

Abstract
Postsynaptic injection of Ca2+/calmodulin (Ca2+/CaM) into hippocampal CA1 pyramidal neurons induces synaptic potentiation, which can occlude tetanus-induced potentiation (Wang and Kelly, 1995). Because Ca2+/CaM activates the major forms of nitric oxide synthase (NOS) to produce nitric oxide (NO), NO may play a role during Ca2+/CaM-induced potentiation. Here we show that extracellular application of the NOS inhibitorNG-nitro-l-arginine methyl ester (l-NAME) or postsynaptic co-injection ofl-NAME with Ca2+/CaM blocked Ca2+/CaM-induced synaptic potentiation. Thus, NO is necessary for Ca2+/CaM-induced synaptic potentiation. In contrast, extracellular perfusion of membrane-impermeable NO scavengersN-methyl-d-glucamine dithiocarbamate/ferrous sulfate mixture (MGD-Fe) or 2-(4-carboxyphenyl)-4,4,5,5-tetramethylimidazoline-1-oxyl-3-oxide (carboxy-PTIO) did not attenuate Ca2+/CaM-induced synaptic potentiation, even though MGD-Fe or carboxy-PTIO blocked tetanus-induced synaptic potentiation. This result indicates that NO is not a retrograde messenger in Ca2+/CaM-induced synaptic potentiation. However, postsynaptic co-injection of carboxy-PTIO with Ca2+/CaM blocked Ca2+/CaM-induced potentiation. Postsynaptic injection of carboxy-PTIO alone blocked tetanus-induced synaptic potentiation without affecting basal synaptic transmission. Our results suggest that NO works as a postsynaptic (intracellular) messenger during Ca2+/CaM-induced synaptic potentiation.
Keywords: nitric oxide, calmodulin, hippocampus, synaptic plasticity, synaptic potentiation, nitric oxide scavengers, nitric oxide synthase inhibitors
The role of nitric oxide (NO) as a retrograde messenger in hippocampal long-term potentiation (LTP) has been suggested by many studies (O’Dell et al., 1991; Schuman and Madison, 1991, 1994a,b; Haley et al., 1992; Bredt and Snyder, 1994;Garthwaite and Boulton, 1995; Arancio et al., 1996). Extracellular application of NO donors facilitates LTP induction (Malen and Chapman, 1997), whereas extracellular administration of nitric oxide synthase (NOS) inhibitors or NO scavengers or postsynaptic injection of NOS inhibitors attenuates and/or blocks LTP induced by tetanus or pairing protocols (Haley et al., 1992; Williams et al., 1993a; Schuman and Madison, 1994a; Arancio et al., 1996) (but see Chetkovitch et al., 1993; Cummings et al., 1994). Two major forms of constitutive NOS in brain are neuronal NOS and endothelial NOS (nNOS and eNOS, respectively) (Bredt and Snyder, 1994), and both are present in the hippocampus (Dinerman et al., 1994; Doyle and Slater, 1997; Eliasson et al., 1997). Mutant mice lacking nNOS and eNOS display significantly attenuated hippocampal LTP (Son et al., 1996).
Previous results have shown that postsynaptic injection of Ca2+/calmodulin (Ca2+/CaM) induces synaptic potentiation in hippocampal CA1 neurons (Wang and Kelly, 1995). Ca2+/CaM-induced synaptic potentiation is similar to tetanus-induced LTP because it is blocked by co-injecting a calmodulin binding peptide or pseudosubstrate inhibitors of Ca2+/CaM-dependent kinase II (CaMKII) or protein kinase C (PKC) (Wang and Kelly, 1995). Tetanus-induced LTP and Ca2+/CaM-induced synaptic potentiation reciprocally occlude each other, so their underlying mechanisms may be similar (Wang and Kelly, 1995). Because mechanisms responsible for tetanus-induced LTP expression are believed to be in part presynaptic (Malinow and Tsien, 1990a; Bolshakov and Siegelbaum, 1995), it is important to determine whether Ca2+/CaM-induced synaptic potentiation involves a presynaptic mechanisms(s), particularly because the latter is induced by an apparently restricted postsynaptic manipulation. For example, postsynaptic injection of Ca2+/CaM could activate n/eNOS and elevate NO in neurons (Bredt and Snyder, 1990; Bredt et al., 1992; Brenman et al., 1996), and NO could act as a retrograde messenger during Ca2+/CaM-induced synaptic potentiation. Alternatively, NO could contribute to Ca2+/CaM-induced synaptic potentiation by acting locally in postsynaptic neurons to directly nitrosylate and/or oxidize proteins (Lei et al., 1992; Lipton et al., 1993, 1996; Li et al., 1999) or indirectly via the activation of guanylyl cyclase/cyclic GMP-dependent protein kinase and/or ADP-ribosyl transferase pathways (Boulton et al., 1995; Schuman et al., 1994; Zhou et al., 1994a,b;Kleppisch et al., 1999).
We examined the role of NOS and NO in synaptic potentiation induced by postsynaptic injection of Ca2+/CaM. We also investigated whether NO acted as a retrograde messenger and/or a postsynaptic signaling molecule in Ca2+/CaM-induced synaptic potentiation. Tetanus-induced synaptic potentiation was monitored as an index for the effectiveness of an NOS inhibitor or NO scavengers. Here we show that postsynaptic NO is involved in Ca2+/CaM-induced potentiation but not as a retrograde messenger. On the other hand, we observed that tetanus-induced potentiation appears to require NO-dependent retrograde signaling, consistent with previous observations (O’Dell et al., 1991;Schuman and Madison, 1991, 1994a; Haley et al., 1992; Arancio et al., 1996; Malen and Chapman, 1997).
MATERIALS AND METHODS
Male and female Sprague Dawley rats (35–55 days old; from Harlan Sprague Dawley, Indianapolis, IN; and Charles River, Wilmington, MA) were used in this study. Animals were group-housed and maintained in a temperature-controlled environment with a 12 hr light/dark cycle. Transverse hippocampal slices (400 μm) were prepared using a McIlwain tissue chopper with ice-cold modified artificial CSF (ACSF). Slices were incubated at 25 °C in ACSF over 1 hr and transferred to a submersion chamber (31.8 ± 0.5 °C; 2.5–3 ml/min perfusion rate) for electrophysiological recordings. ACSF contained (in mm): 124 NaCl, 3 KCl, 1.3 NaH2PO4, 26 NaHCO3, 2.0 MgCl2, 2.4 CaCl2, 10 glucose, and 10 HEPES, pH 7.3. A modified ACSF used for slice preparation contained 4.0 mmMgCl2 and 1.2 mmCaCl2. All media were bubbled with a 95%O2-5%CO2 mixture. Experiments were conducted in “standard” ACSF containing bicuculline (5 μm), and the concentration of MgCl2 was 2.4 mm. Isolated CA1 slices were made by cutting presynaptic axons in stratum radiatum but not in oriens/alveus at the CA1–CA3 border. Under these conditions, seizure activity was never observed during basal synaptic transmission. Complex waveforms only occurred after tetanic stimulation in some experiments. In designated experiments, the perfusion medium also contained 100 μmNG-nitro-l-arginine methyl ester (l-NAME), a mixture of 150 μmN-methyl-d-glucamine dithiocarbamate and 75 μm FeSO4. 7 H2O (150/75 μm MGD-Fe), or 30 μm2-(4-carboxyphenyl)-4,4,5,5-tetramethylimidazoline-1-oxyl-3-oxide (carboxy-PTIO). Constant flow rates were maintained throughout experiments especially during media exchanges.
Glass microelectrodes (60–85 MΩ) filled with 2 mpotassium acetate (KAc), carboxy-PTIO in 2 m KAc, Ca2+/CaM/KAc, or Ca2+/CaM/KAc plus various agents [i.e.,l-NAME,NG-nitro-d-arginine methyl ester (D-NAME), or carboxy-PTIO] were used for intracellular recordings in CA1 pyramidal neurons in bridge mode. Ca2+/CaM/KAc was prepared from CaCl2, CaM, and KAc stocks to obtain final concentrations of 80 and 20 μm and 2m, respectively. Input resistance was estimated by injecting negative current (0.12 nA) for 50 msec before each evoked stimulus and monitored throughout recordings. Results were only collected from neurons in which stable recordings were obtained within the initial 2–5 min after impalement with resting membrane potentials between −65 and −73 mV, and in which input resistance changed <20% throughout the entire experiment. Extracellular field EPSPs were recorded using glass pipettes (containing 0.9% NaCl) placed below the intracellular recording site (halfway between stratum pyramidale and the hippocampal fissure). One bipolar tungsten stimulating electrode (∼12 mΩ) was placed in CA1 stratum radiatum for orthodromic stimulation of Schaffer collateral/commissural fibers. Test stimuli were given at 0.05 Hz, and stimulus intensity was adjusted to evoke about one-half to three-fifths of maximal synaptic responses. Tetanic stimulation was delivered at 100 Hz (five trains of 25 pulses at 5 sec intervals) and the same stimulus intensity used to evoke baseline responses. Intracellular and extracellular recordings, data acquisition, and analysis were performed using an AxoClamp 2B amplifier with Axoscope and Clampfit softwares (Axon Instruments). Initial baseline values were averaged from EPSP slopes obtained during the first 1–2 min after intracellular recordings stabilized and defined as 100%. Values of tetanus- or Ca2+/CaM-induced synaptic potentiation, or different drug treatments, were obtained from data points averaged over a 2 min period at the time indicated (e.g., 45 min after beginning intracellular injection). Student’s t tests were used for comparisons within the same experimental groups at different times (paired t test) and for comparisons between different groups at comparable experimental times (nonpaired t test). Values were expressed as mean ± SEM; significant differences were determined at the p < 0.05 level.
Bicuculline methbromide was obtained from Research Biochemicals (Natick, MA), CaM from Calbiochem (La Jolla, CA), l-NAME and d-NAME from Sigma (St. Louis, MO), carboxy-PTIO from Cayman (Ann Arbor, MI); MGD and FeSO4.7H2O were gifts from Dr. Yashige Kotake (Oklahoma Medical Research Foundation, Oklahoma City, OK); chemicals for ACSF were from Fisher Scientific (Fair Lawn, NJ).
RESULTS
Postsynaptic injection of Ca2+/CaM induces synaptic potentiation
Intracellular recordings using sharp microelectrodes with simultaneous extracellular field recordings were used to monitor synaptic transmission of hippocampal CA1 neurons. Postsynaptic injections of Ca2+/CaM (80 and 20 μm, respectively) into CA1 pyramidal neurons by passive diffusion from microelectrodes induced a gradual increase in the initial slopes of EPSPs (Fig.1A). Initial baseline values were averaged from six consecutive EPSPs during the first 1–2 min after intracellular recordings stabilized and defined as 100%. Figure 1A shows that postsynaptic injection of Ca2+/CaM into CA1 pyramidal neurons for 45 min induced significant synaptic potentiation of EPSP slopes (184 ± 19%; n = 5; 45 min after beginning intracellular injections) compared with initial baseline values (p < 0.05). These results are consistent with previous studies showing that postsynaptic injection of Ca2+/CaM enhanced excitatory synaptic transmission (Wang and Kelly, 1995). Simultaneous extracellular field recordings showed a stable baseline during the pretetanus period (105 ± 6%; n = 5; 45 min after beginning intracellular injections; Fig. 1B), whereas intracellular injections displayed Ca2+/CaM induced potentiation. The magnitude of Ca2+/CaM-induced potentiation decreased after 100 Hz tetanic stimulation (25 pulses, five trains at 5 sec intervals). This depotentiation was previously observed; however, the underlying mechanism is not known (Wang and Kelly, 1995). In contrast, field recordings showed significant potentiation induced by tetanus (155 ± 15% at 30 min after tetanus; n = 5, Fig. 1B; 148 ± 21% at 60 min after tetanus; results not shown). A control group was included, in which microelectrodes were filled only with 2 m KAc. After recording stable baselines for 20 min, tetanus was delivered, which induced significant synaptic potentiation in both intracellular (198 ± 16% at 30 min after tetanus; n = 4; Fig.1C; p < 0.05) and field EPSP slopes (167 ± 11% at 30 min after tetanus; n = 4; Fig.1D; p < 0.05) compared with pretetanus baseline values.
Fig. 1.

Postsynaptic injection of Ca2+/CaM induces synaptic potentiation.A, B, Ca2+/CaM was injected for 45 min, and then a 100 Hz tetanus was delivered.A, Synaptic potentiation induced by postsynaptic Ca2+/CaM injections (n = 5).B, Simultaneous field recordings displayed stable baseline EPSPs followed by tetanus-induced synaptic potentiation (n = 5). C, D, Tetanic stimulation under control conditions induced synaptic potentiation. C, Intracellular recordings, microelectrodes filled with 2 m KAc (n= 4). D, Simultaneous field recordings of control group (n = 4).
Extracellular NOS/NO modulators block tetanus-induced synaptic potentiation
Because our ultimate goal was to determine whether NO signaling pathways contribute to Ca2+/CaM-induced synaptic potentiation, we needed an independent measurement of the efficacy of NO modulators in our experiments. Previous studies indicated that extracellular application of NOS inhibitors or NO scavengers blocked tetanus-induced potentiation in hippocampal CA1 area (Schuman and Madison, 1991, 1994a; Haley et al., 1992). To investigate whether NOS/NO modulators would affect basal synaptic transmission or block tetanus-induced potentiation, the following experiments were conducted using extracellular field recordings. Stable baselines (field EPSPs) were recorded for at least 20–30 min, and then extracellular perfusions of an NOS inhibitor or NO scavenger were initiated. We used recently developed NO scavengers MGD and carboxy-PTIO instead of hemoglobin. Extracellular perfusion of hemoglobin can depolarize CA1 neurons and suppress EPSPs and IPSPs in the presence of the NOS inhibitor N-ω-nitro-l-arginine, suggesting that hemoglobin has other effects that are independent of its NO scavenging activity (Yip et al., 1996). MGD mixed with reduced iron (Fe2+) forms a stable and water-soluble complex (MGD-Fe; Komarov et al., 1993), which is cell membrane-impermeable (Y. Kotake, personal communication) and has been used for in vivo spin trapping of NO in mice (Lai and Komarov, 1994; Komarov and Lai, 1995; Kotake et al., 1995). MGD-Fe has also been used to scavenge NO produced by cells in culture (Kotake, 1996; Kotake et al., 1996) or in vitro cardiovascular preparations (Pieper and Lai, 1996). Perfusion of freshly mixed MGD and FeSO4 (i.e., MGD-Fe) at different concentrations was tested (data not shown). MGD-Fe mixtures of 100/25, 100/37.5, and 100/50 μm did not block tetanus-induced potentiation; however, MGD-Fe at 150/75 μmsuccessfully blocked tetanus-induced potentiation without affecting basal synaptic transmission (see below).
We also tested carboxy-PTIO, which is water-soluble and scavenges NO without affecting NOS activity (Az-ma et al., 1994; Maeda et al., 1994;Yoshida et al., 1994; Amano and Noda, 1995; Hogg et al., 1995a,b). Preliminary results in cultured cells indicate that carboxy-PTIO is cell membrane-impermeable (T. Akaike, personal communication). Extracellular perfusions of carboxy-PTIO at 5, 7.5, 10, or 15 μm failed to block tetanus-induced potentiation (data not shown), whereas 30 μm carboxy-PTIO reliably blocked tetanus-induced potentiation without affecting basal synaptic transmission (see below).
The concentration chosen for the NOS inhibitor l-NAME was based on previous studies in which l-NAME attenuated tetanus-LTP under certain conditions (Williams et al., 1993a;Nicolarakis et al., 1994). l-NAME (100 μm,n = 10) was perfused for 60 min, and NO scavenger MGD-Fe (150/75 μm, n = 7) or carboxy-PTIO (30 μm, n = 7) was perfused for 30 min before tetanus (Fig.2A). Drug containing ACSFs were switched to standard ACSF 5 min after tetanus.l-NAME, MGD-Fe, or carboxy-PTIO was applied separately. Both MGD-Fe and carboxy-PTIO significantly blocked tetanus-induced potentiation (111 ± 4 and 113 ± 13%, respectively, at 30 min after tetanus; Fig. 2A,B) compared with controls (standard ACSF, 189 ± 12%, at 30 min after tetanus; n = 11; p < 0.05). After perfusion of l-NAME for 60 min, there was a slight increase in field EPSP slopes (108 ± 5%;n = 10), which was not significantly different from the pretreatment baseline (104 ± 4% immediately beforel-NAME perfusion). l-NAME attenuated tetanus-induced potentiation at 30 min after tetanus (146 ± 10%; n = 10) compared with controls (p < 0.05) and virtually blocked tetanus-induced potentiation when examined at 60 min after tetanus (113 ± 6%; no significant difference from the pretetanus baseline).
Fig. 2.
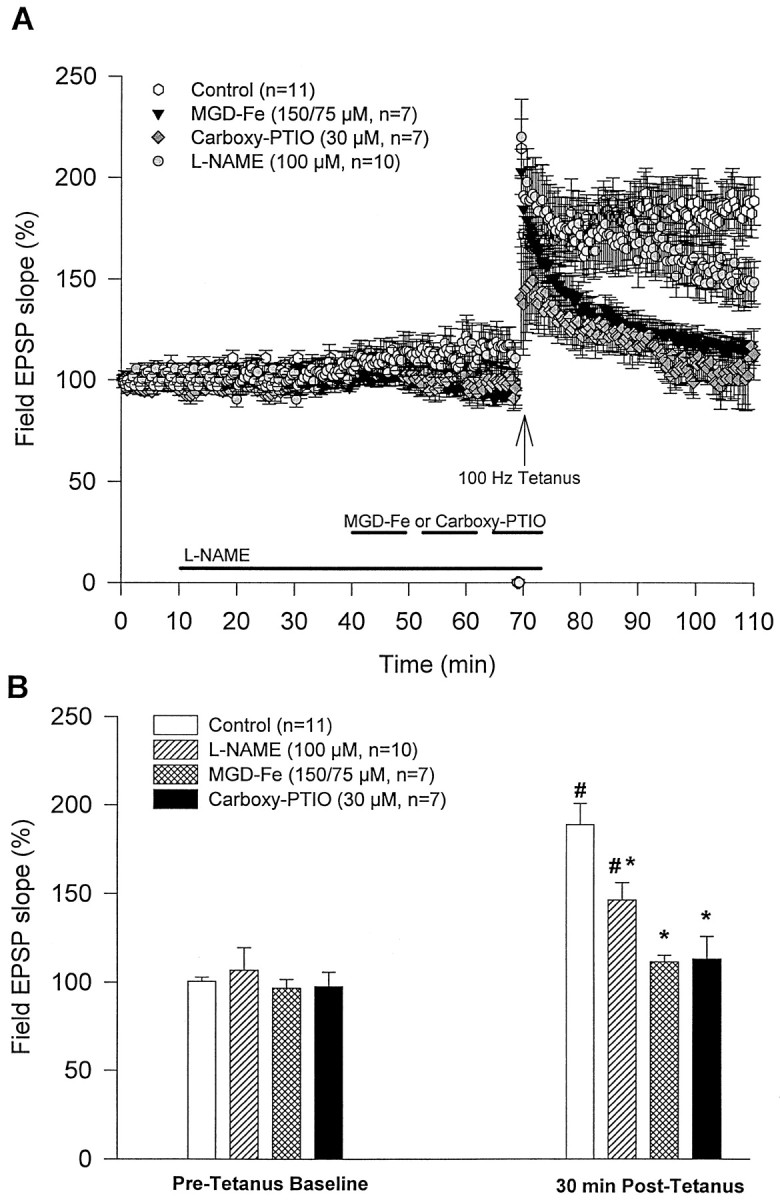
Extracellular perfusion of an NOS inhibitor (l-NAME) or NO scavengers (MGD-Fe and carboxy-PTIO) attenuate tetanus-induced synaptic potentiation. A, After stable baseline recordings, media containing the NOS inhibitor or NO scavengers were perfused for different durations (see Results) followed by tetanic stimulation. Five minutes after tetaus, perfusion media were switched to standard ACSF. Control slices (n = 11) were perfused with standard ACSF.B, l-NAME (100 μm;n = 10), MGD-Fe (150/75 μm;n = 7) or carboxy-PTIO (30 μm;n = 7) did not significantly affect basal synaptic transmission compared with controls. Thirty minutes after tetanus, synaptic potentiation was induced in both control andl-NAME groups, which was significantly different from the pretetanus baseline within the same group (#p < 0.05). However, tetanus-induced potentiation in l-NAME, MGD-Fe, and carboxy-PTIO groups at 30 min after tetanus was significantly attenuated compared with controls (*p < 0.05).
NOS inhibitor blocks Ca2+/CaM-induced potentiation
To examine the possibility that NOS activity might contribute to synaptic potentiation induced by postsynaptic injection of Ca2+/CaM, hippocampal slices were preincubated with l-NAME (100 μm) for 1 hr before being transferred to the recording chamber. Stable field recordings (field EPSPs) were established for at least 20 min, and then postsynaptic injections of Ca2+/CaM with microelectrodes were performed. l-NAME (100 μm) was also present in the perfusate until 5 min after tetanus. Under these conditions, l-NAME significantly blocked Ca2+/CaM-induced synaptic potentiation (104 ± 13%; n = 10, 45 min after beginning injections; Fig. 3A) compared with Ca2+/CaM alone (184 ± 19%; n = 5, 45 min after beginning injections; Fig.1A; p < 0.05).l-NAME also blocked tetanus-induced synaptic potentiation in Ca2+/CaM-injected neurons (114 ± 29%; n = 10, 30 min after tetanus; Fig.3A) compared with controls (at 30 min after tetanus; Fig.1C; p < 0.05). In addition,l-NAME attenuated tetanus-induced potentiation of field EPSPs at 30 min after tetanus (138 ± 8%; n= 10; Fig. 3B) and blocked potentiation at 60 min after tetanus (115 ± 8%; n = 10; results not shown) compared with field EPSPs recorded during Ca2+/CaM injections alone (148 ± 21%; n = 5, 60 min after tetanus; results not shown; p < 0.05).
Fig. 3.
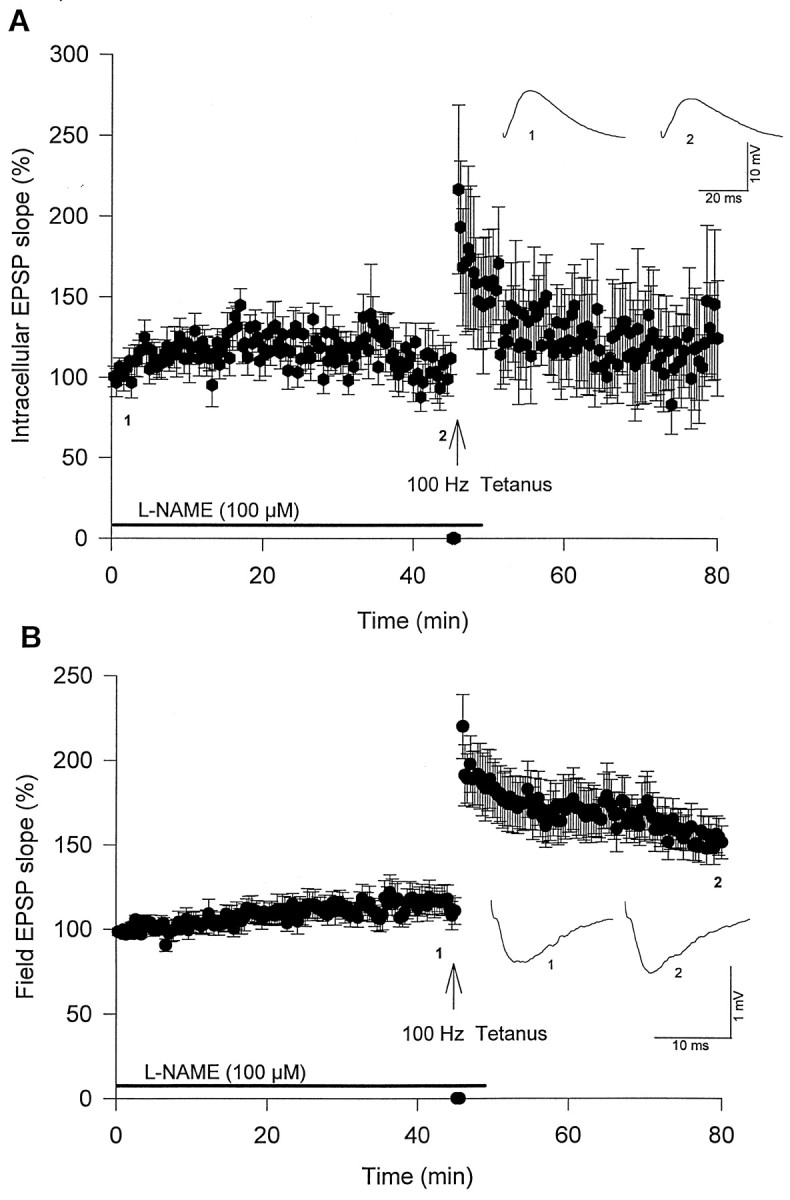
Extracellular l-NAME blocks Ca2+/CaM-induced synaptic potentiation (n = 10). Slices were pretreated withl-NAME (100 μm) for ≥1 hr before beginning postsynaptic injections of Ca2+/CaM. Five minutes after tetanus, slices were perfused with standard ACSF withoutl-NAME. A, Synaptic potentiation induced by postsynaptic Ca2+/CaM injections was blocked byl-NAME. B, Simultaneous field recordings showed that tetanus-induced synaptic potentiation was attenuated byl-NAME.
To investigate whether postsynaptic NOS activity contributes to Ca2+/CaM-induced synaptic potentiation,l-NAME was co-injected with Ca2+/CaM into CA1 neurons. The addition of 100 mml-NAME to Ca2+/CaM (80/20 μm) in microelectrodes blocked Ca2+/CaM-induced potentiation (98 ± 17%; n = 8, 45 min after beginning injections; Fig.4A) compared with Ca2+/CaM injections alone (184 ± 19%; n = 5, 45 min after beginning injections; Fig.1A; p < 0.05). Co-injection ofl-NAME and Ca2+/CaM also blocked tetanus-induced synaptic potentiation (113 ± 30%;n = 5, 30 min after tetanus; Fig. 4A) compared with neurons injected with 2 m KAc alone (30 min after tetanus; Fig. 1C; p < 0.05). In contrast, co-injection of Ca2+/CaM andd-NAME (67 or 100 mm), a stereoisomer much less potent than l-NAME, did not block Ca2+/CaM-induced potentiation (178 ± 16%; n = 6, 45 min after beginning injections; Fig. 4B). Tetanic stimulation of neurons co-injected with d-NAME and Ca2+/CaM resulted in depotentiation (Fig.4B), which was consistent with previous results (Fig.1A). Simultaneous extracellular field recordings in the same slices showed stable baselines for 45 min [99 ± 4%;n = 14 (l-NAME andd-NAME groups combined); Fig. 4C], and tetanus induced significant potentiation at 30 min (135 ± 7%; n = 14; Fig. 4C) or 60 min after tetanus (133 ± 10%; n = 14; results not shown) compared with pretetanus baseline (p < 0.05). Although l-NAME is a membrane-permeable NOS inhibitor, these results suggest that postsynaptic NOS activity is important for Ca2+/CaM-induced synaptic potentiation.
Fig. 4.
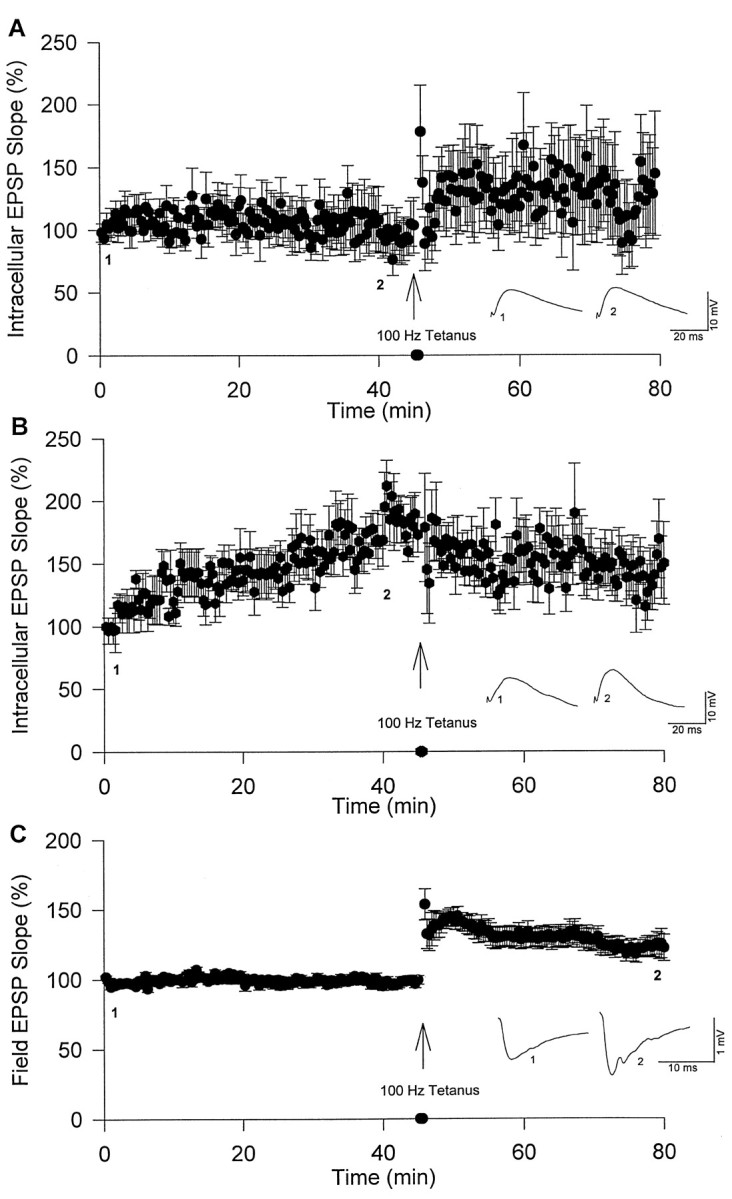
Postsynaptic co-injection of l-NAME, but not d-NAME, blocks Ca2+/CaM-induced synaptic potentiation. A, Synaptic potentiation induced by postsynaptic co-injections of Ca2+/CaM andl-NAME (100 mm;n = 8).B, Synaptic potentiation induced by postsynaptic co-injection of Ca2+/CaM and d-NAME (67 or 100 mm; n = 6). C,Simultaneous field recordings showed synaptic potentiation induced by tetanus (l-NAME and d-NAME groups combined;n = 14).
Extracellular NO scavengers block tetanus-induced potentiation but not Ca2+/CaM-induced potentiation
Since previous studies indicated that NO acted as a retrograde messenger at hippocampal synapses (O’Dell et al., 1991;Schuman and Madison, 1991, 1994a; Haley et al., 1992; Garthwaite and Boulton, 1995; Arancio et al., 1996), and our recent results showed the importance of NOS activity in Ca2+/CaM-induced potentiation (see above;Ko and Kelly, 1998), we examined the role of NO as a retrograde messenger in Ca2+/CaM-induced potentiation using extracellular applications of MGD-Fe or carboxy-PTIO to scavenge NO. Hippocampal slices were pretreated in the recording chamber with either MGD-Fe (150/75 μm;n = 7) for 60 min or carboxy-PTIO (30 μm;n = 4) for 30 min before obtaining extracellular recordings. Thirty minutes after establishing stable extracellular field recordings, postsynaptic injections of Ca2+/CaM were initiated. MGD-Fe or carboxy-PTIO was present in the perfusate until 5 min after tetanus. Both MGD-Fe and carboxy-PTIO effectively blocked tetanus-induced synaptic potentiation (109 ± 7 and 115 ± 14%, respectively, 30 min after tetanus; Fig.5B,D) compared with field recordings of Ca2+/CaM injections alone (30 min after tetanus; Fig. 1B; p < 0.05). In contrast, extracellular perfusion of MGD-Fe or carboxy-PTIO did not block synaptic potentiation induced by postsynaptic injections of Ca2+/CaM (170 ± 23 and 201 ± 44%, respectively, 45 min after beginning injections; Fig.5A,C). Tetanic stimulation of Ca2+/CaM-injected neurons treated with MGD-Fe or carboxy-PTIO resulted in depotentiation (Fig.5A,C), which was consistent with previous results (Figs.1A, 4B). These results suggest that NO does not act as a retrograde messenger in Ca2+/CaM-induced synaptic potentiation, but NO still works as a retrograde messenger in tetanus-induced potentiation, which is consistent with previous reports (O’Dell et al., 1991; Schuman and Madison, 1991, 1994a; Haley et al., 1992;Arancio et al., 1996; Malen and Chapman, 1997).
Fig. 5.

Extracellular applications of NO scavengers MGD-Fe and carboxy-PTIO block tetanus- but not Ca2+/CaM-induced synaptic potentiation.A,B, Slices were pretreated with MGD-Fe (150/75 μm) for 30–60 min before postsynaptic injections of Ca2+/CaM (n = 7). Five minutes after tetanus the medium was switched to standard ACSF.A, Synaptic potentiation induced by postsynaptic Ca2+/CaM injections was not blocked by MGD-Fe.B, MGD-Fe blocked tetanus-induced synaptic potentiation in field recordings. C, D, Slices were pretreated with carboxy-PTIO (30 μm) for 30–60 min before postsynaptic injections of Ca2+/CaM (n = 4). Three minutes after tetanus the medium was switched to standard ACSF.C, Synaptic potentiation induced by postsynaptic Ca2+/CaM injections was not blocked by carboxy-PTIO.D, Carboxy-PTIO blocked tetanus-induced synaptic potentiation in field recordings.
Postsynaptic injection of NO scavenger blocks Ca2+/CaM-induced potentiation
We have shown that NOS activity is essential for synaptic potentiation induced by postsynaptic injections of Ca2+/CaM (Figs. 3A,4A). On the other hand, extracellular administration of NO scavengers MGD-Fe or carboxy-PTIO did not block Ca2+/CaM-induced potentiation (Fig.5A,C). Taken together, these results suggest that NO produced by postsynaptic NOS, which is important for Ca2+/CaM-induced potentiation, may not function as a retrograde messenger but acts directly at postsynaptic sites. To test this hypothesis, we co-injected Ca2+/CaM and carboxy-PTIO (30 mm) into postsynaptic CA1 neurons. Co-injections of carboxy-PTIO significantly blocked Ca2+/CaM-induced potentiation (120 ± 10%; n = 6, 45 min after beginning injections; Fig.6A) compared with Ca2+/CaM injections alone (45 min after beginning injections; Fig. 1A; p < 0.05). Postsynaptic injections of carboxy-PTIO alone (30 mm; Fig. 6C) did not affect basal synaptic transmission (114 ± 16%; n = 4, 45 min after beginning injections) but did block tetanus-induced potentiation (87 ± 10% at 30 min after tetanus). Simultaneous field recordings in these experiments showed stable baselines and tetanus-induced potentiation at 30 min after tetanus (145 ± 10 and 154 ± 14%, respectively; Fig. 6B,D) compared with their baseline values obtained 1–2 min before tetanus (both p < 0.05). These results indicate that NO acts as a postsynaptic and intracellular messenger during Ca2+/CaM-induced synaptic potentiation but not as a retrograde messenger. On the other hand, NO functions, at least in part, as a retrograde messenger in tetanus-induced synaptic potentiation.
Fig. 6.

Postsynaptic injection of carboxy-PTIO blocks Ca2+/CaM- and tetanus-induced synaptic potentiation.A, Postsynaptic co-injection of carboxy-PTIO (30 mm) blocked Ca2+/CaM-induced synaptic potentiation (n = 6). B,Simultaneous field recordings showed synaptic potentiation induced by tetanus (n = 6). C, Postsynaptic injection of carboxy-PTIO (30 mm) alone blocked tetanus-induced synaptic potentiation (n = 4).D, Simultaneous field recordings showed synaptic potentiation induced by tetanus (n = 4).
DISCUSSION
Our results show that postsynaptic injection of Ca2+/CaM into hippocampal CA1 pyramidal neurons induces synaptic potentiation, which is consistent with previous reports (Wang and Kelly, 1995, 1996). In the nervous system, Ca2+/CaM regulates many enzymes and channels (Rhoads and Friedberg, 1997), including adenylyl cyclase (Sunahara et al., 1996; Smit and Iyengar, 1998), Ca2+/CaM-dependent protein kinases (Braun and Shulman, 1995), phosphodiesterases (Sharma, 1995; Zhao et al., 1997), NOS (Bredt and Snyder, 1994; Lee and Stull, 1998), calcineurin (Guerini, 1997; Klee et al., 1998), NMDA receptors (Ehlers et al., 1996; Hisatsune et al., 1997; Zhang et al., 1998), ryanodine receptors (Ikemoto et al., 1995; Guerrini et al., 1995), and cyclic nucleotide-gated channels (Molday, 1996; Zagotta and Siegelbaum, 1996). Many of these signaling molecules, including CaMKII and NOS, are believed to be involved in LTP (Malenka et al., 1989; Ocorr and Schulman, 1991; O’Dell et al., 1991; Schuman and Madison, 1991,1994a,b; Lledo et al., 1995).
Postsynaptic injection of Ca2+/CaM significantly decreases paired-pulse facilitation (PPF) (Wang and Kelly, 1996). Mechanisms responsible for changing PPF are believed to be presynaptic (Magleby, 1987; Zucker, 1989; but see Wang and Kelly, 1996, 1997b); therefore, changes in PPF are often interpreted to be attributable to presynaptic changes in transmitter release (Creager et al., 1980; Muller and Lynch, 1989; Manabe et al., 1993; Schulz et al., 1994). Postsynaptic injections of Ca2+/CaM could activate NOS to produce NO (Bredt and Snyder, 1990, 1994; Bredt et al., 1992; Brenman et al., 1996), which may act like a retrograde messenger that contributes to PPF attenuation. NO is believed to function as an intercellular (retrograde) messenger in synaptic potentiation induced by tetanus or pairing protocols (O’Dell et al., 1991; Schuman and Madison, 1991, 1994a; Haley et al., 1992; Grathwaite and Boulton, 1995; Arancio et al., 1996; Malen and Chapman, 1997) (but see Chetkovitch et al., 1993; Williams et al., 1993a; Cummings et al., 1994).
Our results show that Ca2+/CaM-induced potentiation can be blocked by the NOS inhibitor l-NAME through either extracellular perfusion or postsynaptic co-injection with Ca2+/CaM. Thus, synaptic potentiation induced by postsynaptic injection of Ca2+/CaM is NOS-dependent. To our surprise, extracellular perfusion of membrane-impermeable NO scavengers MGD-Fe and carboxy-PTIO did not decrease Ca2+/CaM-induced potentiation, even though these NO scavengers strongly attenuated tetanus-induced potentiation. Therefore, NO appears not to be a retrograde messenger in Ca2+/CaM-induced synaptic potentiation. In contrast, postsynaptic co-injection of carboxy-PTIO with Ca2+/CaM blocked Ca2+/CaM-induced synaptic potentiation. These results suggest that NO produced by the activation of NOS acts locally at a postsynaptic site(s) and is required during Ca2+/CaM-induced synaptic potentiation.
Previous results indicate that NO might act at postsynaptic sites. First, NO-related species modulate NMDA receptor function by modulating its redox status and thereby decreasing Ca2+ influx (Lei et al., 1992; Lipton et al., 1993; Lipton and Wang, 1996; Lipton et al., 1996). In addition, Ca2+/CaM can bind to NMDA receptors and reduce channel open probability (Ehlers et al., 1996). Thus, the dual actions of NO and Ca2+/CaM on reducing NMDA-mediated Ca2+ influx could occur during Ca2+/CaM-induced synaptic potentiation, suggesting that increased Ca2+ influx via NMDA receptors may not be important for this synaptic plasticity. Second, NO enhances Ca2+/CaM-dependent phosphorylation of proteins in isolated postsynaptic density (PSD) fractions (Wu et al., 1996). The NO-stimulated enhancement of Ca2+/CaM-dependent phosphorylation in PSDs may be important during Ca2+/CaM-induced synaptic potentiation. Both CaMKII and AMPA receptors are present in hippocampal PSDs (Kelly et al., 1984, 1985; Riquelme et al., 1993; Rao et al., 1998). The apparent phosphorylation of AMPA receptors by CaMKII is enhanced during LTP (Barria et al., 1997), and this phosphorylation increases AMPA receptor conductance (Derkach et al., 1998). Therefore, NO might increase the phosphorylation and conductance of AMPA receptors by CaMKII during Ca2+/CaM-induced potentiation. Third, NO oxidizes neurogranin/RC3 (Mahoney et al., 1996;Sheu et al., 1996; Li et al., 1999). Neurogranin is a postsynaptic PKC substrate, which binds calmodulin in the absence of Ca2+ (Baudier et al., 1991; Huang et al., 1993; Gerendasy et al., 1995; Sato et al., 1995). Compared with reduced neurogranin, oxidized neurogranin binds CaM with lower affinity and is a poorer substrate for PKC (Mahoney et al., 1996; Sheu et al., 1996; Li et al., 1999). Thus, if neurogranin undergoes substantial NO-dependent oxidation during postsynaptic injections of Ca2+/CaM, then more CaM and PKC could be available to enhance synaptic transmission.
Postsynaptic NO may activate additional signaling pathways, which could contribute to Ca2+/CaM-induced synaptic potentiation. NO activates soluble guanylyl cyclase, which produces cGMP that then activates protein kinase G (PKG) (Garthwaite and Boulton, 1995). In rat hippocampus, the expression of guanylyl cyclase and CaM mRNAs are high in pyramidal neurons and dentate granule cells (Matsuoka et al., 1992). Thus, postsynaptic injections of Ca2+/CaM could activate NOS and produce NO, which then stimulates guanylyl cyclase to enhance synaptic transmission through the activation of PKG. Inhibitors of guanylyl cyclase and PKG block the induction of LTP (Zhou et al., 1994a; Boulton et al., 1995), whereas cGMP analogs that activate PKG lower the threshold of LTP induction (Zhou et al., 1994b; Arancio et al., 1995). However, there is evidence that does not support a role of NO–cGMP–PKG pathways in synaptic potentiaton (Schuman et al., 1994;Selig et al., 1996; Wu et al., 1998; Kleppisch et al., 1999). Moreover, LTP is normal in mice lacking PKG-I and/or PKG-II but can be attenuated by an NOS inhibitor (Kleppisch et al., 1999). Thus, understanding the involvement of NO–cGMP–PKG signaling pathways in LTP and/or Ca2+/CaM-induced potentiation awaits further investigation.
Another potential target for NO is ADP-ribosyltransferase (ADPRT;Schuman et al., 1994; Willmott et al., 1996). Extracellular application of ADPRT inhibitors block tetanus-induced potentiation (Schuman et al., 1994). Mice lacking PKG-I and/or -II display normal tetanus-LTP, but LTP is blocked by an ADPRT inhibitor (Kleppisch et al., 1999). NO can indirectly activate ADP-ribose cyclase to produce cyclic ADP-ribose (Willmott et al., 1996), which can enhance Ca2+ release from intracellular ryanodine-sensitive Ca2+ stores (Willmott et al., 1996). Calcium release from ryanodine-sensitive Ca2+ stores has been shown to contribute to tetanus-LTP in hippocampal slices (Obenaus et al., 1989; Wang et al., 1996; Wang and Kelly, 1997a). An additional intracellular target for NO is p21ras (Ras) (Yun et al., 1998). NO can activate immunoprecipitated neuronal Ras (Yun et al., 1998). Ras activation leads to phosphorylation and activation of mitogen-activated protein kinases (MAPKs), which regulate gene transcription and modulate long-term synaptic plasticity (Thomas et al., 1992; Wood et al., 1992;Moodie et al., 1993; Williams et al., 1993b; Yun et al., 1998). MAPKs are also involved in LTP induction in the hippocampal CA1 region (English and Sweatt, 1996, 1997). In summary, the ability of NO to modulate these additional pathways could contribute to Ca2+/CaM-induced synaptic potentiation (Wang and Kelly, 1995).
Postsynaptic injections of Ca2+/CaM (Wang and Kelly, 1995) or CaMKII (Lledo et al., 1995) induce synaptic potentiation. In both cases, potentiation induced by these postsynaptic manipulations occludes tetanus-LTP and vice versa (Lledo et al., 1995;Wang and Kelly, 1995, 1996). The expression of a constitutively active recombinant CaMKII in CA1 neurons potentiated synaptic transmission, which occluded tetanus-LTP (Pettit et al., 1994). Mutation studies with mice lacking α-CaMKII, or expressing an altered autophosphorylation phenotype of α-CaMKII indicated that CaMKII is required for tetanus-LTP (Silva et al., 1992; Giese et al., 1998). Ca2+/CaM-induced potentiation and tetanus-LTP require postsynaptic Ca2+/CaM-dependent protein kinase activities (Malenka et al., 1989; Malinow et al., 1989; Malinow and Tsien, 1990b; Lledo et al., 1995; Wang and Kelly, 1995). Thus, Ca2+/CaM- and CaMKII-induced potentiation share common mechanisms with tetanus-LTP (Lledo et al., 1995; Wang and Kelly, 1995).
Here we report that even though both Ca2+/CaM-induced potentiation and tetanus-LTP are NO-dependent, they are not the same. NO appears to function primarily as a retrograde messenger in LTP, because NOS inhibitors and extracellular NO scavengers block tetanus- or pairing-induced synaptic potentiation (O’Dell et al., 1991, 1994;Schuman and Madison, 1991, 1994a; Haley et al., 1992; Hawkins et al., 1994; Garthwaite and Boulton, 1995; Arancio et al., 1996; Malen and Chapman, 1997). However, these studies indicate that NO may also work at postsynaptic sites, because postsynaptic injections of a NOS inhibitor or NO scavenger blocked tetanus-LTP (Schuman and Madison, 1994a; Arancio et al., 1996). Similar to tetanus-LTP, Ca2+/CaM-induced potentiation is blocked by extracellular application or postsynaptic co-injection of an NOS inhibitor. In contrast, Ca2+/CaM-induced potentiation is blocked by postsynaptic co-injection of an NO scavenger, but not by extracellular applications of NO scavengers. Thus, we believe that NO acts at a postsynaptic site(s) during Ca2+/CaM-induced potentiation. It is possible that during high-frequency stimulation or pairing protocols, presynaptic as well as postsynaptic components are activated through a variety of signal transduction cascades, so NO could react with its targets at both presynaptic and postsynaptic sites. Potentiation induced by injecting Ca2+/CaM might activate postsynaptic NO targets without activating presynaptic targets. In conclusion, NO serves as a postsynaptic intracellular signaling molecule but not a retrograde messenger during Ca2+/CaM-induced potentiation.
Footnotes
This work was supported by National Institutes of Health Grant NS 32470 and National Institutes of Health Training Grant NS 07373. We thank Dr. Yashige Kotake (Oklahoma Medical Research Foundation, Oklahoma City, OK) for MGD and FeSO4 and Drs. Jaroslaw Aronowski (University of Texas-Houston Medical School, Houston, TX) and Owen Griffith (Medical College of Wisconsin, Milwaukee, WI) for fruitful comments and discussion. We also thank Drs. Y. Kotake and Takaaki Akaike (Kumamoto University School of Medicine, Kumamoto, Japan) for making available unpublished results.
Correspondence should be addressed to Paul T. Kelly, Department of Molecular Biosciences, 7042 Haworth Hall, The University of Kansas, Lawrence, KS 66045-2106. E-mail:ptkelly@eagle.cc.ukans.edu
Dr. Ko’s present address: Department of Biology and Biochemistry, Science and Research 2 Building, University of Houston, Houston, TX 77204.
REFERENCES
- 1.Amano F, Noda T. Improved detection of nitric oxide radical (NO⋅) production in an activated macrophage culture with a radical scavenger, carboxy-PTIO and Griess reagent. FEBS Lett. 1995;368:425–428. doi: 10.1016/0014-5793(95)00700-j. [DOI] [PubMed] [Google Scholar]
- 2.Arancio O, Kandel ER, Hawkins RD. Activity-dependent long-term enhancement of transmitter release by presynaptic 3′-5′-cyclic GMP in cultured hippocampal neurons. Nature. 1995;376:74–80. doi: 10.1038/376074a0. [DOI] [PubMed] [Google Scholar]
- 3.Arancio O, Kiebler M, Lee C, Lev-Ram V, Tsien R, Kandel E, Hawkins R. Nitric oxide acts directly in the presynaptic neuron to produce long-term potentiation in cultured hippocampal neurons. Cell. 1996;87:1025–1035. doi: 10.1016/s0092-8674(00)81797-3. [DOI] [PubMed] [Google Scholar]
- 4.Az-ma T, Fujii K, Yuge O. Reaction between imidazolineoxil N-oxide (carboxy-PTIO) and nitric oxide released from cultured endothelial cells: quantitative measurement of nitric oxide by ESR spectrometry. Life Sci. 1994;54:PL185–PL190. doi: 10.1016/0024-3205(94)90166-x. [DOI] [PubMed] [Google Scholar]
- 5.Barria A, Muller D, Griffith LC, Soderling TR. Phosphorylation of AMPA-type glutamate receptors by Ca2+/calmodulin-dependent protein kinase II during long-term potentiation. Science. 1997;276:2042–2045. doi: 10.1126/science.276.5321.2042. [DOI] [PubMed] [Google Scholar]
- 6.Baudier J, Deloulme JC, Van Dorsselear A, Black D, Matthes HWD. Purification and characterization of a brain-specific protein kinase C substrate, neurogranin (p 17). J Biol Chem. 1991;266:229–237. [PubMed] [Google Scholar]
- 7.Bliss TVP, Collingridge GL. A synaptic model of memory: LTP in the hippocampus. Nature. 1993;361:31–39. doi: 10.1038/361031a0. [DOI] [PubMed] [Google Scholar]
- 8.Bolshakov VY, Siegelbaum SA. Regulation of hippocampal transmitter release during development and long-term potentiation. Science. 1995;269:1730–1734. doi: 10.1126/science.7569903. [DOI] [PubMed] [Google Scholar]
- 9.Boulton C, Southam E, Garthwaite J. Nitric oxide-dependent long-term potentiation is blocked by a specific inhibitor of soluble guanylyl cyclase. Neuroscience. 1995;69:699–703. doi: 10.1016/0306-4522(95)00349-n. [DOI] [PubMed] [Google Scholar]
- 10.Braun AP, Shulman H. The multifunctional calcium/calmodulin-dependent protein kinase: from form to function. Annu Rev Physiol. 1995;57:417–445. doi: 10.1146/annurev.ph.57.030195.002221. [DOI] [PubMed] [Google Scholar]
- 11.Bredt D, Snyder S. Nitric oxide: a physiologic messenger molecule. Annu Rev Biochem. 1994;63:175–195. doi: 10.1146/annurev.bi.63.070194.001135. [DOI] [PubMed] [Google Scholar]
- 12.Bredt DS, Snyder SH. Isolation nitric oxide synthetase, a calmodulin-requiring enzyme. Proc Natl Acad Sci USA. 1990;87:682–685. doi: 10.1073/pnas.87.2.682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bredt DS, Ferris CD, Snyder SH. Nitric oxide synthase regulatory sites. J Biol Chem. 1992;267:10976–10981. [PubMed] [Google Scholar]
- 14.Brenman J, Chao D, Gee S, McGee A, Craven S, Santillano D, Wu Z, Huang F, Xia H, Peters M, Froehner S, Bredt D. Interaction of nitric oxide synthase with the postsynaptic density protein PSD-95 and alpha1-syntrophin mediated by PDZ domains. Cell. 1996;84:757–767. doi: 10.1016/s0092-8674(00)81053-3. [DOI] [PubMed] [Google Scholar]
- 15.Chetkovitch D, Klann E, Sweatt J. Nitric oxide synthase-independent long-term potentiation in area CA1 of hippocampus. NeuroReport. 1993;4:919–922. doi: 10.1097/00001756-199307000-00020. [DOI] [PubMed] [Google Scholar]
- 16.Creager R, Dunwiddie T, Lynch G. Paired-pulse and frequency facilitation in the CA1 region of the in vitro rat hippocampus. J Physiol (Lond) 1980;299:409–424. doi: 10.1113/jphysiol.1980.sp013133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cummings J, Nicola S, Malenka R. Induction in the rat hippocampus of long-term potentiation and long-term depression in the presence of a nitric oxide synthase inhibitor. Neurosci Lett. 1994;176:110–114. doi: 10.1016/0304-3940(94)90883-4. [DOI] [PubMed] [Google Scholar]
- 18.Derkach V, Barria A, Soderling TR. CaM-KII increases single channel conductance of recombinant AMPA glutamate receptor. Soc Neurosci Abstr. 1998;24:1826. [Google Scholar]
- 19.Dinerman JL, Dawson TM, Schell MJ, Snowman A, Snyder SH. Endothelial nitric oxide synthase localized to hippocampal pyramidal cells: implication for synaptic plasticity. Proc Natl Acad Sci USA. 1994;91:4214–4218. doi: 10.1073/pnas.91.10.4214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Doyle C, Slater P. Localization of neuronal and endothelial nitric oxide synthase isoforms in human hippocampus. Neuroscience. 1997;76:387–395. doi: 10.1016/s0306-4522(96)00297-7. [DOI] [PubMed] [Google Scholar]
- 21.Ehlers M, Zhang S, Bernhardt J, Huganir R. Inactivation of NMDA receptors by direct interaction of calmodulin with the NR1 subunit. Cell. 1996;84:745–755. doi: 10.1016/s0092-8674(00)81052-1. [DOI] [PubMed] [Google Scholar]
- 22.Eliasson MJ, Blackshaw S, Schell MJ, Snyder SH. Neuronal nitric oxide synthase alternatively spliced forms: prominent functional localizations in the brain. Proc Natl Acad Sci USA. 1997;94:3396–3401. doi: 10.1073/pnas.94.7.3396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.English JD, Sweatt JD. Activation of p42 mitogen-activated protein kinase in hippocampal long term potentiation. J Biol Chem. 1996;271:24329–24332. doi: 10.1074/jbc.271.40.24329. [DOI] [PubMed] [Google Scholar]
- 24.English JD, Sweatt JD. A requirement for the mitogen-activated protein kinase cascade in hippocampal long term potentiation. J Biol Chem. 1997;272:19103–19106. doi: 10.1074/jbc.272.31.19103. [DOI] [PubMed] [Google Scholar]
- 25.Garthwaite J, Boulton C. Nitric oxide signaling in the central nervous system. Annu Rev Physiol. 1995;57:683–706. doi: 10.1146/annurev.ph.57.030195.003343. [DOI] [PubMed] [Google Scholar]
- 26.Gerendasy DD, Herron SR, Jennings PA, Sutcliffe JG. Calmodulin stabilizes an amphiphilic alpha-helix within RC3/neurogranin and GAP-43/neuromodulin only when Ca2+ is absent. J Biol Chem. 1995;270:6741–6750. doi: 10.1074/jbc.270.12.6741. [DOI] [PubMed] [Google Scholar]
- 27.Giese KP, Fedorov NB, Filipkowski RK, Silva AJ. Autophosphorylation at Thr286 of the alpha calcium-calmodulin kinase II in LTP and learning. Science. 1998;279:870–873. doi: 10.1126/science.279.5352.870. [DOI] [PubMed] [Google Scholar]
- 28.Guerini D. Calcineurin: not just a simple protein phosphatase. Biochem Biophys Res Commun. 1997;235:271–275. doi: 10.1006/bbrc.1997.6802. [DOI] [PubMed] [Google Scholar]
- 29.Guerrini R, Menegazzi P, Anacardio R, Marastoni M, Tomatis R, Zorzato F, Treves S. Calmodulin binding sites of the skeletal, cardiac, and brain ryanodine receptor Ca2+ channels: modulation by the catalytic subunit of cAMP-dependent protein kinase? Biochemistry. 1995;34:5120–5129. doi: 10.1021/bi00015a024. [DOI] [PubMed] [Google Scholar]
- 30.Haley J, Wilcox G, Chapman P. The role of nitric oxide in hippocampal long-term potentiation. Neuron. 1992;8:211–216. doi: 10.1016/0896-6273(92)90288-o. [DOI] [PubMed] [Google Scholar]
- 31.Hawkins RD, Zhuo M, Arancio O. Nitric oxide and carbon monoxide as possible retrograde messengers in hippocampal long-term potentiation. J Neurobiol. 1994;25:652–665. doi: 10.1002/neu.480250607. [DOI] [PubMed] [Google Scholar]
- 32.Hisatsune C, Umemori H, Inoue T, Michikawa T, Kohda K, Mikoshiba K, Yamamoto T. Phosphorylation-dependent regulation of N-methyl-d-aspartate receptors by calmodulin. J Biol Chem. 1997;272:20805–20810. doi: 10.1074/jbc.272.33.20805. [DOI] [PubMed] [Google Scholar]
- 33.Hogg N, Singh R, Joseph J, Neese F, Kalyanaraman B. Reactions of nitric oxide with nitronyl nitroxides and oxygen: prediction of nitrite and nitrate formation by kinetic simulation. Free Radic Res. 1995a;22:47–56. doi: 10.3109/10715769509147527. [DOI] [PubMed] [Google Scholar]
- 34.Hogg N, Struck A, Goss S, Santanam N, Joseph J, Parthasarathy S, Kalyanaraman B. Inhibition of macrophage-dependent low density lipoprotein oxidation by nitric oxide donors. J Lipid Res. 1995b;36:1756–1762. [PubMed] [Google Scholar]
- 35.Huang K-P, Huang FL, Chen H-C. Characterization of a 7.5-kDa protein kinase C substrate (RC3 protein, neurogranin) from rat brain. Arch Biochem Biophys. 1993;305:570–580. doi: 10.1006/abbi.1993.1463. [DOI] [PubMed] [Google Scholar]
- 36.Ikemoto T, Iino M, Endo M. Enhancing effect of calmodulin on Ca2+-induced Ca2+ release in the sarcoplasmic reticulum of rabbit skeletal muscle fibres. J Physiol (Lond) 1995;487:573–582. doi: 10.1113/jphysiol.1995.sp020901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kelly PT, McGuinness TL, Greengard P. Evidence that the major postsynaptic density protein is a component of a Ca2+/calmodulin-dependent protein kinase. Proc Natl Acad Sci USA. 1984;81:945–949. doi: 10.1073/pnas.81.3.945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kelly PT, Yip RK, Shields SM, Hay M. Calmodulin-dependent protein phosphorylation in synaptic junctions. J Neurochem. 1985;45:1620–1634. doi: 10.1111/j.1471-4159.1985.tb07235.x. [DOI] [PubMed] [Google Scholar]
- 39.Klee CB, Ren H, Wang X. Regulation of the calmodulin-stimulated protein phosphatase, calcineurin. J Biol Chem. 1998;273:13367–13370. doi: 10.1074/jbc.273.22.13367. [DOI] [PubMed] [Google Scholar]
- 40.Kleppisch T, Pfeifer A, Klatt P, Ruth P, Montkowski A, Fassler R, Hofmann F. Long-term potentiation in the hippocampal CA1 region of mice lacking cGMP-dependent kinases is normal and susceptible to inhibition of nitric oxide synthase. J Neurosci. 1999;19:48–55. doi: 10.1523/JNEUROSCI.19-01-00048.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ko G, Kelly P. A role of nitric oxide synthase in Ca2+/calmodulin-induced synaptic potentiation in hippocampal CA1 neurons. Soc Neurosci Abstr. 1998;24:1074. [Google Scholar]
- 42.Komarov A, Lai C. Detection of nitric oxide production in mice by spin-trapping electron paramagnetic resonance spectroscopy. Biochim Biophys Acta. 1995;1272:29–36. doi: 10.1016/0925-4439(95)00061-8. [DOI] [PubMed] [Google Scholar]
- 43.Komarov A, Maattson D, Jones M, Singh P, Lai C. In vivo spin trapping of nitric oxide in mice. Biochem Biophys Res Commun. 1993;195:1191–1198. doi: 10.1006/bbrc.1993.2170. [DOI] [PubMed] [Google Scholar]
- 44.Kotake Y. Continuous and quantitative monitoring of rate of cellular nitric oxide generation. Methods Enzymol. 1996;268:222–229. doi: 10.1016/s0076-6879(96)68024-0. [DOI] [PubMed] [Google Scholar]
- 45.Kotake Y, Tanigawa T, Tanigawa M, Ueno I. Spin trapping isotopically-labelled nitric oxide produced from [15N]l-arginine and [17O]dioxygen by activated macrophages using a water soluble Fe 2+-dithiocarbamate spin trap. Free Radic Res. 1995;23:287–295. doi: 10.3109/10715769509064041. [DOI] [PubMed] [Google Scholar]
- 46.Kotake Y, Tanigawa T, Tanigawa M, Ueno I, Allen D, Lai C. Continuous monitoring of cellular nitric oxide generation by spin trapping with an iron-dithiocarbamate complex. Biochim Biophys Acta. 1996;1289:362–368. doi: 10.1016/0304-4165(95)00172-7. [DOI] [PubMed] [Google Scholar]
- 47.Lai C, Komarov A. Spin trapping of nitric oxide produced in vivo in septic-shock mice. FEBS Lett. 1994;345:120–124. doi: 10.1016/0014-5793(94)00422-6. [DOI] [PubMed] [Google Scholar]
- 48.Lee S-J, Stull JT. Calmodulin-dependent regulation of inducible and neuronal nitric oxide synthase. J Biol Chem. 1998;273:27430–27437. doi: 10.1074/jbc.273.42.27430. [DOI] [PubMed] [Google Scholar]
- 49.Lei SZ, Pan Z-H, Aggarwal SK, Chen H-S, Hartman J, Sucher NJ, Lipton SA. Effect of nitric oxide production on the redox modulatory site of the NMDA receptor-channel complex. Neuron. 1992;8:1087–1099. doi: 10.1016/0896-6273(92)90130-6. [DOI] [PubMed] [Google Scholar]
- 50.Li J, Pak J, Huang F, Huang K-P. N-Methyl-d-aspartate induces neurogranin/RC3 oxidation in rat brain slices. J Biol Chem. 1999;274:1294–1300. doi: 10.1074/jbc.274.3.1294. [DOI] [PubMed] [Google Scholar]
- 51.Lipton SA, Wang YF. NO-related species can protect from focal cerebral ischemia/reperfusion. In: Krieglstein J, editor. Pharmacology of cerebral ischemia 1996. Medpharm Scientific; Stuttgart: 1996. pp. 183–191. [Google Scholar]
- 52.Lipton SA, Choi Y-B, Pan Z-H, Lei SZ, Chen H-S, Sucher NJ, Loscalzo J, Singel DJ, Stamler JS. A redox-based mechanism for the neuroprotective and neurodestructive effects of nitric oxide and related nitroso-compounds. Nature. 1993;364:626–632. doi: 10.1038/364626a0. [DOI] [PubMed] [Google Scholar]
- 53.Lipton SA, Choi Y-B, Sucher NJ, Pan Z-H, Stamier JS. Redox state, NMDA receptors and NO-related species. Trends Pharmacol. 1996;17:186–187. doi: 10.1016/0165-6147(96)20028-8. [DOI] [PubMed] [Google Scholar]
- 54.Lledo PM, Hjelmstad GO, Mukherji S, Soderling TR, Malenka RC, Nicoll RA. Calcium/calmodulin-dependent kinase II and long-term potentiation enhance synaptic transmission by the same mechanism. Proc Natl Acad Sci USA. 1995;92:11175–11179. doi: 10.1073/pnas.92.24.11175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Maeda H, Akaike T, Yoshida M, Suga M. Multiple functions of nitric oxide in pathophysiology and microbiology: analysis by a new nitric oxide scavenger. J Leukocyte Biol. 1994;56:588–592. doi: 10.1002/jlb.56.5.588. [DOI] [PubMed] [Google Scholar]
- 56.Magleby KL. Short-term changes in synaptic efficacy. In: Edelman GM, Gall WE, Cowan WM, editors. Synaptic function. Wiley; New York: 1987. pp. 21–57. [Google Scholar]
- 57.Mahoney C, Pak J, Huang K-P. Nitric oxide modification of rat brain neurogranin. J Biol Chem. 1996;46:28798–28804. doi: 10.1074/jbc.271.46.28798. [DOI] [PubMed] [Google Scholar]
- 58.Malen P, Chapman P. Nitric oxide facilitates long-term potentiation, but not long-term depression. J Neurosci. 1997;17:2645–2651. doi: 10.1523/JNEUROSCI.17-07-02645.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Malenka RC, Kauer JA, Perkel DJ, Mauk MD, Kelly PT, Nicoll RA, Waxham MN. An essential role for postsynaptic calmodulin and protein kinase activity in long-term potentiation. Nature. 1989;340:554–557. doi: 10.1038/340554a0. [DOI] [PubMed] [Google Scholar]
- 60.Malinow R, Tsien RW. Presynaptic enhancement shown by whole-cell recordings of long-term potentiation in hippocampal slices. Nature. 1990a;346:177–180. doi: 10.1038/346177a0. [DOI] [PubMed] [Google Scholar]
- 61.Malinow R, Tsien RW. Identifying and localizing protein kinases necessary for LTP. Adv Exp Med Biol. 1990b;268:301–305. doi: 10.1007/978-1-4684-5769-8_33. [DOI] [PubMed] [Google Scholar]
- 62.Malinow R, Schulman H, Tsien RW. Inhibition of postsynaptic PKC or CaMKII blocks induction but not expression of LTP. Science. 1989;245:862–866. doi: 10.1126/science.2549638. [DOI] [PubMed] [Google Scholar]
- 63.Manabe T, Wyllie DJA, Perkel DJ, Nicoll RA. Modulation of synaptic transmission and long-term potentiation: effect on paired-pulse facilitation and EPSC variance in the CA1 region of the hippocampus. J Neurophysiol. 1993;70:1451–1459. doi: 10.1152/jn.1993.70.4.1451. [DOI] [PubMed] [Google Scholar]
- 64.Matsuoka I, Giuili G, Poyard M, Stengel D, Parma J, Guellaen G, Hanoune J. Localization of adenylyl and guanylyl cyclase in rat brain by in situ hybridization: comparison with calmodulin mRNA distribution. J Neurosci. 1992;12:3350–3360. doi: 10.1523/JNEUROSCI.12-09-03350.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Molday RS. Calmodulin regulation of cyclic-nucleotide-gated channels. Curr Opin Neurobiol. 1996;6:445–452. doi: 10.1016/s0959-4388(96)80048-1. [DOI] [PubMed] [Google Scholar]
- 66.Moodie SA, Willumsen BM, Weber MJ, Wolfman A. Complexes of ras.GTP with raf-1 and mitogen-activated protein kinase kinase. Science. 1993;260:1658–1661. doi: 10.1126/science.8503013. [DOI] [PubMed] [Google Scholar]
- 67.Muller D, Lynch G. Evidence that changes in presynaptic calcium currents are not responsible for long-term potentiation in hippocampus. Brain Res. 1989;479:290–299. doi: 10.1016/0006-8993(89)91631-4. [DOI] [PubMed] [Google Scholar]
- 68.Nicolarakis PJ, Lin YQ, Bennett MR. Effect of nitric oxide synthase inhibition on long-term potentiation at associational-commissural and mossy fibre synapses on CA3 pyramidal neurones. Br J Pharmacol. 1994;111:521–524. doi: 10.1111/j.1476-5381.1994.tb14768.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Obenaus A, Mody I, Baimbridge KG. Dantrolene-Na (Dantrium) blocks induction of long-term potentiation in hippocampal slices. Neurosci Lett. 1989;98:172–178. doi: 10.1016/0304-3940(89)90505-3. [DOI] [PubMed] [Google Scholar]
- 70.Ocorr KA, Schulman H. Activation of multifunctional Ca++/calmodulin-dependent kinase in intact hippocampal slices. Neuron. 1991;6:907–914. doi: 10.1016/0896-6273(91)90231-n. [DOI] [PubMed] [Google Scholar]
- 71.O’Dell TJ, Hawkins RD, Kandel ER, Arancio O. Tests of the roles of two diffusible substances in long-term potentiation: evidence for nitric oxide as a possible early retrograde messenger. Proc Natl Acad Sci USA. 1991;88:11285–11289. doi: 10.1073/pnas.88.24.11285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.O’Dell TJ, Huang PL, Dawson TM, Dinerman JL, Snyder SH, Kandel ER, Fishman MC. Endothelial NOS and the blockade of LTP by NOS inhibitors in mice lacking neuronal NOS. Science. 1994;265:542–546. doi: 10.1126/science.7518615. [DOI] [PubMed] [Google Scholar]
- 73.Pettit DL, Periman R, Malinow R. Potentiated transmission and prevention of further LTP by increased CaMKII activity in postsynaptic hippocampal slice neurons. Science. 1994;266:1881–1885. doi: 10.1126/science.7997883. [DOI] [PubMed] [Google Scholar]
- 74.Pieper G, Lai C. Evaluation of vascular actions of the nitric oxide-trapping agent, N-methyl-d-glucamine dithiocarbamate-Fe2+, on basal and agonist-stimulated nitric oxide activity. Biochem Biophys Res Commun. 1996;219:584–590. doi: 10.1006/bbrc.1996.0277. [DOI] [PubMed] [Google Scholar]
- 75.Rao A, Kim E, Sheng M, Craig AM. Heterogeneity in the molecular composition of excitatory postsynaptic sites during development of hippocampal neurons in culture. J Neurosci. 1998;18:1217–1229. doi: 10.1523/JNEUROSCI.18-04-01217.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Rhoads A, Friedberg F. Sequence motifs for calmodulin recognition. FASEB J. 1997;11:331–340. doi: 10.1096/fasebj.11.5.9141499. [DOI] [PubMed] [Google Scholar]
- 77.Riquelme G, Wyneken U, Villanueva S, Orrego F. Recordings of glutamate receptor channels in isolated postsynaptic densities. NeuroReport. 1993;4:1163–1166. [PubMed] [Google Scholar]
- 78.Sato T, Xiao D-M, Li H, Huang FL, Huang K-P. Structure and regulation of the gene encoding the neuron-specific protein kinase C substrate neurogranin (RC3 protein). J Biol Chem. 1995;270:10314–10322. doi: 10.1074/jbc.270.17.10314. [DOI] [PubMed] [Google Scholar]
- 79.Schulz PE, Cook EP, Johnston D. Changes in paired-pulse facilitation suggest presynaptic involvement in long-term potentiation. J Neurosci. 1994;14:5325–5337. doi: 10.1523/JNEUROSCI.14-09-05325.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Schuman E, Madison D. Locally distributed synaptic potentiation in the hippocampus. Science. 1994a;263:532–36. doi: 10.1126/science.8290963. [DOI] [PubMed] [Google Scholar]
- 81.Schuman E, Madison D. Nitric oxide and synaptic function. Annu Rev Neurosci. 1994b;17:153–183. doi: 10.1146/annurev.ne.17.030194.001101. [DOI] [PubMed] [Google Scholar]
- 82.Schuman E, Meffert M, Schulman H, Madison D. An ADP-ribosyltransferase as a potential target for nitric oxide action in hippocampal long-term potentiation. Proc Natl Acad Sci USA. 1994;91:11958–11962. doi: 10.1073/pnas.91.25.11958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Schuman EM, Madison DV. A requirement for the intercellular messenger nitric oxide in long-term potentiation. Science. 1991;254:1503–1506. doi: 10.1126/science.1720572. [DOI] [PubMed] [Google Scholar]
- 84.Selig DK, Segal MR, Liao D, Malenka RC, Malinow R, Nicoll RA, Lisman JE. Examination of the role of cGMP in long-term potentiation in the CA1 region of the hippocampus. Learn Mem. 1996;3:42–48. doi: 10.1101/lm.3.1.42. [DOI] [PubMed] [Google Scholar]
- 85.Sharma RK. Signal transduction: regulation of cAMP concentration in cardiac muscle by calmodulin-dependent cyclic nucleotide phosphodiesterase. Mol Cell Biochem. 1995;149–150:241–247. doi: 10.1007/BF01076583. [DOI] [PubMed] [Google Scholar]
- 86.Sheu F-S, Mahoney C, Seki K, Huang K-P. Nitric oxide modification of rat brain neurogranin affects its phosphorylation by protein kinase C and affinity for calmodulin. J Biol Chem. 1996;271:22407–22413. doi: 10.1074/jbc.271.37.22407. [DOI] [PubMed] [Google Scholar]
- 87.Silva AJ, Stevens CF, Tonegawa S, Wang Y. Deficient hippocampal long-term potentiation in alpha-calcium-calmodulin kinase II mutant mice. Science. 1992;257:201–206. doi: 10.1126/science.1378648. [DOI] [PubMed] [Google Scholar]
- 88.Smit MJ, Iyengar R. Mammalian adenylyl cyclases. Adv Second Messenger Phosphoprotein Res. 1998;32:1–21. doi: 10.1016/s1040-7952(98)80003-7. [DOI] [PubMed] [Google Scholar]
- 89.Son H, Hawkins R, Martin K, Kiebler M, Huang P, Fishman M, Kandel E. Long-term potentiation is reduced in mice that are doubly mutant in endothelial and neuronal nitric oxide synthase. Cell. 1996;87:1015–1023. doi: 10.1016/s0092-8674(00)81796-1. [DOI] [PubMed] [Google Scholar]
- 90.Sunahara RK, Dessauer CW, Gilman AG. Complexity and diversity of mammalian adenylyl cyclases. Annu Rev Pharmacol Toxicol. 1996;36:461–480. doi: 10.1146/annurev.pa.36.040196.002333. [DOI] [PubMed] [Google Scholar]
- 91.Thomas SM, DeMarco M, D’Arcangelo G, Halegoua S, Brugge JS. Ras is essential for nerve-growth factor- and phorbol ester- induced tyrosine phosphorylation of MAP kinase. Cell. 1992;68:1031–1040. doi: 10.1016/0092-8674(92)90075-n. [DOI] [PubMed] [Google Scholar]
- 92.Wang JH, Kelly PT. Postsynaptic injection of Ca2+/CaM induces synaptic potentiation requiring CaM-KII and PKC activity. Neuron. 1995;15:443–452. doi: 10.1016/0896-6273(95)90048-9. [DOI] [PubMed] [Google Scholar]
- 93.Wang J-H, Kelly P. Regulation of synaptic facilitation by postsynaptic Ca2+/CaM pathways in hippocampal CA1 neurons. J Neurophysiol. 1996;76:276–286. doi: 10.1152/jn.1996.76.1.276. [DOI] [PubMed] [Google Scholar]
- 94.Wang J-H, Kelly P. Developmental changes of postsynaptic calcineurin and IP3 receptors regulating synaptic transmission. Soc Neurosci Abstr. 1997a;23:1128. [Google Scholar]
- 95.Wang J-H, Kelly PT. Attenuation of paired-pulse facilitation associated with synaptic potentiation mediated by postsynaptic mechanisms. J Neurophysiol. 1997b;78:2707–2716. doi: 10.1152/jn.1997.78.5.2707. [DOI] [PubMed] [Google Scholar]
- 96.Wang Y, Wu J, Rowan MJ, Anwyl R. Ryanodine produces a low frequency stimulation-induced NMDA receptor-independent long-term potentiation in the rat dentate gyrus in vitro. J Physiol (Lond) 1996;495.3:755–767. doi: 10.1113/jphysiol.1996.sp021631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Williams JH, Li Y-G, Nayak A, Errington ML, Murphy KPSJ, Bliss TVP. The suppression of long-term potentiation in rat hippocampus by inhibitors of nitric oxide synthase is temperature and age dependent. Neuron. 1993a;11:877–884. doi: 10.1016/0896-6273(93)90117-a. [DOI] [PubMed] [Google Scholar]
- 98.Williams MG, Paradis H, Agarwal S, Charest DL, Pelech SL, Roberts TM. Raf-1 and p21v-ras cooperate in the activation of mitogen-activated protein kinase. Proc Natl Acad Sci USA. 1993b;90:5772–5776. doi: 10.1073/pnas.90.12.5772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Willmott N, Sethi JK, Walseth TF, Lee HC, White AM, Galione A. Nitric oxide-induced mobilization of intracellular calcium via the cyclic ADP-ribose signaling pathway. J Biol Chem. 1996;271:3699–3705. doi: 10.1074/jbc.271.7.3699. [DOI] [PubMed] [Google Scholar]
- 100.Wood KW, Sarneck C, Roberts TM, Blenis J. Ras mediates nerve growth factor receptor modulation of three signal-transducing protein kinases: MAP kinase, raf-1 and RSK. Cell. 1992;68:1041–1050. doi: 10.1016/0092-8674(92)90076-o. [DOI] [PubMed] [Google Scholar]
- 101.Wu J, Wang Y, Rowan M, Anwyl R. Evidence for involvement of the cGMP-protein kinase G signaling system in the induction of long-term depression, but not long-term potentiation, in the dentate gyrus in vitro. J Neurosci. 1998;18:3589–3596. doi: 10.1523/JNEUROSCI.18-10-03589.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Wu K, Xu J, Suen P, Huang Y, Mount HTJ. Nitric oxide increases calcium/calmodulin-dependent phosphorylation of proteins in the postsynaptic density of adult rat cerebral cortex. Mol Brain Res. 1996;40:22–26. doi: 10.1016/0169-328x(96)00028-9. [DOI] [PubMed] [Google Scholar]
- 103.Yip S, Ip J, Sastry B. Electrophysiological actions of hemoglobin on rat hippocampal CA1 pyramidal neurons. Brain Res. 1996;713:134–142. doi: 10.1016/0006-8993(95)01499-3. [DOI] [PubMed] [Google Scholar]
- 104.Yoshida M, Akaike T, Wada Y, Sato K, Ikeda K, Ueda S, Maeda H. Therapeutic effects of imidazolineoxyl N-oxide against endotoxin shock through its direct nitric oxide-scavenging activity. Biochem Biophys Res Commun. 1994;202:923–930. doi: 10.1006/bbrc.1994.2018. [DOI] [PubMed] [Google Scholar]
- 105.Yun H-Y, Gonzalez-Zulueta M, Dawson VL, Dawson TM. Nitric oxide mediates N-methyl-d-aspartate receptor-induced activation of p21ras. Proc Natl Acad Sci USA. 1998;95:5773–5778. doi: 10.1073/pnas.95.10.5773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Zagotta W, Siegelbaum S. Structure and function of cyclic nucleotide-gated channels. Annu Rev Neurosci. 1996;19:235–263. doi: 10.1146/annurev.ne.19.030196.001315. [DOI] [PubMed] [Google Scholar]
- 107.Zhang S, Ehler M, Bernhardt J, Su C, Huganir R. Calmodulin mediates calcium-dependent inactivation of N-methyl-d-aspartate receptors. Neuron. 1998;21:443–453. doi: 10.1016/s0896-6273(00)80553-x. [DOI] [PubMed] [Google Scholar]
- 108.Zhao AZ, Yan C, Sonnenburg WK, Beavo JA. Recent advances in the study of Ca2+/CaM-activated phosphodiesterases: expression and physiological functions. Adv Second Messenger Phosphoprotein Res. 1997;31:237–251. [PubMed] [Google Scholar]
- 109.Zhou M, Hu Y, Schultz C, Kandel E, Hawkins R. Role of guanylyl cyclase and cGMP-dependent protein kinase in long-term potentiation. Nature. 1994a;368:635–639. doi: 10.1038/368635a0. [DOI] [PubMed] [Google Scholar]
- 110.Zhou M, Kandel E, Hawkins R. Nitric oxide and cGMP can produce either synaptic depression or potentiation depending on the frequency of presynaptic stimulation in the hippocampus. NeuroReport. 1994b;5:1033–1036. doi: 10.1097/00001756-199405000-00004. [DOI] [PubMed] [Google Scholar]
- 111.Zucker RS. Short-term synaptic plasticity. Annu Rev Neurosci. 1989;12:13–31. doi: 10.1146/annurev.ne.12.030189.000305. [DOI] [PubMed] [Google Scholar]