

Abstract
Volume-sensitive organic anion channels (VSOACs) in astrocytes are activated by cell swelling and are permeable to organic anions, such as glutamate and taurine. We have examined the release of glutamate through VSOACs during the propagation of spreading depression (SD). SD was induced by bath application of ouabain in hippocampal brain slices and was monitored by imaging intrinsic optical signals, a technique that provides a measure of cellular swelling. The onset of SD was associated with increased light transmittance, confirming previous studies that cellular swelling occurs during SD. NMDA receptor antagonists, either noncompetitive (MK-801, 10–50 μm) or competitive (CGS-17355, 100 μm), reduced the rate of propagation of SD, indicating that glutamate release contributes to SD onset. SD still occurred in zero Ca2+–EGTA (0-Ca2+–EGTA) solution, a manipulation that depresses synaptic transmission. HPLC measurements indicated that, even in this solution, there was significant glutamate release. Two lines of experiments indicated that glutamate was released through VSOACs during SD. First, 5-nitro-2-(3-phenylpropylamino) benzoic acid (NPPB), a blocker of VSOACs, depressed the rate of propagation of SD in a manner similar to NMDA antagonists. Second, NPPB inhibited the release of glutamate during SD in 0-Ca2+–EGTA external solution. These results indicate that cellular swelling during SD causes the activation of VSOACs and the release of glutamate by permeation through this channel. Cellular swelling is a result of neuronal activity and is observed during excitotoxicity. Therefore, glutamate release from VSOAC activation could occur under conditions of cell swelling and contribute to excitotoxic damage.
Keywords: volume-sensitive organic anion channels, astrocytes, volume-sensitive chloride channels, NMDA receptors, intrinsic optical imaging, ischemia, CLC-3, swelling
Volume-sensitive organic anion channels (VSOACs) [also called volume-sensitive chloride channels (Icl,vol)] are a potential source of glutamate efflux in the CNS because they are permeable to glutamate (Strange et al., 1996). Volume changes in astrocytes activate VSOACs in a process involving mitogen-activated protein and tyrosine kinases (Crepel et al., 1998) and the cytoskeleton (Lascola et al., 1998). VSOACs have also been described recently in cerebellar neurons (Patel et al., 1998). Efflux of Cl− and anions, such as glutamate, aspartate, and taurine, through VSOACs helps to restore volume through a process called regulatory volume decrease (RVD) that also involves K+ channels (Pasantes-Morales and Schousboe, 1989; Pasantes-Morales et al., 1990,1994a,b). We designed these experiments to test the hypothesis that glutamate efflux through volume-activated channels occurs during spreading depression (SD).
SD is a wave of glial and neuronal depolarization and cellular swelling that slowly propagates through gray matter of the CNS (Van Harreveld and Khattab, 1967; Leao, 1944; Somjen et al., 1992; Jing et al., 1994;Nicholson and Sykova, 1998). Intrinsic optical signals, which measure the changes in light transmittance in tissue, provide an indirect measure of cell volume changes because increased light transmittance through brain slices is associated with cellular swelling (MacVicar and Hochman, 1991; Andrew and MacVicar, 1994; Holthoff and Witte, 1996). We previously recorded intrinsic optical signals during SD and found that the propagation of SD could be mapped by a wave of swelling (Basarsky et al., 1998). Activation of glutamate-permeable VSOACs by swelling of astrocytes and possibly neurons could contribute to glutamate release during SD.
In our previous study, we mapped SD by imaging intrinsic optical signals and simultaneously monitored intracellular calcium dynamics by measuring fura-2 fluorescence (Basarsky et al., 1998). In normal artificial CSF (aCSF) containing calcium, an intracellular calcium wave was associated with SD. However, SD still propagated in 0 external Ca with 2 mm EGTA with no detectable changes in intracellular calcium. Because glutamate release is thought to contribute to the propagation of SD (Mody et al., 1987; Lauritzen et al., 1988; Marrannes et al., 1988; Lauritzen and Hansen, 1992), we hypothesized that at least some of the glutamate efflux during SD could be attributable to VSOAC activation not involving calcium-dependent release from neurons and/or astrocytes. If glutamate release through VSOAC was contributing to SD, then we predict the following. First, NMDA receptor antagonists should depress the propagation of SD in isolated brain slices (Marrannes et al., 1988; Lauritzen and Hansen, 1992). Second, 5-nitro-2-(3-phenylpropylamino) benzoic acid (NPPB), an antagonist for VSOACs (Crepel et al., 1998), should depress the propagation of SD. Third, glutamate should still be released from brain slices during SD, even after depressing synaptic transmission in solutions containing 0 external calcium and 2 mm EGTA. Finally, NPPB should depress the Ca-insensitive release of glutamate from brain slices during SD. We have used intrinsic optical imaging of ouabain-induced SD and HPLC measurement of glutamate efflux to demonstrate that glutamate release from VSOACs contributes to the propagation of SD.
MATERIALS AND METHODS
Experiments were performed as described previously (Basarsky et al., 1998). A brief description of these methods is given below.
Slices. Hippocampal slices (400 μm) were prepared from 16- to 23-d-old Sprague Dawley rats. Slices were maintained in aCSF aerated with 95% O2–5% CO2 for a minimum of 1 hr after preparation before experiments were performed. For all experiments, the slices were transferred to a superfusion chamber mounted to the imaging setup described below. Slices were maintained at 33–34°C and held in place with platinum wires during the experiment. Control experiments were always performed each day on slices obtained from the same animal from which the experimental slices were prepared. Control experiments were alternated with experiments in which transmitter antagonists were applied to ensure that the slices were healthy and that there was no rundown in the quality of the tissue.
Imaging. The intrinsic optical imaging system was composed of a COHU 4982 charge-coupled device camera connected to an Axon Image Lightning 2000 frame grabber (Axon Instruments, Foster City, CA) that was driven by Axon Imaging Workbench (version 2.1; Axon Instruments). The illumination source was a standard Zeiss (Oberkochen, Germany) tungsten bulb whose output was directed through a 750DF20 discriminating filter. Typically, four frames were averaged for each image. This approach allowed the visualization of SD at a sampling frequency of 1 Hz, which was sufficiently fast given the relatively slow propagation rate of SD. The intrinsic optical signals were recorded and presented as subtracted images, with the first image acquired during acquisition serving as the reference image, which was then subtracted from all subsequent images during acquisition. The intrinsic optical signals were acquired at a frequency of 1 Hz but were saved to disk at a variable frequency of between 0.008 and 1.0 Hz to reduce data storage requirements. We described the rate of change of intrinsic optical signals during spreading depression as the change in the intensity in a specified zone region of the subtracted image per second (ΔT/T%). Quantification of intrinsic optical signals during SD was performed as described by Basarsky et al. (1998), their Figure 3.
Solutions. Regular aCSF contained (in mm): NaCl, 124; KCl, 5; MgCl2, 1.3; CaCl2, 2; glucose, 10; and NaHCO3, 26.2. For the zero calcium aCSF (0-Ca2+ aCSF), calcium was replaced with magnesium, and 2 mm EGTA was added, yielding a 0-Ca2+ aCSF that contained (in mm): NaCl, 114; KCl, 5; MgCl2, 3.3; glucose, 10; NaHCO3, 26.2; and EGTA, 2. The pH was adjusted to 7.37 for both of these solutions. NPPB was purchased from Biomol(Plymouth Meeting, PA).
HPLC. The methods described by Saleh et al. (1997) were used to measure amino acid content in the superfusate, with the following modifications. One milliliter samples were collected at 1 min intervals. These samples were lyophilized and reconstituted into 50 μl aliquots for HPLC analysis. This ensured that the concentration of glutamate was well within detection limits and also allowed for the perfusion of the slice at flow rates that minimized slice deterioration. Although taurine is often measured during cellular swelling experiments, it was not possible to always discern the taurine peak from the arginine peak in our HPLC experiments. Consequently, taurine was not analyzed.
Statistics. Unless otherwise stated, all statistics were performed with the Mann–Whitney U test. GB-STAT version 3.53 (Dynamic Microsystems) was used for all statistical calculations.
RESULTS
Glutamate is involved in the initial phase of spreading depression
In hippocampal brain slices in normal aCSF, ouabain induced spreading depression, which propagated throughout the hippocampus, and was measured as a transient increase in tissue transmittance (Fig.1A). Previous reports (Marrannes et al., 1988; Gill et al., 1992; McLachlan, 1992; Nellgard and Wieloch, 1992; Martin et al., 1994; Willette et al., 1994;Obrenovitch and Zilkha, 1996; Tatlisumak et al., 1998) have demonstrated that spreading depression in vivo is delayed or inhibited by glutamate receptor antagonists. In hippocampal brain slices, application of the noncompetitive NMDA receptor antagonist MK-801 (50 μm) inhibited the onset of spreading depression (Fig. 1B). In the presence of MK-801, there was a slow and uniformly distributed increase in tissue transmittance throughout the CA1 region. This widespread increase was followed by a propagating decrease in light transmittance (Fig.1B). In contrast, in control conditions, there was a rapidly propagating increase in light transmittance that was followed by a decrease in transmittance (Fig. 1A).
Fig. 1.

Time course of ouabain-induced spreading depression in hippocampal slices. A, Spreading depression in control conditions. SD rapidly initiated in CA1 and propagated throughout the entire CA1 region as a wave of increased light transmittance followed by a rapid decrease. B, Spreading depression in the presence of 50 μm MK-801, after preincubation for 20 min in MK-801. The waveform of SD was different in that the initial increase in light transmittance was slow, but this was still followed by a rapidly propagating wave of decreased transmittance. C, Bright-field image of the slice used in A. The purple dot denotes a typical region used for measurements, and the CA1 region has been shown inA and B. Ouabain (100 μm) was added at 1 minute 30 seconds. All times are given in minutes:seconds. Scale bar, 100 μm.
The time course of the light transmittance changes during SD at a single region in the stratum radiatum of CA1 is shown from single experiments in Figure 2 and is summarized in Figure 3. In addition to the noncompetitive antagonist MK-801, we also examined the effects of the competitive NMDA receptor antagonist CGS-19755 (100 μm). Both MK-801 and CGS-197555 inhibit the onset of spreading depression. Furthermore, the effect of MK-801 was approximately fourfold greater at 50 μm than at 10 μm. Quantification of the kinetics of spreading depression revealed that MK-801 and CGS-19755 greatly inhibited the onset slope of spreading depression (Fig.3B) and that MK-801 at the highest concentration attenuated the peak change in transmittance (Fig. 3A). In paired control slices, the onset slope was typically 10 ΔT/sec. After bath application of antagonist, the onset slope decreased to 0.4 ± 0.02 ΔT/sec (n = 21) in 10 μm MK-801, 0.4 ± 0.04 ΔT/sec (n = 21) in 100 μm CGS-19755, and 0.1 ± 0.01 ΔT/sec (n = 18) in 50 μm MK-801. In contrast, CNQX, an antagonist to the AMPA receptor, did not alter SD. We preapplied CNQX at 30 μm, which we have observed completely blocks AMPA-mediated synaptic potentials (data not shown) and found it had no significant effect on the peak amplitude of SD (65 ± 8 ΔT; n = 5; vs CNQX, 63 ± 5 ΔT; n = 5) or the maximum rate of rise (control, 2.2 ± 0.7; n = 5; vs CNQX, 2.1 ± 0.4;n = 5). Together, these data indicate that glutamate plays an important role the initial phase of spreading depression; inhibition of NMDA-type glutamate receptors suppresses the onset of spreading depression.
Fig. 2.

Time course of spreading depression in the presence of NMDA receptor antagonists. Each tracerepresents the change in transmittance in a single region of interest (as denoted in Fig. 1) recorded in separate brain slices. Thetop trace is a profile of spreading depression in a control slice. The bottom three traces are profiles of spreading depression in separate brain slices incubated in the indicated NMDA receptor antagonist. Both the competitive antagonist (CGS-19755) and the noncompetitive antagonist (MK-801) reduced the onset of SD, and the effects of MK-801 were dose-dependent.Solid bar represents the time of application of ouabain (100 μm).
Fig. 3.

Quantification of the effects of NMDA receptor antagonists on spreading depression. A, The amplitude of the peak response was only reduced significantly by 50 μmMK-801 (p < 0.01) but was not significantly affected by CGS-19755 (100 μm) or MK-801 (10 μm) (p > 0.05).B, The onset slope was reduced by both the competitive (CGS-19755) and noncompetitive (MK-801) NMDA receptor antagonists. Note that the scale in B is split to enable the display of both control and antagonist measurements.
Inhibition of VSOACs inhibits the initial phase of spreading depression
We examined the effects of the VSOAC antagonist NPPB (100 μm) under normal conditions and under conditions in which synaptic release of neurotransmitter is inhibited. In normal Ca external solution, the addition of NPPB slowed the decline of the SD waveform. The decline at 30 sec is reduced from 47 ± 6% (n = 14) in control solution with calcium to 2 ± 0.5% (n = 14) in control with NPPB. This is consistent with the role for Cl channels in regulatory volume decrease after the swelling during SD. In external 0-Ca2+, spreading depression occurs in a normal manner, albeit with slower kinetics (Jing et al., 1993; Herreras et al., 1994) (Figure4B) as we have shown previously (Basarsky et al., 1998). Under these conditions, inhibition of VSOACs by NPPB further reduced the onset slope of spreading depression (Fig. 4A,B). Incubation of slices with 100 μm NPPB caused a significant reduction in the onset slope from 1.43 ± 0.4 ΔT/sec in paired controls in 0-Ca2+ to 0.53 ± 0.05 ΔT/sec in NPPB (p < 0.01; n = 7). The decline of the SD signal in 0-Ca2+ was also slowed from 35 ± 2% (n = 21) to 19 ± 2% (n = 18) in NPPB. We also examined the effects of threo-hydroxy-β-aspartate (THBA), which has been shown to block glutamate release caused by reverse transport in astrocytes after extracellular application (Rutledge and Kimelberg, 1996). A 40 min preapplication of 1 mm THBA had no effect on the maximum signal in SD (control, 77 ± 5%; vs THBA, 80 ± 8%;n = 10) nor on the maximum rate of rise of the SD signal (control, 4.1 ± 0.6%/sec; vs THBA, 4.5 ± 0.6%/sec).
Fig. 4.
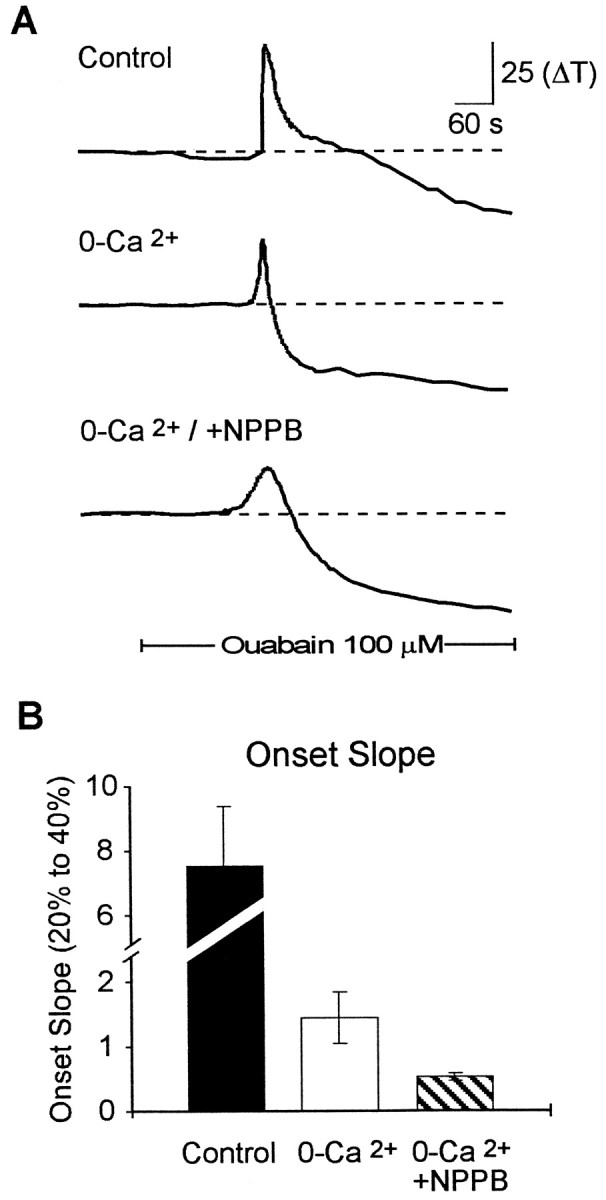
NPPB, a VSOAC antagonist, inhibited the onset of spreading depression. A, Representativetraces of the light transmittance changes in the presence and absence of external calcium and in the presence of the VSOAC antagonist NPPB (100 μm). B, Summary data for the effects of 0-Ca2+ and NPPB on the onset slope of spreading depression. In the absence of calcium, NPPB significantly inhibited the onset slope. Solid bardenotes the time of application of ouabain (100 μm).
Inhibition of VSOACs inhibits the release of glutamate during spreading depression
We next determined whether VSOACs are involved in the release of glutamate during spreading depression when synaptic transmission was greatly depressed in 0-Ca2+–EGTA solution. To do this, we used HPLC to measure glutamate levels in the superfusate and examined the effect of the VSOAC antagonist NPPB on glutamate release during spreading depression. Not surprisingly, HPLC measurements showed that, during SD in control solution with calcium, there was substantial increase in glutamate efflux to 2668 ± 1444% (n= 3) of control levels. When SD was observed in the brain slice in 0-Ca2+–EGTA solution, HPLC measurement showed that there was still a substantial increase in the release of glutamate, GABA, and glutamine, although it was approximately eightfold less (Fig.5). Incubation of slices with 100 μm NPPB resulted in a significant inhibition of the release of glutamate (Fig. 5A,B). However the release of both GABA and glutamine was not depressed by NPPB, consistent with previous observations that these amino acids do not significantly permeate VSOACs. Together, these data are consistent with the model that activation of VSOACs during spreading depression results in the calcium-independent release of glutamate, which contributes to the rapid onset of spreading depression. Inhibition of these channels inhibits the release of glutamate and delays the onset of spreading depression.
Fig. 5.

Inhibition of VSOACs inhibited the calcium-independent release of glutamate. All data were collected from brain slices incubated in 0-Ca2+–EGTA solution for at least 20 min before the start of the experiment. A, Representative HPLC measurements of glutamate release in the superfusate before and during SD in two separate brain slices. The initial onset of SD was measured using intrinsic optical signals and is taken as t = 0. SD propagated throughout the brain slice during the collection of superfusate and induced the release of glutamate, even in the 0-Ca2+–EGTA solution. Preincubation with NPPB (100 μm) reduced the release of glutamate during spreading depression. B, Effect of NPPB on glutamate, GABA, and glutamine release during spreading depression. Each point represents the increase in the indicated amino acid 7 min after the initiation of spreading depression from at least n = 5 separate brain slices. All numbers are expressed normalized to the initial levels before spreading depression. NPPB significantly depressed the release of only glutamate during SD (*p < 0.01) and had no effect on the release of either GABA or glutamine.
DISCUSSION
These results provide the first evidence that glutamate release through VSOACs in the hippocampus contributes to the propagation of spreading depression. We confirmed the importance of glutamate release in spreading depression by describing the inhibition of spreading depression by NMDA receptor antagonists. Using HPLC, we measured a significant release of glutamate during spreading depression, even in 0-Ca2+ aCSF in which conventional synaptic release is blocked. NPPB, an antagonist to VSOAC channels, depressed this calcium-insensitive release of glutamate and inhibited the rate of propagation of spreading depression.
This study continues our previous work using intrinsic optical imaging to map spreading depression in the hippocampal slice (Basarsky et al., 1998). Intrinsic optical signals arise from the dynamics of light transmittance. For example, cellular swelling decreases light scattering, resulting in an increase in light transmittance through the slice, whereas cellular shrinkage does the reverse (MacVicar and Hochman, 1991; Andrew and MacVicar, 1994; Holthoff and Witte, 1996). The pattern of increased light transmittance during the onset of spreading depression indicates that there is substantial cellular swelling during spreading depression. In our previous study (Basarsky et al., 1998), we simultaneously imaged intrinsic optical signals and intracellular calcium changes using fura-2 fluorescence measurements and found that a calcium wave was normally associated with spreading depression. However, we made the surprising discovery that, in 0-Ca2+ aCSF, spreading depression still occurred, and there were no associated changes in intracellular calcium. These results, combined with the volume changes suggested by the intrinsic optical signals, indicated that spreading depression is a calcium-independent phenomenon involving cellular swelling.
In astrocytes and other cell types, swelling activates VSOACs that are permeant to chloride ions and organic anions, including glutamate (Jackson and Strange, 1993; Sanchez-Olea et al., 1993; Pasantes-Morales et al., 1994a,b; Roy, 1994, 1995; Gonzalez et al., 1995; Lascola and Kraig, 1996; Strange et al., 1996; Crepel et al., 1998). The efflux of organic anions and Cl− through VSOACs can contribute to the active process called RVD to restore the initial cellular volume (Pasantes-Morales et al., 1994a,b). The efflux of K+ through other channels also contributes to RVD. VSOACs have not been widely investigated in neurons but have recently been described in cerebellar granule cells (Patel et al., 1998). CLC-3, a cloned Cl-selective ion channel, is a good candidate for the VSOAC (Duan et al., 1997) and is highly expressed in the CNS and the hippocampus (Kawasaki et al., 1994). Therefore, VSOACs are present in the hippocampus and are likely found in both astrocytes and neurons.
We used NPPB to block the VSOAC for two reasons. First, NPPB has been reported to inhibit the VSOAC and the efflux of amino acids activated by cell swelling in astrocytes and other cell types (Rutledge et al., 1998). Second, NPPB inhibits the VSOAC in a nonvoltage-dependent manner (Crepel et al., 1998), which suggests that NPPB will effectively inhibit the VSOAC in astrocytes at both the resting potential and the depolarized levels reached during SD.
The profile of the amino acid efflux during SD and the selective inhibition of glutamate release by NPPB are consistent with release of glutamate through VSOACs. The HPLC data showed that glutamate and GABA are both released from hippocampal brain slices during SD in 0-Ca2+ aCSF. Glutamine was released to a lesser extent. Under these conditions, synaptic transmission is negligible in brain slices (Schweitzer et al., 1992), and we have shown previously that there are no detectable changes in intracellular calcium during SD (Basarsky et al., 1998). Therefore, we conclude that both glutamate and GABA can be released in high levels by non-Ca2+-dependent mechanisms. The relative contribution of glutamate release during SD through these mechanisms appears to be less than that caused by calcium-dependent exocytosis judging by the HPLC measurements of glutamate release. Only the glutamate efflux was depressed by NPPB, which is consistent with studies that have shown that glutamine does not permeate the VSOAC (Pasantes-Morales et al., 1994a,b; Rutledge et al., 1998). We have not investigated the mechanisms by which GABA efflux occurs, but it is presumably because of another transporter.
The potential roles for reversed glutamate transport are difficult to unequivocally rule out because most inhibitors are competitive and would not be expected to act on reverse transport (Kanai et al., 1993). However, the glial transporter is inhibited by external application of THBA (Rutledge and Kimelberg, 1996). We have found no effect of THBA on SD.
Previous studies have shown that NMDA receptor antagonists depress the activation of spreading depression in vivo (Marrannes et al., 1988; Gill et al., 1992; McLachlan, 1992; Nellgard and Wieloch, 1992; Martin et al., 1994; Willette et al., 1994; Obrenovitch and Zilkha, 1996; Tatlisumak et al., 1998). Our results point to a pivotal role for NMDA receptor activation in the initial onset of SD. NMDA receptor antagonists decreased the rate of onset of SD. This was observed with both the competitive antagonist CGS-197555 and the noncompetitive antagonist MK-801. The similarity of action of both types of NMDA antagonists is reassuring because MK-801 can affect other ion channels at higher concentrations (>100 μm) (ffrench-Mullen and Rogawski, 1989).
The results in this study suggest a new addition to the cellular mechanisms contributing to SD. The original hypothesis stated that increased external K+ caused depolarization of both postsynaptic processes and presynaptic terminals (Leao, 1944; Kraig and Nicholson, 1978; Van Harreveld, 1978; Nicholson and Kraig, 1981; Somjen et al., 1992). Presynaptic depolarization of terminals evoked calcium-dependent synaptic transmission and the release of glutamate, which in turn further depolarized postsynaptic elements and caused further increases in external K+. This was thought to result in positive feedback between K+accumulation and glutamate release leading to regenerative depolarizations. SD was thought to be a propagating wave of high K+-induced glutamate release and depolarization. However, studies in brain slices in which synaptic transmission could be blocked demonstrated that SD still occurred in calcium-free solution (Herreras et al., 1994). Our results further illustrate the dichotomy between the calcium studies and the glutamate hypothesis and provide a partial although incomplete explanation. We found that spreading depression propagated normally in external solutions in which synaptic transmission would be blocked or at least greatly depressed. However, the propagation of SD under these conditions was still influenced by the activation of NMDA receptors because the propagation rate of SD was reduced in the presence of either competitive or noncompetitive NMDA receptor antagonists. Therefore, this study indicates that swelling, which has long been known to be associated with SD (Van Harreveld and Khattab, 1967; Jing et al., 1994), is an important factor in its generation. Previously, glutamate was thought to be the factor that caused swelling during SD. However, we suggest that swelling is a contributing factor in causing the release of glutamate.
We suggest the following addition to the mechanisms underlying spreading depression. Swelling, perhaps as a result of intense neuronal activity (Nicholson and Sykova, 1998), opens VSOACs that are permeable to glutamate. The efflux of glutamate activates NMDA receptors on neurons, which increases extracellular K+leading to further astrocyte swelling. The swelling attributable to K+ accumulation causes additional activation of VSOACs and further release of glutamate through this channel. The net result is positive feedback with a central role for swelling-induced glutamate release.
Conclusion
Spreading depression may represent an extreme example of a normal process that occurs regularly on a less dramatic scale. In normal conditions, cellular swelling is known to occur in conjunction with neuronal activity (Nicholson and Sykova, 1998). Our results suggest that activity-induced cellular swelling may activate VSOACs and thereby cause glutamate release from these channels. A similar mechanism may contribute to ischemic-induced excitotoxicity because of the associated cellular swelling. Interestingly, NPPB has been reported to decrease the efflux of glutamate from ischemic brain regions (Phillis et al., 1997). Therefore, the release of glutamate through volume-activated channels could have widespread implications in the CNS.
Footnotes
This work was supported by the Medical Research Council of Canada (MRC). T.A.B. is a postdoctoral fellow of the Alberta Heritage Foundation for Medical Research (AHFMR). B.A.M. is an MRC Senior Scientist and an AHFMR Scientist. We thank Lorenzo Bauce for performing the HPLC measurements and Drs. J. Armstrong and V. Parpura for useful comments on earlier versions of this manuscript.
Correspondence should be addressed to Dr. B. A. MacVicar, Department of Physiology and Biophysics, Neuroscience Research Group, University of Calgary, 3330 Hospital Drive N.W., Calgary, Alberta T2N 4N1, Canada.
REFERENCES
- 1.Andrew RD, MacVicar BA. Imaging cell volume changes and neuronal excitation in the hippocampal slice. Neuroscience. 1994;62:371–383. doi: 10.1016/0306-4522(94)90372-7. [DOI] [PubMed] [Google Scholar]
- 2.Basarsky TA, Duffy SN, Andrew RD, MacVicar BA. Imaging spreading depression and associated intracellular calcium waves in brain slices. J Neurosci. 1998;18:7189–7199. doi: 10.1523/JNEUROSCI.18-18-07189.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Crepel V, Panenka W, Kelly ME, MacVicar BA. Mitogen-activated protein and tyrosine kinases in the activation of astrocyte volume-activated chloride current. J Neurosci. 1998;18:1196–1206. doi: 10.1523/JNEUROSCI.18-04-01196.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Duan D, Winter C, Cowley S, Hume JR, Horowitz B. Molecular identification of a volume-regulated chloride channel. Nature. 1997;390:417–421. doi: 10.1038/37151. [DOI] [PubMed] [Google Scholar]
- 5.ffrench-Mullen JM, Rogawski MA. Interaction of phencyclidine with voltage-dependent potassium channels in cultured rat hippocampal neurons: comparison with block of the NMDA receptor-ionophore complex. J Neurosci. 1989;9:4051–4061. doi: 10.1523/JNEUROSCI.09-11-04051.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gill R, Andine P, Hillered L, Persson L, Hagberg H. The effect of MK-801 on cortical spreading depression in the penumbral zone following focal ischaemia in the rat. J Cereb Blood Flow Metab. 1992;12:371–379. doi: 10.1038/jcbfm.1992.54. [DOI] [PubMed] [Google Scholar]
- 7.Gonzalez E, Sanchez-Olea R, Pasantes-Morales H. Inhibition by Cl− channel blockers of the volume-activated, diffusional mechanism of inositol transport in primary astrocytes in culture. Neurochem Res. 1995;20:895–900. doi: 10.1007/BF00970734. [DOI] [PubMed] [Google Scholar]
- 8.Herreras O, Largo C, Ibarz JM, Somjen GG, Martin del Rio R. Role of neuronal synchronizing mechanisms in the propagation of spreading depression in the in vivo hippocampus. J Neurosci. 1994;14:7087–7098. doi: 10.1523/JNEUROSCI.14-11-07087.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Holthoff K, Witte OW. Intrinsic optical signals in rat neocortical slices measured with near-infrared dark-field microscopy reveal changes in extracellular space. J Neurosci. 1996;16:2740–2749. doi: 10.1523/JNEUROSCI.16-08-02740.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jackson PS, Strange K. Volume-sensitive anion channels mediate swelling-activated inositol and taurine efflux. Am J Physiol. 1993;265:C1489–C1500. doi: 10.1152/ajpcell.1993.265.6.C1489. [DOI] [PubMed] [Google Scholar]
- 11.Jing J, Aitken PG, Somjen GG. Role of calcium channels in spreading depression in rat hippocampal slices. Brain Res. 1993;604:251–259. doi: 10.1016/0006-8993(93)90376-x. [DOI] [PubMed] [Google Scholar]
- 12.Jing J, Aitken PG, Somjen GG. Interstitial volume changes during spreading depression (SD) and SD-like hypoxic depolarization in hippocampal tissue slices. J Neurophysiol. 1994;71:2548–2551. doi: 10.1152/jn.1994.71.6.2548. [DOI] [PubMed] [Google Scholar]
- 13.Kaai Y, Smith CP, Hediger MA. The elusive transporters with a high affinity for glutamate. Trends Neurosci. 1993;16:365–370. doi: 10.1016/0166-2236(93)90094-3. [DOI] [PubMed] [Google Scholar]
- 14.Kawasaki M, Uchida S, Monkawa T, Miyawaki A, Mikoshiba K, Marumo F, Sasaki S. Cloning and expression of a protein kinase C-regulated chloride channel abundantly expressed in rat brain neuronal cells. Neuron. 1994;12:597–604. doi: 10.1016/0896-6273(94)90215-1. [DOI] [PubMed] [Google Scholar]
- 15.Kraig RP, Nicholson C. Extracellular ionic variations during spreading depression. Neuroscience. 1978;3:1045–1059. doi: 10.1016/0306-4522(78)90122-7. [DOI] [PubMed] [Google Scholar]
- 16.Lascola CD, Kraig RP. Whole-cell chloride currents in rat astrocytes accompany changes in cell morphology. J Neurosci. 1996;16:2532–2545. doi: 10.1523/JNEUROSCI.16-08-02532.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lascola CD, Nelson DJ, Kraig RP. Cytoskeletal actin gates a Cl− channel in neocortical astrocytes. J Neurosci. 1998;18:1679–1692. doi: 10.1523/JNEUROSCI.18-05-01679.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lauritzen M, Hansen AJ. The effect of glutamate receptor blockade on anoxic depolarization and cortical spreading depression. J Cereb Blood Flow Metab. 1992;12:223–229. doi: 10.1038/jcbfm.1992.32. [DOI] [PubMed] [Google Scholar]
- 19.Lauritzen M, Rice ME, Okada Y, Nicholson C. Quisqualate, kainate and NMDA can initiate spreading depression in the turtle cerebellum. Brain Res. 1988;475:317–327. doi: 10.1016/0006-8993(88)90620-8. [DOI] [PubMed] [Google Scholar]
- 20.Leao AAP. Spreading depression of activity in the cerebral cortex. J Neurophysiol. 1944;7:359–390. doi: 10.1152/jn.1947.10.6.409. [DOI] [PubMed] [Google Scholar]
- 21.MacVicar BA, Hochman D. Imaging of synaptically evoked intrinsic optical signals in hippocampal slices. J Neurosci. 1991;11:1458–1469. doi: 10.1523/JNEUROSCI.11-05-01458.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Marrannes R, Willems R, De Prins E, Wauquier A. Evidence for a role of the N-methyl-d-aspartate (NMDA) receptor in cortical spreading depression in the rat. Brain Res. 1988;457:226–240. doi: 10.1016/0006-8993(88)90690-7. [DOI] [PubMed] [Google Scholar]
- 23.Martin H, Warner DS, Todd MM. Effects of glycine receptor antagonism on spreading depression in the rat. Neurosci Lett. 1994;180:285–289. doi: 10.1016/0304-3940(94)90540-1. [DOI] [PubMed] [Google Scholar]
- 24.McLachlan RS. Suppression of spreading depression of Leao in neocortex by an N-methyl-d-aspartate receptor antagonist. Can J Neurol Sci. 1992;19:487–491. [PubMed] [Google Scholar]
- 25.Mody I, Lambert JD, Heinemann U. Low extracellular magnesium induces epileptiform activity and spreading depression in rat hippocampal slices. J Neurophysiol. 1987;57:869–888. doi: 10.1152/jn.1987.57.3.869. [DOI] [PubMed] [Google Scholar]
- 26.Nellgard B, Wieloch T. NMDA-receptor blockers but not NBQX, an AMPA-receptor antagonist, inhibit spreading depression in the rat brain. Acta Physiol Scand. 1992;146:497–503. doi: 10.1111/j.1748-1716.1992.tb09451.x. [DOI] [PubMed] [Google Scholar]
- 27.Nicholson C, Kraig RP. The behaviour of extracellular ions during spreading depression. In: Zeuthen T, editor. The application of ion-selective electrodes. Elsevier; Amsterdam: 1981. pp. 217–238. [Google Scholar]
- 28.Nicholson C, Sykova E. Extracellular space analysis revealed by diffusion analysis. Trends Neurosci. 1998;21:207–215. doi: 10.1016/s0166-2236(98)01261-2. [DOI] [PubMed] [Google Scholar]
- 29.Obrenovitch TP, Zilkha E. Inhibition of cortical spreading depression by L-701,324, a novel antagonist at the glycine site of the N-methyl-d-aspartate receptor complex. Br J Pharmacol. 1996;117:931–937. doi: 10.1111/j.1476-5381.1996.tb15283.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Pasantes-Morales H, Schousboe A. Release of taurine from astrocytes during potassium-evoked swelling. Glia. 1989;2:45–50. doi: 10.1002/glia.440020105. [DOI] [PubMed] [Google Scholar]
- 31.Pasantes-Morales H, Moran J, Schousboe A. Volume-sensitive release of taurine from cultured astrocytes: properties and mechanism. Glia. 1990;3:427–432. doi: 10.1002/glia.440030514. [DOI] [PubMed] [Google Scholar]
- 32.Pasantes-Morales H, Murray RA, Lilja L, Moran J. Regulatory volume decrease in cultured astrocytes. I. Potassium- and chloride-activated permeability. Am J Physiol. 1994a;266:C165–C171. doi: 10.1152/ajpcell.1994.266.1.C165. [DOI] [PubMed] [Google Scholar]
- 33.Pasantes-Morales H, Murray RA, Sanchez-Olea R, Moran J. Regulatory volume decrease in cultured astrocytes. II. Permeability pathway to amino acids and polyols. Am J Physiol. 1994b;266:C172–C178. doi: 10.1152/ajpcell.1994.266.1.C172. [DOI] [PubMed] [Google Scholar]
- 34.Patel AJ, Lauritzen I, Lazdunski M, Honore E. Disruption of mitochondrial respiration inhibits volume-regulated anion channels and provokes neuronal cell swelling. J Neurosci. 1998;18:3117–3123. doi: 10.1523/JNEUROSCI.18-09-03117.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Phillis JW, Song D, O’Regan MH. Inhibition by anion channel blockers of ischemia-evoked release of excitotoxic and other amino acids from rat cerebral cortex. Brain Res. 1997;758:9–16. doi: 10.1016/s0006-8993(97)00155-8. [DOI] [PubMed] [Google Scholar]
- 36.Roy G. Channels for amino acids and metabolites activated by cell volume regulation. Jpn J Physiol [Suppl 44] 1994;2:S37–S42. [PubMed] [Google Scholar]
- 37.Roy G. Amino acid current through anion channels in cultured human glial cells. J Membr Biol. 1995;147:35–44. doi: 10.1007/BF00235396. [DOI] [PubMed] [Google Scholar]
- 38.Rutledge EM, Kimelberg HK. Release of [3H]-d-aspartate from primary astrocyte cultures in response to raised external potassium. J Neurosci. 1996;16:7803–7811. doi: 10.1523/JNEUROSCI.16-24-07803.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Rutledge EM, Aschner M, Kimelberg HK. Pharmacological characterization of swelling-induced d-[3H]aspartate release from primary astrocyte cultures. Am J Physiol. 1998;274:C1511–C1520. doi: 10.1152/ajpcell.1998.274.6.C1511. [DOI] [PubMed] [Google Scholar]
- 40.Saleh TM, Bauce LG, Pittman QJ. Glutamate release in parabrachial nucleus and baroreflex alterations after vagal afferent activation. Am J Physiol. 1997;272:R1631–R1640. doi: 10.1152/ajpregu.1997.272.5.R1631. [DOI] [PubMed] [Google Scholar]
- 41.Sanchez-Olea R, Pena C, Moran J, Pasantes-Morales H. Inhibition of volume regulation and efflux of osmoregulatory amino acids by blockers of Cl− transport in cultured astrocytes. Neurosci Lett. 1993;156:141–144. doi: 10.1016/0304-3940(93)90458-w. [DOI] [PubMed] [Google Scholar]
- 42.Schweitzer JS, Patrylo PR, Dudek FE, Snow RW, Taylor CP. Prolonged field bursts in the dentate gyrus: dependence on low calcium, high potassium, and nonsynaptic mechanisms. J Neurophysiol. 1992;68:2016–2025. doi: 10.1152/jn.1992.68.6.2016. [DOI] [PubMed] [Google Scholar]
- 43.Somjen GG, Aitken PG, Czeh GL, Herreras O, Jing J, Young JN. Mechanism of spreading depression: a review of recent findings and a hypothesis. Can J Physiol Pharmacol. 1992;70:S248–S254. doi: 10.1139/y92-268. [DOI] [PubMed] [Google Scholar]
- 44.Strange K, Emma F, Jackson PS. Cellular and molecular physiology of volume-sensitive anion channels. Am J Physiol. 1996;270:C711–C730. doi: 10.1152/ajpcell.1996.270.3.C711. [DOI] [PubMed] [Google Scholar]
- 45.Tatlisumak T, Takano K, Meiler MR, Fisher M. A glycine site antagonist, ZD9379, reduces number of spreading depressions and infarct size in rats with permanent middle cerebral artery occlusion. Stroke. 1998;29:190–195. doi: 10.1161/01.str.29.1.190. [DOI] [PubMed] [Google Scholar]
- 46.Van Harreveld A. Two mechanisms for spreading depression in the chicken retina. J Neurobiol. 1978;9:419–431. doi: 10.1002/neu.480090602. [DOI] [PubMed] [Google Scholar]
- 47.Van Harreveld A, Khattab FI. Changes in cortical extracellular space during spreading depression investigated with the electron microscope. J Neurophysiol. 1967;30:911–929. doi: 10.1152/jn.1967.30.4.911. [DOI] [PubMed] [Google Scholar]
- 48.Willette RN, Lysko PG, Sauermelch CF. A comparison of (+)SK&F 10047 and MK-801 on cortical spreading depression. Brain Res. 1994;648:347–351. doi: 10.1016/0006-8993(94)91140-1. [DOI] [PubMed] [Google Scholar]