

Abstract
Cannabinoids, the active constituents of marijuana, are known to impair learning and memory. Receptors for cannabinoids are highly expressed in the hippocampus, a brain region that is believed to play an important role in certain forms of learning and memory. To investigate the possible contribution of cannabinoid receptor-mediated deficits in hippocampal function to the learning and memory impairments produced by marijuana, we studied the effects of cannabinoid receptor activation on two models of learning and memory, long-term potentiation (LTP) and long-term depression (LTD), in hippocampal slices. Although LTP and LTD of CA1 field potentials were blocked by cannabinoid receptor activation in the presence of Mg2+, they could be induced after Mg2+ was removed. Similarly, LTP and LTD of whole-cell EPSCs were unimpaired in the presence of cannabinoid receptor agonist when the postsynaptic membrane was depolarized during the LTP or LTD induction protocol. Cannabinoid receptor activation also reduced EPSCs and enhanced paired-pulse facilitation, while having no effect on the amplitude of spontaneous miniature EPSCs. Finally, as with cannabinoid receptor activation, inhibition of LTP by adenosine receptor activation could be overcome by removal of Mg2+ or depolarization of the postsynaptic membrane during tetanus. Our results indicate that cannabinoid receptor activation does not directly inhibit the molecular mechanisms responsible for long-term synaptic plasticity but instead impairs LTP and LTD by reducing presynaptic neurotransmitter release to a level below that required to depolarize the postsynaptic membrane to relieve Mg2+ blockade of NMDA receptors.
Keywords: cannabinoids, cannabinoid receptors, hippocampus, synaptic transmission, long-term potentiation, long-term depression, learning and memory
Because the hippocampus is believed to play an important role in learning and memory and cannabinoid receptors are highly expressed in the hippocampus (Herkenham et al., 1990, 1991; Matsuda et al., 1990; Buckley et al., 1998; Tsou et al., 1998), the learning and memory impairments produced by marijuana (Miller, 1984; Howlett, 1995) may be caused by its effects in the hippocampus.
Memory storage in the mammalian brain is generally believed to involve modifications of synaptic connections between neurons. In particular, long-term potentiation (LTP) of synaptic connections in the hippocampus is a compelling candidate for a cellular mechanism that underlies certain types of learning and memory (Goda and Stevens, 1996; Cain, 1997; Chen and Tonegawa, 1997). However, it is thought that information storage requires mechanisms for weakening, as well as strengthening, synaptic efficacy. Long-term depression (LTD), the functional inverse of LTP, is thus also a candidate mechanism for memory formation in the hippocampus (Christie et al., 1994; Bear and Abraham, 1996). If either LTP or LTD is impaired by cannabinoid receptor activation, learning and memory may also be impaired.
Indeed, previous studies have shown that cannabinoid receptor activation blocks field potential LTP in the hippocampus (Nowicky et al., 1987; Collins et al., 1994, 1995; Terranova et al., 1995). In addition, two endogenous ligands of cannabinoid receptors, anandamide (Devane et al., 1992) and sn-2 arachidonylglycerol (Stella et al., 1997), inhibit LTP of hippocampal field potentials (Terranova et al., 1995; Stella et al., 1997). These results suggest that cannabinoids may act to inhibit a protein (or proteins) required for LTP and LTD induction, perhaps via activation of the G-protein to which cannabinoid receptors are coupled (Howlett, 1995). However, cannabinoid receptor activation has also been shown to inhibit glutamatergic synaptic transmission presynaptically in cultured hippocampal neurons (Shen et al., 1996; Sullivan, 1999). Because hippocampal LTP and LTD require the depolarization of the postsynaptic membrane to relieve magnesium blockade of NMDA receptors and allow the entry of calcium (Malenka and Nicoll, 1993; Nicoll and Malenka, 1995), a reduction in neurotransmitter release could impair LTP and LTD by failing to depolarize the postsynaptic cell to a level that relieves magnesium blockade.
Using whole-cell patch-clamp and field potential recordings in hippocampal slices, we found that cannabinoid receptor activation inhibited EPSCs via a presynaptic inhibition of neurotransmitter release and, in agreement with previous studies, blocked LTP of field potentials (Nowicky et al., 1987; Collins et al., 1994, 1995; Terranova et al., 1995). Here, we show for the first time that hippocampal field potential LTD was also prevented by cannabinoid receptor activation. However, holding cells at depolarized levels during LTP and LTD induction protocols allowed normal levels of LTP and LTD to be induced. Therefore, cannabinoid receptor activation does not directly inhibit the molecular mechanisms of synaptic plasticity but rather acts to reduce the probability of neurotransmitter release, which in turn prevents depolarization of the postsynaptic cell and the subsequent entry of calcium through NMDA receptors.
MATERIALS AND METHODS
Slice preparation. Mice were anesthetized by Metofane (Mallinckrodt Veterinary, Mundelein, IL) and decapitated. The brain was removed and placed in ice-cold solution (120 mm NaCl, 3.5 mm KCl, 0.7 mm CaCl2, 4 mm MgCl2, 1.25 mmNaH2PO4, 26 mmNaHCO3, and 10 mm glucose) bubbled with 95% O2/5% CO2. The same cold high Mg2+, low Ca2+ solution was used throughout the dissection procedure to prevent transmitter release and minimize injury to the cells. The hindbrain was cut away, and the flat surface of the forebrain was glued to the pan of a DSK microslicer vibratome with cyanoacrylate glue. The hippocampus was dissected out and placed in a chamber containing the same dissecting buffer perfused with 95% O2/5% CO2. Slices (350 μm) were allowed to incubate at room temperature in the holding chamber for at least 1 hr before recording. Field potential experiments were performed on mice between 4 and 6 weeks of age. Whole-cell patch-clamp experiments were performed on 13- to 19-d-old mice. Because the distribution of cannabinoid receptors at birth is very similar to that in the adult (Buckley et al., 1998), there is no reason to suspect that there would be any age-related differences in the response to cannabinoids at these different ages.
Electrophysiology. All experiments were performed at room temperature. Individual slices were placed in a submerged recording chamber, held by a net made with flattened platinum wire and nylon threads. Slices were perfused with solution (120 mm NaCl, 3.5 mm KCl, 2.6 mm CaCl2, 1.3 mm MgCl2, 1.25 mmNaH2PO4, 26 mmNaHCO3, and 10 mm glucose) saturated with 95% O2/5% CO2, at a rate of 2 ml/min. Picrotoxin (25–50 μm; Calbiochem, La Jolla, CA) was included during whole-cell recordings to block inhibitory GABAergic currents (except where noted), and a cut was generally made between the CA1 and CA3 region to prevent recurrent excitation. Bovine serum albumin (1 mg/ml; Boehringer Mannheim, Indianapolis, IN) was run through the tubing before and after all experiments to reduce the nonspecific binding of WIN55,212-2 and other drugs. WIN55,212-2 (Research Biochemicals, Natick, MA), WIN55,212-3 (Research Biochemicals), SR141716A (a generous gift from Dr. K. Mackie, University of Washington), and 2-chloroadenosine (Research Biochemicals) were applied by bath application. WIN55,212-2, WIN55,212-3, and SR141716A solutions were made up as 10 mm stock solutions in DMSO, stored at −20°C, and used at a final DMSO concentration of ≤0.05%. 2-Chloroadenosine was made up as a 10 mm stock solution in water and stored at −20°C. Pertussis toxin (List Biologic, Campbell, CA) was used at a concentration of 5 μg/ml, and slices were incubated with the toxin for at least 12 hr before recording (Hsu, 1996).
Schaeffer collateral–commissural fibers were stimulated by ultrasmall concentric bipolar electrodes (FHC, Bowdoinham, ME) delivering 0.1 msec pulses. Stimulus intensities were adjusted to evoke similarly sized baseline responses. Field EPSPs (fEPSPs) were recorded in CA1 with glass electrodes (2–3 MΩ resistance) filled with perfusing buffer. Whole-cell patch-clamp recordings were performed in CA1 pyramidal neurons according to standard techniques. The pipettes (3.5–5 MΩ resistance) were filled with a solution containing 170 mm K-gluconate, 10 mm HEPES, 10 mmNaCl, 2 mm MgCl2, 0.2 mmEGTA, 3.5 mm Mg-ATP, and 1.0 mm GTP-Li, with pH adjusted to 7.2 and 290–300 mOsm. Access resistance was monitored, and only cells with stable access resistance were included in the data analysis. In whole-cell recordings, the membrane potential of the postsynaptic cell was held at −70 mV, except during LTP and LTD experiments, at which time they were held at −20 and −50 mV, respectively. Recordings were performed using an Axopatch 200 amplifier, filtered at 2 kHz, and analyzed with programs written in Visual Basic. Field EPSPs and EPSCs were evoked every 20 sec. The initial slopes of fEPSPs and peak amplitudes of EPSCs were measured for field potential and whole-cell recordings, respectively.
The fiber volley was recorded as a field potential in the presence of 20 μm 6-cyano-7-nitroquinoxaline-2,3-dione (CNQX; Cambridge Research Biochemicals, Worcester, MA) and 10 μmd-(−)-2-amino-5-phosphopentanoic acid (AP-5; Research Biochemicals).
The amplitude and frequency of spontaneous miniature EPSCs (mEPSCs) were studied by recording continuously over 300 sec in the presence of 1 μm tetrodotoxin (Calbiochem) before and after WIN55,212-2 application. The peak amplitudes of the mEPSCs were measured off-line semiautomatically using an adjustable amplitude threshold. All deflections from baseline that were greater than the threshold were detected. Selected events were then visually examined, and any spurious events were manually rejected, whereas any missed events were flagged for inclusion in the mean amplitude and frequency calculations. Frequencies were calculated by dividing the total number of mEPSC events by the total time sampled.
Effects of WIN55,212-2 on paired-pulse responses were studied by applying pairs of stimuli at varying interpulse intervals (20–200 msec). The responses to single pulses were used to generate a template that could be subtracted from the second response after 20 or 50 msec interpulse intervals to yield an accurate measurement of the response. At longer intervals, subtraction was not required to measure the peak current amplitude accurately. The peak amplitude of the response to the second pulse was averaged over 5–10 trials and divided by the averaged peak amplitude of the response to the first pulse to give a paired-pulse ratio (P2/P1) before and after WIN55,212-2 application at each interpulse interval.
Field potential and whole-cell LTP were induced by a tetanus consisting of five trains of 100 Hz stimulation lasting 200 msec at an intertrain interval of 10 sec. Cells were held at −20 mV during tetanus. LTD was evoked by 900 stimuli at a frequency of 1 Hz for field potentials and at 2 Hz for whole-cell recordings. Cells were held at −50 mV during low-frequency stimulation. All data were normalized to the baseline, and the percentage of LTP and LTD was calculated by averaging the response 25–35 min after induction. For these experiments, slices were incubated with 1 μm WIN55,212-2 for 1–8 hr.
RESULTS
Cannabinoid receptor activation decreases the size of EPSCs in hippocampal slices
Two subtypes of G-protein–coupled cannabinoid receptors have been identified to date: CB1, which is found in the CNS and testes (Matsuda et al., 1990), and CB2, which is found in the peripheral nervous system (Munro et al., 1993). The effects of cannabinoid receptor activation on glutamatergic synaptic transmission in the CNS were studied by applying the selective and potent CB1 cannabinoid receptor agonist WIN55,212-2 (Compton et al., 1992;D’Ambra et al., 1992) to hippocampal CA1 pyramidal cells in intact slices. WIN55,212-2 (1–5 μm) reduced EPSC amplitude by 54.3 ± 3.1% (n = 26; Fig.1). The degree of WIN55,212-2–mediated inhibition varied from 30 to 81%, although cells from slices made on the same day generally exhibited the same amount of inhibition. This inhibition by WIN55,212-2 was observed in both the presence and absence of the GABA receptor antagonist picrotoxin, indicating that cannabinoid receptor agonist directly inhibits glutamatergic transmission (also seePaton et al., 1998). No appreciable change in the amount of inhibition by WIN55,212-2 was observed when picrotoxin was excluded from the recording solution (data not shown), although small changes could be missed because of the variability of inhibition from slice to slice.
Fig. 1.

WIN55,212-2 reduces EPSCs in hippocampal CA1 neurons by activation of CB1 cannabinoid receptors. a, Data traces of EPSCs before (gray) and after (black) application of 1 μmWIN55,212-2. The stimulus artifact is not shown. b, Bar graph showing the average percent inhibition of EPSC size (mean ± SEM) after application of 1–5 μm WIN55,212-2 (54.3 ± 3.1%; n = 26), 1 μm WIN55,212-2 in the presence of the selective CB1 receptor antagonist SR141716A (−6.3 ± 8.9%; n = 4), and 1 μm WIN55,212-2 after pretreatment with PTX (−4.0 ± 5.4%; n = 3). The average inhibition of field EPSPs after application of the inactive cannabinoid receptor agonist WIN55,212-3 (5 μm) is also shown (−4.2 ± 1.8%; n = 7).
To establish that the reduction in EPSC amplitude was attributable to cannabinoid receptor activation rather than to some nonspecific effect, we applied the inactive form of WIN55,212-2 (Pacheco et al., 1991) (WIN55,212-3) to hippocampal slices. WIN 55,212-3 does not activate cannabinoid receptors, but at concentrations ≥3 μm, this drug can directly block calcium channels in cultured hippocampal neurons (Shen and Thayer, 1998). WIN55,212-3 (5 μm) had no effect on field EPSP size (n = 7; Fig.1b), indicating that WIN55,212-2 had no direct effect on calcium channels mediating neurotransmitter release in these slices. In addition, the effects of 1 μm WIN55,212-2 were blocked by coapplication with 1 μm SR141716A, a cannabinoid receptor antagonist (Rinaldi-Carmona et al., 1994) (n = 4; Fig. 1b), indicating that the antagonist competitively inhibited the effects of WIN55,212-2 via the cannabinoid receptor. Finally, 1 μm WIN55,212-2 did not change the amplitude of the fiber volley of the field potential recorded in the presence of 20 μm CNQX and 10 μm AP-5 (103.0 ± 6.7%;n = 4), indicating that cannabinoid receptor activation did not alter the excitability of hippocampal neurons.
An inhibitor of Gi/o proteins, pertussis toxin (PTX), was used to verify that G-proteins mediated the effects of cannabinoid receptor activation. Slices were made ≥12 hr before recording, with one-half of the slices incubated in the normal recording solution and the other one-half incubated with PTX (5 μg/ml) (Hsu, 1996). WIN55,212-2 (1 μm) was bath applied to the slices incubated with PTX, and the EPSC was monitored. No appreciable change in the size of the EPSC was observed (104.0 ± 5.4%; n = 3; Fig. 1b). To ensure that the lack of inhibition by WIN55,212-2 was not an artifact of the slices, we applied WIN55,212-2 (1 μm) to control slices after every PTX slice, and an appreciable inhibition of EPSC size was observed (45.1 ± 3.0%; n = 3). These experiments indicate that the inhibition of EPSC size after cannabinoid receptor activation is mediated by inhibitory G-proteins coupled to the receptors.
The effect of cannabinoid receptor activation is not caused by a change in postsynaptic responsiveness to glutamate
The decrease in EPSC amplitude caused by WIN55,212-2 could be caused by changes in the amount of neurotransmitter released presynaptically and/or by changes in the postsynaptic responsiveness to glutamate. The amplitude of spontaneous mEPSCs was used to monitor changes in postsynaptic responsiveness to glutamate resulting from cannabinoid receptor activation. Because spontaneous mEPSCs are believed to be the postsynaptic response to a single spontaneously released synaptic vesicle, any change in postsynaptic sensitivity to glutamate should be reflected as a change in the amplitude of mEPSCs.
Cannabinoid receptor activation decreased mEPSC frequency but had no effect on mEPSC amplitude. mEPSCs were measured before and after application of 1 μm WIN55,212-2 in the presence of tetrodotoxin (1 μm) to inhibit spontaneous action potential firing. The mean amplitude of mEPSCs remained unchanged after drug application (9.93 ± 0.22 pA before drug and 10.04 ± 0.23 pA after drug; n = 6; Fig.2a,b), and the distribution of mEPSC amplitudes was indistinguishable from that of the control (p > 0.05, Kolmogorov–Smirnov two-sample test). These results suggest that WIN55,212-2 does not change the sensitivity of postsynaptic receptors to glutamate, in agreement with previous work in the cerebellum (Levenes et al., 1998). WIN55,212-2 (1 μm) did decrease the frequency of mEPSCs, from 0.41 ± 0.08 Hz before drug to 0.23 ± 0.06 Hz after drug (p < 0.05, paired t test;n = 6; Fig. 2d), also consistent with the previous report using cerebellar slices (Levenes et al., 1998). Altogether, these results indicate that the decrease in EPSC amplitude caused by cannabinoid receptor activation is attributable to a decrease in the amount of neurotransmitter released presynaptically (Shen et al., 1996; Sullivan, 1999) and not to any decrease in postsynaptic sensitivity to glutamate.
Fig. 2.
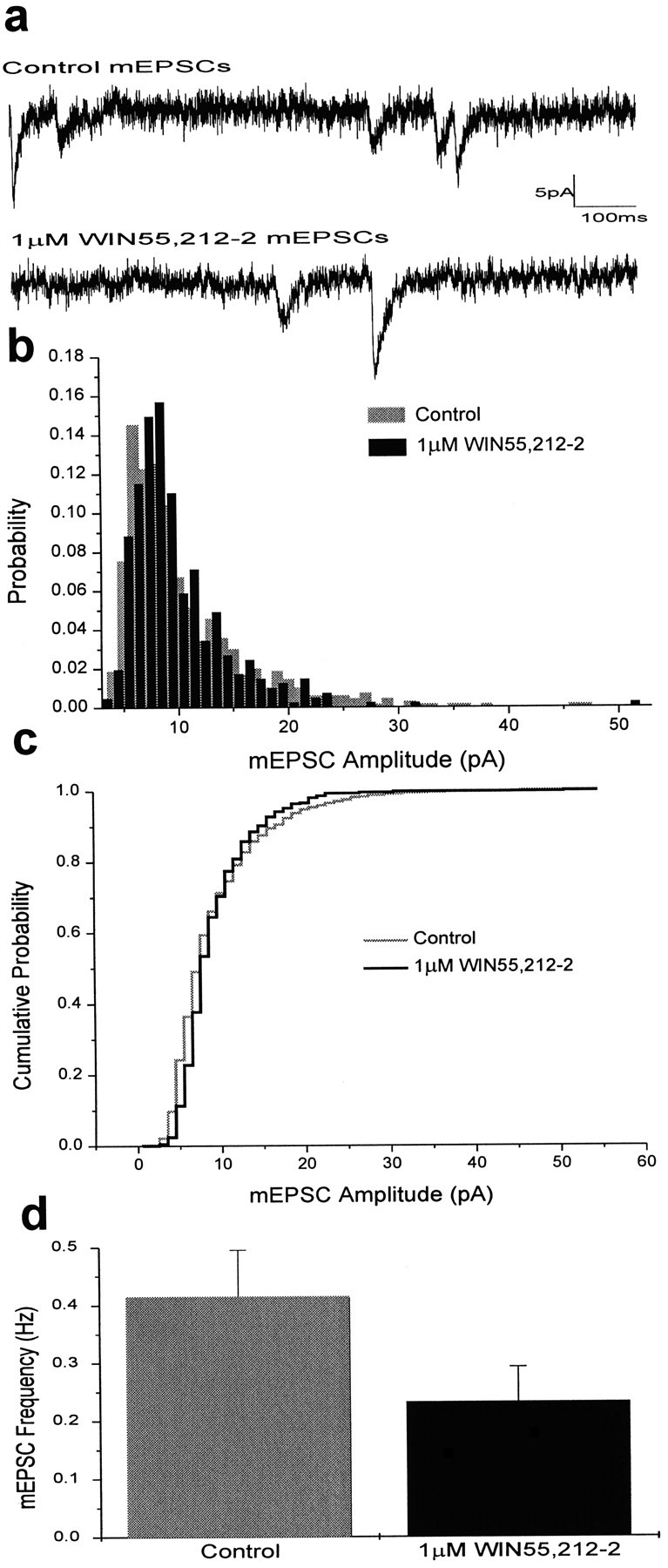
Cannabinoid receptor activation does not change the postsynaptic sensitivity to glutamate but does reduce mEPSC frequency. a, Sample traces of mEPSCs before and after application of 1 μm WIN55,212-2.b, Amplitude histogram of spontaneous mEPSCs. The threshold for peak detection was set between 5 and 10 pA. Data were binned in 1 pA intervals. The mean amplitude of mEPSCs was 9.93 ± 0.22 pA before (gray) and 10.04 ± 0.23 pA after (black) application of 1 μm WIN55,212-2 (n = 6). c, Cumulative probability histogram of mEPSC amplitude before (gray) and after (black) application of 1 μm WIN55,212-2. d, Bar graph showing average mEPSC frequency (mean ± SEM) before (gray) and after (black) application of 1 μm WIN55,212-2. The mean frequency of mEPSCs was 0.41 ± 0.08 Hz before WIN55,212-2 and 0.23 ± 0.06 Hz after WIN55,212-2 (p < 0.05, paired t test; n = 6).
Cannabinoid receptor activation increases paired-pulse facilitation by lowering the probability of neurotransmitter release
To confirm that cannabinoids act to regulate the amount of presynaptic neurotransmission, paired-pulse facilitation (PPF) was measured before and after cannabinoid receptor activation. PPF is an enhancement of the synaptic response to the second of two closely spaced action potentials and is believed to reflect the effects of residual calcium from the first action potential on release triggered by the second action potential (Zucker, 1989). PPF is enhanced if the probability of release is lowered, as in experiments with lower levels of calcium (Creager et al., 1980; Manabe et al., 1993; Mennerick and Zorumski, 1995; Debanne et al., 1996).
The amount of PPF increased after activation of cannabinoid receptors. PPF was measured in whole-cell patch-clamped neurons at intervals between 20 and 200 msec in the postsynaptic cell before and after application of 5 μm WIN55,212-2. We found that PPF increased after application of drug at all intervals (Fig.3a). Because 5 μm WIN55,212-2 has been shown to block calcium channels directly in cultured hippocampal neurons (Shen and Thayer, 1998), the effect of 1 μm WIN55,212-2 on PPF at 50 msec intervals was also investigated. Again, the PPF after drug application was significantly increased over that of control cells (2.11 ± 0.17 compared with 1.70 ± 0.08, respectively;p < 0.01, paired t test; n= 9; Fig. 3b). These data are consistent with those of Paton et al. (1998) and indicate that WIN55,212-2 enhances paired-pulse facilitation via a cannabinoid receptor-mediated decrease in the amount of neurotransmitter released presynaptically.
Fig. 3.

Cannabinoid receptor activation increases paired-pulse facilitation. a, Paired-pulse facilitation (mean ± SEM) before (gray circles;n = 4) and after (black squares) 5 μm WIN55,212-2. The ratio of the second EPSC (P2) to the first EPSC (P1) is shown as a function of the interpulse interval. At all intervals, paired-pulse facilitation increased after application of 5 μm WIN55,212-2. b, Top, Sample traces showing paired-pulse facilitation at 50 msec before (left) and after (right) application of 1 μm WIN55,212-2. Bottom, Bar graph showing paired-pulse facilitation at 50 msec (mean ± SEM) before (1.70 ± 0.08) and after (2.11 ± 0.17) application of 1 μm WIN55,212-2 (p < 0.01, paired t test;n = 9).
Long-term potentiation of field potentials is only moderately impaired by cannabinoid receptor activation in the absence of Mg2+
Previous studies have shown that WIN55,212-2 prevents the induction of LTP of field EPSPs recorded from the CA1 region of hippocampal slices (Nowicky et al., 1987; Collins et al., 1994, 1995;Terranova et al., 1995). This inhibition could be attributable to a direct effect on proteins involved in long-term plasticity. However, it is also possible that LTP is inhibited by WIN55,212-2 simply because tetanic stimulation fails to release enough excitatory neurotransmitter to depolarize the postsynaptic neurons and relieve Mg2+ blockade. If this latter hypothesis is correct, then eliminating Mg2+ from the extracellular solution during tetanus should allow the induction of LTP in the presence of WIN55,212-2.
To test this hypothesis, tetanic stimulation was used to induce LTP of field potentials in the presence of WIN55,212-2 (5 μm) both with and without Mg2+ present. In slices with Mg2+ present (1.3 mm) during tetanic stimulation (five trains of 100 Hz stimuli lasting 200 msec), the induction of LTP was prevented by the presence of WIN55,212-2 (0.98 ± 0.03; n = 7; Fig.4b), as reported previously (Nowicky et al., 1987; Collins et al., 1994, 1995; Terranova et al., 1995). However, in slices in which Mg2+was washed out before and during tetanus, the induction of LTP was reduced but still present 25–35 min after induction (1.21 ± 0.06; n = 10; control = 1.55 ± 0.12;n = 4; Fig. 4b). Application of the CB1 receptor antagonist SR141716A (1 μm) before application of WIN55,212-2 blocked the inhibition of LTP by cannabinoid receptor activation (1.45 ± 0.24; n = 3; data not shown).
Fig. 4.

Field potential LTP is only moderately impaired by cannabinoid receptor activation in the absence of Mg2+. a, Bottom, Summary of field potential recordings in the presence of 5 μm WIN55,212-2. Data are expressed as normalized field EPSPs (± SEM). The initial slopes of fEPSPs were normalized to the baseline value preceding the induction of LTP. Testing stimuli were given every 20 sec. Mean values of LTP 25–35 min after tetanus were 1.00 ± 0.04 with Mg2+ present (n = 5) and 1.22 ± 0.08 after washout of Mg2+.Top, Sample traces showing fEPSPs recorded before LTP induction (left), after tetanus delivered in the presence of normal concentrations of Mg2+ (middle), and after tetanus delivered after washout of Mg2+(right). b, Cumulative probability histogram of the magnitude of field potential LTP 25–35 min after tetanus. Cumulative probability distributions are shown for control fEPSPs (1.55 ± 0.12; n = 4; solid gray), fEPSPs recorded in 5 μm WIN55,212-2 with Mg2+ present (0.98 ± 0.03;n = 7; dotted black), fEPSPs recorded in 5 μm WIN55,212-2 after washout of Mg2+ (1.21 ± 0.06; n = 10;solid black), and fEPSPs recorded in 5 μmWIN55,212-2 and low Mg2+ (1.34 ± 0.12;n = 6; dotted gray).
To ascertain that the effect of Mg2+washout on LTP was not attributable to variability among slices, we first induced LTP in the presence of WIN55,212-2 and normal Mg2+ concentrations and then induced LTP again in the same slice after washing out the Mg2+ before and during tetanus. As before, LTP was blocked in the presence of WIN55,212-2 and normal Mg2+ concentrations, but when Mg2+ was removed from the bath, LTP was successfully induced, although at a reduced level (1.22 ± 0.08;n = 5; Fig. 4a). The degree of LTP may have been reduced because of incomplete magnesium washout. Therefore, LTP was induced continuously in slices bathed in low magnesium (0.5 mm Mg2+ and 3.5 mm Ca2+) in the presence of WIN55,212-2 (5 μm). As our hypothesis would predict, the degree of LTP was increased (1.34 ± 0.12; n = 6) under these conditions, suggesting that more complete removal of magnesium further relieved magnesium blockade of NMDA receptors. These results indicate that the machinery involved in the induction of LTP is not completely inhibited by WIN55,212-2.
Long-term depression of field potentials is unimpaired by cannabinoid receptor activation in the absence of Mg2+
Cannabinoid receptor activation also blocked LTD of hippocampal field EPSPs in the presence of Mg2+. LTD induced by low-frequency (1 Hz) stimulation for 15 min in the presence of 5 μm WIN55,212-2 and normal concentrations of Mg2+ (1.3 mm) was completely inhibited (1.01 ± 0.01; n = 7; Fig.5b). To determine whether this inhibition of LTD was attributable to a direct effect on LTD induction machinery, we induced LTD in the absence of Mg2+. In Mg2+-free conditions, LTD in the presence of WIN55,212-2 was unimpaired and long lasting (0.76 ± 0.04;n = 9; Fig. 5b). This was not an artifact of slice-to-slice variability, because low-frequency stimulation in the presence of WIN55,212-2 produced robust LTD after washout of Mg2+ (0.75 ± 0.03; n= 5) in slices that failed to produce LTD after low-frequency stimulation in the presence of Mg2+(1.01 ± 0.01; Fig. 5a). Thus WIN55,212-2 does not directly inhibit the proteins involved in LTD.
Fig. 5.

Field potential LTD is unimpaired by cannabinoid receptor activation in the absence of Mg2+.a, Bottom, Summary of field potential recordings in the presence of 5 μm WIN55,212-2. Data are expressed as normalized field EPSPs (± SEM). The initial slopes of fEPSPs were normalized to the baseline value preceding the induction of LTD. Testing stimuli were given every 20 sec. Mean values of LTD 25–35 min after low-frequency stimulation (LFS) were 1.01 ± 0.01 with Mg2+ present (n = 5) and 0.75 ± 0.03 after washout of Mg2+.Top, Sample traces showing fEPSPs recorded before LTD induction (left), after low-frequency stimulation delivered in the presence of normal concentrations of Mg2+ (middle), and after low-frequency stimulation delivered after washout of Mg2+ (right). b, Cumulative probability histogram of the magnitude of field potential LTD 25–35 min after low-frequency stimulation. Cumulative probability distributions are shown for control fEPSPs (0.78 ± 0.08;n = 6; solid gray), fEPSPs recorded in 5 μm WIN55,212-2 with Mg2+ present (1.01 ± 0.01; n = 7; dotted black), and fEPSPs recorded in 5 μm WIN55,212-2 after washout of Mg2+ (0.76 ± 0.04;n = 9; solid black).
Long-term potentiation is unimpaired by cannabinoid receptor activation when the postsynaptic membrane is depolarized during tetanic stimulation
To test further our hypothesis that cannabinoids act simply to decrease neurotransmitter release, with no direct effect on the proteins involved in synaptic plasticity, we next investigated the effects of cannabinoid receptor activation on the induction of LTP in individual neurons, using whole-cell patch clamping. In the presence of 1 μm WIN55,212-2, LTP was fully induced in neurons depolarized by whole-cell voltage clamp. LTP was induced by tetanic stimulation (five trains of 100 Hz stimuli lasting 200 msec) of the Schaeffer collaterals while holding the cell at −20 mV. Because washout is a factor in whole-cell LTP experiments, a 10 min baseline was recorded immediately after breaking into the cell, followed by tetanus. Cells that failed to produce LTP were included with cells that did show LTP for consistency. The levels of LTP attained in this manner were indistinguishable from those of control cells in the absence of WIN55,212-2 (1.62 ± 0.27 with drug; n = 7; 1.56 ± 0.11 without drug; n = 8;p > 0.05, Kolmogorov–Smirnov two-sample test; Fig.6a). This whole-cell LTP was effectively blocked by the coapplication of an NMDA receptor antagonist, AP-5 (50 μm), with 1 μm WIN55,212-2 (1.03 ± 0.02;n = 3), indicating that this potentiation was mediated by NMDA receptors. Thus, depolarization of the postsynaptic cell was sufficient to overcome the effects of reduced neurotransmitter release and permit normal LTP expression. These data further support the hypothesis that the proteins involved in LTP induction are unaffected by cannabinoid receptor activation.
Fig. 6.
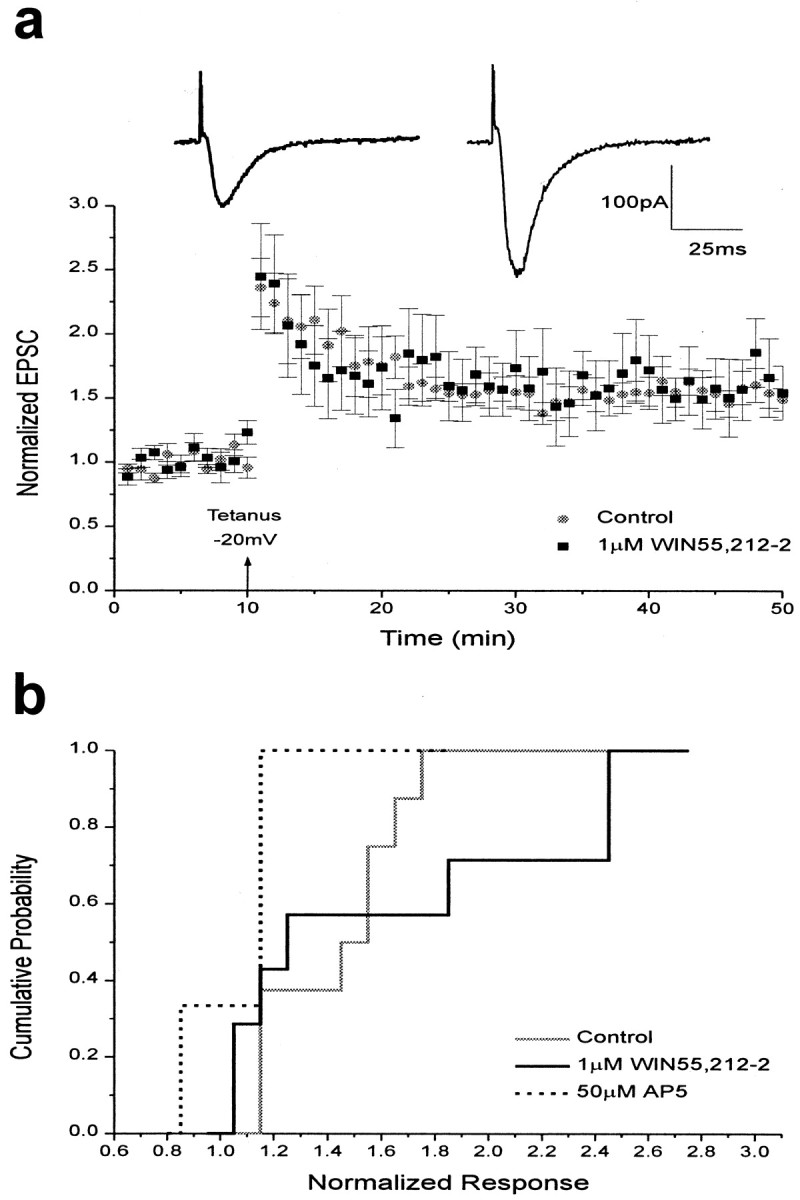
LTP is unimpaired by cannabinoid receptor activation when the postsynaptic membrane is depolarized during tetanus. a, Bottom, Summary of control and 1 μm WIN55,212-2 whole-cell experiments. Data are expressed as normalized EPSC amplitudes (± SEM). The amplitudes of EPSCs were normalized to the baseline value preceding the induction of LTP. Testing stimuli were given every 20 sec. Mean values of LTP 25–35 min after tetanus when the postsynaptic cell was depolarized to −20 mV were 1.56 ± 0.11 for controls (n = 8;gray circles) and 1.62 ± 0.27 in the presence of 1 μm WIN55,212-2 (n = 7; black squares). Top, Sample tracesshowing EPSCs recorded in the presence of WIN55,212-2 before (left) and after (right) the induction of LTP. b, Cumulative probability histogram of the magnitude of whole-cell LTP 25–35 min after tetanus. Cumulative probability distributions are shown for control EPSCs (solid gray), EPSCs recorded in the presence of 1 μmWIN55,212-2 (solid black), and EPSCs recorded in the presence of 50 μm AP-5 (dotted black). The control and 1 μm WIN55,212-2 distributions were not significantly different (Kolmogorov–Smirnov two-sample test,p > 0.05).
Long-term depression is unimpaired by cannabinoid receptor activation when the postsynaptic membrane is depolarized during low-frequency stimulation
Finally, the effects of cannabinoid receptor activation on hippocampal whole-cell LTD were investigated. Because our experiments studying field potentials showed that normal levels of LTD were attainable in the absence of Mg2+, we hypothesized that in whole-cell experiments holding the postsynaptic cell at a slightly depolarized potential would be sufficient to allow normal induction of LTD during cannabinoid receptor activation. Indeed, LTD was fully induced in neurons depolarized by whole-cell voltage clamp in the presence of WIN55,212-2. As before, 1 μmWIN55,212-2 was added to the holding chamber after dissection. Cells were held at −50 mV during low-frequency (2 Hz) stimulation, and the LTD that ensued was both robust and long lasting (0.56 ± 0.05 with drug; n = 9; 0.62 ± 0.05 without drug;n = 9; Fig.7a) and was undistinguishable from that of controls (p > 0.05, Kolmogorov–Smirnov two-sample test). Because previous work showed that cerebellar LTD—which, in contrast to hippocampal LTD, does not require activation of NMDA receptors (Linden and Connor, 1993)—was impaired under current-clamp conditions (Levenes et al., 1998), we attempted to determine whether the same held true in hippocampal slices. When LTD was induced in the presence of 1 μm WIN55,212-2 under current-clamp conditions and then again under slightly depolarized conditions, we found that LTD was indeed impaired when the cell was held in current clamp during low-frequency stimulation (0.77 ± 0.07; n = 4), as reported in cerebellar slices, but increased to normal levels when the cell was depolarized during low-frequency stimulation (0.51 ± 0.07; n= 4; Fig. 7c). As with whole-cell LTP, LTD was blocked in the presence of AP-5 (50 μm) and 1 μm WIN55,212-2 (0.99 ± 0.02;n = 3), demonstrating that this depression was mediated via NMDA receptors. Thus, the induction of LTD is possible in the presence of cannabinoid receptor agonists when the postsynaptic neuron is weakly depolarized, confirming that cannabinoid receptor activation does not directly affect the mechanisms of long-term synaptic plasticity in the hippocampus.
Fig. 7.
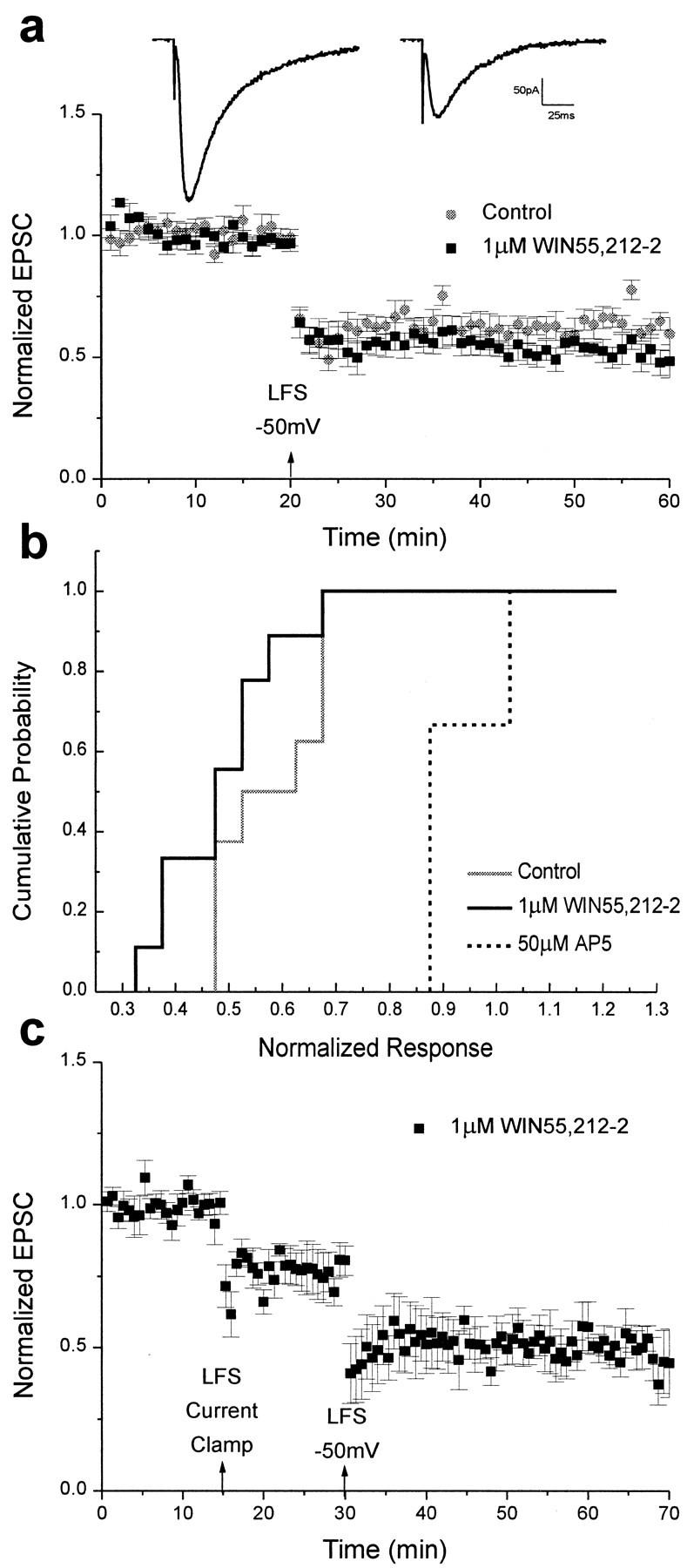
LTD is unimpaired by cannabinoid receptor activation when the postsynaptic membrane is depolarized during low-frequency stimulation. a, Bottom, Summary of control and 1 μm WIN55,212-2 whole-cell experiments. Data are expressed as normalized EPSC amplitudes (± SEM). The amplitudes of EPSCs were normalized to the baseline value preceding the induction of LTD. Testing stimuli were given every 20 sec. Mean values of LTD 25–35 min after low-frequency stimulation (LFS) when the postsynaptic cell was held at −50 mV were 0.62 ± 0.05 for controls (n = 9; gray circles) and 0.56 ± 0.05 in the presence of 1 μm WIN55,212-2 (n = 9; black squares). Top, Sample tracesshowing EPSCs recorded in the presence of WIN55,212-2 before (left) and after (right) the induction of LTD. b, Cumulative probability histogram of the magnitude of whole-cell LTD 25–35 min after low-frequency stimulation. Cumulative probability distributions are shown for control EPSCs (solid gray), EPSCs recorded in the presence of 1 μm WIN55,212-2 (solid black), and EPSCs recorded in the presence of 50 μm AP-5 (dotted black). The control and 1 μm WIN55,212-2 distributions were not significantly different (Kolmogorov–Smirnov two-sample test, p > 0.05). c, Summary of whole-cell LTD recordings in the presence of 1 μm WIN55,212-2 when the cell was held in current clamp (0.77 ± 0.07; n = 4) and then at −50 mV (0.51 ± 0.07) during low-frequency stimulation.
LTP can be induced after adenosine receptor activation if magnesium blockade of NMDA receptors is experimentally relieved
Previous studies have shown that adenosine receptor agonists prevent the induction of LTP of field EPSPs recorded from the CA1 region of hippocampal slices (de Mendonça and Ribeiro, 1990). Like WIN55,212-2, adenosine is an inhibitor of presynaptic neurotransmitter release (Scholz and Miller, 1992). Therefore, as with WIN55,212-2, eliminating Mg2+ from the extracellular solution or simply depolarizing the postsynaptic membrane during tetanus should allow the induction of LTP in the presence of adenosine receptor agonist.
To test this hypothesis, we first induced LTP by tetanic stimulation delivered in the presence of an adenosine receptor agonist, 2-chloroadenosine (CADO; 10 μm), and normal Mg2+ concentrations; then LTP was induced again in the same slice after washing out the Mg2+ before and during tetanus. As reported previously, LTP was partially blocked in the presence of CADO and normal Mg2+ concentrations (1.17 ± 0.05; n = 4; Fig.8a), but when Mg2+ was removed from the bath, LTP was enhanced (1.33 ± 0.10). This result suggests that inhibition of LTP by CADO is likely caused by decreased levels of neurotransmitter released presynaptically. The average reduction of EPSC size by 10 μm CADO was 39.7 ± 4.4% (n = 4; data not shown), indicating that inhibition of presynaptic neurotransmitter release by CADO (10 μm) was not as complete as with WIN55,212-2 (5 μm) in this preparation, which may explain why inhibition of LTP was also not as complete using CADO.
Fig. 8.
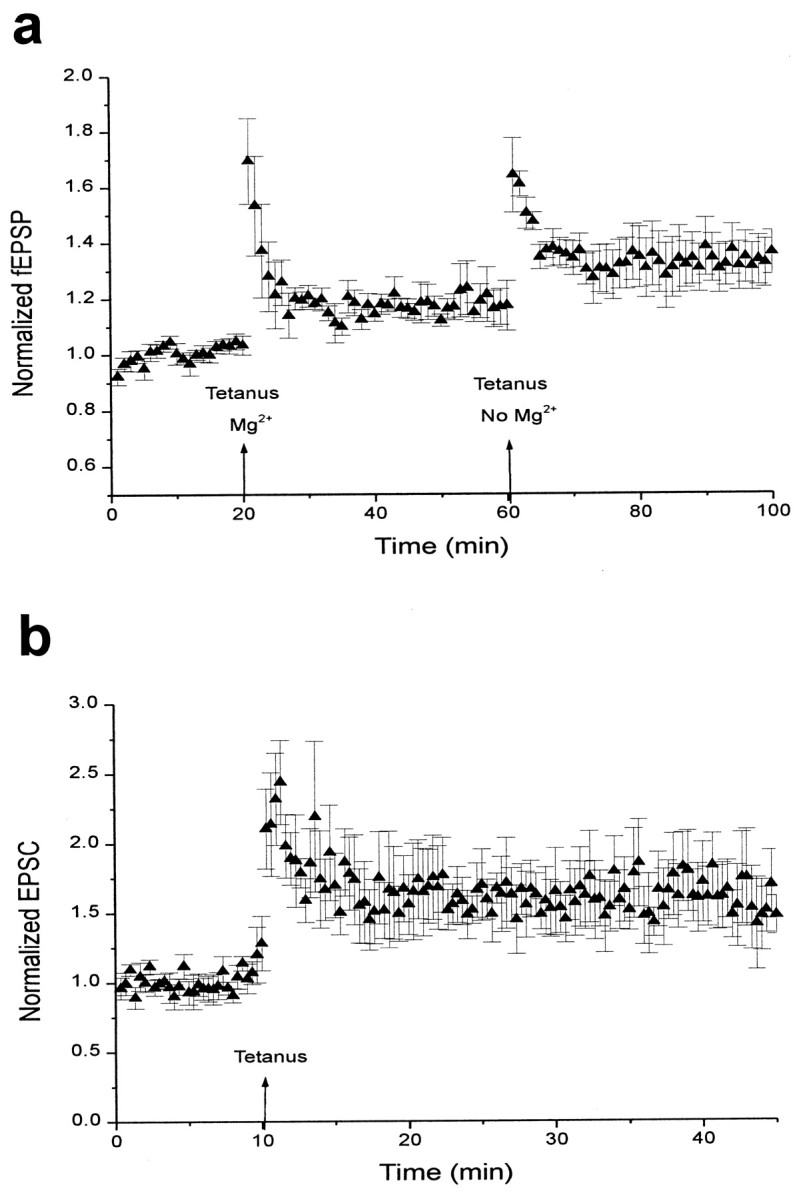
LTP can be induced after adenosine receptor activation if magnesium blockade of NMDA receptors is experimentally relieved. a, Summary of field potential recordings in the presence of 10 μm CADO. Data are expressed as normalized field EPSPs (± SEM). The initial slopes of fEPSPs were normalized to the baseline value preceding the induction of LTP. Testing stimuli were given every 20 sec. Mean values of LTP 25–35 min after tetanus were 1.17 ± 0.05 with Mg2+present (n = 4) and 1.33 ± 0.10 after washout of Mg2+. b, Summary of whole-cell recordings in the presence of 10 μm CADO. Data are expressed as normalized EPSC amplitudes (± SEM). The amplitudes of EPSCs were normalized to the baseline value preceding the induction of LTP. Testing stimuli were given every 20 sec. Mean values of LTP 25–35 min after tetanus were 1.62 ± 0.25 (n = 5).
We next investigated the effects of adenosine receptor activation on the induction of LTP in individual neurons, using whole-cell patch clamping. In the presence of 10 μm CADO, LTP was fully induced in neurons depolarized by whole-cell voltage clamp. For these experiments, slices were recorded in solution containing 10 μm CADO, and LTP was induced by tetanic stimulation while holding the cell at −20 mV. A 10 min baseline was recorded immediately after breaking into the cell, followed by tetanus. The levels of LTP attained in this manner were similar to those of control cells (1.62 ± 0.25; n = 5; Fig. 8b). Thus, depolarization of the postsynaptic cell was sufficient to overcome the effects of adenosine receptor activation and permit normal LTP expression. These data further support the hypothesis that long-term synaptic plasticity can be impaired by reduced presynaptic neurotransmitter release.
DISCUSSION
Previous experiments have shown that LTP of field potential recordings in the hippocampus is blocked by cannabinoid receptor activation (Nowicky et al., 1987; Collins et al., 1994, 1995; Terranova et al., 1995). This inhibition could be caused by a direct effect on the protein(s) involved in long-term synaptic plasticity. Activation of cannabinoid receptors has been found to inhibit the production of cAMP via activation of a G-protein to which the receptor is coupled (Bidaut-Russell et al., 1990; Deadwyler et al., 1995). This decrease in cAMP could, in turn, reduce the activity of cAMP-dependent protein kinase, which has been identified as an important modulator of LTP (Brandon et al., 1997). Other direct effects on proteins involved in LTP and LTD are possible. However, cannabinoid receptor-mediated G-protein activation is also known to inhibit release of neurotransmitter in cultured hippocampal neurons (Shen et al., 1996). To determine whether cannabinoid receptor activation inhibits long-term synaptic plasticity in hippocampal CA1 pyramidal cells directly by affecting proteins involved in LTP and LTD or simply by inhibiting neurotransmitter release or both, a series of experiments was performed using whole-cell patch-clamp and field potential recordings in hippocampal slices.
Cannabinoid receptor activation reduced EPSC size in CA1 hippocampal neurons to approximately one-half of its original value. Two separate experiments confirmed that these effects were attributable to cannabinoid receptor activation, rather than to some nonspecific action of the drug (such as direct blockade of calcium channels): (1) the inactive cannabinoid receptor agonist WIN55,212-3 had no effect on EPSC size, and (2) the selective CB1 cannabinoid receptor antagonist SR141716A competitively blocked the action of the agonist at the CB1 receptor. The effects of WIN55,212-2 were also blocked by pretreatment with pertussis toxin. These results indicate that the reduction in EPSC size caused by WIN55,212-2 is mediated via an inhibitory G-protein coupled to the CB1 receptor. Recent studies have shown that CB1 receptors are not expressed in the cell bodies of CA1 neurons but are strongly expressed in surrounding fibers, an expression pattern similar to that of GABAergic interneurons (Tsou et al., 1998). The presence of a GABAergic blocker did not notably alter WIN55,212-2 inhibition of EPSC size, however, consistent with a direct effect of cannabinoid receptor activation on glutamatergic transmission (Paton et al., 1998).
The failure of WIN55,212-2 to affect the amplitude of mEPSCs is a strong indication that cannabinoids reduce the size of EPSCs not by decreasing the postsynaptic sensitivity to glutamate but by decreasing the probability of transmitter release presynaptically. Interestingly, mEPSC frequency was significantly decreased after cannabinoid receptor activation, an effect observed with other agonists of presynaptic receptors that inhibit neurotransmitter release (Thompson et al., 1993). Finally, paired-pulse facilitation was consistently higher after cannabinoid receptor activation, confirming that cannabinoid receptor activation directly inhibits transmitter release.
To understand how the inhibition of neurotransmitter release might play a role in cannabinoid receptor-mediated impairment of long-term synaptic plasticity, we studied the effect of WIN55,212-2 on LTP in hippocampal slices. In agreement with previous studies (Nowicky et al., 1987; Collins et al., 1994, 1995; Terranova et al., 1995), WIN55,212-2 blocked LTP of hippocampal field potentials under normal recording conditions. However, field potential LTP was induced when Mg2+ was removed from the extracellular solution during tetanus. The removal of extracellular Mg2+ presumably relieved the magnesium block of NMDA receptors, allowing LTP induction in the presence of WIN55,212-2. This result suggests that, in the presence of WIN55,212-2, glutamate release was insufficient to depolarize the postsynaptic neuron fully and relieve the Mg2+ block of NMDA receptors, thus preventing the glutamate-mediated entry of postsynaptic calcium that is necessary for LTP induction.
Although LTP was achieved in the presence of WIN55,212-2 when Mg2+ was removed, the level of potentiation of field potentials was below that in control slices. One possible explanation for this reduction in field potential LTP is that some residual Mg2+ remained, even after washout. If there were some residual Mg2+present, then the cannabinoid receptor-mediated reduction in transmitter release might not allow full relief from Mg2+ blockade in all postsynaptic neurons, resulting in a reduced level of LTP. This idea is consistent with the findings that recording in low magnesium throughout resulted in increased levels of LTP, although these levels were still below those of controls. An alternative explanation is that WIN55,212-2 inhibited voltage-dependent L-type calcium channels, which do not contribute to neurotransmitter release but have been implicated in the induction of certain forms of LTP (Teyler et al., 1994). Although cannabinoid receptor activation does not inhibit L-type voltage-dependent calcium channels (Twitchell et al., 1997), it remains possible that 5 μm WIN55,212-2 blocks these channels directly (Shen and Thayer, 1998). However, our finding that preincubation with the cannabinoid receptor antagonist SR141716A allowed the induction of normal LTP in the presence of cannabinoid receptor agonist argues against a direct effect of WIN55,212-2 on L-type calcium channels. In addition, cannabinoid receptor activation has been found to activate inwardly rectifying and transient A-type potassium channels (Deadwyler et al., 1995; Henry and Chavkin, 1995; Mackie et al., 1995). Although the lack of effect of WIN55,212-2 on the amplitude of the fiber volley argues against a cannabinoid receptor-mediated modulation of potassium channels in these slices, an enhancement of potassium currents may contribute to the reduction in field potential LTP observed in the presence of WIN55,212-2. However, in whole-cell experiments, in which the postsynaptic cell was experimentally depolarized to −20 mV during tetanic stimulation, the level of LTP in the presence of WIN55,212-2 was indistinguishable from control. Therefore, the molecular mechanisms responsible for LTP induction are most likely unaffected by cannabinoid receptor activation.
In contrast to LTP, LTD of hippocampal field potentials in the presence of WIN55,212-2 reached normal levels when Mg2+ was removed from the extracellular solution. If WIN55,212-2 did have any effects on voltage-dependent calcium or potassium channels, this was evidently not great enough to block the induction of LTD. However, LTD requires lower levels of calcium influx than does LTP (Bear, 1995). In whole-cell experiments, LTD was impaired in the presence of WIN55,212-2 when the postsynaptic cell was held in current clamp during low-frequency stimulation but reached normal levels when the cell was held at a slightly depolarized level (−50 mV). As with the LTP experiments, this suggests that cannabinoid receptor activation affects synaptic plasticity by inhibiting the amount of neurotransmitter released presynaptically, leading to insufficient depolarization to permit calcium influx into the postsynaptic neuron. By depolarizing the cell during low-frequency and tetanic stimulation, this effect is essentially bypassed, and synaptic plasticity functions normally.
Finally, experimental relief of magnesium blockade of NMDA receptors, either by removing magnesium or by depolarizing the postsynaptic cell during tetanus, resulted in the expression of LTP in the presence of another inhibitor of neurotransmitter release, 2-chloroadenosine. Our finding that inhibition of LTP by adenosine receptor activation could be rescued by experimental relief of magnesium blockade of NMDA receptors further supports our hypothesis that cannabinoid receptor-mediated inhibition of LTP is caused by decreased presynaptic neurotransmitter release.
In conclusion, cannabinoid receptor activation does indeed block the induction of hippocampal LTP and LTD, candidate mechanisms for learning and memory. However, cannabinoid receptor agonists do not directly inhibit the molecular mechanisms underlying long-term synaptic plasticity but rather act presynaptically to reduce neurotransmitter release. This study provides an important step toward the goal of exploiting cannabinoids’ potential benefits by identifying the mechanism of their effects on two model systems of learning and memory.
Footnotes
This work was supported by grants from the National Alliance for Research on Schizophrenia and Depression and the National Institute on Drug Abuse to J.M.S. and from the National Institutes of Health to C. F. Stevens for D.L.M. We are indebted to Dr. C. F. Stevens for his generous support. We also thank Drs. A. K. McAllister and A. M. Zador for critical reading of this manuscript, Dr. E. P. Huang for editorial assistance, M. A. Pilla for excellent technical assistance, and Dr. K. Mackie for his gift of SR141716A.
Correspondence should be addressed to Dr. Jane M. Sullivan, Molecular Neurobiology Laboratory, The Salk Institute, 10010 North Torrey Pines Road, La Jolla, CA 92037.
REFERENCES
- 1.Bear MF. Mechanism for a sliding synaptic modification threshold. Neuron. 1995;15:1–4. doi: 10.1016/0896-6273(95)90056-x. [DOI] [PubMed] [Google Scholar]
- 2.Bear MF, Abraham WC. Long-term depression in hippocampus. Annu Rev Neurosci. 1996;19:437–462. doi: 10.1146/annurev.ne.19.030196.002253. [DOI] [PubMed] [Google Scholar]
- 3.Bidaut-Russell M, Devane WC, Howlett AC. Cannabinoid receptors and modulation of cyclic AMP accumulation in the rat brain. J Neurochem. 1990;55:21–26. doi: 10.1111/j.1471-4159.1990.tb08815.x. [DOI] [PubMed] [Google Scholar]
- 4.Brandon EP, Idzerda RL, McKnight GS. PKA isoforms, neural pathways, and behaviour: making the connection. Curr Opin Neurobiol. 1997;7:397–403. doi: 10.1016/s0959-4388(97)80069-4. [DOI] [PubMed] [Google Scholar]
- 5.Buckley NE, Hansson S, Harta G, Mezey E. Expression of the CB1 and CB2 receptor messenger RNAs during embryonic development in the rat. Neuroscience. 1998;82:1131–1149. doi: 10.1016/s0306-4522(97)00348-5. [DOI] [PubMed] [Google Scholar]
- 6.Cain DP. LTP, NMDA, genes and learning. Curr Opin Neurobiol. 1997;7:235–242. doi: 10.1016/s0959-4388(97)80012-8. [DOI] [PubMed] [Google Scholar]
- 7.Chen C, Tonegawa S. Molecular genetic analysis of synaptic plasticity, activity-dependent neural development, learning, and memory in the mammalian brain. Annu Rev Neurosci. 1997;20:157–184. doi: 10.1146/annurev.neuro.20.1.157. [DOI] [PubMed] [Google Scholar]
- 8.Christie BR, Kerr DS, Abraham WC. Flip side of synaptic plasticity: long-term depression mechanisms in the hippocampus. Hippocampus. 1994;4:127–135. doi: 10.1002/hipo.450040203. [DOI] [PubMed] [Google Scholar]
- 9.Collins DR, Pertwee RG, Davies SN. The action of synthetic cannabinoids on the induction of long-term potentiation in the rat hippocampal slice. Eur J Pharmacol. 1994;259:R7–R8. doi: 10.1016/0014-2999(94)90666-1. [DOI] [PubMed] [Google Scholar]
- 10.Collins DR, Pertwee RG, Davies SN. Prevention by the cannabinoid antagonist, SR141716A, of cannabinoid-mediated blockade of long-term potentiation in the rat hippocampal slice. Br J Pharmacol. 1995;115:869–870. doi: 10.1111/j.1476-5381.1995.tb15889.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Compton DR, Gold LH, Ward SJ, Balster RL, Martin BR. Aminoalkylindole analogs: cannabimimetic activity of a class of compounds structurally distinct from delta 9-tetrahydrocannabinol. J Pharmacol Exp Ther. 1992;263:1118–1126. [PubMed] [Google Scholar]
- 12.Creager R, Dunwiddie TV, Lynch G. Paired-pulse and frequency facilitation in the CA1 region of the in vitro rat hippocampus. J Physiol (Lond) 1980;299:409–424. doi: 10.1113/jphysiol.1980.sp013133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.D’Ambra TE, Estep KG, Bell MR, Eissenstat MA, Josef KA, Ward SJ, Haycock DA, Baizman ER, Casiano FM, Beglin NC, Chippari SM, Grego JD, Kullnig RK, Daley GT. Conformationally restrained analogues of pravadoline: nanomolar potent, enantioselective, (aminoalkyl)indole agonists of the cannabinoid receptor. J Med Chem. 1992;35:124–135. doi: 10.1021/jm00079a016. [DOI] [PubMed] [Google Scholar]
- 14.Deadwyler SA, Hampson RE, Mu J, Whyte A, Childers S. Cannabinoids modulate voltage sensitive potassium A-current in hippocampal neurons via a cAMP-dependent process. J Pharmacol Exp Ther. 1995;273:734–743. [PubMed] [Google Scholar]
- 15.Debanne D, Guérineau NC, Gähwiler BH, Thompson SM. Paired-pulse facilitation and depression at unitary synapses in rat hippocampus: quantal fluctuation affects subsequent release. J Physiol (Lond) 1996;491:163–176. doi: 10.1113/jphysiol.1996.sp021204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.de Mendonça A, Ribeiro JA. 2-Chloroadenosine decreases long-term potentiation in the hippocampal CA1 area of the rat. Neurosci. Lett. 1990;118:107–111. doi: 10.1016/0304-3940(90)90260-g. [DOI] [PubMed] [Google Scholar]
- 17.Devane WA, Hanus L, Breuer A, Pertwee RG, Stevenson LA, Griffin G, Gibson D, Mandelbaum A, Etinger A, Mechoulam R. Isolation and structure of a brain constituent that binds to the cannabinoid receptor [see comments]. Science. 1992;258:1946–1949. doi: 10.1126/science.1470919. [DOI] [PubMed] [Google Scholar]
- 18.Goda Y, Stevens CF. Synaptic plasticity: the basis of particular types of learning. Curr Biol. 1996;6:375–378. doi: 10.1016/s0960-9822(02)00499-2. [DOI] [PubMed] [Google Scholar]
- 19.Henry DJ, Chavkin C. Activation of inwardly rectifying potassium channels (GIRK1) by coexpressed rat brain cannabinoid receptors in Xenopus oocytes. Neurosci Lett. 1995;186:91–94. doi: 10.1016/0304-3940(95)11289-9. [DOI] [PubMed] [Google Scholar]
- 20.Herkenham M, Lynn AB, Little MD, Johnson MR, Melvin LS, De Costa BR. Cannabinoid receptor localization in brain. Proc Natl Acad Sci USA. 1990;87:1932–1936. doi: 10.1073/pnas.87.5.1932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Herkenham M, Lynn AB, Johnson MR, Melvin LS, de Costa BR, Rice KC. Characterization and localization of cannabinoid receptors in rat brain: a quantitative in vitro autoradiographic study. J Neurosci. 1991;11:563–583. doi: 10.1523/JNEUROSCI.11-02-00563.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Howlett AC. Pharmacology of cannabinoid receptors. Annu Rev Pharmacol Toxicol. 1995;35:607–634. doi: 10.1146/annurev.pa.35.040195.003135. [DOI] [PubMed] [Google Scholar]
- 23.Hsu KS. Characterization of dopamine receptors mediating inhibition of excitatory synaptic transmission in the rat hippocampal slice. J Neurophysiol. 1996;76:1887–1895. doi: 10.1152/jn.1996.76.3.1887. [DOI] [PubMed] [Google Scholar]
- 24.Levenes C, Daniel H, Soubrie P, Crepel F. Cannabinoids decrease excitatory synaptic transmission and impair long- term depression in rat cerebellar Purkinje cells. J Physiol (Lond) 1998;510:867–879. doi: 10.1111/j.1469-7793.1998.867bj.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Linden DJ, Connor JA. Cellular mechanisms of long-term depression in the cerebellum. Curr Opin Neurobiol. 1993;3:401–406. doi: 10.1016/0959-4388(93)90133-j. [DOI] [PubMed] [Google Scholar]
- 26.Mackie K, Lai Y, Westenbroek R, Mitchell R. Cannabinoids activate an inwardly rectifying potassium conductance and inhibit Q-type calcium currents in AtT20 cells transfected with rat brain cannabinoid receptor. J Neurosci. 1995;15:6552–6561. doi: 10.1523/JNEUROSCI.15-10-06552.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Malenka RC, Nicoll RA. NMDA-receptor-dependent synaptic plasticity: multiple forms and mechanisms. Trends Neurosci. 1993;16:521–527. doi: 10.1016/0166-2236(93)90197-t. [DOI] [PubMed] [Google Scholar]
- 28.Manabe T, Wyllie DJ, Perkel DJ, Nicoll RA. Modulation of synaptic transmission and long-term potentiation: effects on paired pulse facilitation and EPSC variance in the CA1 region of the hippocampus. J Neurophysiol. 1993;70:1451–1459. doi: 10.1152/jn.1993.70.4.1451. [DOI] [PubMed] [Google Scholar]
- 29.Matsuda LA, Lolait SJ, Brownstein MJ, Young AC, Bonner TI. Structure of a cannabinoid receptor and functional expression of the cloned cDNA. Nature. 1990;346:561–564. doi: 10.1038/346561a0. [DOI] [PubMed] [Google Scholar]
- 30.Mennerick S, Zorumski CF. Paired-pulse modulation of fast excitatory synaptic currents in microcultures of rat hippocampal neurons. J Physiol (Lond) 1995;488:85–101. doi: 10.1113/jphysiol.1995.sp020948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Miller LL. Marijuana: acute effects on human memory. In: Agurell S, Dewey WL, Willette RE, editors. The cannabinoids: chemical, pharmacological and therapeutic aspects. Academic; New York: 1984. pp. 21–46. [Google Scholar]
- 32.Munro S, Thomas KL, Abu-Shaar M. Molecular characterization of a peripheral receptor for cannabinoids. Nature. 1993;365:61–65. doi: 10.1038/365061a0. [DOI] [PubMed] [Google Scholar]
- 33.Nicoll RA, Malenka RC. Contrasting properties of two forms of long-term potentiation in the hippocampus. Nature. 1995;377:115–118. doi: 10.1038/377115a0. [DOI] [PubMed] [Google Scholar]
- 34.Nowicky AV, Teyler TJ, Vardaris RM. The modulation of long-term potentiation by delta-9-tetrahydrocannabinol in the rat hippocampus, in vitro. Brain Res Bull. 1987;19:663–672. doi: 10.1016/0361-9230(87)90052-9. [DOI] [PubMed] [Google Scholar]
- 35.Pacheco M, Childers SR, Arnold R, Casiano F, Ward SJ. Aminoalkylindoles: actions on specific G-protein-linked receptors. J Pharmacol Exp Ther. 1991;257:170–183. [PubMed] [Google Scholar]
- 36.Paton GS, Pertwee RG, Davies SN. Correlation between cannabinoid mediated effects on paired pulse depression and induction of long term potentiation in the rat hippocampal slice. Neuropharmacology. 1998;37:1123–1130. doi: 10.1016/s0028-3908(98)00096-3. [DOI] [PubMed] [Google Scholar]
- 37.Rinaldi-Carmona M, Barth F, Heaulme M, Shire D, Calandra B, Congy C, Martinez S, Maruani J, Neliat G, Caput D, Ferrara P, Soubrie P, Breliere JC, LeFur G. SR141716A, a potent and selective antagonist of the brain cannabinoid receptor. FEBS Lett. 1994;350:240–244. doi: 10.1016/0014-5793(94)00773-x. [DOI] [PubMed] [Google Scholar]
- 38.Scholz KP, Miller RJ. Inhibition of quantal transmitter release in the absence of calcium influx by a G protein-linked adenosine receptor at hippocampal synapses. Neuron. 1992;8:1139–1150. doi: 10.1016/0896-6273(92)90134-y. [DOI] [PubMed] [Google Scholar]
- 39.Shen M, Thayer SA. The cannabinoid agonist WIN55,212-2 inhibits calcium channels by receptor-mediated and direct pathways in cultured rat hippocampal neurons. Brain Res. 1998;783:77–84. doi: 10.1016/s0006-8993(97)01195-5. [DOI] [PubMed] [Google Scholar]
- 40.Shen M, Piser TM, Seybold VS, Thayer SA. Cannabinoid receptor agonists inhibit glutamatergic synaptic transmission in rat hippocampal cultures. J Neurosci. 1996;16:4322–4334. doi: 10.1523/JNEUROSCI.16-14-04322.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Stella N, Schweitzer P, Piomelli D. A second endogenous cannabinoid that modulates long-term potentiation. Nature. 1997;388:773–778. doi: 10.1038/42015. [DOI] [PubMed] [Google Scholar]
- 42.Sullivan JM (1999) Cannabinoid receptor-mediated inhibition of synaptic transmission in cultured hippocampal pyramidal neurons-mechanisms of action. J. Neurophysiol, in press. [DOI] [PubMed]
- 43.Terranova JP, Michaud JC, Le Fur G, Soubrie P. Inhibition of long-term potentiation in rat hippocampal slices by anandamide and WIN55212-2: reversal by SR141716 A, a selective antagonist of CB1 cannabinoid receptors. Naunyn Schmiedebergs Arch Pharmacol. 1995;352:576–579. doi: 10.1007/BF00169393. [DOI] [PubMed] [Google Scholar]
- 44.Teyler TJ, Cavus I, Coussens C, DiScenna P, Grover L, Lee YP, Little Z. Multideterminant role of calcium in hippocampal synaptic plasticity. Hippocampus. 1994;4:623–634. doi: 10.1002/hipo.450040602. [DOI] [PubMed] [Google Scholar]
- 45.Thompson SM, Capogna M, Scanziani M. Presynaptic inhibition in the hippocampus. Trends Neurosci. 1993;16:222–226. doi: 10.1016/0166-2236(93)90160-n. [DOI] [PubMed] [Google Scholar]
- 46.Tsou K, Brown S, Sanudo-Pena MC, Mackie K, Walker JM. Immunohistochemical distribution of cannabinoid CB1 receptors in the rat central nervous system. Neuroscience. 1998;83:393–411. doi: 10.1016/s0306-4522(97)00436-3. [DOI] [PubMed] [Google Scholar]
- 47.Twitchell W, Brown S, Mackie K. Cannabinoids inhibit N- and P/Q-type calcium channels in cultured rat hippocampal neurons. J Neurophysiol. 1997;78:43–50. doi: 10.1152/jn.1997.78.1.43. [DOI] [PubMed] [Google Scholar]
- 48.Zucker RS. Short-term synaptic plasticity. Annu Rev Neurosci. 1989;12:13–31. doi: 10.1146/annurev.ne.12.030189.000305. [DOI] [PubMed] [Google Scholar]