

Abstract
The symptoms of Parkinson disease are thought to result in part from increased burst activity in globus pallidus neurons. To gain a better understanding of the factors governing this activity, we studied delayed rectifier K+ conductances in acutely isolated rat globus pallidus (GP) neurons, using whole-cell voltage-clamp and single-cell RT-PCR techniques. From a holding potential of −40 mV, depolarizing voltage steps in identified GP neurons evoked slowly inactivating K+ currents. Analysis of the tail currents revealed rapidly and slowly deactivating currents of similar amplitude. The fast component of the current deactivated with a time constant of 11.1 ± 0.8 msec at −40 mV and was blocked by micromolar concentrations of 4-AP and TEA (KD ∼140 μm). The slow component of the current deactivated with a time constant of 89 ± 10 msec at −40 mV and was less sensitive to TEA (KD = 0.8 mm) and 4-AP (KD ∼6 mm). Organic antagonists of Kv1 family channels had little or no effect on somatic currents. These properties are consistent with the hypothesis that the rapidly deactivating current is attributable to Kv3.1/3.2 channels and the slowly deactivating current to Kv2.1-containing channels. Semiquantitative single-cell RT-PCR analysis of Kv3 and Kv2 family mRNAs supported this conclusion. An alteration in the balance of these two channel types could underlie the emergence of burst firing after dopamine-depleting lesions.
Keywords: globus pallidus, delayed rectifier, Kv2.1, Kv3.1/3.2, voltage clamp, single-cell RT-PCR, TEA, 4-AP, potassium channels
Parkinson’s disease (PD) is a common neurodegenerative disease characterized by rigidity, tremor, and bradykinesia (Bergman et al., 1998). The symptoms of disease are attributable to the loss of dopaminergic neurons in the substantia nigra pars compacta (Albin et al., 1989). These dopaminergic neurons richly innervate basal ganglia nuclei, particularly the striatum (Graybiel, 1990). Studies in primate models of PD surprisingly have found few changes in striatal activity (Nisenbaum et al., 1988). However, clear changes have been detected in the activity of one of the major targets of the striatum—the globus pallidus (GP). FollowingN-methyl-phenyl-1,2,3,6-tetrahydropyridine (MPTP) lesions of dopaminergic neurons, neurons in the external segment of the GP exhibit a greater degree of synchrony and burst firing (Filion et al., 1991;Hutchison et al., 1994; Nini et al., 1995). The bursting activity patterns found in this model are correlated with muscle tremor (Bergman et al., 1998). Neural recordings in humans suffering from PD have noted similar alterations in GP activity (Lozano et al., 1996; Taha et al., 1996). The proposition that this pathophysiology plays an important role in determining the motor symptoms of PD is consistent with the success of pallidotomy (Lozano and Lang, 1998).
The intrinsic mechanisms that permit the emergence of burst firing in GP neurons are poorly defined. Although several factors are likely to be involved, voltage-dependent K+ channels must be key enablers of this anomalous activity pattern. For example, delayed rectifier channels regulate spike repolarization and afterpotentials that determine refractory periods and the ability to discharge at high frequencies. At least five gene families are known to code for subunits having delayed rectifier-like properties (Kv1, Kv2, Kv3, eag, KCNQ) (Pongs, 1992; Shi et al., 1997, 1998; H. Wang et al., 1998). Of these, Kv3.1/3.2 channels are unique in their ability to deactivate within 1 msec at −60 mV (Vega-Saenz de Miera et al., 1994; Robertson, 1997). Several studies have concluded that these channels underlie a “fast-spiking” pattern found primarily in GABAergic interneurons (Du et al., 1996; Massengill et al., 1997;Martina et al., 1998; L. Wang et al., 1998). For example, blocking a rapidly deactivating Kv3-like K+ current reduces the ability of interneurons to discharge at high frequencies. On the other hand, Kv2.1/2.2 channels are more slowly deactivating (Hwang et al., 1992; Kirsch et al., 1993). The prolonged deactivation of these channels at negative membrane potentials should increase the relative refractory period, effectively slowing discharge rates. By regulating the coexpression of these Kv3 and Kv2 family channels, neurons may tune their discharge patterns effectively. This view is consistent with recent studies of hippocampal pyramidal neurons and interneurons that used combined patch-clamp and mRNA profiling techniques (Martina et al., 1998).
It is not clear whether globus pallidus neurons use similar mechanisms to control discharge patterning. As a first step toward evaluating this possibility, acutely isolated rat GP neurons were studied with voltage-clamp and single-cell reverse transcription-PCR (scRT-PCR) techniques. These studies revealed the presence of both a rapidly deactivating current attributable to Kv3.1/3.2 channels and a slowly deactivating current attributable to Kv2.1/2.2 channels. The amplitude of these currents at depolarized potentials was nearly the same, suggesting that modest changes in the relative expression of either subunit could have substantial effects on critical current properties and discharge patterning in GP neurons.
MATERIALS AND METHODS
Tissue preparation. Globus pallidus, neostriatal, and basal forebrain neurons from young adult rats were dissociated acutely, using procedures similar to those we have described previously (Surmeier et al., 1995). In brief, the rats were anesthetized with methoxyflurane and decapitated; brains were removed quickly, iced, and blocked for slicing. Sagittal slices (300 μm) were cut with a Microslicer (Dosaka, Kyoto, Japan) while bathed in zero Ca2+ solution containing (in mm): 140 sodium isethionate, 2 KCl, 4 MgCl2, 23 glucose, and 15 HEPES, pH 7.4 (300–305 mOsm/l). Slices were incubated for 1–6 hr at room temperature (20–22°C) in NaHCO3-buffered saline bubbled with 95% O2/5% CO2 containing (in mm): 126 NaCl, 2.5 KCl, 2 CaCl2, 2 MgCl2, 26 NaHCO3, 1.25 NaH2PO4, 1 pyruvic acid, 0.2 ascorbic acid, 0.1NG-nitro-l-arginine, 1 kynurenic acid, and 10 glucose, pH 7.4 (300–305 mOsm/l). Then the slices were removed into the zero Ca2+ solution, and regions of globus pallidus or dorsal neostriatum were dissected with the aid of a dissecting microscope and placed in an oxygenated Cell-Stir chamber (Wheaton, Millville, NJ) containing Pronase (1–2 mg/ml; Sigma protease type XIV, St Louis, MO) in HEPES-buffered HBSS (Sigma) at 35°C. After 20–40 min of enzyme digestion, the tissue was rinsed three times in zero Ca2+ buffer and dissociated mechanically with a graded series of fire-polished Pasteur pipettes. Then the cell suspension was plated into a 35 mm Lux Petri dish containing HEPES-buffered HBSS saline, and the dish was mounted on the stage of an inverted microscope. All reagents were obtained from Sigma.
Electrophysiological methods. Whole-cell recordings used standard techniques (Hamill et al., 1981; Song and Surmeier, 1996). Electrodes were pulled from Corning (Corning, NY) 7052 glass and were fire-polished before use. The internal solution consisted of (in mm): 30–60 K2SO4, 30–60N-methyl-d-glucamine, 2 MgCl2, 40 HEPES, 5 EGTA, 12 phosphocreatine, 2 Na2ATP, 0.2 Na3GTP, and 0.1 leupeptin, pH 7.2, with H2SO4 (osmolarity, 260–270 mOsm/l). The external solution consisted of (in mm): 140 Na-isethionate, 2 KCl, 4 MgCl2, 10 HEPES, 12 glucose, and 0.001 TTX, pH 7.35, with NaOH (osmolarity, 295–305 mOsm/l).
In all experiments Na+ currents were blocked with TTX, and Ca2+ currents were eliminated by replacing the calcium with magnesium in the external solution. In some experiments, 4-aminopyridine (4-AP; Sigma) or tetraethylammonium chloride (TEA; Sigma) was applied. When 4-AP was included in the extracellular solutions, the pH was adjusted to 7.35 by using H2SO4. When 4-AP and TEA were applied at concentrations >1 mm, the osmolarity was adjusted by reducing the concentration of Na-isethionate. Solutions were applied by a gravity-fed sewer pipe system. An array of application capillaries (∼400 μm inner diameter) was positioned a few hundred micrometers from the cell under study. Solution changes were effected by altering the position of the array with a DC drive system controlled by a microprocessor-based controller (Newport, Irvine, CA). Solution changes were complete within <1 sec.
Recordings were obtained with an Axon Instruments 200 patch-clamp amplifier and controlled and monitored with a PC running pClamp software (version 7.0) with a 125 kHz interface (Axon Instruments, Foster City, CA). Electrode resistance was typically 1.5–2.2 MΩ in the bath. After seal rupture the series resistance (4–10 MΩ) was compensated (75–90%) and monitored periodically. Potentials were not corrected for the liquid junction potential, which was estimated to be 1–2 mV. All averaged data are presented as an average ± SEM. All data fits were obtained with Igor Pro (version 3.12, WaveMetrics, Lake Oswego, OR) software by the least-squares method. Activation data were fit with a Boltzmann equation of the form: 1/(1 + exp [(Vh −V)/Vc], where Vstands for membrane potential, Vh is the half-activation voltage, and Vc is the slope constant. Dose–response data were fit with a Langmuir isotherm of the form: C/(C + IC50), whereC stands for a concentration of the blocking agent. Statistical analyses were run with SYSTAT (version 5.2; Evanston, IL); small, nonmatched samples were analyzed with Kruskal–Wallis ANOVA.
Single-cell RT-PCR (scRT-PCR). Two types of scRT-PCR profiling were performed. To maximize mRNA yields, we aspirated some neurons without recording. Isolated neurons were patched in the cell-attached mode and lifted into a stream of control solution. Neurons then were aspirated into the electrode. Electrodes contained ∼5 μl of sterile water. In other experiments the neurons briefly were subjected to whole-cell voltage-clamp recordings before aspiration. In these cases the electrode recording solution was made nominally RNase-free, and the total volume was kept near 5 μl. In both sets of experiments the capillary glass used for making the electrodes was autoclaved and heated to 200°C for 1 hr. Sterile gloves were worn during the procedure to minimize RNase contamination. After aspiration of the neuron the electrode was removed from the holder and broken; the contents were ejected into a 0.5 ml Eppendorf tube containing 3.6 μl of diethyl pyrocarbonate-treated water, 0.7 μl of RNasin (28,000 U/ml), and 0.7 μl of oligo-dT (0.5 μg/μl). The mixture was heated to 70°C and incubated on ice for 1 min. Single-strand cDNA was synthesized from the cellular mRNA by the addition of SuperScript II RT (1 μl, 200 U/μl), 10× PCR buffer, MgCl2 (2 μl, 25 mm), DTT (2 μl, 0.1m), and mixed dNTPs (1 μl, 10 mm), followed by incubation at 42°C for 50 min. The reaction was terminated by heating the mixture at 70°C for 15 min and then icing it. The RNA strand in the RNA–DNA hybrid was removed by adding 1 μl of RNase H (2 U/μl) and was incubated for 20 min at 37°C. All reagents except RNasin (Promega, Madison, WI) were obtained from Life Technologies (Grand Island, NY). The cDNA from the reverse transcription (RT) of RNA in a single neuron was subjected to PCR to detect the expression of various mRNAs. Conventional PCR was performed with a thermal cycler (MJ Research, Watertown, MA). PCR primers were developed from GenBank with the commercially available software OLIGO (National Biosciences, Plymouth, MN). Primers for choline acetyltransferase (ChAT), GAD67, Kv3.4, and parvalbumin (PV) were described previously (Song and Surmeier, 1996; Yan and Surmeier, 1996;Tkatch et al., 1998; Vysokanov et al., 1998). Kv2.1 mRNA (GenBank accession X16476) was detected with a pair of primers 5′-CAACTTCGAGGCGGGAGTC (position 2244) and 5′-TCCAGTCAACCCTTCTGAGGAGTA (position 2449), which give a PCR product of 229 bp. Kv2.2 mRNA (GenBank accession M77482) was detected with a pair of primers 5′-ACCAGGAGGTTAGCCAAAAAGACT (position 1861) and 5′-AGGCCCCTTATCTCTGCTTAGTGT (position 2283), which give a PCR product of 446 bp. Kv3.1 mRNA (GenBank accession X62840) was detected with a pair of primers 5′-CCAACAAGGTGGAGTTCATCAAG (position 1089) and 5′-TGGTGTGGAGAGTTTACGACAGATT (position 1704), which give a PCR product of 640 bp. Kv3.2 mRNA (GenBank accession X62839) was detected with a pair of primers 5′-ACCTAATGATCCCTCAGCGAGTGA (position 1417) and 5′-CAAAATGTAGGTGAGCTTGCCAGAG (position 1692), which give a PCR product of 302 bp.
PCR procedures were performed following procedures designed to minimize the chance of cross-contamination (Cimino et al., 1990). Negative controls for contamination from extraneous and genomic DNA were run for every batch of neurons. To ensure that genomic DNA did not contribute to the PCR products, we aspirated and processed neurons in the normal manner, except that the reverse transcriptase was omitted. Contamination from extraneous sources was checked by replacing the cellular template with water. Both controls were consistently negative in these experiments.
RESULTS
Our previous work has shown that ∼15–20% of the neurons dissociated from the globus pallidus region express ChAT mRNA—a marker of cholinergic neurons (Tkatch et al., 1998). Because it is believed that all globus pallidus projection neurons are exclusively GABAergic (Rouzaire-Dubois et al., 1980; Gritti et al., 1993), these neurons are considered to be displaced cholinergic neurons from the adjacent basal forebrain nucleus of Meynert (Armstrong et al., 1983; Tkatch et al., 1998). Unless otherwise noted, all of the GP neurons reported here expressed mRNA for glutamate decarboxylase (67 kDa isoform) (GAD67), but not ChAT. Of these, 24 GP neurons were subject to physiological analysis, and an additional sixty-three were sampled for scRT-PCR alone.
Tail currents were bi-exponential
Delayed rectifier currents were evoked by depolarizing voltage steps from a holding potential of −40 mV. Previous work had shown that A- and D-type K+ currents inactivate within seconds at this potential (Stefani et al., 1992, 1995; our unpublished observations). The currents evoked by this protocol were either noninactivating or slowly inactivating (Fig.1A). To estimate the voltage dependence of activation and deactivation kinetics, we measured tail currents at −40 mV immediately after the test voltage step. As shown in Figure 1B, tail current amplitudes saturated for test steps above ∼0 mV. Kinetic analysis of the tails clearly indicated that there were fast and slow components of the tail currents (see the semilog plot inset, Fig.1B). The time constant of each component was estimated from a bi-exponential fit of the tail currents after a step to +20 mV. The fast component had a time constant of 11.1 ± 0.8 msec, and the slow component had a time constant of 89 ± 10 msec (n = 10; at −40 mV). These time constants then were used to constrain the bi-exponential fit of tail currents after steps to less depolarized potentials. The fast and slow components of the tail separated in this way are shown in Figure 1, C andD, respectively. Plots of the fast and slow tail amplitude as a function of test voltage were fit with a Boltzmann function (Fig.1E). Although there was a tendency for the slow component to activate at more negative potentials (Vh = −18 mV;Vc = 7 mV) than the fast component (Vh = −13 mV;Vc = 5 mV), this difference was not statistically significant (p > 0.05;n = 4; Kruskal–Wallis ANOVA). In addition to being similar in voltage dependence, these two components were similar in size. The amplitude of the rapidly deactivating current was, on average, 56 ± 3% of the total current (n = 10). The conductance density of the rapidly deactivating currents was, on average, 3.8 ± 0.9 nS/pF, whereas the slowly deactivating current was 3.0 ± 0.8 nS/pF (n = 10).
Fig. 1.

Delayed rectifier currents in GP neurons have rapidly and slowly deactivating components that differ in activation voltage dependence. A, Currents evoked during voltage-clamp experiment by protocol shown at the top of each panel. B, Tail currents obtained during experiment shown in A at −40 mV membrane potential after increasingly more depolarized voltage steps. C, Rapidly deactivating component of tail currents shown in Bobtained by subtraction of steady-state and slow components of double-exponential fit. D, Slowly deactivating component of tail currents obtained by subtraction of steady-state and fast components of double-exponential fit. E, Plot of rapidly (open circles) and slowly (filled circles) deactivating tail current amplitudes as a function of the preceding voltage step for the cell shown in A. Thelines show Boltzmann fits of the experimental data;Vh is the half-activation voltage, andVc is the slope constant.
Because previous studies have suggested that PV expression may be an important predictor of functional properties of GP neurons (Kita, 1994;Kawaguchi et al., 1995), GAD-expressing GP neurons having detectable levels of PV mRNA were compared with those that lacked such expression. Although these two populations did not differ significantly in their current amplitudes, the ratio of the rapidly to slowly deactivating current was significantly larger in neurons having detectable levels of PV mRNA (median = 1.5; n = 4) than in neurons lacking detectable PV mRNA (median = 0.9; n = 5;p < 0.02; Kruskal–Wallis ANOVA).
Rapidly and slowly deactivating currents were pharmacologically distinguishable
The differences in the voltage dependence of the rapidly and slowly deactivating currents suggest that they are attributable to distinct channel proteins. To test this hypothesis, we used pharmacological tools.
First, the sensitivity of each component of the tail current to TEA was examined. TEA commonly is found to block delayed rectifier currents (Rudy, 1988). At the lowest concentration tested in this cell (100 μm), TEA significantly reduced currents evoked by a test step to +40 mV from a holding potential of −60 mV (Fig.2A). Increasing the concentration of TEA progressively blocked more of the current, but a component was left unblocked by the highest concentration tested (10 mm) in this cell (similar results were obtained with up to 50 mm TEA in other cells). Analysis of the fast and slow components of the tail current revealed a differential sensitivity to TEA. The fast component of the tail was very sensitive to TEA (Fig.2C), having an IC50 of 0.14 mm(Fig. 2E). On the other hand, the slow component of the tail current was blocked significantly at higher concentrations (Fig. 2D), having an IC50 (0.80 mm) nearly six times greater than the fast component (Fig.2E).
Fig. 2.

TEA differentially blocks rapidly and slowly deactivating currents. A, Currents evoked in the presence of different concentrations of TEA by the voltage-clamp protocol shown above the traces. B, Tail currents at −40 mV obtained during the experiment shown inA. C, Rapidly deactivating tail currents in increasing concentrations of TEA obtained by the subtraction method described in Figure 1. D, Slowly deactivating tail currents in increasing concentrations of TEA. E, Plot of rapidly and slowly deactivating tail current amplitudes as a function of TEA concentration. The lines represent Langmuir first-order fits of experimental data.
Second, the sensitivity of each component of the tail current to 4-AP was examined. In many vertebrate neurons, 4-AP preferentially blocks A-like potassium current (Rudy, 1988), but recently it has been shown that some types of delayed rectifier current are also sensitive to submillimolar concentrations of 4-AP (Du et al., 1996; Massengill et al., 1997; Martina et al., 1998; L. Wang et al., 1998). These 4-AP-sensitive delayed rectifiers typically deactivate rapidly (Du et al., 1996), much like Kv3.1/3.2 channels (Vega-Saenz de Miera et al., 1994). In GP neurons, low concentrations (0.1 mm) of 4-AP significantly reduced the total current (Fig.3A). As predicted, the rapidly deactivating component of the current was more sensitive to 4-AP than the slowly deactivating component (Fig. 3C,D). The estimated IC50 for the fast component was 0.14 mm, whereas it was >1 mm for the slow component of the tail current (Fig. 3E).
Fig. 3.

4-AP differentially blocks rapidly and slowly deactivating currents. A, Currents evoked in the presence of different concentrations of 4-AP by the voltage-clamp protocol shown above the traces. B, Tail currents at −40 mV obtained during the experiment shown inA. C, Rapidly deactivating tail currents in increasing concentrations of 4-AP obtained by the subtraction method described in Figure 1. D, Slowly deactivating tail currents in increasing concentrations of 4-AP. E, Plot of rapidly and slowly deactivating tail current amplitudes as a function of 4-AP concentration. The lines represent Langmuir first-order fits of experimental data.
Last, the sensitivity of each component of the tail current to margatoxin (MgTX) and α-dendrotoxin (DTX) was examined. These two toxins block Kv1 family channels. DTX blocks Kv1.1, Kv1.2, and Kv1.6 channels, and MgTx blocks Kv1.2 and Kv1.3 channels (Grupe et al., 1990;Grissmer et al., 1994; Vega-Saenz de Miera et al., 1994). Neither toxin substantially reduced whole-cell currents. MgTx (5 nm) reduced peak current by only 9 ± 3% at +40 mV (n= 4; data not shown). DTX (100 nm) did not have any detectable effect in seven of nine cells tested (data not shown). In two cells it reduced peak current by ∼15% at +40 mV (data not shown). These results argue that Kv1 family channels do not make a major contribution to proximal somatodendritic K+currents and are consistent with immunocytochemical studies suggesting an axonal and terminal location in globus pallidus neurons (Sheng et al., 1992; Rhodes et al., 1997) and other cell types (Sheng et al., 1993; Martina et al., 1998; Song et al., 1998) (but seeMaletic-Savatic et al., 1995).
Single-cell RT-PCR analysis revealed Kv2.1 and Kv3.1/3.2 coexpression
The biophysical and pharmacological properties of two components of the delayed rectifier suggest that they are attributable to two distinct channel types. Rapid deactivation and high affinity for both TEA and 4-AP are characteristics typical of channels containing Kv3.1/3.2 subunits (Vega-Saenz de Miera et al., 1994). The insensitivity to MgTx and DTX is also consistent with this identification. In agreement with this conclusion, scRT-PCR analysis detected Kv3.1 mRNA in 21 of 31 GP neurons that were tested. Kv3.2 mRNA was detected in a smaller percentage of GP neurons (6/13). To gain a better understanding of the less than perfect detection probabilities, we undertook semiquantitative analyses of Kv3.1 and Kv3.2 mRNA abundance (Song et al., 1998; Tkatch et al., 1998). These studies used a serial dilution technique to determine what fraction of the total cellular cDNA was required to reach a standard detection threshold with PCR. The higher the abundance of a particular template, the smaller will be the fraction of the cellular cDNA required to reach the detection threshold. Because this approach controls for differences in reverse transcription and amplification efficiency, a transcript can be compared across a single population or between populations. In the case at hand, if two populations of GP neuron were present—one expressing high levels of Kv3.1/3.2 and another expressing little or none—the distributions of detection thresholds should be bimodal. In fact, the distributions of both Kv3.1 and Kv3.2 were mainly unimodal (Fig.4B,C), arguing that, insofar as these mRNAs were concerned, GP neurons were approximately similar (these results do not exclude the possibility that there are smaller quantitative differences between GP neurons). A comparison of neurons having detectable levels of PV with those lacking detectable levels failed to reveal any significant differences in Kv3.1 or Kv3.2 expression levels. This conclusion is consistent with the electrophysiological data showing the presence of a rapidly deactivating component of the tail current in all GP neurons, regardless of PV detection.
Fig. 4.
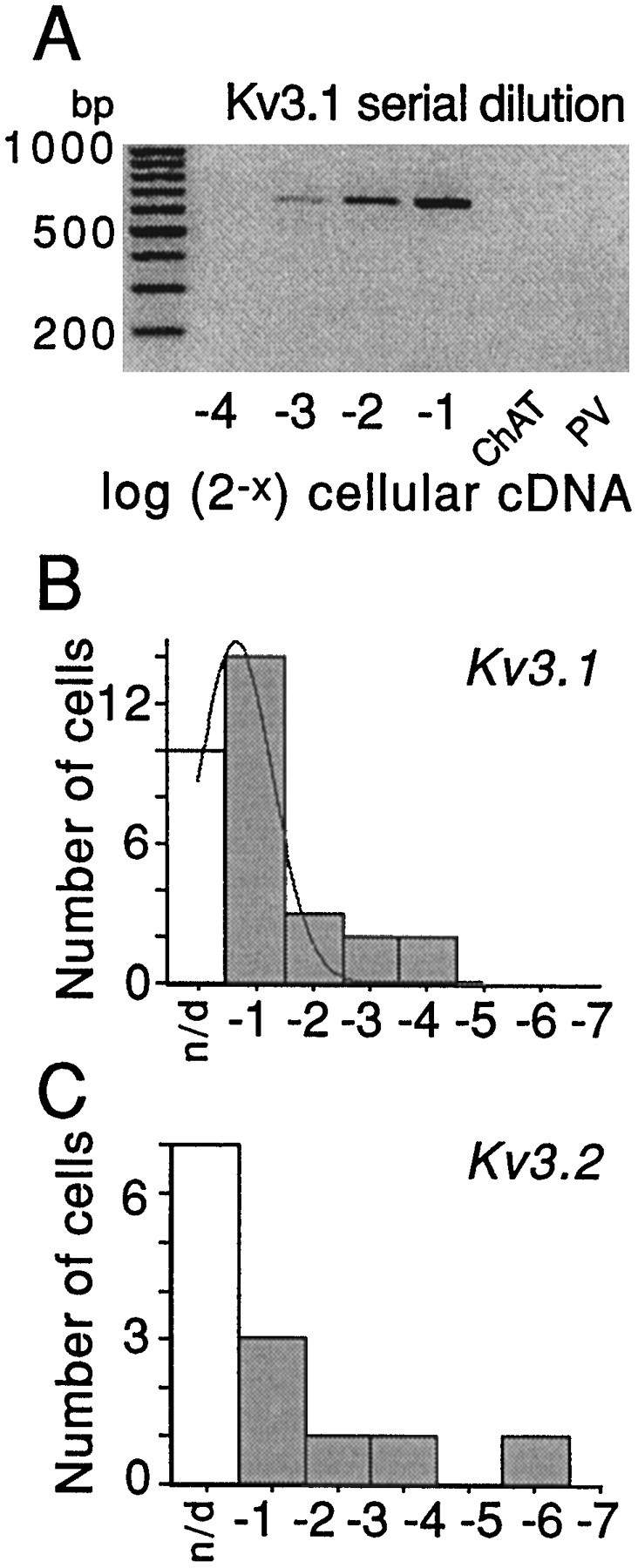
Serial dilution experiments demonstrate that Kv3.1 and Kv3.2 mRNAs are expressed by GP neurons and that variation in detection frequency is not likely to reflect prominent subpopulations.A, Representative serial dilution gel for Kv3.1 mRNA detection in GP neuron. The first lane on theleft of gel is the marker. The first laneon the right is for PV mRNA detection with no visible PCR product band; hence, the cell was PV mRNA-negative. Thesecond band on the right is for ChAT mRNA detection with no visible PCR product band; hence the neuron was not ChAT-positive. The four lanes in betweenwere obtained after the use of an increasing (from leftto right) amount of total cellular cDNA (expressed in log2 units) to detect Kv3.1 mRNA. Note that one-eighth of the total cDNA was enough to detect Kv3.1 transcripts in this cell.B, Summary of detection thresholds for Kv3.1 mRNA detection in GP neurons. The thin line represents a Gaussian fit. C, Summary of detection thresholds for Kv3.2 mRNA detection in GP neurons.
Less clear is the identity of the slowly deactivating component of the delayed rectifier currents. Kv2.1 channels are slowly deactivating, with time constants similar to those seen here. They are also relatively insensitive to 4-AP and insensitive to MgTx and DTX—as is the slowly deactivating current in GP neurons. The one notable difference between the reported properties of Kv2.1 channels and the GP current is their sensitivity to TEA. In heterologous expression systems, rat Kv2.1 channels have a very low affinity for TEA, having IC50 values between 3 and 10 mm (Frech et al., 1989; Taglialatela et al., 1991; Ikeda et al., 1992). In contrast, our estimated IC50 was <1 mm.
Two additional experiments were performed to test the linkage between the slowly deactivating current and Kv2.1 channels. First, in addition to slow deactivation, Kv2.1 channels inactivate slowly at depolarized potentials (VanDongen et al., 1990). To determine whether the slowly deactivating current also inactivated slowly, we isolated this component of the current by recording in the presence of 0.3 mm TEA (approximately twice the IC50 of the rapidly deactivating component). The inactivating component of the remaining current was isolated by subtraction. As shown in Figure5A, currents were evoked first by a step to +40 mV from a holding potential of −60 mV. The noninactivating part of the current was isolated by stepping to 0 mV for 10 sec before the test step to +40 mV. Subtraction of the currents evoked with the prepulse from those evoked without the prepulse currents yielded an estimate of the inactivating current (Fig.5B). The initial component of the current isolated by this procedure inactivated more rapidly than currents attributable to Kv2.1 (VanDongen et al., 1990). Based on our profiling data (not shown), this rapidly inactivating component of the current is attributable mainly to channels containing Kv3.4 subunits (Schroter et al., 1991; Vega-Saenz de Miera et al., 1994). These channels inactivate almost completely in the first 500 msec of the step at +40 mV and should not contribute to tail currents. So, in the presence of 0.3 mm TEA, currents late in the response should be attributable mainly to Kv2.1-like channels. In agreement with this conclusion, tail currents at the end of the step were mainly slow (slow tail amplitude was ∼80% of the total), in contrast to the control condition in which the rapidly deactivating component was ∼60% of the peak current. Semilog plots of evoked currents also showed that the late component of the currents decayed with a time constant near 3 sec at +40 mV (Fig. 5B), similar to that described for Kv2.1 current in heterologous systems (VanDongen et al., 1990; Klemic et al., 1998). To determine TEA sensitivity of this component, we recorded currents in increasing concentrations of TEA (Fig. 5A,C). A plot of the fraction of the inactivating current (measured at the end of the step) blocked as a function of TEA concentration is shown in Figure 5D. The IC50 estimated from a Langmuir isotherm fit of these data was 0.8 mm—the same value obtained for the slowly deactivating tail currents.
Fig. 5.

Slowly inactivating currents have pharmacological properties similar to those of the slowly deactivating currents. A, Currents evoked by the voltage-clamp protocol shown at the top. The protocol was applied each 40 sec. The thin lines show currents evoked with a prepulse to 0 mV. The protocol was applied in the presence of 0.3, 1, and 10 mm TEA. B, Semilogarithmic plot of the current, obtained by stepping from −60 to 0 mV. The solid line shows the exponential fit of the slow component of the data; the time constant was 3.4 sec. C, Inactivating fraction of the current at +40 mV. This current was obtained by subtracting the currents evoked by the step to +40 mV without and with the 10 sec prepulse to 0 mV. D, A graph depicts the normalized amplitude of the block by TEA of the inactivating current shown in C. It was assumed that 0.3 mm TEA leaves the same fraction of inactivating current as seen Figure 2,D and E.
Next, scRT-PCR experiments were performed to determine whether GP neurons expressed Kv2.1 mRNA. In 12 of 16 GP neurons that were examined, Kv2.1 mRNA was detected. Kv2.2 mRNA also was detected, but at much lower frequencies (two of eight). Multiplex amplification consistently found coexpression of Kv2.1 and Kv3.1 mRNAs (data not shown). Serial dilution experiments showed that Kv2.1 detection thresholds were distributed unimodally, arguing for a single population of GP neurons insofar as Kv2.1 expression was concerned. This again was consistent with the electrophysiological data showing that all GP neurons expressed the slowly deactivating current. Then an attempt was made to correlate Kv2.1 detection thresholds in individual GP neurons to current amplitudes recorded in that neuron. In these experiments the amplitude of the inactivating current was measured (Fig. 5) rather than the tail currents to avoid the possible contribution of KCNQfamily genes—these genes encode a noninactivating channel with a pharmacological profile and deactivation kinetics similar to that of Kv2.1 channels (H. Wang et al., 1998). In our sample of GP neurons the amplitude of the slowly inactivating current measured in the presence of 0.3 mm TEA and Kv2.1 detection thresholds were positively correlated, but the correlation was not strong. For example, at a given current amplitude the detection thresholds could vary by a factor of two or more. Because the scRT-PCR was performed after extended electrophysiological recording, the weakness of the correlation is not particularly surprising. Variation in mRNA degradation during recording, aspiration, or RT efficiency all could have contributed to the failure to detect a correlation.
To overcome this obstacle, we altered our experimental strategy in two ways. First, to increase the range of Kv2.1 mRNA expression in our sample, we studied two additional populations of neuron. Basal forebrain cholinergic neurons and striatal cholinergic interneurons both have Kv2.1-like currents but in very different relative amplitudes. Initial profiling work suggested that basal forebrain neurons expressed high levels of Kv2.1 mRNA, and cholinergic interneurons expressed low levels. Second, rather than trying to profile neurons after prolonged recording that could compromise our ability to estimate Kv2.1 mRNA abundance accurately, we divided neurons into two groups. One group was studied electrophysiologically to determine the amplitude of the Kv2.1-like current. These neurons were profiled only for phenotyping markers (GAD67, ChAT, PV). The other group was simply aspirated and profiled without recording. Because basal forebrain and striatal cholinergic neurons could be identified visually in the dissociated cell preparation, sampling from them was relatively straightforward. Then these neurons were subjected to serial dilution analysis of Kv2.1 mRNA abundance.
The serial dilution experiments are summarized in Figure6. On the left side of each panel is a representative gel from an individual neuron showing the serial dilution of cellular cDNA. On the right of each panel is a histogram showing the distribution of detection thresholds in our sample of neurons. Each histogram is fit with a Gaussian function. Note that, as with Kv3.1 mRNA, the detection thresholds in GP neurons were distributed unimodally, arguing that there were not pronounced differences in expression within this population. Explicit comparisons of neurons having detectable levels of PV mRNA with those lacking detectable levels failed to resolve a clear difference in Kv2.1 mRNA abundance. As can be seen by comparing the distributions in the other neuronal populations, Kv2.1 mRNA was of intermediate abundance in GP neurons relative to that in basal forebrain cholinergic neurons and striatal cholinergic interneurons.
Fig. 6.
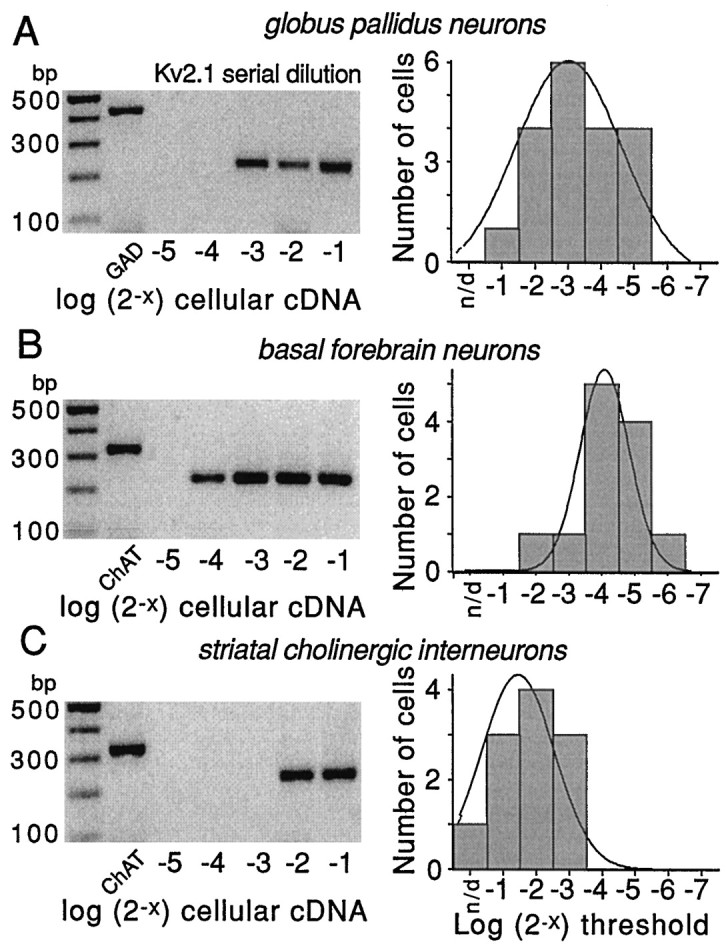
Serial dilution experiments demonstrate differences in Kv2.1 mRNA abundance in three cell types. Representative serial dilution gels for each cell type are shown on the left side of each panel. The first lane on theleft of gels is the marker; the second lane is a phenotyping transcript (GAD67 or ChAT). Thelast five lanes are Kv2.1 amplicons produced by runs with increasing fractions of total cellular cDNA (expressed in log2 units). Note that in all three cells the use of one-half of the total cellular cDNA resulted in detection. However, only 1/16th of the total cDNA was sufficient in basal forebrain cholinergic neurons. On the right side of each panel is the distribution of detection thresholds for Kv2.1 mRNA in that cell type. Note that Kv2.1 mRNA appeared to be most abundant in the basal forebrain cholinergic neurons, of intermediate abundance in GP neurons, and least abundant in striatal cholinergic interneurons. Thethin line is a Gaussian fit of the distribution.
Next, the amplitude of the inactivating current in the presence of 0.3 mm TEA and 100 nm DTX was measured with a protocol like that described in Figure 5. Again, currents were measured at the end of the test step to minimize the contribution of Kv3.4 and any other rapidly inactivating channel. A representative set of current traces from a GP neuron is shown in Figure7A. The mean Kv2.1-like current measured in each population then was plotted against the mean detection threshold (expressed as a fraction of total cDNA) for Kv2.1 mRNA in that same population. The result is shown in Figure7B. The data were very well fit with a straight line (R2 = 0.98) having a zero intercept. The strength of the correlation argues that the slowly inactivating, slowly deactivating current in GP neurons is attributable to K+ channels containing Kv2.1 subunits.
Fig. 7.
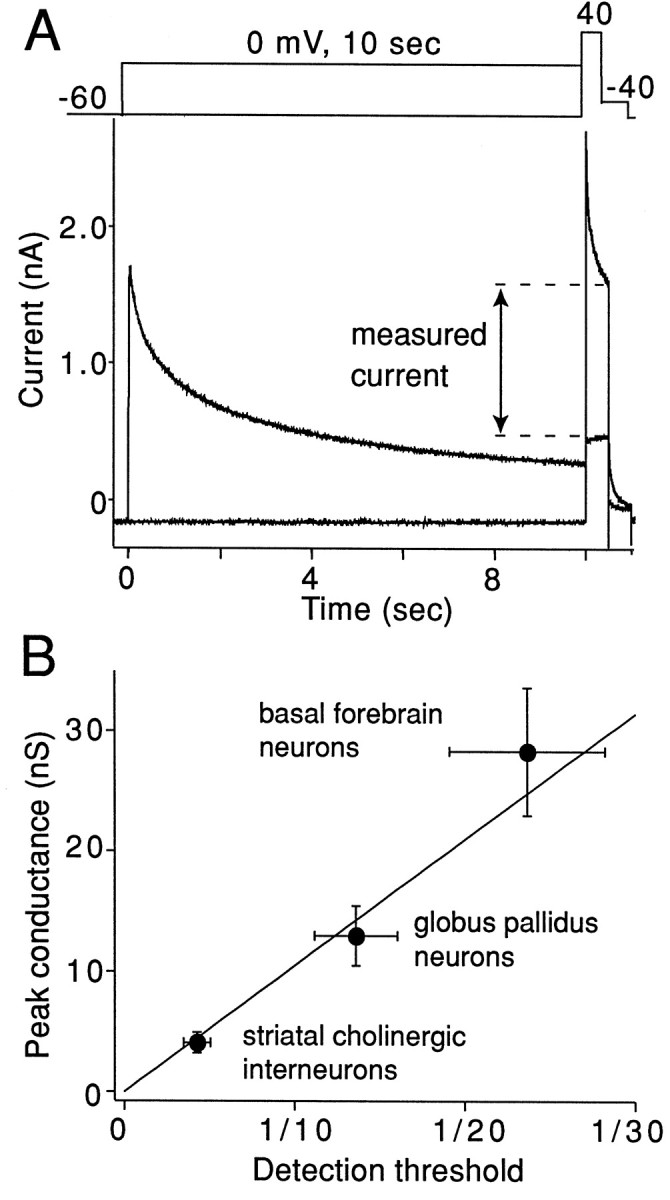
Kv2.1-like currents are related linearly to estimates of Kv2.1 mRNA abundance. A, K+ currents were evoked by the voltage-clamp protocol shown above the traces. Currents evoked with and without a prepulse to 0 mV are superimposed. Protocols were separated by a 40 sec recovery period. B, Average amplitudes of the measured current in three cell types (filled circles) are plotted against the average of detection threshold obtained from the data shown in Figure 6(right panels). The line represents the regression line fit to the data points (R2 = 0.98). The thin lines from the filled circles represent SEM for the current amplitude (vertical lines) and for the detection threshold (horizontal lines).
DISCUSSION
Our results show that delayed rectifier currents in rat GP neurons have rapidly and slowly deactivating components that are approximately equal in size. In addition to deactivation kinetics, these two currents differed in their activation voltage dependence and their sensitivity to the inorganic K+ channel blockers TEA and 4-AP. These differences were consistent with their attribution to distinct channel types. Although tail currents arising from the deinactivation of rapidly inactivating A-like K+ channels have been described (Demo and Yellen, 1991; Ruppersberg et al., 1991), these currents are unlikely to have made a significant contribution to the measurements here. These currents arise primarily from channels passing through an open state in recovering from N-type inactivation. Two types of A-like current were observed in GP neurons: a high-threshold, TEA-sensitive current attributable to Kv3.4 channels (see Fig. 5) and a low-threshold, TEA-insensitive current attributable to Kv4 family channels (our unpublished observations). At −40 mV (where tail currents were measured), Kv4 family channels are inactivated completely (Serodio et al., 1994; Song et al., 1998), eliminating any concern about the contribution of these channels to the tail currents. Although potential contributors, the absence of a slow TEA-sensitive component of the tail current argues that deinactivation of Kv3.4 channels was not a significant factor in our paradigm (Ruppersberg et al., 1991).
Rapidly deactivating currents are attributable to Kv3.1/3.2 channels
The rapidly deactivating current was very sensitive to TEA and 4-AP, having IC50 values of 140 μm for both blockers. On the other hand, this current was insensitive to DTX and MgTX. In addition, this component of the current activated at relatively depolarized potentials (Vh = −13 mV). All of these properties are similar to those described for channels containing Kv3.1/3.2 subunits (Vega-Saenz de Miera et al., 1994), with the exception of a slightly more negative activation voltage dependence, which may be attributed to channel phosphorylation (Murakoshi et al., 1997) or minor differences in recording conditions. In agreement with this attribution, both Kv3.1 and Kv3.2 mRNA were detected readily in GP neurons. This finding is in agreement with previous studies localizing Kv3.1 mRNA in the globus pallidus (Weiser et al., 1994). Serial dilution via scRT-PCR revealed that the abundance of these mRNAs was mainly unimodal, suggesting that there were not prominent subpopulations of the GP neuron, based on expression levels of either mRNA. There also did not appear to be any significant correlation between Kv3.1/3.2 mRNA expression and parvalbumin mRNA expression. Additional studies will be required to determine whether the expression of Kv3 family subunits or the properties of currents arising from these channels are correlated with other features of GP neurons, such as axonal projection site.
Slowly deactivating currents are attributable to Kv2.1 channels
The slowly deactivating delayed rectifier currents were relatively insensitive to 4-AP (IC50 ∼6 mm) but possessed a moderate sensitivity to TEA (IC50 = 0.8 mm). These currents were insensitive to DTX and only marginally sensitive to MgTX. This pharmacological profile suggests that channels containing Kv1.1–3 and Kv1.6 subunits do not make a major contribution to this somatodendritic current (Stuhmer et al., 1989; Swanson et al., 1990; Leonard et al., 1992; Garcia-Calvo et al., 1993; Grissmer et al., 1994; Tytgat et al., 1995), despite the fact that several of these mRNAs (Kv1.1, 1.2, 1.5, and 1.6) were detected by scRT-PCR analysis of GP neurons. The failure to detect substantial Kv1 family currents in the soma and proximal dendrites of GP neurons is consistent with a number of other studies suggesting that these channels are either in axonal or in distal dendritic regions (Sheng et al., 1992; Song et al., 1998) (cf. Maletic-Savatic et al., 1995).
In contrast, Kv2 family channels possess a pharmacological profile similar to that of the slowly deactivating current. Kv2.1 channels are insensitive to DTX and MgTx (Knaus et al., 1995; Swartz and MacKinnon, 1995) and relatively insensitive to 4-AP (Frech et al., 1989; Kirsch et al., 1993). In addition, Kv2.1 currents are slowly inactivating between −60 and 0 mV, much like the currents producing the slow tail current (VanDongen et al., 1990). These properties differentiate Kv2.1 currents from those attributable to either Kv1-, Kv3-, KCNQ-, or eag-family channels. As discussed above, Kv1 family channels are unlikely to have made a major contribution to the slowly deactivating delayed rectifier current. A contribution of Kv3-family channels, including the slowly inactivating Kv3.3 channel, can be excluded on the basis of their sensitivity to 4-AP (Schroter et al., 1991; Vega-Saenz de Miera et al., 1994). KCNQ-family channels are unlikely to have contributed to any significant extent because they exhibit very little inactivation at potentials below +10 mV (Pusch et al., 1998; Tristani-Firouzi and Sanguinetti, 1998; Yang et al., 1998) and are very insensitive to 4-AP (Yang et al., 1998). Last, eag-family channels can be discarded as important determinants of the observed currents because of their relative insensitivity to both TEA and 4-AP (Faravelli et al., 1996;Shi et al., 1998). These factors all point to Kv2 family channels as being responsible for the slowly deactivating, slowly inactivating delayed rectifier in GP neurons.
Another compelling argument that the kinetically slower delayed rectifier current is attributable to channels containing Kv2.1 subunits comes from the scRT-PCR experiments. First, semiquantitative scRT-PCR profiles showed that Kv2.1 mRNA was expressed by GP neurons. On a cell-by-cell basis, Kv2.1 mRNA abundance estimates were correlated positively with Kv2.1-like current amplitudes. When the range of Kv2.1 mRNA expression was expanded and profiling was performed only on neurons that had not been subjected to extended dialysis, a very linear relationship was found between Kv2.1 mRNA abundance and Kv2.1-like conductance over a broad range. This relationship resembles that reported between shal gene expression and A-current magnitude in lobster stomatogastric neurons (Baro et al., 1997). It is difficult to imagine how this strong correlation could have arisen in our case if Kv2.1 subunits were not participants in the channels underlying these currents. This conclusion does not exclude the possibility of heteromeric channels, but the available evidence suggests that Kv2.1/Kv2.2 heteromultimeric channels are unlikely to be important (Blaine and Ribera, 1998).
The only significant difference between the properties of the slowly deactivating K+ current and those of Kv2.1 channels in heterologous expression systems is TEA sensitivity. Kv2.1 channels have been reported to have a TEA IC50 of 3–10 mm (Frech et al., 1989; Taglialatela et al., 1991; Ikeda et al., 1992). In GP neurons the slowly deactivating current had a TEA IC50 of 0.8 mm. The Kv2.1-like current in striatal cholinergic interneurons and basal forebrain neurons possessed a similar TEA sensitivity (data not shown). There are several possible explanations for this discrepancy. Post-translational modification of the extracellular face of the channel (e.g., glycosylation) could alter the apparent affinity for TEA (Kavanaugh et al., 1991). Another possibility is that auxiliary subunits alter TEA sensitivity (Zagotta et al., 1989). Auxiliary subunits are known to prolong deactivation time constants (Kramer et al., 1998) and may be responsible for a somewhat slower deactivation time constant in GP neurons at −40 mV (∼89 msec) than found in Xenopus oocytes (∼45 msec). A definitive test of the hypothesis that Kv2.1 subunits contribute to the slowly deactivating channels will require alterations in Kv2.1 expression or function in situ. Negative-dominant strategies that use viral vectors currently are being explored to this end. Another means of providing a molecular identification would be the application of Kv2-specific antibodies that disrupt channel function (Murakoshi and Trimmer, 1999).
The ratio of rapidly to slowly deactivating currents may govern the ability to discharge at high rates
Rapidly deactivating K+ currents like those attributable to Kv3.1/3.2 channels have been associated with “fast spiking” or the capacity to discharge at high rates for a sustained period of time in a variety of brain neurons, including interneurons (Du et al., 1996; Massengill et al., 1997; Martina et al., 1998) and auditory neurons (L. Wang et al., 1998). The speed with which these channels deactivate minimizes hyperpolarizing K+currents after spike repolarization, effectively shortening the relative refractory period. However, these rapidly deactivating currents typically are coexpressed with more slowly deactivating currents that can moderate their impact on discharge patterning and rates. For example, Martina et al. (1998) found that the ratio of rapidly to slowly deactivating currents in fast spiking hippocampal interneurons was ∼2, whereas it was <0.5 in slowly spiking hippocampal pyramidal neurons. In GP neurons having detectable levels of PV mRNA, this ratio was close to that in hippocampal interneurons (∼1.5), whereas it was significantly lower in neurons lacking detectable levels of PV mRNA (∼0.9). This difference is consistent with reports that approximately three-fourths of the GP population (presumably PV-expressing neurons) in rat are capable of discharging repetitively at high rates (Kita and Kitai, 1991). A similar behavior is found in primates, for which a high, regular discharge rate is thought to be functionally important (Wichmann and DeLong, 1996;Bergman et al., 1998). Although our scRT-PCR profiling was not able to resolve differences in Kv3.1 and Kv2.1 expression that could explain the differences in the ratio of rapidly to slowly deactivating currents, it is our working hypothesis that this simply reflects current technical limitations in estimating small differences in mRNA abundance in single cells.
After the loss of dopaminergic neurons innervating the basal ganglia, many GP neurons begin to discharge more frequently in bursts and at higher rates within bursts (Filion and Tremblay, 1991; Hutchison et al., 1994). This aberrant high-frequency discharge is thought to be a critical determinant in the emergence of tremor and rigidity in Parkinson’s disease (Taha et al., 1996; Wichmann and DeLong, 1996). In rats that followed similar manipulations in dopamine levels, Kv3.1 mRNA is upregulated in GP neurons (Chesselet et al., 1998). This upregulation can be expected to alter the ratio of rapidly deactivating (Kv3.1/3.2) channels to slowly deactivating Kv2.1 channels in GP neurons. One consequence of this change would be an enhanced ability to discharge at high frequencies, as observed after dopamine depletion. Although other ionic conductances are likely to contribute to the emergence of this altered pattern of activity, genetic manipulations that serve to normalize the Kv3.1/Kv2.1 ratio should assist in suppressing unwanted high-frequency activity.
Footnotes
This work was supported by National Institute of Neurological Diseases and Stroke Grants NS 26473 and NS 34696 to D.J.S.
Correspondence should be addressed to Dr. D. James Surmeier, Department of Physiology/Northwestern University Institute for Neuroscience, Northwestern University Medical School, Searle 5-474, 320 East Superior Street, Chicago, IL 60611.
REFERENCES
- 1.Albin RL, Young AB, Penney JB. The functional anatomy of basal ganglia disorders [see comments]. Trends Neurosci. 1989;12:366–375. doi: 10.1016/0166-2236(89)90074-x. [DOI] [PubMed] [Google Scholar]
- 2.Armstrong DM, Saper CB, Levey AI, Wainer BH, Terry RD. Distribution of cholinergic neurons in rat brain: demonstrated by the immunocytochemical localization of choline acetyltransferase. J Comp Neurol. 1983;216:53–68. doi: 10.1002/cne.902160106. [DOI] [PubMed] [Google Scholar]
- 3.Baro DJ, Levini RM, Kim MT, Willms AR, Lanning CC, Rodriguez HE, Harris-Warrick RM. Quantitative single-cell reverse transcription-PCR demonstrates that A-current magnitude varies as a linear function of shal gene expression in identified stomatogastric neurons. J Neurosci. 1997;17:6597–6610. doi: 10.1523/JNEUROSCI.17-17-06597.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bergman H, Feingold A, Nini A, Raz A, Slovin H, Abeles M, Vaadia E. Physiological aspects of information processing in the basal ganglia of normal and parkinsonian primates. Trends Neurosci. 1998;21:32–38. doi: 10.1016/s0166-2236(97)01151-x. [DOI] [PubMed] [Google Scholar]
- 5.Blaine JT, Ribera AB. Heteromultimeric potassium channels formed by members of the Kv2 subfamily. J Neurosci. 1998;18:9585–9593. doi: 10.1523/JNEUROSCI.18-23-09585.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chesselet MF, Delfs JM, Mackenzie L. Dopamine control of gene expression in basal ganglia nuclei: striatal and nonstriatal mechanisms. Adv Pharmacol. 1998;42:674–677. doi: 10.1016/s1054-3589(08)60838-8. [DOI] [PubMed] [Google Scholar]
- 7.Cimino GD, Metchette K, Isaacs ST, Zhu YS. More false-positive problems [letter and comment]. Nature. 1990;345:773–774. doi: 10.1038/345773b0. [DOI] [PubMed] [Google Scholar]
- 8.Demo SD, Yellen G. The inactivation gate of the Shaker K+ channel behaves like an open-channel blocker. Neuron. 1991;7:743–753. doi: 10.1016/0896-6273(91)90277-7. [DOI] [PubMed] [Google Scholar]
- 9.Du J, Zhang L, Weiser M, Rudy B, McBain CJ. Developmental expression and functional characterization of the potassium-channel subunit Kv3.1b in parvalbumin-containing interneurons of the rat hippocampus. J Neurosci. 1996;16:506–518. doi: 10.1523/JNEUROSCI.16-02-00506.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Faravelli L, Arcangeli A, Olivotto M, Wanke E. A HERG-like K+ channel in rat F-11 DRG cell line: pharmacological identification and biophysical characterization. J Physiol (Lond) 1996;496:13–23. doi: 10.1113/jphysiol.1996.sp021661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Filion M, Tremblay L. Abnormal spontaneous activity of globus pallidus neurons in monkeys with MPTP-induced parkinsonism. Brain Res. 1991;547:142–151. [PubMed] [Google Scholar]
- 12.Frech GC, VanDongen AM, Schuster G, Brown AM, Joho RH. A novel potassium channel with delayed rectifier properties isolated from rat brain by expression cloning. Nature. 1989;340:642–645. doi: 10.1038/340642a0. [DOI] [PubMed] [Google Scholar]
- 13.Garcia-Calvo M, Leonard RJ, Novick J, Stevens SP, Schmalhofer W, Kaczorowski GJ, Garcia ML. Purification, characterization, and biosynthesis of margatoxin, a component of Centruroides margaritatus venom that selectively inhibits voltage-dependent potassium channels. J Biol Chem. 1993;268:18866–18874. [PubMed] [Google Scholar]
- 14.Graybiel AM. Neurotransmitters and neuromodulators in the basal ganglia. Trends Neurosci. 1990;13:244–254. doi: 10.1016/0166-2236(90)90104-i. [DOI] [PubMed] [Google Scholar]
- 15.Grissmer S, Nguyen AN, Aiyar J, Hanson DC, Mather RJ, Gutman GA, Karmilowicz MJ, Auperin DD, Chandy KG. Pharmacological characterization of five cloned voltage-gated K+ channels, types Kv1.1, 1.2, 1.3, 1.5, and 3.1, stably expressed in mammalian cell lines. Mol Pharmacol. 1994;45:1227–1234. [PubMed] [Google Scholar]
- 16.Gritti I, Mainville L, Jones BE. Codistribution of GABA- with acetylcholine-synthesizing neurons in the basal forebrain of the rat. J Comp Neurol. 1993;329:438–457. doi: 10.1002/cne.903290403. [DOI] [PubMed] [Google Scholar]
- 17.Grupe A, Schroter KH, Ruppersberg JP, Stocker M, Drewes T, Beckh S, Pongs O. Cloning and expression of a human voltage-gated potassium channel. A novel member of the RCK potassium channel family. EMBO J. 1990;9:1749–1756. doi: 10.1002/j.1460-2075.1990.tb08299.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hamil OP, Marty A, Neher E, Sakmann B, Sigworth FJ. Improved patch-clamp techniques for high resolution current recording from cells and cell free membrane patches. Pflügers Arch. 1981;391:85–100. doi: 10.1007/BF00656997. [DOI] [PubMed] [Google Scholar]
- 19.Hutchison WD, Lozano AM, Davis KD, Saint-Cyr JA, Lang AE, Dostrovsky JO. Differential neuronal activity in segments of globus pallidus in Parkinson’s disease patients. NeuroReport. 1994;5:1533–1537. doi: 10.1097/00001756-199407000-00031. [DOI] [PubMed] [Google Scholar]
- 20.Hwang PM, Glatt CE, Bredt DS, Yellen G, Snyder SH. A novel K+ channel with unique localizations in mammalian brain: molecular cloning and characterization. Neuron. 1992;8:473–481. doi: 10.1016/0896-6273(92)90275-i. [DOI] [PubMed] [Google Scholar]
- 21.Ikeda SR, Soler F, Zuhlke RD, Joho RH, Lewis DL. Heterologous expression of the human potassium channel Kv2.1 in clonal mammalian cells by direct cytoplasmic microinjection of cRNA. Pflügers Arch. 1992;422:201–203. doi: 10.1007/BF00370422. [DOI] [PubMed] [Google Scholar]
- 22.Kavanaugh MP, Varnum MD, Osborne PB, Christie MJ, Busch AE, Adelman JP, North RA. Interaction between tetraethylammonium and amino acid residues in the pore of cloned voltage-dependent potassium channels. J Biol Chem. 1991;266:7583–7587. [PubMed] [Google Scholar]
- 23.Kawaguchi Y, Wilson CJ, Augood SJ, Emson PC. Striatal interneurones: chemical, physiological, and morphological characterization. Trends Neurosci. 1995;18:527–535. doi: 10.1016/0166-2236(95)98374-8. [DOI] [PubMed] [Google Scholar]
- 24.Kirsch GE, Shieh CC, Drewe JA, Vener DF, Brown AM. Segmental exchanges define 4-aminopyridine binding and the inner mouth of K+ pores. Neuron. 1993;11:503–512. doi: 10.1016/0896-6273(93)90154-j. [DOI] [PubMed] [Google Scholar]
- 25.Kita H. Parvalbumin-immunopositive neurons in rat globus pallidus: a light and electron microscopic study. Brain Res. 1994;657:31–41. doi: 10.1016/0006-8993(94)90950-4. [DOI] [PubMed] [Google Scholar]
- 26.Kita H, Kitai ST. Intracellular study of rat globus pallidus neurons: membrane properties and responses to neostriatal, subthalamic, and nigral stimulation. Brain Res. 1991;564:296–305. doi: 10.1016/0006-8993(91)91466-e. [DOI] [PubMed] [Google Scholar]
- 27.Klemic KG, Shieh CC, Kirsch GE, Jones SW. Inactivation of Kv2.1 potassium channels. Biophys J. 1998;74:1779–1789. doi: 10.1016/S0006-3495(98)77888-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Knaus HG, Koch RO, Eberhart A, Kaczorowski GJ, Garcia ML, Slaughter RS. [125I]margatoxin, an extraordinarily high-affinity ligand for voltage-gated potassium channels in mammalian brain. Biochemistry. 1995;34:13627–13634. doi: 10.1021/bi00041a043. [DOI] [PubMed] [Google Scholar]
- 29.Kramer JW, Post MA, Brown AM, Kirsch GE. Modulation of potassium channel gating by coexpression of kv2.1 with regulatory kv5.1 or kv6.1 α-subunits. Am J Physiol. 1998;274:C1501–C1510. doi: 10.1152/ajpcell.1998.274.6.C1501. [DOI] [PubMed] [Google Scholar]
- 30.Leonard RJ, Garcia ML, Slaughter RS, Reuben JP. Selective blockers of voltage-gated K+ channels depolarize human T lymphocytes: mechanism of the antiproliferative effect of charybdotoxin. Proc Natl Acad Sci USA. 1992;89:10094–10098. doi: 10.1073/pnas.89.21.10094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lozano AM, Lang AE. Pallidotomy for Parkinson’s disease. Neurosurg Clin N Am. 1998;9:325–336. [PubMed] [Google Scholar]
- 32.Lozano A, Hutchison W, Kiss Z, Tasker R, Davis K, Dostrovsky J. Methods for microelectrode-guided posteroventral pallidotomy [see comments]. J Neurosurg. 1996;84:194–202. doi: 10.3171/jns.1996.84.2.0194. [DOI] [PubMed] [Google Scholar]
- 33.Maletic-Savatic M, Lenn NJ, Trimmer JS. Differential spatiotemporal expression of K+ channel polypeptides in rat hippocampal neurons developing in situ and in vitro. J Neurosci. 1995;15:3840–3851. doi: 10.1523/JNEUROSCI.15-05-03840.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Martina M, Schultz JH, Ehmke H, Monyer H, Jonas P. Functional and molecular differences between voltage-gated K+ channels of fast-spiking interneurons and pyramidal neurons of rat hippocampus. J Neurosci. 1998;18:8111–8125. doi: 10.1523/JNEUROSCI.18-20-08111.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Massengill JL, Smith MA, Son DI, O’Dowd DK. Differential expression of K4-AP currents and Kv3.1 potassium channel transcripts in cortical neurons that develop distinct firing phenotypes. J Neurosci. 1997;17:3136–3147. doi: 10.1523/JNEUROSCI.17-09-03136.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Murakoshi H, Trimmer JS. Identification of the Kv2.1 K+ channel as a major component of the delayed rectifier K+ current in rat hippocampal neurons. J Neurosci. 1999;19:1728–1735. doi: 10.1523/JNEUROSCI.19-05-01728.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Murakoshi H, Shi G, Scannevin RH, Trimmer JS. Phosphorylation of the Kv2.1 K+ channel alters voltage-dependent activation. Mol Pharmacol. 1997;52:821–828. doi: 10.1124/mol.52.5.821. [DOI] [PubMed] [Google Scholar]
- 38.Nini A, Feingold A, Slovin H, Bergman H. Neurons in the globus pallidus do not show correlated activity in the normal monkey, but phase-locked oscillations appear in the MPTP model of parkinsonism. J Neurophysiol. 1995;74:1800–1805. doi: 10.1152/jn.1995.74.4.1800. [DOI] [PubMed] [Google Scholar]
- 39.Nisenbaum ES, Stricker EM, Zigmond MJ, Berger TW. Spontaneous activity of type II but not type I striatal neurons is correlated with recovery of behavioral function after dopamine-depleting brain lesions. Brain Res. 1988;473:389–393. doi: 10.1016/0006-8993(88)90872-4. [DOI] [PubMed] [Google Scholar]
- 40.Pongs O. Molecular biology of voltage-dependent potassium channels. Physiol Rev. 1992;72:S69–S88. doi: 10.1152/physrev.1992.72.suppl_4.S69. [DOI] [PubMed] [Google Scholar]
- 41.Pusch M, Magrassi R, Wollnik B, Conti F. Activation and inactivation of homomeric KvLQT1 potassium channels. Biophys J. 1998;75:785–792. doi: 10.1016/S0006-3495(98)77568-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Rhodes KJ, Strassle BW, Monaghan MM, Bekele-Arcuri Z, Matos MF, Trimmer JS. Association and colocalization of the Kvβ1 and Kvβ2 β-subunits with Kv1 α-subunits in mammalian brain K+ channel complexes. J Neurosci. 1997;17:8246–8258. doi: 10.1523/JNEUROSCI.17-21-08246.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Robertson B. The real life of voltage-gated K+ channels: more than model behaviour. Trends Pharmacol Sci. 1997;18:474–483. doi: 10.1016/s0165-6147(97)01140-1. [DOI] [PubMed] [Google Scholar]
- 44.Rouzaire-Dubois B, Hammond C, Hamon B, Feger J. Pharmacological blockade of the globus palidus-induced inhibitory response of subthalamic cells in the rat. Brain Res. 1980;200:321–329. doi: 10.1016/0006-8993(80)90923-3. [DOI] [PubMed] [Google Scholar]
- 45.Rudy B. Diversity and ubiquity of K channels. Neuroscience. 1988;25:729–749. doi: 10.1016/0306-4522(88)90033-4. [DOI] [PubMed] [Google Scholar]
- 46.Ruppersberg JP, Frank R, Pongs O, Stocker M. Cloned neuronal IKA channels reopen during recovery from inactivation [see comments]. Nature. 1991;353:657–660. doi: 10.1038/353657a0. [DOI] [PubMed] [Google Scholar]
- 47.Schroter KH, Ruppersberg JP, Wunder F, Rettig J, Stocker M, Pongs O. Cloning and functional expression of a TEA-sensitive A-type potassium channel from rat brain. FEBS Lett. 1991;278:211–216. doi: 10.1016/0014-5793(91)80119-n. [DOI] [PubMed] [Google Scholar]
- 48.Serodio P, Kentros C, Rudy B. Identification of molecular components of A-type channels activating at subthreshold potentials. J Neurophysiol. 1994;72:1516–1529. doi: 10.1152/jn.1994.72.4.1516. [DOI] [PubMed] [Google Scholar]
- 49.Sheng M, Tsaur ML, Jan YN, Jan LY. Subcellular segregation of two A-type K+ channel proteins in rat central neurons. Neuron. 1992;9:271–284. doi: 10.1016/0896-6273(92)90166-b. [DOI] [PubMed] [Google Scholar]
- 50.Sheng M, Liao YJ, Jan YN, Jan LY. Presynaptic A-current based on heteromultimeric K+ channels detected in vivo. Nature. 1993;365:72–75. doi: 10.1038/365072a0. [DOI] [PubMed] [Google Scholar]
- 51.Shi W, Wymore RS, Wang HS, Pan Z, Cohen IS, McKinnon D, Dixon JE. Identification of two nervous system-specific members of the erg potassium channel gene family. J Neurosci. 1997;17:9423–9432. doi: 10.1523/JNEUROSCI.17-24-09423.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Shi W, Wang HS, Pan Z, Wymore RS, Cohen IS, McKinnon D, Dixon JE. Cloning of a mammalian elk potassium channel gene and EAG mRNA distribution in rat sympathetic ganglia. J Physiol (Lond) 1998;511:675–682. doi: 10.1111/j.1469-7793.1998.675bg.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Song WJ, Surmeier DJ. Voltage-dependent facilitation of calcium channels in rat neostriatal neurons. J Neurophysiol. 1996;16:2290–2306. doi: 10.1152/jn.1996.76.4.2290. [DOI] [PubMed] [Google Scholar]
- 54.Song WJ, Tkatch T, Baranauskas G, Ichinohe N, Kitai ST, Surmeier DJ. Somatodendritic depolarization-activated potassium currents in rat neostriatal cholinergic interneurons are predominantly of the A type and attributable to coexpression of Kv4.2 and Kv4.1 subunits. J Neurosci. 1998;18:3124–3137. doi: 10.1523/JNEUROSCI.18-09-03124.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Stefani A, Calabresi P, Mercuri NB, Bernardi G. A-current in rat globus pallidus: a whole-cell voltage-clamp study on acutely dissociated neurons. Neurosci Lett. 1992;144:4–8. doi: 10.1016/0304-3940(92)90703-a. [DOI] [PubMed] [Google Scholar]
- 56.Stefani A, Pisani A, Bonci A, Stratta F, Bernardi G. Outward potassium currents activated by depolarization in rat globus pallidus. Synapse. 1995;20:131–136. doi: 10.1002/syn.890200206. [DOI] [PubMed] [Google Scholar]
- 57.Stuhmer W, Ruppersberg JP, Schroter KH, Sakmann B, Stocker M, Giese KP, Perschke A, Baumann A, Pongs O. Molecular basis of functional diversity of voltage-gated potassium channels in mammalian brain. EMBO J. 1989;8:3235–3244. doi: 10.1002/j.1460-2075.1989.tb08483.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Surmeier DJ, Bargas J, Hemmings HC, Jr, Nairn AC, Greengard P. Modulation of calcium currents by a D1 dopaminergic protein kinase/phosphatase cascade in rat neostriatal neurons. Neuron. 1995;14:385–397. doi: 10.1016/0896-6273(95)90294-5. [DOI] [PubMed] [Google Scholar]
- 59.Swanson R, Marshall J, Smith JS, Williams JB, Boyle MB, Folander K, Luneau CJ, Antanavage J, Oliva C, Buhrow SA, Bennett C, Stein RB, Kaczmarek LK. Cloning and expression of cDNA and genomic clones encoding three delayed rectifier potassium channels in rat brain. Neuron. 1990;4:929–939. doi: 10.1016/0896-6273(90)90146-7. [DOI] [PubMed] [Google Scholar]
- 60.Swartz KJ, MacKinnon R. An inhibitor of the Kv2.1 potassium channel isolated from the venom of a Chilean tarantula. Neuron. 1995;15:941–949. doi: 10.1016/0896-6273(95)90184-1. [DOI] [PubMed] [Google Scholar]
- 61.Taglialatela M, Vandongen AM, Drewe JA, Joho RH, Brown AM, Kirsch GE. Patterns of internal and external tetraethylammonium block in four homologous K+ channels. Mol Pharmacol. 1991;40:299–307. [PubMed] [Google Scholar]
- 62.Taha JM, Favre J, Baumann TK, Burchiel KJ. Characteristics and somatotopic organization of kinesthetic cells in the globus pallidus of patients with Parkinson’s disease. J Neurosurg. 1996;85:1005–1012. doi: 10.3171/jns.1996.85.6.1005. [DOI] [PubMed] [Google Scholar]
- 63.Tkatch T, Baranauskas G, Surmeier DJ. Basal forebrain neurons adjacent to the globus pallidus coexpress GABAergic and cholinergic marker mRNAs. NeuroReport. 1998;9:1935–1939. doi: 10.1097/00001756-199806220-00004. [DOI] [PubMed] [Google Scholar]
- 64.Tristani-Firouzi M, Sanguinetti MC. Voltage-dependent inactivation of the human K+ channel KvLQT1 is eliminated by association with minimal K+ channel (minK) subunits. J Physiol (Lond) 1998;510:37–45. doi: 10.1111/j.1469-7793.1998.037bz.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Tytgat J, Debont T, Carmeliet E, Daenens P. The α-dendrotoxin footprint on a mammalian potassium channel. J Biol Chem. 1995;270:24776–24781. doi: 10.1074/jbc.270.42.24776. [DOI] [PubMed] [Google Scholar]
- 66.VanDongen AM, Frech GC, Drewe JA, Joho RH, Brown AM. Alteration and restoration of K+ channel function by deletions at the N- and C-termini. Neuron. 1990;5:433–443. doi: 10.1016/0896-6273(90)90082-q. [DOI] [PubMed] [Google Scholar]
- 67.Vega-Saenz de Miera E, Weiser M, Kentros C, Lau D, Moreno H, Serodio P, Rudy B. Handbook of membrane channels. Academic; New York: 1994. Shaw-related K+ channels in mammals. pp. 41–78. [Google Scholar]
- 68.Vysokanov A, Flores-Hernandez J, Surmeier DJ. mRNAs for clozapine-sensitive receptors colocalize in rat prefrontal cortex neurons. Neurosci Lett. 1998;258:179–182. doi: 10.1016/s0304-3940(98)00882-9. [DOI] [PubMed] [Google Scholar]
- 69.Wang HS, Pan Z, Shi W, Brown BS, Wymore RS, Cohen IS, Dixon JE, McKinnon D. KCNQ2 and KCNQ3 potassium channel subunits: molecular correlates of the M-channel [see comments]. Science. 1998;282:1890–1893. doi: 10.1126/science.282.5395.1890. [DOI] [PubMed] [Google Scholar]
- 70.Wang LY, Gan L, Forsythe ID, Kaczmarek LK. Contribution of the Kv3.1 potassium channel to high-frequency firing in mouse auditory neurones. J Physiol (Lond) 1998;509:183–194. doi: 10.1111/j.1469-7793.1998.183bo.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Weiser M, Vega-Saenz de Miera E, Kentros C, Moreno H, Franzen L, Hillman D, Baker H, Rudy B. Differential expression of Shaw-related K+ channels in the rat central nervous system. J Neurosci. 1994;14:949–972. doi: 10.1523/JNEUROSCI.14-03-00949.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Wichmann T, DeLong MR. Functional and pathophysiological models of the basal ganglia. Curr Opin Neurobiol. 1996;6:751–758. doi: 10.1016/s0959-4388(96)80024-9. [DOI] [PubMed] [Google Scholar]
- 73.Yan Z, Surmeier DJ. Muscarinic (m2/m4) receptors reduce N- and P-type Ca2+ currents in rat neostriatal cholinergic interneurons through a fast, membrane-delimited, G-protein pathway. J Neurosci. 1996;16:2592–2604. doi: 10.1523/JNEUROSCI.16-08-02592.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Yang WP, Levesque PC, Little WA, Conder ML, Ramakrishnan P, Neubauer MG, Blanar MA. Functional expression of two KvLQT1-related potassium channels responsible for an inherited idiopathic epilepsy. J Biol Chem. 1998;273:19419–19423. doi: 10.1074/jbc.273.31.19419. [DOI] [PubMed] [Google Scholar]
- 75.Zagotta WN, Germeraad S, Garber SS, Hoshi T, Aldrich RW. Properties of ShB A-type potassium channels expressed in Shaker mutant Drosophila by germline transformation. Neuron. 1989;3:773–782. doi: 10.1016/0896-6273(89)90246-8. [DOI] [PubMed] [Google Scholar]