

Abstract
We have shown that physiological levels of estradiol exert profound protective effects on the cerebral cortex in ischemia induced by permanent middle cerebral artery occlusion. The major goal of this study was to begin to elucidate potential mechanisms of estradiol action in injury. Bcl-2 is a proto-oncogene that promotes cell survival in a variety of tissues including the brain. Because estradiol is known to promote cell survival via Bcl-2 in non-neural tissues, we tested the hypothesis that estradiol decreases cell death by influencingbcl-2 expression in ischemic brain injury. Furthermore, because estradiol may protect the brain through estrogen receptor-mediated mechanisms, we examined expression of both receptor subtypes ERα and ERβ in the normal and injured brain. We analyzed gene expression by RT-PCR in microdissected regions of the cerebral cortex obtained from injured and sham female rats treated with estradiol or oil. We found that estradiol prevented the injury-induced downregulation of bcl-2expression. This effect was specific to bcl-2, as expression of other members of the bcl-2 family (bax, bcl-xL, bcl-xS, and bad) was unaffected by estradiol treatment. We also found that estrogen receptors were differentially modulated in injury, withERβ expression paralleling bcl-2expression. Finally, we provide the first evidence of functional ERβ protein that is capable of binding ligand within the region of the cortex where estradiol-mediated neuroprotection was observed in cerebral ischemia. These findings indicate that estradiol modulates the expression of bcl-2 in ischemic injury. Furthermore, our data suggest that estrogen receptors may be involved in hormone-mediated neuroprotection.
Keywords: estradiol, estrogen, neuroprotection, cerebral ischemia, stroke, menopause, bcl-2, bcl-2 family, estrogen receptors, ERβ, ERα, receptor binding, RT-PCR, in situ hybridization
Increasing evidence demonstrates that estradiol is a pleiotropic hormone that acts beyond the scope of its reproductive functions. Clinical studies indicate that estradiol reduces the risk or severity of neurodegenerative conditions such as Alzheimer’s disease (Henderson et al., 1996; Kawas et al., 1997) and stroke (Paganini-Hill, 1995; Schmidt et al., 1996). There is a cellular and molecular basis for these clinical observations. Estradiol is a neuroprotective factor that decreases cell death in a variety ofin vitro (Goodman et al., 1996; Singer et al., 1996; Green et al., 1997; Gridley et al., 1997; Weaver et al., 1997) and in vivo (Hall et al., 1991; Simpkins et al., 1997; Alkayed et al., 1998; Dubal et al., 1998) paradigms of brain injury. Recently, we (Dubal et al., 1998) and other investigators (Simpkins et al., 1997;Alkayed et al., 1998) have shown that estradiol attenuates stroke-related injury using animal models of cerebral ischemia.
We have shown that low, physiological levels of estradiol are sufficient to exert profound protective effects in the ischemic brain of rats, specifically in the cortex (Dubal et al., 1998). Several potential mechanisms may underlie the effects of estradiol. Our previous data indicate that estradiol may act through classic or nonclassic genomic mechanisms to protect against ischemic injury because, in our model, the actions of estradiol are not rapid and require pretreatment. Furthermore the actions of estradiol do not appear to be mediated via changes in blood flow or other major physiological parameters (Dubal et al., 1998).
The major goal of this study was to begin to elucidate potential mechanisms of estradiol-mediated neuroprotection in cerebral ischemia. Estradiol is known to promote cell survival via Bcl-2 in non-neuronal tissues (Teixeira et al., 1995; Kandouz et al., 1996; Huang et al., 1997). We, therefore, hypothesized that estradiol may decrease brain injury, in part, by influencing bcl-2 expression in cerebral ischemia. Bcl-2 is a survival factor that can block both necrotic and apoptotic cell death (Bredesen, 1995), two modes of cell death that contribute to ischemic injury (MacManus and Linnik, 1997; Namura et al., 1998). Bcl-2 acts upstream to prevent the activation of caspases, inhibits free radical formation, regulates calcium sequestration (MacManus and Linnik, 1997), and blocks the pro-apoptotic actions of other members of the Bcl-2 family such as Bax and Bad (Merry and Korsmeyer, 1997). To assess whether estradiol influences these cell survival and cell death factors, we examined expression ofbcl-2 and other members of the bcl-2 family in our paradigm of hormone replacement and neurodegeneration.
Because estradiol acts, and may protect, through receptor-mediated mechanisms, we also examined the expression of both estrogen receptor subtypes ERα and ERβ in normal and injured rat brain. Evidence from a study that used ERα knock-out mice suggests that ERβ may mediate the protective effects of estradiol in non-neural tissue (Iafrati et al., 1997). Although it is intriguing to speculate that a similar role for ERβ may exist in the brain, the presence of a functional ERβ protein in cortex has not yet been demonstrated. We, therefore, performed in vivo binding studies to determine whether functional ERβ protein exists in the rat cerebral cortex.
Our findings demonstrate that low, physiological levels of estradiol modulate bcl-2 expression in ischemic injury. Furthermore, our data suggest that estrogen receptors, specifically ERβ, may be involved in hormone-mediated neuroprotection.
MATERIALS AND METHODS
Cerebral ischemia
Female Sprague Dawley rats (225–275 gm) were maintained in a 14/10 light/dark cycle with ad libitum access to food and water. Under methoxyflurane anesthesia, rats were bilaterally ovariectomized (n = 8–11 per experimental group) to eliminate endogenous estradiol production and then implanted with a SILASTIC capsule containing oil or 17β-estradiol (180 μg/ml). This paradigm of estradiol treatment produces levels of estradiol equivalent to basal circulating levels (Dubal et al., 1998) observed during the rat estrous cycle (DePaolo et al., 1979). Seven days after ovariectomy and treatment, rats underwent cerebral ischemia or sham surgery. Rats were anesthetized with ketamine/acepromazine (80.0/0.52 mg/kg, i.p.). Body temperature was monitored with a rectal probe and maintained at normothermia (36.5–37.5°C). The right femoral artery was cannulated for monitoring major physiological parameters (blood gases, MABP, pH, glucose). The right middle cerebral artery was permanently occluded using previously described methods (Dubal et al., 1998). Briefly, a 4/0 monofilament suture was inserted through the internal carotid artery to the base of the middle cerebral artery, thus occluding blood flow to the cortex and striatum. Brains were collected 24 hr after the onset of ischemia and used for RT-PCR or in situ hybridization studies.
RT-PCR studies
Microdissection. Alternating 1 mm sections of brain were collected using a brain matrix (Activational Systems) and then stained in 2% triphenyltetrazolium chloride (TTC) to visualize injury (Bederson et al., 1986) or frozen at −80°C to monitor gene expression. The area of the cortex analyzed for gene expression was selected using the following criteria. We first examined tissue from a 1 mm TTC-stained coronal section, corresponding to the middle of the infarct. Then, the adjacent fresh, frozen 1 mm section (∼0.3 mm anterior to the bregma) was used to analyze gene expression. A region apposed to the infarct in ovariectomized oil-treated rats and the equivalent region on the contralateral side of the brain were microdissected using the Palkovits (1978) method. Anatomically matched regions were microdissected in (1) injured ovariectomized estradiol-treated, (2) sham ovariectomized oil-treated, and (3) sham ovariectomized estradiol-treated rats. For all samples, the microdissected regions were anatomically similar while remaining in noninfarcted tissue.
cDNA preparation. Total RNA was isolated from microdissected samples by the method of Chomczynski and Sacchi (1987). For each sample, we reverse transcribed 0.5 μg of total RNA to produce cDNA in a final reaction volume of 40 μl, containing 2.5 μmrandom hexamers (Perkin-Elmer, Branchburg, NJ), 100 U murine leukemia virus reverse transcriptase (Perkin-Elmer), 1 mmdNTP mix (Life Technologies, Gaithersburg, MD), 80 U RNAsin (Promega, Madison, WI), 5 mm MgCl2 (Life Technologies), and 1× reaction buffer (Life Technologies). Each sample was incubated for reverse transcription at room temperature for 15 min, 37°C for 2 min, 42°C for 1 hr, and 99°C for 5 min. The same procedure was performed on samples using a reaction solution without reverse transcriptase to check for genomic contamination.
PCR amplification. We used well characterized RT-PCR methods to determine relative changes in gene expression at the mRNA level (Estus, 1996). For each gene examined, we generated standard curves of input RNA and cycle number to determine the optimum cycle number within the linear range for PCR amplification (data not shown); for all genes examined, this was determined to be between 25 and 30 cycles. These methods have been validated in studies showing that, within the optimal range of amplification, yields of PCR product are linear with respect to input RNA (Estus, 1996).
For each gene, stock solutions were prepared containing 1.5 mm MgCl2, 1× reaction buffer, 10 μCi of [32P]dCTP (3000 Ci/mmol) (NEN, Boston, MA), 1 μm each primer, and 1.5 U of Taq polymerase (Life Technologies). For ERα and ERβ PCR, 1.5 U of Taq antibody (Life Technologies) was included in each reaction. The stock solution was aliquoted (49 μl/tube), and 1/30 of synthesized cDNA (from reverse transcription reaction) was added to each sample tube. Samples were then thermocycled for PCR amplification (Touchdown thermocycler; Hybaid, Middlesex, UK) according to the following reaction conditions: 1 min 95°C, 1 min 55°C, and 2 min 72°C (25–30 cycles). After amplification, PCR products were resolved by PAGE. The gels were dried, and the products were visualized and quantified using a PhosphorImager (Molecular Dynamics, Sunnyvale, CA).
All the oligonucleotide sequence pairs used for gene amplification in this study generated PCR products of expected sizes that have been sequenced to verify their identities: L27A sense primer, 5′-ATGCTAACTGTCCAAGTCTA-3′ and antisense primer, 5′-GGGAGCAACTCCATTCTTGT-3′ (214 bp) (Hoshimaru et al., 1996); neuron-specific enolase (NSE) sense primer, 5′-ATCTTGGACTCCCGTGGGAA-3′ and antisense primer, 5′-TTTGGCAGTATGGAGATCCA-3′ (54 bp) (Estus et al., 1997);bcl-2 sense primer, 5′-CTGTACGGCCCCAGCATGCG-3′ and antisense primer, 5′-GCTTTGTTTCATGGTACATC-3′ (231 bp) (Greenlund et al., 1995); bax sense primer, 5′-GGGAATTCTGGAGCTGCAGAGGATGATT-3′ and antisense primer, 5′-GCGGATCCAAGTTGCCATCAGCAAACAT-3′ (96 bp) (Greenlund et al., 1995);bcl-x (L & S) sense primer, 5′-AGGCTGGCGATGAGTTTGAA-3′ and antisense primer, 5′ CGGCTCTCGGCTGCTGCATT-3′ (bcl-xL 337 bp;bcl-xS 150 bp) (Greenlund et al., 1995);bad sense primer, 5′-CACTCCCTAGGCTTGAGGAA-3′ and antisense primer, 5′-TCCTGCTCACTCGGCTCAAA-3′ (209 bp); ERα sense primer, 5′-AATTCTGACAATCGACGCCAG-3′ and antisense primer, 5′-GTGCTTCAACATTCTCCCTCCTC-3′ (344bp) (Kuiper et al., 1997); and ERβ sense primer, 5′-TTCCCGGCAGCCCAGTAACC-3′ and antisense primer, 5′-TCCCTCTTTGCGTTGGACTA-3′ (262 bp) (Kuiper et al., 1997).
In situ hybridization studies
Brains were collected from female Sprague Dawley rats to examine the cellular localization of ERα and ERβ mRNA expression in normal and injured cerebral cortex. Brains were removed, frozen on dry ice, and stored at −80°C. The cortical distributions of ERβ mRNA in normal rats (n = 5) and ofERβ and ERα mRNA in ovariectomized, oil-treated rats that underwent ischemia (n = 3) were evaluated with in situ hybridization histochemistry as previously described (Shughrue et al., 1997). Briefly, coronal sections (20 μm) were sliced in a cryostat, mounted on gelatin-coated slides, and processed for in situ hybridization. They were hybridized with 200 μl of an antisense or sense (control)35S-labeled riboprobe (6 × 106 dpm per probe per slide) 50% formamide hybridization mixture containing a cocktail of two unique riboprobes for ERβ mRNA (ERβ 285 and ERβ 558) or one unique riboprobe for ERα mRNA (ERα 800) (Shughrue et al., 1997). The section-mounted slides were incubated overnight at 55°C in a humidified chamber, treated with RNase A, and stringently washed at 67°C in 0.1× SSC to remove nonspecific labeling. Slides were then dehydrated, apposed to x-ray film for 3 d, and dipped in NTB2 nuclear emulsion (Eastman Kodak, Rochester, NY). The slides were exposed for 4–8 weeks, photographically processed, stained with cresyl violet, and coverslipped. The studies described in this paper were reviewed and approved by the Radnor Animal Care and Use Committee (RACUC) at Wyeth-Ayerst Research.
In vivo binding studies
On postnatal day 21, female Sprague Dawley rats (n = 4) were ovariectomized. Seven days after surgery, rats were injected subcutaneously in the dorsal cervical region with 2 μg/kg BW of 17α-iodovinyl-11β- methoxyestradiol ([125I]estrogen; specific activity, 2200 Ci/mm) in 200 μl of vehicle (50% DMSO and 50% PBS). The [125I]estrogen was obtained from the iodination (NEN, custom iodination) of E-17α-tributylstannyvinyl-11β-methoxy estradiol (RAXL Enterprises, Newton, MA) as described previously (Hughes et al., 1997). The [125I]estrogen ligand has been characterized in vitro and in vivo for its specificity and affinity to estrogen receptors (Shughrue et al., 1999). Four hours after injection of [125I]estrogen, the brains were collected, frozen and 20 μm coronal sections were sliced in a cryostat and thaw-mounted onto gelatin-coated slides. Section-mounted slides were apposed to x-ray film (Eastman Kodak; BMR-1) for 48 hr and then post-fixed and processed before emulsion coating (Brown et al., 1995). Briefly, section-mounted slides were washed for 5 min in 4°C PM buffer (3 mm MgCl2, 1 mmKH2PO4, pH 6.8), post-fixed for 5 min in ice-cold 4% paraformaldehyde, washed 3 times for 5 min in 4°C PMTX buffer (PM buffer containing 0.1% Triton X-100), washed 2 times for 5 min in 4°C PM buffer, dipped in water, and allowed to dry at room temperature. The slides were then dipped in liquid nuclear emulsion (NTB-2; Eastman Kodak; diluted 1:1 with water), air-dried, and stored at 4°C in light-tight desiccator slide boxes. After 10–20 d of exposure, the slides were developed, stained with cresyl violet, and coverslipped.
Data analysis
All data are expressed as mean ± SE. To determine whether estradiol influenced the expression of genes in normal or injured brains, two-way mixed factorial (two treatment × two hemisphere) ANOVAs were performed. Significant interactions were probed using one-way ANOVAs. To determine whether ischemic values were different from sham values, two-way mixed factorial ANOVAs were performed, and interactions were probed with one-way ANOVAs. Differences were considered significant at p < 0.05.
RESULTS
Figure 1 shows representative TTC-stained brain sections from an ovariectomized oil-treated and an ovariectomized estradiol-treated rat that have undergone permanent cerebral ischemia. Physiological levels of estradiol protected against ischemic injury, an effect that was most pronounced in the cerebral cortex. The microdissected cortical regions were analyzed for changes in gene expression by semiquantitative RT-PCR. Figure2 is a composite of RT-PCR results showing representative mRNA expression patterns for control genes (L27A and NSE), bcl-2 family members (bcl-2, bax, bcl-xL, bcl-xS, and bad), and the estrogen receptors (ERα and ERβ).
Fig. 1.

Representative TTC-stained brain sections from an oil-treated (left) and an estradiol-treated (right) rat that underwent permanent cerebral ischemia. Infarcted tissue is white, whereas live tissue is darkly stained by TTC. An adjacent 1-mm-thick frozen coronal section was microdissected in anatomically equivalent regions on the ipsilateral and contralateral cortex from oil- and estradiol-treated, ischemic and sham (data not shown) rats, according to the method of Palkovits (1978). The microdissected regions, which are represented by holes, were analyzed for gene expression.
Fig. 2.

Composite of PCR results showing representative expression of control genes (L27A, NSE),bcl-2 family members (bcl-2, bax, bcl-xL, bcl-xS), and estrogen receptor subtypes (ERα and ERβ) in the ipsilateral and contralateral cortex of oil-treated (labeled O) and estradiol-treated (labeled E), ischemic and sham rats.
Baseline cellular expression among the experimental groups was determined by quantification of cellular markers, ribosomalL27A and NSE. Neither L27A norNSE gene expression changed with injury or estradiol treatment (Table 1), indicating that the numbers of live cells, particularly live neurons, were represented equally among the ischemic and sham, oil- and estradiol-treated, ipsilateral and contralateral experimental groups (n = 8–11 per group). Because neither L27A nor NSEgene expression changed with injury or estradiol treatment, data for each gene were normalized to the control L27A (Table2). Values shown in Table 2 indicate that estradiol treatment and/or ischemia selectively altered gene expression after the induction of injury. However, estradiol did not alter expression of any genes in sham animals. Statistical analyses of injury and sham values are indicated in Figures3-5.
Table 1.
Gene expression of cellular markers
| Injury | Sham | |||||||
|---|---|---|---|---|---|---|---|---|
| Ipsi | Contra | Ipsi | Contra | |||||
| Oil | E | Oil | E | Oil | E | Oil | E | |
| L27A | 1.28 ± 0.13 | 1.32 ± 0.09 | 1.29 ± 0.04 | 1.35 ± 0.05 | 1.39 ± 0.07 | 1.36 ± 0.05 | 1.31 ± 0.06 | 1.39 ± 0.10 |
| NSE | 1.22 ± 0.15 | 1.31 ± 0.06 | 1.33 ± 0.09 | 1.40 ± 0.06 | 1.38 ± 0.07 | 1.38 ± 0.06 | 1.37 ± 0.06 | 1.47 ± 0.06 |
Baseline levels of L27A and NSE mRNA were not affected by estradiol treatment or by ischemia (n = 8–11 per group), indicating that the overall number of live cells, including live neurons, were represented equally among the experimental groups. Data are expressed as arbitrary units of quantification (mean ± SE).
Table 2.
Gene expression of bcl-2 family members and ER subtypes
| Injury | Sham | |||||||
|---|---|---|---|---|---|---|---|---|
| Ipsi | Contra | Ipsi | Contra | |||||
| Oil | E | Oil | E | Oil | E | Oil | E | |
| bcl-2 | 0.62 ± 0.09 | 0.97 ± 0.11 | 1.21 ± 0.12 | 1.12 ± 0.08 | 0.99 ± 0.07 | 0.92 ± 0.04 | 1.06 ± 0.13 | 1.01 ± 0.16 |
| bax | 0.81 ± 0.12 | 0.87 ± 0.09 | 0.89 ± 0.10 | 0.86 ± 0.06 | 1.00 ± 0.10 | 0.92 ± 0.13 | 0.95 ± 0.07 | 0.91 ± 0.04 |
| bcl-xL | 0.17 ± 0.03 | 0.19 ± 0.02 | 0.20 ± 0.01 | 0.23 ± 0.02 | 0.22 ± 0.01 | 0.24 ± 0.03 | 0.22 ± 0.01 | 0.27 ± 0.03 |
| bcl-xS | 0.27 ± 0.05 | 0.30 ± 0.05 | 0.21 ± 0.05 | 0.20 ± 0.02 | 0.18 ± 0.02 | 0.20 ± 0.03 | 0.24 ± 0.09 | 0.18 ± 0.02 |
| bad | 0.73 ± 0.10 | 0.96 ± 0.07 | 1.06 ± 0.07 | 1.21 ± 0.04 | 1.06 ± 0.06 | 1.07 ± 0.05 | 1.07 ± 0.09 | 1.06 ± 0.06 |
| ERα | 2.52 ± 0.65 | 2.09 ± 0.42 | 0.48 ± 0.01 | 0.39 ± 0.04 | 0.42 ± 0.09 | 0.44 ± 0.08 | 0.49 ± 0.12 | 0.42 ± 0.06 |
| ERβ | 0.50 ± 0.17 | 0.97 ± 0.25 | 1.33 ± 0.25 | 1.16 ± 0.15 | 1.08 ± 0.15 | 0.88 ± 0.23 | 1.09 ± 0.10 | 0.88 ± 0.06 |
Gene expression of bcl-2 family members and estrogen receptor subtypes in sham and injured rat cortex (n = 7–11 per group) were normalized to the control L27A. After injury, the expression of specific genes was altered by estradiol and/or ischemia. However, estradiol did not alter the expression of any genes in sham animals. Statistical analyses of the injury and sham values are indicated in Figures 3-5. Data are represented as mean ± SE.
Fig. 3.

Estradiol-modulated bcl-2 gene expression in cerebral ischemia. In injury, estradiol (n = 11) significantly prevented the injury-induced downregulation of bcl-2 mRNA in the ipsilateral cortex, compared with oil-treated rats (n = 10) (*p < 0.02). In the absence of estradiol,bcl-2 gene expression declined significantly below constitutive bcl-2 expression (#p < 0.01). Injury values are graphed as a percentage of sham values. These results were repeated four times, using two independent experimental groups. Data are represented as mean ± SE.
Figure 3 shows bcl-2 gene expression on the ipsilateral and contralateral side of oil- and estradiol-treated rats that have undergone ischemia. Injury values are expressed as a percentage of sham values to reflect changes in injury with respect to normal expression. Two-way ANOVA revealed a significant interaction between treatment and hemisphere (F(1,19) = 5.70;p < 0.03). On the ipsilateral side of the brain, estradiol prevented the injury-induced downregulation ofbcl-2 (p < 0.02) (Fig. 3). In the absence of estradiol, bcl-2 expression declined to ∼60% of sham values in the injured hemisphere (p < 0.01). These results were repeated four times, using two independent experimental groups.
The action of estradiol was specific to bcl-2, as expression of other members of the bcl-2 family, bax (Fig.4A), bcl-xL (Fig.4B), bcl-xS (Fig.4C), and bad (Fig. 4D) was not affected by estrogen treatment. However, two-way ANOVA analysis indicated that the injury-induced expression ofbcl-xS was higher in the ipsilateral cortex, as compared with the contralateral cortex (F(1,11) = 8.82; p < 0.02) (Fig. 4C). In contrast to bcl-xL, the expression of bad in ischemia was lower in the ipsilateral cortex, as compared with the contralateral cortex (F(1,15) = 20.93; p < 0.01) (Fig. 4D). Althoughbcl-xS and bad exhibited laterality, these changes were not significantly different from sham controls.
Fig. 4.

Estradiol did not affect expression of otherbcl-2 family members, bax(A), bcl-xL(B), bcl-xS(C), or bad(D). bcl-xS gene expression was higher in the ipsilateral cortex, compared with the contralateral cortex (†p < 0.02). Levels ofbad expression were higher in the contralateral cortex, compared with the ipsilateral cortex (†p < 0.01). Data (n = 7–11 per experimental group) are graphed as a percentage of sham values. Data are represented as mean ± SE.
Figure 5 shows differential modulation of estrogen receptors in microdissected cortical regions after ischemia. In injury,ERα was dramatically upregulated in the ipsilateral cortex of oil- and estradiol-treated brains, as compared with the contralateral cortex (F(1,13) = 39.17;p < 0.001) (Fig. 5A). Two-way ANOVA analysis revealed an interaction between injury and hemisphere for oil- (F(1,12) = 6.78; p < 0.02) and estradiol-treated (F(1,13) = 24.04;p < 0.001) rats. The injury-induced upregulation ofERα was significantly higher than sham expression in both oil-treated (p < 0.02) and estradiol-treated (p < 0.01) rats. Estradiol did not influence the expression of ERα. These data were repeated four times, using two independent experimental groups.
Fig. 5.

Estrogen receptors were differentially modulated in ischemic injury. A, In injury, ERαmRNA was dramatically upregulated in the ipsilateral cortex of oil- and estradiol- treated rats, as compared with the contralateral cortex (†p < 0.001). The injury-induced upregulation ofERα gene expression was significantly higher than constitutive levels in both oil- (#p < 0.02) and estradiol-treated (#p < 0.01) rats.B, Estradiol prevented the injury-induced downregulation of ERβ mRNA in the ipsilateral cortex (*p < 0.01). In the absence of estradiol,ERβ expression in injury declined significantly below constitutive levels (#p < 0.01). Data (n = 7–11 per group) are graphed as a percentage of sham expression. These results were repeated four times, using two independent experimental groups. Data are represented as mean ± SE.
In contrast to ERα, estradiol influenced the expression ofERβ after injury (Fig. 5B). Two-way ANOVA revealed a significant interaction between treatment and hemisphere (F(1,14) = 5.05; p < 0.05). On the ipsilateral side of the brain, estradiol prevented the injury-induced downregulation of ERβ(p < 0.01) (Fig. 5B). In the absence of estradiol, ERβ expression declined to ∼50% of sham values in the injured hemisphere (p < 0.01). The estradiol-mediated modulation of ERβ expression is strikingly similar to the estradiol-mediated modulation ofbcl-2 expression. These results were repeated four times, using two independent experimental groups.
Figure 6 shows representative cellular mRNA expression of estrogen receptor subtypes after cerebral ischemia. The expression of ERα and ERβ was examined byin situ hybridization in the cerebral cortex of ovariectomized, oil-treated rats after injury and confirmed the differential modulation of ERα and ERβinduced by ischemia. In response to injury, the cellular expression ofERα mRNA was dramatically upregulated in the ipsilateral cortex (Fig. 6A), but was not detected in the contralateral cortex (Fig. 6B). In contrast toERα, the cellular expression of ERβ mRNA declined in the ipsilateral cortex after injury (Fig. 6C), but remained strong in the contralateral cortex (Fig.6D).
Fig. 6.
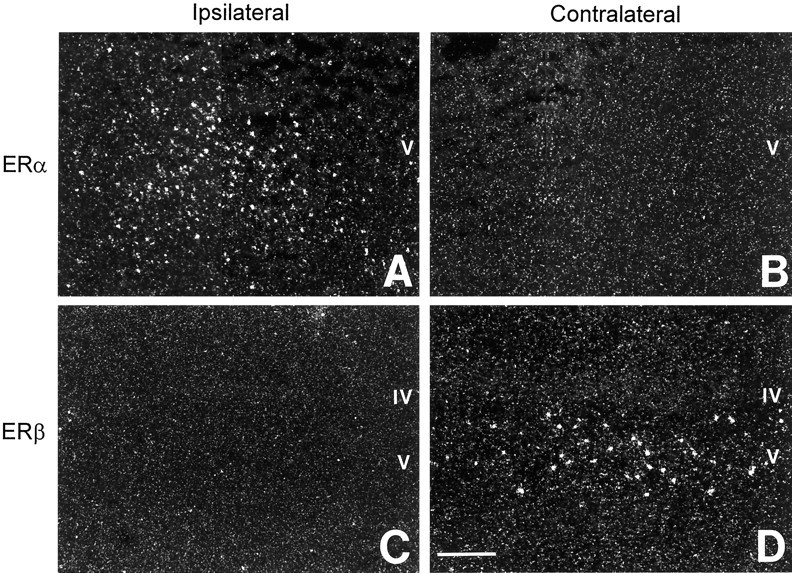
Representative photomicrographs showing differential modulation of ERα (A,B) and ERβ (C,D) mRNA by in situ hybridization in the cerebral cortex of ovariectomized, oil-treated rats after cerebral ischemia. In response to injury, the cellular expression ofERα was dramatically upregulated in ipsilateral cerebral cortex (A); ERα was not detected in contralateral cortex (B). In contrast to ERα, ERβ expression was downregulated in the ipsilateral cortex after injury (C), whereas its cellular expression remained strong in contralateral cortex (D). Scale bar, 300 μm.
The ratio of ERβ/ERα expression in the area of estradiol-mediated neuroprotection is shown in Figure7. Estradiol increased the ratio ofERβ/ERα in the ipsilateral, injured cortex. Because both estrogen receptors are differentially modulated in injury, a ratio of their relative levels may be crucial to understanding estradiol action in cerebral ischemia. It should be noted that Figure 7 reflects only relative, and not absolute, changes in the expression of ERs.
Fig. 7.

Estradiol increased the ratio ofERβ/ERα expression in the ipsilateral cortex of ischemic rat brains. The mean level of ERβ mRNA in the ipsilateral side of the ischemic cortex was divided by the mean level of ERα mRNA in the same region. Values for the ratio of ERβ/ERα expression in injury were obtained from the RT-PCR studies. This ratio represents relative changes ofERβ and ERα in ischemic injury.
ERβ mRNA and ERβ binding in the cerebral cortex are shown in Figure 8. The cellular expression of ERβ mRNA, examined by in situhybridization, demonstrated that numerous strongly labeled cells were scattered throughout laminae IV and V of the cerebral cortex (Fig.8A,B). The distribution ofERβ mRNA was similar to the localization of cells with a nuclear uptake and retention of [125I]estrogen in the cortex. Because ERα appears to be absent from the normal female cortex (Shughrue et al., 1997; Laflamme et al., 1998), these data indicate that [125I]estrogen is binding to ERβ in the cerebral cortex (Fig. 8C,D). The cortical region analyzed for ERβ mRNA by in situ hybridization and [125I]estrogen binding corresponds to the area where we observed estradiol-mediated neuroprotection and estradiol-mediated changes in gene expression in permanent cerebral ischemia.
Fig. 8.
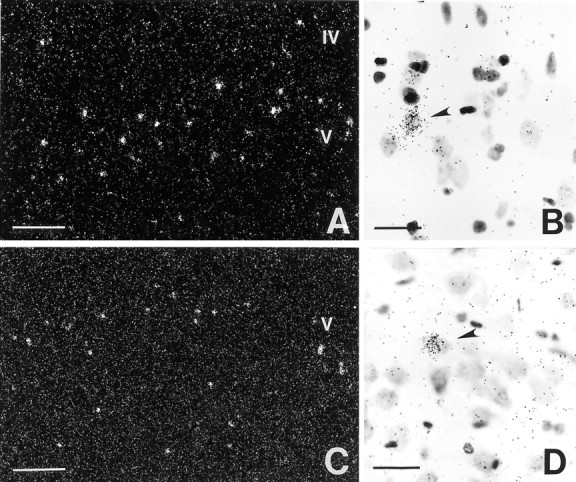
Representative photomicrographs ofERβ mRNA by in situ hybridization (A, B) and [125I]estrogen binding in the rat cerebral cortex at the level where estradiol-mediated neuroprotection was observed (C, D). A, The distribution of ERβ mRNA reveals strongly labeled cells throughout laminae IV and V of the isocortex. B, High-power magnification shows the high level of ERβexpression by certain neurons, whereas many other cells are unlabeled.C, A similar distribution of [125I]estrogen binding was also found in the cerebral cortex. D, High-power magnification shows neurons in lamina V with a nuclear uptake and retention of radiolabeled ligand. Scale bars: A, C, 200 μm;B, D, 40 μm.
DISCUSSION
This study demonstrates three important findings. First, physiological levels of estradiol, which protect the cerebral cortex against ischemic brain injury, correlate with changes in a specific member of the bcl-2 family. Second, estradiol and injury induce the differential expression of ERβ andERα, respectively, in the cerebral cortex. Third, we observed that the number and distribution of cells that bind [125I]estrogen in the cortex correspond to the distribution of ERβ mRNA.
Several potential mechanisms, genomic and nongenomic, may underlie the trophic and protective effects of estradiol. Our previous findings strongly suggest that estradiol-induced changes in gene expression may explain, in part, the protective actions of estradiol in our experimental model. When estradiol was administered at the time of injury, no protection was afforded; instead, pretreatment was required. In addition, the protective effect of estradiol in this model does not appear to involve changes in blood flow or glucose (Dubal et al., 1998). However, under some circumstances, estradiol may act through rapid, nongenomic actions, such as modulation of the NMDA(R) (Weaver et al., 1997) and reduction of lipid peroxidation (Goodman et al., 1996;Behl et al., 1997) to attenuate neural injury. Simpkins et al. (1997)reported that acute or delayed treatment with high doses of estradiol can decrease injury induced by in vivo cerebral ischemia, suggesting nongenomic mechanisms of estradiol action. Under other circumstances, estradiol may induce phosphorylation of one or more second messengers of transcription factors and thereby influence neuroprotection (Gu and Moss, 1996; Gu et al., 1996; Zhou et al., 1996;Murphy and Segal, 1997; Wang and Dow, 1998; Singer et al., 1999). To what extent estrogen receptors are involved and/or whether activation of transcription is required are unclear. It should be emphasized that estradiol may act by multiple mechanisms and that the predominant mechanisms may depend on multiple factors, including the dose of hormone or the type of injury. In general, at physiological levels, estradiol requires a period of pretreatment to exert neuroprotective effects (Green et al., 1996; Gridley et al., 1997; Dubal et al., 1998), suggesting that its physiological effects are mediated genomically through classic intracellular estrogen receptors and that transcription of hormone-responsive genes plays a critical role.
Bcl-2, a cell survival factor, has been identified as an estrogen-responsive gene in reproductive tissues (Teixeira et al., 1995; Kandouz et al., 1996; Huang et al., 1997). Estradiol may directly upregulate this survival factor through receptor-mediated interactions with regions of the bcl-2 promoter, which contains several putative estrogen-responsive sites, or by indirect pathways (Teixeira et al., 1995). Garcia-Segura et al. (1998) recently demonstrated that in the hypothalamus, Bcl-2 is elevated in estradiol-treated and estrous rats. Furthermore, Singer et al. (1998) reported that Bcl-2 is elevated with estradiol treatment in a neuronal cell line. We now report that, in injury, estradiol enhances the cortical expression ofbcl-2 when compared with oil-treated animals. Specifically, we report that estradiol prevents the ischemia-induced downregulation of bcl-2 mRNA. In ischemia, the loss of Bcl-2 is associated with exacerbated injury (Krajewski et al., 1995; Sato et al., 1998), whereas overexpression of this factor protects against injury induced by a variety of toxic stimuli (Martinou et al., 1994; Choi, 1996;Kitagawa et al., 1998; Yang et al., 1998). Thus, our finding that estradiol pretreatment prevents ischemia-induced downregulation ofbcl-2 gene expression strongly suggests that estradiol protects against cell death by affecting the balance between cell viability and apoptotic cell death. Interestingly, we did not observe constitutive regulation of bcl-2 in the cerebral cortex by estradiol, as sham and contralateral expression remained constant regardless of hormone treatment. Normal, constitutive regulation of Bcl-2 by estradiol in the arcuate nucleus of the hypothalamus (Garcia-Segura et al., 1998) and not in the cerebral cortex could reflect regional differences in estrogen receptor subtype and/or receptor densities in transynaptic pathways of communication, or in the cell types present, because estradiol is known to regulate the normal expression of genes in some areas of the brain and not in others (Stone et al., 1998).
Bcl-2 acts to counter cell death by inhibiting free radical production, suppressing caspase activation, regulating calcium sequestration (MacManus and Linnik, 1997), and/or by preventing the pro-apoptotic actions of Bax, Bcl-xS, or Bad, other members of the Bcl-2 family (Merry and Korsmeyer, 1997). In our studies, the effect of estradiol was specific to bcl-2, because expression of other members of the bcl-2 gene family (bax,bcl-xL,bcl-xS, and bad) were unaffected by hormone treatment. In contrast to a recent report (Pike, 1999), we did not detect any effects of estradiol on the expression ofbcl-xL. It is difficult to draw parallels between our studies because the former study used hippocampal cell cultures, a different injury paradigm, and did not examine the expression of bcl-2 or other bcl-2 family members. Nevertheless, it is possible that differential effects on various members of the bcl-2 family may depend on the dose of estradiol that is used, the stage in the evolution of ischemic injury that is examined, the mechanism of neural injury, and/or the region of the brain analyzed.
Estradiol influenced the cortical expression of the newly discovered estrogen receptor ERβ, and this effect was strikingly parallel to its effect on bcl-2 gene expression. That is, estradiol prevented the injury-induced downregulation ofERβ. The function of this novel estrogen receptor subtype is not clear. However, the discovery of ERβ in 1996 (Kuiper et al., 1996), and the subsequent localization of its mRNA in the regions where ERα is sparse or absent, including the cerebral cortex (Shughrue et al., 1997), suggest that estradiol acts through ERβ to improve cognitive function and decrease neurodegeneration associated with Alzheimer’s disease and stroke in hormone-replaced postmenopausal women. Indeed, ERβ is thought to mediate the protective actions of estradiol in non-neural tissue (Iafrati et al., 1997; Lindner et al., 1998). It is interesting to speculate that estradiol may act through ERβ to influence expression of the survival factor Bcl-2. However, further studies are necessary to definitively link Bcl-2 with ERβ-targeted gene expression.
Our finding that estrogen receptors are differentially modulated in the injured cerebral cortex suggests novel and unique roles for estrogen receptors in the injured brain. We were surprised to discover that the expression of ERα was dramatically upregulated after injury. Although estradiol did not influence the extent of this increase, the presence of elevated levels of ERα in the injured cortex may contribute to the ability of estradiol to protect. The increase in ERα expression is reminiscent of its expression during early postnatal development, during the interval of sex-specific differentiation of the cortex and extensive neurogenesis and neuritogenesis (Shughrue et al., 1990; Toran-Allerand et al., 1992). It is possible that the injury-induced upregulation ofERα is a component of a de-differentiation, recapitulation of this stage of development, and attempt to re-enter the cell cycle, which is hypothesized to occur in response to injury (Heintz, 1993). The repercussions of ERα upregulation in injury are not clear. However, in a preliminary study conducted in ERα knock-out mice, the absence of ERα did not affect the ability of estradiol to protect in stroke-related injury (Hurn et al., 1998), suggesting that ERα, alone, may not mediate the neuroprotective actions of estradiol.
It has been difficult to prove that ERβ mRNA is translated into protein because the quality and specificity of antibodies has been controversial. We demonstrate, for the first time, thatERβ mRNA is translated into a functional ERβ protein that is capable of binding ligand in regions of the cerebral cortex where we observed estradiol-induced protection against ischemic injury. These data may explain past observations of estrogen receptor binding in the cerebral cortex (Loy et al., 1988) because this binding is unaccounted for by normal ERα distribution (Shughrue et al., 1997; Laflamme et al., 1998). Our findings clearly demonstrate that functional ERβ receptors exist in the rat cerebral cortex and may, therefore, play an important role in the protective effects of estradiol.
Our results indicate a potentially intriguing role for ERβ in the ability of estradiol to protect against ischemic injury. We found that estradiol increases the ratio of ERβ/ERα in ischemia. The ratio of receptor subtype expression may be crucial to understanding how estradiol acts because each receptor can differentially activate certain response elements (Paech et al., 1997). The estradiol-mediated increase in the ERβ/ERα ratio suggests that ERβ-dependent signaling is linked with neuroprotection.
In summary, our results begin to elucidate potential mechanisms by which physiological levels of estradiol protect in cerebral ischemia. The striking parallelism between the protective effects of estradiol onbcl-2 and ERβ gene expression and our demonstration of functional ERβ binding suggest that estradiol decreases the extent of cell death by an estrogen receptor-β-mediated effect on Bcl-2.
Footnotes
This work was supported by the Glenn Foundation and an American Federation for Aging Research Scholarship (D.B.D.), and National Institutes of Health Training Grants AG00242 (D.B.D.), AG05818 (M.E.W.), and AG02224 (P.M.W.). We thank Dr. Steve Estus and his lab for insightful discussions and for invaluable assistance with gene expression methods.
Correspondence should be addressed to Dr. Phyllis M. Wise, Department of Physiology, University of Kentucky College of Medicine, 800 Rose Street, Lexington, KY 40536-0084.
REFERENCES
- 1.Alkayed NJ, Harukuni I, Kimes AS, London ED, Traystman RJ, Hurn PD. Gender-linked brain injury in experimental stroke. Stroke. 1998;29:159–166. doi: 10.1161/01.str.29.1.159. [DOI] [PubMed] [Google Scholar]
- 2.Bederson JB, Pitts LH, Germano SM, Nishimura MC, Davis RL, Bartkowski HM. Evaluation of 2, 3, 5-triphenyltetrazolium chloride as a stain for detection and quantification of experimental cerebral infarction in rats. Stroke. 1986;17:1304–1308. doi: 10.1161/01.str.17.6.1304. [DOI] [PubMed] [Google Scholar]
- 3.Behl C, Skutella T, Lezoualch F, Post A, Widmann M, Newton CJ, Holsboer F. Neuroprotection against oxidative stress by estrogens: structure-activity relationship. Mol Pharmacol. 1997;51:535–541. [PubMed] [Google Scholar]
- 4.Bredesen DE. Neural apoptosis. Ann Neurol. 1995;38:837–851. doi: 10.1002/ana.410380604. [DOI] [PubMed] [Google Scholar]
- 5.Brown TJ, Sharma M, Heisler LE, Karsan N, Walters MJ, MacLusky NJ. In vitro labeling of gonadal steroid hormone receptors in brain tissue sections. Steroids. 1995;60:726–737. doi: 10.1016/0039-128x(95)00107-2. [DOI] [PubMed] [Google Scholar]
- 6.Choi DW. Ischemia-induced neuronal apoptosis. Curr Opin Neurobiol. 1996;6:667–672. doi: 10.1016/s0959-4388(96)80101-2. [DOI] [PubMed] [Google Scholar]
- 7.Chomczynski P, Sacchi N. Single-step methods of RNA isolation by acid guanidinium thiocyanate-phenol-chloroform extraction. Anal Biochem. 1987;162:156–159. doi: 10.1006/abio.1987.9999. [DOI] [PubMed] [Google Scholar]
- 8.DePaolo LV, Shander D, Wise PM, Barraclough CA, Channing CP. Identification of inhibin-like activity in ovarian venous plasma of rats during the estrous cycle. Endocrinology. 1979;105:647–654. doi: 10.1210/endo-105-3-647. [DOI] [PubMed] [Google Scholar]
- 9.Dubal DB, Kashon ML, Pettigrew LC, Ren JM, Finklestein SP, Rau SW, Wise PM. Estradiol protects against ischemic injury. J Cereb Blood Flow Metab. 1998;18:1253–1258. doi: 10.1097/00004647-199811000-00012. [DOI] [PubMed] [Google Scholar]
- 10.Estus S. Optimization and validation of RT-PCR as a tool to analyze gene expression during apoptosis. In: Poirler J, editor. Apoptosis techniques and protocols. Humana; Totowa, NJ: 1996. pp. 67–84. [Google Scholar]
- 11.Estus S, Tucker HM, van Rooyen C, Wright S, Brigham EF, Wogulis M, Rydel RE. Aggregated amyloid-b protein induces cortical neuronal apoptosis and concomitant “apoptotic” pattern of gene induction. J Neurosci. 1997;17:7736–7745. doi: 10.1523/JNEUROSCI.17-20-07736.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Garcia-Segura LM, Cardona-Gomez P, Naftolin F, Chowen JA. Estradiol upregulates Bcl-2 expression in adult brain neurons. NeuroReport. 1998;9:593–597. doi: 10.1097/00001756-199803090-00006. [DOI] [PubMed] [Google Scholar]
- 13.Goodman Y, Bruce AJ, Cheng B, Mattson MP. Estrogens attenuate and corticosterone exacerbates excitotoxicity, oxidative injury, and amyloid β-peptide toxicity in hippocampal neurons. J Neurochem. 1996;66:1836–1844. doi: 10.1046/j.1471-4159.1996.66051836.x. [DOI] [PubMed] [Google Scholar]
- 14.Green SG, Gridley KE, Simpkins JW. Estradiol protects against β-amyloid (25–35)-induced toxicity in SK-N-SH human neuroblastoma cells. Neurosci Lett. 1996;218:165–168. doi: 10.1016/s0304-3940(96)13148-7. [DOI] [PubMed] [Google Scholar]
- 15.Green PS, Bishop J, Simpkins JW. 17α -Estradiol exerts neuroprotective effects on SK-N-SH cells. J Neurosci. 1997;17:511–515. doi: 10.1523/JNEUROSCI.17-02-00511.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Greenlund LJS, Korsmeyer SJ, Johnson EM., Jr Role of bcl-2 in the survival and function of developing and mature sympathetic neurons. Neuron. 1995;15:649–661. doi: 10.1016/0896-6273(95)90153-1. [DOI] [PubMed] [Google Scholar]
- 17.Gridley KE, Green PS, Simpkins JW. Low concentrations of estradiol reduce β-amyloid (25–35)-induced toxicity, lipid peroxidation and glucose utilization in human SK-N-SH neuroblastoma cells. Brain Res. 1997;778:158–165. doi: 10.1016/s0006-8993(97)01056-1. [DOI] [PubMed] [Google Scholar]
- 18.Gu G, Rojo AA, Zee MC, Yu J, Simerly RB. Hormonal regulation of CREB phosphorylation in the anteroventral periventricular nucleus. J Neurosci. 1996;16:3035–3044. doi: 10.1523/JNEUROSCI.16-09-03035.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gu Q, Moss RL. 17b-estradiol potentiates kainate-induced currents via activation of the cAMP cascade. J Neurosci. 1996;16:3620–3629. doi: 10.1523/JNEUROSCI.16-11-03620.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hall ED, Pazara KE, Linseman KL. Sex differences in postischemic neuronal necrosis in gerbils. J Cereb Blood Flow Metab. 1991;11:292–298. doi: 10.1038/jcbfm.1991.61. [DOI] [PubMed] [Google Scholar]
- 21.Heintz N. Cell death and the cell cycle: a relationship between transformation and neurodegeneration? Trends Biochem Sci. 1993;18:157–159. doi: 10.1016/0968-0004(93)90103-t. [DOI] [PubMed] [Google Scholar]
- 22.Henderson VW, Watt L, Buckwalter JG. Cognitive skills associated with estrogen replacement in women with Alzheimer’s disease. Psychoneuroendocrinology. 1996;21:421–430. doi: 10.1016/0306-4530(95)00060-7. [DOI] [PubMed] [Google Scholar]
- 23.Hoshimaru M, Ray J, Sah DWY, Gage FH. Differentiation of the immortalized adult neuronal progenitor cell line HC2S2 into neurons by regulatable suppression of the v-myc oncogene. Proc Natl Acad Sci USA. 1996;963:1518–1523. doi: 10.1073/pnas.93.4.1518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Huang Y, Ray S, Reed JC, Ibrado AM, Tang C, Nawabi A, Bhalla K. Estrogen increases intracellular p26Bcl-2 to p21Bax ratios and inhibits taxol-induced apoptosis of human breast cancer MCF-7 cells. Breast Cancer Res Treat. 1997;42:73–81. doi: 10.1023/a:1005777219997. [DOI] [PubMed] [Google Scholar]
- 25.Hughes A, Larson SM, Hanson RN, DeSombre ER. Uptake and interconversion of the Z and E isomers of 17α-iodovinyl-11β methoxyestradiol in the immature female rat. Steroids. 1997;62:244–252. doi: 10.1016/s0039-128x(96)00206-1. [DOI] [PubMed] [Google Scholar]
- 26.Hurn PD, Sampei K, Sawada M, Goto S, Crain BJ, Alkayed NJ, Korach KS, Traystman RJ, Demas GE, Nelson RJ. Stroke in mice deficient in classical estrogen receptors (ERKOα). Soc Neurosci Abstr. 1998;24:207.2. [Google Scholar]
- 27.Iafrati MD, Karas RH, Aronovitz M, Kim S, Sullivan TR, Lubahn DB, O’Donnell TF, Jr, Korach KS, Mendelsohn ME. Estrogen inhibits the vascular injury response in estrogen receptor α-deficient mice. Nat Med. 1997;3:545–548. doi: 10.1038/nm0597-545. [DOI] [PubMed] [Google Scholar]
- 28.Kandouz M, Siromachkova M, Jacob D, Marquet BC, Therwath A, Gompel A. Antagonism between estradiol and progestin on bcl-2 expression in breast-cancer cells. Int J Cancer. 1996;68:120–125. doi: 10.1002/(SICI)1097-0215(19960927)68:1<120::AID-IJC21>3.0.CO;2-E. [DOI] [PubMed] [Google Scholar]
- 29.Kawas C, Resnick S, Morrison A, Brookmeyer R, Corrada M, Zonderman A, Bacal C, Lingle D, Metter E. A prospective study of estrogen replacement therapy and the risk of developing Alzheimer’s disease: the Baltimore longitudinal study of aging. Neurology. 1997;48:1517–1521. doi: 10.1212/wnl.48.6.1517. [DOI] [PubMed] [Google Scholar]
- 30.Kitagawa K, Matsumoto M, Tsujimoto Y, Ohtsuki T, Kuwabara K, Matsushita K, Yang G, Tanabe H, Martinou J-C, Hori M, Yanagihara T. Amelioration of hippocampal neuronal damage after global ischemia by neuronal overexpression of BCL-2 in transgenic mice. Stroke. 1998;29:2616–2621. doi: 10.1161/01.str.29.12.2616. [DOI] [PubMed] [Google Scholar]
- 31.Krajewski S, Mai JK, Krajewska M, Sikorska M, Mossakowski MJ, Reed JC. Upregulation of bax protein levels in neurons following cerebral ischemia. J Neurosci. 1995;15:6364–6375. doi: 10.1523/JNEUROSCI.15-10-06364.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kuiper GGJM, Enmark E, Pelto-Huikko M, Nilsson S, Gustafsson J-A. Cloning of a novel estrogen receptor expressed in rat prostate and ovary. Proc Natl Acad Sci USA. 1996;93:1–6. doi: 10.1073/pnas.93.12.5925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kuiper GGJM, Carlsson B, Grandien K, Enmark E, Haggblad J, Nilsson S, Gustafsson J-A. Comparison of the ligand binding specificity and transcript distribution of estrogen receptors α and β. Endocrinol. 1997;138:863–870. doi: 10.1210/endo.138.3.4979. [DOI] [PubMed] [Google Scholar]
- 34.Laflamme N, Nappi RE, Drolet G, Labrie C, Rivest S. Expression and neuropeptidergic characterization of estrogen receptors (ERα and ERβ) throughout the rat brain: anatomical evidence of distinct roles for each subtype. J Neurobiol. 1998;36:357–378. doi: 10.1002/(sici)1097-4695(19980905)36:3<357::aid-neu5>3.0.co;2-v. [DOI] [PubMed] [Google Scholar]
- 35.Lindner V, Kim SK, Karas RH, Kuiper GGJM, Gustafsson J-A, Mendelsohn ME. Increased expression of estrogen receptor-β mRNA in male blood vessels after vascular injury. Circ Res. 1998;83:224–229. doi: 10.1161/01.res.83.2.224. [DOI] [PubMed] [Google Scholar]
- 36.Loy R, Gerlach JL, McEwen BS. Autoradiographic localization of estradiol-binding neurons in the rat hippocampal formation and entorhinal cortex. Brain Res. 1988;467:245–251. doi: 10.1016/0165-3806(88)90028-4. [DOI] [PubMed] [Google Scholar]
- 37.MacManus JP, Linnik MD. Gene expression induced by cerebral ischemia: an apoptotic perspective. J Cereb Blood Flow Metab. 1997;17:815–832. doi: 10.1038/aj.jcbfm.9590266. [DOI] [PubMed] [Google Scholar]
- 38.Martinou J-C, Dubois-Dauphin M, Staple JK, Rodriguez I, Frankowski H, Missotten M, Albertini P, Talabot D, Catsicas S, Pietra C, Huarte J. Overexpression of bcl-2 in transgenic mice protects neurons from naturally occurring cell death and experimental ischemia. Neuron. 1994;13:1017–1030. doi: 10.1016/0896-6273(94)90266-6. [DOI] [PubMed] [Google Scholar]
- 39.Merry DE, Korsmeyer SJ. Bcl-2 gene family in the nervous system. Annu Rev Neurosci. 1997;20:245–267. doi: 10.1146/annurev.neuro.20.1.245. [DOI] [PubMed] [Google Scholar]
- 40.Murphy DD, Segal M. Morphological plasticity of dendritic spines in central neurons is mediated by activation of cAMP response element binding protein. Proc Natl Acad Sci USA. 1997;94:1482–1487. doi: 10.1073/pnas.94.4.1482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Namura S, Zhu J, Fink K, Endres M, Srinivasan A, Tomaselli KJ, Yuan J, Moskowitz MA. Activation and cleavage of caspase-3 in apoptosis induced by cerebral ischemia. J Neurosci. 1998;18:3659–3668. doi: 10.1523/JNEUROSCI.18-10-03659.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Paech K, Webb P, Kuiper GG, Nilsson S, Gustafsson J, Kushner PJ, Scanlan TS. Differential ligand activation of estrogen receptors ERα and ERβ at AP1 sites. Science. 1997;277:1508–1510. doi: 10.1126/science.277.5331.1508. [DOI] [PubMed] [Google Scholar]
- 43.Paganini-Hill A. Estrogen replacement therapy and stroke. Prog Cardiovasc Dis. 1995;38:223–242. doi: 10.1016/s0033-0620(95)80014-x. [DOI] [PubMed] [Google Scholar]
- 44.Palkovits M. Topography of chemically identified neurons in the central nervous system: a review. Acta Morphol Acad Sci Hun. 1978;26:211–290. [PubMed] [Google Scholar]
- 45.Pike CJ. Estrogen modulates neuronal Bcl-xL expression and β-amyloid-induced apoptosis: relevance to Alzheimer’s disease. J Neurochem. 1999;72:1552–1563. doi: 10.1046/j.1471-4159.1999.721552.x. [DOI] [PubMed] [Google Scholar]
- 46.Sato S, Gobbel GT, Honkaniemi J, Li Y, Kondo T, Murakami K, Sato M, Copin J-C, Sharp FR, Chan PH. Decreased expression of bcl-2 and bcl-x mRNA coincides with apoptosis following intracerebral administration of 3-nitropropionic acid. Brain Res. 1998;808:56–64. doi: 10.1016/s0006-8993(98)00784-7. [DOI] [PubMed] [Google Scholar]
- 47.Schmidt R, Fazekas F, Reinhart B, Kapeller P, Fazekas G, Offenbacher H, Eber B, Schumacher M, Freidl W. Estrogen replacement therapy in older women: a neuropsychological and brain MRI study. J Amer Geriat Soc. 1996;44:1307–1313. doi: 10.1111/j.1532-5415.1996.tb01400.x. [DOI] [PubMed] [Google Scholar]
- 48.Shughrue PJ, Stumpf WE, MacLusky NJ, Zielinski JE, Hochberg RB. Developmental changes in estrogen receptors in mouse cerebral cortex between birth and postweaning: studied by autoradiography with 11β-methoxy-16α-[125I]Iodoestradiol. Endocrinol. 1990;126:1112–1124. doi: 10.1210/endo-126-2-1112. [DOI] [PubMed] [Google Scholar]
- 49.Shughrue PJ, Lane MV, Merchenthaler I. Comparative distribution of estrogen receptor-α and -β mRNA in the rat central nervous system. J Comp Neurol. 1997;388:507–525. doi: 10.1002/(sici)1096-9861(19971201)388:4<507::aid-cne1>3.0.co;2-6. [DOI] [PubMed] [Google Scholar]
- 50.Shughrue PJ, Lane MV, Merchenthaler I. Biologically active estrogen receptor-β: evidence from in vivo autoradiographic studies with estrogen receptor α-knockout mice. Endocrinology. 1999;140:2613–2620. doi: 10.1210/endo.140.6.6876. [DOI] [PubMed] [Google Scholar]
- 51.Simpkins JW, Rajakumar G, Zhang Y-Q, Simpkins CE, Greenwald D, Yu CJ, Bodor N, Day AL. Estrogens may reduce mortality and ischemic damage caused by middle cerebral artery occlusion in the female rat. J Neurosurg. 1997;87:724–730. doi: 10.3171/jns.1997.87.5.0724. [DOI] [PubMed] [Google Scholar]
- 52.Singer CA, Rogers KL, Strickland TM, Dorsa DM. Estrogen protects primary cortical neurons from glutamate toxicity. Neurosci Lett. 1996;212:13–16. doi: 10.1016/0304-3940(96)12760-9. [DOI] [PubMed] [Google Scholar]
- 53.Singer CA, Rogers KL, Dorsa DM. Modulation of Bcl-2 expression: a potential component of estrogen protection in NT2 neurons. NeuroReport. 1998;9:2565–2568. doi: 10.1097/00001756-199808030-00025. [DOI] [PubMed] [Google Scholar]
- 54.Singer CA, Figueroa-Masot XA, Batchelor RH, Dorsa DM. The mitogen-activated protein kinase pathway mediates estrogen neuroprotection after glutamate toxicity in primary cortical cultures. J Neurosci. 1999;19:2455–2463. doi: 10.1523/JNEUROSCI.19-07-02455.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Stone DJ, Song Y, Anderson CP, Krohn KK, Finch CE, Rozovsky I. Bidirectional transcriptional regulation of glial fibrillary acidic protein by estradiol in vivo and in vitro. Endocrinol. 1998;139:3202–3209. doi: 10.1210/endo.139.7.6084. [DOI] [PubMed] [Google Scholar]
- 56.Teixeira C, Reed JC, Pratt MAC. Estrogen promotes chemotherapeutic drug resistance by a mechanism involving bcl-2 proto-oncogene expression in human breast cancer cells. Cancer Res. 1995;55:3902–3907. [PubMed] [Google Scholar]
- 57.Toran-Allerand CD, Miranda RC, Hochberg RB, MacLusky NJ. Cellular variations in estrogen receptor mRNA translation in the developing brain: evidence from combined [125I]estrogen autoradiography and non-isotopic in situ hybridization histochemistry. Brain Res. 1992;576:25–41. doi: 10.1016/0006-8993(92)90606-a. [DOI] [PubMed] [Google Scholar]
- 58.Wang W, Dow KE. Quantitative analysis of mRNA expression of neuron-specific growth-associated genes in rat primary neurons by competitive PCR. Brain Res Protoc. 1998;2:199–208. doi: 10.1016/s1385-299x(97)00043-3. [DOI] [PubMed] [Google Scholar]
- 59.Weaver CE, Jr, Park-Chung M, Gibbs TT, Farb DH. 17-β-Estradiol protects against NMDA-induced excitotoxicity by direct inhibition of NMDA receptors. Brain Res. 1997;761:338–341. doi: 10.1016/s0006-8993(97)00449-6. [DOI] [PubMed] [Google Scholar]
- 60.Yang L, Matthews RT, Schulz JB, Klockgether T, Liao AW, Martinou J-C, Penney JB, Jr, Hyman BT, Beal MF. 1-methyl-4-phenyl-1,2,3,6-tetrahydropyride neurotoxicity is attenuated in mice overexpressing Bcl-2. J Neurosci. 1998;18:8145–8152. doi: 10.1523/JNEUROSCI.18-20-08145.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Zhou Y, Watters JJ, Dorsa DM. Estrogen rapidly induces the phosphorylation of the cAMP response element binding protein in rat brain. Endocrinol. 1996;137:2163–2166. doi: 10.1210/endo.137.5.8612562. [DOI] [PubMed] [Google Scholar]