

Abstract
The prion protein (PrPC) is a copper-binding protein of unknown function that plays an important role in the etiology of transmissible spongiform encephalopathies. Using morphological techniques and synaptosomal fractionation methods, we show that PrPC is predominantly localized to synaptic membranes. Atomic absorption spectroscopy was used to identify PrPC-related changes in the synaptosomal copper concentration in transgenic mouse lines. The synaptic transmission in the presence of H2O2, which is known to be decomposed to highly reactive hydroxyl radicals in the presence of iron or copper and to alter synaptic activity, was studied in these animals. The response of synaptic activity to H2O2 was found to correlate with the amount of PrPC expression in the presynaptic neuron in cerebellar slice preparations from wild-type, Prnp0/0, and PrP gene-reconstituted transgenic mice. Thus, our data gives strong evidence for the predominantly synaptic location of PrPC, its involvement in the regulation of the presynaptic copper concentration, and synaptic activity in defined conditions.
Keywords: prion protein, synaptosomes, synaptic transmission, hydrogen peroxide, copper, cerebellum
The prion protein (PrP) is a sialoglycoprotein localized at the cell surface (Stahl et al., 1987). It is mainly expressed in the CNS, particularly in neurons (Kretzschmar et al., 1986; Moser et al., 1995) and to a lesser extent in extraneural tissues (Bendheim et al., 1992; Manson et al., 1992b). In prion diseases, a post-translational modification of the cellular prion protein (PrPC) leads to the accumulation of an abnormal, conformationally altered isoform, PrPSc (Prusiner et al., 1996), particularly in the neuropil (Kitamoto et al., 1992).
The localization of PrPC within neurons, however, is not clear. The majority of immunohistochemical studies describe a somatic expression of PrPC in neurons with no or only a minor signal in the neuropil (DeArmond et al., 1987; Piccardo et al., 1990; Safar et al., 1990; Bendheim et al., 1992). Data obtained using the histoblot technique (Taraboulos et al., 1992) and electron microscopy (Fournier et al., 1995; Salès et al., 1998), however, indicate a synaptic localization of PrPC. Moreover, colocalization of PrPC with the presynaptic vesicle protein synaptophysin was observed, and it has been speculated that PrPC may be a constituent of the synaptic vesicle membrane or a product stored within vesicles (Fournier et al., 1995). However, the immunoelectron microscopic procedures used in these studies resulted in a destruction of cellular membranes leading to a loss of exact morphological PrPClocation.
Attempts to identify the role of PrPC in synaptic transmission have yielded controversial results (Collinge et al., 1994; Herms et al., 1995; Manson et al., 1995; Whittington et al., 1995; Lledo et al., 1996). Altered GABAAreceptor-mediated synaptic transmission in the hippocampus of Prnp0/0 mice was observed, and postsynaptic mechanisms were implicated (Collinge et al., 1994). Outside-out membrane patches of cerebellar Purkinje cells, however, did not reveal any alteration of the GABAA receptor in Prnp0/0 mice (Herms et al., 1995). Also, the described alteration in the rise time of IPSCs in hippocampal CA1 neurons (Collinge et al., 1994) could not be reproduced in either cerebellar Purkinje cells (Herms et al., 1995) or hippocampal CA1 neurons (Lledo et al., 1996).
In this study, we used synaptosomal fractionation methods to determine the subcellular neuronal localization of PrPC. These analyses were complemented by a more detailed morphological investigation of PrPC expression using immunohistochemical techniques. Our results support the notion that PrPC is predominantly expressed at the presynaptic membrane. Because PrPC is known to bind copper, we used atomic absorption spectroscopy to show a reduction in the copper concentration in synaptosomal preparations of Prnp0/0 mice.
For a functional analysis of PrPC and the copper concentration at the presynapse, we performed patch-clamp measurements on cerebellar slice preparations of wild-type, Prnp0/0, and PrP-reconstituted transgenic mice. For these experiments, we used hydrogen peroxide, which is known to alter synaptic vesicle release by reacting with metal ions, such as iron and copper at the presynapse (Pellmar et al., 1994), by increasing the presynaptic calcium concentration. Using transgenic lines that express PrPC in anatomically defined neurons, we observed that the effect of H2O2 on the frequency of spontaneous IPSCs (sIPSCs) in cerebellar Purkinje cells correlates with the amount of PrPC expressed in the presynaptic neuron.
Thus, our data show that PrPC is localized to the synaptic membrane and that PrPC is involved in regulating the presynaptic copper concentration and synaptic transmission under biochemically defined conditions.
MATERIALS AND METHODS
Experimental animals. The animals used for these experiments were Prnp0/0 mice (Büeler et al., 1992) and wild-type C57Bl/6J×129/sv(ev) hybrids. Also included in the analysis were mice overexpressing the mouse Prnp-b allele (tg35) and the mouse Prnp-a allele (tg20) on a Prnp0/0 background, as described previously (Fischer et al., 1996).
Subcellular fractionation. Synaptic plasma membranes (SPMs), synaptic vesicle (SV) fractions, and cytosolic synaptosomal (CS) fractions were prepared following standard protocols (Huttner et al., 1983; Brose et al., 1989). All procedures were performed at 4°C. Twenty brains of adult wild-type, Prnp0/0, and tg35 mice were collected in 80 ml of ice-cold 0.32m sucrose and homogenized using a glass–Teflon homogenizer. The resulting homogenate was centrifuged at 800 × g for 10 min. The supernatant was pelleted again at 9200 × g for 15 min. The resulting crude synaptosomal pellet was washed by resuspension and recentrifugation (9200 × g for 15 min), resuspended in 15 ml of 0.32m sucrose, and homogenized in 9 vol of H2O containing 0.2 mm PMSF, 0.5 μg/ml leupeptin, and 1 μm pepstatin. The suspension was stirred at 4°C for 15 min. Centrifugation at 25,000 × g for 20 min produced a pellet with lysed synaptosomal membranes. After ultracentrifugation of the supernatant at 230,000 × g for 2 hr, the supernatant contained the CS fraction, and the pellet contained the crude SV fraction. The lysed synaptosomal membrane fraction was resuspended in 5 ml of H2O and loaded on a sucrose density gradient containing 15 ml of 1.2 m sucrose, 15 ml of 0.8m sucrose, and 5 ml of 0.3m sucrose. After centrifugation at 113,000 × g for 150 min in a Beckman Instruments (Fullerton, CA) SW 28 rotor, the SPM fraction band between 0.8 and 1.2m sucrose was collected.
Postsynaptic densities. Triton X-100 was used to separate postsynaptic densities (PSDs) from other synaptic elements based on insolubility in the detergent (Carlin et al., 1980). The prepared crude synaptosomal fraction was resuspended in buffer B (0.32m sucrose and 1 mmNaHCO3) and loaded onto a sucrose density gradient containing 15 ml each of 1.4 and 1.0 msucrose. After centrifugation at 82,500 × g for 60 min in a Beckman SW 28 rotor, the band between 1.4 and 1.0 sucrose was collected and diluted in buffer B to a final concentration of 4 mg/ml. After adding an equal volume of 1% (v/v) Triton X-100, the suspension was stirred for 15 min at 4°C and centrifuged at 32,800 ×g for 10 min. The resulting pellet was resuspended in buffer B and loaded on another sucrose density gradient containing 4 ml of 2.0m sucrose, 3 ml of 1.5 msucrose and 3 ml of 1.0 m sucrose. After centrifugation at 201,800 × g for 120 min in the Beckman SW 40 rotor, the PSD fraction band between 1.5 and 2.0m sucrose was collected, diluted in buffer B to a volume of 9 ml, mixed with an equal volume of 1% Triton X-100 in 150 mm KCl, loaded onto a further sucrose density gradient containing 1.5 ml each of 2.1 and 1.5 m sucrose, and spun at 113,000 × g for 10 min using the Beckmann SW 40 rotor. This procedure was repeated once. The PSD band was diluted in HEPES–KOH and pelleted at 113,500 × g for 10 min. The resulting pellet containing PSDs was collected in HEPES–KOH buffer.
Preparation of synaptosomes. Synaptosomes were prepared by a modification of the procedure of Nicholls (McMahon et al., 1992). Five brains, excluding brainstem of female, 2-month-old wild-type, Prnp0/0, and tg20 mice were collected in 30 ml of ice-cold 0.32 m sucrose and homogenized using a glass–Teflon homogenizer. The resulting homogenate was centrifuged at 3440 × g for 2 min. The supernatant was pelleted again at 16,700 × g for 12 min. The resulting crude synaptosomal pellet was resuspended in 4 ml of 0.32m sucrose and put on a three-step Ficoll (Amersham Pharmacia Biotech, München, Germany) gradient (6, 9, and 13%). After centrifugation at 64,000 × g for 35 min, the interfaces were collected and pooled. The integrity of the synaptosomes was confirmed by electron microscopy.
Atomic absorption spectroscopy. Synaptosomes were dried at 150°C overnight and ashed at low temperature for 24 hr using plasma processor TePLa 100-e (Technics Plasma). The residue was absorbed in nitric acid. Cu measurements were performed using a Zeeman 3030 (Perkin-Elmer, Emeryville, CA) flameless atomic absorption spectrophotometer at a wavelength of 325.2 nm after rapid atomization at 2000°C in a graphite-tube cuvette HGA-70 (Perkin-Elmer).
Antibodies. Murine PrPC was detected using either a rabbit antiserum (Ra5) raised against a synthetic peptide corresponding to amino acid residues 107–122 (Groschup et al., 1994) or the monoclonal antibody 3B5 raised by DNA-mediated immunization of Prnp0/0 mice and mapped to amino acids 54–69 of human PrP (Krasemann et al., 1996). Polyclonal antibodies to synaptotagmin I (Perin et al., 1990) were a gift from T. C. Südhof (Dallas, TX). Monoclonal antibodies to NMDA-R1 were raised against a fusion protein of NMDA-R1 with glutathione S-transferase (Brose et al., 1994).
SDS-PAGE and Western blot analysis. Protein samples were quantified by standard procedures (BCA Protein Assay; Sigma, Deisenhofen, Germany) with bovine serum albumin as a standard. SDS-PAGE was performed using either 10 or 15% gels. Proteins were transferred to a nitrocellulose membrane (Schleicher & Schuell, Göttingen, Germany) using a semi-dry electrotransfer system (Phase, Lübeck, Germany). The blots were blocked with 0.2% I-Block (Tropix, Bedford, MA) in PBS–0.1% Tween 20 for 30 min and were then incubated with primary antibodies for 1 hr at room temperature, followed by an incubation with alkaline phosphatase (AP)-conjugated goat anti-rabbit or anti-mouse antibodies from Dako (Hamburg, Germany) (1:500) for 1 hr at room temperature. After the incubation steps, the membranes were washed with PBS–0.1% Tween 20. Immunopositive signals were detected using the chemiluminescent substrate CDP-StarTM (Tropix) following the manufacturer’s instructions. The light signals were monitored, documented, and quantified using a CCD-video system and the accompanying software (Raytest, Straubenhardt, Germany). Blots were developed using nitroblue-tetrazolium-chloride (NBT) and 5-brom-4-chlor-indolyl-phosphate (BCIP) (Boehringer Mannheim, Mannheim, Germany).
Immunohistochemistry of cryostat sections. Animals were killed by an overdose of 7% chloral hydrate, and their eyes were immediately removed from the sockets. The eyes were mounted in Tissue Tek (OCT compound; Miles Inc., Elkhart, IN) and rapidly frozen in precooled isopentane; the same procedure was performed for the brain after removal from the skull. The samples were kept at −80°C until sectioning. Sections of 8 μm thickness were made at −20°C, air-dried, fixed in ethanol for 10 min, and kept at −20°C. Frozen sections were thawed, rinsed in 0.1 PBS, pH 7.4, and blocked with 2% bovine serum albumin. The sections were incubated with an undiluted medium supernatant containing the monoclonal antibody 3B5. After rinsing, the slides were incubated with a fluorescent goat anti-mouse antibody (Oregon Green 488; 1:200; Molecular Probes, Eugene, OR). For synaptophysin immunohistochemistry, sections were treated as described above and incubated overnight with a monoclonal antibody against synaptophysin (1:10; Boehringer Mannheim). A tetramethylrhodamine isothiocyanate-coupled goat anti-mouse IgG was used as a secondary antibody (1:200; Dianova, Hamburg, Germany). Sections were examined using the Leica (Heidelberg, Germany) TCS NT laser scanning system mounted on an Olympus Opticals (Tokyo, Japan) BX50 WI microscope.
Histoblots. Histoblots for the detection of PrPC were performed using a modification of the method described previously (Taraboulos et al., 1992). Sections of frozen brain tissue were cut at 10 μm and immediately transferred onto nitrocellulose membranes that had been wetted in lysis buffer (0.5% NP-40, 0.5% sodium desoxycholate, 100 mmNaCl, 10 mm EDTA, and 10 mmTris-HCl, pH 7.8). The membranes were air-dried thoroughly and rehydrated for 1 hr in TBST-buffer (100 mm NaCl, 0.05% Tween 20, and 10 mm Tris-HCl, pH 7.8). Blots were rinsed three times in TBST, then blocked with 5% nonfat dry milk, rinsed in TBST again, and incubated for 12 hr at 4°C with antiserum Ra5 in TBST and 1% nonfat milk. Rabbit antiserum was used at a dilution of 1:500. Secondary goat anti-rabbit AP-conjugated antibody (Dako) was used at a dilution of 1:1000 in TBST and 1% nonfat milk. Blots were developed using NBT and BCIP (Boehringer Mannheim).
Whole-cell voltage-clamp recordings of cerebellar Purkinje cells. Patch-clamp experiments were performed on Purkinje cells in thin slices of the cerebellum following standard procedures (Hamill et al., 1981; Edwards et al., 1989). Sagittal slices of cerebellum (150 μm) were prepared from 9- to 13-d-old mice as described previously (Edwards et al., 1989; Herms et al., 1995) and maintained at 37°C in a continuously bubbled (95% O2 and 5% CO2) solution (125 mm NaCl, 2.5 mm KCl, 1.25 mmNaH2PO4, 26 mm NaHCO3, 2 mm CaCl2, 1 mm MgCl2, and 25 mm glucose). After 60 min recovery, the slices were placed in the recording chamber and superfused with the above solution at room temperature. A Purkinje cell was selected using an upright microscope with a 63× water-immersion lens (Zeiss, Göttingen, Germany). Electrodes were pulled from borosilicate glass capillaries and filled with a solution containing (in mm140 CsCl, 10 HEPES, 10 EGTA, 1 CaCl2, 2 MgCl2, 4 Na2-ATP, and 0.4 Na3-GTP (adjusted to pH 7.3 with CsOH). Single-electrode voltage-clamp recordings (Edwards et al., 1989) were performed with a patch-clamp amplifier (EPC-9; Heka Elektronik, Lambrecht, Germany) using optimal series resistance compensation as recommended (Llano et al., 1991). The whole-cell capacitance and series resistance (Rs) adjustment of the amplifier were used to compensate the initial portion of the capacitance transient elicited by 10 mV hyperpolarizing pulses and to estimate the value of the series resistance (Llano et al., 1991). The series resistance of Purkinje cells before compensation was typically 3–10 MΩ. No significant differences were observed here between the different transgenes studied and wild-type Purkinje cells. Series-resistance compensation was set at between 60 and 70%.Rs was monitored throughout the experiments by hyperpolarizing pulses applied every 30 sec. The experiment was discontinued if the series resistance increased above 15 MΩ or >20% of the initial value. Our results are derived from a data set of 96 Purkinje cells.
H2O2 was diluted daily from a 30% stock solution and applied by bath perfusion. Given the perfusion rate of 1.0–1.5 ml/min and the volume of 1.1 ml of the chamber, exchange of the external solution was achieved within a very short time. The bath solutions contained 10 μm of the antagonist of ionotropic glutamate receptors 6-cyano-7-nitroquinoxaline-2,3-dione, as well as in some experiments 0.5 μm tetrodotoxin (TTX) purchased from Biotrend (Köln, Germany). All other chemicals used were purchased from Sigma.
RESULTS
Detection of PrPC in histoblots
Histoblots of brain sections from wild-type and PrPC-overexpressing mice (tg35) were immunostained to analyze the pattern of PrPC expression. Only a very weak, non-definitive signal was detected in wild-type mice (Fig.1A). No signal was seen in Prnp0/0 mice, which served as negative controls (Fig. 1B). However, strong staining was seen on brain sections from tg35 mice overexpressing PrPC (Fig. 1C). In these mice, strong staining was found in regions of high synaptic density, i.e., in hippocampal stratum oriens, stratum radiatum, stratum lacunosum moleculare (Fig. 1D), and in the molecular layer of the cerebellum (Fig. 1F). The signal was absent in regions primarily consisting of nerve cell somata, i.e., in the granular layer of the hippocampal dentate gyrus and the stratum pyramidale of the hippocampal CA1 region (Fig. 1E, hematoxilin–eosin stain of a parallel section). Intermediate-to-faint staining was observed in the cerebral cortex and other gray matter regions without localization to specific cortical regions or layers.
Fig. 1.
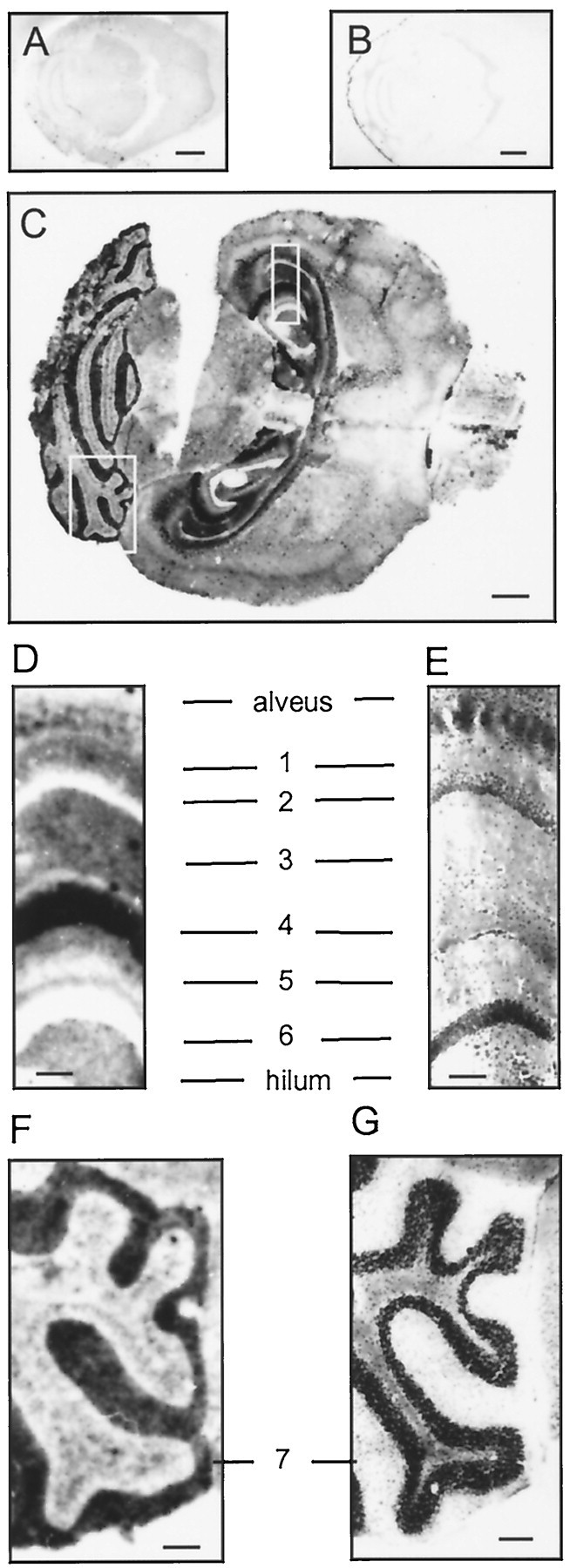
PrPC is preferentially localized to areas of high synaptic density. Histoblots of wild-type (A), Prnp0/0(B), and PrPC-overexpressing reconstituted Prnp0/0 (tg35) (C) mice. The blots were probed with anti-PrP antiserum Ra5. No PrPC was detected in Prnp0/0 mice (B) or in wild-type mouse brains (A). D and F show enlargements from boxes in C.E and G show consecutive hematoxilin and eosin stains. D, E, PrPC is strongly expressed in areas of high synaptic density of the hippocampal stratum orients (1), stratum radiatum (3), stratum lacunosum moleculare (4), and the molecular layer (5). No signal was detected in the hippocampal pyramidal cell layer (2) or the granular layer of the dentate gyrus (6). F,G, High PrPC expression in the cerebellar molecular layer (7). Scale bars:A, 1 mm; B, C, 2.5 mm;D–G, 250 μm.
Localization of PrPC in mouse cerebellum and retina of transgenic animals
Further immunohistochemical examination using confocal laser microscopy revealed highest expression of PrPC in the cerebellar molecular layer in both tg20 mice and tg35 mice (Fig.2A,B). Here, PrPC was found to be coexpressed with synaptophysin (data not shown). In tg35 mice, the Purkinje cell bodies also showed immunoreactivity, whereas in tg20 mice, we observed no signal in the Purkinje cell layer (Fig. 2B), consistent with the finding that tg20 Purkinje cells do not express PrPC (Fischer et al., 1996).
Fig. 2.
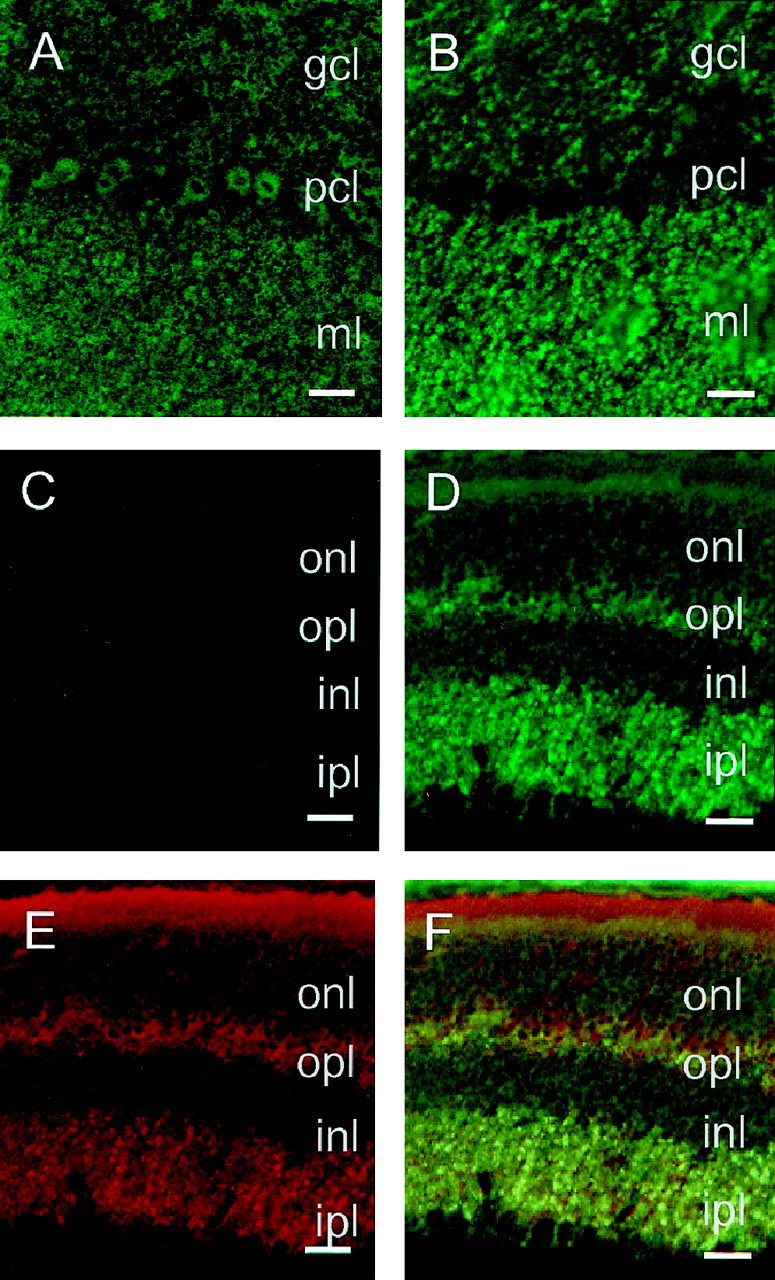
Laser scanning confocal images of PrPC in the cerebellar cortex and retina of wild-type and PrPC-overexpressing Prnp0/0-reconstituted mice. A,B, Cerebellar cortex of tg35 and tg20 animals analyzed using PrP antibody 3B5. In tg35 (A), PrPC was detected over the granule cell layer (gcl), the Purkinje cell bodies (pcl), and the molecular layer (ml). In tg20 (B), a strong PrPC expression was observed over the molecular layer and granule cell layer but not in the Purkinje cell layer.C–F, Retina of wild-type (C) and PrPC-overexpressing tg20 (D–F) mice. Using PrP antibody 3B5, no signal was detected in wild-type mice (C), whereas the tg20 retina (D) showed a strong PrPC expression in the inner plexiform layer (ipl), as well as the outer plexiform layer (opl). In tg20 mice, synaptophysin (E) was found to be coexpressed with PrPC in the outer and inner plexiform layers (F). PrPC expression is low in the outer (onl) and inner nuclear layers (inl). Scale bars: A–F, 150 μm.
Both transgenic lines showed strong immunoreactivity in the molecular layer, which is primarily composed of granule cell axons (parallel fibers) and Purkinje cell dendrites. Because Purkinje cells of tg20 mice do not express PrPC, the strong PrPC immunoreactivity in the molecular layer of these animals does not originate from the Purkinje cell dendrites, but mostly from granule cell axons.
No signal was detected in wild-type retina (Fig. 2C). Within the retina of a transgenic line overexpressing PrPC (tg20), immunohistochemistry revealed strong staining in the inner and outer plexiform layers (Fig.2D), which are regions of high synaptic density, as shown by synaptophysin immunohistochemistry (Fig.2E). There was only low immunoreactivity in the internal or external granule cell layers. Very low PrPC immunoreactivity was observed in the inner segments of the photoreceptor cells. No PrPC immunopositivity was detected in the outer segments. Figure 2F clearly shows that PrPC is colocated with the synaptic protein synaptophysin.
Preferential localization of PrPC in the synaptic membrane fraction
Biochemical preparation of subcellular fractions of wild-type mice and PrPC-overexpressing mice was used to more clearly define the localization of PrPC in wild-type neurons and to compare these with PrPC-overexpressing neurons. Whole-brain homogenates, crude SV fractions, CS fractions, and SPM fractions from adult mice were isolated. The synaptic plasma membrane fraction from Prnp0/0 mice served as control. Using the antibody 3B5 (Fig.3A,C) and the antiserum Ra5 (Fig. 3B), we observed a strong PrPC signal in the SPM fraction in wild-type mice (Fig. 3A,B) and in tg35 (Fig. 3C). The two main glycosylated forms (33–35 and 30 kDa) and one unglycosylated form (27 kDa) of PrPC were clearly distinguishable. A small band was seen in the SPM fraction of Prnp0/0 mice (Fig. 3A), which was unspecific because it was not detectable in the blot probed with a different PrP antibody (Fig. 3B). PrPC was also detected in the crude synaptic vesicle fraction in both wild-type and PrPC-overexpressing mice (Fig.3A–C, SV). In contrast to the synaptic vesicle protein synaptotagmin (Fig. 3D), there was a stronger band of PrPC in the SPM fraction than in the crude synaptic vesicle fraction (Fig. 3A–C). Neither synaptotagmin nor PrPC were detected in the CS fraction (Fig. 3A–D). A comparison of the PrPC signal of the brain homogenate with the PrPC signal from the SPM fraction showed an enrichment factor of 3 to 4 in both wild-type mice and PrPC-overexpressing mice. As shown in a Western blot with an antibody directed against NMDA receptor subunit 1, the synaptic vesicle fraction is not pure (Fig. 3E). Although these results show that the major amount of PrPC is localized to the synaptic plasma membrane, it remains unclear what proportion of PrPC is in the synaptic vesicle fraction.
Fig. 3.
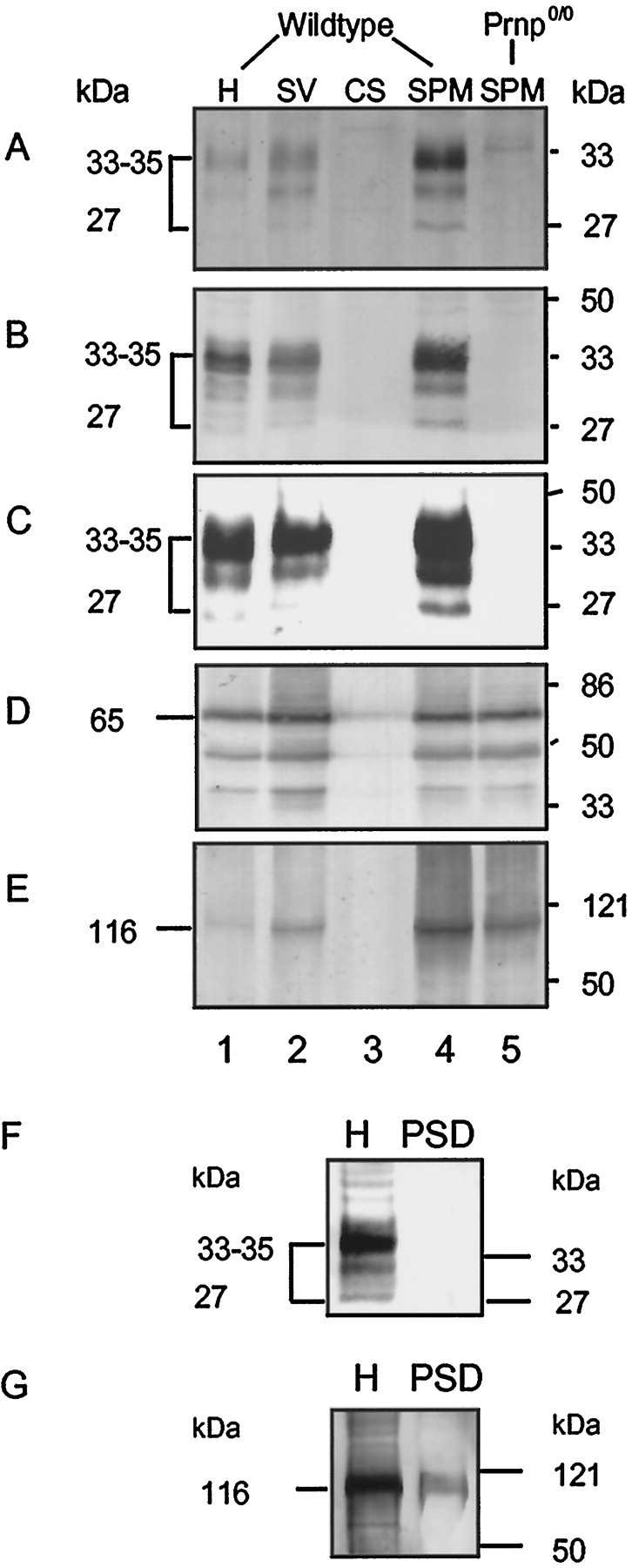
Enrichment of PrPC in the synaptic plasma membrane fraction but not in the postsynaptic density fraction isolated from mouse brains. Equal amounts (100 μg/lane) of subcellular fractions from wild-type mouse brain and prion protein-deficient mouse brain (Prnp0/0) were analyzed by immunoblotting with anti-PrP antibody 3B5 (A) and antiserum Ra5 (B). For comparison, subcellular fractions of PrPC-overexpressing tg 35 mice (30 μg/lane) were also examined using antibody Ra5 (C). The immunostaining for synaptotagmin (D) and NMDA receptor subunit NMDA-R1 (E) was used as a control for synaptic vesicle proteins and postsynaptic membrane proteins, respectively. Subcellular fractions are designated as follows: H, homogenate; SV, crude synaptic vesicle fraction; CS, cytosolic synaptosomal fraction; SPM, synaptic plasma membranes.F, In the PSD fraction, PrPC is not detectable (Western blot analysis in homogenates and PSD with antibody 3B5; 30 μg of protein were loaded in each lane). G, Immunostaining for NMDA-R1 was used as a control for postsynaptic membrane proteins.
To address the subcellular localization of PrPC in synapses more directly, we quantified the relative amounts of PrPCon the cell surface and on transport–synaptic vesicles. For that purpose, we isolated Ficoll-purified synaptosomes and treated these with trypsin. This treatment should lead to complete degradation of the cell surface–plasma membrane pool of PrPC but leave intracellular vesicular pools unaffected. We found that trypsin digestion of Ficoll-purified synaptosomes leads to complete degradation of all detectable synaptosomal PrPC, indicating that very little or no PrPC is present on synaptic vesicles. In contrast, the synaptic vesicle protein synaptotagmin remained intact under these conditions (data not shown). This finding further supports the notion that PrPC is located predominantly on the synaptic plasma membrane rather than on the synaptic vesicle membrane, as proposed by Fournier et al. (1995).
For a clearer location of PrPC at the presynaptic or postsynaptic membrane, we isolated PSDs. In contrast to the NMDA receptor subunit 1 (Fig. 3G), a protein known to be present in this fraction, PrPC was not detected in the postsynaptic density fraction of wild-type mice (Fig. 3F, using the Ra5 PrP antibody as inB).
Copper concentration in synaptosomes is related to PrPC
A strong correlation of the prion protein expression and the copper content of crude membrane-enriched fractions from brain homogenates has been described previously (Brown et al., 1997a). Whole-brain homogenates, however, do not show any significant difference in the copper content between wild-type, Prnp0/0, and PrPC-overexpressing mice (Fig.4A). However, differences were observed in synaptosomal preparations, fractions in which PrPC is enriched (Fig.4B). Here, the copper concentration was significantly lower in Prnp0/0 (23 ± 6 ng of Cu/mg of protein) than in wild-type mouse preparations (39 ± 9 ng of Cu/mg of protein). In tg20 mice, which overexpress PrPC on a Prnp0/0 background, the copper concentration was found to be in the same range as in wild-type animals (40 ± 13 ng of Cu/mg of protein).
Fig. 4.
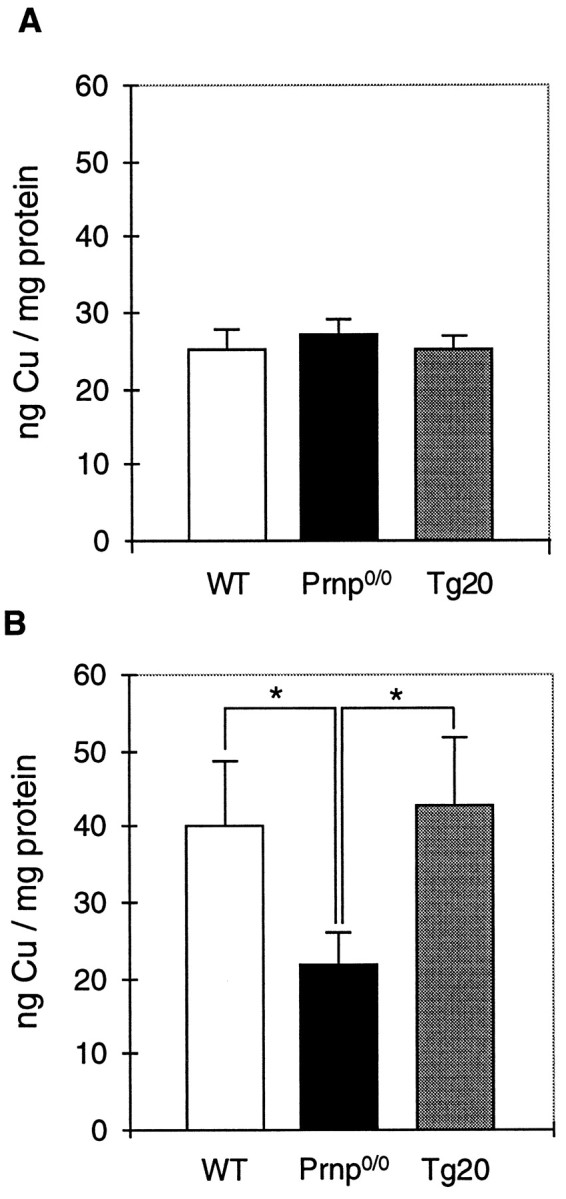
Copper concentration in synaptosomes correlates with PrPC expression. The copper concentration in whole-brain homogenates (A) and synaptosomal fractions (B) of wild-type (WT), Prnp0/0, and Prnp-reconstituted Prnp0/0 mice (tg20) was studied by atomic absorption spectroscopy. The copper concentration was not significantly different in the whole-brain homogenates of the three lines tested. The synaptosomal preparations reveal a significantly reduced copper concentration in Prnp0/0 mice compared with wild-type mice (Student’s t test;p < 0.05). In PrPCgene-reconstituted Prnp0/0 mice (tg20), the synaptosomal copper concentration was similar to wild-type mice. Shown are the mean and SE for four to six independent preparations of five age- and sex-matched brains. Asterisks indicate that the observed differences were found to be statistically significant.
Effect of hydrogen peroxide on inhibitory synaptic transmission in wild-type, Prnp0/0, and PrPC-overexpressing mice
Biochemical investigation has shown a predominant location of PrPC at the synaptic plasma membrane, and atomic absorption measurements have revealed that synaptosomal copper concentration is related to PrPC. Copper binding by PrPC may therefore have functional consequences for synaptic transmission (Brown et al., 1997a). Here, we studied the effect of hydrogen peroxide, which is known to react with metal ions such as copper to form highly reactive oxygen species. These have been shown to modulate synaptic transmission predominantly on a presynaptic level (Gilman et al., 1992; Palmeira et al., 1993; Pellmar et al., 1994; Pellmar, 1995).
Figure 5 illustrates the effect of 0.01% H2O2 on the inhibitory synaptic activity of a Purkinje cell in wild-type (A–G) and Prnp0/0(H–N) mice. A and H display a sample of a continuous recording of sIPSCs in control saline 5 min before the application of H2O2, as indicated in Figure 5, C and J. Spontaneous IPSCs were recorded as inward currents because of the fact that the Nernst equilibrium potential for Cl− ions is close to 0 mV. B andI correspond to samples of sIPSCs taken from the same cells 8 min after addition of 0.01% H2O2 to the external solution. The application led to a marked enhancement of synaptic activity in wild-type mice (B), whereas there was no comparable effect in Prnp0/0 mice (I). The time course of the H2O2 effect on the sIPSC frequency, mean amplitude, and mean charge is shown in C–Efor the wild-type Purkinje cells and in J–L for the Prnp0/0 mice Purkinje cells. In wild-type mice, the enhancement of the sIPSC frequency after the application of H2O2 (0.01%) develops progressively, reaching a peak value 15 min after the application has been seeded (Fig. 5C). In Prnp0/0 mice, the sIPSC increases only very transient during the H2O2 application and decays during the following 10 min to values slightly lower than the frequency before the application (Fig. 5J).
Fig. 5.

Hydrogen peroxide enhances inhibitory synaptic activity in wild-type (A–G), but not Prnp0/0 (H–N) mouse Purkinje cells. Samples of the continuous recording of inhibitory synaptic currents before (A, H) and after (B, I) bath perfusion with 0.01% H2O2 at time points indicated inC and J. C andJ show plots of the number of sIPSCs detected in 30 sec sample intervals as a function of time. The barindicates the time of H2O2 application.D and K, as well as E andL, show the mean amplitudes and the mean charge of sIPSCs, calculated for 30 sec intervals, as a function of time.F and M show the amplitude histograms for the time periods (2.5 min) indicated in C andJ. G and N show the cumulative amplitude distributions of the histograms shown inF and M. No shift in the amplitude distribution was observed in either the wild type (G) or Prnp0/0(N). The holding potential was −70 mV in this and all experiments shown in the following figures.
The plots of the mean IPSC amplitude (Fig. 5D) and the mean charge (E) showed no significant effect of H2O2 in wild-type mice on these parameters. In the Prnp0/0 mouse Purkinje cell, the mean IPSC amplitude and charge decreases slightly (K, L).
No significant changes were observed when comparing the amplitude histograms of the IPSCs (Fig.5F,M) and the cumulative amplitude distributions (G, N) recorded from equivalent time periods (2.5 min), as indicated in Figure 5,C and J, before the application of H2O2 (Fa,Ma) and 10 min after the application of H2O2 (Fb,Mb).
Given that the major effect of H2O2 is an increase in the frequency of synaptic events, a presynaptic mechanism is very likely involved. It was therefore of interest to determine whether this effect could be observed in the absence of Na+-dependent spikes in presynaptic neurons. In Purkinje cells studied here, the addition of TTX (0.5 μm) to the extracellular fluid did not alter the increase of miniature IPSCs (mIPSCs) after treatment with H2O2(255 ± 53% of the basal frequency; n = 4). Because H2O2 increases the frequency of mIPSCs, this might result from an increased multiquantal release of transmitter caused by the discharge of Ca2+ spikes in the presynaptic inhibitory neurons. Experiments were therefore performed under conditions designed to eliminate Ca2+ entry. However, a similar increase in mIPSC frequency (215 ± 43%;n = 4) was observed in experiments in which Ca2+ was omitted from saline containing TTX and 200 μm EGTA.
No significant differences were observed in the resting membrane potentials and input resistances of Purkinje cells from Prnp0/0 and wild-type mice, as described previously (Herms et al., 1995). H2O2 in the concentration used in this study was found to have no effect on either parameter, similar to the observation made by Pellmar et al.(1994) on hippocampal CA1 neurons. To elucidate whether there is a general alteration in Prnp0/0 mouse synapses, which does not allow an increase of the sIPSC frequency because of a presynaptic stimulus, we studied the effect of the adenylyl cyclase activator forskolin. This is known to increase sIPSC, as well as mIPSC, frequency (Llano and Gerschenfeld, 1993). With the application of 10 μm forskolin, a similar increase of sIPSC frequency in both wild-type mice and Prnp0/0 mice was observed (170 ± 50% compared with 165 ± 48% in Prnp0/0 mouse Purkinje cells;n = 4).
To elucidate whether the differences observed between Prnp0/0 and wild-type cells are caused by expression of PrPC, we also examined transgenic mice, which overexpress PrPC on a Prnp0/0 background (tg35). Figure6A–C shows a typical cell of a tg35 mouse. In Figure 6A, the sIPSC frequency, calculated for 30 sec intervals, is plotted as a function of time. As shown by the plot, the sIPSC frequency increased over sixfold when the response to H2O2was maximal. No change in the mean sIPSC amplitude (Fig.6B) or a significant shift in the cumulative amplitude distribution (Fig. 6C) were observed.
Fig. 6.

Hydrogen peroxide enhances inhibitory synaptic activity in two lines of Prnp-reconstituted Prnp0/0mice. PrPC is overexpressed in all neurons in tg35 mice (A–C) and in all neurons except Purkinje cells in tg20 mice (D–F). A,D, Plots of the sIPSC frequency in 30 sec sample intervals against time in tg35 (A) and tg20 (D) mice. The bar indicates the time of H2O2 application. B,E, Plots of the mean sIPSCs amplitude, calculated for 30 sec intervals, as a function of time in tg35 (B) and tg20 (E) mice. C,G, Cumulative amplitude distributions for the time periods indicated in A and B. Whereas in tg35 (C) no significant shift can be observed, there is a shift to higher values in tg20 (F).
To elucidate whether the overexpression of PrPC in either Purkinje cells or presynaptic neurons, i.e., stellate or basket cells, is the cause of the restored sIPSC frequency increase in PrPC gene-reconstituted mice, we examined tg20 mice, which express PrPC in cerebellar interneurons but not Purkinje cells (Fischer et al., 1996) and are therefore ideally suited for the distinction of presynaptic and postsynaptic effects when compared with Prnp0/0 mice with no PrPC expression at all and with tg35 and wild-type mice, which express PrPC in interneurons and Purkinje cells as well. Figure 6D–Fshows the response of a tg20 mouse Purkinje cell. The dramatic increase in the sIPSC frequency (Fig. 6D) after H2O2 application indicates that presynaptic PrPC expression is important for the observed effect, whereas postsynaptic PrPC does not seem to be necessary. The plot of the mean sIPSC amplitude (E) showed a small increase after H2O2application, as well as a shift of the cumulative amplitude distribution to higher values (F), which indicates that H2O2 also modulates the sIPSC amplitude in tg20 mice.
Figure 7 illustrates cumulative data from 14 wild-type, 21 Prnp0/0, 14 tg35, and 4 tg20 mouse Purkinje cells. In wild-type mice, the sIPSC frequency increased up to 250% of the level before H2O2 application (A). In Prnp0/0 mice, the mean sIPSC frequency increased only transiently, resuming its initial level 5 min after H2O2 application (A). In tg35 Purkinje cells, which overexpress PrPC on a Prnp0/0 background, an increase of the sIPSC frequency was observed (B). Moreover this increase was significantly stronger than in wild-type mice, indicating that the amount of PrPC expressed is critical for the effect of H2O2 on synaptic transmission. A similar increase of the sIPSC frequency was observed in tg20 mice (C). In wild-type and tg35 mice, the sIPSC amplitude was not altered by H2O2, whereas the mean values decreased in Prnp0/0 mice. In tg20 mice, the mean sIPSC amplitude increased slightly after application of H2O2(D).
Fig. 7.

Pooled data of the effects of 0.01% H2O2 on the frequency and mean amplitude of Purkinje cell inhibitory synaptic currents in wild-type (●), Prnp0/0 (○), tg35 (■), and tg20 (▵) mice. Each point represents the mean ± SEM of frequency or amplitude of sIPSCs in 30 sec intervals normalized to the values before H2O2 application. Because of the tendency for amplitudes and frequencies of sIPSCs to decay slowly during the control period, only the last 3 min before the application were chosen for calculating the baseline (broken line at the 100% level). The threshold for detection of sIPSCs was set to −30 pA.A, Plots of the mean values of sIPSC frequency in wild-type (n = 14) and Prnp0/0(n = 21) mouse Purkinje cells, calculated for 30 sec intervals, as a function of time. The bar indicates the time during which H2O2 was applied. Significant differences between the two mouse strains are marked byasterisks. p < 0.01;t test. B, C, Plots of the mean values of the frequency of sIPSCs in tg35 (B;n = 15) and tg20 (C;n = 4) compared with wild-type and Prnp0/0 mouse data. D, Plots of the mean sIPSC amplitudes normalized to values before H2O2 application in wild-type, Prnp0/0, tg35, and tg20 mouse Purkinje cells. The amplitudes did not change in wild-type and tg35 cells after the application of H2O2. It increased slightly in tg20 mouse Purkinje cells and decreased significantly in Prnp0/0 mouse cells.
DISCUSSION
Light microscopic studies on the cellular and subcellular location of PrPC have yielded conflicting results. The majority of immunohistochemical investigations have indicated a somatic staining pattern of PrPC (DeArmond et al., 1987; Piccardo et al., 1990; Safar et al., 1990; Bendheim et al., 1992; Manson et al., 1992a). In the cerebellum, a strong staining of Purkinje cell bodies, but not of the neuropil, has been described (DeArmond et al., 1987; Manson et al., 1992a). In contrast, using the histoblot technique, Taraboulos et al. (1992) found a neuropil staining pattern in hamster brain. The cerebellum was not examined in that study. More recently, an immunoelectron microscopic study (Fournier et al., 1995) demonstrated colocalization of PrPC with the presynaptic vesicle protein synaptophysin, which led to the speculation that PrPC might be a constituent of the synaptic vesicle membrane or a product stored within vesicles. Also,Salès et al. (1998) described synaptic staining in hamster brain using a highly sensitive free-floating immunohistochemical procedure. However, the electron microscopic data did not give convincing evidence of a preferential expression of PrPC at the synaptic membrane because the tissue preservation in that study was not of sufficient quality as a result of the preembedding staining technique that was used. On the functional level, previous physiological data on Prnp0/0 mice have also yielded contradictory results concerning the direct involvement in the synaptic function of PrPC (Collinge et al., 1994; Herms et al., 1995; Manson et al., 1995; Whittington et al., 1995; Lledo et al., 1996).
For a clarification of these questions, we performed biochemical, immunohistochemical, and electrophysiological experiments using various mouse lines. All investigations were performed on wild-type, PrPC gene-ablated (Prnp0/0), and PrPC gene-reconstituted Prnp0/0 (tg20 and tg35) mice. Because these transgenic lines differ in their expression pattern of PrPC, they were particularly suitable for our experiments. Tg35 mice express PrPC in all neurons, whereas tg20 mice express PrPC in all neurons except Purkinje cells (Fischer et al., 1996). These mice are therefore ideal for a functional analysis of presynaptic and postsynaptic factors in the synaptic transmission of Purkinje cells depending on the PrPC expression.
The histoblot data of PrPC-overexpressing mice presented here clearly show that PrPCimmunostaining is most intense in regions of dense neuropil, such as the stratum radiatum and stratum oriens of the CA1 region, and is virtually absent from the granule cell layer of the dentate gyrus and the pyramidal cell layer throughout the Ammon’s horn. Immunohistochemistry of the retina shows the predominant location of PrPC in regions of high synaptic density, such as the inner and outer plexiform layers. The observed colocalization of PrPC with the synaptic vesicle protein synaptophysin in the inner and outer plexiform layers is another strong indication of the synaptic location of PrPC.
Immunohistochemistry had not shown detectable levels of PrPC in the brains of wild-type mice. Therefore, a Western blot analysis of synaptic fractions of wild-type mouse brains was performed and was compared with preparations of PrPC-overexpressing transgenic animals. PrPC was found to be enriched in the synaptic plasma membrane fraction in both wild-type and PrPC-overexpressing mice. This was similar to the distribution of other synaptic proteins, such as synaptotagmin and the NMDA-R1 receptor subunit. In contrast to the synaptic vesicle protein synaptotagmin, which is more strongly enriched in the synaptic vesicle fraction than in the synaptic plasma membrane fraction, PrPC was more strongly enriched in the synaptic plasma membrane fraction, indicating that a predominant location of PrPC on the synaptic vesicle membrane as supposed recently (Fournier et al., 1995) does not seem very likely. Also, the finding that trypsinization of synaptosomes leads to a complete degradation of PrPC while a synaptic vesicle protein remains intact further supports the notion that PrPC is predominantly, if not exclusively, located on the surface of the synaptic plasma membrane.
On the functional level, our previous electrophysiological investigations of inhibitory and excitatory synaptic transmission in the cerebellum of Prnp0/0 mice did not support the notion that PrPC is involved in synaptic transmission under normal conditions (Herms et al., 1995). The findings we present here indicate that prominent differences in synaptic transmission between wild-type and Prnp0/0 mice can be observed in certain biochemically defined conditions. The key result of our experiments is that bath application of H2O2 induces a strong and sustained increase in the frequency of sIPSCs in Purkinje cells of normal mice but not of Prnp0/0 mice. In transgenic mouse strains in which the prion protein gene had been reintroduced on a Prnp0/0 background, i.e., tg 35 and tg20 (PrPCgene-reconstituted mice), all tested cells presented a strong and lasting increase of sIPSC frequency after H2O2 application. Therefore, the effect elicited by hydrogen peroxide is not unspecific but is related to PrPC expression in these animals (functional reconstitution). The fact that the increase in sIPSC frequency was also observed in Purkinje cells of tg20 mice, which have been shown to overexpress PrPC in neuronal cells excluding Purkinje cells, gives strong evidence that it is the expression on the presynaptic plasma membrane of the interneuron that is responsible for the observed differences in the response to H2O2 in the different transgenic lines.
How is the effect of H2O2related to PrPC expression? We propose that the different H2O2responses of wild-type, Prnp0/0, and transgenic mice that overexpress PrPC(tg20 and tg35) are caused by the different amounts of copper located at the presynaptic plasma membrane. Metal ions, i.e., copper and iron, decompose H2O2 to highly reactive oxygen radicals at the site at which the metal ions are located (Halliwell, 1992). At the synapse, these radicals alter synaptic vesicle release through an increase in the presynaptic calcium concentration (Pellmar et al., 1994; Pellmar, 1995) probably because of an alteration of intracellular calcium pools that are located close to the presynaptic membrane. Because an increase in the free calcium concentration at the presynaptic membrane is known to increase the probability of synaptic vesicle release (Heidelberger et al., 1994), this is the most likely explanation for the observed sIPSC and mIPSC frequency increase after the application of H2O2 in wild-type mouse cerebellar Purkinje cells of the present study. The sIPSC frequency increase after the application of H2O2 is significantly lower in Prnp0/0 mice in which the amount of copper was found to be reduced in synaptosomes than in wild-type mice. In the transgenic mice in which PrPC had been reintroduced, however, both the synaptosomal copper content and the H2O2 effect on sIPSC frequency were found to be rescued.
Also, the IPSC amplitude was found to be altered by H2O2 application, but only in Prnp0/0 mice and the transgenic mice, which highly overexpress PrPC on the presynaptic side (tg20). Although a decrease of the IPSC amplitude was observed in Prnp0/0 mice compared with wild-type mice, an increased was found in the transgenic mice that overexpress PrPC on the presynaptic side. Because tg20 mice do not express PrPC in Purkinje cells, these two mouse lines differ only in the PrPC expression on the presynaptic side of the interneuron–Purkinje cell synapse. A presynaptic cause therefore seems very likely.
However, copper-binding by PrPC at the presynaptic plasma membrane is probably not the only cause of the observed differences in the synaptosomal copper content of wild-type and Prnp0/0 mice. Synaptosomes include the presynaptic plasma membrane, as well as parts of the postsynaptic plasma membrane, and they also include high amounts of presynaptic cytosol and organelles such as mitochondria. Considering that several proteins in synaptosomes bind copper, the observed reduction of 50% in the copper concentration of Prnp0/0 mouse synaptosomes is too high to be only caused by a loss of copper that is bound to PrPC. Indeed, we suppose that the copper bound to PrPC contributes only very little to the overall copper content measured in synaptosomes. This is supported by the finding that the transgenic line that overexpresses PrPC (tg20) shows an increased response to H2O2 compared with wild-type mice but does not show a higher synaptosomal copper concentration.
It therefore seems that the strong differences in the synaptosomal copper content of Prnp0/0 and wild-type mice are caused by an alteration of copper uptake into synaptosomes by loss of PrPC. Our working hypothesis is that PrPC recaptures copper that is released into the synaptic cleft by synaptic vesicle fusion (Hartter and Barnea, 1988). In Prnp0/0 mice, copper is lost into the extracellular space and cannot be taken up as effectively into the presynaptic cytosol in which the copper content is lastingly reduced. This may be the cause of the reduced activity of a cytosolic copper-dependent enzyme, the superoxide dismutase, observed in Prnp0/0 mice (Brown et al., 1997b).
In addition, the loss of copper binding at the synaptic cleft through the loss of PrPC at the synaptic membrane, which might lead to a slightly higher copper concentration in the extracellular fluid in Prnp0/0 mice, may well be a relevant factor in explaining alterations in the properties of the GABAA receptor, as well as in hippocampal long-term potentiation (LTP), as has been described for Prnp0/0 mice (Collinge et al., 1994;Manson et al., 1995; Whittington et al., 1995). The GABAA receptor, as well as long-term potentiation in the hippocampus, have been shown recently to be affected by copper concentrations of only 0.1–1 μm (Doreulee et al., 1997;Sharonova et al., 1998). Therefore, minor differences in the extracellular copper concentration would cause alterations of the GABAA receptor and of LTP. Because the physiological constitution of the extracellular fluid using brain slices in electrophysiological experiments may vary considerably, differences in the experimental setup, such as slice thickness, the position of the cell in the studied slice, the buffer used, and, in particular, higher temperatures with increased oxidative stress, quite clearly influence results, depending on the amount of PrPC and copper present in the synapse in wild-type and Prnp0/0 animals. Such differences may therefore explain why an alteration of the GABAA receptor, as well as a diminished LTP in Prnp0/0 mouse neurons, was not reproduced by other observers using slightly different protocols, including lower temperatures (Herms et al., 1995; Lledo et al., 1996).
In conclusion, morphological and biochemical investigations of different PrPC-transgenic mouse lines provide strong evidence of a predominantly synaptic location of PrPC. Electrophysiological studies indicate that PrPC is a prominent copper-binding protein at the presynaptic plasma membrane. Loss of PrPC in Prnp0/0 mice strongly affects the copper content in synaptosomes, indicating that PrPC is involved in synaptic copper homeostasis. The exact function of copper binding by PrPC at the synaptic plasma membrane and its role in synaptic copper metabolism, as well as in synaptic transmission, remain to be clarified.
Footnotes
This work was supported by BMBF (German Federal Ministry of Science and Technology) Grant KI 9461/8, Wilhelm-Sander-Stiftung Grant 9343008, and Deutsche Forschungsgemeinschaft Grant KR 1561/2–1 and Sonderforschungsbereich 406. We thank Charles Weissmann (University of Zürich) for Prnp0/0, tg35, and tg20 mice, Dr. Christophe Pouzat for the program for analysis of synaptic activity, and Dr. Bernhard Keller for helpful comments on this manuscript.
Correspondence should be addressed to Dr. H. A. Kretzschmar, Georg-August Universität Göttingen, Robert-Koch-Strasse 40, 37075 Göttingen, Germany.
REFERENCES
- 1.Bendheim PE, Brown HR, Rudelli RD, Scala LJ, Goller NL, Wen GY, Kascsak RJ, Cashman NR, Bolton DC. Nearly ubiquitous tissue distribution of the scrapie agent precursor protein. Neurology. 1992;42:149–156. doi: 10.1212/wnl.42.1.149. [DOI] [PubMed] [Google Scholar]
- 2.Brose N, Halpain S, Suchanek C, Jahn R. Characterization and partial purification of a chloride- and calcium-dependent glutamate-binding protein from rat brain. J Biol Chem. 1989;264:9619–9625. [PubMed] [Google Scholar]
- 3.Brose N, Huntley GW, Stern-Bach Y, Sharma G, Morrison JH, Heinemann SF. Differential assembly of coexpressed glutamate receptor subunits in neurons of rat cerebral cortex. J Biol Chem. 1994;269:16780–16784. [PubMed] [Google Scholar]
- 4.Brown DR, Qin K, Herms JW, Madlung A, Manson J, Strome R, Fraser P, Kruck T, von Bohlen A, Schulz-Schaeffer W, Giese A, Westaway D, Kretzschmar H. The cellular prion protein binds copper in vivo. Nature. 1997a;390:684–687. doi: 10.1038/37783. [DOI] [PubMed] [Google Scholar]
- 5.Brown DR, Schulz-Schaeffer WJ, Schmidt B, Kretzschmar HA. Prion protein-deficient cells show altered response to oxidative stress due to decreased SOD-1 activity. Exp Neurol. 1997b;146:104–112. doi: 10.1006/exnr.1997.6505. [DOI] [PubMed] [Google Scholar]
- 6.Büeler H, Fischer M, Lang Y, Bluethmann H, Lipp H-P, DeArmond SJ, Prusiner SB, Aguet M, Weissmann C. Normal development and behaviour of mice lacking the neuronal cell-surface PrP protein. Nature. 1992;356:577–582. doi: 10.1038/356577a0. [DOI] [PubMed] [Google Scholar]
- 7.Carlin RK, Grab DJ, Cohen RS, Siekevitz P. Isolation and characterization of postsynaptic densities from various brain regions: enrichment of different types of postsynaptic densities. J Cell Biol. 1980;86:831–843. doi: 10.1083/jcb.86.3.831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Collinge J, Whittington MA, Sidle KCL, Smith CJ, Palmer MS, Clarke AR, Jefferys JGR. Prion protein is necessary for normal synaptic function. Nature. 1994;370:295–297. doi: 10.1038/370295a0. [DOI] [PubMed] [Google Scholar]
- 9.DeArmond SJ, Mobley WC, DeMott DL, Barry RA, Beckstead JH, Prusiner SB. Changes in the localization of brain prion proteins during scrapie infection. Neurology. 1987;37:1271–1280. doi: 10.1212/wnl.37.8.1271. [DOI] [PubMed] [Google Scholar]
- 10.Doreulee N, Yanovsky Y, Haas HL. Suppression of long-term potentiation in hippocampal slices by copper. Hippocampus. 1997;7:666–669. doi: 10.1002/(SICI)1098-1063(1997)7:6<666::AID-HIPO8>3.0.CO;2-C. [DOI] [PubMed] [Google Scholar]
- 11.Edwards FA, Konnerth A, Sakmann B, Takahashi T. A thin slice preparation for patch-clamp recordings from neurones of the mammalian central nervous system. Pflügers Arch. 1989;414:600–612. doi: 10.1007/BF00580998. [DOI] [PubMed] [Google Scholar]
- 12.Fischer M, Rülicke T, Raeber A, Sailer A, Moser M, Oesch B, Brandner S, Aguzzi A, Weissmann C. Prion protein (PrP) with amino-proximal deletions restoring susceptibility of PrP knockout mice to scrapie. EMBO J. 1996;15:1255–1264. [PMC free article] [PubMed] [Google Scholar]
- 13.Fournier J-G, Escaig-Haye F, De Villemeur TB, Robain O. Ultrastructural localization of cellular prion protein (PrPC) in synaptic boutons of normal hamster hippocampus. C R Acad Sci III. 1995;318:339–344. [PubMed] [Google Scholar]
- 14.Gilman SC, Bonner MJ, Pellmar TC. Peroxide effects on [H-3]L-glutamate release by synaptosomes isolated from the cerebral cortex. Neurosci Lett. 1992;140:157–160. doi: 10.1016/0304-3940(92)90091-k. [DOI] [PubMed] [Google Scholar]
- 15.Groschup MH, Langeveld J, Pfaff E. The major species specific epitope in prion proteins of ruminants. Arch Virol. 1994;136:423–431. doi: 10.1007/BF01321071. [DOI] [PubMed] [Google Scholar]
- 16.Halliwell B. Reactive oxygen species and the central nervous system. J Neurochem. 1992;59:1609–1623. doi: 10.1111/j.1471-4159.1992.tb10990.x. [DOI] [PubMed] [Google Scholar]
- 17.Hamill O, Marty A, Neher E, Sakmann B, Sigworth FJ. Improved patch-clamp techniques for high resolution current recording from cells and cell-free membrane patches. Pflügers Arch. 1981;391:85–100. doi: 10.1007/BF00656997. [DOI] [PubMed] [Google Scholar]
- 18.Hartter DE, Barnea A. Evidence for release of copper in the brain: depolarization-induced release of newly taken-up 67copper. Synapse. 1988;2:412–415. doi: 10.1002/syn.890020408. [DOI] [PubMed] [Google Scholar]
- 19.Heidelberger R, Heinemann C, Neher E, Matthews G. Calcium dependence of the rate of exocytosis in a synaptic terminal. Nature. 1994;371:513–515. doi: 10.1038/371513a0. [DOI] [PubMed] [Google Scholar]
- 20.Herms JW, Kretzschmar HA, Titz S, Keller BU. Patch-clamp analysis of synaptic transmission to cerebellar Purkinje cells of prion protein knockout mice. Eur J Neurosci. 1995;7:2508–2512. doi: 10.1111/j.1460-9568.1995.tb01049.x. [DOI] [PubMed] [Google Scholar]
- 21.Huttner WB, Schiebler W, Greengard P, De Camilli P. Synapsin I (protein I), a nerve terminal-specific phosphoprotein. III. Its association with synaptic vesicles studied in a highly purified synaptic vesicle preparation. J Cell Biol. 1983;96:1374–1388. doi: 10.1083/jcb.96.5.1374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kitamoto T, Shin R-W, Doh-ura K, Tomokane N, Miyazono M, Muramoto T, Tateishi J. Abnormal isoform of prion proteins accumulates in the synaptic structures of the central nervous system in patients with Creutzfeldt-Jakob disease. Am J Pathol. 1992;140:1285–1294. [PMC free article] [PubMed] [Google Scholar]
- 23.Krasemann S, Groschup M, Hunsmann G, Bodemer W. Induction of antibodies against human prion proteins (PrP) by DNA-mediated immunization of PrP0/0 mice. J Immunol Methods. 1996;199:109–118. doi: 10.1016/s0022-1759(96)00165-2. [DOI] [PubMed] [Google Scholar]
- 24.Kretzschmar HA, Prusiner SB, Stowring LE, DeArmond SJ. Scrapie prion proteins are synthesized in neurons. Am J Pathol. 1986;122:1–5. [PMC free article] [PubMed] [Google Scholar]
- 25.Llano I, Gerschenfeld HM. β-adrenergic enhancement of inhibitory synaptic activity in rat cerebellar stellate and Purkinje cells. J Physiol (Lond) 1993;468:201–224. doi: 10.1113/jphysiol.1993.sp019767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Llano I, Marty A, Armstrong CM, Konnerth A. Synaptic- and agonist-induced excitatory currents of Purkinje cells in rat cerebellar slices. J Physiol (Lond) 1991;434:183–213. doi: 10.1113/jphysiol.1991.sp018465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lledo PM, Tremblay P, DeArmond SJ, Prusiner SB, Nicoll RA. Mice deficient for prion protein exhibit normal neuronal excitability and synaptic transmission in the hippocampus. Proc Natl Acad Sci USA. 1996;93:2403–2407. doi: 10.1073/pnas.93.6.2403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Manson J, McBride P, Hope J. Expresson of the PrP gene in the brain of sinc congenic mice and its relationship to the development of scrapie. Neurodegeneration. 1992a;1:45–52. [Google Scholar]
- 29.Manson J, West JD, Thomson V, McBride P, Kaufman MH, Hope J. The prion protein gene: a role in mouse embryogenesis? Development. 1992b;115:117–122. doi: 10.1242/dev.115.1.117. [DOI] [PubMed] [Google Scholar]
- 30.Manson JC, Hope J, Clarke AR, Johnston A, Black C, MacLeod N. PrP gene dosage and long term potentiation. Neurodegeneration. 1995;4:113–115. doi: 10.1006/neur.1995.0014. [DOI] [PubMed] [Google Scholar]
- 31.McMahon HT, Foran P, Dolly JO, Verhage M, Wiegant V, Nicholls DG. Tetanus toxin and botulinum toxins type A and B inhibit glutamate, γ-aminobutyric acid, aspartate, and metenkephalin release from synaptosomes. Clues to the locus of action. J Biol Chem. 1992;267:21338–21343. [PubMed] [Google Scholar]
- 32.Moser M, Colello RJ, Pott U, Oesch B. Developmental expression of the prion protein gene in glial cells. Neuron. 1995;14:509–517. doi: 10.1016/0896-6273(95)90307-0. [DOI] [PubMed] [Google Scholar]
- 33.Palmeira CM, Santos MS, Carvalho AP, Oliveira CR. Membrane lipid peroxidation induces changes in γ-[3H]aminobutyric acid transport and calcium uptake by synaptosomes. Brain Res. 1993;609:117–123. doi: 10.1016/0006-8993(93)90863-i. [DOI] [PubMed] [Google Scholar]
- 34.Pellmar TC. Use of brain slices in the study of free-radical actions. J Neurosci Methods. 1995;59:93–98. doi: 10.1016/0165-0270(94)00198-p. [DOI] [PubMed] [Google Scholar]
- 35.Pellmar TC, Gilman SC, Keyser DO, Lee KH, Lepinski DL, Livengood D, Meyers LS., Jr Reactive oxygen species on neural transmission. Ann NY Acad Sci. 1994;738:121–129. doi: 10.1111/j.1749-6632.1994.tb21797.x. [DOI] [PubMed] [Google Scholar]
- 36.Perin MS, Fried VA, Mignery GA, Jahn R, Südhof TC. Phospholipid binding by a synaptic vesicle protein homologous to the regulatory region of protein kinase C. Nature. 1990;345:260–263. doi: 10.1038/345260a0. [DOI] [PubMed] [Google Scholar]
- 37.Piccardo P, Safar J, Ceroni M, Gajdusek DC, Gibbs CJ., Jr Im- munohistochemical localization of prion protein in spongiform encephalopathies and normal brain tissue. Neurology. 1990;40:518–522. doi: 10.1212/wnl.40.3_part_1.518. [DOI] [PubMed] [Google Scholar]
- 38.Prusiner SB, Telling G, Cohen FE, DeArmond SJ. Prion diseases of humans and animals. Semin Virol. 1996;7:159–173. [Google Scholar]
- 39.Safar J, Ceroni M, Piccardo P, Liberski PP, Miyazaki M, Gajdusek DC, Gibbs CJ., Jr Subcellular distribution and physicochemical properties of scrapie-associated precursor protein and relationship with scrapie agent. Neurology. 1990;40:503–508. doi: 10.1212/wnl.40.3_part_1.503. [DOI] [PubMed] [Google Scholar]
- 40.Salès N, Rodolfo K, Hässig R, Faucheux B, Di Giamberardino L, Moya KL. Cellular prion protein localization in rodent and primate brain. Eur J Neurosci. 1998;10:2464–2471. doi: 10.1046/j.1460-9568.1998.00258.x. [DOI] [PubMed] [Google Scholar]
- 41.Sharonova IN, Vorobjev VS, Haas HL. High-affinity copper block of GABAA-receptor-mediated currents in acutely isolated cerebellar Purkinje cells of the rat. Eur J Neurosci. 1998;10:522–528. doi: 10.1046/j.1460-9568.1998.00057.x. [DOI] [PubMed] [Google Scholar]
- 42.Stahl N, Borchelt DR, Hsiao K, Prusiner SB. Scrapie prion protein contains a phospatidylinositol glycolipid. Cell. 1987;51:229–240. doi: 10.1016/0092-8674(87)90150-4. [DOI] [PubMed] [Google Scholar]
- 43.Taraboulos A, Jendroska K, Serban D, Yang S-L, DeArmond SJ, Prusiner SB. Regional mapping of prion proteins in brain. Proc Natl Acad Sci USA. 1992;89:7620–7624. doi: 10.1073/pnas.89.16.7620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Whittington MA, Sidle KCL, Gowland I, Meads J, Hill AF, Palmer MS, Jefferys JGR, Collinge J. Rescue of neurophysiological phenotype seen in PrP null mice by transgene encoding human prion protein. Nat Genet. 1995;9:197–207. doi: 10.1038/ng0295-197. [DOI] [PubMed] [Google Scholar]