

Abstract
Alzheimer’s disease (AD) is a progressive neurodegenerative disorder that is thought to be caused in part by the age-related accumulation of amyloid β-protein (Aβ). The presence of neuritic plaques containing abundant Aβ-derived amyloid fibrils in AD brain tissue supports the concept that fibril accumulation per se underlies neuronal dysfunction in AD. Recent observations have begun to challenge this assumption by suggesting that earlier Aβ assemblies formed during the process of fibrillogenesis may also play a role in AD pathogenesis. Here, we present the novel finding that protofibrils (PF), metastable intermediates in amyloid fibril formation, can alter the electrical activity of neurons and cause neuronal loss. Both low molecular weight Aβ (LMW Aβ) and PF reproducibly induced toxicity in mixed brain cultures in a time- and concentration-dependent manner. No increase in fibril formation during the course of the experiments was observed by either Congo red binding or electron microscopy, suggesting that the neurotoxicity of LMW Aβ and PF cannot be explained by conversion to fibrils. Importantly, protofibrils, but not LMW Aβ, produced a rapid increase in EPSPs, action potentials, and membrane depolarizations. These data suggest that PF have inherent biological activity similar to that of mature fibrils. Our results raise the possibility that the preclinical and early clinical progression of AD is driven in part by the accumulation of specific Aβ assembly intermediates formed during the process of fibrillogenesis.
Keywords: Alzheimer, amyloid β-protein, neurotoxicity, electrophysiology, fibrillogenesis, neurodegeneration
Alzheimer’s disease (AD) is a slowly progressive disorder at both the histopathological and clinical levels. Early symptoms of mild memory loss and minimal cognitive impairment lead gradually over 5–15 years to profound dementia and death. Biochemical and morphological studies suggest that clinical impairment in AD involves early synaptic dysfunction (Anderton et al., 1998; Cummings et al., 1998), followed by more severe neuronal changes that include increased synaptic loss, widespread neuritic dystrophy, neurofibrillary tangles, and frank neuronal death (Terry et al., 1991;Gómez-Isla et al., 1996; Sze et al., 1997; Anderton et al., 1998). The mechanism underlying the initiation of this progressive pathophysiology is thought to involve the age-related accumulation of amyloid β-protein (Aβ), which can form the abundant amyloid fibrils observed in neuritic plaques at autopsy (Esiri et al., 1997). The observation of these end-stage lesions in postmortem brain tissue has led to an assumption that accumulation of fibrils per se underlies the progression of AD. This impression has been supported in part by studies of neuronal cultures, in which progressive neurodegeneration can be induced by highly aggregated, fibrillar Aβ but not by equivalent concentrations of Aβ monomers (Mattson et al., 1993; Pike et al., 1993; Lorenzo and Yankner, 1994).
Aβ is thought to start accumulating in vivo as low molecular weight species (LMW Aβ) consisting principally of monomers that are constitutively secreted from brain cells. These may, under certain circumstances, progress to oligomers and ultimately to mature 7- to 10-nm-wide amyloid fibrils. Aβ oligomers (dimers, trimers, tetramers, and possibly larger assemblies) have been identified in the conditioned media of certain cell lines that constitutively secrete Aβ (Podlisny et al., 1995, 1998; Xia et al., 1997), and recently, in CSF (Pitschke et al., 1998). Fibril formation by synthetic Aβ peptides is believed to proceed in vitro via a transition of LMW Aβ to intermediate species that go on to form fibrils (Harper and Lansbury, 1997; Teplow, 1998). Recently, two laboratories identified such intermediates in the formation of synthetic Aβ fibrils that are referred to as protofibrils (PF) (Harper et al., 1997a,b; Walsh et al., 1997).
In this paper, the electrophysiology and neurotoxicity of LMW Aβ (monomers/dimers) and PF and their relationship to fibrillar Aβ was investigated. We found that both of these earlier species reproducibly induce toxicity in cultured primary cortical neurons over a period of days. Furthermore, we show that submicromolar concentrations of PF can acutely increase the electrical activity of cortical neurons, whereas LMW Aβ at the same concentrations elicits no electrophysiological response. Based on these and other data, we hypothesize that neuronal dysfunction is initiated by the formation of PF and that the PF can trigger neuronal loss directly and/or via their transition to higher MW species, including fibrils. Our model raises the possibility that the preclinical and early clinical progression of AD is driven, in part, by temporal changes in specific Aβ assemblies formed during the process of fibrillogenesis. Elucidating the biological activity of PF should help to determine the role of early Aβ intermediates in the mechanism of neuronal dysfunction in AD, with attendant therapeutic implications.
MATERIALS AND METHODS
Aβ preparation. Synthetic Aβ peptides prepared as trifluoroacetic acid (TFA) salts are highly acidic, and care was taken to properly buffer the peptide to stabilize the effects of pH on morphology and neurotoxicity. Aβ1–40 (TFA salt) was purchased from Bachem (King of Prussia, PA; lot number ZN-571; 73.3% peptide). Aβ at a concentration of 1 mm(4.3 mg/ml, based on total weight of peptide) was dissolved in 1 mm NaOH plus phenol red (0.1 mg/ml to monitor pH), pH ∼3. To minimize isoelectric precipitation of Aβ (pI 5.5), 10 mm NaOH (120–145 μl NaOH/mg of peptide) was added to achieve a rapid transition to a pH of ∼7.0–7.5. The peptide was then further diluted to 500 μm in water and PBS (final concentration (in mm): 70 NaCl, 1.35 KCl, and 5 NaH2PO4/Na2HPO4). Fibrils were generated by incubating the peptide in a sealed tube at 37°C for 2–3 d. PF and LMW Aβ were generated by incubating Aβ peptide at room temperature (RT) for 2–3 d, centrifuging at 16,000 ×g for 10 min, and then fractionated by size-exclusion chromatography (SEC) (see below), as previously described (Walsh et al., 1997).
Size-exclusion chromatography to isolate LMW Aβ and PF.These species were isolated on a Superdex 75 SEC column using a Waters 650 Advanced Protein Purification System, as described (Walsh et al., 1997). However, for cell culture purposes, this procedure was modified by eluting the peaks with 70 mm NaCl and 5 mmTris, pH 7.4 (TBS). Characterization by quasielastic light scattering and negative contrast electron microscopy (EM) indicated that LMW Aβ and PF produced in this manner were indistinguishable from those previously reported (Walsh et al., 1997). As the LWM Aβ and the PF peaks eluted from the column, each was collected in ∼450 μl fractions, yielding four or five fractions per peak. Peptide content in individual fractions was quantified by amino acid analysis. The following tissue culture components were added to each fraction before applying them to cultures: MEM (1×); glucose (10 mm), penicillin streptomycin (500 U/ml and 500 μg/ml, respectively), HEPES (20 mm), and NaHCO3 (26 mm) (all final concentrations).
Primary mixed brain cultures. Cultures were prepared according to Hartley et al. (1993), with slight modifications for rat tissue. Briefly, brain cells were isolated from the neocortex of embryonic day 15 (E15)–E17 rat embryos, plated at 200,000 cells/ml (or 1.3 × 105cells/cm2) in plating medium containing DMEM, 10% fetal bovine serum, Ham’s F-12 (10%), HEPES (20 mm), glutamine (2 mm), and penicillin streptomycin (500 U/ml and 500 μg/ml, respectively) onto glial feeder layers in 48 well plates. Cultures were fed twice a week with this plating medium. After 9–10 d, cultures were inhibited with 10 μm ARA-C to stop glial growth and then changed into reduced-serum medium (plating medium with only 5% bovine calf serum). Upon initiation of all Aβ toxicity experiments, the medium was completely removed and replaced with serum-free medium containing either no Aβ, LMW Aβ, PF, or fibrils (see previous section for final composition of toxicity medium).
The cultures, which contained neurons, astroglia, and microglia, were used after 3–4 weeks in vitro. Phase-contrast microscopy of typical cultures revealed phase-bright neurons resting on a darker, granular layer of astroglia. Neurons stained positively for the neuron-specific marker, microtubule-associated protein-2 (MAP-2), whereas the astroglia were positive for glial fibrillary acidic protein (GFAP). Microglia were identified by their uptake of rhodamine-conjugated low-density lipoprotein. These mixed brain cultures provided a useful model of cortical tissue, in that all major cell types were present, and neurons displayed appropriate electrophysiological activity (see Results).
Unless otherwise indicated, chemicals and tissue culture supplies were purchased from Sigma (St. Louis, MO) and Life Technologies (Gaithersburg, MD), respectively. Serum was purchased from Hyclone (Logan, UT).
Immunocytochemistry. Cultures were fixed with 4% paraformaldehyde in 0.15 mNa2PO4:KH2PO4, pH 7.4, for 20 min, washed, treated with 0.3% H2O2 in methanol for 30 min, and then blocked in blocking buffer (10% FBS, 0.3% Triton X-100, and 0.15 mNa2PO4:KH2PO4, pH 7.4). Primary antibodies were applied in this blocking buffer for 1 hr at RT using the following concentrations: anti-MAP2, 1:1000 (monoclonal, Sigma M4403); R1282, 1:750 (polyclonal) (Lemere et al., 1996). Primary antibody was visualized with anti-mouse or anti-rabbit biotinylated secondary antibodies bound to horseradish peroxidase or alkaline phosphatase, respectively, using an avidin–biotin kit (ABC Elite; Vector Laboratories, Burlingame, CA).
Cell death. Cell death was assessed visually by phase-contrast microscopy and quantitatively by measuring the release of the cytosolic enzyme, lactate dehydrogenase (LDH), into the medium, as previously described (Hartley et al., 1993).
Congo red binding. Congo red binding (CRB) was measured as described by Klunk et al. (1989), with slight modifications for reading in a microplate reader. Samples (25 μl) were mixed with 225 μl of 20 μm Congo red in 20 mmKH2PO4 buffer, pH 7.4, and incubated at RT for 30 min. Absorbance at 480 and 540 nm were determined, and the amount of Congo red bound to fibrils was determined using the equation CRB (μmol/l) = A540/25,295-A480/46,306.
Search for fibrils by centrifugation and electron microscopy. Media of cultures treated with each of the three Aβ assembly forms for 5 d were centrifuged for 20 min at 16,000 × g, and the pellets were resuspended in 10 μl of H20 and applied to EM grids. The grids were incubated with the samples for 20 min, rinsed with TBS, and blocked with 0.1% egg albumin for 30 min, all at RT. Polyclonal antibody R1282 to human Aβ1–40 was added at 1:400 to 1:800 dilution, incubated for 30 min at RT, and followed by washing. Goat anti-rabbit secondary antibodies conjugated to 10 nm gold particles were incubated for 30 min at RT, followed by rinsing. The proteins were then fixed to the grids with 2.5% glutaraldehyde for 5 min, and the grids were negatively stained with 2% uranyl acetate and then examined in a JEOL CX100 or a 1200LX electron microscope.
SDS-PAGE/Western blotting. Conditioned medium (15 μl) was mixed with 2× Tricine–SDS sample buffer (Novex, San Diego, CA), boiled for 5 min, and loaded onto 10–20% Tricine gels (Novex) and electrophoresed. Proteins were electroblotted for 2 hr at 400 mA (4°C) onto 0.2 μm polyvinylidene difluoride membranes (Bio-Rad, Hercules, CA). Membranes were blocked for 1 hr at RT with 5% milk in TBST, washed, and probed with Aβ antibody R1282 (1:3000) in TBST for 1 hr (RT). After washing, a goat anti-rabbit secondary antibody conjugated to HRP was applied for 1 hr (RT). Immunopositive bands were visualized by chemiluminescence (ECL+Plus; Amersham, Arlington, IL), per manufacturer’s instruction.
Electrophysiological studies. Patch-clamp electrophysiology in the cell-attached voltage-clamp (−50 mV) mode was used for measuring EPSCs. Whole-cell and current-clamp mode was used for recording action potentials and membrane depolarizations. Mixed brain cultures (above) were perfused at RT in an extracellular bath solution containing (in mm): NaCl 140; KCl 4; MgCl2 1; CaCl2 1; HEPES 10; and Tris 5, pH 7.4. For cell-attached recordings, the pipette solution contained (in mm): NaCl 87; KCl 55; MgCl2 0.5; CaCl2 0.5; glucose 10; and HEPES 5, pH 7.4. The internal solution for whole-cell recordings contained (in mm): KCl 150; EGTA 10; MgCl2 1; HEPES 10; and Mg-ATP 3, pH 7.2 (using KOH). Patch pipettes (catalog #7052; Corning-Garner) were pulled and fire-polished to a tip diameter of <1 μm. High resistance (>10 GΩ) seals were used, and currents were measured with an integrating patch-clamp amplifier (catalog #3900; Dagan). Freshly isolated LMW Aβ and PF (see SEC section above) were used for electrophysiology experiments. To achieve the required sample volumes, the LMW Aβ and PF peaks were not fractionated as described for neurotoxicity experiments (above); rather, the whole peak was collected as a single fraction. To these LMW Aβ or PF peaks, stock electrophysiological buffer components were added to achieve the concentrations listed above (i.e., extracellular bath solution). Recordings were completed within 4–5 hr of peptide collection from the SEC. Aβ preparations were held at 4°C until applied. Baseline recordings were monitored for 3–5 min before applying the Aβ preparations, which were applied by adding an equal volume of the Aβ preparation directly into the bath solution (no perfusion system was used).
RESULTS
Structural and biochemical characterization of LMW Aβ and PF preparations
Two laboratories have previously isolated and characterized intermediate structures, termed protofibrils (PF), that are formed during the assembly of synthetic Aβ into typical 4–8 nm amyloid fibrils (Harper et al., 1997a,b; Walsh et al., 1997). Using SEC, we separated and collected the PF as well as the LMW Aβ1–40 peptides from which the PF are derived (Fig. 1a). No structures could be detected in the LMW Aβ fraction by EM. Quasielastic light-scattering spectroscopy has shown that LMW Aβ has a hydrodynamic radius of 1.5–2.0 nm, too small to be resolved by EM (Walsh et al., 1997). EM of the PF showed individual, curvilinear structures of 4–11 nm diameter and <200 nm length, as previously described (Walsh et al., 1997) (Fig. 1b, PF). This appearance contrasted with that of mature fibrils, which were straighter and much longer (indeterminate length with a width of 6–10 nm) and often found to occur in dense mats of multiple fibrils (Fig.1b, F).
Fig. 1.
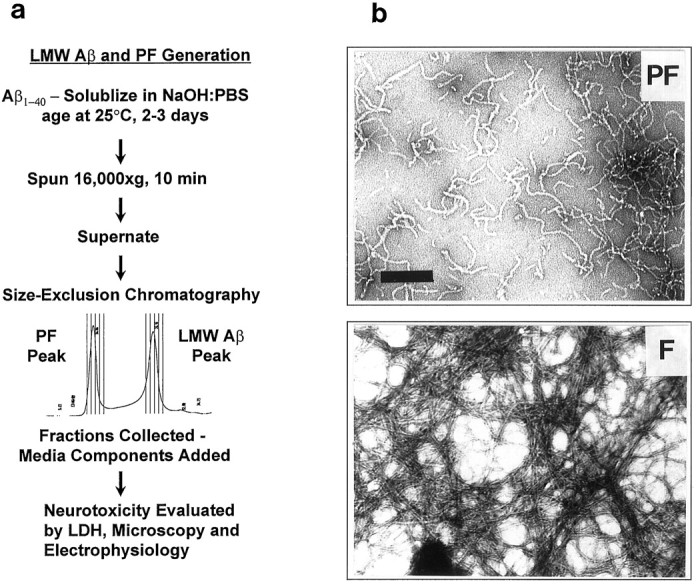
Flow diagram depicting generation of LMW Aβ and PF and their structural differences. a, Aβ was dissolved in NaOH:PBS, incubated for 2–3 d, centrifuged, and then injected onto a size-exclusion column. As the PF or LMW Aβ peak emerged from the column, it was collected in fractions, to which tissue culture reagents were added and then applied to mixed brain cultures. Neuronal viability was monitored by phase-contrast microscopy and by measuring the release of LDH into the medium. b,Protofibrils (PF) were present as individual, dispersed, curvilinear structures of 4–11 nm diameter and <200 nm length. Mature fibrils (F) were 6–10 nm in diameter, much longer (indeterminate length; mean diameter, ≅8 nm), and often occurred in dense mats of multiple fibrils.
In addition to their EM characterization, LMW Aβ, PF, and fibrils were analyzed by SDS-PAGE. Freshly prepared LMW Aβ, PF, and fibrils were each diluted into the buffer used for neurotoxicity experiments (see Materials and Methods) and subjected to Western blotting with the Aβ antibody R1282 (Lemere et al., 1996). Significant differences were observed among the preparations. The LMW Aβ material showed only an ∼4 kDa band (monomer) (Fig.2a), whereas this band and two or three higher molecular weight bands (oligomers between 6–12 kDa) were detected in the PF material (Fig. 2b). The fibrillar material produced bands corresponding to monomer and oligomers and an additional band at the very top of the gel (Fig. 2b). Thus, a portion of fibrils does not enter the gel or forms a smear after entering the gel. Centrifuging the fibril preparation at 16,000 ×g for 10 min yielded a pellet that contained solely the band at the top of the gel (Pike et al., 1993); no 4 kDa monomer or oligomers were observed (data not shown). It is important to note that the LMW Aβ and PF are purified by SEC, whereas our standard Aβ fibril preparations are mixed fractions similar to those used in numerous published Aβ neurotoxicity studies, i.e., they were prepared by incubating (“aging”) high concentrations of Aβ (100–500 μm) at 37°C for several days. After denaturing in SDS, the latter preparations showed not only gel-excluded Aβ-reactive material [i.e., SDS-insoluble fibrils (Pike et al., 1993)], but also Aβ monomers and oligomers (Fig. 2c), demonstrating that fibril preparations contain a mixture of species. Our unfractionated fibril preparations thus contained LMW Aβ, PF, and higher MW species and are similar, in this regard, to Aβ preparations used by others for neurotoxicity experiments.
Fig. 2.

Western blotting reveals electrophoretic differences between LMW Aβ, PF, and fibril preparations. Freshly collected fractions of LMW Aβ (a) and PF (b) from the size-exclusion column and preformed fibrils (c) were subjected to SDS-PAGE. Proteins were immunoblotted with an anti-Aβ1–40 polyclonal antibody, R1282, and immunopositive bands were visualized by chemiluminescence. The numbers under each lane represent the concentrations of Aβ in individual SEC fractions for LMW Aβ and PF (and similar amounts of fibrils), as determined by amino acid analysis. Molecular weight of protein standards (×1000) are atright. This gel depicts one of three independent experiments that gave similar results.
Neuronal injury as a function of Aβ assembly state
As the LMW Aβ and PF peaks emerged from the SEC column, each peak was collected in four or five fractions of 450 μl. The Aβ concentrations in these fractions varied from 5–53 μm, as determined by amino acid analysis, and these were usually within the concentration range of our fibril preparations, i.e., 7–42 μm. The PF and LMW SEC fractions were each individually added to wells of mixed brain cultures and incubated for up to 6 d. At ∼24 hr intervals, aliquots of the conditioned media were collected and assayed for the release of intracellular LDH (see below). After termination of the experiment, the cultures were fixed and immunostained to assess Aβ deposition and neuronal loss. Control cultures receiving no Aβ showed little neuronal loss after 5 d, as judged by MAP2 immunostaining, (Fig.3a, blue stain). In contrast, substantial neuronal loss was consistently observed after 5 d exposure to LMW Aβ (Fig. 3b; 18 μm in experiment illustrated). The neuronal loss as judged by MAP2 immunostaining was concentration-dependent (data not shown; however, see LDH data below as a quantitative marker of cell injury). Similar results were obtained with PF; a majority of the neurons were lost in the presence of PF for 5 d (Fig.3c; 21 μm in this experiment). The degree of cell death from either the LMW Aβ or PF preparations approached that observed with fibrils (Fig. 3d; 28 μm in this experiment). With each of the three preparations, Aβ deposition could be observed on and around the cells at day 5, as detected by the Aβ-specific antibody, R1282 (Fig.3b–d, red stain). However, no detectable difference in the light microscopic pattern of this Aβ immunoreactivity could be observed among the three preparations. Cell death was primarily neuronal, as indicated by the clear loss of MAP2-stained neurons compared to the control (untreated) cultures, whereas the glial monolayer showed little or no cell loss by phase-contrast microscopy (Fig. 3). Neuronal loss in controls (untreated) cultures over the 5–6 d toxicity experiments was usually <10%, whereas the Aβ-induced neuronal loss was usually >80%.
Fig. 3.
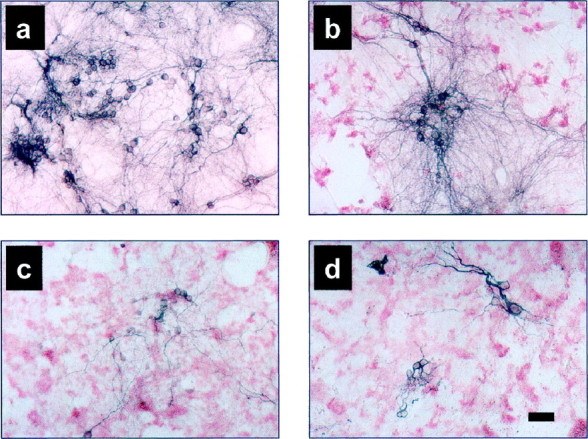
Immunocytochemistry of neurons and of Aβ in primary mixed cortical cultures exposed to various Aβ preparations. Mixed cortical cultures were exposed to medium only (a), LMW Aβ (b), PF (c), or fibrils (d) for 5 d, fixed and double-labeled for Aβ deposition (R1282) (red staining) and neurons (MAP2) (blue staining). Significant neuron loss is observed with each of the Aβ preparations (LMW Aβ, 18 μm; PF, 21 μm; fibrils, 28 μm). Scale bar, 50 μm.
We quantified LDH release induced by each of the three Aβ preparations for all 6 d in vitro (Fig. 4). Consistent with the MAP2 immunostaining (above), concentrations of any of LMW Aβ, PF, or fibrils at concentrations >7 μmall induced LDH release. The amount of LDH released was both time- and concentration-dependent. In general, day 3 (Fig. 4, red area) was the first day that a substantial rise in LDH could be detected, and on this day, LDH levels were significantly (p < 0.05) increased by the LMW Aβ, PF, and fibril preparations versus controls (Table1). The principal difference among the LMW Aβ, PF, and fibril preparations was the earlier and more rapid induction of LDH release by the fibrils. In four separate experiments, the relative degree of LDH release by day 3 was similar for both the LMW Aβ and PF cultures (each significantly higher than controls;p < 0.05), whereas that of fibrils was significantly (p < 0.05) higher than those of the LMW Aβ and PF as well as the control cultures (Table 1).
Fig. 4.

Comparison of neurotoxicity induced by LMW Aβ, PF, and fibrils. Various concentrations of LMW Aβ, PF, and fibrils [μm = concentration of Aβ applied (top numbers, x-axis)] were applied to mixed cortical cultures established 3–4 weeks earlier. Fraction number refers to the order the fractions were collected from either the LMW Aβ or PF peak (bottom numbers,x-axis)(also see Fig. 1). After each day of treatment (z-axis), an aliquot of conditioned medium was analyzed for LDH release (y-axis). Values were normalized to blanks (0 μm Aβ), which were given a value of 1. The graph represents one experiment, with each bar being the mean of duplicate assays; this graph is representative of four independent experiments in which four separate brain dissections were used (see Table 1 for comparison of the relative neurotoxicity of the preparations using LDH values from day 3).
Table 1.
LDH release from mature mixed brain cultures on day 3 ofin vitro treatment with different Aβ assemblies
| Preparation | [Aβ]1-a | LDH | n |
|---|---|---|---|
| Control | 0 μm | 1.00 + 0.101-b | 8 |
| LMW Aβ | 25 μm | 1.79 + 0.26* | 6 |
| PF | 20 μm | 1.76 + 0.17* | 8 |
| Fibrils | 28 μm | 2.85 + 0.20*,** | 8 |
Mean concentration of Aβ added at time 0.
Control LDH values were normalized to 1.
*Statistically different (p < 0.05) from control.
**Statistically different (p < 0.05) from LMW Aβ or PF samples [one-way ANOVA, multiple comparison (Tukey Test)].
Assaying for fibrillar Aβ in the culture media by Congo red binding and EM
We performed several different kinds of experiments to determine whether the reproducible neurotoxicity induced by the LMW Aβ and PF preparations (above) was caused by their gradual conversion to fibrils during the culture experiment or whether intermediate forms of Aβ may themselves induce cell injury. One approach involved the use of CRB, which has been used to identify both synthetic and endogenous Aβ fibrils (Klunk et al., 1989, 1999; Wood et al., 1996; Dickson, 1997). Conditioned media were collected at the end of the experiment (day 6) and assayed for CRB (Klunk et al., 1989). Fibril-containing cultures showed a concentration-dependent rise in CRB in their media: no binding was observed at ≤6 μm, whereas significant increases were detected at 13, 26, and 39 μm (Fig.5a). In contrast, LMW Aβ and PF at the highest concentrations used in these cultures (25 and 19 μm, respectively), produced no significant CRB. These results were observed consistently in three separate experiments examined for CRB: a clear dose response was observed for fibrillar Aβ, but no significant CRB occurred in cultures treated with the SEC fractions containing LMW Aβ or PF, despite their producing definite and significant neurotoxicity. Thus, the neuronal injury and loss produced by PF did not correlate with the development of significant CRB-positive fibrillar material during our experiments.
Fig. 5.
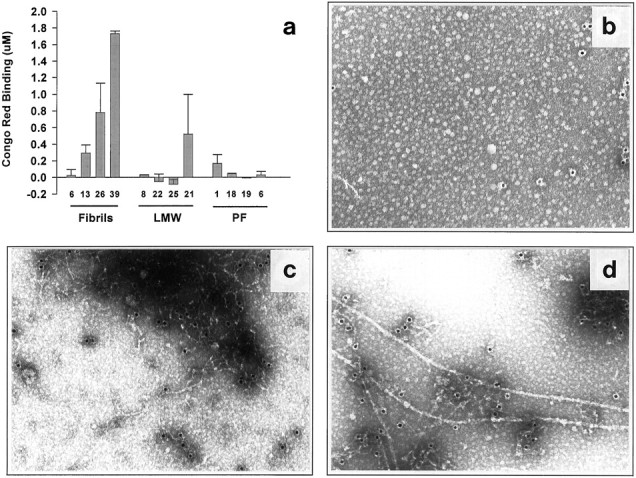
Lack of detectable fibril formation in mixed brain cultures treated for 5–6 d with LMW Aβ or PF. a,Mixed cortical cultures were exposed to LMW, PF, or preformed fibrils for 6 d, at which time the medium was removed and analyzed for fibril formation by Congo red binding. Each column is the mean of duplicate assays. Graph represents one of three identical experiments yielding similar results. b–d, Lack of fibril formation as determined by immuno-EM in 5 d conditioned media of LMW Aβ (b), PF (c), or fibril (d) preparation. PF-treated media (c) predominantly contained large electron-dense mats of distinct protofibrils. In contrast, distinct fibrils (d) could readily be detected in the media of cultures treated with preformed fibrils. Results represent one of two experiments, in both of which few or no fibrils were detected in the media of LMW Aβ and PF cultures.
Because the CRB assay may lack the sensitivity to detect small amounts of fibrillar Aβ, we performed additional assays for the development of insoluble, fibrillar aggregates in the conditioned media using centrifugation and EM. The culture media of cultures conditioned for 5 d in the presence of LMW Aβ, PF, or fibrils were centrifuged (16,000 × g, 20 min), and the pellets were examined by EM for fibril content. No fibrils were detected by EM in the cultures exposed to control (Aβ-free) medium (data not shown) or LWM Aβ (Fig. 5b). However, large mats of PF were readily observed in the pellets of the media of the PF-treated cultures, with occasional fibrils evident (Fig. 5c). Although evaluation by EM is only semiquantitative, there was a large difference in the ease with which fibrils were detected in the media pellets of cultures treated with fibrils (Fig. 5d) versus those treated with LMW Aβ or PF. These data, in combination with the lack of Congo red binding (Fig.5a), suggest that the consistent neurotoxicity obtained with the LMW Aβ and PF is caused by intermediate forms of Aβ, not to any substantial conversion to mature fibrils during our experiments.
Electrophysiological evidence of early neuronal alterations caused by protofibrils
In view of the above evidence that intermediate forms of Aβ may have biological activity that can induce neuronal injury, we sought to confirm such activity at a time point so early and at concentrations so low that any progressive changes in the physical state of the Aβ material were very unlikely to have occurred. To this end, we undertook electrophysiological recordings of our mixed brain cultures in the absence versus presence of either LMW Aβ, PF, or fibrillar Aβ. Three- to 4-week-old cultures containing both neurons and glia (indistinguishable from those used for the toxicity studies above) were examined in electrophysiological studies using the patch-clamp technique (see Materials and Methods). For these experiments, LMW Aβ and PF were isolated by SEC just before their addition to the culture being recorded. The interval between the generation of LMW Aβ and PF and their addition to the cultures was as brief as 30 min and always <5 hr. Once a stable baseline recording was established in the voltage-clamp mode, EPSCs were measured. In each case, the baseline was established for 4 min before the addition of an Aβ preparation. The addition of LMW Aβ at 3 μm produced either no increase or a very transient increase in EPSCs followed by a rapid return to baseline (Fig. 6a, LMW). Concentrations approaching 20 μm (i.e., neurotoxic) of LMW Aβ still showed no increase in EPSCs (data not shown). In sharp contrast, addition of either PF or fibrils at ∼3 μm concentrations invariably produced a rapid and sustained increase in electrical activity (Fig. 6a, PF, Fibrils). Each trace represents a recording from a single cell. Traces in Figure6a were then graphed as the number of EPSCs per minute versus time (Fig. 6b). The normalized mean values (±SEM) of the EPSCs per minute for the first and the eighth minute of treatment with PF (6.4 ± 1.4 and 7.0 ± 1.3, respectively;n = 8) and fibrils (6.2 ± 1.8 and 7.4 ± 1.5, respectively; n = 5) were found to be approximately sixfold greater (p < 0.05) than that of LMW Aβ (1.0 ± 0.34; n = 7) (Fig.6c). PF and fibrils were both statistically different from medium only and LMW Aβ, but no difference was found between the PF and fibril preparations. This neuronal activation was concentration-dependent for both the PF and fibrils, with EC50 values of 760 and 560 nm, respectively (data not shown). Interestingly, at concentrations >2 μm, no further increase in activation was observed. The lowest Aβ concentrations that caused an increase in EPSCs during an 8–10 min monitoring period were ∼144 nm for PF and ∼300 nm for fibrils.
Fig. 6.

Sustained increases in EPSCs caused by PF and fibrils but not LMW Aβ. Cell-attached patch-clamp mode was used to record inward EPSCs in voltage-clamp mode. a,Examples of current traces are shown in left panels. Baseline activities were recorded for 4 min (the ends of which periods are shown as dotted lines). Addition of LMW Aβ (a, LMW, solid line) at a concentration of 3 μm caused only a brief, transient increase in EPSCs compared with the preceding control period. In contrast, addition of PF (3–5 μm) (a, PF) or fibrils (3–5 μm) (a, Fibrils, solid lines) caused a rapid, sustained increase in EPSCs. b, The number of EPSCs per minute plotted versus time for the experiment shown in a. Each plot inb graphs data from a representative single cell exposed to one of the three Aβ preparations. Data from multiple experiments (c) were then compiled by normalizing the EPSCs per minute to the basal activity, which was given a value of one. The normalized mean EPSC values (5–8 experiments; mean ± SEM) for the basal, first, and eighth minute revealed a significant increase in EPSCs with PF and fibril application compared with the basal or LMW Aβ activity (c). PF and fibrils were found to be statistically different from both medium alone and LMW Aβ (p < 0.05); no significant difference between the PF and fibril preparations was observed.
To further evaluate the effects of PF on the electrical activity of neurons, we investigated the relative change of action potentials (APs) and membrane depolarizations (MDs) in the presence of LMW Aβ, PF, and fibrils. Whole-cell recording in current-clamp mode was used to monitor the baseline activity for 3–5 min. Freshly prepared LMW Aβ (3.9 μm), PF (2.2 μm), or preformed fibrils (0.75 μm) were then applied to the mixed brain cultures (Fig. 7). Application of either PF or fibrils increased the number of APs per minute by 2.59 ± 0.24-fold (n = 6) or 3.04 ± 1.03-fold (n = 8), respectively, versus the values of controls [i.e., no Aβ (normalized to 1)] or LMW Aβ (1.15 ± 0.10,n = 4) (Fig. 7). The values for PF and fibrils were found to be statistically different from control and LMW Aβ (p < 0.05), but not from each other. Furthermore, increased frequencies and larger sizes of MDs were observed after application of PF or fibrils compared to controls or LMW Aβ (Fig. 7, arrows; compare MDs of control or LMW Aβ vs PF or fibrils). These data suggest that a consequence of the increase in EPSCs is an increase in the rates of APs and large membrane depolarizations, which should have a significant impact on the physiology of the cell because of the large ion fluxes that occur.
Fig. 7.

Increased frequency of action potentials and membrane depolarizations caused by PF but not LMW Aβ. Whole-cell recordings in current-clamp mode were used to measure APs (single sharp deflections) and MDs (arrows). Baseline activities were recorded for 3–5 min (dotted lines). Addition of LMW Aβ (solid line) produced no significant increase in APs and MDs, whereas PF and fibrils (solid lines) increased the frequency of APs and the amplitude of MDs.
DISCUSSION
In this study, we have compared the biological effects of different Aβ assemblies on primary mixed brain cultures to determine whether nonfibrillar or immature, prefibrillar forms of Aβ may be neurotoxic. This potentially important source of neuronal injury has been overlooked until very recently, perhaps because the neuropathological diagnosis of AD requires the presence of abundant fibrillar amyloid in the form of myriad neuritic plaques in postmortem brain tissue. Because mature amyloid plaques surrounded by dystrophic neurites, activated microglia, and reactive astrocytes are composed principally of Aβ fibrils, it has been generally assumed that fibrillar Aβ is the form most likely to be responsible for neuronal and glial injury in AD. The apparent importance of amyloid fibrils has been reinforced by cell culture studies that consistently show that the aggregation state of Aβ, most notably the formation of amyloid fibrils, is associated with neuronal alteration and loss (Pike et al., 1991, 1993; Mattson et al., 1993; Lorenzo and Yankner, 1994; Estus et al., 1997; Seilheimer et al., 1997). Therefore, in vitrotoxicity and human neuropathological studies have heretofore not provided clear evidence that intermediate species formed during the Aβ fibrillogenesis process play a significant role in neuronal injury and death.
Based on the results of the experiments reported here, we hypothesize that nonfibrillar or immature fibrillar Aβ species have specific biological activities that may underlie, at least in part, the slowly progressive neuronal/synaptic changes that occur in AD. In support of this hypothesis, we present the novel finding that a recently described protofibrillar intermediate formed during fibrillogenesis of synthetic Aβ (Harper et al., 1997b; Walsh et al., 1997), consistently and significantly alters the electrical activity of neurons as measured by EPSCs, action potentials, and membrane depolarization at biologically relevant concentrations. Interestingly, electrophysiological effects of PF and mature fibrils were similar, because both increased membrane conductance and depolarizations. In contrast, LMW Aβ, consisting solely of monomers and/or dimers, had no significant electrophysiological effect at the same concentration. These results suggest that PFs have biological activity that mimics in part the ability of mature amyloid fibrils to alter ion fluxes across membranes (Mark et al., 1992; Mattson et al., 1992; Arispe et al., 1993;Wu et al., 1995; Kawahara et al., 1997; Ye et al., 1997).
The work reported here is consistent with the recent observation that another soluble, diffusible form of synthetic Aβ, referred to as Aβ-derived diffusible ligands (ADDLs), can alter the electrical activity of neurons, observed as an attenuation of long-term potentiation (LTP) (Lambert et al., 1998). However, our rapid increase in electrical activity (e.g., EPSCs), would not necessarily be associated with a reduction in LTP. These differences in effects are currently being investigated, but it is of interest to note that the structural and biochemical features of ADDLs appear to be distinct from those of PF. First, the ADDLs are much smaller, globular structures (∼5 nm diameter), compared to the curvilinear PF that have an average length of 25 nm and lengths as high as 200 nm. Second, SDS-PAGE of fresh preparations suggests that ADDLs contain two principal Aβ species, 17 and 22 kDa, whereas our PF yield two or three oligomeric bands of ∼6, 8, and 12 kDa. This physical disparity between PF and ADDLs supports the concept that different Aβ assemblies have distinct neurobiological activities, which may be manifested differently using an electrophysiological readout.
In support of the hypothesis that intermediate species formed during fibrillogenesis may play an important role in the disease process are the findings that some transgenic mice overexpressing human APP have shown altered behavior and/or electrophysiological responses before or during the initiation of Aβ plaque formation, before any substantial histological lesions or neuronal pathology were observed (Holcomb et al., 1998; Chapman et al., 1999; Hsia et al., 1999). The decline in synaptic activity in many of these animal models appears to take several months to develop (Hsiao et al., 1996; Holcomb et al., 1998;Chapman et al., 1999; Hsia et al., 1999), perhaps reflecting a gradual increase in Aβ levels to a concentration that allows formation of intermediates that noticeably alter neuronal activity. In humans, alterations in neuronal physiology caused by PF or other Aβ oligomeric intermediates could underlie the earliest mild amnesiac symptoms observed in elderly subjects with minimal cognitive impairment (Morris et al., 1996), as observed in the above mouse models (Holcomb et al., 1998; Chapman et al., 1999).
Recent data also suggest that intermediate species formed during Aβ fibrillogenesis could not only be responsible for early synaptic dysfunction, but may contribute to frank cell death. For example, an Aβ complex sedimenting more slowly than amyloid fibrils was shown to have a neurotoxic effect on cultured PC12 cells, as quantified by the MTT conversion assay (Oda et al., 1995). In addition, α-1-antichymotrypsin is capable of blocking fibril formation without decreasing Aβ-induced neurotoxicity (Aksenova et al., 1996). A soluble Aβ species isolated from AD cerebral cortex that kills neurons in mixed brain cultures has been identified (Giulian et al., 1996; Roher et al., 1996). Furthermore, the ADDLs mentioned above can also induce neuronal death (Lambert et al., 1998).
These various data are consistent with our findings that neurons exposed to LMW Aβ or PF show evidence of progressive injury leading to frank cell loss. This effect was similar to that observed with Aβ fibrils, although fibrillar preparations generally produced a more rapid injury. Interestingly, we found that LMW Aβ induced neuronal death at similar concentrations and time points. LMW Aβ could either progress in vitro to PF that cause the neurotoxicity or it could induce neurotoxicity by a different mechanism than PF, as supported by the fact that LMW Aβ did not acutely activate ion channels (this study) or cause the reduction of MTT (Walsh et al., 1999).
Because the induction of neurotoxicity in our cultures took 3–5 din vitro, a major concern was that LMW Aβ and PF were transitioning to fibrils, and these caused the neuronal injury. However, several observations suggest that neither of these Aβ assembly forms principally converted to fibrils. Conditioned media from the LMW Aβ and PF cultures (day 5–6) showed no rise in Congo red binding, whereas media exposed to fibrils from time 0 showed a dose-dependent recovery of Congo red-positive (fibrillar) Aβ. EM analysis of day 5–6 media that were centrifuged to concentrate any fibrils revealed few or no detectable fibrils in the LMW Aβ and PF cultures. These data clearly suggest that it is not the formation of fibrils in our LMW Aβ and PF experiments that explains the reproducible and profound neurotoxicity that these preparations induce. Furthermore, we have found by another relatively rapid biological readout, MTT reduction, that PF can directly alter cortical neurons biochemically, presumably before significant formation of fibrils occurs (Walsh et al., 1999). Finally, and most importantly, the almost instantaneous and highly reproducible enhancement in electrical activity of neurons by freshly prepared PF strongly suggests that PF can initiate a cell death process or render neurons more vulnerable to further insult without having to age to mature fibrils.
An important goal of our future work will be to search for the existence of protofibril-like assemblies of Aβ in vivo. In this regard, we have observed stable oligomers (dimers, trimers) of natural Aβ in the conditioned media of some cells overexpressing APP (Podlisny et al., 1995, 1998). Their potential pathological relevance was shown by observing an increase in the amounts of such Aβ1–42 oligomers when AD-causing presenilin 1 or 2 mutations, which increase Aβ1–42secretion, are coexpressed with APP (Xia et al., 1997). These findings suggest that natural Aβ under physiological conditions (nanomolar concentrations) can form oligomeric species, which could then seed the aggregation process as cerebral Aβ levels begin to rise with age in AD (Harper and Lansbury, 1997; Teplow, 1998). This hypothesis is consistent with fluorescence correlation spectroscopy study that identified soluble higher order, Aβ aggregates in the CSF of AD patients (Pitschke et al., 1998). Furthermore, soluble oligomeric Aβ, primarily Aβ1–42, has been shown to be elevated in AD cortex compared to age-matched control subjects (Kuo et al., 1996). Similarly, specific increases principally in Aβ1–42 rather than Aβ1–40 have been observed in other studies of AD brain tissue (Gravina et al., 1995) or transgenic mice expressing mutant human APP (Johnson-Wood et al., 1997). Even though the Aβ aggregation state was not determined in the latter studies, the documented rise in Aβ1–42 would increase the probability of aggregation occurring.
In summary, we have shown that soluble, intermediate forms of Aβ assemblies, particularly PF, can induce acute as well as delayed neurotoxic effects in the absence of significant conversion to fibrils. If oligomeric Aβ species resembling PF are present in human brain tissue before or during neuritic plaque (i.e., amyloid fibril) formation, the PF-like assemblies could well have a greater neuropathological impact than amyloid fibrils. As fibrils clump and become sequestered in glial-rich neuritic plaques, their ability to interact with surrounding cells may decrease. Soluble assemblies could have a more detrimental effect than mature fibrils because of their diffusibility. Based on the results reported here, identifying the presence of PF in vivo and determining their concentrations and half-life will be the next critical step in assessing the importance of prefibrillar species in AD. From a therapeutic point of view, blocking formation of mature fibrils could be harmful if toxic intermediates resembling those described here are allowed to accumulate.
Footnotes
This work was supported by National Institutes of Health Grants AG12749 and AG06173 (D.J.S.), AG00891 (C.P.Y.), and AG14266 and NS38328 (D.B.T.). We thank Margaret Condron for expert technical assistance.
Correspondence should be addressed to Dean M. Hartley or Dennis J. Selkoe, Center for Neurologic Diseases, Harvard Institutes of Medicine, Room 740, 77 Avenue Louis Pasteur, Boston, MA 02115.
REFERENCES
- 1.Aksenova MV, Aksenov MY, Butterfield DA, Carney JM. α-1-antichymotrypsin interaction with Aβ (1–40) inhibits fibril formation but does not affect the peptide toxicity. Neurosci Lett. 1996;211:45–48. doi: 10.1016/0304-3940(96)12717-8. [DOI] [PubMed] [Google Scholar]
- 2.Anderton BH, Callahan L, Coleman P, Davies P, Flood D, Jicha GA, Ohm T, Weaver C. Dendritic changes in Alzheimer’s disease and factors that may underlie these changes. Prog Neurobiol. 1998;55:595–609. doi: 10.1016/s0301-0082(98)00022-7. [DOI] [PubMed] [Google Scholar]
- 3.Arispe N, Pollard HB, Rojas E. Giant multilevel cation channels formed by Alzheimer disease amyloid β-protein [Aβ P-(1–40)] in bilayer membranes. Proc Natl Acad Sci USA. 1993;90:10573–10577. doi: 10.1073/pnas.90.22.10573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chapman PF, White GL, Jones MW, Cooper-Blacketer D, Marshall VJ, Irizarry M, Younkin L, Good MA, Bliss TV, Hyman BT, Younkin SG, Hsiao KK. Impaired synaptic plasticity and learning in aged amyloid precursor protein transgenic mice. Nat Neurosci. 1999;2:271–276. doi: 10.1038/6374. [DOI] [PubMed] [Google Scholar]
- 5.Cummings JL, Vinters HV, Cole GM, Khachaturian ZS. Alzheimer’s disease: etiologies, pathophysiology, cognitive reserve, and treatment opportunities. Neurology. 1998;51:S2–S17. doi: 10.1212/wnl.51.1_suppl_1.s2. [DOI] [PubMed] [Google Scholar]
- 6.Dickson DW. The pathogenesis of senile plaques. J Neuropathol Exp Neurol. 1997;56:321–339. doi: 10.1097/00005072-199704000-00001. [DOI] [PubMed] [Google Scholar]
- 7.Esiri M, Hyman B, Beyreuther K, Masters C. Aging and dementia. In: Graham D, Lantos P, editors. Greenfield’s neuropathology, Vol 2. Arnold; London: 1997. pp. 153–233. [Google Scholar]
- 8.Estus S, Tucker HM, van Rooyen C, Wright S, Brigham EF, Wogulis M, Rydel RE. Aggregated amyloid-β protein induces cortical neuronal apoptosis and concomitant apoptotic pattern of gene induction. J Neurosci. 1997;17:7736–7745. doi: 10.1523/JNEUROSCI.17-20-07736.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Giulian D, Haverkamp LJ, Yu JH, Karshin W, Tom D, Li J, Kirkpatrick J, Kuo LM, Roher AE. Specific domains of β-amyloid from Alzheimer plaque elicit neuron killing in human microglia. J Neurosci. 1996;16:6021–6037. doi: 10.1523/JNEUROSCI.16-19-06021.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gómez-Isla T, Price JL, McKeel DW, Jr, Morris JC, Growdon JH, Hyman BT. Profound loss of layer II entorhinal cortex neurons occurs in very mild Alzheimer’s disease. J Neurosci. 1996;16:4491–4500. doi: 10.1523/JNEUROSCI.16-14-04491.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gravina SA, Ho L, Eckman CB, Long KE, Otvos L, Jr, Younkin LH, Suzuki N, Younkin SG. Amyloid β protein (Aβ) in Alzheimer’s disease brain. Biochemical and immunocytochemical analysis with antibodies specific for forms ending at Aβ 40 or Aβ 42(43). J Biol Chem. 1995;270:7013–7016. doi: 10.1074/jbc.270.13.7013. [DOI] [PubMed] [Google Scholar]
- 12.Harper JD, Lansbury PT., Jr Models of amyloid seeding in Alzheimer’s disease and scrapie: mechanistic truths and physiological consequences of the time-dependent solubility of amyloid proteins. Annu Rev Biochem. 1997;66:385–407. doi: 10.1146/annurev.biochem.66.1.385. [DOI] [PubMed] [Google Scholar]
- 13.Harper JD, Wong SS, Lieber CM, Lansbury PT., Jr Observation of metastable Aβ amyloid protofibrils by atomic force microscopy. Chem Biol. 1997a;4:119–125. doi: 10.1016/s1074-5521(97)90255-6. [DOI] [PubMed] [Google Scholar]
- 14.Harper JD, Lieber CM, Lansbury PT., Jr Atomic force microscopic imaging of seeded fibril formation and fibril branching by the Alzheimer’s disease amyloid-β protein. Chem Biol. 1997b;4:951–959. doi: 10.1016/s1074-5521(97)90303-3. [DOI] [PubMed] [Google Scholar]
- 15.Hartley DM, Kurth MC, Bjerkness L, Weiss JH, Choi DW. Glutamate receptor-induced 45Ca2+ accumulation in cortical cell culture correlates with subsequent neuronal degeneration. J Neurosci. 1993;13:1993–2000. doi: 10.1523/JNEUROSCI.13-05-01993.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Holcomb L, Gordon MN, McGowan E, Yu X, Benkovic S, Jantzen P, Wright K, Saad I, Mueller R, Morgan D, Sanders S, Zehr C, O’Campo K, Hardy J, Prada CM, Eckman C, Younkin S, Hsiao K, Duff K. Accelerated Alzheimer-type phenotype in transgenic mice carrying both mutant amyloid precursor protein and presenilin 1 transgenes. Nat Med. 1998;4:97–100. doi: 10.1038/nm0198-097. [DOI] [PubMed] [Google Scholar]
- 17.Hsia A, Masliah E, McConlogue L, Yu G, Tatsuno g, Hu K, Kholodenko D, Malenka R, Nicoll R, Mucke L. Plaque-independent disruption of neural circuits in Alzheimer’s disease mouse models. Proc Natl Acad Sci USA. 1999;96:3228–3233. doi: 10.1073/pnas.96.6.3228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hsiao K, Chapman P, Nilsen S, Ekman C, Harigaya Y, Younkin S, Yang F, Cole G. Correlative memory deficits, Aβ elevation, and amyloid plaques in transgenic mice. Science. 1996;274:99–102. doi: 10.1126/science.274.5284.99. [DOI] [PubMed] [Google Scholar]
- 19.Johnson-Wood K, Lee M, Motter R, Hu K, Gordon G, Barbour R, Khan K, Gordon M, Tan H, Games D, Lieberburg I, Schenk D, Seubert P, McConlogue L. Amyloid precursor protein processing and Aβ42 deposition in a transgenic mouse model of Alzheimer disease. Proc Natl Acad Sci USA. 1997;94:1550–1555. doi: 10.1073/pnas.94.4.1550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kawahara M, Arispe N, Kuroda Y, Rojas E. Alzheimer’s disease amyloid β-protein forms Zn(2+)-sensitive, cation-selective channels across excised membrane patches from hypothalamic neurons. Biophys J. 1997;73:67–75. doi: 10.1016/S0006-3495(97)78048-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Klunk WE, Pettegrew JW, Abraham DJ. Quantitative evaluation of congo red binding to amyloid-like proteins with a β-pleated sheet conformation. J Histochem Cytochem. 1989;37:1273–1281. doi: 10.1177/37.8.2666510. [DOI] [PubMed] [Google Scholar]
- 22.Klunk WE, Jacob RF, Mason RP. Quantifying amyloid β-peptide (Aβ) aggregation using the Congo red-Aβ (CR-aβ) spectrophotometric assay. Anal Biochem. 1999;266:66–76. doi: 10.1006/abio.1998.2933. [DOI] [PubMed] [Google Scholar]
- 23.Kuo Y-M, Emmerling MR, Vigo-Pelfrey C, Kasunic TC, Kirkpatrick JB, Murdoch GH, Ball MJ, Roher AE. Water-soluble Aβ (N-40, N-42) oligomers in normal and Alzheimer disease brains. J Biol Chem. 1996;271:4077–4081. doi: 10.1074/jbc.271.8.4077. [DOI] [PubMed] [Google Scholar]
- 24.Lambert MP, Barlow AK, Chromy BA, Edwards C, Freed R, Iosatos M, Morgan TE, Rozovsky I, Trommer B, Viola KL, Wals P, Zhang C, Finch CE, Krafft GA, Klein WL. Diffusible, nonfibrillar ligands derived from Aβ1–42 are potent central nervous system neurotoxins. Proc Natl Acad Sci USA. 1998;95:6448–6453. doi: 10.1073/pnas.95.11.6448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lemere CA, Blustzjan JK, Yamaguchi H, Wisniewski T, Saido TC, Selkoe DJ. Sequence of deposition of heterogeneous amyloid β-peptides and Apo E in Down syndrome: implications for initial events in amyloid plaque formation. Neurobiol Dis. 1996;3:16–32. doi: 10.1006/nbdi.1996.0003. [DOI] [PubMed] [Google Scholar]
- 26.Lorenzo A, Yankner B. β-amyloid neurotoxicity requires fibril formation and is inhibited by Congo red. Proc Natl Acad Sci USA. 1994;91:12243–12247. doi: 10.1073/pnas.91.25.12243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mark RJ, Hensley K, Butterfield DA, Mattson MP. Amyloid β-peptide impairs ion-motive ATPase activities: evidence for a role in loss of neuronal Ca2+ homeostasis and cell death. J Neurosci. 1992;15:6239–6249. doi: 10.1523/JNEUROSCI.15-09-06239.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mattson MP, Cheng B, Davis D, Bryant K, Lieberburg I, Rydel RE. β-Amyloid peptides destabilize calcium homeostasis and render human cortical neurons vulnerable to excitotoxicity. J Neurosci. 1992;12:379–389. doi: 10.1523/JNEUROSCI.12-02-00376.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mattson MP, Tomaselli KJ, Rydel RE. Calcium-destabilizing and neurodegenerative effects of aggregated β-amyloid peptide are attenuated by basic FGF. Brain Res. 1993;621:35–49. doi: 10.1016/0006-8993(93)90295-x. [DOI] [PubMed] [Google Scholar]
- 30.Morris JC, Storandt M, McKeel DW, Jr, Rubin EH, Price JL, Grant EA, Berg L. Cerebral amyloid deposition and diffuse plaques in “normal” aging: evidence for presymptomatic and very mild Alzheimer’s disease. Neurology. 1996;46:707–719. doi: 10.1212/wnl.46.3.707. [DOI] [PubMed] [Google Scholar]
- 31.Oda T, Wals P, Osterburg HH, Johnson SA, Pasinetti GM, Morgan TE, Rozovsky I, Stine WB, Snyder SW, Holzman TF, Krafft GA, Finch CE. Clusterin (apoJ) alters the aggregation of amyloid β-peptide (Aβ 1–42) and forms slowly sedimenting Aβ complexes that cause oxidative stress. Exp Neurol. 1995;136:22–31. doi: 10.1006/exnr.1995.1080. [DOI] [PubMed] [Google Scholar]
- 32.Pike CJ, Walencewicz AJ, Glabe CG, Cotman CW. In vitro aging of β-amyloid protein causes peptide aggregation and neurotoxicity. Brain Res. 1991;563:311–314. doi: 10.1016/0006-8993(91)91553-d. [DOI] [PubMed] [Google Scholar]
- 33.Pike CJ, Burdick D, Walencewicz AJ, Glabe CG, Cotman CW. Neurodegeneration induced by β-amyloid peptides in vitro: the role of peptide assembly state. J Neurosci. 1993;13:1676–1687. doi: 10.1523/JNEUROSCI.13-04-01676.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Pitschke M, Prior R, Haupt M, Riesner D. Detection of single amyloid B-protein aggregates in the cerebrospinal fluid of Alzheimer’s patients by fluorescence correlation spectroscopy. Nat Med. 1998;4:832–834. doi: 10.1038/nm0798-832. [DOI] [PubMed] [Google Scholar]
- 35.Podlisny MB, Ostaszewski BL, Squazzo SL, Koo EH, Rydel RE, Teplow DB, Selkoe DJ. Aggregation of secreted amyloid β-protein into SDS-stable oligomers in cell culture. J Biol Chem. 1995;270:9564–9570. doi: 10.1074/jbc.270.16.9564. [DOI] [PubMed] [Google Scholar]
- 36.Podlisny MB, Walsh DM, Amarante P, Ostaszewski BL, Stimson ER, Maggio JE, Teplow DB, Selkoe DJ. Oligomerization of endogenous and synthetic amyloid β-protein at nanomolar levels in cell culture and stabilization of monomer by Congo red. Biochemistry. 1998;37:3602–3611. doi: 10.1021/bi972029u. [DOI] [PubMed] [Google Scholar]
- 37.Roher AE, Chaney MO, Kuo Y-M, Webster SD, Stine WB, Haverkamp LJ, Woods AS, Cotter RJ, Tuohy JM, Krafft GA, Bonnell BS, Emmerling MR. Morphology and toxicity of Aβ-(1–42) dimer derived from neuritic and vascular amyloid deposits of Alzheimer’s disease. J Biol Chem. 1996;271:20631–20635. doi: 10.1074/jbc.271.34.20631. [DOI] [PubMed] [Google Scholar]
- 38.Seilheimer B, Bohrmann B, Bondolfi L, Müller F, Stüber D, Döbeli H. The toxicity of the Alzheimer’s β-amyloid peptide correlates with a distinct fiber morphology. J Struct Biol. 1997;119:59–71. doi: 10.1006/jsbi.1997.3859. [DOI] [PubMed] [Google Scholar]
- 39.Sze CI, Troncoso JC, Kawas C, Mouton P, Price DL, Martin LJ. Loss of the presynaptic vesicle protein synaptophysin in hippocampus correlates with cognitive decline in Alzheimer disease. J Neuropathol Exp Neurol. 1997;56:933–944. doi: 10.1097/00005072-199708000-00011. [DOI] [PubMed] [Google Scholar]
- 40.Teplow DB. Structural and kinetic features of amyloid β-protein fibrillogenesis. Amyloid. 1998;5:121–42. doi: 10.3109/13506129808995290. [DOI] [PubMed] [Google Scholar]
- 41.Terry RD, Masliah E, Salmon DP, Butters N, DeTeresa R, Hill R, Hansen LA, Katzman R. Physical basis of cognitive alterations in Alzheimer’s disease: synapse loss is the major correlate of cognitive impairment. Ann Neurol. 1991;30:572–580. doi: 10.1002/ana.410300410. [DOI] [PubMed] [Google Scholar]
- 42.Walsh DM, Hartley DM, Kusumoto Y, Fezoui Y, Condron MM, Lomakin A, Benedek GB, Selkoe DJ, Teplow DB. Amyloid β-protein fibrillogenesis: structure and biological activity of protofibrillar intermediates. J Biol Chem. 1999;274:25945–25952. doi: 10.1074/jbc.274.36.25945. [DOI] [PubMed] [Google Scholar]
- 43.Walsh DM, Lomakin A, Benedek GB, Maggio JE, Condron MM, Teplow DB. Amyloid β-protein fibrillogenesis: detection of a protofibrillar intermediate. J Biol Chem. 1997;272:22364–22374. doi: 10.1074/jbc.272.35.22364. [DOI] [PubMed] [Google Scholar]
- 44.Wood SJ, Maleeff B, Hart T, Wetzel R. Physical, morphological and functional differences between pH 5.8 and 7.4 aggregates of the Alzheimer’s amyloid peptide Aβ. J Mol Biol. 1996;256:870–877. doi: 10.1006/jmbi.1996.0133. [DOI] [PubMed] [Google Scholar]
- 45.Wu J, Anwyl R, Rowan MJ. β-Amyloid selectively augments NMDA receptor-mediated synaptic transmission in rat hippocampus. NeuroReport. 1995;6:2409–2413. doi: 10.1097/00001756-199511270-00031. [DOI] [PubMed] [Google Scholar]
- 46.Xia W, Zhang J, Kholodenko D, Citron M, Podlisny MB, Teplow DB, Haas C, Seubert P, Koo EH, Selkoe DJ. Enhanced production and oligomerization of the 42-residue amyloid β-protein by Chinese hamster ovary cells stably expressing mutant presenilins. J Biol Chem. 1997;272:7977–7982. doi: 10.1074/jbc.272.12.7977. [DOI] [PubMed] [Google Scholar]
- 47.Ye C, Ho-Pao CL, Kanazirska M, Quinn S, Rogers K, Seidman CE, Seidman JG, Brown EM, Vassilev PM. Amyloid-β proteins activate Ca(2+)-permeable channels through calcium-sensing receptors. J Neurosci Res. 1997;47:547–554. doi: 10.1002/(sici)1097-4547(19970301)47:5<547::aid-jnr10>3.0.co;2-v. [DOI] [PubMed] [Google Scholar]