD. Effects of repeated injections of cocaine on catecholamine receptor binding sites, dopamine transporter binding sites and behavior in rhesus monkey. Brain Res. 1992;578:235–243. doi: 10.1016/0006-8993(92)90252-5. [DOI] [PubMed] [Google Scholar]
- 22.Fischman MW. Behavioral pharmacology of cocaine. J Clin Psychiatry. 1988;49:7–10. [PubMed] [Google Scholar]
- 23.Friedman E, Yadin E, Wang H-Y. Effect of prenatal cocaine on dopamine receptor-G protein coupling in mesocortical regions of the rabbit brain. Neuroscience. 1996;70:739–747. doi: 10.1016/s0306-4522(96)83011-9. [DOI] [PubMed] [Google Scholar]
- 24.Gerfen CR, Wilson CJ. The basal ganglia. In: Swanson LW, Björklund A, Hökfelt T, editors. Handbook of chemical neuroanatomy, Vol 12, Integrated systems of the CNS, Pt III. Elsevier; Amsterdam: 1996. pp. 371–468. [Google Scholar]
- 25.Glenthoj BY. The brain dopaminergic system: pharmacological, behavioural and electrophysiological studies. Dan Med Bull. 1995;42:1–21. [PubMed] [Google Scholar]
- 26.Goldman-Rakic P. Prenatal formation of cortical input and development of cytoarchitectonic compartments in the neostriatum of the rhesus monkey. J Neurosci. 1981;1:721–735. doi: 10.1523/JNEUROSCI.01-07-00721.1981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gribnau AAM, Geijsberts LGM. Developmental stages in the rhesus monkey (Macaca mulatta). In: Brodal A, Hild W, Limborgh J, Ortmann R, Pauly JE, Schiebler TH, Wolff E, editors. Advances in anatomy, embryology and cell biology. Springer; New York: 1981. pp. 1–84. [DOI] [PubMed] [Google Scholar]
- 28.Haber SN, Ryoo H, Cox C, Lu W. Subset of midbrain dopaminergic neurons in monkeys are distinguished by different levels of mRNA for the dopamine transporter: comparison with the mRNA for the D2 receptor, tyrosine hydroxylase and calbindin immunoreactivity. J Comp Neurol. 1995;362:400–410. doi: 10.1002/cne.903620308. [DOI] [PubMed] [Google Scholar]
- 29.Hökfelt T, Mårtensson R, Björklund A, Kleinau S, Goldstein M. Distributional maps of tyrosine-hydroxylase-immunoreactive neurons in the rat brain. In: Björklund A, Hökfelt T, editors. Handbook of chemical neuroanatomy. Elsevier; Amsterdam: 1984. pp. 277–379. [Google Scholar]
- 30.Hunt RD, Hoehn R, Stephens KJ, Osten C. Clinical patterns of ADHD: a treatment model based on brain functioning. Compr Ther. 1994;20:106–112. [PubMed] [Google Scholar]
- 31.Ikemoto K, Nagatsu I, Kitahama K, Jouvet A, Nishimura A, Nishi K, Maeda T, Arai R. A dopamine-synthesizing cell group demonstrated in the human basal forebrain by dual labeling immunohistochemical technique of tyrosine hydroxylase and aromatic l-amino acid decarboxylase. Neurosci Lett. 1998;243:129–132. doi: 10.1016/s0304-3940(98)00103-7. [DOI] [PubMed] [Google Scholar]
- 32.Itoh K, Akiva K, Nomura S, Migano N, Nakamura Y, Sugimoto T. Application of coupled oxidation reaction to electron microscope demonstration of horseradish peroxidase: cobalt-glucose oxidase method. Brain Res. 1979;175:341–346. doi: 10.1016/0006-8993(79)91013-8. [DOI] [PubMed] [Google Scholar]
- 33.Kaufman MJ, Spealman RD, Madras BK. Distribution of cocaine recognition sites in monkey brain: I. in vitro autoradiography with [3H]CFT. Synapse. 1991;9:177–187. doi: 10.1002/syn.890090304. [DOI] [PubMed] [Google Scholar]
- 34.Keller RW, Jr, Johnson KS, Snyder-Keller AM, Carlson JN, Glick SD. Effects of prenatal cocaine exposure on the mesocorticolimbic dopamine system: an in vivo microdialysis study in the rat. Brain Res. 1996;742:71–79. doi: 10.1016/s0006-8993(96)00959-6. [DOI] [PubMed] [Google Scholar]
- 35.Knable MB, Egan MF, Heinz A, Gorey J, Lee KS, Coppola R, Weinberger DR. Altered dopaminergic function and negative symptoms in drug-free patients with schizophrenia: [123I]-iodobenzamide SPECT study. Br J Psychiatry. 1997;171:574–577. doi: 10.1192/bjp.171.6.574. [DOI] [PubMed] [Google Scholar]
- 36.Koob GF. Drugs of abuse: anatomy, pharmacology and function of reward pathways. Trends Pharmacol Sci. 1992;13:177–184. doi: 10.1016/0165-6147(92)90060-j. [DOI] [PubMed] [Google Scholar]
- 37.Koob GF, LeMoal M. Drug abuse: hedonic homeostatic dysregulation. Science. 1997;278:52–58. doi: 10.1126/science.278.5335.52. [DOI] [PubMed] [Google Scholar]
- 38.Leslie CA, Robertson MW, Jung AB, Liebermann J, Bennett JP., Jr Effects of prenatal cocaine exposure upon postnatal development of neostriatal dopaminergic function. Synapse. 1994;17:210–215. doi: 10.1002/syn.890170311. [DOI] [PubMed] [Google Scholar]
- 39.Levitt P, Rakic P. The time of genesis, embryonic origin and differentiation of the brain stem monoamine neurons in the Rhesus monkey. Dev Brain Res. 1982;4:35–57. doi: 10.1016/0165-3806(82)90095-5. [DOI] [PubMed] [Google Scholar]
- 40.Little KY, Zhang L, Desmond T, Frey KA, Dalack GW, Cassin BJ. Striatal dopaminergic abnormalities in human cocaine users. Am J Psychiatry. 1999;156:238–245. doi: 10.1176/ajp.156.2.238. [DOI] [PubMed] [Google Scholar]
- 41.Mayes LC, Bornstein MH, Chawarska K, Granger RH. Information processing and developmental assessment in 3-month-old infants exposed prenatally to cocaine. Pediatrics. 1995;95:539–545. [PubMed] [Google Scholar]
- 42.Mayes LC, Grillon C, Granger R, Schottenfeld R. Regulation of arousal and attention in preschool children exposed to cocaine prenatally. In: Harvey JA, Kosofsky BE, editors. Annals of the New York Academy of Sciences: cocaine: effects on the developing brain. The New York Academy of Sciences; New York: 1998. pp. 126–143. [PubMed] [Google Scholar]
- 43.Minabe Y, Ashby CR, Jr, Heyser C, Spear LP, Wang RY. The effects of prenatal cocaine exposure on spontaneously active midbrain dopamine neurons in adult male offspring: an electrophysiological study. Brain Res. 1992;586:152–156. doi: 10.1016/0006-8993(92)91387-t. [DOI] [PubMed] [Google Scholar]
- 44.Moody CA, Granneman JG, Bannon MJ. Dopamine transporter binding in rat striatum and nucleus accumbens is unaltered following chronic changes in dopamine levels. Neurosci Lett. 1996;217:55–57. doi: 10.1016/0304-3940(96)13048-2. [DOI] [PubMed] [Google Scholar]
- 45.Nestler EJ. Hard target: understanding dopaminergic neurotransmission. Cell. 1994;79:923–926. doi: 10.1016/0092-8674(94)90022-1. [DOI] [PubMed] [Google Scholar]
- 46.Nestler EJ, Aghajanian GK. Molecular and cellular basis of addiction. Science. 1997;278:58–63. doi: 10.1126/science.278.5335.58. [DOI] [PubMed] [Google Scholar]
- 47.Parsons LH, Schad CA, Justice JB., Jr Co-administration of the D2 antagonist pimozide inhibits up-regulation of dopamine release and uptake induced by repeated cocaine. J Neurochem. 1993;60:376–379. doi: 10.1111/j.1471-4159.1993.tb05864.x. [DOI] [PubMed] [Google Scholar]
- 48.Pilotte NS, Sharpe LG, Kuhar MJ. Withdrawal of repeated intravenous infusions of cocaine persistently reduces binding to dopamine transporters in the nucleus accumbens of Lewis rats. J Pharmacol Exp Ther. 1994;269:963–969. [PubMed] [Google Scholar]
- 49.Pumford KM, Russell JA, Leng G. Effects of the selective kappa-opioid agonist U50,488 upon the electrical activity of supraoptic neurones in morphine- tolerant and morphine-naive rats. Exp Brain Res. 1993;94:237–246. doi: 10.1007/BF00230291. [DOI] [PubMed] [Google Scholar]
- 50.Reader TA, Ase AR, Huang N, Hébert C, Van Gelder NM. Neuroleptics and dopamine trasporters. Neurochem Res. 1998;23:73–80. doi: 10.1023/a:1022405621365. [DOI] [PubMed] [Google Scholar]
- 51.Reith MEA, Sershen H, Lajtha A. Binding of [3H]cocaine in mouse brain: kinetics and saturability. J Receptor Res. 1981;2:233–243. doi: 10.3109/10799898109038802. [DOI] [PubMed] [Google Scholar]
- 52.Ritz MC, Lamb RJ, Goldberg SR, Kuhar MJ. Cocaine receptors on dopamine transporters are related to self administration of cocaine. Science. 1987;237:1219–1223. doi: 10.1126/science.2820058. [DOI] [PubMed] [Google Scholar]
- 53.Rønnekleiv OK, Naylor BR. Chronic cocaine exposure in the fetal rhesus monkey: consequences for early development of dopamine neurons. J Neurosci. 1995;15:7330–7343. doi: 10.1523/JNEUROSCI.15-11-07330.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Rønnekleiv OK, Fang Y, Choi WS, Chai L. Changes in the midbrain-rostral forebrain dopamine circuitry in the cocaine-exposed primate fetal brain. Ann NY Acad Sci. 1998;846:165–181. [PubMed] [Google Scholar]
- 55.Rowe DC, Stever C, Gard JMC, Cleveland HH, Sanders ML, Abramowitz A, Kozol ST, Mohr JH, Sherman SL, Waldman ID. The relation of the dopamine transporter gene (DAT1) to symptoms of internalizing disorders in children. Behav Genet. 1998;28:215–225. doi: 10.1023/a:1021427314941. [DOI] [PubMed] [Google Scholar]
- 56.Saadouni S, Refahi-Lyamani F, Costentin J, Bonnet J-J. Cocaine and GBR 12783 recognize nonidentical, overlapping binding domains on the dopamine neuronal carrier. Eur J Pharmacol. 1994;268:187–197. doi: 10.1016/0922-4106(94)90188-0. [DOI] [PubMed] [Google Scholar]
- 57.Sagvolden T, Sergeant JA. Attention deficit/hyperactivity disorder—from brain dysfunctions to behaviour. Behav Brain Res. 1998;94:1–10. [PubMed] [Google Scholar]
- 58.Scalzo FM, Ali SF, Frambes NA, Spear LP. Weanling rats exposed prenatally to cocaine exhibit an increase in striatal D2 dopamine binding associated with an increase in ligand affinity. Pharmacol Biochem Behav. 1990;37:371–373. doi: 10.1016/0091-3057(90)90350-q. [DOI] [PubMed] [Google Scholar]
- 59.Searight HR, Nahlik JE, Campbell DC. Attention-deficit/hyperactivity disorder: assessment, diagnosis, and management. J Fam Pract. 1995;40:270–279. [PubMed] [Google Scholar]
- 60.Self DW, Nestler EJ. Molecular mechanisms of drug reinforcement and addiction. Annu Rev Neurosci. 1995;18:463–495. doi: 10.1146/annurev.ne.18.030195.002335. [DOI] [PubMed] [Google Scholar]
- 61.Sharpe LG, Pilotte NS, Mitchell WM, De Souza EB. Withdrawal of repeated cocaine decreases autoradiographic [3H]mazinol-labelling of dopamine transporter in rat nucleus accumbens. Eur J Pharmacol. 1991;203:141–144. doi: 10.1016/0014-2999(91)90804-y. [DOI] [PubMed] [Google Scholar]
- 62.Shearman LP, Collins LM, Meyer JS. Characterization and localization of [125I]RTI-55-labeled cocaine binding sites in fetal and adult rat brain. J Pharmacol Exp Ther. 1996;277:1770–1783. [PubMed] [Google Scholar]
- 63.Simon JR, Bare DJ, Ghetti B, Richter JA. A possible role for tyrosine kinases in the regulation of the neuronal dopamine transporter in mouse striatum. Neurosci Lett. 1997;224:201–205. doi: 10.1016/S0304-3940(97)13479-6. [DOI] [PubMed] [Google Scholar]
- 64.Solanto MV. Neuropsychopharmacological mechanisms of stimulant drug action in attention-deficit hyperactivity dissorder: a review and integration. Behav Brain Res. 1998;94:127–152. doi: 10.1016/s0166-4328(97)00175-7. [DOI] [PubMed] [Google Scholar]
- 65.Spear LP, Campbell J, Snyder K, Silveri M, Katovic N. Animal behavior models: increased sensitivity to stresses and other environmental experiences after prenatal cocaine exposure. Ann NY Acad Sci. 1998;846:76–88. [PubMed] [Google Scholar]
- 66.Stadlin A, Choi HL, Tsang D. Postnatal changes in [3H]mazindol-labelled dopamine uptake sites in the rat striatum following prenatal cocaine exposure. Brain Res. 1994;637:345–348. doi: 10.1016/0006-8993(94)91259-9. [DOI] [PubMed] [Google Scholar]
- 67.Staley JK, Hearn WL, Ruttenber AJ, Wetli CV, Mash DC. High affinity cocaine recognition sites on the dopamine transporter are elevated in fatal cocaine overdose victims. J Pharmacol Exp Ther. 1994;271:1678–1685. [PubMed] [Google Scholar]
- 68.Staley JK, Boja JW, Carroll FI, Seltzman HH, Wyrick CD, Lewin AH, Abraham P, Mash DC. Mapping dopamine transporters in the human brain with novel selective cocaine analog [125I]RTI-121. Synapse. 1995;21:364–372. doi: 10.1002/syn.890210412. [DOI] [PubMed] [Google Scholar]
- 69.Stevenson J. Evidence for a genetic etiology in hyperactivity in children. Behav Genet. 1992;22:337–344. doi: 10.1007/BF01066665. [DOI] [PubMed] [Google Scholar]
- 70.Struthers JM, Hansen RL. Visual recognition memory in drug-exposed infants. Dev Behav Pediatr. 1992;13:108–111. doi: 10.1097/00004703-199204000-00005. [DOI] [PubMed] [Google Scholar]
- 71.Taylor E. Clinical foundations of hyperactivity research. Behav Brain Res. 1998;94:11–24. doi: 10.1016/s0166-4328(97)00165-4. [DOI] [PubMed] [Google Scholar]
- 72.Tella SR, Ladenheim B, Andrews AM, Goldberg SR, Cadet JL. Differential reinforcing effects of cocaine and GBR-12909: biochemical evidence for divergent neuroadaptive changes in the mesolimbic dopaminergic system. J Neurosci. 1996;16:7416–7427. doi: 10.1523/JNEUROSCI.16-23-07416.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Tronick EZ, Frank DA, Cabral H, Mirochnick M, Zuckerman B. Late dose-response effects of prenatal cocaine exposure on newborn neurobehavioral performance. Pediatrics. 1996;98:76–83. [PMC free article] [PubMed] [Google Scholar]
- 74.Unis AS, Roberson MD, Robinette R, Ha J, Dorsa DM. Ontogeny of human brain dopamine receptors I. Differential expression of [3H]-SCH23390 and [3H]-YM09151–2. Dev Brain Res. 1998;106:109–117. doi: 10.1016/s0165-3806(97)00202-2. [DOI] [PubMed] [Google Scholar]
- 75.Vaughan RA, Huff RA, Uhl G, Kuhar MJ. Protein kinase C-mediated phosphorylation and functional regulation of dopamine transporters in striatal synaptosomes. J Biol Chem. 1997;272:15541–15546. doi: 10.1074/jbc.272.24.15541. [DOI] [PubMed] [Google Scholar]
- 76.Verney C, Zecevic N, Nikolic B, Alvarez C, Berger B. Early evidence of catecholaminergic cell groups in 5- and 6-week-old human embryos using tyrosine hydroxylase and dopamine-β-hydroxylase immunocytochemistry. Neurosci Lett. 1991;131:121–124. doi: 10.1016/0304-3940(91)90351-s. [DOI] [PubMed] [Google Scholar]
- 77.Waldman ID, Rowe DC, Abramowitz A, Kozel ST, Mohr JH, Sherman SL, Cleveland HH, Sanders ML, Gard JMC, Stever C. Association and linkage of the dopamine transporter gene and attention-deficit hyperactivity disorder in children: heterogeneity owing to diagnostic subtype and severity. Am J Hum Genet. 1998;63:1767–1776. doi: 10.1086/302132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Wang L, Pitts DK. Perinatal cocaine exposure decreases the number of spontaneously active midbrain dopamine neurons in neonatal rats. Synapse. 1994;17:275–277. doi: 10.1002/syn.890170408. [DOI] [PubMed] [Google Scholar]
- 79.Wickens J. Striatal dopamine in motor actrivation and reward-mediated learning: steps towards a unifying model. J Neural Transm. 1990;80:9–31. doi: 10.1007/BF01245020. [DOI] [PubMed] [Google Scholar]
- 80.Wilson JM, Nobrega JN, Carroll ME, Niznik HB, Shannak K, Lac ST, Pristupa ZB, Dixon LM, Kish SJ. Heterogeneous subregional binding patterns of 3H-WIN 35,428 and 3H-GBR 12,935 are differentially regulated by chronic cocaine self-administration. J Neurosci. 1994;14:2966–2979. doi: 10.1523/JNEUROSCI.14-05-02966.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]