

Abstract
Transgenic mice with a point mutation in the light neurofilament gene develop amyotrophic lateral sclerosis–like motor neuron disease characterized by selective spinal motor neuron loss, neurofilamentous accumulations, and severe muscle atrophy. To test whether the large motor neurons at risk in this disease could be protected from mutant neurofilament-mediated killing, these mice were bred to mice overexpressing the human Bcl-2proto-oncogene. Elevated levels of Bcl-2 increased the numbers of motor and sensory axons surviving after the developmental period of naturally occurring cell death but did not greatly reduce the number of degenerating axons or protect the large motor neurons from mutant neurofilament-mediated death.
Keywords: Bcl-2, amyotrophic lateral sclerosis, transgenic mice, motor neuron, cell death, neurofilament
Amyotrophic lateral sclerosis (ALS) is a progressive neurodegenerative disease characterized by the loss of large spinal motor neurons that results in paralysis and death. A small proportion of human familial ALS cases can be attributed to mutations in the free radical–scavenging enzyme copper zinc superoxide dismutase 1 (SOD1), but the majority of ALS cases are sporadic and of unknown origin. It has been proposed that the etiology of ALS may involve abnormal neurofilaments since a prominent pathological feature of this disease includes neurofilamentous accumulations in cell bodies and axons of both humans (Carpenter, 1968; Chou and Fakadej, 1971;Hirano et al., 1984a,b) and mice (Gurney et al., 1994; Lee et al., 1994; Wong et al., 1995; Tu et al., 1996). Additional evidence for the involvement of neurofilaments in motor neuron disease comes from work by two groups who demonstrated that the overexpression of neurofilaments in mice can cause ALS-like pathology (Cote et al., 1993;Xu et al., 1993). Neurofilaments (NFs) are composed of the three subunits [heavy NF-H (115 kDa), medium NF-M (95 kDa), light NF-L (65 kDa)] and constitute the major intermediate filament component of mature axons. Neurofilaments are thought to be important not just for structural support of long axons (up to 1 m in humans) but also for determining the diameter (Yamasaki et al., 1991; Zhu et al., 1997) and therefore the conduction velocity of axons (Gasser and Grundfest, 1939; Sakaguchi et al., 1993). Of the two size classes of motor axons, the larger axons are distinguished by faster conduction velocity and higher neurofilament content. In both humans (Kawamura et al., 1981; Murakami, 1990) and mouse models (Lee et al., 1994; Bruijn et al., 1997) of ALS, it is the large neurofilament-rich motor neurons that are at risk, further implicating neurofilaments in this disease.
To date, the only mouse model successfully using a mutant neurofilament subunit to cause motor neuron disease is a series of transgenic lines expressing a mutant NF-L gene (Lee et al., 1994). This mutant NF-L gene that carries a Leu-to-Pro substitution at amino acid 394 (in the conserved LLEGE sequence within the rod domain as well as a C-terminal 12 amino acid epitope tag) disrupts neurofilament network assembly and organization in transfected cultured cells (Lee et al., 1994). When expressed at moderate levels in mice, this mutant NF-L polypeptide causes degeneration and loss of motor neurons, neurofilamentous accumulations, muscle atrophy, and neuronal death like that seen in both human sporadic (Carpenter, 1968; Hirano et al., 1984a) and SOD1-mediated familial ALS (Hirano et al., 1984b; Rouleau et al., 1996;Shibata et al., 1996).
To test the mechanism of neuronal death arising from abnormal neurofilaments and the methods by which this disease might be alleviated, we chose to exploit the endogenous protective machinery already in place within motor neurons (Merry et al., 1994), namely, the Bcl-2 family of cell death regulatory proteins. The Bcl-2proto-oncogene was initially recognized as a regulator of cell death by its ability to prevent the death of cultured neurons deprived of neurotrophic factors (Garcia et al., 1992; Allsopp et al., 1993; Mah et al., 1993). Since then, Bcl-2 has been shown to prevent both apoptotic and necrotic cell death induced by a variety of stimuli in several different systems (for review, see Allen et al., 1998; Chao and Korsmeyer, 1998). Although its exact mechanism of action is still unclear, Bcl-2 does not appear to act by reducing intracellular calcium levels, increasing glutathione levels, or altering oxygen consumption or ATP concentrations (Kane et al., 1993; Zhong et al., 1993), but its expression is correlated with reduced amounts of intracellular reactive oxygen species and lipid peroxidation (Hockenbery et al., 1993; Kane et al., 1993). This finding was especially intriguing for motor neuron disease considering that 15% of inherited human ALS cases result from mutations in the free radical–scavenging enzyme SOD1.
To determine whether Bcl-2 protection could be effective in an ALS-like disease caused by disorganized neurofilament arrays, we have examined disease progression and pathology in mice expressing a neurofilament mutant in the presence or absence of chronically elevated levels of Bcl-2.
MATERIALS AND METHODS
Mouse lines used. Mice expressing a transgene comprised of the murine sarcoma virus promoter linked to the murine NF-L gene mutated at amino acid position 394 (Leu-to-Pro substitution) with a C-terminal 12 amino acid epitope tag from human myc [mouse line 61 (Lee et al., 1994)] were mated to mice overexpressing the human Bcl-2 gene under control of the neuron-specific enolase promoter [line 73 (Martinou et al., 1994)]. The single-step breeding scheme produced doubly transgenic mice and all the necessary controls. Mice carrying the NF-L (Pro) transgene were scored by genomic DNA blot as described (Lee et al., 1994). Bcl-2–overexpressing mice were scored by a PCR strategy that amplified the human Bcl-2 sequence using a human sequence-specific primer and a primer to the transgene promoter.
Bcl-2 and mutant NF-L (Pro) protein detection. Eleven percent polyacrylamide gels were loaded with 30 μg of sciatic nerve total homogenate and transferred to nitrocellulose. The myc-tagged mutant NF-L (Pro) subunit was detected using anti-myc polyclonal antibodies (Gill et al., 1991) and 125I-conjugated protein A (1:2000; Amersham, Arlington Heights, IL). Wild-type NF-L was detected using a mouse monoclonal antibody (Sigma, St. Louis, MO) with a rabbit anti-mouse bridging antibody (1:100; Sigma) and125I-conjugated protein A (1:2000; Amersham). The human Bcl-2 transgene protein product was detected using monoclonal hamster anti-human Bcl-2 antisera (1:100; PharMingen, San Diego, CA) with a rabbit anti-hamster bridging antibody (1:100; Sigma) and125I-conjugated protein A (1:2000; Amersham). Immunoreactive bands were visualized by autoradiography using Kodak Biomax MS film (Eastman Kodak, Rochester, NY).
Tissue preparation and morphological analysis. Mice were transcardially perfused with 4% paraformaldehyde and 2.5% glutaraldehyde in 0.1 m sodium phosphate, pH 7.6, and post-fixed overnight in the same solution. Samples were treated with 2% osmium tetroxide, washed, dehydrated, and embedded in Epon-Araldite resin. Thick sections (0.75 μm) for light microscopy were stained with toluidine blue. Axons were counted from the L5 ventral and dorsal roots of three to five mice from each genotype and age. The number of degenerating axons in a root was counted from three to five mice from each genotype and age. Axon diameters from two mice of each genotype and age were measured using the Integrated Morphometry function from the Image 1 Metamorph System (Universal Imaging Corporation, West Chester, PA). Entire roots were imaged, and imaging thresholds were chosen individually. The cross-sectional area of each axon was calculated and reported as the diameter of a circle of equivalent area, and these diameters were grouped into 0.5 μm bins.
Immunocytochemistry. Mouse tissues perfused with 4% paraformaldehyde as described above were paraffin embedded. Deparaffinized sections were reacted with monoclonal hamster anti-human Bcl-2 antisera (PharMingen) using the peroxidase-antiperoxidase method (Vector Laboratories, Burlingame, CA) and counterstained with hematoxylin (Bruijn et al., 1997).
RESULTS
Bcl-2 overexpression does not markedly alter the phenotype or life span of mutant NF-L mice
As early as 2–3 weeks of age, mutant NF-L (Pro) mice can be distinguished from wild-type littermates by their runted appearance, muscle atrophy, limb quivering, and limb retraction when held by the tail (Fig. 1A). NF-L (Pro) mice display variable disease severity and variable mortality. On average, approximately one-half of the NF-L (Pro)–positive pups in a litter die before 1 month of age, whereas the one-half that survive this critical period continue to live without a reduced life span. Overexpressing Bcl-2 in these NF-L (Pro) mice does not change the disease course, and at ages up to 15 months, NF-L (Pro) mice with or without the Bcl-2 transgene are virtually indistinguishable. Bcl-2 overexpression may increase the body weights of mutant NF-L (Pro) mice slightly but does not restore them to wild-type levels (Fig.1B). Bcl-2 overexpression in NF-L (Pro) mice does not alter the fraction of animals that survives the initial critical period.
Fig. 1.

Mutant mice are easily identified by retracted limb posture and decreased weight. A, Wild-type mice (left) extend their hindlimbs when held by the tail, whereas mutant NF-L (Pro) mice with either normal (middle) or increased (right) levels of Bcl-2 retract their hindlimbs. Mice shown are 1-month-old littermates.B, Overexpression of Bcl-2 (▴) does not drastically alter the body weight of mice when compared with that of wild-type littermates (●). Overexpression of Bcl-2 in mutant NF-L (Pro) mice (♦) slightly increases the body weight of mice over that of singly transgenic NF-L (Pro) mice (▪) at most ages.Points represent averages from four mice from each genotype. Error bars represent the SEM. C, Total sciatic nerve homogenates from 2-, 4-, and 24-week-old mice from NF-L (Pro) mice and doubly transgenic NF-L (Pro)/Bcl-2 mice were resolved on an 11% Coomassie blue–stained polyacrylamide gel. Two mice from each age are shown in adjacent lanes. Parallel gels were used for immunoblotting. Molecular weight markers are shown on theleft. Levels of myc-tagged NF-L transgene in the sciatic nerves of NF-L (Pro) [lanes 1–6 (lane 1 to the right of the molecular weight markers)] and doubly transgenic NF-L (Pro)/Bcl-2 (lanes 7–12) mice are shown. Levels of endogenous mouse NF-L in the sciatic nerves of mutant NF-L (Pro) (lanes 1–6) and doubly transgenic NF-L (Pro)/Bcl-2 (lanes 7–12) mice of each age are shown. Levels of human Bcl-2 (hBCL) transgene protein in the sciatic nerves of mutant NF-L (Pro) (lanes 1–6) and doubly transgenic NF-L (Pro)/Bcl-2 (lanes 7–12) mice of each age are shown.D–F, Immunocytochemical detection of the human Bcl-2 transgene product in the ventral spinal cord of 3-week-old mice is shown. Bcl-2−/NF-L (Pro)− mice (D), Bcl-2+/NF-L (Pro)− (E), and Bcl-2+/NF-L (Pro)+ (F) mice are shown. Bcl-2+/NF-L (Pro)+ cell bodies undergoing degeneration also contain the human Bcl-2 transgene product (arrows). Scale bar, 30 μm. BCLand BCL2, Bcl-2; PRO, NF-L (Pro).
This unusual survival pattern in NF-L (Pro) mice arises because the expression of the NF-L (Pro) transgene is silenced beginning at ∼4 weeks of age. An initial transgene expression level (50–75% of endogenous NF-L) is present up to 2 weeks of age but declines with age (Lee et al., 1994). To demonstrate that this was indeed the case and to verify that Bcl-2 was continuously expressed in the presence and absence of the NF-L mutant, sciatic nerve total homogenates from 2-, 4-, and 24-week-old singly transgenic NF-L (Pro) and doubly transgenic Bcl-2/NF-L (Pro) animals were subjected to quantitative immunoblot. Sciatic nerve homogenates from two mice from each genotype and age were resolved on polyacrylamide gels and stained by Coomassie blue (Fig.1C) to verify equal loading. Parallel gels were immunoblotted with antibodies to the epitope tag on the NF-L (Pro) transgene subunit, with antibodies to the endogenous NF-L subunit, and with antisera specific for human, but not mouse, Bcl-2. This demonstrated that, whereas mutant NF-L transgene levels decrease markedly after 2 weeks of age in both singly and doubly transgenic mice, wild-type NF-L levels do not change after the initial phase of transgene-mediated neuronal death occurring between 2 and 4 weeks of age (see below). As expected, human Bcl-2 levels are not affected by the mutant NF-L transgene at any age.
To verify that the Bcl-2 transgene was expressed in the motor neuron cell bodies at risk during the early phase of the disease, immunocytochemistry was performed using human-specific anti-Bcl-2 antibodies (Fig. 1D–F). Three-week-old wild-type mice express no detectable human Bcl-2 (Fig.1D), whereas the large motor neuron cell bodies of both singly transgenic Bcl-2+/NF-L (Pro)− (Fig.1E) and doubly transgenic Bcl-2+/NF-L (Pro)+ (Fig.1F) mice are immunoreactive for the Bcl-2 transgene product. Therefore, the introduced Bcl-2 is present in the cells at risk in this disease, and expression of the NF-L (Pro) transgene does not affect human Bcl-2 expression or localization.
Degeneration of mutant NF-L (Pro) motor axons is an early and transient event
Although Bcl-2 overexpression failed to affect the mutant NF-L–mediated disease onset or progression, it remained possible that increased Bcl-2 could support more efficient neuronal survival after the initial insult. To test this, the most severely affected L5 ventral (motor) roots were examined at several ages. Wild-type mice exhibited the expected pattern of axonal radial growth from 2 weeks to 15 months of age (Fig. 2A), usually with no degenerating axons observed. When compared with wild-type mice (Fig. 2A), an obvious initial difference was that the Bcl-2–overexpressing mice possessed more small axons at each age examined (Fig. 2B), whereas the number of large axons and the size of these large axons did not appear to differ from that of wild type. The increased number of axons in the Bcl-2 transgenic mice must reflect Bcl-2–dependent inhibition of naturally occurring programmed cell death that would normally occur during development. This finding is not unexpected given the previous evidence that the transgene in this line of Bcl-2–overexpressing mice is activated very early in embryonic development (Martinou et al., 1994).
Fig. 2.

Effects of mutant NF-L (Pro) transgene expression and Bcl-2 overexpression on axonal morphology. Cross sections of L5 motor (ventral root) from 2-week-old and 1-, 2-, 6-, and 15-month-old mice are shown. A–D, Representative sections from each genotype are as indicated. Arrows indicate degenerating axons and invading macrophages. Scale bar, 10 μm.
The NF-L (Pro) mutant axons showed only minor changes at 2 weeks of age; the number and sizes of axons appeared essentially normal with very few macrophages and degenerating axonal profiles visible (Fig. 2D). Degeneration began soon thereafter and was rampant by 1 month of age, accompanied by a high degree of macrophage invasion and deposition of cellular debris. In addition, the nerves contained many fewer intact axons, with the loss of the largest diameter axons as the most obvious feature. By 2 months of age [when NF-L (Pro) transgene levels are significantly reduced], the NF-L (Pro) mice that survive this early crisis period displayed very few macrophages, degenerating axons, or cellular debris. Six-month-old axonal profiles also looked reasonably healthy but were severely reduced in size when compared with similarly aged wild-type axons. By 15 months of age, the NF-L (Pro) axons were still markedly smaller than wild-type axons, but a partial recovery of larger diameter axons was evident.
Not unexpectedly, doubly transgenic Bcl-2/NF-L (Pro) axons at 2 weeks of age were similar in appearance and number to the other three genotypes (Fig. 2C). The amount and severity of degeneration displayed at 1 month of age were slightly less obvious in NF-L (Pro) mice with the Bcl-2 transgene than in those without it. The roots of doubly transgenic mice had fewer macrophages present, and smaller axons filled these spaces instead of debris. At 2 months of age, the doubly transgenic mouse axons were more shrunken than their NF-L (Pro) counterparts, and small amounts of debris and scattered macrophages were still observable. This was in contrast to the absence of pathology seen in the NF-L (Pro) axons at this same age. This result is consistent with either a delayed onset of axonal degeneration or a lengthened degeneration period caused by overexpression of Bcl-2 in NF-L (Pro) mice. Also of note is the fact that the doubly transgenic 6- and 15-month-old axons had increased in diameter to become larger than the singly transgenic NF-L (Pro) axons but were still considerably smaller than the wild-type axons.
Increases in initial motor axon numbers and a delay in axon degeneration upon Bcl-2 overexpression in NF-L (Pro) mice
To assess more accurately any protective effects of Bcl-2 overexpression in NF-L (Pro) mice, the number of axons within the L5 ventral and dorsal roots of mice from each genotype were counted at several ages (Fig. 3). The selectivity of the mutant NF-L (Pro)–mediated cell killing for motor neurons is demonstrated by the 50% loss of the motor axons between weeks 2 and 4 (Fig. 3A), with only a 15% loss of sensory axons (Fig.3B) when compared with respective wild-type axon levels. Initially obvious by qualitative inspection of Figure 2, the number of motor axons in Bcl-2–overexpressing mice was considerably greater (33–71% of wild type) than that in wild-type mice at all ages examined. The doubly transgenic Bcl-2/NF-L (Pro) mice initially possessed the same increased number of axons as the Bcl-2–overexpressing mice alone but experienced an early loss of motor axons at 3 weeks of age similar to that of the NF-L (Pro) mice. However, this loss was different with respect to duration and severity, as suggested previously by the degenerating axonal profiles still evident in the 2-month-old doubly transgenic mice but absent in the NF-L (Pro) mice (Fig. 2). Whereas the doubly transgenic axons either recovered or were replaced by spouting axons to regain initial axon numbers eventually by 4 months of age, the NF-L (Pro) axons did not, demonstrating that Bcl-2 overexpression is capable of changing some aspects of the disease course.
Fig. 3.
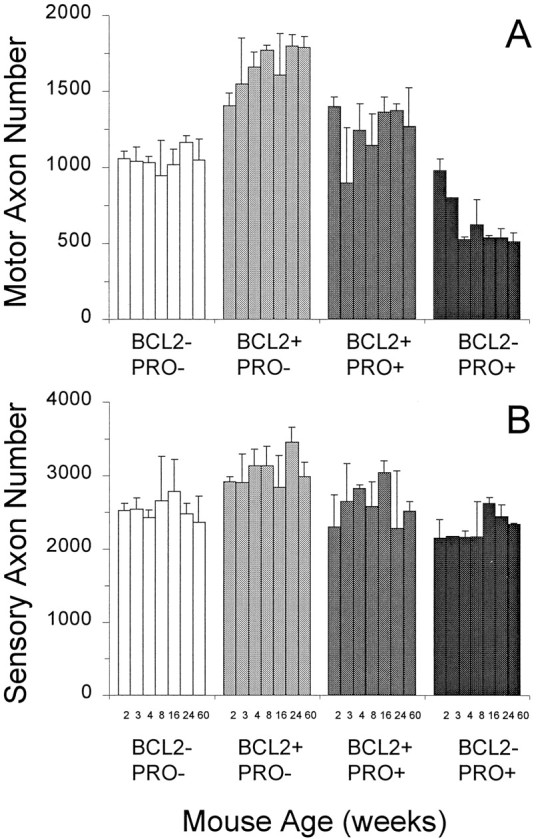
Bcl-2 overexpression increases the number of motor and sensory axons in mutant NF-L (Pro) mice to above wild-type levels. Axon counts from L5 motor (A) and sensory (B) roots are indicated for the various mouse genotypes at ages of 2, 3, 4, 8, 16, 24, and 60 weeks.A, NF-L (Pro) mutant mice have considerably fewer motor axons than wild-type mice do. Overexpression of Bcl-2 alone results in mice with many more axons than in wild type. Bcl-2 overexpression in mutant NF-L (Pro) mice restores the total number of axons to greater than wild-type levels at all ages examined. Counts are averages from three to five mice of each genotype and age with the exception of the 15-month-old animals (n = 2). B, Sensory axons are relatively spared from mutant NF-L (Pro) transgene–mediated killing. Overexpression of Bcl-2 may increase the numbers of NF-L (Pro) axons early in life, but a common final number of axons is reached in NF-L (Pro) mutants regardless of Bcl-2 expression levels. Counts are averages from three to five mice of each genotype and age with the exception of the 15-month-old animals (n = 2). Error bars represent the SD of the data.
To further compare the temporal degeneration patterns among the four genotypes, the number of degenerating axonal profiles within L5 ventral (Fig. 4A) and dorsal (Fig. 4B) roots was determined at different ages. As expected, wild-type and Bcl-2–overexpressing mice experienced little to no degeneration at any age in either motor or sensory roots. However, >40 motor axons per NF-L (Pro) ventral root were visibly degenerating at 3 weeks of age (Fig. 4A,arrow). At the same age, only ∼7 motor axons per root were degenerating in NF-L (Pro) mice overexpressing Bcl-2. This initial delay in degeneration was short-lived because, by 1 week later (Fig.4A, arrowhead), the peak amount of degeneration in the doubly transgenic roots equaled that in the NF-L (Pro) mice. By 8 weeks of age, degeneration had nearly ceased in mice of both genotypes. Although the peak number of degenerating axons was the same with or without the Bcl-2 transgene, the overall amount of degeneration was decreased upon overexpression of Bcl-2. When summed over the first 2 months of age, NF-L (Pro) mice displayed an average of 88 degenerating axons per root, whereas the NF-L (Pro) mice overexpressing Bcl-2 had only 57 axons per root degenerating. Since the degeneration in NF-L (Pro) axons started earlier and remained at peak degeneration levels longer than the doubly transgenic axons did, overexpressing Bcl-2 was able to delay the onset of degeneration and reduce the amount of degeneration caused by transient neurofilamentous insult.
Fig. 4.

Overexpression of Bcl-2 in NF-L (Pro) mice delays onset and decreases the amount of motor axon degeneration caused by NF-L (Pro) transgene–mediated damage. A, L5 motor axon degeneration is an early and transient event in the lifetime of the NF-L (Pro) mice (▪). The total number of motor axons degenerating in the NF-L (Pro) roots is much higher at 3 weeks of age than is that in the NF-L (Pro)/Bcl-2 overexpressor mice (♦). By 4 weeks of age the levels of degeneration are comparable between the singly and doubly transgenic animals, and by 8 weeks of age most of the degeneration has ceased. The arrow indicates degeneration at 3 weeks of age, and the arrowhead indicates degeneration at 4 weeks of age. Very few degenerating axons are observed in wild-type (▴) and Bcl-2–overexpressing (●) mice. Counts are averages from three to five mice from each genotype and age. B, The amount of NF-L (Pro) transgene–mediated sensory axon degeneration exhibited is less than that seen in the motor axons. L5 sensory axon degeneration is an early and transient event in the lifetime of the NF-L (Pro) mice (▪). The total number of sensory axons degenerating in mutant NF-L (Pro) roots is decreased upon Bcl-2 overexpression (♦). Very few degenerating axons are observed in wild-type (▴) and Bcl-2–overexpressing (●) mice. Counts are averages from three to five mice from each genotype and age. Error bars represent the SD of the data.
The number of degenerating sensory axons was reduced by Bcl-2 overexpression (Fig. 4B) from 24 in NF-L (Pro) mice to <10 in Bcl-2/NF-L (Pro) mice at 4 weeks of age. In addition, the period of degeneration appeared to be lengthened in the doubly transgenic mice. These findings indicate that the effects of Bcl-2 overexpression apparently act in a similar manner in both motor and sensory neurons.
Bcl-2 overexpression in NF-L (Pro) mice selectively increases the number of small, but not large, motor axons
To determine whether the beneficial effects of Bcl-2 overexpression are conferred on one axonal size class or another, the distribution of axon sizes was measured in wild-type mice, Bcl-2–overexpressing mice, and NF-L (Pro) mice with or without the Bcl-2 transgene. At 2 weeks of age, wild-type mice displayed a typical bimodal distribution of axon sizes (average size class diameters are 1.5 and 4 μm) with a majority of axons in the larger class (Fig.5A). Bcl-2–overexpressing mice also had two axon size classes with average diameters similar to that of wild type, but in addition, these nerves contain two times more small axons than does wild type (Fig. 5A). Even at 2 weeks of age, before significant axonal degeneration and loss, NF-L (Pro) mice have only one axon size class centered on 2 μm, indicating that the mutant neurofilament-mediated damage has already affected normal axonal radial growth (Fig. 5B). Overexpression of Bcl-2 in the NF-L (Pro) mutant mice yields 30% more small axons than in NF-L (Pro) mutant mice alone (Fig. 5C) and 2.5 times more small axons than in wild-type mice (Fig. 5A). In 2-week-old doubly transgenic nerves, the large axon class (normally centered on 4 μm in wild-type mice) is already reduced but may contain slightly more axons than the NF-L (Pro) mutants. The average size of wild-type large motor axons is indicated in Figure 5C by an arrow.
Fig. 5.

Increased expression of Bcl-2 selectively increases the number of small, not large, motor axons in mutant NF-L (Pro) mice. Axonal areas were calculated for all axons within an entire L5 motor root using a computer-imaging program and plotted as the frequency of appearance of diameters corresponding to circles of equivalent areas. Distributions of 2-week-old (A–C), 2-month-old (D–F), and 6-month-old (G–I) animals from all four genotypes are shown. Wild-type mice (▴) have the expected bimodal distribution of small and large motor axons. Bcl-2–overexpressing mice (●) also have two axon size classes but exhibit an increased number of small axons. Mutant NF-L (Pro) mice (▪) selectively lose the large motor axon population. Mutant NF-L (Pro) mice overexpressing Bcl-2 (♦) have greatly increased numbers of small motor axons but still lose large motor axons. Points represent the averaged axon size distributions of two mice for each genotype and age.Arrows indicate the mean size of the population of large motor neurons found in normal, age-matched animals.
As with the 2-week-old mice, both the wild-type and Bcl-2–overexpressing mice at 2 months of age have two axonal size classes, although normal radial growth of axons has increased the diameters significantly (Fig. 5D). Not only do Bcl-2–overexpressing mice have more than three times the wild-type number of small motor axons, but the Bcl-2–overexpressing axons are reduced in size (peaks centered on 1.5 and 9.0 μm) when compared with the wild-type axon classes (2.5 and 9.5 μm). Bcl-2–overexpressing mice also have slightly more large motor neurons than wild-type mice do. By this age, compared with normal mice the mutant NF-L (Pro) mice (Fig. 5E) have lost completely the very largest class of neurons but retain slightly elevated levels of small axons. Doubly transgenic mice have exceedingly high numbers (3.5 times that of wild type) of small axons but almost no large axons (Fig.5F). These findings reveal that overexpression of Bcl-2 in NF-L (Pro) mice does not rescue the large motor neurons from death at this age.
By 6 months of age, normal radial growth increases the wild-type small axon diameters to ∼3 μm and the large axon diameters to 12.5 μm (Fig. 5G). Bcl-2 overexpression results in a much larger population of small axons centered on 1.5 μm and a slightly elevated large axon population with diameters of ∼12 μm (Fig.5G). NF-L (Pro) mice have an increased number of small axons ∼4 μm in diameter but almost no axons larger than 7 μm (Fig.5H). Overexpression of Bcl-2 in the mutant NF-L (Pro) mice is not able to rescue the axons larger than 7 μm (Fig.5I; the arrow marks the mean diameter of the group of large axons in normal wild-type mice) but does result in more than four times the wild-type levels of small axons. The increased population of small axons in doubly transgenic mice maintains diameters slightly smaller than those of wild-type axons but larger than those of the Bcl-2 overexpressors alone. These analyses show that at 6 months of age, even though Bcl-2 overexpression has the capability to produce increased numbers of both small and large motor neurons, when it is overexpressed in NF-L (Pro) mice, it does not exert a protective effect on the large motor axons at risk in this disease.
Motor neuron cell body degeneration in NF-L (Pro) mice is largely unchanged by overexpression of Bcl-2
There are a number of pathological changes that occur in the spinal cords of NF-L (Pro) mutant mice in addition to the almost complete loss of large motor axons described previously. In ALS patients and in NF-L (Pro) mutant mice, considerable degeneration is evident in the ventral column of the lumbar spinal cord that houses the cell bodies of axons that project to muscles of the lower limbs. Although Bcl-2 overexpression was unable to prevent the loss of large NF-L (Pro) motor axons, it remained a possibility that their cell bodies might benefit from Bcl-2 overexpression and persist long enough to send out new axons and reinnervate targets. To assess this possibility, the lumbar spinal regions from NF-L (Pro) mutant mice and doubly transgenic Bcl-2/NF-L (Pro) mice were examined at several ages.
The motor neuron cell bodies of wild-type mice are distinguished by their large asymmetric appearance, prominent nucleolus, extended projections, and position within the ventral portion of the spinal cord (Fig. 6A). The cell bodies of Bcl-2–overexpressing mice are morphologically identical to wild-type cell bodies, except for an apparent increase in number (Fig.6B). NF-L (Pro) mutant mice display motor neuron cell body pathology as early as 2 weeks of age and at least as late as 15 months (Fig. 6D). This pathology includes distended cell bodies, cytoplasmic neurofilament accumulations, and axonal swellings (Lee et al., 1994). The nonuniform cytoplasm of NF-L (Pro) mutant cells contains neurofilamentous accumulations that stain lightly with toluidine blue but stain darkly by silver-staining methods. Overexpression of Bcl-2 in mutant NF-L (Pro) mice does not eliminate motor neuron cell body degeneration (Fig. 6C). At all ages examined, doubly transgenic motor neuron cell bodies exhibited features of degeneration similar to that observed in NF-L (Pro) mice. Comparable numbers of degenerating cell bodies were observed in NF-L (Pro) mice and Bcl-2/NF-L (Pro) mice at ages up to 15 months.
Fig. 6.
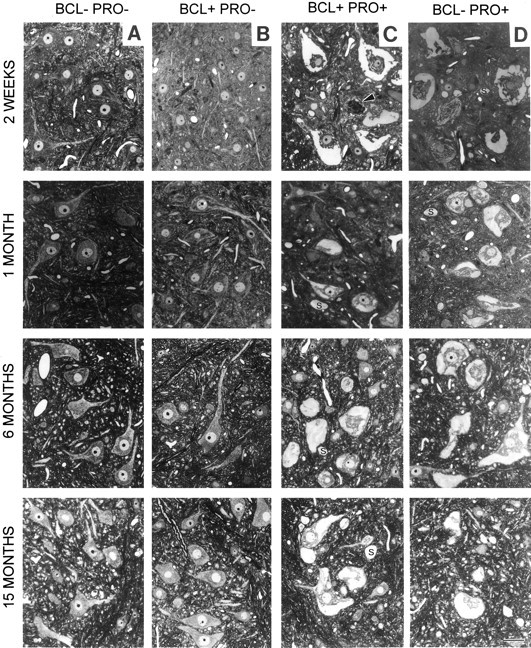
Spinal cord anterior horn motor neuron pathology is a prominent feature of both NF-L (Pro) mutant and doubly transgenic Bcl-2/NF-L (Pro) mutant mice. Cross sections of lumbar spinal cord from 2-week-old and 1-, 6-, and 15-month-old mice are shown.A–D, Genotypes are as indicated.Arrowheads indicate degenerating motor neuron cell bodies. Axonal swellings are marked with an S. Scale bar, 50 μm.
DISCUSSION
In testing whether mutant NF-L–induced motor neuron loss could be alleviated by Bcl-2, we have determined that Bcl-2 overexpression was effective in preventing the death of many motor neurons during the developmental period of naturally occurring cell death but that these additional neurons did not grow to replace the motor neurons with the largest axonal calibers that were selectively killed by mutant neurofilament-mediated mechanisms. The degree of large motor axon loss exhibited by these mice was only slightly reduced, and there were no measurable phenotypic changes in disease onset or progression, despite the ability of Bcl-2 to delay and slightly moderate the degeneration period. By these measures, the overexpression of human Bcl-2 did not protect the at-risk large motor axons from mutant neurofilament-mediated degeneration. Moreover, comparison of the motor neuron cell bodies within the lumbar spinal cord of singly and doubly transgenic mice revealed that overexpression of Bcl-2 afforded no significant protection from mutant neurofilament-mediated degeneration at ages up to 15 months. Thus, increased Bcl-2 seems to delay and then prolong the period of neurofilament-mediated damage for large motor neurons without markedly changing the clinical disease course.
There are several potential reasons why the overexpression of human Bcl-2 failed to rescue the large motor axons affected by mutant NF-L–mediated disease. It may be that the NF-L–mediated damage had progressed to an advanced state too rapidly, before the action of Bcl-2 could counteract the deleterious effects. Alternatively, the levels of Bcl-2 produced in the motor neurons might not have been high enough to counteract such a powerful insult. This is implausible, however, since this level of transgenic Bcl-2 not only promotes a large increase in the numbers of small caliber axons in these mice but also has been shown to be expressed very early and at high levels in the cell bodies of large α motor neurons in the ventral spinal cord (Martinou et al., 1994; Kostic et al., 1997).
A recent study (Kostic et al., 1997) showed that the onset of ALS-like motor neuron disease in SOD1 mutant mice (G93A line) could be delayed slightly (37 d) by overexpressing human Bcl-2 in neurons, thereby causing a corresponding increase in life span. However, the duration of disease after onset was not lengthened. Because these authors did not report any increased number of axons in the nerve type they counted, these findings suggested that Bcl-2 overexpression offered a degree of initial antiapoptotic protection from mutant SOD1-mediated toxicity, but after the neurons reached a threshold disease state, the Bcl-2 action was no longer sufficient to slow disease progression. A similar study that produced seemingly conflicting results used mice expressing a dominant-negative interleukin-1B–converting enzyme (ICE) crossed to the same SOD1 mutant mouse line (G93A) (Friedlander et al., 1997). Although ICE itself has not been clearly linked to apoptosis except in thymocytes (Kuida et al., 1995), the inhibition of ICE-like proteases is thought to reduce cell death mediated by a variety of different stimuli. Interpretation of the Friedlander et al. (1997) study is further complicated by the discordance in reported disease duration in the G93A mice. Despite reports by other investigators of disease durations of 48 d (Gurney et al., 1994) or 72 and 70 d with and without the Bcl-2 transgene, respectively (Kostic et al., 1997), the dominant-negative ICE was reported to increase disease duration from a much reduced 12 to 27 d without effect on the age of disease onset (Friedlander et al., 1997). In contrast to the Kostic et al. (1997) study, these results argued that the inhibition of apoptosis acted to keep the diseased neurons alive longer instead of preventing them from becoming diseased at an earlier time.
These same Bcl-2 mice were also used in a previous attempt to protect the facial nucleus motor neurons that degenerate in progressive motor neuropathy (pmn) mice. In this case, the pmn facial motor neuron cell bodies were spared by Bcl-2 overexpression, but the facial motor axons and phrenic motor axons were not (Sagot et al., 1995). In addition, Bcl-2 overexpression in wobbler mutant mice did not prevent the degeneration of facial motor neurons or musculocutaneous nerve axons (Coulpier et al., 1996). Nor were the onset and duration of disease in the pmnand wobbler mutant mice altered by Bcl-2 overexpression, although the onset of SOD1-mediated ALS-like disease was delayed. These three examples demonstrate the ability of Bcl-2 overexpression to alter some aspects of various motor neuron diseases, but the variability of protection offered indicates that other potent upstream influences may be producing more damage or more apoptotic or necrotic messengers than the downstream Bcl-2 can overcome.
As to the relevance of using these antiapoptotic strategies to reduce the type of motor neuron death in both sporadic and inherited forms of ALS, morphological evidence (Sendtner et al., 1994; Troost et al., 1995) has suggested that the form of cell death occurring in ALS is indeed apoptotic, although no consensus has yet emerged. Several groups have demonstrated the presence of terminal deoxynucleotidyl transferase–mediated dUTP–biotin nick end labeling (TUNEL)-positive staining in muscle cells (Tews et al., 1997) and in spinal cord motor neurons (Yoshiyama et al., 1994) of ALS patients but not in controls. Expression of several different ALS-linked SOD1 protein mutants in cultured neuronal cells resulted in apoptotic cell death (as judged by TUNEL staining), whereas expression of wild-type SOD1 conferred protection from apoptosis (Rabizadeh et al., 1995; Durham et al., 1997). The finding that Bcl-2 mRNA is selectively reduced and proapoptotic Bax mRNA is increased in motor neurons of sporadic ALS patients (Mu et al., 1996) further supports the idea that cell death occurs via apoptotic mechanisms in ALS and strengthens the argument for using Bcl-2 to counteract the cell death pathways that may be responsible for motor neuron degeneration in ALS.
The proposed mechanism of disorganized neurofilaments choking axonal transport as the primary cause of selective death of motor neurons in the NF-L (Pro) mice has recently been called into question by a study using a similar NF-L transgene that reported enteric nervous system abnormalities (accompanied by death within a few days of birth for two transgenic founder mice), stunted growth (in progeny from a third founder), and pathology comprised of vacuolar inclusions (Canete-Soler et al., 1999). Because insertion of a C-terminal myc epitope tag into the NF-L gene was shown to stabilize the resultant mRNA by twofold and to alter the association of mRNA-binding proteins when expressed in a neuroblastoma cell line, the authors proposed that the defects in their transgenic mice may arise from a dominant effect of the transgenic mRNA on the metabolism of other RNAs. (This hypothesis remains to be tested directly by analysis of similar transgenic animals bearing an identical transgene with the myc-coding sequence out of frame with that of NF-L.) There are several important differences between our mutant NF-L (Pro) mice and these newly reported mice. Specifically, different promoters were used for the transgenes, and the levels of transgene expression required to provoke disease differed markedly [the mutant NF-L (Pro) causes disease with between 50 and 70% of endogenous NF-L levels, whereas the Canete-Soler et al. (1999) transgene requires levels much higher than those of wild type]. Moreover, there are striking differences in the time course of the disease, the neurons at risk, and the pathology developed in those neurons. Instead of neurofilamentous accumulations and axonal swellings found in our NF-L (Pro) mice (which are also characteristic of human ALS), the Canete-Soler et al. (1999)mice were reported to have vacuolar changes without neurofilamentous abnormalities. On the basis of these distinct differences, we conclude that it is unlikely that the pathology and resultant cell death observed in our NF-L (Pro) mice are a result of C-terminal myc tag–induced alterations in mRNA metabolism.
In any event, we emphasize that the mutant neurofilament mouse model used here recapitulates many of the pathological neurofilamentous misaccumulations observed in sporadic human disease (Carpenter, 1968;Chou and Fakadej, 1971; Hirano et al., 1984a), in SOD1 mutant–mediated familial disease (Hirano et al., 1984b; Rouleau et al., 1996; Shibata et al., 1996), and in SOD1 mutant mice (Gurney et al., 1994; Wong et al., 1995; Tu et al., 1996). Since neurofilament misaccumulation is a hallmark of human disease, the finding that neurofilament disorganization arising solely from damage to a neurofilament subunit can selectively kill motor neurons offers strong evidence that neurofilaments are a part of pathogenesis in sporadic and inherited disease. Indeed, in mice, complete removal of axonal neurofilaments through disruption of the NF-L gene slows disease onset mediated by familial ALS-linked mutant SOD1G85R by 5 weeks (Williamson et al., 1998). Similarly, trapping most neurofilaments in the perikarya of motor neurons extends the life span of mutant SOD1G37R mice by 6 months (Couillard-Despres et al., 1998). Combined with the known neurofilament-dependent slowing of slow axonal transport and the finding that the earliest defect to arise in SOD1G37R and SOD1G85R mice is a slowing of selected cargoes of slow transport (Williamson and Cleveland 1999), disorganized axonal neurofilaments must represent an important aspect of pathogenesis in ALS.
All of this evidence combine to suggest that an effective therapeutic strategy would be one that could selectively diminish neurofilament synthesis and accumulation in axons. This might be achieved by disruption of the signaling pathway that normally upregulates neurofilament synthesis after myelination by nearly an order of magnitude relative to synthesis levels during axon elongation or during recovery from axotomy (Hoffman et al., 1987). Furthermore, the inability of chronically increased Bcl-2 levels to protect adequately the large caliber neurons at risk in the previously described mouse models of motor neuron disease argues that therapies based on apoptotic inhibitors might be most useful in combination with an agent lowering axonal neurofilament accumulation.
Footnotes
This work has been supported by National Institutes of Health Grant NS 27093 to D.W.C. Salary support for D.W.C. was provided by the Ludwig Institute for Cancer Research. We thank Dr. Jean-Claude Martinou for his gift of the human Bcl-2 overexpressor mouse used in this study. We appreciate the efforts of Janet Folmer and Karen Anderson in sectioning and staining slides for light microscopy, without which this work would not have been possible.
Correspondence should be addressed to Dr. D. W. Cleveland, Ludwig Institute for Cancer Research, University of California at San Diego, 9500 Gilman Drive, La Jolla, CA 92093.
REFERENCES
- 1.Allen RT, Cluck MW, Agrawal DK. Mechanisms controlling cellular suicide: role of Bcl-2 and caspases. Cell Mol Life Sci. 1998;54:427–445. doi: 10.1007/s000180050171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Allsopp T, Wyatt S, Paterson H, Davies A. The proto-oncogene bcl-2 can selectively rescue neurotrophic factor-dependent neurons from apoptosis. Cell. 1993;73:295–307. doi: 10.1016/0092-8674(93)90230-n. [DOI] [PubMed] [Google Scholar]
- 3.Bruijn LI, Becher MW, Lee MK, Anderson KL, Jenkins NA, Copeland NG, Sisodia SS, Rothstein JD, Borchelt DR, Price DL, Cleveland DW. ALS-linked SOD1 mutant G85R mediates damage to astrocytes and promotes rapidly progressive disease with SOD1-containing inclusions. Neuron. 1997;18:327–338. doi: 10.1016/s0896-6273(00)80272-x. [DOI] [PubMed] [Google Scholar]
- 4.Canete-Soler R, Silberg DG, Gershon MD, Schlaepfer WW. Mutation in neurofilament transgene implicates RNA processing in the pathogenesis of neurodegenerative disease. J Neurosci. 1999;19:1273–1283. doi: 10.1523/JNEUROSCI.19-04-01273.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Carpenter S. Proximal axonal enlargement in motor neuron disease. Neurology. 1968;18:841–851. doi: 10.1212/wnl.18.9.841. [DOI] [PubMed] [Google Scholar]
- 6.Chao D, Korsmeyer SJ. BCL-2 family: regulators of cell death. Annu Rev Immunol. 1998;16:395–419. doi: 10.1146/annurev.immunol.16.1.395. [DOI] [PubMed] [Google Scholar]
- 7.Chou SM, Fakadej AV. Ultrastructure of chromatolytic motoneurons and anterior spinal roots in a case of Werdnig-Hoffmann disease. J Neuropathol Exp Neurol. 1971;30:368–379. doi: 10.1097/00005072-197107000-00005. [DOI] [PubMed] [Google Scholar]
- 8.Cote F, Collard JF, Julien JP. Progressive neuronopathy in transgenic mice expressing the human neurofilament heavy gene: a mouse model of amyotrophic lateral sclerosis. Cell. 1993;73:35–46. doi: 10.1016/0092-8674(93)90158-m. [DOI] [PubMed] [Google Scholar]
- 9.Couillard-Despres S, Zhu Q, Wong PC, Price DL, Cleveland DW, Julien JP. Protective effect of neurofilament heavy gene overexpression in motor neuron disease induced by mutant superoxide dismutase. Proc Natl Acad Sci USA. 1998;95:9626–9630. doi: 10.1073/pnas.95.16.9626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Coulpier M, Junier M-P, Peschanski M, Dreyfus PA. Bcl-2 sensitivity differentiates two pathways for motoneuronal death in the wobbler mutant mouse. J Neurosci. 1996;16:5897–5904. doi: 10.1523/JNEUROSCI.16-19-05897.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Durham HD, Roy J, Dong L, Figlewicz DA. Aggregation of mutant Cu/Zn superoxide dismutase proteins in a culture model of ALS. J Neuropathol Exp Neurol. 1997;56:523–530. doi: 10.1097/00005072-199705000-00008. [DOI] [PubMed] [Google Scholar]
- 12.Friedlander RM, Brown RH, Gagliardini V, Wang J, Yuan J. Inhibition of ICE slows ALS in mice. Nature. 1997;388:6637. doi: 10.1038/40299. [DOI] [PubMed] [Google Scholar]
- 13.Garcia I, Martinou I, Tsujimoto Y, Martinou JC. Prevention of programmed cell death of sympathetic neurons by the bcl-2 proto-oncogene. Science. 1992;258:302–304. doi: 10.1126/science.1411528. [DOI] [PubMed] [Google Scholar]
- 14.Gasser HS, Grundfest H. Axon diameters in relation to the spike dimensions and the conduction velocity in mammalian A fibers. Am J Physiol. 1939;127:393–414. [Google Scholar]
- 15.Gill SR, Wong PC, Montiero MJ, Cleveland DW. Assembly properties of dominant and recessive mutations in the small mouse neurofilament (NF-L) subunit. J Cell Biol. 1991;111:2005–2019. doi: 10.1083/jcb.111.5.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gurney ME, Pu H, Chiu AY, Dal Canto MC, Polchow CY, Alexander DD, Caliendo J, Hentati A, Kwon YW, Deng HX, Siddique T. Motor neuron degeneration in mice that express a human Cu, Zn superoxide dismutase mutation. Science. 1994;164:1772–1775. doi: 10.1126/science.8209258. [DOI] [PubMed] [Google Scholar]
- 17.Hirano A, Donnenfeld H, Sasaki S, Nakano I. Fine structural observations of neurofilamentous changes in amyotrophic lateral sclerosis. J Neuropathol Exp Neurol. 1984a;43:461–470. doi: 10.1097/00005072-198409000-00001. [DOI] [PubMed] [Google Scholar]
- 18.Hirano A, Nakano I, Kurland LT, Mulder DW, Holley PW, Saccomanno G. Fine structural study of neurofibrillary changes in a family with amyotrophic lateral sclerosis. J Neuropathol Exp Neurol. 1984b;43:471–480. doi: 10.1097/00005072-198409000-00002. [DOI] [PubMed] [Google Scholar]
- 19.Hockenbery DM, Oltvai ZN, Yin XM, Milliman CL, Korsmeyer SJ. Bcl-2 functions in an antioxidant pathway to prevent apoptosis. Cell. 1993;75:241–251. doi: 10.1016/0092-8674(93)80066-n. [DOI] [PubMed] [Google Scholar]
- 20.Hoffman PN, Cleveland DW, Griffin JW, Landes PW, Cowan NJ, Price DL. Neurofilament gene expression: a major determinant of axonal caliber. Proc Natl Acad Sci USA. 1987;84:3472–3476. doi: 10.1073/pnas.84.10.3472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kane DJ, Sarafian TA, Anton R, Hahn H, Gralla EB, Valentine JS, Ord T, Bredesen DE. Bcl-2 inhibition of neural death: decreased generation of reactive oxygen species. Science. 1993;262:1274–1277. doi: 10.1126/science.8235659. [DOI] [PubMed] [Google Scholar]
- 22.Kawamura Y, Dyck PJ, Shimono M, Okazaki H, Tateishi J, Doi H. Morphometric comparison of the vulnerability of peripheral motor and sensory neurons in amyotrophic lateral sclerosis. J Neuropathol Exp Neurol. 1981;40:667–675. doi: 10.1097/00005072-198111000-00008. [DOI] [PubMed] [Google Scholar]
- 23.Kostic V, Jackson-Lewis V, deBilbao F, Dubois-Dauphin M, Przedborski S. Bcl-2 prolonging life in a transgenic mouse model of familial amyotrophic lateral sclerosis. Science. 1997;277:559–562. doi: 10.1126/science.277.5325.559. [DOI] [PubMed] [Google Scholar]
- 24.Kuida K, Lippke JA, Ku G, Harding MW, Livingston DJ, Su M-S, Flavell RA. Altered cytokine export and apoptosis in mice deficient in interleukin-1 beta converting enzyme. Science. 1995;267:2000–2003. doi: 10.1126/science.7535475. [DOI] [PubMed] [Google Scholar]
- 25.Lee MK, Marszalek J, Cleveland DW. Expression of a mutant neurofilament subunit causes massive, selective motor neuron death and ALS-like motor neuron disease. Neuron. 1994;13:975–988. doi: 10.1016/0896-6273(94)90263-1. [DOI] [PubMed] [Google Scholar]
- 26.Mah SP, Zhong LT, Liu Y, Roghani A, Edwards RH, Bredesen DE. The proto-oncogene bcl-2 inhibits apoptosis in PC12 cells. J Neurochem. 1993;60:1183–1186. doi: 10.1111/j.1471-4159.1993.tb03275.x. [DOI] [PubMed] [Google Scholar]
- 27.Martinou JC, Dubois-Dauphin M, Staple JK, Rodriguez I, Frankowski H, Missotten M, Albertini P, Talabot D, Pietra C. Overexpression of BCL-2 in transgenic mice protects neurons from naturally occurring cell death and experimental ischemia. Neuron. 1994;13:1017–1030. doi: 10.1016/0896-6273(94)90266-6. [DOI] [PubMed] [Google Scholar]
- 28.Merry DE, Veis DJ, Hickey WF, Korsmeyer SJ. Bcl-2 protein expression is widespread in the developing nervous system and retained in the adult PNS. Development. 1994;120:301–311. doi: 10.1242/dev.120.2.301. [DOI] [PubMed] [Google Scholar]
- 29.Mu X, He J, Anderson D, Trojanowski J, Springer J. Altered expression of bcl-2 and bax in amyotrophic lateral sclerosis spinal cord motor neurons. Ann Neurol. 1996;40:379–386. doi: 10.1002/ana.410400307. [DOI] [PubMed] [Google Scholar]
- 30.Murakami T. Motor neuron disease: quantitative morphological and microdensitophotometric studies of neurons of anterior horn and ventral root of cervical spinal cord with special reference to the pathogenesis. J Neurol Sci. 1990;99:101–115. doi: 10.1016/0022-510x(90)90204-z. [DOI] [PubMed] [Google Scholar]
- 31.Rabizadeh S, Gralla EB, Borchelt DR, Gwinn R, Valentine JS, Sisodia S, Wong P, Lee M, Hahn H, Bredesen DE. Mutations associated with amyotrophic lateral sclerosis convert superoxide dismutase from an antiapoptotic gene to a proapoptotic gene: studies in yeast and neural cells. Proc Natl Acad Sci USA. 1995;92:3024–3028. doi: 10.1073/pnas.92.7.3024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rouleau GA, Clark AW, Rooke K, Pramatarova A, Krizus A, Suchowersky O, Julien J-P, Figlewicz D. SOD1 mutation is associated with accumulation of neurofilaments in amyotrophic lateral sclerosis. Ann Neurol. 1996;29:128–131. doi: 10.1002/ana.410390119. [DOI] [PubMed] [Google Scholar]
- 33.Sagot Y, Dubois-Dauphin M, Tan SA, de Bilbao F, Aebischer P, Martinou JC, Kato AC. Bcl-2 overexpression prevents motoneuron cell body loss but not axonal degeneration in a mouse model of a neurodegenerative disease. J Neurosci. 1995;15:7727–7733. doi: 10.1523/JNEUROSCI.15-11-07727.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sakaguchi T, Okada M, Kitamura T, Kawasaki K. Reduced diameter and conduction velocity of myelinated fibers in the sciatic nerve of a neurofilament-deficient mutant quail. Neurosci Lett. 1993;153:65–68. doi: 10.1016/0304-3940(93)90078-y. [DOI] [PubMed] [Google Scholar]
- 35.Sendtner M, Dittrich F, Hughes RA, Thoenen H. Actions of CNTF and neurotrophins on degenerating motor neurons: preclinical studies with clinical implications. J Neurol Sci [Suppl] 1994;124:77–83. doi: 10.1016/0022-510x(94)90187-2. [DOI] [PubMed] [Google Scholar]
- 36.Shibata NA, Hirano A, Kobayashi M, Siddique T, Deng H-X, Hung W-Y, Kato T, Asayama K. Intense superoxide dismutase-1 immunoreactivity in intracytoplasmic hyaline inclusions of familial amyotrophic lateral sclerosis with posterior column involvement. J Neuropathol Exp Neurol. 1996;55:481–490. doi: 10.1097/00005072-199604000-00011. [DOI] [PubMed] [Google Scholar]
- 37.Tews DS, Goebel HH, Meinck HM. DNA-fragmentation and apoptosis-related proteins of muscle cells in motor neuron disorders. Acta Neurol Scand. 1997;96:380–386. doi: 10.1111/j.1600-0404.1997.tb00302.x. [DOI] [PubMed] [Google Scholar]
- 38.Troost D, Aten J, Morsink F, de Jong JMBV. Apoptosis in amyotrophic lateral sclerosis is not restricted to motor neurons. Bcl-2 expression is increased in unaffected post-central gyrus. Neuropathol Appl Neurobiol. 1995;21:498–504. doi: 10.1111/j.1365-2990.1995.tb01096.x. [DOI] [PubMed] [Google Scholar]
- 39.Tu PH, Raju P, Robinson KA, Gurney ME, Trojanowski JQ, Lee VM. Transgenic mice carrying a mutant superoxide dismutase transgene develop neuronal cytoskeletal pathology resembling human amyotrophic lateral sclerosis lesions. Proc Natl Acad Sci USA. 1996;93:3155–3160. doi: 10.1073/pnas.93.7.3155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Williamson TL, Cleveland DW. Slowing of axonal transport is a very early event in the toxicity of ALS-linked SOD1 mutants to motor neurons. Nat Neurosci. 1999;2:50–56. doi: 10.1038/4553. [DOI] [PubMed] [Google Scholar]
- 41.Williamson TL, Bruijn LI, Zhu Q, Anderson KL, Anderson SD, Julien JP, Cleveland DW. Absence of neurofilaments reduces the selective vulnerability of motor neurons and slows disease caused by a familial amyotrophic lateral sclerosis-linked superoxide dismutase 1 mutant. Proc Natl Acad Sci USA. 1998;95:9631–9636. doi: 10.1073/pnas.95.16.9631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wong PC, Pardo CA, Borchelt DR, Lee MK, Copeland NG, Jenkins NA, Sisodia SS, Cleveland DW, Price DL. An adverse property of a familial ALS-linked SOD1 mutation causes motor neuron disease characterized by vacuolar degeneration of mitochondria. Neuron. 1995;14:1105–1116. doi: 10.1016/0896-6273(95)90259-7. [DOI] [PubMed] [Google Scholar]
- 43.Xu Z, Cork LC, Griffin JW, Cleveland DW. Increased expression of neurofilament subunit NF-L produces morphological alterations that resemble the pathology of human motor neuron disease. Cell. 1993;73:23–33. doi: 10.1016/0092-8674(93)90157-l. [DOI] [PubMed] [Google Scholar]
- 44.Yamasaki H, Itakura C, Mizutani M. Hereditary hypotrophic axonopathy with neurofilament deficiency in a mutant strain of the Japanese quail. Acta Neuropathol (Berl) 1991;82:427–434. doi: 10.1007/BF00293376. [DOI] [PubMed] [Google Scholar]
- 45.Yoshiyama Y, Yamada T, Asanuma K, Asahi T. Apoptosis related antigen, LeY and nick-end labeling are positive in spinal motor neurons in amyotrophic lateral sclerosis. Acta Neuropathol (Berl) 1994;88:207–211. doi: 10.1007/BF00293395. [DOI] [PubMed] [Google Scholar]
- 46.Zhong LT, Sarafian T, Kane DJ, Charles AC, Mah SP, Edwards RH, Bredesen DE. Bcl-2 inhibits death of central neural cells induced by multiple agents. Proc Natl Acad Sci USA. 1993;90:4533–4537. doi: 10.1073/pnas.90.10.4533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zhu Q, Couillard-Despres S, Julien JP. Delayed maturation of regenerating myelinated axons in mice lacking neurofilaments. Exp Neurol. 1997;148:299–316. doi: 10.1006/exnr.1997.6654. [DOI] [PubMed] [Google Scholar]