

Abstract
Here, we investigate the involvement of caveolins in the pathophysiology of Alzheimer’s disease (AD). We show dramatic upregulation of caveolin-3 immunoreactivity in astroglial cells surrounding senile plaques in brain tissue sections from authentic AD patients and an established transgenic mouse model of AD. In addition, we find that caveolin-3 physically interacts and biochemically colocalizes with amyloid precursor protein (APP) both in vivo and in vitro. Interestingly, recombinant overexpression of caveolin-3 in cultured cells stimulated β-secretase–mediated processing of APP. Immunoreactivities of APP and presenilins were concomitantly increased in caveolin-3–positive astrocytes. Because the presenilins also form a physical complex with caveolin-3, caveolin-3 may provide a common platform for APP and the presenilins to associate in astrocytes. In AD, augmented expression of caveolin-3 and presenilins in reactive astrocytes may alter APP processing, leading to the overproduction of its toxic amyloid metabolites.
Keywords: Alzheimer’s disease, caveolin, presenilin, amyloid precursor protein, secretase, astrocyte
Alzheimer’s disease (AD) is the most common cause of dementia in patients over 60 years of age. Senile plaques and paired helical filaments are the two hallmarks of the brain pathology of AD (Selkoe, 1994a). Currently, AD research is focused on understanding the pathophysiological roles of these senile plaques and paired helical filaments.
The β-amyloid peptide, a major protein component of the senile plaque, is generated from its precursor protein termed the amyloid precursor protein (APP) by enzymatic digestion involving β- and γ-secretase activities. Another cleavage enzymatic activity, α-secretase, cuts APP in the middle of amyloid region (Sisodia et al., 1990; Haass et al., 1992; Sisodia, 1992), thereby precluding amyloid production. In non-neuronal cells, α-secretase–mediated APP shedding plays a major role in APP metabolism, whereas in neurons, β- and γ-secretase–mediated processing is the main metabolic pathway for APP processing. However, the molecular identities of these α-, β-, and γ-secretases remain unknown.
Our recent findings provide clear evidence that APP is enriched within caveolae, where caveolin-1 provides a direct means for APP to be concentrated in this microdomain of the plasma membrane. Caveolin-1 expression also regulates APP processing by promoting α-secretase activity in cultured kidney epithelial cell lines (Ikezu et al., 1998a).
Caveolae are flask-shaped plasma membrane invaginations with a diameter of ∼50–100 nm. The principal protein components of caveolae are the caveolin family of proteins. Three caveolin gene family members have been identified and cloned thus far and are termed caveolin-1, -2, and -3 (Kurzchalia et al., 1992; Scherer et al., 1995, 1996; Tang et al., 1996). Recent studies have provided direct evidence that caveolins are also expressed within cells of the nervous system, including astrocytes (Cameron et al., 1997; Ikezu et al., 1998b) and neurons (Galbiati et al., 1998).
Caveolae are biochemically characterized by their detergent insolubility, because caveolae have a unique lipid composition with a high content of both cholesterol and glycosphingolipids. In the brain, caveolae-like microdomains of the plasma membrane that have a similar lipid composition have been termed detergent-insoluble glycolipid membrane complexes (or DIGs) (Simons and Ikonen, 1997). It has been reported recently that DIGs isolated from whole brain contain not only APP (Bouillot et al., 1996) but also presenilin-1 and -2 together with the Aβ-amyloid peptide (Lee et al., 1998). Therefore, it has been suggested that brain DIGs are the site where amyloid biogenesis or transport takes place. In support of this notion, cholesterol depletion, which leads to the loss of DIG integrity, efficiently inhibits Aβ-amyloid peptide secretion in cultured hippocampal neurons (Simons et al., 1998).
Because there is now an established link between caveolin-1 and APP processing and because known caveolin family members have been detected in nervous systems (Cameron et al., 1997; Galbiati et al., 1998; Ikezu et al., 1998a), our current study was aimed at investigating the hypothesis that caveolins contribute to the pathophysiology of AD.
MATERIALS AND METHODS
Materials. Caveolin antibodies [caveolin-1 (rabbit polyclonal IgG; mouse monoclonal antibodies, clones 2234 and 2297), caveolin-2 (mouse monoclonal antibody, clone 65), and caveolin-3 (rabbit polyclonal antibody; mouse monoclonal antibody, clone 26)] were as we described previously (Scherer et al., 1995, 1997; Song et al., 1996b). The glial fibrillary acidic protein (GFAP) antibody was from Dako (Glostrup, Denmark). Anti-myc epitope IgG (monoclonal, 9E10) was from Santa Cruz Biotechnology (Tebu, France); anti-hemagglutinin (HA) epitope IgG [rat and mouse (12CA5) monoclonal] was from Boehringer Mannheim (Indianapolis, IN). Anti-glutamate receptor (NR1) monoclonal antibody was from PharMingen (San Diego, CA). Presenilin antibodies were as described previously (Zhang et al., 1998). A variety of other reagents were purchased commercially: fetal bovine serum (JRH Biologicals, Lenexa, KA) and prestained protein markers [Bio-Rad (Hercules, CA) and NOVEX].
Antibodies against APP are well characterized and include (1) monoclonal [22C11 specific for the extracellular domain of APP and Alz90 specific for APP511-608 (Boehringer Mannheim); 4G8 specific for Aβ residues 18–24 and 6E10 specific for Aβ residues 1–17 (Senetek) (Kim et al., 1990); and BAN50 specific for human Aβ1–16, BA27 specific for the C terminal of Aβ40, and BC05 specific for the C terminal of Aβ42 (Iwatsubo et al., 1994)] and (2) polyclonal [Aβ (Zymed, San Francisco, CA), 369 (kindly provided by Dr. Samuel E. Gandy) (Buxbaum et al., 1990), and AC-1 (kindly provided by Dr. Kazuaki Yoshikawa) (Hayashi et al., 1992)] antibodies, both specific for the cytoplasmic region.
The cDNAs for caveolin-1 and -3 were as we described previously (Song et al., 1997; Ikezu et al., 1998a). The APP cDNA dually epitope tagged with HA within its extracellular domain and with FLAG at its C terminal was constructed by PCR using a pair of primers: CTGACCGAGGACTGACCAC and GCTCTAGACTACTTGTCATCGTCGTCCTTGTAGTCTCCTCCGTTCTGCATTTGCTCAAAG. We used the human APP695 cDNA as a template for PCR amplification (Yamada et al., 1987). We then digested the APP695 cDNA in the pcDNA-1 vector that contained the HA epitope tag sequence in the extracellular domain (Ikezu et al., 1998a) with BglII and XbaI (APP-pcDNA). Finally, the amplified PCR fragment was similarly digested and subcloned into the APP-pcDNA. The correct sequence of the amplified portion of APP was confirmed by DNA sequencing. A schematic diagram (see Fig. 5E) summarizes the construction of caveolin-3 and APP.
Fig. 5.

Recombinant overexpression of caveolin-3 stimulates the β-secretase processing of APP. A–D, The medium and/or cell lysates from COS-7 cells cotransfected with APP and caveolin-3 were subjected to immunoprecipitation and immunoblot analysis or ELISA. Mock-transfected cells and/or cells transfected with APP alone were processed in parallel. A,Top, The total amount of sAPP in the medium was measured by immunoprecipitation and immunoblotting using an anti-HA antibody. Note that sAPP was significantly increased in cells cotransfected with APP and caveolin-3. Middle, Cell lysates were probed with anti-HA antibodies to assess the total amount of APP expressed. Note that the amount of APP expression is similar in cells transfected with APP alone or in cells cotransfected with APP and caveolin-3 (compare lanes 2, 3).Bottom, Cell lysates were also probed with anti-myc to detect expression of caveolin-3. B, Top, The amount of sAPPα in the medium was measured by immunoprecipitation with the anti-Aβ antibody and subsequent immunoblot analysis with the anti-HA antibody. Note that sAPPα was detected only in medium harvested from cells that were transfected with APP alone but not in medium from cells cotransfected with APP and caveolin-3. Bottom, Cell lysates were probed with anti-HA antibodies to detect intact APP. Note that the amount of intact APP is similar in cells transfected with APP alone or in cells cotransfected with APP and caveolin-3 (compare lanes 1,2). C, Top, The C-terminal fragments of APP generated by α-secretase cleavage (αAPPct) or β-secretase cleavage (βAPPct) were detected by immunoprecipitation with the antibody 4G8 and immunoblot analysis with antibodies directed against the C-terminal FLAG epitope attached to APP. Note that in cells cotransfected with APP and caveolin-3 that the levels of bothβAPPct and αAPPct were increased.Bottom, Cell lysates were immunoprecipitated with and later probed with anti-FLAG antibodies to detect intact APP. Note that the amount of intact APP is similar in cells transfected with APP alone or in cells cotransfected with APP and caveolin-3 (compare lanes 1, 2). D, Top, The amount of Aβ-amyloid secreted into the medium was measured with a double-sandwich ELISA that uses the antibodies 4G8 and 6E10. Note that a significant reduction of Aβ-amyloid secretion was observed in cells cotransfected with APP and caveolin-3 (p < 0.05) compared with that in cells transfected with APP alone.Bottom, A double-sandwich ELISA used the antibodies BAN50 and BA27 or antibodies BAN50 and BC05 to measure separately the amounts of Aβ40 and Aβ42, respectively. Note that the ratio of Aβ1-42/(Aβ1-40 + Aβ1-42) was not significantly altered by cotransfection of APP with caveolin-3. E, Schematic diagram of caveolin-3 and APP cDNAs is shown.
Immunohistochemistry of human and mouse specimens. Eight cases of sporadic Alzheimer’s disease [age (years), 79.3 ± 7.3 (mean ± SE)]and four normal controls [age (years), 73.3 ± 11.4 (mean ± SE)] were used for immunohistochemical analyses, which were described previously (Mochizuki et al., 1996). Specimens were fixed in 4% paraformaldehyde and frozen after cryoprotection in 30% sucrose.
Five transgenic mice (ages of 29, 26, 21, 16, and 14 months old) that overexpress human APP with the Swedish mutation (Hsiao et al., 1996) and four normal control mice (ages of 27, 16, 14, and 14 months old) were used for immunohistochemistry.
For DAB staining, the sections pretreated with microwaving were treated with a buffer containing 10% Triton X-100 and 1% (v/v) H2O2 for 30 min, followed by 2% (v/v) normal goat serum for 1 hr. Sections were incubated with the primary antibodies for 12 hr at 4°C, followed by sequential incubation with biotinylated secondary antibody and the avidin–biotin complex reagent (Vectastain Elite ABC Kit; Vector Laboratories, Burlingame, CA) at room temperature for 30 min, and the color was developed using 3–3′-diaminobenzidine tetrahydrochloride.
For double immunofluorescence staining, 30-μm-thick sections from human and mouse specimens were pretreated with microwaving (three times for 1 min each in citrate buffer), followed by incubation in a buffer containing 10% Triton X-100 for 30 min and subsequently in 2% (w/v) normal goat serum for 1 hr. Sections were then incubated with the primary antibodies overnight at 4°C. This incubation was followed by sequential incubation with secondary antibodies containing FITC-conjugated or Texas Red–conjugated secondary antibodies (Jackson ImmunoResearch, West Grove, PA) at room temperature for 60 min. The immunostained sections were mounted with antifading medium and microscopically observed under a confocal immunofluorescence microscope (TCS-NT; Leica, Nussloch, Germany). To control for antibody specificity, we immunostained neighboring sections in the same way except that anti-caveolin-3 antibody was preincubated with antigen peptide.
Detergent-free purification of caveolin-rich membrane domains. Cells were used to prepare caveolin-enriched membrane fractions, as described previously (Song et al., 1996a). After two washes with ice-cold PBS, cultured cells (two confluent 150 mm dishes) were scraped into 2 ml of 500 mm sodium carbonate, pH 11.0, and homogenized sequentially with a loose-fitting Dounce homogenizer (10 strokes), a Polytron tissue grinder (three 10 sec bursts; Kinematica GmbH; Brinkmann Instruments), and a sonicator (three 20 sec bursts; Branson Sonifier 250; Branson, Danbury, CT). The homogenate was then adjusted to 45% sucrose by the addition of 2 ml of 90% sucrose in Mes-buffered saline (25 mm Mes, pH 6.5, and 0.15 m NaCl) and placed at the bottom of an ultracentrifuge tube. A 5–35% discontinuous sucrose gradient was formed above (4 ml of 5% sucrose/4 ml of 35% sucrose; both in MBS containing 250 mm sodium carbonate) and centrifuged at 39,000 rpm for 16–20 hr in an SW41 rotor (Beckman Instruments). A light-scattering band confined to the 5–35% sucrose interface contained caveolin but excluded most other cellular proteins. The caveolae-rich fractions were diluted threefold with MBS and centrifuged at 15,000 rpm for 20 min at 4°C. The pellets were used as “purified caveolae-enriched membranes.” This protocol separated caveolin from the bulk of cellular membranes and cytosolic proteins. By the use of this scheme, endogenous caveolin was not only recovered almost quantitatively in fractions 4 and 5 while excluding most cellular proteins but also was separated from the glycosylphosphatidylinositol-linked plasma membrane marker carbonic anhydrase IV (Song et al., 1996a).
As an alternative approach to purify caveolae, a protocol developed bySmart et al. (1995) was used. A plasma membrane fraction was prepared from 10 100 mm dishes of confluent tissue culture cells. Each dish was washed twice with 5 ml of buffer A (0.25 m sucrose, 1 mm EDTA, and 20 mm Tricine, pH 7.8). Cells were collected by centrifugation at 1400 × g for 5 min (Sorvall RT6000; 3000 rpm), resuspended in 1 ml of buffer A, and homogenized 15 times with a Teflon glass homogenizer. Homogenized cells were centrifuged at 1000 × g for 10 min (Sorvall RT6000; 2500 rpm), and the supernatant was subjected to Percoll gradient centrifugation. It was overlayed on top of 23 ml of 30% Percoll solution in buffer A and ultracentrifuged at 83,000 ×g (30,000 rpm) for 30 min in a Beckman 60Ti. The plasma membrane fraction was collected, and the volume was adjusted to 2 ml in buffer A. Fifty percent Optiprep (1.84 ml) in buffer B (0.25m sucrose, 6 mm EDTA, and 120 mmTricine, pH 7.8) was added to 0.16 ml of buffer A (23% Optiprep solution), which was mixed with the sonicated plasma membrane fraction. The entire solution was placed at the bottom of the Beckman SW41Ti tube, overlayed onto a linear 20–10% Optiprep gradient (prepared by diluting 50% Optiprep in buffer A and B), and centrifuged at 52,000 × g (18,000 rpm) for 90 min using SW41Ti (Beckman Instruments). The bottom fraction was collected (noncaveolae membrane). The top 5 ml of the gradient (fractions 1–6) was collected and mixed with 50% Optiprep in buffer B, which was then placed on the bottom of a SW41Ti tube and overlayed by 2 ml of 5% Optiprep in buffer A. The membrane fractions were centrifuged at 52,000 ×g for 90 min. An opaque band located just above the 5% interface was designed the “caveolae fraction.” In this approach, the bulk of the protein that was recovered in fractions 7–13, termed “plasma membrane fractions,” was separated from the “caveolae membrane fractions” (fractions 1–6). The enrichment of caveolae membrane fractions versus plasma membrane fractions was assessed by the amount of protein recovered in the former fraction divided by that in the latter fraction.
Immunoblotting of gradient fractions. From the top of each gradient, 1 ml gradient fractions were collected to yield a total of 12 fractions. Caveolin migrates mainly in fractions 4 and 5 of these sucrose density gradients (Song et al., 1996a). Gradient fractions were separated by SDS-PAGE and transferred to Immobilon-PSQ sheets (Millipore, Bedford, MA). After transfer, sheets were stained with Ponceau S to visualize protein bands and subjected to immunoblotting. For immunoblotting using ECL, incubation conditions were as described by the manufacturer (Amersham, Arlington Heights, IL), except we supplement our blocking solution with 1% bovine serum albumin, 3% nonfat dry milk, and 0.02% sodium azide.
Immunoprecipitation of caveolins from tissue samples and cultured cells. Tissue (∼1 gm) was lysed in a 10-fold volume (∼10 ml) of solubilization buffer (10 mm Tris-HCl, pH 7.4, 1 mm EDTA, 150 mm NaCl, 60 mm octyl glycoside, 1% Triton X-100, 1 mm PMSF, 5 μmleupeptin, 1 μm pepstatin, and 0.3 μmaprotinin) using a Polytron homogenizer. After centrifugation at 15,000 rpm for 30 min, the supernatant (20 mg) was subjected to immunoprecipitation (2 μg of antibody). After washing, beads were boiled in SDS sample buffer and subjected to Western blot analysis.
For cultured cells, ∼1 × 107 cells (CRT, COS-7, or primary cultured astrocytes) were grown on a 100 mm dish. After being washed twice with 1× PBS, pH 7.2, the cells were collected by centrifugation. Cell pellets were solubilized in 10 volumes of solubilizing buffer (10 mm Tris, 1 mm EDTA, 150 mm NaCl, 60 mm octyl glycoside, 1% Triton X-100, 1 mm PMSF, 5 μmleupeptin, 1 μm pepstatin, and 0.3 μmaprotinin). Cells were homogenized using a Polytron homogenizer for 10 sec, followed by sonication (power 3.5, duty cycle 70, and stroke 10) and centrifugation at 15,000 rpm for 30 min. The protein concentration of the supernatant was determined by the Bradford method.
Five hundred micrograms of protein were used for immunoprecipitation and incubated with 2 μg of normal IgG or specific primary antibody IgG at 4°C overnight. Immunoprecipitates were mixed with sample buffer containing β-mercaptoethanol, denatured at 100°C for 5 min, and subjected to SDS-PAGE.
The following procedures were performed as described previously: human astrocytoma (CRT) cell cultures (Ransohoff et al., 1991; Shrikant et al., 1996); primary astrocyte cell cultures, COS-7 cell cultures and cDNA transfection, and immunoprecipitation of the soluble extracellular domain of APP (sAPP) (Ikezu et al., 1998a,b); and a double sandwich ELISA to measure Aβ-amyloid peptide (Kim et al., 1990; Iwatsubo et al., 1994).
RESULTS
Caveolin-3 expression is upregulated in reactive astrocytes surrounding senile plaques in the brains of AD patients and a transgenic mouse model of AD
To investigate the possibility that caveolin expression is altered in AD brain tissue, we immunostained AD brain sections from patients with a well-characterized panel of isoform-specific caveolin antibody probes that recognize either caveolin-1, -2, or -3 selectively. We detected caveolin-1 and -2 expression in endothelial cells equally well in both AD and normal brain sections (data not shown), which is consistent with a recent report that caveolin-1 and -2 are coexpressed in brain endothelial cells in rodents (Ikezu et al., 1998b).
Only very low levels of caveolin-3 immunostaining were barely detected in normal human brain sections (n = 4), although caveolin-3 immunoreactivity is clearly detected in brain astrocytes in rodents (Ikezu et al., 1998b) (data not shown). We speculate that the difference of fixation conditions hampered caveolin-3 staining in human brains. In fact, we detected caveolin-3 immunoreactivity by immunoprecipitation and immunoblot analysis using normal human brain tissue (see Fig. 3A) that was obtained immediately postmortem, indicating that low levels of caveolin-3 are indeed present in normal human brain tissue.
Fig. 3.

Caveolin-3 physically interacts with APP and the presenilins in human brain, primary cultures of astrocytes, CRT astrocytoma cells, and COS-7 cells. A, Human brain. Extracts were prepared and immunoprecipitated with a caveolin-3–specific antibody (left,center) or with an NR1-specific antibody (right). Left, Center, After SDS-PAGE and transfer to nitrocellulose, these immunoprecipitates were probed by immunoblot analysis with the following antibodies: 4G8 to detect APP [left, top; lane 2 (in this and subsequent figures, lane 1 is theleft-hand lane)], anti-presenilin-1 (-PS-1) directed against the N-terminal fragment of presenilin-1 (center, top; lane 2), anti-PS-2 (1209) directed against the N-terminal fragment of presenilin-2 (center,second from top; lane 2), and anti-tubulin (center, third fromtop; lane 2). Immunoprecipitation with normal mouse IgG (NMG) was performed in parallel as a negative control and did not show any association with APP, PS-1, or PS-2, as expected (all; lane 1). Tubulin was clearly detected in the starting material by immunoblot analysis with anti-tubulin antibody (center,bottom). Right, In the NR1 immunoprecipitates, NR1 was clearly detected by immunoblot analysis with NR1 antibody (top); however no APP, PS-1, PS-2, or caveolin-3 was detected by immunoblot analysis (middle,bottom). B, Primary cultured astrocytes. Extracts were prepared and immunoprecipitated with a caveolin-3–specific monoclonal antibody (bottom;lane 2). After SDS-PAGE and transfer to nitrocellulose, these immunoprecipitates were probed by immunoblot analysis with 4G8 to detect APP (top; lane 2). Immunoprecipitation with NMG was performed in parallel as a negative control and did not show any association with APP, as expected (lane 1). C, CRT astrocytoma cell line. Extracts were prepared and immunoprecipitated with a caveolin-3–specific monoclonal antibody (left,center) or with an NR1-specific monoclonal antibody (right). Left, Center, After SDS-PAGE and transfer to nitrocellulose, these immunoprecipitates were probed by immunoblot analysis with the following antibodies: 4G8 to detect APP (left, top; lane 2), anti-PS-1 directed against the N-terminal fragment of presenilin-1 (center,top; lane 2), anti-PS-2 directed against the N-terminal fragment of presenilin-2 (center, second fromtop; lane 2), and anti-tubulin (center, third from top;lane 2). Immunoprecipitation with NMG was performed in parallel as a negative control and did not show any association with APP, PS-1, or PS-2, as expected (all;lane 1). Tubulin was clearly detected in the starting material by immunoblot analysis with anti-tubulin antibody (center, bottom). Right, In the NR1 immunoprecipitates, NR1 was clearly detected by immunoblot analysis with NR1 antibody (top); however no APP, PS-1, PS-2, or caveolin-3 was detected by immunoblot analysis (middle, bottom). D, COS-7 cells. Cells were cotransfected with HA-tagged APP and myc-tagged caveolin-3 and analyzed by immunoprecipitation and Western blotting. Cells were immunoprecipitated with anti-myc IgG (mAb 9E10), and these immunoprecipitates were analyzed by Western blotting with antibodies directed against HA (to detect APP; top) and myc (to detect caveolin-3; bottom). Under these conditions, immunoprecipitation of caveolin-3 shows that APP coprecipitates. Immunoprecipitation with NMG was performed in parallel as a negative control and showed little or no association with APP.cav-3, Caveolin-3; IP, immunoprecipitation; pAb, polyclonal antibody;MM, molecular marker; N-TF, N-terminal fragment.
Surprisingly, in brain tissue sections from AD patients, we detected strong caveolin-3 immunoreactivity via DAB staining (Fig.1h). The cells that stained positive for caveolin-3 had the morphological and anatomical characteristics of reactive astrocytes, because they displayed spikes on their cell surface and surrounded the senile plaques. Double immunostaining demonstrated that caveolin-3–positive cells were indeed GFAP-positive astrocytes (Fig. 1a–c). An Aβ-amyloid antibody was next used to detect amyloid plaques (Fig.1e), around which caveolin-3–positive astrocytes were found to be localized (Fig. 1d,f). Amyloid plaques without dense cores were also surrounded by caveolin-3–positive astrocytes (Fig. 1g). Congo Red–negative and Aβ-amyloid–positive plaques were also surrounded by caveolin-3–positive astrocytes (data not shown). Essentially identical results were obtained in eight independent AD brain tissue sections. The observed caveolin-3 immunostaining in astrocytes was completely absorbed in the presence of its corresponding epitope peptide (Fig. 1i), indicating that the staining was specific for caveolin-3. Caveolin-1 and -2 were not detected in the reactive astrocytes (data not shown).
Fig. 1.
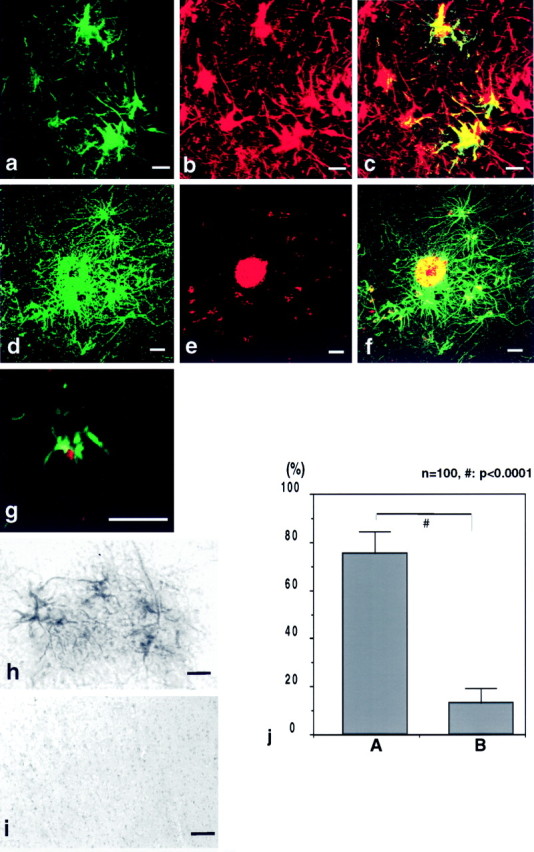
Caveolin-3 is upregulated in reactive astrocytes surrounding senile plaques in brain tissue from AD patients. Brain tissue sections from AD patients were probed with antibodies directed against caveolin-3, GFAP, and Aβ-amyloid. a–c, Confocal image of double immunostaining of caveolin-3 and GFAP showed that caveolin-3–positive (a; green) cells were GFAP-positive (b; red) astrocytes. The merged image is shown in c.d–f, Confocal image of double-immunofluorescent staining of caveolin-3 (d; green) and Aβ-amyloid (e; red) revealed that caveolin-3–positive cells surrounded senile plaques. The merged image is shown in f. g, Confocal image of double-immunofluorescent staining of caveolin-3 (green) and Aβ-amyloid (red) revealed that caveolin-3–positive cells surrounded a senile plaque without a core. h, i, Caveolin-3 peptide competition experiments (h, i) revealed that caveolin-3 immunostaining in reactive astrocytes (h) was completely abolished by preincubation of purified anti-caveolin-3 IgG with an excess amount of the corresponding epitope peptide used to generate this caveolin-3 antibody probe (i). Scale bars, 10 μm. j. Densitometric analysis of the caveolin-3 staining intensity in reactive astrocytes in AD brain tissue sections (i) revealed an ∼sixfold increase of caveolin-3–positive staining (vertical bar A) over that in nonreactive astrocytes (vertical bar B).
Importantly, caveolin-3 immunostaining was only marginally detected in astrocytes irrelevant to senile plaques. Quantitation revealed that ∼96% of caveolin-3–positive astrocytes were associated with senile plaques. Caveolin-3–positive reactive astrocytes accounted for ∼50% of the total reactive astrocytes associated with senile plaques.
Additionally, we assessed the intensity of caveolin-3 DAB staining by densitometric analysis (Fig. 1j). Caveolin-3 immunoreactivity showed an ∼sixfold increase compared with that seen in astrocytes that were not associated with senile plaques (Fig.1j). These results establish that caveolin-3 is highly upregulated in reactive astrocytes associated with senile plaques.
Astrogliosis is also observed in other human neurological diseases, such as amyotrophic lateral sclerosis (ALS). To investigate the possibility that caveolin-3 upregulation in reactive astrocytes is specific to the pathology of the AD brain, we stained spinal cord sections from ALS patients. Caveolin-3 immunostaining was not detected in these reactive astrocytes (data not shown), indicating that caveolin-3 upregulation might be relatively specific to AD.
To substantiate these observations further, we next immunostained brain sections derived from a transgenic mouse model of AD. These AD mice are transgenically engineered to overexpress the human APP695 containing the Swedish mutation. Again, we detected caveolin-3 immunostaining in reactive astrocytes associated with amyloid plaques with dense cores (Fig. 2a–c). Amyloid plaques without dense cores were also surrounded by caveolin-3–positive astrocytes (Fig. 2g). Caveolin-1 and -2 were not detected in these astrocytes (data not shown). Double labeling of caveolin-3 and Aβ-amyloid peptide revealed that caveolin-3–positive astrocytes surrounded these senile plaques (Fig. 2d–f).
Fig. 2.
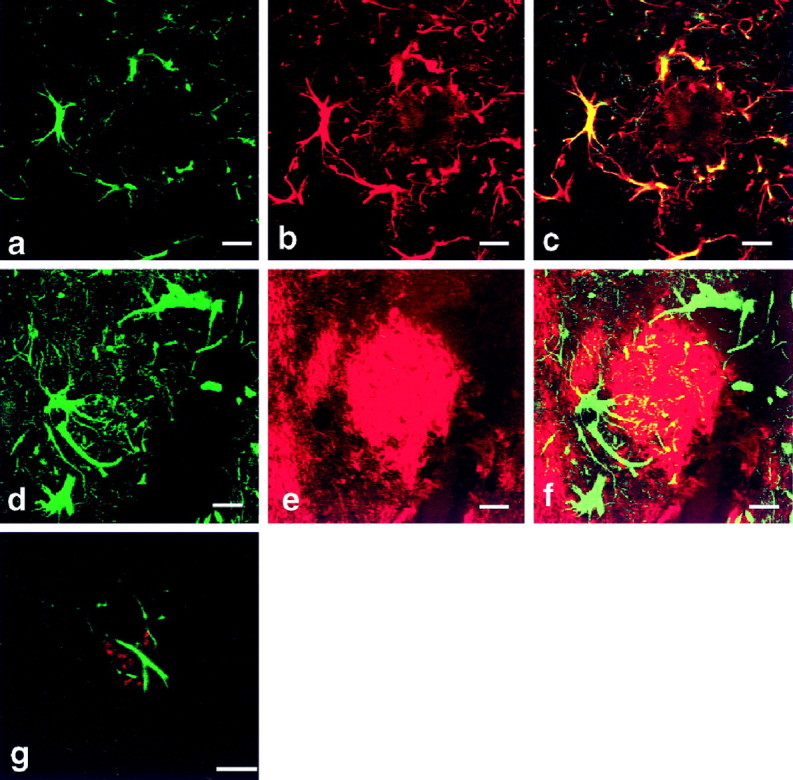
Caveolin-3 expression is dramatically upregulated in reactive astrocytes surrounding senile plaques in brain tissue from a transgenic mouse model of AD. Brain tissue sections from AD transgenic mice were probed with antibodies directed against caveolin-3, GFAP, and Aβ-amyloid. a–c, Confocal image of double immunostaining of caveolin-3 and GFAP showed that caveolin-3–positive (a; green) cells were GFAP-positive (b; red) astrocytes. The merged image is shown in c. d–f, Confocal image of double-immunofluorescent staining of caveolin-3 (d; green) and Aβ-amyloid (e; red) revealed that caveolin-3–positive cells surrounded senile plaques with cores. The merged image is shown in f. g, Confocal image of double-immunofluorescent staining of caveolin-3 (green) and Aβ-amyloid (red) revealed that caveolin-3–positive cells surrounded a senile plaque without a core. Scale bars, 10 μm.
As a negative control, brain sections from Wobbler mice, which are a well-established mouse model of ALS (Pioro and Mitsumoto, 1995), were also stained. Caveolin-3 immunoreactivity was not detected in reactive astrocytes in spinal cord sections from Wobbler mice (data not shown), indicating that caveolin-3 is relatively specifically upregulated in reactive astrocytes associated with senile plaques in AD transgenic mice.
Taken together, these results establish that reactive astrocytes associated with senile plaques specifically overexpress caveolin-3 both in the brains of AD patients and AD transgenic mice.
Caveolin-3 forms a physical complex with APPin vivo
Because caveolin-1 provides a physical means for the association of APP with caveolae (Ikezu et al., 1998a), we next investigated the possibility that caveolin-3 also forms a physical complex with APP. We prepared cell lysates from normal human brain tissue and immunoprecipitated caveolin-3 with an anti-caveolin-3–specific monoclonal antibody. Immunoblot analysis of these immunoprecipitates demonstrated that they contain APP immunoreactivity (Fig.3A, left), indicating that APP and caveolin-3 form a physical complex in vivo. Importantly, immunoprecipitation with normal mouse IgG or with anti-NR1 antibody (glutamate receptor) did not coimmunoprecipitate either caveolin-3 or APP. NR-1 was reported not to be present in the caveolae-like membrane domain in brain (Wu et al., 1997). In addition, although tubulin was clearly detected in cell lysates (starting material), tubulin was not detected in caveolin-3 immunoprecipitates, a further indication that the observed interaction between caveolin-3 and APP is specific.
The physical interaction of endogenous APP with caveolin-3 was also observed in primary cultures of astrocytes (Fig. 3B) and in an astrocytoma cell line (termed CRT) (Fig. 3C). Because we used immunoblotting with an anti-Aβ–specific antibody (4G8), the band detected is not amyloid precursor-like protein 2 but APP itself. Importantly, control immunoprecipitates with normal IgG did not coprecipitate APP immunoreactivity. In CRT cells, anti-NR1 antibody did not coimmunoprecipitate either caveolin-3 or APP. These results clearly indicate that endogenous APP and caveolin-3 form a physical complex in astrocytes and in a cell line derived from the astrocyte lineage.
We evaluated the physical interaction between APP and caveolin-3 further using a heterologous expression system, i.e., transient transfection of COS-7 cells. We transiently transfected the cDNAs encoding APP (tagged with the HA epitope) and caveolin-3 (tagged with the c-myc epitope) into COS-7 cells. Next, cell lysates were prepared, and caveolin-3 was immunoprecipitated with an antibody that recognizes the myc-epitope. These immunoprecipitates were then subjected to immunoblot analysis with an antibody that recognizes the HA epitope to detect APP.
As shown in Figure 3D, lane 2, caveolin-3 immunoprecipitates recovered a significant amount of APP immunoreactivity, indicating that recombinant caveolin-3 forms a physical complex with recombinant APP during transient expression in COS-7 cells. In contrast, immunoprecipitates using normal mouse IgG (a negative control for these experiments) showed only a very minor amount of nonspecifically associated APP immunoreactivity (Fig. 3D,lane 1). These results clearly demonstrate that caveolin-3 and APP form a specific physical complex in intact cells. However, the possibility still remains that caveolin-3 might interact indirectly with APP. For example, APP was more efficiently associated with caveolin-3 immunoprecipitates from brain homogenates, as compared with heterologous expression in COS-7 cells.
We next tested the hypothesis that APP and caveolin-3 are coenriched within caveolae membranes in an astrocytic cell line, CRT cells. We purified caveolae microdomains using an established detergent-free sucrose density gradient approach (Song et al., 1996a). As shown in Figure 4A, caveolin-3 was recovered within the caveolae-enriched fraction derived from CRT astrocytoma cells. Most of the APP immunoreactivity was also recovered in the caveolae-enriched membrane fraction (Fig. 4A,left), suggesting that APP is localized within caveolin-3–positive caveolae membrane domains. In COS-7 cells, APP and caveolin-3 were also recovered in the caveolae-enriched membrane fraction using this method (Fig. 4B).
Fig. 4.

Caveolin-3, APP, and the presenilins are colocalized within caveolae membranes in cultured cells.A, CRT astrocytoma cell line.Left, CRT cells were subjected to detergent-free subcellular fractionation that separates caveolae from the bulk of cellular and cytosolic proteins, as described by Song et al. (1996s).Fraction 5 represents the caveolae-rich fraction, andfractions 8–12 contain ∼99% of the total cellular protein. Note that APP (top, middle) and caveolin-3 (bottom) were highly enriched within the caveolae fraction. APP was detected by immunoblotting using AC-1 and 4G8 antibodies. Right, CRT cells were subjected to a second independent detergent-free approach used to purify caveolae membranes (the Optiprep method). Each lane contains 2 μg of protein. APP, PS-1, PS-2, and caveolin-3 were all detected primarily in the caveolae membrane fraction (lane 4), whereas they were barely detectable in the noncaveolar fraction of the plasma membrane (lane 3). B, COS-7 cells. Cells cotransfected with APP and caveolin-3 were subjected to subcellular fractionation as described in A(left). Note that APP and caveolin-3 were confined primarily to the caveolae membrane fraction (seefraction 5).
As an alternative approach, we used a second independent detergent-free method to purify caveolae membranes (the Optiprep method) (Smart et al., 1995). Using this method, we could biochemically enrich caveolae membranes by ∼95-fold as compared with the noncaveolae plasma membrane fraction (total protein amount of purified caveolae membrane fraction = 1.6 μg vs total protein amount of purified noncaveolae membrane fraction = 150.4 μg). As shown in Figure4A, right, APP immunoreactivity was recovered selectively in the caveolae membrane fraction using this methodology. These data clearly indicate that APP forms a physical complex with caveolin-3 in caveolae membranes.
Recombinant overexpression of caveolin-3 activates β-secretase activity
Because caveolin-1 overexpression enhances the activity of α-secretase, resulting in increased secretion of the soluble extracellular domain of APP (sAPPα) (Ikezu et al., 1998a), we next examined the possibility that caveolin-3 overexpression alters APP processing. We cotransfected APP695 (tagged with HA in the extracellular domain and FLAG at its C terminal) and myc-tagged caveolin-3 into COS-7 cells. Then, we assessed the amount of total sAPP in the medium by immunoprecipitation and immunoblotting with an anti-HA antibody.
In mock-transfected cells, we did not detect any sAPP in the medium (Fig. 5A, top,lane 1). In contrast, we detected sAPP in the medium from cells transfected with APP alone (Fig. 5A, top,lane 2). Interestingly, the amount of sAPP detected in the medium from cells cotransfected with APP and caveolin-3 was significantly increased compared with that in medium recovered from cells singly transfected with APP alone (Fig. 5A,top, lane 3). Because we detected similar amounts of total recombinant APP expression in cells transfected with APP alone or APP plus caveolin-3 (Fig. 5A, middle,bottom), caveolin-3 coexpression clearly augmented sAPP secretion into the medium.
We next directly measured the amount of sAPPα in the medium by immunoprecipitation with an anti-Aβ antibody and immunoblot analysis with the anti-HA antibody. Surprisingly, sAPPα was not detected in media of cells cotransfected with APP and caveolin-3, although significant amounts of sAPPα were recovered in the medium of cells singly transfected with APP alone (Fig. 5B, top). Because the amount of APP expressed in total cell lysates was similar between the two independent transfections (Fig. 5B,bottom), these results indicate that β-secretase activity (rather than α-secretase activity) was augmented by caveolin-3 overexpression.
Consistent with this conclusion, immunoprecipitation (of the cell lysates derived from cells cotransfected with caveolin-3 and APP) with 4G8 and immunoblot analysis with anti-FLAG antibody demonstrated a significant accumulation of the C-terminal APP fragment known to be derived from β-secretase processing of APP (Fig. 5C,top). Because the total amount of intact APP was similar among different transfections (Fig. 5C, bottom), caveolin-3 clearly promoted β-secretase–mediated cleavage of APP.
We next explored the possibility that caveolin-3 overexpression might also increase γ-secretase activity. To test this hypothesis, we measured the amount of Aβ-amyloid peptide in the medium by a double-sandwich ELISA technique using two independent anti-Aβ-amyloid peptide antibodies, 6E10 and 4G8. The amount of Aβ-amyloid peptide was moderately, but significantly, decreased in COS-7 cells cotransfected with APP and caveolin-3 compared with that in cells singly transfected with APP alone (Fig. 5D,top).
Aβ40 and Aβ42 were also separately measured by ELISA using antibodies that specifically recognize the C-terminal regions of these Aβ-amyloid peptides. The results showed that the ratio of Aβ1–42/(Aβ1–40 + Aβ1–42) was not altered (Fig.5D, bottom). These data clearly indicate that caveolin-3 overexpression selectively stimulates β-secretase activity but does not affect γ-secretase activity.
APP and the presenilins are upregulated in reactive astrocytes in AD brain tissue
Astrogliosis is known to be associated with increased expression of APP. Therefore, we assessed the amount of APP in reactive astrocytes surrounding senile plaques in brain sections from AD patients. We stained AD brain tissue sections with the antibody 22C11 and observed a significant increase of 22C11 immunoreactivity in reactive astrocytes (Fig. 6g).
Fig. 6.

Presenilin-1 and -2 and APP are concomitantly increased in reactive astrocytes in brain tissue from AD patients. Brain tissue sections from AD patients were probed with antibodies directed against presenilins, APP, and GFAP. Presenilin-1 was detected using the PS-1 antibody (a), presenilin-2 was detected using the PS-2 antibody (d), and APP was detected using 22C11 (g), Alz90 (j), 6E10 (m), and Aβ (p). AD brain astrocytes were identified by double labeling with GFAP antibodies (b,e, h, k, n,q). Merged images are also presented (a +b = c, d +e = f, g +h = i, j +k = l, m +n = o, and p +q = r). Scale bars:a–c, d–f, g–i,j–l, m–o, p–r, 10 μm.
As shown in Figure 6h, these cells were also positive for GFAP staining. The merged image of Figure 6, g andh, clearly shows the strong immunoreactivity of 22C11 in GFAP-positive astrocytes (Fig. 6i). The antibody Alz90 also detected strong APP immunoreactivity within reactive astrocytes (Fig.6j–l). Antibodies specific for the Aβ-amyloid region (4G8 and Aβ) also detected Aβ immunoreactivity within these reactive astrocytes (Fig.6m–o,p–r). Taken together, these results show that the amount of APP is increased within reactive astrocytes of the AD brain.
We next investigated the possibility that presenilin immunoreactivity is altered in reactive astrocytes surrounding amyloid plaques in AD brain sections. As shown in Figure 6a, presenilin-1 immunoreactivity was detected in reactive astrocytes as well as in neurons. We confirmed that these cells were indeed astrocytes, because these presenilin-1–positive cells were also positive for GFAP (Fig.6b,c). Presenilin-2 immunoreactivity was also detected in these reactive astrocytes (Fig. 6d–f). These data demonstrate that the expression levels of presenilin-1 and -2 are increased in reactive astrocytes in AD brain concomitantly with the upregulation of caveolin-3 and APP.
Caveolin-3 forms a physical complex with the presenilins
Because APP and caveolin-3 were coenriched within caveolae microdomains, we next investigated the possibility that presenilin-1 and -2 are also localized within caveolin-3–positive caveolae membranes using detergent-free purification of caveolae. In CRT astrocytoma cells, purified caveolae membrane fractions contained both presenilin-1 and -2, together with caveolin-3 and APP (Fig.4A, right).
We then tested the hypothesis that caveolin-3 forms a physical complex, not only with APP, but also with the presenilins. Immunoprecipitation experiments show that caveolin-3 physically associates with presenilin-1 and -2 in CRT astrocytoma cells (Fig. 3C), which is also the case with lysates prepared from human brain tissue (Fig. 3A). To create a negative control, we immunoblotted caveolin-3 immunoprecipitates with an anti-tubulin antibody. Although we could clearly detect tubulin immunoreactivity in cell lysates (starting material) with simple Western blotting, no tubulin immunoreactivity was detected in these immunoprecipitates. In addition, immunoprecipitation with anti-NR1 antibody did not coprecipitate presenilin-1, presenilin-2, or caveolin-3, confirming the specificity of the interaction between caveolin-3 and the presenilins. These data indicate that caveolin-3 forms a physical complex with both APP and the presenilins in vitro and in vivo. These results suggest a novel role for caveolin-3 as a common platform for APP and presenilin interactions in brain astrocytes.
DISCUSSION
For the first time, our current data establish that caveolins contribute to the pathophysiology of AD. We show that senile plaques are surrounded by caveolin-3–positive astrocytes in brain tissue sections from patients with AD. In addition, virtually identical results were obtained with brain tissue sections from mice engineered to overexpress the Swedish APP mutant as a transgene.
Here, we also provide multiple independent lines of evidence that caveolin-3 forms a physical complex with APP in vivo. It is likely that the underlying mechanism by which caveolin-3 forms a physical complex with APP is similar to that of the caveolin-1 and APP interaction because caveolin-1 directly binds APP by its scaffolding domain (Ikezu et al., 1998a) and the homologous region in caveolin-3 recognizes similar sequence motifs for direct interaction with its binding partners (Couet et al., 1997).
We have reported recently that not only APP but also presenilin-1 directly binds heterotrimeric GTP binding (G)-proteins (Okamoto et al., 1995; Smine et al., 1998). G-proteins and their coupled receptors are highly enriched in caveolae microdomains, in which caveolins may provide a direct physical means for them to be concentrated (Okamoto et al., 1998). We have reported recently that caveolin-3 is the major caveolin family member present in astrocytes (Ikezu et al., 1998b). Our current data that APP and presenilins are highly enriched in caveolin-3–positive caveolae in astrocytes and an astrocytic cell line in which caveolin-3 forms a physical complex with APP and presenilins fit well with these recent findings.
Caveolin-3 directly interacts with the α subunits of heterotrimeric G-proteins and negatively regulates their activities (Tang et al., 1996; Okamoto et al., 1998), by inhibiting GDP release and activating GTP hydrolysis of G-proteins. Caveolin-3 binding to APP and presenilins may activate β-secretase activity via G-protein inactivation, because G-protein signaling leading to protein kinase C activation is pivotal for regulating secretase activity (Selkoe, 1994b).
Our findings also provide evidence that APP is upregulated within caveolin-3–positive astrocytes. It has been shown that APP is expressed in astrocytes (Gegelashvili et al., 1994) and these isoforms are predominantly APP751 and APP770 that contain the Kunitz protease inhibitor (KPI) domain in their extracellular region. The fact that a soluble extracellular domain of APP that contains the KPI region is increased in AD brain (Kitaguchi et al., 1990; Palmert et al., 1990;Moir et al., 1998) may simply reflect the upregulation of caveolin-3 and APP in reactive astrocytes, because we show here that caveolin-3 overexpression promotes APP shedding by activating β-secretase activity.
Most recently, it has been reported that the β-amyloid peptide is generated mainly in cultured primary astrocytes by transfection experiments using Semliki Forest virus that encodes the Swedish APP mutation (Forman et al., 1997), suggesting an important role for astrocytes in Aβ-amyloid production in patients bearing the Swedish mutations in APP. Caveolin-3 may play a key role in augmenting Aβ-amyloid peptide formation in astrocytes by stimulating β-secretase activity together with that of other molecule(s) such as presenilins (Tomita et al., 1997; De Strooper et al., 1998) that are involved in γ-secretase activation.
Cholesterol homeostasis plays a pivotal role in APP metabolism (Howland et al., 1998; Simons et al., 1998), which may simply reflect that regulators of APP metabolism are localized within caveolae. Indeed, cholesterol depletion reduces the activity of β-secretase. This fits well with our current data that caveolin-3–containing caveolae are involved in β-secretase–mediated cleavage of APP. In support of this hypothesis, it has been shown recently that APP and presenilin-1 and -2 together with the Aβ-amyloid peptide cofractionate in detergent-insoluble cholesterol-rich complexes purified from brain tissue (Lee et al., 1998).
sAPPβ induces cytokine production from microglial cells (Barger and Harmon, 1997). Microglial activation is one of the hallmark pathological changes observed in the AD brain (Dickson et al., 1993). In AD model mice used in this study, microglial activation was also documented (Frautschy et al., 1998). Large-scale statistical analysis of clinical data showed that anti-inflammatory agents are an effective means to prevent the progression of dementia seen in AD patients (McGeer and McGeer, 1996). Thus, the involvement of inflammation and microglial activation is an important aspect of AD pathophysiology. Our results showing that caveolin-3 upregulation or overexpression augments secretion of sAPPβ suggest that caveolin-3 is a potential therapeutic target to block further activation of microglial cells and inflammation pertinent to AD dementia.
Because it has been shown using genetically altered mice that presenilin-1 is essential for γ-secretase activity in vivo(De Strooper et al., 1998), it is possible that upregulation of caveolin-3 and presenilins may coordinately enhance the production of Aβ-amyloid peptide in AD brain. Upregulation of secretase activators (caveolin-3 for β-secretase and presenilins for γ-secretase) and secretase substrate (APP) in AD brain astrocytes indicates that astrocytes are a previously unrecognized and important source of Aβ-amyloid production in AD patients. The data that early plaques are surrounded by caveolin-3–positive astrocytes further support the hypothesis that caveolin-3 upregulation contributes to the genesis of senile plaques. In addition, caveolin-3–positive astrocytes are minor populations of the whole reactive astrocytes, indicating that caveolin-3 upregulation in astrocytes is mediated via a mechanism distinct from that for generation of reactive astrocytes. Thus, the identification of caveolin-3 upregulation in AD astrocytes provides us with new avenues to explore in the complex mechanism of altered APP metabolism and amyloid biogenesis in the AD brain.
Footnotes
This work was supported by United States Public Health Service Grant R29-MH56036, the Prentiss Foundation, and The John and Margaret Knupa Charitable Foundation (to T.O.). B.D.T. was supported by National Institutes of Health (NIH) Grants NS29818 and NS35058. T.I. was supported by NIH National Research Service Award Fellowship 1F32 AG0586-01. M.P.L. was supported by NIH National Cancer Institute Grant R01-CA-80250 and grants from the Charles E. Culpeper Foundation, the G. Harold and Leila Y. Mathers Charitable Foundation, and the Sidney Kimmel Foundation for Cancer Research. We greatly appreciate Ed Koo for providing presenilin antibodies and Sam Sisodia, Samuel Gandy, and Kazuaki Yoshikawa for providing APP antibodies.
Correspondence should be addressed to Dr. Takashi Okamoto, Department of Neurosciences, Cleveland Clinic Foundation, 9500 Euclid Avenue, Cleveland, OH 44195.
REFERENCES
- 1.Barger SW, Harmon AD. Microglial activation by Alzheimer amyloid precursor protein and modulation by apolipoprotein E. Nature. 1997;388:878–881. doi: 10.1038/42257. [DOI] [PubMed] [Google Scholar]
- 2.Bouillot C, Prochiantz A, Rougon G, Allinquant B. Axonal amyloid precursor protein expressed by neurons in vitro is present in a membrane fraction with caveolae-like properties. J Biol Chem. 1996;271:7640–7644. doi: 10.1074/jbc.271.13.7640. [DOI] [PubMed] [Google Scholar]
- 3.Buxbaum JD, Gandy SE, Cicchetti P, Ehrlich ME, Czernk AJ, Fracasso RP, Ramabhadran TV, Unterbeck AJ, Greengard P. Processing of Alzheimer beta/A4 amyloid precursor protein: modulation by agents that regulate protein phosphorylation. Proc Natl Acad Sci USA. 1990;87:6003–6006. doi: 10.1073/pnas.87.15.6003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cameron PL, Ruffin JW, Bollag R, Rasmussen H, Cameron RS. Identification of caveolin and caveolin-related proteins in the brain. J Neurosci. 1997;17:9520–9535. doi: 10.1523/JNEUROSCI.17-24-09520.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Couet J, Li S, Okamoto T, Ikezu T, Lisanti MP. Identification of peptide and protein ligands for the caveolin-scaffolding domain. Implications for the interaction of caveolin with caveolae-associated proteins. J Biol Chem. 1997;272:6525–6533. doi: 10.1074/jbc.272.10.6525. [DOI] [PubMed] [Google Scholar]
- 6.De Strooper B, Saftig P, Craessaerts K, Vanderstichele H, Guhde G, Annaert W, Von Figura K, Van Leuven F. Deficiency of presenilin-1 inhibits the normal cleavage of amyloid precursor protein. Nature. 1998;391:387–390. doi: 10.1038/34910. [DOI] [PubMed] [Google Scholar]
- 7.Dickson DW, Lee SC, Mattiace LA, Yen SH, Brosnan C. Microglia and cytokines in neurological disease, with special reference to AIDS and Alzheimer’s disease. Glia. 1993;7:75–83. doi: 10.1002/glia.440070113. [DOI] [PubMed] [Google Scholar]
- 8.Forman MS, Cook DG, Leight S, Doms RW, Lee VMY. Differential effects of the Swedish mutant amyloid precursor protein on beta-amyloid accumulation and secretion in neurons and nonneuronal cells. J Biol Chem. 1997;272:32247–32253. doi: 10.1074/jbc.272.51.32247. [DOI] [PubMed] [Google Scholar]
- 9.Frautschy SA, Yang F, Irrizarry M, Hyman B, Saido TC, Hsiao K, Cole GM. Microglial response to amyloid plaques in APPsw transgenic mice. Am J Pathol. 1998;152:307–317. [PMC free article] [PubMed] [Google Scholar]
- 10.Galbiati F, Volonte D, Gil O, Zanazzi G, Salzer JL, Sargiacomo M, Scherer PE, Engelman JA, Schlegel A, Parenti M, Okamoto T, Lisanti MP. Expression of caveolins 1 and 2 in differentiating PC12 cells and dorsal root ganglion cells. Proc Natl Acad Sci USA. 1998;95:10257–10262. doi: 10.1073/pnas.95.17.10257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gegelashvili G, Schousboe A, Linnemann D. Expression of amyloid precursor protein (APP) in rat brain and cultured neural cells. Int J Dev Neurosci. 1994;12:703–708. doi: 10.1016/0736-5748(94)90050-7. [DOI] [PubMed] [Google Scholar]
- 12.Haass C, Koo EH, Mellon A, Hung AY, Selkoe DJ. Targeting of cell-surface beta-amyloid precursor protein to lysosomes: alternative processing into amyloid-bearing fragments. Nature. 1992;357:500–503. doi: 10.1038/357500a0. [DOI] [PubMed] [Google Scholar]
- 13.Hayashi Y, Kashiwagi K, Yoshikawa K. Protease inhibitors generate cytotoxic fragments from Alzheimer amyloid protein precursor in cDNA-transfected glioma cells. Biochem Biophys Res Commun. 1992;187:1249–1255. doi: 10.1016/0006-291x(92)90437-p. [DOI] [PubMed] [Google Scholar]
- 14.Howland DS, Trusko SP, Savage MJ, Reaume AG, Lang DM, Hirsch JD, Maeda N, Siman R, Greenberg BD, Scott RW, Flood DG. Modulation of secreted beta-amyloid precursor protein and amyloid beta-peptide in brain by cholesterol. J Biol Chem. 1998;273:16576–16582. doi: 10.1074/jbc.273.26.16576. [DOI] [PubMed] [Google Scholar]
- 15.Hsiao K, Chapman P, Nilsen S, Eckman C, Harigaya Y, Younkin S, Yang F, Cole G. Correlative memory deficits, Abeta elevation, and amyloid plaques in transgenic mice. Science. 1996;274:99–102. doi: 10.1126/science.274.5284.99. [DOI] [PubMed] [Google Scholar]
- 16.Ikezu T, Trapp BD, Song KS, Schlegel A, Lisanti MP, Okamoto T. Caveolae, plasma membrane microdomains for alpha-secretase-mediated processing of the amyloid protein precursor. J Biol Chem. 1998a;273:10485–10495. doi: 10.1074/jbc.273.17.10485. [DOI] [PubMed] [Google Scholar]
- 17.Ikezu T, Ueda H, Trapp BD, Nishiyama K, Sha JF, Volonte D, Galbiati F, Byrd AL, Bassell G, Serizawa H, Lane WS, Lisanti MP, Okamoto T. Affinity-purification and characterization of caveolins from the brain. Brain Res. 1998b;804:177–192. doi: 10.1016/s0006-8993(98)00498-3. [DOI] [PubMed] [Google Scholar]
- 18.Iwatsubo T, Odaka A, Suzuki N, Mizusawa H, Nukina N, Ihara Y. Visualization of A beta 42(43) and A beta 40 in senile plaques with end-specific A beta monoclonals: evidence that an initially deposited species is A beta 42(43). Neuron. 1994;13:45–53. doi: 10.1016/0896-6273(94)90458-8. [DOI] [PubMed] [Google Scholar]
- 19.Kim KS, Wen GY, Bancher CM, Chen J, Sapeenza VJ, Hong H, Wisniewski HM. Detection of quantitation of amyloid b-peptide with 2 monoclonal antibodies. Neurosci Res Commun. 1990;7:118–122. [Google Scholar]
- 20.Kitaguchi N, Tokushima Y, Oishi K, Takahashi Y, Shiojiri S, Nakamura S, Tanaka S, Kodaira R, Ito H. Determination of amyloid beta protein precursors harboring active form of proteinase inhibitor domains in cerebrospinal fluid of Alzheimer’s disease patients by trypsin-antibody sandwich ELISA. Biochem Biophys Res Commun. 1990;166:1453–1459. doi: 10.1016/0006-291x(90)91030-v. [DOI] [PubMed] [Google Scholar]
- 21.Kurzchalia T, Dupree P, Parton RG, Kellner R, Virta H, Lehnert M, Simons K. VIP 21, a 21-kD membrane protein, is an integral component of trans-Golgi-network-derived transport vesicles. J Cell Biol. 1992;118:1003–1014. doi: 10.1083/jcb.118.5.1003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lee S-J, Liyanage U, Bickel PE, Xia W, Lansbury PT, Kosik KS. A detergent-insoluble membrane compartment contains Abeta in vivo. Nat Med. 1998;4:730–734. doi: 10.1038/nm0698-730. [DOI] [PubMed] [Google Scholar]
- 23.McGeer PL, McGeer EG. Anti-inflammatory drugs in the fight against Alzheimer’s disease. Ann NY Acad Sci. 1996;777:213–220. doi: 10.1111/j.1749-6632.1996.tb34421.x. [DOI] [PubMed] [Google Scholar]
- 24.Mochizuki A, Peterson JW, Mufson EJ, Trapp BD. Amyloid load and neuronal elements in Alzheimer’s disease and non-demented individuals with high amyloid plaque density. Exp Neurol. 1996;142:89–102. doi: 10.1006/exnr.1996.0181. [DOI] [PubMed] [Google Scholar]
- 25.Moir RD, Lynch T, Bush AI, Whyte S, Henry A, Portbury S, Multhaup G, Small DH, Tanzi RE, Beyreuther K, Masters CL. Relative increase in Alzheimer’s disease of soluble forms of cerebral Abeta amyloid protein precursor containing the Kunitz protease inhibitory domain. J Biol Chem. 1998;273:5013–5019. doi: 10.1074/jbc.273.9.5013. [DOI] [PubMed] [Google Scholar]
- 26.Okamoto T, Takeda S, Murayama Y, Ogata E, Nishimoto I. Ligand-dependent G protein-coupling of amyloid transmembrane precursor. J Biol Chem. 1995;270:4205–4208. doi: 10.1074/jbc.270.9.4205. [DOI] [PubMed] [Google Scholar]
- 27.Okamoto T, Schlegel A, Scherer PE, Lisanti MP. Caveolins, a family of scaffolding proteins for organizing “preassembled signaling complexes” at the plasma membrane. J Biol Chem. 1998;273:5419–5422. doi: 10.1074/jbc.273.10.5419. [DOI] [PubMed] [Google Scholar]
- 28.Palmert MR, Usiak M, Mayeux R, Raskind M, Tourtellotte WW, Younkin SG. Soluble derivatives of the beta amyloid protein precursor in cerebrospinal fluid: alterations in normal aging and in Alzheimer’s disease. Neurology. 1990;40:1028–1034. doi: 10.1212/wnl.40.7.1028. [DOI] [PubMed] [Google Scholar]
- 29.Pioro EP, Mitsumoto H. Animal models of ALS. Clin Neurosci. 1995;3:375–385. [PubMed] [Google Scholar]
- 30.Ransohoff RM, Devajyothi C, Estes ML, Babcock G, Rudick RA, Frohman EM, Barna BP. Interferon-beta specifically inhibits interferon-gamma-induced class II major histocompatibility complex gene transcription in a human astrocytoma cell line. J Neuroimmunol. 1991;33:103–112. doi: 10.1016/0165-5728(91)90054-b. [DOI] [PubMed] [Google Scholar]
- 31.Scherer PE, Tang Z-L, Chun MC, Sargiacomo M, Lodish HF, Lisanti MP. Caveolin isoforms differ in their N-terminal protein sequence and subcellular distribution: identification and epitope mapping of an isoform-specific monoclonal antibody probe. J Biol Chem. 1995;270:16395–16401. doi: 10.1074/jbc.270.27.16395. [DOI] [PubMed] [Google Scholar]
- 32.Scherer PE, Okamoto T, Chun M, Nishimoto I, Lodish HF, Lisanti MP. Identification, sequence, and expression of caveolin-2 defines a caveolin gene family. Proc Natl Acad Sci USA. 1996;93:131–135. doi: 10.1073/pnas.93.1.131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Scherer PE, Lewis RY, Volonté D, Engelman JA, Galbiati F, Couet J, Kohtz DS, van Donselaar E, Peters P, Lisanti MP. Cell-type and tissue-specific expression of caveolin-2. Caveolins 1 and 2 co-localize and form a stable hetero-oligomeric complex in vivo. J Biol Chem. 1997;272:29337–29346. doi: 10.1074/jbc.272.46.29337. [DOI] [PubMed] [Google Scholar]
- 34.Selkoe DJ. Cell biology of the amyloid beta-protein precursor and the mechanism of Alzheimer’s disease. Annu Rev Cell Biol. 1994a;10:373–403. doi: 10.1146/annurev.cb.10.110194.002105. [DOI] [PubMed] [Google Scholar]
- 35.Selkoe DJ. Normal and abnormal biology of the beta-amyloid precursor protein. Annu Rev Neurosci. 1994b;17:489–517. doi: 10.1146/annurev.ne.17.030194.002421. [DOI] [PubMed] [Google Scholar]
- 36.Shrikant P, Lee SJ, Kalvakolanu I, Ransohoff RM, Benveniste EN. Stimulus-specific inhibition of intracellular adhesion molecule-1 gene expression by TGF-beta. J Immunol. 1996;157:892–900. [PubMed] [Google Scholar]
- 37.Simons K, Ikonen E. Functional rafts in cell membranes. Nature. 1997;387:569–572. doi: 10.1038/42408. [DOI] [PubMed] [Google Scholar]
- 38.Simons M, Keller P, De Strooper B, Beyreuther K, Dotti CG, Simons K. Cholesterol depletion inhibits the generation of beta-amyloid in hippocampal neurons. Proc Natl Acad Sci USA. 1998;95:6460–6464. doi: 10.1073/pnas.95.11.6460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sisodia SS. Beta-amyloid precursor protein cleavage by a membrane-bound protease. Proc Natl Acad Sci USA. 1992;89:6075–6079. doi: 10.1073/pnas.89.13.6075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sisodia SS, Koo EH, Beyreuther K, Unterbeck A, Price DL. Evidence that beta-amyloid protein in Alzheimer’s disease is not derived by normal processing. Science. 1990;248:492–495. doi: 10.1126/science.1691865. [DOI] [PubMed] [Google Scholar]
- 41.Smart EJ, Ying Y, Mineo C, Anderson RGW. A detergent free method for purifying caveolae membrane from tissue cultured cells. Proc Natl Acad Sci USA. 1995;92:10104–10108. doi: 10.1073/pnas.92.22.10104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Smine A, Xu X, Nishiyama K, Katada T, Gambetti P, Yadav SP, Wu X, Shi Y-C, Yasuhara S, Homburger V, Okamoto T. Regulation of brain G-protein, Go, by Alzheimer disease gene presenilin-1. J Biol Chem. 1998;273:16281–16288. doi: 10.1074/jbc.273.26.16281. [DOI] [PubMed] [Google Scholar]
- 43.Song KS, Li S, Okamoto T, Quilliam L, Sargiacomo M, Lisanti MP. Copurification and direct interaction of Ras with caveolin, an integral membrane protein of caveolae microdomains. Detergent free purification of caveolae membranes. J Biol Chem. 1996a;271:9690–9697. doi: 10.1074/jbc.271.16.9690. [DOI] [PubMed] [Google Scholar]
- 44.Song KS, Scherer PE, Tang Z-L, Okamoto T, Li S, Chafel M, Chu C, Kohtz DS, Lisanti MP. Expression of caveolin-3 in skeletal, cardiac, and smooth muscle cells. Caveolin-3 is a component of the sarcolemma and co-fractionates with dystrophin and dystrophin-associated glycoproteins. J Biol Chem. 1996b;271:15160–15165. doi: 10.1074/jbc.271.25.15160. [DOI] [PubMed] [Google Scholar]
- 45.Song KS, Tang Z, Li S, Lisanti MP. Mutational analysis of the properties of caveolin-1. A novel role for the C-terminal domain in mediating homo-typic caveolin-caveolin interactions. J Biol Chem. 1997;272:4398–4403. doi: 10.1074/jbc.272.7.4398. [DOI] [PubMed] [Google Scholar]
- 46.Tang Z, Scherer PE, Okamoto T, Song K, Chu C, Kohtz DS, Nishimoto I, Lodish HF, Lisanti MP. Molecular cloning of caveolin-3, a novel member of the caveolin gene family expressed predominantly in muscle. J Biol Chem. 1996;271:2255–2261. doi: 10.1074/jbc.271.4.2255. [DOI] [PubMed] [Google Scholar]
- 47.Tomita T, Maruyama K, Saido TC, Kume H, Shinozaki K, Tokuhiro S, Capell A, Walter J, Grunberg J, Haass C, Iwatsubo T, Obata K. The presenilin 2 mutation (N141I) linked to familial Alzheimer disease (Volga German families) increases the secretion of amyloid beta protein ending at the 42nd (or 43rd) residue. Proc Natl Acad Sci USA. 1997;94:2025–2030. doi: 10.1073/pnas.94.5.2025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wu C, Butz S, Ying YS, Anderson RGW. Tyrosine kinase receptors concentrated in caveolae-like domains from neuronal plasma membrane. J Biol Chem. 1997;272:3554–3559. doi: 10.1074/jbc.272.6.3554. [DOI] [PubMed] [Google Scholar]
- 49.Yamada T, Sasaki H, Furuya H, Miyata T, Goto I, Sakaki Y. Complementary DNA for the mouse homolog of the human amyloid β protein precursor. Biochem Biophys Res Commun. 1987;149:665–671. doi: 10.1016/0006-291x(87)90419-0. [DOI] [PubMed] [Google Scholar]
- 50.Zhang J, Kang DE, Xia W, Okochi M, Mori H, Selkoe DJ, Koo EH. Subcellular distribution and turnover of presenilins in transfected cells. J Biol Chem. 1998;273:12436–12442. doi: 10.1074/jbc.273.20.12436. [DOI] [PubMed] [Google Scholar]