

Abstract
Neuropeptide receptors couple via G-proteins to two principal signaling pathways that elevate cAMP through adenylate cyclase (AC) or mobilize intracellular Ca2+ through phospholipase C (PLC)-stimulated inositol phosphate (IP) turnover and production of inositol 1,4,5-trisphosphate (IP3). We showed previously that high-affinity receptors for pituitary adenylate cyclase-activating polypeptide (PACAP) are present on chick ciliary ganglion neurons and that receptor occupation increases cAMP production, resulting in enhanced acetylcholine sensitivity. After we suppressed AC activity and cAMP production with 2′-5′ dideoxyadenosine, however, PACAP no longer increased acetylcholine sensitivity but instead reduced it, suggesting that an AC-independent signal pathway activated by PACAP inhibits some nicotinic acetylcholine receptors (AChRs). We now use fast-perfusion, imaging, and biochemical methods to identify the AChRs modulated by PACAP and to characterize the signal pathway responsible for their inhibition. Without previous AC block, both the rapidly desensitizing, α-bungarotoxin (αBgt)-sensitive α7-AChRs and the slowly desensitizing, αBgt-insensitive α3*-AChRs on the neurons were potentiated by PACAP. After AC blockade, however, PACAP inhibited α7-AChRs but left α3*-AChRs unaffected. The selective inhibition of α7-AChRs appeared to use a PLC signaling pathway because it was not seen after lowering PLC activity or buffering intracellular Ca2+ and was mimicked by dialyzing neurons with an IP3 receptor agonist. PACAP also induced IP turnover and increased [Ca2+]i assessed directly with Fluo-3AM imaging. Given our previous findings that PACAP receptors couple to AC, the present results demonstrate a remarkable ability of a single neuropeptide to activate two signaling pathways and in so doing selectively regulate two classes of downstream ion channel targets.
Keywords: neuropeptide, acetylcholine, ion channel, modulation, fast perfusion, Ca2+ imaging, whole-cell recording
Neuropeptides serve as trophic factors, co-transmitters, and modulators of ion channel function. An important route of neuropeptide action involves binding to a cell-surface receptor that couples via a membrane-associated G-protein (e.g., Gs, Gq, Gi) to effector enzymes such as adenylate cyclase (AC) or phospholipase C (PLC) (Ross, 1989; Hille, 1992; Exton, 1996). The resulting changes in second messenger and kinase activities can then exert potent effects on downstream targets, including ion channels (Swope et al., 1993; Levitan, 1994). Our work has explored the regulation of nicotinic acetylcholine receptor (AChR) channels on chick ciliary ganglion neurons. We reported previously that AChR function is enhanced after intracellular cAMP is elevated by application of either cAMP analogs (Margiotta et al., 1987) or AC-stimulating neuropeptides [vasoactive intestinal peptide (VIP) or pituitary adenylate cyclase-activating polypeptide (PACAP)] that increase endogenous cAMP production via PACAP type I receptors (Gurantz et al., 1994; Margiotta and Pardi, 1995). To link the cAMP produced by PACAP to subsequent AChR modulation, neurons were pretreated with AC inhibitors (Margiotta and Pardi, 1995). The pretreatments blocked the ability of PACAP to increase cAMP but, quite surprisingly, reduced the subsequent peak ACh response below control levels. The results suggested that a second PACAP-activated intracellular pathway inhibits AChR function, and the present studies were undertaken to characterize this pathway and identify the AChRs that are affected.
Ciliary ganglion neurons express two AChR classes that differ in subunit composition, pharmacological, and electrophysiological properties. One class contains α7 subunits, which are recognized with high affinity by α-bungarotoxin (αBgt). Membrane currents generated by native α7-AChRs on ciliary or hippocampal neurons or by recombinant chick or rat α7 homo-oligomers expressed inXenopus oocytes activate and desensitize rapidly and are blocked by αBgt (Couturier et al., 1990; Alkondon and Albuquerque, 1993; Zhang et al., 1994). The other AChR class on ciliary ganglion neurons (α3*-AChRs) contains α3, β4, α5, and (sometimes) β2 subunits, but not α7 subunits, and is not recognized by αBgt (Vernallis et al., 1993; Conroy and Berg, 1995). Whole-cell currents generated by rapidly perfusing the neurons with 20 μmnicotine feature two distinct components. The large, initial component is mediated by α7-AChRs because it displays rapid onset and desensitization kinetics and is αBgt sensitive, whereas the smaller component is mediated by α3*-AChRs because it activates and desensitizes more slowly and is predominantly αBgt insensitive (Zhang et al., 1994; Blumenthal et al., 1999).
Evidence from other systems demonstrates that PACAP type I receptors can couple to both AC and PLC (Rawlings, 1994). PLC-dependent signaling would be expected to produce inositol phosphate (IP) turnover culminating in inositol 1,4,5-trisphosphate (IP3)-dependent mobilization of [Ca2+]i (Hille, 1992). Thus the observation that AC block unmasks an inhibition of ACh sensitivity suggests that AC and PLC signals normally interact to achieve balanced ACh sensitivity. The present findings confirm that PACAP type I receptors couple to both AC- and PLC-dependent pathways and demonstrate that the AC pathway produces PKA-dependent enhancement of both α3*- and α7-AChRs, whereas the PLC pathway produces a Ca2+-dependent inhibition of α7-AChRs.
A preliminary account of these results has been published previously (Pardi and Margiotta, 1996).
MATERIALS AND METHODS
Cell and substrate preparation. Neurons were dissociated from embryonic chick ciliary ganglia using collagenase A treatment (0.3 mg/ml for 20 min at 37°C) and mechanical trituration as described previously (Margiotta and Gurantz, 1989) and plated on glass or plastic substrates coated with poly-d-lysine. Embryonic day 13 (E13) or E14 neurons were used in most experiments because they can be isolated with >80% recovery and express high levels of both AChRs and PACAP type I receptors (Margiotta and Gurantz, 1989; Margiotta and Pardi, 1995). For electrophysiological and imaging experiments, the neurons were dissociated and plated at a density of one ganglion equivalent (∼3–4 × 103neurons) per glass coverslip (12 mm diameter) (Fisher Scientific, Houston, TX) in the following recording solution (in mm): 145.0 NaCl, 5.3 KCl, 5.4 CaCl2, 0.8 MgSO4, 5.6 glucose, and 5.0 HEPES, pH 7.4, containing 10% (v/v) heat-inactivated horse serum. Neurons attached to the substrate within 30 min and were maintained at 37°C for 1–2 hr before use. For IP turnover measurements, dissociated neurons from E12 embryos were plated on coated tissue culture wells (24 mm diameter; Falcon) at two to six ganglion equivalents per well and maintained at 37°C in 95% air/5% CO2 in culture medium containingmyo-[3H]inositol for 16–24 hr (see below). The culture medium consisted of MEM (no. 11090–08) supplemented with 100 U/ml penicillin, 100 μg/ml streptomycin, 2 mm glutamine, and 10% (vol/vol) heat-inactivated horse serum (all components from Life Technologies-BRL, Rockville, MD).
To coat glass coverslips, they were first acid-washed, then treated with poly-d-lysine in 0.13 m borate buffer, pH 8.5, washed four times with distilled water, and air-dried. For electrophysiological experiments, 70–150 kDa poly-d-lysine (P-0899; Sigma, St. Louis, MO) was applied to coverslips at 1 μg/ml for 1 min (21–23°C). With this minimal coating protocol, the neuron cell bodies became loosely attached, which allowed them to be lifted above the substrate for agonist exposure during fast perfusion experiments (see below). For Ca2+ imaging and IP turnover experiments, stronger neuron attachment was achieved by coating glass coverslips or tissue culture wells with ≥300 kDa poly-d-lysine (P-1024, Sigma) at 1 mg/ml for 12–16 hr (4°C).
Electrophysiology. Coverslips were placed in a lucite chamber (model RC-25F, Warner Instruments, Hamden, CT) on the stage of an Olympus CK2 inverted microscope, and the neurons were viewed with Hoffman modulation contrast optics using a 40× (0.5 NA) long working distance objective. The chamber volume was maintained at ∼200 μl and continuously perfused at ∼2 ml/min from one of two 30 ml syringes that contained recording solution or recording solution plus test reagent (e.g., αBgt, PACAP, VIP) at room temperature (21–23°C). The syringes were controlled with stopcocks and connected to the chamber via a perfusion manifold (Model MP-4, Warner Instruments). Patch recording pipettes were pulled from Corning 8161 glass tubing [1.5 mm outer diameter (o.d.)] and had tip impedances of 1–2 MΩ when filled with internal solution containing (in mm): 145.6 CsCl, 1.2 CaCl2, 2 EGTA, 15.4 glucose, 1 ATP, and 5 Na-HEPES, pH 7.3. Nicotine was dissolved in recording solution and applied by fast microperfusion (Zhang et al., 1994; Jonas, 1995) using solution streams delivered by laminar gravity flow from the channels of glass θ-tubing (1.6 mm o.d.; BT-150–10, Sutter Instruments, Novato, CA) pulled to an overall o.d. of ∼100–120 μm. The θ-tubing was mounted to a piezoelectric device (Burleigh Instruments, model LSS-3100) that provided rapid movement steps and was controlled by an amplifier/driver (Burleigh Instruments, model PZ-150M) and triggered by the recording software. To achieve fast perfusion, an “on-cell” seal with resistance ≥10 GΩ was first formed on a loosely attached neuron, which was then gently lifted off the substrate. A whole-cell configuration (Hamill et al., 1981) was then established, and the neuron was manipulated into the control recording solution stream flowing from the θ-tubing. After capacitance compensation and testing for sodium currents (see below), the stream bathing the neuron was rapidly switched to 20 μm nicotine for 3 sec and then switched back to recording solution. With use of these devices, junction current experiments with open tip patch pipettes containing 150 mm CsCl reveal that movement of the interface separating streams of 150 and 75 mm NaCl occurred in <1 msec.
Neuronal responses induced by fast microperfusion with nicotine were assessed in whole-cell mode at a holding potential (Vh) of −70 mV as described previously (Margiotta and Gurantz, 1989; Zhang et al., 1994). Membrane currents were collected with an Axopatch 1B amplifier (Axon Instruments, Burlingame, CA) and digitized using a TL-1 interface controlled by pClamp 6.0 software (Axon Instruments). For capacity compensation, series resistance measurement, and evaluation of sodium channels, the currents were filtered and digitized at 10 kHz. AChR currents were filtered at 1 kHz and digitized at 1–2 kHz. After achieving the whole-cell configuration, membrane capacitance compensation was achieved by eliminating the capacitive current transient in response to a −10 mV pulse using the amplifier series resistance (Rs) and capacitance (Cm) controls, thereby obtaining a measure of Rs. For most cells,Rs was measured both before and after fast perfusion with nicotine. Data from cells in which the averageRs exceeded 4 MΩ were not analyzed further. Because Rs was usually ≤3 MΩ, peak nicotine-induced currents (approximately −7000 pA) generated a voltage deviation (Vs) from the applied holding potential no larger than −21 mV. In such cases, the actual holding potential (Vh −Vs = −49 mV) was well below that for activating sodium currents (see below). Recordings were corrected forRs errors using one of two approaches, both of which gave similar results. For some recordings, 60–80% compensation was achieved with the patch-clamp controls, just before the nicotine trial. In other cases, current values were adjusted off-line for errors in holding potential as described previously (Margiotta and Gurantz, 1989), assuming an AChR reversal potential (Er) of −11 mV. Briefly stated, each measured current value was multiplied by a factor ω representing the ratio of the voltage gradient across the AChR under conditions of zero and finite series resistance voltage error (ω = (Vh −Er)/(Vh −Er − Vs)). Although the Rs correction allowed more accurate measurement of individual nicotine-induced whole-cell currents, the general conclusions presented here were not significantly changed when the results were reanalyzed without the correction. To test for adequate access, sodium currents were induced by applying a family of test depolarizations from −40 to +30 mV in 5 mV increments. Sodium current activation occurred in a voltage range from −25 to 0 mV. Cells in which sodium currents activated late or displayed discontinuities in their rising or falling phases were not studied further.
Whole-cell AChR currents induced by fast nicotine perfusion activated rapidly to a peak value (Ip) and then decayed with complex kinetics caused by the contribution of distinct AChR classes having different rates of desensitization (Zhang et al., 1994; Vijayaraghavan et al., 1995). The nicotine-induced current at time t (It) fromIp (t = 0) was well characterized by fitting to the data the sum of two to three exponential functions given by It =Af exp(−t/τf) +Ai exp(−t/τf) +As exp(−t/τf) +C using a Simplex algorithm included in pClamp.Af, Ai, andAs indicate the amplitudes of the fast, intermediate, and slow current components, τf, τi, and τs represent their respective decay time constants, and C represents the amplitude of the nondecaying current. The algorithm usually converged within 103 iterations and produced fits with associated errors <80 pA (typically <2% ofIp). Because we were most interested here in the fast component, its amplitude (If) was estimated both from the fit value (Af) and by subtracting the summed slower component amplitudes (Is =Ai + As + C) from the decaying peak current (Ip).If values presented here were obtained using the latter method and were within 10% of those obtained using the former approach. Component current values were normalized for differences in cell size by dividing Ip,If, and Is values by the membrane capacitance (Cm) determined from the patch-clamp controls. The change in agonist response for neurons in the test condition was determined by comparing normalized current amplitude values with neurons in control conditions from the same experiment. The statistical significance of any changes (p < 0.05) was determined using Student’s unpaired t test.
Inositol phosphate release assay. The coupling of type I PACAP receptors to IP turnover was assessed using an approach similar to that described by Rathouz et al. (1995). Neurons were incubated at 37°C in culture medium containing 2 μCi/mlmyo-[3H]inositol (NEN/DuPont, Wilmington, DE) (specific activity ∼19 mCi/mmol) for 16–24 hr, washed three times in MEM containing 50 mm LiCl, and equilibrated a final 20 min in the same buffer at 37°C. Test peptides were then added from frozen stocks to duplicate or quadruplicate wells, and the cells were incubated for 50–60 min at 37°C. In some cases neurons were preincubated for 20 min with indicated concentrations of AC or PLC inhibitor before peptide addition. Reactions were stopped by placing the cultures on ice, followed by addition of 0.5 vol ice-cold methanol. Cells were scraped from the wells, the lipids were extracted with chloroform and water (0.5:0.4, v/v), and an aliquot of the chloroform phase was taken to determine the extent of incorporation of label into IPs. The aqueous layer was transferred to a tube containing 300 mg of the anion exchange resin AG-1X8 (formate form), vortexed briefly, and incubated at room temperature for 20 min. After rapid centrifugation, the liquid phase was removed, and an aliquot was counted in a scintillation counter to determine the amount of label remaining as free inositol. The resin was rinsed three times with water and then incubated for 30 min with 1 ml of ice-cold 2 mammonium formate/0.1 m formic acid, the tubes were centrifuged briefly, and 10 ml of scintillation fluid was added to the eluate for counting. Specific counts from each individual sample well for a given condition were averaged. For each peptide dose, the change in total IPs was determined as the average of the counts in the presence of peptide minus the average of counts in the absence of peptide, and the data were fit by nonlinear regression using Prism version 2.0 (GraphPad Software, San Diego, CA).
Calcium imaging. After a 1 hr incubation at 37°C, cells were loaded with the calcium indicator dye Fluo-3AM (1 μm; Molecular Probes, Eugene, OR) (Kao et al., 1989) in recording solution (see description of solutions) for 45 min at room temperature in the dark. Cells were then washed three times with recording solution and maintained in darkness until use. Neurons were examined with reflected light fluorescence optics using an Olympus BX50 microscope equipped with a 40× water immersion objective (Uplan Fl 40×, 0.8 N.A.). Excitation light from a 100W mercury lamp was passed through a 450–480 nm bandpass filter, reflected from a 500 nm dichroic mirror, and focused through the objective onto the neurons. The emitted light passed through the objective and dichroic mirror and was observed using a standard 515 nm barrier filter set. The bath was continuously perfused by gravity flow at ∼2 ml/min, and solutions were switched manually between control and peptide tests just after the shutter was opened. In some cases, neurons were clamped in whole-cell mode using patch pipettes filled with intracellular solution containing 200 μm 2′-5′ dideoxyadenosine (ddA), with or without other test compounds. For each trial, a sequence of 12–15 eight-bit images covering time before, during, and after application of test solutions was captured using a cooled digital CCD camera (SENSYS; Photometrics, Tucson AZ) under the control of IP Lab software (v 3.0 Scanalytics, Reading, PA). For analysis and quantitation, the Fluo-3AM fluorescence signals were corrected for variability in dye loading and path length by normalizing to the baseline fluorescence (F0) as described previously (Cornell-Bell et al., 1990; Vijayaraghavan et al., 1992). The change in fluorescence intensity (ΔF = F −F0) relative to F0was determined from ellipsoid regions of interest (ROIs) overlaid on each neuron somata in a field (one to five neurons per field). As a criterion, neurons displaying ΔF/F0≥0.15 during the 30–90 sec of peptide application were scored as responsive. By this criterion, 85% of 67 neurons tested responded to application of 100 nm PACAP38 with an increase in [Ca2+]i. For other test conditions, the sample size required for the proportion of responsive neurons to be significantly different (p < 0.05) from 0.85 was estimated using the equation for comparing two independent proportions (Motulsky, 1995), with α = 0.05 and β = 0.2. Digitized image frames were manipulated for presentation using Photoshop (v 4.0.1; Adobe Systems, Inc.) after conversion to TIFF format.
Materials. Fertilized White Leghorn chicken eggs were obtained from Hertzfeld Poultry Farm (Waterville, OH) and maintained at 37°C in a forced-air draft incubator at 100% humidity. PACAP38, PACAP27, and VIP were obtained from the American Peptide Company (San Diego, Ca). Glucagon was obtained from Peninsula Laboratories (Belmont, CA). αBgt, ddA, d-myo-inositol 1,4,5,-trisphosphate, 3-deoxy-, hexasodium salt (HA-IP3), d-myo-inositol 1,4,5,-trisphosphate, 2,3,6-trideoxy-, hexasodium salt (LA-IP3), {N-[2-((p-bromocinnamyl)amino)ethyl]-5-isoquinolinesulfonamide, HCl} (H-89), Ruthenium Red, and {1-[6-((17β-3-methoxyestra-1,3,5(10)-trien-17-yl)amino)hexyl]-1H-pyrrole-2,5-dione} (U-73122) were all purchased from Calbiochem (La Jolla, CA). All other reagents were obtained from Sigma.
RESULTS
PACAP differentially regulates α7- and α3*-AChRs
We previously reported a dual action for PACAP on neuronal nicotinic AChR function (Margiotta and Pardi, 1995). Application of the peptide to ciliary ganglion neurons normally increased cAMP production and subsequent ACh sensitivity but led to reduced ACh sensitivity when the neurons were pretreated with AC inhibitors (ddA or SQ-5536) to limit cAMP production. To identify the AChR subtypes modulated by exposure to PACAP and to further characterize the relevant signaling mechanisms, we incorporated fast nicotine perfusion, a method that allows whole-cell currents activated by the two major classes of AChRs on the neurons to be distinguished on the basis of differences in component amplitudes and desensitization kinetics (Zhang et al., 1994;Blumenthal et al., 1999). Neurons pretreated with ddA displayed a large inward current in response to fast perfusion with 20 μmnicotine that activated to peak value (Ip) within 2–5 msec and subsequently desensitized with kinetics that could be described by the sum of two or three exponential functions (Fig.1A) (see Materials and Methods). The large-amplitude initial current (If) decayed rapidly (τf = 1–10 msec) and was accompanied by one to two smaller components that decayed more slowly (τi = 0.1–0.4 sec; τs = 0.4–12.0 sec). The fast current component of the nicotine response (If) is likely to represent activation of α7-AChRs. Exposure to αBgt, a toxin that specifically recognizes α7-AChRs on ciliary ganglion neurons and blocks their function, abolishedIf/Cm in all of the 13 neurons tested (Fig. 1B), reducingIp/Cm by >80%. In contrast, the slow current components are likely to primarily represent activation of α3*-AChRs becauseIs/Cm was not detectably different (p > 0.1) in 22 control and 9 αBgt-treated neurons (−60 ± 6 and −51 ± 5 pA/pF, respectively). In addition, the slow current desensitization kinetics (τi, τs) were not detectably changed after exposure to αBgt (p > 0.1 for each). Thus the whole-cell fast perfusion method permits identification of currents produced by activation of α7- and α3*-AChRs, the two major nicotinic receptor classes on chick ciliary ganglion neurons.
Fig. 1.

PACAP selectively inhibits α7-AChR currents when AC activity is suppressed. A–C, Representative whole-cell current records are displayed from separate E13 and E14 ciliary ganglion neurons held at −70 mV and exposed to 20 μm nicotine for ∼3 sec by fast perfusion, as indicated by the solid bar. All neurons were pretreated with 200 μm ddA for 1–7 min to inhibit AC as described in Materials and Methods. Arrows labeledIf and Isindicate peak values of the α7- and α3*-AChR current components, respectively. Calibration bars in C apply toA–C. A, Response to 20 μmnicotine application from a control neuron (Cm = 23 pF) features a rapidly activating current reaching a peak value (Ip) of −6121 pA and decaying with complex kinetics. The rapidly activating and desensitizing part of the response is shown on an expanded time scale in theinset, showing the superimposed fit to the equation in Materials and Methods (dots) and the associated values of τf and τi (τs shown above). Similar nicotine responses were obtained in the presence of 1 μm TTX and 10 μm CdCl2 (J. Margiotta, unpublished observations), indicating that they arise from nicotinic AChRs rather than from voltage-dependent Na+ or Ca2+ channels (Zhang et al., 1994). B, Nicotine response from a neuron (Cm = 28 pF) incubated with 60 nm αBgt beginning 2 hr before, and continuing throughout, the ddA pretreatment and recording (dashed bar). Note that the fast component is abolished by αBgt, indicating that it arises from α7-AChRs. C, Nicotine response from a neuron (Cm = 25 pF) treated with 100 nm PACAP38 for 6 min after the ddA pretreatment. Note that the PACAP38 treatment reduced the α7-AChR component (If), leaving the α3*-AChR component (Is) unaffected.D, Summary of the percentage change (mean ± SEM) in the α7-AChR (If/Cm) and α3*-AChR (Is/Cm) response components after treatment with neuropeptides. Each bar is based on nicotine responses like those in A andC where the individual fast and slow current components indicated by the black and gray bars, respectively, were determined as described above for the indicated peptide treatment (n = 6–19 for each concentration). Results are expressed as a percentage of the component nicotine responses obtained from the same number of control neurons tested in parallel that were treated with ddA alone. Thebars labeled none depict results from neurons treated with ddA alone, compared with untreated controls.Asterisks indicate a significant change (p < 0.05 by Student’s ttest) in the relevant current component associated with the indicated peptide treatments. Note that both PACAP38 and PACAP27 selectively reduced the α7-AChR component without affecting the α3*-AChR component.
Whole-cell nicotine response parameters obtained from neurons pretreated with ddA (200 μm, 2–5 min) (Table1) were indistinguishable from those obtained from untreated neurons tested in parallel (Fig.1D). When the ddA pretreatment was followed by treatment with PACAP38 (100 nm, 10 min) to maximally occupy PACAP type I receptors, however, the subsequent nicotine-induced α7-AChR current was reduced compared with control neurons pretreated with ddA alone (Fig. 1, compare A, C). The inhibition was specific for α7-AChRs becauseIf/Cm was significantly reduced, whereasIs/Cm representing α3*-AChRs was unchanged (Table 1). Percentage changes inIf/Cm for neurons treated with high- or low-affinity type I receptor ligands relative to ddA controls tested in parallel are compiled in Figure1D. PACAP38 and PACAP27 significantly reducedIf/Cm by similar amounts (55 ± 10% and 40 ± 9%, respectively;p < 0.05 for both), whereas neither peptide detectably changed the slower α3*-AChR current componentIs/Cm(p > 0.1 for both). No significant inhibition was observed for VIP, a low-affinity PACAP type I receptor ligand (IC50 ∼1 μm).If/Cm was unchanged in neurons treated with 100 nm VIP, and although 1 μm VIP produced an apparent 20% inhibition, the effect was not statistically significant (p > 0.1;n = 4 neurons). Neither of the VIP concentrations that was tested detectably alteredIs/Cm (Fig.1D). PACAP38 appeared to reduce τfsomewhat (Table 1); however, the kinetics of desensitization reflected in values of τf, τi, and τs were not significantly altered in neurons treated with either of the PACAPs when compared with control neurons tested in parallel (p > 0.05 for each; n= 4–17 treated and control neurons). These findings indicate that occupation of PACAP type I receptors by high-affinity ligands leads to selective inhibition of α7-AChRs and suggest that the inhibition does not involve a change in AChR desensitization.
Table 1.
Effect of PACAP38 on neuronal nicotinic response parameters
| Condition | Cm(pF) | If/Cm(pA/pF) | τf(msec) | Is/Cm(pA/pF) | τi (msec) | τs (sec) |
|---|---|---|---|---|---|---|
| ddA (all) | 30 ± 1 (43) | −198 ± 16 (43) | 9 ± 1 (43) | −81 ± 10 (43) | 173 ± 25 (29) | 1.4 ± 0.3 (37) |
| ddA + PACAP38 | 29 ± 1 (19) | −90 ± 13 (19)* | 6 ± 2 (19) | −63 ± 8 (19) | 153 ± 32 (16) | 1.1 ± 0.2 (17) |
Chick ciliary ganglion neurons were challenged with 20 μm nicotine delivered by fast microperfusion after treatments with ddA and PACAP38 as described in Materials and Methods and Figure 1. Nicotine-induced whole-cell currents were fit to the equation in Materials and Methods to determine the relative contributions of the peak α7-AChR currents (If) and α3*-AChR currents (Is) and their respective desensitization kinetics (τf, τi, and τs). Each peak current value shown was corrected for differences in cell size by normalizing to membrane capacitance (I/Cm). Values presented for each parameter are mean ± SEM with the number of neurons tested for each condition (n). The asterisk indicates that PACAP38 specifically reduced the peak α7-AChR response size (If/Cm; p< 0.002) without affecting its desensitization kinetics (τf; p > 0.05) or the α3*-AChR response size (Is/Cm) or desensitization properties (τi and τs;p > 0.1 for each). Note that not all neurons displayed both intermediate and slow decay kinetics.
A similar approach was used to examine the ability of PACAP to regulate α7- and α3*-AChR currents when the ddA pretreatment was omitted, allowing AC activity to remain intact and PACAP receptor activation to maximally stimulate cAMP production (Margiotta and Pardi, 1995). Under such conditions, and in accord with previous observations (Margiotta et al., 1987; Margiotta and Pardi, 1995), PACAP38 treatment (100 nm, 5 min) significantly increased both α7- and α3*-AChR current components of the subsequent response to 20 μm nicotine by approximately twofold (Fig.2). In addition to involving an increase in cAMP, the ability of PACAP to enhance both the α7- and α3*-AChR currents is likely to require downstream activation of protein kinase A (PKA), because 5 min preincubation with the PKA inhibitor H-89 (10 μm) blocked the increases in bothIf/Cm andIs/Cm. The upregulation of α3*- and α7-AChRs produced by PACAP in ciliary ganglion neurons is consistent with relatively slow peptide receptor activation of a cAMP- and PKA-dependent signaling cascade rather than with rapid AChR modulation expected from membrane-delimited direct G-protein interaction, as reported previously for rat intracardiac neurons (Cuevas and Adams, 1996). We nevertheless tested for rapid modulatory effects by comparing α3*- and α7-AChR responses induced by nicotine (20 μm) plus PACAP38 (100 nm) with those induced by nicotine alone. In such experiments, however, the α3*- and α7-AChR responses were not significantly different under the two conditions (Fig. 3, left). Negative results were also obtained after pretreating the neurons with ddA (200 μm, 10 min) to block AC (Fig. 3,right). Taken together, these findings indicate that PACAP receptor activation leads to differential regulation of α7- and α3*-AChRs, occurring over the course of minutes, that depends on the availability of the AC signaling pathway. PACAP enhances the function of both α7- and α3*-AChRs when AC is active, causing cAMP to increase and subsequently activate PKA but selectively inhibits α7-AChR function after AC activity is suppressed. We next focused on defining the latter, cAMP-independent signaling pathway activated by PACAP that leads to α7-AChR inhibition.
Fig. 2.
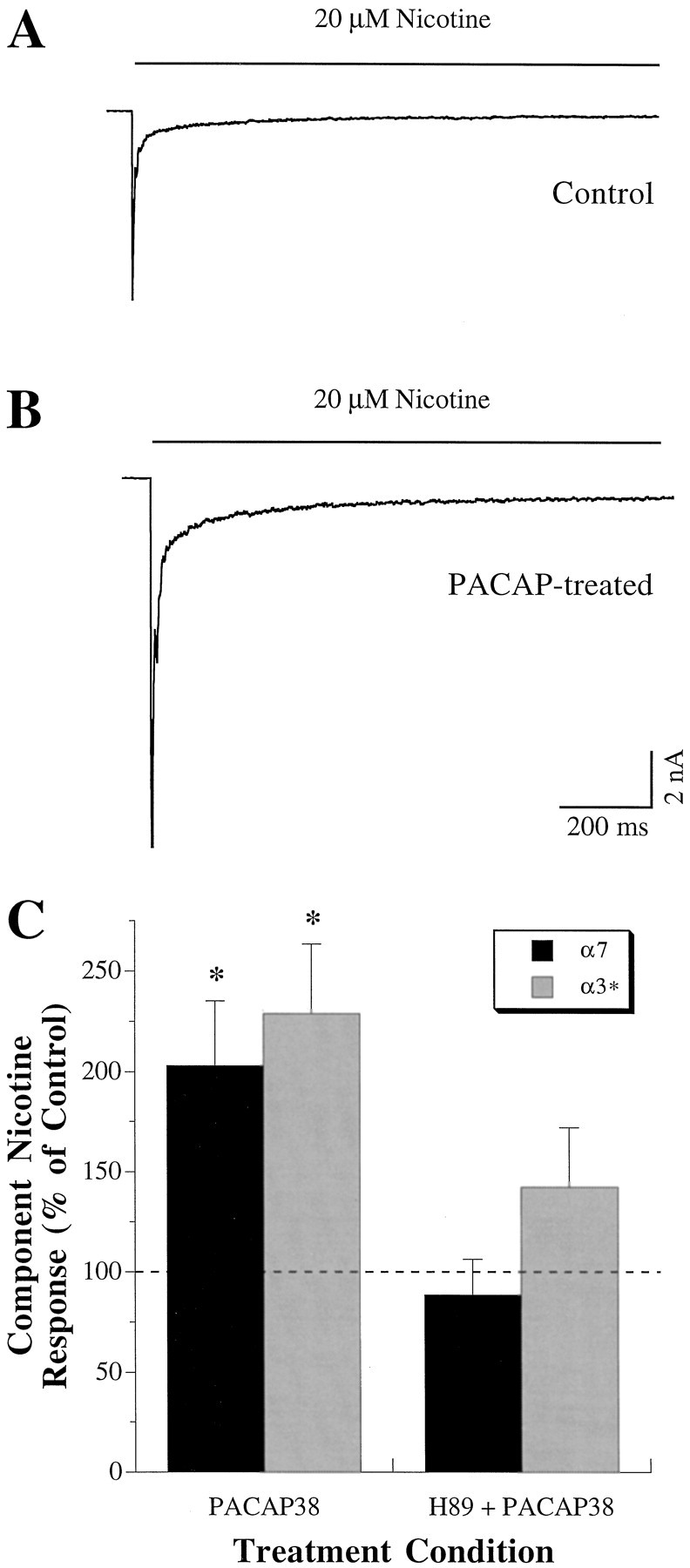
When AC activity is left intact, PACAP enhances α7- and α3*-AChR currents by a mechanism requiring PKA. Representative whole-cell currents induced by fast perfusion with 20 μm nicotine are shown from a control neuron (A) (Cm = 29 pF) and a neuron treated with 100 nm PACAP38 for 8 min before testing (B) (Cm = 27 pF). Neurons were held at −70 mV and not pretreated with ddA. Calibration bars in B apply to both records. The values of If/Cm(α7-AChRs) andIs/Cm(α3*-AChRs) were −174 and −40 pA/pF, respectively, for the neuron in A, and −288 and −133 pA/pF, respectively, for the neuron in B. C, Summary of α7- and α3*-AChR component responses to 20 μm nicotine (black and gray bars, respectively) after 10 min incubation in 100 nm PACAP38 (left). PACAP treatment enhanced both α7- and α3*-AChR currents by approximately twofold (asterisks abovebars indicate p < 0.02). When neurons were pretreated for 5 min with 10 μm H-89 to inhibit PKA activity (right), however, treatment with PACAP38 failed to change subsequent α7- or α3*-AChR components (p > 0.1). Results were obtained for eight neurons for each condition and are expressed as a percentage (mean ± SEM) of the α7- and α3*-AChR component nicotine responses obtained from control neurons (exposed to neither PACAP nor H-89) tested in parallel (dashed line).
Fig. 3.

Absence of any immediate AChR modulation by PACAP. α7- and α3*-AChR responses (black and gray bars, respectively) to 20 μm nicotine coapplied with 100 nm PACAP38 (Nic + PACAP) are compiled relative to responses from neurons tested in parallel that were challenged with 20 μm nicotine alone (dashed line). Under such conditions, PACAP38 failed to detectably alter the AChR responses (p > 0.1 for each bar) either when AC was left intact (−) or after AC inhibition (+) by 10 min pretreatment with 200 μm ddA (n = 7–10 neurons tested with Nic + PACAP or with Nic alone for each condition).
The ability of PACAP to inhibit α7-AChR function is Ca2+ dependent
A previous report suggested that free intracellular calcium [Ca2+]i might play a role in inhibiting α7-AChR function (Vijayaraghavan et al., 1995). We therefore examined the involvement of [Ca2+]i in PACAP38-mediated α7-AChR inhibition by including BAPTA in the patch pipette to buffer [Ca2+]i. The presence of BAPTA specifically abolished PACAP’s ability to subsequently inhibit α7-AChRs (Fig. 4). As in Figure 1 and Table 1, α7-AChR currents from ddA-pretreated/PACAP-treated neurons dialyzed with standard intracellular solution were attenuated compared with neurons not exposed to peptide. When ddA-pretreated neurons were dialyzed with intracellular solution containing 10 mm BAPTA and then treated with 100 nm PACAP38, however, subsequent α7-AChR responses were indistinguishable from those obtained from neurons not exposed to PACAP38. As reported previously (Zhang et al., 1994), including BAPTA in the recording pipette did not alter nicotine-evoked currents in control neurons not exposed to ddA or PACAP (n = 3 neurons) but did reduce Ca2+elevation in response to application of 20 μm nicotine or 60 mm KCl (data not shown). These findings indicate that the signaling pathway, recruited after PACAP receptor activation, increases free [Ca2+]i as a requirement for subsequent α7-AChR inhibition.
Fig. 4.

PACAP inhibits α7-AChRs by a mechanism involving [Ca2+]i mobilization. Ciliary ganglion neurons were pretreated with ddA (200 μm) for 2–7 min to block AC activity, subjected to the indicated treatments, and subsequently tested for nicotine responses as described in the legend for Figure 1. Control neurons received no PACAP38 treatment (unfilled bar) and displayed α7-AChR responses (If/Cm) of 217 ± 18 pA/pF (n = 6). PACAP38 treatment (100 nm, 10 min) inhibited α7-AChRs (black bar), reducingIf/Cm in this experiment by nearly 70% (*p < 0.001). When the same PACAP38 treatment was preceded by dialysis with 10 mm BAPTA, however, it failed to inhibit α7-AChRs (gray bar). Each bar represents the mean (±SEM) α7-AChR component nicotine response for each condition (n = 6–8 neurons), expressed as a percentage of that obtained from ddA controls from the same two experiments. α7-AChR responses obtained from three neurons dialyzed with BAPTA but not treated with PACAP (striped bar) were not detectably different from control neurons dialyzed with normal intracellular solution. α3*-AChR responses were not significantly changed by the treatments (p > 0.1; data not shown).
PACAP releases Ca2+ from an IP3-sensitive intracellular store
Many G-protein-coupled receptors are known to increase [Ca2+]i by stimulating PLC, thereby initiating IP turnover that culminates in the generation of IP3, a soluble intracellular messenger (Hille, 1992;Exton, 1996). IP3 can then bind to a receptor on the endoplasmic reticulum membrane, stimulating the release of calcium into the cytoplasm (Berridge, 1993, 1997). Because elevated [Ca2+]i was required for PACAP to inhibit α7-AChRs, the ability of PACAP38 and related peptides to raise [Ca2+]i was tested using imaging methods after loading the neurons with a calcium indicator dye (Fluo-3AM). In typical experiments, the whole-cell recording configuration was established on dye-loaded neurons using patch pipettes filled with intracellular solution containing ddA, and the bath then was perfused with recording solutions containing PACAP38 (Fig. 5A). Neurons loaded with Fluo-3AM before peptide exposure displayed a pale blue-green fluorescence associated with basal calcium levels. During bath perfusion with 100 nm PACAP38, the fluorescence intensity gradually increased in 85% of neurons tested (Fig. 5A, Table 2), reaching a plateau approximately 1.5-fold above basal levels within 30–90 sec. No change was detected in neurons exposed to recording solution lacking the peptide (data not shown). The increased fluorescence response to PACAP38 application was indicative of elevated [Ca2+]i because it was absent in most neurons dialyzed with 10 mm BAPTA to buffer [Ca2+]i. PACAP-dependent activation of AC was not a requirement for elevation of [Ca2+]i levels because the proportion of neurons responding to PACAP application was indistinguishable in neurons dialyzed with ddA (89%, n = 9) and in adjacent neurons not exposed to the inhibitor (84%, n = 58). In contrast, PACAP-dependent activation of PLC was required because the fraction of PACAP-responsive neurons was reduced to 25% when neurons were dialyzed with U-73122 to block PLC (Basille et al., 1995; Barnhart et al., 1997) and subsequent IP3 formation. PACAP-induced elevation of [Ca2+]i similar to that obtained in normal perfusion solution was observed in the absence of added Ca2+ (Fig. 5B), indicating that the [Ca2+]i arose from an intracellular store. To localize the source of the Ca2+ released by PACAP application, we examined the effects of IP3 or ryanodine receptor blockers applied to neurons via the patch pipette. Inclusion of heparin in the patch pipette to block IP3receptors (Ghosh et al., 1988; Rawlings et al., 1994) reduced the fraction of neurons responding to PACAP with a detectable increase in [Ca2+]i to 11%, whereas Ruthenium Red, a ryanodine receptor blocker (Kano et al., 1995), was without effect (Table 2). Thus consistent with its Ca2+-dependent ability to inhibit α7-AChRs, imaging experiments indicate that PACAP38 mobilizes [Ca2+]i from an IP3-sensitive store by using a PLC-dependent signaling pathway.
Fig. 5.

PACAP mobilizes intracellular Ca2+ stores. A, Application of PACAP38 elevates [Ca2+]i in both ddA-dialyzed and intact ciliary ganglion neurons. A1, Image obtained using bright-field optics that depicts a field of three neurons bathed in normal recording solution. A whole-cell recording has been established on the center neuron, causing its intracellular contents to be dialyzed with pipette solution containing 200 μm ddA to inhibit AC. A2,A3, The same field as in A1 showing images obtained using epifluorescence optics taken 0 and 90 sec after initiating bath perfusion with normal recording solution containing 100 nm PACAP38. Note the increased Fluo-3AM fluorescence in each of the three neurons. B, PACAP38 elevates [Ca2+]i in ciliary ganglion neurons bathed in recording solution lacking added Ca2+.B1, Bright-field optics. B2,B3, Epifluorescence images taken 0 and 90 sec after initiating perfusion with 0 Ca2+ recording solution containing 100 nm PACAP38. Scale bar (shown inB3 for all panels): 20 μm.
Table 2.
Properties of [Ca2+]imobilization induced by PACAP38
| Intracellular dialysis | Control (±ddA) | BAPTA | U-73122 | Heparin | Ruthenium Red |
|---|---|---|---|---|---|
| Detectable [Ca2+]i increase (% of neurons) | 85 | 29* | 25* | 11* | 86 |
| 95% confidence interval (±%) | 8 | 33 | 30 | 20 | 26 |
| n | 67 | 7 | 8 | 9 | 7 |
PACAP38-induced increases in [Ca2+]iwere detected using Fluo-3AM imaging as described in Materials and Methods and Figure 5. For each condition, the percentage of ciliary ganglion neurons (n = 7–67) responding to PACAP38 application (±95% confidence interval) with a detectable increase in Fluo-3AM fluorescence (∼15% above basal) is indicated. The percentage of responsive control neurons not subjected to dialysis was indistinguishable from that for control neurons dialyzed with 200 μm ddA (see Results), and results for both data sets are pooled (±ddA). Test neurons were dialyzed with intracellular solution containing BAPTA (10 mm), U-73122 (100 nm), heparin (300 μm), or Ruthenium Red (10 μm) for 1–2 min before bath perfusion with 100 nm PACAP38. Asterisks indicate a significant difference in the percentage of PACAP38-responsive neurons (p< 0.05) in the test populations compared with controls (±ddA).
PACAP increases inositol phosphate turnover via PLC
We directly tested the ability of PACAP38 and related peptides to activate the PLC-dependent signaling pathway in ciliary ganglion neurons using IP turnover assays (Fig.6). After 16–24 hr incubation with3H-inositol, exposure to PACAP38 or PACAP27 caused a dose-dependent release of 3H-IPs (Fig.6A). Maximal 3H-IP release (approximately two- to threefold above basal) was obtained with 100 nmPACAP38 or PACAP27, the same concentration that inhibited α7-AChR currents in the electrophysiological assays (Fig. 1, Table 1). Peptide potencies were determined from the concentrations required to produce 50% of maximum 3H-IPs release (EC50). EC50 values were 0.2 ± 0.1 nm(n = 5) for PACAP38 and 2.4 ± 1.1 nm(n = 3) for PACAP27. The 12-fold higher potency of PACAP38 over PACAP27 (p < 0.02) for stimulating IP turnover in ciliary ganglion neurons contrasts with their similar potencies for stimulating cAMP production in the neurons (EC50 = 0.5 nm and 1.1 nm, respectively (Margiotta and Pardi, 1995). Consistent with our electrophysiological findings (Fig. 1), 100 nm VIP failed to stimulate IP turnover and did so only marginally even when applied at 1 μm. Glucagon, a PACAP-related peptide with low affinity for the type I receptor, was ineffective even at 1 μm. A similar pharmacology indicative of PACAP specificity and differential coupling of PACAP38 and PACAP27 to AC and PLC transduction pathways is observed for native type I receptors on PC12 cells (Deutsch and Sun, 1992) and for cloned type I receptors expressed in LLC-PK1 cells (Spengler et al., 1993). Because of this ability to couple to two transduction cascades, it was important to determine the relative importance of the PLC and AC effectors for increasing IP production. When neurons were preincubated with U-73122 to inhibit PLC and then challenged with PACAP, there was a 50% reduction in the specific peptide-induced release of 3H-IPs (Fig. 6B). In contrast, preincubation with ddA to inhibit AC did not detectably alter basal or PACAP-induced release of3H-IPs. These findings demonstrate that PACAP38 increases IP turnover in ciliary ganglion neurons at the same concentration that inhibits α7-AChRs and increases [Ca2+]i levels. The similar pharmacological profile of both the increased IP turnover and [Ca2+]i levels indicates that PACAP type I receptors couple via PLC to increase IP turnover and Ca2+ release from an IP3-sensitive internal store.
Fig. 6.
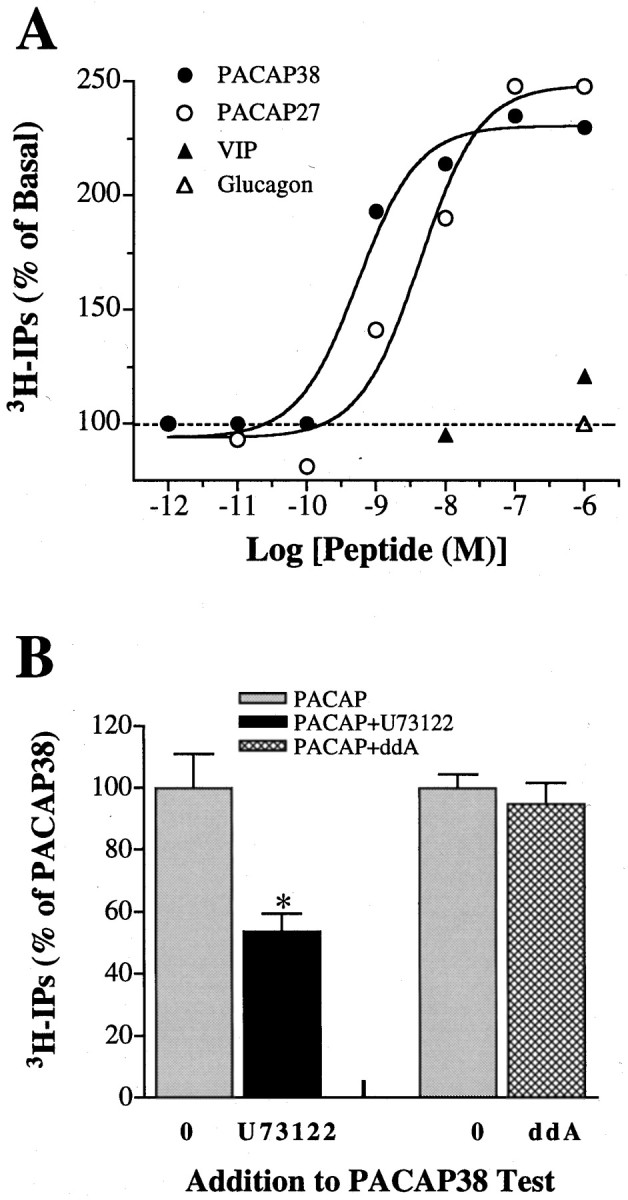
PACAP increases IP turnover via PLC. A, PACAP38 (●) and PACAP27 (○) stimulate IP production in a dose-dependent manner. Ciliary ganglion neurons were incubated overnight in media containing 2 μCi/mlmyo-[3H]inositol and then challenged with the indicated concentrations of PACAP for 40–60 min. Membrane lipids were then extracted, and 3H-IPs were measured as described in Materials and Methods. For each peptide concentration, the results are expressed as the percentage increase in3H-IPs for treated neurons above the basal levels obtained from untreated neurons. The assays were performed in duplicate, with each curve representing a single experiment, and the results were fitted to a sigmoidal dose–response curve (solid lines) by nonlinear regression (r2 > 0.97). For the PACAP38 and PACAP27 data depicted, EC50values representing the concentrations required for half-maximal IP production were 0.5 and 4.2 nm, and maximal peptide-induced3H-IP release above basal levels were 230 and 249%, respectively. Similar results were obtained in four other experiments with PACAP38 and in two experiments with PACAP27. VIP (▴) failed to increase 3H-IP release at 100 nm and induced only a nominal 20% increase (p < 0.1;n = 3) at 1 μm, whereas 1 μm Glucagon (▵) produced no detectable change (n = 2). B, PACAP38 couples via PLC signaling to stimulate IP turnover. IPs were measured from neuron cultures after preincubation withmyo-[3H]inositol and treatment with 1 μm PACAP38 as in A (gray bars) and compared with parallel cultures treated with 1 μm PACAP38 plus 200 μm ddA or 1 μm U-73122 to block AC or PLC (black andhatched bars), respectively. Results depicted were obtained from quadruplicate well assays from two to three experiments and indicate 3H-IP release (mean ± SEM) as a percentage of that obtained after treatment with PACAP38 alone. Note that U-73122 significantly reduced PACAP38-induced IP turnover (*p < 0.01), whereas ddA was ineffective (p > 0.1).
IP turnover-dependent Ca2+ release is required for α7-AChR inhibition
We next sought to determine whether activation of the PLC-dependent pathway producing IP turnover and [Ca2+]i release is required for the α7-AChR inhibition produced by PACAP. This was accomplished by dialyzing neurons with reagents that interfere with or mimic activation of the PLC signaling pathway via the patch pipette before and during exposure to PACAP38 (Fig. 7). To examine a requirement for PLC, neurons were pretreated with ddA, then dialyzed with intracellular solution containing the PLC inhibitor U-73122. Because it prevented the PACAP-induced increase in [Ca2+]i (Table 2), dialysis with 100 nm U-73122 also prevented PACAP38-induced inhibition of α7-AChRs. The PACAP-induced, PLC-dependent inhibition of α7-AChRs is likely to require increased IP turnover, leading to IP3production. Thus dialysis with a high-affinity IP3 receptor agonist (HA-IP3) mimicked the inhibition produced by PACAP38, whereas application of a weak IP3 agonist (LA-IP3) was ineffective in detectably changing α7-AChR currents. Further downstream, IP3-dependent Ca2+ mobilization is also important for inhibition of α7-AChRs. Inclusion of heparin in the patch pipette (300 μm) to block the IP3 receptor-mediated increase in [Ca2+]i (Table 2) also blocked the ability of PACAP38 to inhibit α7-AChRs whereas inclusion of Ruthenium Red was ineffective. These findings demonstrate that PACAP’s Ca2+-dependent inhibition of α7-AChRs involves PACAP type I receptor activation of PLC, IP turnover, and IP3-sensitive Ca2+ release.
Fig. 7.

PLC-dependent signaling is required for PACAP38-induced inhibition of α7-AChRs. Ciliary ganglion neurons were assayed for α7-AChR currents in response to fast perfusion with 20 μm nicotine as described in Materials and Methods and in the legend for Figure 1. Results are expressed as a percentage (mean ± SEM) of the α7-AChR component response (If/Cm) under the indicated test conditions, relative to that from ddA-pretreated control neurons tested in parallel that were not treated with PACAP38 (100%; dashed line). The PACAP38-induced reduction of α7-AChR responses (black bars;p < 0.02) was reversed by dialysis for 1–2 min with U-73122 (100 nm; p > 0.1) or heparin (300 μm; p > 0.1) before PACAP38 treatment (gray bars) but was unaffected by dialysis with Ruthenium Red (10 μm;p < 0.002). Results for each condition were based on measurements from six control neurons, six neurons treated with PACAP38 alone, and six neurons treated with PACAP38 plus (+) the indicated inhibitor. The PACAP38-induced inhibition α7-AChR responses was mimicked by dialysis with HA-IP3 but not by LA-IP3 (each at 2 μm; hatched and unfilled bars, respectively; n = 6 neurons for each). *Significant difference from control neurons tested in parallel (dashed line).
DISCUSSION
The preceding results elucidate dual signal cascades that differentially modulate AChRs on chick ciliary ganglion neurons (Fig.8). Previous experiments demonstrated that PACAP type I receptors couple via increased AC activity and cAMP production to enhance AChR function, but they also suggested a second signaling pathway, unmasked after blocking AC, that reduced AChR function (Margiotta and Pardi, 1995). The goals of the present study were to characterize the inhibitory pathway and determine the relevant AChRs affected. We now report that PACAP type I receptors on the neurons also couple to a parallel PLC-dependent signaling pathway involving IP turnover and IP3-stimulated Ca2+ release that inhibits α7-AChRs without detectably affecting α3*-AChRs. Supportive evidence for these conclusions emerges from three interdependent sets of experiments. First, electrophysiological studies demonstrated that PACAP reduced α7-AChR currents when AC was suppressed. Second, biochemical and Ca2+ imaging experiments revealed that PACAP activates the PLC pathway, eliciting PLC-dependent IP turnover, IP3 production, and Ca2+ release from an IP3-sensitive store. Third, pharmacological experiments demonstrated that reagents that interfere with the PLC cascade block the ability of PACAP to inhibit α7-AChRs.
Fig. 8.

PACAP type I receptors couple through AC- or PLC-dependent signaling cascades to differentially regulate α7- and α3*-AChR function. See Results for details.
Interpreting the electrophysiological results depended on assigning currents induced by fast nicotine perfusion to either α7- or α3*-AChRs. The appropriateness of such assignments has already been established (Zhang et al., 1994; Blumenthal et al., 1999) but warrants brief discussion. First, the fitted α7- and α3*-AChR current amplitudes (If andIs) and associated decay time constants (τf, τi, and τs) obtained in the present study (Fig. 1, Table1) are in good agreement with those previously reported for the same neurons (Zhang et al., 1994). Second, the initial fast α7-AChR component was independently identified becauseIf was selectively abolished by αBgt (Fig.1B). Third, the smaller, more slowly decaying current component (Is) was insensitive to αBgt and therefore unlikely to represent a contribution from α7-AChRs in significant numbers. After identifying If andIs as arising primarily from α7- and α3*-AChRs, respectively, we observed differential effects on the α7- or α3*-AChRs after PACAP receptor occupation that depended on the status of the AC signaling pathway. Because PACAP type I receptors activate two signal cascades in other systems (Rawlings, 1994), the selective inhibition of α7-AChRs (Fig. 1) was presumed to result from unmasked activation of another signal cascade, possibly one requiring PLC. This hypothesis was initially supported by observations that ddA blocks AC-dependent production of cAMP (Margiotta and Pardi, 1995) and that without ddA pretreatment, PACAP enhances α7- and α3*-AChR currents in a PKA-dependent manner (Fig. 2). Further support emerged by documenting a PLC-dependent pathway in the neurons required for PACAP-induced α7-AChR inhibition.
We showed previously that ciliary ganglion neurons express PACAP type I receptors that stimulate cAMP production (Margiotta and Pardi, 1995). The receptors have nanomolar affinities for PACAP38 and PACAP27 and 500- to 1000-fold lower affinity for VIP. The same ligands displayed potencies for stimulating cAMP production that paralleled their relative affinities. Studies with recombinant PACAP receptors have demonstrated that type I receptor isoforms, featuring seven membrane-spanning domains characteristic of G-protein-coupled receptors, can activate PLC-dependent (for review, see Spengler et al., 1993; Rawlings, 1994) as well as AC-dependent (Hashimoto et al., 1993;Pisegna and Wank, 1993) signal cascades. Other G-protein-coupled receptors such as those for calcitonin, parathyroid hormone, and thyrotropin are known to activate both AC and PLC cascades (for review, see Deutsch and Sun, 1992; Spengler et al., 1993). PACAP receptors on ciliary ganglion neurons also couple through PLC, because high-affinity PACAP receptor ligands induced [Ca2+]imobilization (Fig. 5) and IP turnover (Fig. 6) even with AC activity blocked. PACAP38, PACAP27, and VIP had potencies for IP production (EC50 = 0.2, 2.4, ≫1000 nm, respectively) that were comparable to those obtained previously for recombinant type I receptors expressed in cell lines (Spengler et al., 1993; Rawlings, 1994) and native PACAP type I receptors on PC12 cells (Deutsch and Sun, 1992). We presume that the lower potency of PACAP27 in stimulating IP production was not reflected in our electrophysiological assays using PACAP38 or PACAP27 because the 100 nm concentrations used produced maximal IP turnover in both cases.
In the present experiments, VIP (1 μm) did not significantly inhibit α7-AChRs (Fig. 1) or stimulate IP turnover (Fig. 6), although this concentration occupies ∼50% of the type I receptors on the neurons and stimulates substantial cAMP production (Margiotta and Pardi, 1995). The negligible potency of VIP in stimulating IP production is consistent with results from native and recombinant PACAP type I receptors cited above. In such cases, the low-affinity VIP binding to PACAP type I receptors might be inadequate to provide sufficient activation of PLC. Our previous finding that, after ddA pretreatment, 1 μm VIP reduced total ACh sensitivity by 50% (Margiotta and Pardi, 1995) contrasts with its nominal 20% inhibition of α7-AChRs seen here. The difference in results is likely to be quantitative and reflect both the different agonists and application methods used and the marginal ability of VIP to activate PLC. In accord with the ability of PACAP38 to activate a PLC cascade in ciliary ganglion neurons, application of the peptide also led to increased [Ca2+]i levels assessed by direct imaging (Table 2). The [Ca2+]i mobilization was verified as originating from an intracellular store because it persisted with no added Ca2+ in the extracellular solution. As expected for a response arising from IP turnover and subsequent IP3 production, the [Ca2+]i store was verified to be IP3 sensitive because heparin, a classic IP3receptor antagonist, blocked the response, whereas Ruthenium Red, a ryanodine receptor antagonist, did not. In addition, the ability of PACAP38 to increase IP turnover and [Ca2+]i levels in the neurons was specific to the PLC signal pathway because PLC inhibition reduced peptide-induced IP and Ca2+ production, whereas AC inhibition was ineffective (Figs. 5, 6). Taken together, the simplest scheme that can explain our findings is that PACAP receptors on ciliary ganglion neurons belong to a single type I class that can couple to both AC and PLC signaling pathways (Fig. 8).
If a PLC-dependent signal cascade were required for α7-AChR inhibition, then reagents applied via the patch pipette that perturb critical elements or products of the cascade should block the inhibition. All of the tests we performed satisfied this prediction, and support the proposed scheme depicted in Figure 8. First, reducing PLC activity with U-73122 blocked the ability of PACAP38 to not only raise [Ca2+]i but also to inhibit α7-AChRs. Second, the production of IP3 was critical because HA-IP3 mimicked the PACAP-induced inhibition of α7-AChRs, whereas the low-affinity IP3 receptor analog (LA-IP3) was without effect. Third, IP3-sensitive Ca2+ release was critical because dialysis with heparin blocked both PACAP-induced increase in [Ca2+]i and resultant inhibition of α7-AChRs. Last, the [Ca2+]i that is produced is required because chelating Ca2+ with application of BAPTA abolished PACAP’s subsequent ability to inhibit α7-AChRs (Fig. 4). Taken together, these results strongly support the hypothesis that the PLC pathway activated by PACAP inhibits α7-AChRs by a process requiring Ca2+ release from an IP3-dependent intracellular store.
How the PACAP-induced increase in [Ca2+]i might inhibit α7-AChR function is still an open question. A direct [Ca2+]i effect on α7-AChRs, however, such as that implicated for M-type K+ channels (Selyanko and Brown, 1996), seems unlikely. Although previous studies showed that increasing extracellular Ca2+ inhibits neuronal AChRs, the effect does not involve a concomitant increase in [Ca2+]i (Amador and Dani, 1995). In addition, elevating [Ca2+]i in neuron-like chromaffin cells by depolarization did not reduce subsequent nicotinic AChR currents (Khiroug et al., 1998). Thus explanations for the selective inhibition of α7-AChRs are likely to involve indirect Ca2+-dependent mechanisms. One possibility is that increased [Ca2+]ienhances the activity of phospholipase A2, a Ca2+-dependent enzyme (Kennedy, 1989; Mayer and Marshall, 1993), causing release of arachidonic acid (AA), which in turn inhibits α7-AChRs. Others have demonstrated that PACAP potentiates the release of glutamate-induced AA from cortical neurons (Stella and Magistretti, 1996). In addition AA (1 μm) inhibited both native α7-AChRs on ciliary ganglion neurons and recombinant α7-AChRs expressed in Xenopus oocytes (Vijayaraghavan et al., 1995). Interestingly, the AA effect on ciliary ganglion neurons was specific for α7-AChRs because high concentrations were required to detectably inhibit a3*-AChRs. Another possibility may involve activation of a Ca2+-dependent phosphatase. One candidate is calcineurin, which antagonizes the effects of elevations in cAMP (Kurosawa, 1994) by facilitating the dephosphorylation of proteins phosphorylated by PKA (Yakel, 1997). Because α7- and α3*-AChRs are likely to be substrates for PKA-dependent phosphorylation (Vijayaraghavan et al., 1990; Moss et al., 1996) and their upregulation by cAMP is dependent on PKA activity (Fig. 2), α7-AChR inhibition could involve selective dephosphorylation by a Ca2+-dependent phosphatase.
Under normal conditions when PACAP type I receptors can couple to both PLC and AC, cAMP production is increased in the neurons, and both α7- and α3*-AChR currents are enhanced by a process requiring PKA (Figs.2, 8). Whether the modulation is explained by PKA-dependent phosphorylation of AChRs alone, however, or requires additional intermediary steps and/or phosphorylation events still remains to be determined. Because PACAP38 is approximately equipotent for stimulating cAMP production and IP turnover in the neurons, the bias toward AChR enhancement via AC is not likely to involve second messenger production but instead may result from different abilities of the activated cascades to influence their downstream targets. Overall, the results demonstrate a remarkable flexibility in ciliary ganglion neurons for differentially modulating two AChR classes. At one level, the findings are consistent with PACAP’s PLC-dependent dampening of α7-AChR responses being a safety mechanism to counteract the α7- and α3*-AChR enhancement produced by concomitant AC activity. Other scenarios are also possible, however. For example, more selective inhibition of α7-AChRs could be achieved if the G-proteins presumed to couple PACAP type I receptors to their AC and PLC cascades were localized at α3*- versus α7-AChR synaptic sites, respectively, or if receptors for other neuropeptides were present on the neurons that couple specifically with the PLC cascade. Future efforts will be directed toward answering such questions.
Footnotes
This work was supported by National Institutes of Health Grant NS24417 to J.F.M. Special thanks go to Dr. Diomedes Logothetis for support and advice. We also thank Min Chen for expert technical assistance, and Drs. Joan Brown, Kathleen Dunlap, Marthe Howard, Victor May, and Phyllis Pugh for helpful discussions.
Correspondence should be addressed to Dr. Joseph Margiotta, Department of Anatomy and Neurobiology, Medical College of Ohio, 3035 Arlington Avenue, Toledo, OH 43614-5804.
Dr. Pardi’s present address: Department of Medicine, Mount Sinai School of Medicine, 1 Gustave Levy Place, New York, NY 10029.
REFERENCES
- 1.Alkondon M, Albuquerque EX. Diversity of nicotinic acetylcholine receptors in rat hippocampal neurons. I. Pharmacological and functional evidence for distinct structural subtypes. J Pharmacol Exp Ther. 1993;265:1455–1473. [PubMed] [Google Scholar]
- 2.Amador M, Dani JA. Mechanism for modulation of nicotinic acetylcholine receptors that can influence synaptic transmission. J Neurosci. 1995;15:4525–4532. doi: 10.1523/JNEUROSCI.15-06-04525.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Barnhart DC, Sarosi GA, Mullholland MW. PACAP-38 causes phospholipase C-dependent calcium signaling in rat acinar cell line. Surgery. 1997;122:465–475. doi: 10.1016/s0039-6060(97)90040-4. [DOI] [PubMed] [Google Scholar]
- 4.Basille M, Gonzalez BJ, Desrues L, Demas M, Fournier A, Vaudry H. Pituitary adenylate cyclase-activating polypeptide (PACAP) stimulates adenylate cyclase and PLC activity in rat cerebellar neuroblasts. J Neurochem. 1995;65:1318–1324. doi: 10.1046/j.1471-4159.1995.65031318.x. [DOI] [PubMed] [Google Scholar]
- 5.Berridge MJ. Inositol triphosphate and calcium signaling. Nature. 1993;361:315–326. doi: 10.1038/361315a0. [DOI] [PubMed] [Google Scholar]
- 6.Berridge MJ. Elementary and global aspects of calcium signaling. J Physiol (Lond) 1997;499:291–306. doi: 10.1113/jphysiol.1997.sp021927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Blumenthal EM, Shoop RD, Berg DK. Developmental changes in the nicotinic responses of ciliary ganglion neurons. J Neurophysiol. 1999;81:111–120. doi: 10.1152/jn.1999.81.1.111. [DOI] [PubMed] [Google Scholar]
- 8.Conroy WG, Berg DK. Neurons can maintain multiple classes of nicotinic acetylcholine receptors distinguished by different subunit compositions. J Biol Chem. 1995;270:4424–4431. doi: 10.1074/jbc.270.9.4424. [DOI] [PubMed] [Google Scholar]
- 9.Cornell-Bell A, Finkbeiner S, Cooper M, Smith S. Glutamate induces calcium waves in cultured astrocytes: long-range glial signaling. Science. 1990;247:470–473. doi: 10.1126/science.1967852. [DOI] [PubMed] [Google Scholar]
- 10.Couturier S, Bertrand D, Matter J-M, Hernandez M-C, Bertrand S, Millar N, Valera S, Barkas T, Ballivet M. A neuronal nicotinic acetylcholine receptor subunit (α7) is developmentally regulated and forms a homo-oligomeric channel blocked by α-Btx. Neuron. 1990;5:847–496. doi: 10.1016/0896-6273(90)90344-f. [DOI] [PubMed] [Google Scholar]
- 11.Cuevas J, Adams D. VIP modulation of nAChR channels in rat intracardiac neurons. J Physiol (Lond) 1996;493:503–515. doi: 10.1113/jphysiol.1996.sp021399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Deutsch PJ, Sun Y. The 38-amino acid form of pituitary adenylate cyclase-activating polypeptide stimulates dual signaling cascades in PC12 cells and promotes neurite outgrowth. J Biol Chem. 1992;267:5108–5113. [PubMed] [Google Scholar]
- 13.Exton JH. Regulation of phosphoinositide phospholipases by hormones, neurotransmitters, and other agonists linked to G-proteins. Annu Rev Pharmacol Toxicol. 1996;36:481–509. doi: 10.1146/annurev.pa.36.040196.002405. [DOI] [PubMed] [Google Scholar]
- 14.Ghosh TK, Eis PS, Mullaney JM, Ebert CL, Gill DL. Competitive, reversible and potent antagonism of inositol 1,4,5-triphosphate-activated calcium release by heparin. J Biol Chem. 1988;263:11075–11079. [PubMed] [Google Scholar]
- 15.Gurantz D, Harootunian AT, Tsien RY, Dionne VE, Margiotta JF. VIP modulates neuronal nicotinic acetylcholine receptor function by a cyclic AMP-dependent mechanism. J Neurosci. 1994;14:3540–3547. doi: 10.1523/JNEUROSCI.14-06-03540.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hamill OP, Marty A, Neher E, Sakmann B, Sigworth FJ. Improved patch-clamp techniques for high-resolution current recording from cells and cell-free membrane patches. Pflügers Arch. 1981;391:85–100. doi: 10.1007/BF00656997. [DOI] [PubMed] [Google Scholar]
- 17.Hashimoto H, Ishihara T, Shigemoto R, Mori K, Nagata S. Molecular cloning and tissue distribution of a receptor for pituitary adenylate cyclase-activating polypeptide. Neuron. 1993;11:333–342. doi: 10.1016/0896-6273(93)90188-w. [DOI] [PubMed] [Google Scholar]
- 18.Hille B. G protein-coupled mechanisms and nervous signaling. Neuron. 1992;9:187–195. doi: 10.1016/0896-6273(92)90158-a. [DOI] [PubMed] [Google Scholar]
- 19.Jonas P. Fast application of agonists to isolated membrane patches. In: Sakmann B, Necher E, editors. Single channel recording. Plenum; New York: 1995. [Google Scholar]
- 20.Kano M, Garaschuk O, Verkhratsky A, Konnerth A. Ryanodine receptor-mediated intracellular calcium release in rat cerebellar Purkinje neurons. J Physiol (Lond) 1995;487.1:1–16. doi: 10.1113/jphysiol.1995.sp020857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kao JPY, Harootunian AT, Tsien RY. Photochemically generated cytosolic calcium pulses and their detection by Fluo-3. J Biochem. 1989;264:8179–8184. [PubMed] [Google Scholar]
- 22.Kennedy M. Regulation of neuronal function by calcium. Trends Neurosci. 1989;112:417–420. doi: 10.1016/0166-2236(89)90089-1. [DOI] [PubMed] [Google Scholar]
- 23.Khiroug L, Sokolova E, Giniatullin R, Afzalov R, Nistri A. Recovery from desensitization of neuronal nicotinic acetylcholine receptors of rat chromaffin cells is modulated by intracellular calcium through distinct second messengers. J Neurosci. 1998;18:2458–2466. doi: 10.1523/JNEUROSCI.18-07-02458.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kurosawa M. Phosphorylation and dephosphorylation of proteins in regulating cellular function. J Pharmacol Toxicol Methods. 1994;31:135–139. doi: 10.1016/1056-8719(94)90075-2. [DOI] [PubMed] [Google Scholar]
- 25.Levitan IB. Modulation of ion channels by protein phosphorylation and dephosphorylation. Annu Rev Physiol. 1994;56:193–212. doi: 10.1146/annurev.ph.56.030194.001205. [DOI] [PubMed] [Google Scholar]
- 26.Margiotta JF, Gurantz D. Changes in the number, function and regulation of nicotinic acetylcholine receptors during neuronal development. Dev Biol. 1989;135:326–399. doi: 10.1016/0012-1606(89)90183-8. [DOI] [PubMed] [Google Scholar]
- 27.Margiotta JF, Pardi D. Pituitary adenylate cyclase-activating polypeptide type I receptors mediate cyclic AMP-dependent enhancement of neuronal acetylcholine sensitivity. Mol Pharmacol. 1995;48:63–71. [PubMed] [Google Scholar]
- 28.Margiotta JF, Berg DK, Dionne VE. Cyclic AMP regulates the proportion of functional acetylcholine receptors on chick ciliary ganglion neurons. Proc Natl Acad Sci USA. 1987;84:8155–8159. doi: 10.1073/pnas.84.22.8155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mayer RJ, Marshall LA. New insights on mammalian phospholipase A2: comparison of arachidonoyl-selective and nonselective enzymes. FASEB J. 1993;7:339–348. doi: 10.1096/fasebj.7.2.8440410. [DOI] [PubMed] [Google Scholar]
- 30.Moss SJ, McDonald BJ, Rudhard Y, Schoepfer R. Phosphorylation of the predicted major intracellular domains of the rat and chick nicotinic a7 subunit by cAMP-dependent protein kinase. Neuropharmacology. 1996;35:1023–1028. doi: 10.1016/s0028-3908(96)00083-4. [DOI] [PubMed] [Google Scholar]
- 31.Motulsky H. Intuitive Biostatistics. Oxford; New York: 1995. [Google Scholar]
- 32.Pardi D, Margiotta JF. PACAP receptor activation inhibits α7-containing neuronal acetylcholine receptors. Soc Neurosci Abstr. 1996;22:1522. [Google Scholar]
- 33.Pisegna JR, Wank SA. Molecular cloning and functional expression of the pituitary adenylate cyclase-activating polypeptide type I receptor. Proc Natl Acad Sci USA. 1993;90:6345–6349. doi: 10.1073/pnas.90.13.6345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Rathouz MM, Vijayaraghavan S, Berg DK. Acetylcholine differentially affects intracellular calcium via nicotinic and muscarinic receptors on the same population of neurons. J Biol Chem. 1995;270:14366–14375. doi: 10.1074/jbc.270.24.14366. [DOI] [PubMed] [Google Scholar]
- 35.Rawlings SR. At the cutting edge: PACAP, PACAP receptors and intracellular signaling. Mol Cell Endocrinol. 1994;101:C5–C9. doi: 10.1016/0303-7207(94)90212-7. [DOI] [PubMed] [Google Scholar]
- 36.Rawlings SR, Demaurex N, Schlegel W. Pituitary adenylate cyclase-activating polypeptide increases [Ca2+]i in rat gonadotrophs through an inositol triphosphate-dependent mechanism. J Biol Chem. 1994;269:5680–5686. [PubMed] [Google Scholar]
- 37.Ross EM. Signal sorting and amplification through G-protein coupled receptors. Neuron. 1989;3:141–152. doi: 10.1016/0896-6273(89)90027-5. [DOI] [PubMed] [Google Scholar]
- 38.Selyanko AA, Brown DA. Intracellular calcium directly inhibits potassium M channels in excised membrane patches from rat sympathetic neurons. Neuron. 1996;16:151–162. doi: 10.1016/s0896-6273(00)80032-x. [DOI] [PubMed] [Google Scholar]
- 39.Spengler D, Waeber C, Pantaloni C, Holsboer F, Bockaert J, Seeburg P, Journot L. Differential signal transduction by five splice variants of the PACAP receptor. Nature. 1993;365:170–175. doi: 10.1038/365170a0. [DOI] [PubMed] [Google Scholar]
- 40.Stella N, Magistretti PJ. Vasoactive intestinal peptide (VIP) and pituitary adenylate cyclase-activating polypeptide (PACAP) potentiate the glutamate-evoked release of arachidonic acid from mouse cortical neurons. J Biol Chem. 1996;271:23705–23710. doi: 10.1074/jbc.271.39.23705. [DOI] [PubMed] [Google Scholar]
- 41.Swope SL, Moss SJ, Blackstone CD, Huganir RL. Phosphorylation of ligand-gated ion channels: a possible mode of synaptic plasticity. FASEB J. 1993;6:2514–2523. [PubMed] [Google Scholar]
- 42.Vernallis AB, Conroy WG, Berg DK. Neurons assemble AChRs with as many as 3 kinds of subunits while maintaining subunit segregation among subtypes. Neuron. 1993;10:451–464. doi: 10.1016/0896-6273(93)90333-m. [DOI] [PubMed] [Google Scholar]
- 43.Vijayaraghavan D, Schmid HA, Halvorsen SW, Berg DK. Cyclic AMP-dependent phosphorylation of a neuronal acetylcholine receptor α-type subunit. J Neurosci. 1990;10:3255–3262. doi: 10.1523/JNEUROSCI.10-10-03255.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Vijayaraghavan S, Pugh PC, Zhang Z-W, Rathouz MM, Berg DK. Nicotinic receptors that bind α-bungarotoxin on neurons raise intracellular free Ca2+. Neuron. 1992;8:353–362. doi: 10.1016/0896-6273(92)90301-s. [DOI] [PubMed] [Google Scholar]
- 45.Vijayaraghavan S, Huang B, Blumenthal EM, Berg DK. Arachidonic acid as a possible negative feedback inhibitor of nicotinic acetylcholine receptors on neurons. J Neurosci. 1995;15:3679–3687. doi: 10.1523/JNEUROSCI.15-05-03679.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Yakel JL. Calcineurin regulation of synaptic function: from ion channels to transmitter release and gene expression. Trends Pharmacol Sci. 1997;18:124–134. doi: 10.1016/s0165-6147(97)01046-8. [DOI] [PubMed] [Google Scholar]
- 47.Zhang Z-W, Vijayaraghavan S, Berg DK. Neuronal acetylcholine receptors that bind a-bungarotoxin with high affinity function as ligand-gated ion channels. Neuron. 1994;12:167–177. doi: 10.1016/0896-6273(94)90161-9. [DOI] [PubMed] [Google Scholar]