

Abstract
Apolipoprotein E (apoE)-related synthetic peptides, the 22 kDa N-terminal thrombin-cleavage fragment of apoE (truncated apoE), and full-length apoE have all been shown to exhibit neurotoxic activity under certain culture conditions. In the present study, protease inhibitors reduced the neurotoxicity and proteolysis of full-length apoE but did not block the toxicity of truncated apoE or a synthetic apoE peptide, suggesting that fragments of apoE may account for its toxicity. Additional experiments demonstrated that both truncated apoE and the apoE peptide elicit an increase in intracellular calcium levels and subsequent death of embryonic rat hippocampal neurons in culture. Similar effects on calcium were found when the apoE peptide was applied to chick sympathetic neurons. The rise in intracellular calcium and the hippocampal cell death caused by the apoE peptide were significantly reduced by receptor-associated protein, removal of extracellular calcium, or administration of the specific NMDA glutamate receptor antagonist MK-801. These results suggest that apoE may be a source of both neurotoxicity and calcium influx that involves cell surface receptors. Such findings strengthen the hypothesis that apoE plays a direct role in the pathology of Alzheimer’s disease.
Keywords: proteolysis, intracellular calcium, apolipoprotein E, neurotoxicity, LRP, degeneration, Alzheimer’s disease
Apolipoprotein E (apoE) is a lipid-associated protein that binds and transports cholesterol-rich lipoproteins for internalization via receptors of the low density lipoprotein (LDL) receptor family (Mahley, 1988). In addition, apoE has other putative functions that do not seem to involve lipid transport (Weisgraber, 1994). Involvement of apoE in the pathogenesis of late-onset Alzheimer’s disease (AD) was suggested by the association between inheritance of the allele for the E4 isoform of apoE and the increased risk and earlier age of onset of the disease (Corder et al., 1993; Saunders et al., 1993a,b; Strittmatter et al., 1993), as well as immunohistochemical localization of apoE to senile plaques and neurofibrillary tangles (Namba et al., 1991). Several hypotheses have been proposed to account for the isoform-specific association of apoE with AD (for review, see Laskowitz and Roses, 1998). However, there is still no consensus regarding the role played by apoE in this or other neurodegenerative conditions.
The observation that synthetic apoE-related peptides cause degeneration of sympathetic neurites in culture (Crutcher et al., 1994) led to the hypothesis that apoE could be a source of neurotoxic fragments, thus playing a direct role in AD pathology (Crutcher et al., 1997). This hypothesis is consistent with the finding that a 22 kDa thrombin cleavage fragment of apoE (truncated apoE), which may be analogous to a similar fragment found in brain and CSF, exhibits neurotoxicity, and that truncated E4 is significantly more toxic than truncated E3 (Marques et al., 1996). Furthermore, full-length apoE4 has been shown to exhibit greater neurotoxicity than apoE3, an effect that is associated with production of truncated apoE (Marques et al., 1997). However, whether proteolysis of apoE is involved in toxicity and the mechanism by which apoE-related peptides elicit toxic effects are unknown. All of the toxic apoE species include the receptor-binding region (Innerarity et al., 1983; Weisgraber et al., 1983; Lalazar et al., 1988), as well as the overlapping high-affinity heparin-binding region (Weisgraber et al., 1986), suggesting that specific receptors might be involved. In fact, there is some evidence implicating the LDL receptor-related protein (LRP) and heparan sulfate proteoglycan (HSPG) in mediating the neurotoxicity (Tolar et al., 1997).
The fragmentation of neurites and swelling of neuronal cell bodies that occurs after exposure to apoE peptides is similar to the excitotoxic effects of glutamate (Lucas and Newhouse, 1957), which is accompanied by a rapid influx of calcium (Jancso et al., 1984; Choi 1985; Mattson et al., 1995; Tymianski 1996). Although the receptors that mediate lipoprotein uptake have not generally been associated with intracellular signaling pathways, apoE has been reported to affect calcium regulation in nerve cells (Hartmann et al., 1994; Müller et al., 1998). In this study, inhibition of the proteolysis of apoE was found to be associated with a significant reduction of apoE neurotoxicity. In addition, both an apoE peptide and recombinant human truncated apoE4 were found to elicit increases in intracellular calcium as well as neurite degeneration and neuronal death.
MATERIALS AND METHODS
Cell culture. Embryonic day 9 chick lumbar sympathetic ganglia were procured and cultured as described previously (Tolar et al., 1997). Dissociated sympathetic neurons were plated onto poly-dl-ornithine-coated 96-well plates or on polyethyleneimine-coated glass coverslips in 35 mm dishes for calcium studies. The cultures were incubated in a humidified environment with 5% CO2 and 95% 02 in Neurobasal medium overnight. The next day, dissociated chick sympathetic cultures were transferred to F12 medium supplemented with 20 nm progesterone, 100 μm putrescine, 30 nm selenium, 100 μg/ml human transferrin, 1% penicillin/streptomycin and 5 μg/ml bovine insulin or, for some calcium experiments, left in Neurobasal medium until being transferred to Locke’s solution (see below). For studies of proteolysis of full-length apoE, sympathetic neurons were treated with one of the following in the presence or absence of a mixture of protease inhibitors (leupeptin, pepstatin A, aprotinin, antipain and hirudin; each at 8 μ g/ml): apoE peptide, truncated recombinant human apoE4, mouse IgG (which had been subjected to the purification procedure used for apoE), or full-length recombinant human apoE4 diluted in the supplemented F12 medium.
Primary rat hippocampal neuronal cultures were prepared as previously described (Mark et al., 1995; Mattson et al., 1995). Dissociated hippocampal cells were grown on a polyethyleneimine substrate in either plastic (for analyses of neuronal survival) or glass-bottom (fura-2 AM studies) 35 mm dishes. Cultures were maintained for 1–2 days in Eagle’s MEM supplemented with 20 mm KCl, 1 mmpyruvate and 10% fetal bovine serum and then transferred to a serum-free defined medium (Mattson et al., 1993c) for the purpose of arresting the proliferation of non-neuronal cells. Under these culture conditions, non-neuronal cells constitute less than 5% of the total cell number.
Experiments with hippocampal neurons were done in 6–10-d-old cultures when the neurons express both AMPA and NMDA receptors (Mattson et al., 1993c). The apoE peptide or human recombinant truncated apoE4 were added to dissociated rat hippocampal neurons. This apoE peptide has been shown to exert neurotoxic effects that may involve an LRP-like receptor and/or HSPG (Crutcher et al., 1994; Tolar et al., 1997). The ability of various agents to block the calcium influx and/or neurotoxicity associated with the apoE peptide was determined by treating the cultures with one of the following for 30 min before addition of the peptide: recombinant human LRP receptor-associated protein (RAP), EGTA (Sigma, St. Louis, MO), MK801 (Sigma), CNQX (Tocris Cookson), or nifedipine (Sigma).
Western blot analysis. The production of apoE fragments was monitored using Western blotting of medium from cultures that had been exposed to full-length apoE in the presence or absence of protease inhibitors. The medium was collected and electrophoresed using Tris-tricine 10–16% SDS-PAGE, electroblotted onto polyvinylidene difluoride membranes, and blocked with 5% nonfat dry milk (w/v) in 0.156 m Tris-buffered saline, pH 7.5, with 0.1% (w/v) Tween (TBST). After extensive washing with TBST, membranes were incubated with a monoclonal anti-apoE antibody (1D7) followed by incubation with horseradish peroxidase-labeled sheep anti-mouse secondary antibody (Calbiochem, La Jolla, CA) in TBST with 5% nonfat dry milk (w/v) and 10% glycerol (w/v). Horseradish peroxidase activity was visualized with enhanced chemiluminescence (ECL) (Amersham, Buckinghamshire, UK).
Preparation of apoE-related molecules. Synthetic tandem apoE peptide E(141–149)2, consisting of a duplicated sequence of apoE amino acids 141 through 149, was prepared as described previously (Clay et al., 1995). The 22 kDa N-terminal portion of apoE4 (truncated apoE4) was produced using a bacterial expression system. Escherichia coli harboring the expression plasmid E4 22 kDa/pET21 were grown overnight in 500 ml of sterile Luria-Bertani medium (LB) with 100 μg/ml ampicillin at 37°C. The next morning, each liter of fresh LB containing 100 μg/ml ampicillin was inoculated with 50 ml of overnight culture and grown at 37°C in a shaking platform incubator at 250 rpm to an optical density of 0.6 at 600 nm. Expression was induced by the addition of isopropyl-1-thio β-d-galactopyranoside to a final concentration of 0.5 mm and continued shaking for another 3–4 hr. Cells were pelleted by centrifugation for 20 min at 4000 rpm at 4°C, then resuspended in 30 ml of cold sonication buffer containing 150 mm NaCl, 20 mmNa2PO4, 25 mm EDTA containing 1% trasylol, 1% antipain, and 0.1% β-mercaptoethanol. After three sonication cycles of 1 min each, cells were centrifuged at 30,000 × g for 30 min at 4°C. The supernatant was then dialyzed extensively against 0.1mNH4HCO3.
The 22 kDa bacterial protein was purified using a DEAE-5PW HPLC column, 7.5 cm × 7.5 mm (Supelco, Bellefonte, PA) as reported previously (Tolar et al., 1997) or using the following procedure. Supernatant cell lysate was diluted 1:1 in running buffer (150 mm NaCl, 20 mm NaPO4, pH 7.0, 0.02% EDTA, 0.01% NaN3) and loaded onto a Hi-trap heparin Sepharose column (Pharmacia Biotech, Piscataway, NJ) pre-equilibrated with running buffer. N-terminal truncated 22 kDa apoE 4 was eluted with 1m NaCl, 20 mm NaPO4, pH 7.0, 0.02% EDTA, 0.01% NaN3. Samples from several runs from the DEAE HPLC column were concentrated in 0.1m NH4HCO3 using Centricon 10 and then lyophilized. Samples from several runs from the Hi-trap heparin Sepharose column were desalted using a 50 ml bed volume Bio-Gel P-6DG desalting column (Bio-Rad, Richmond, CA) pre-equilibrated with F12 medium. After protein concentration was assessed by the Bradford method (Bio-Rad protein assay), using a monoclonal antibody (Pierce, Rockford, IL) as standard protein, samples were freeze-dried and stored at −20°C.
Purification of recombinant apoE was carried out with medium from human embryonic kidney (HEK) cells stably transfected with the human apoE4 gene. HEK cells were cultured in an artificial capillary system (Cellco), supernatant medium was loaded onto a heparin chromatography column (heparin-agarose beads, Sigma), and eluted proteins were electrophoresed using Tris-tricine 10–16% SDS-PAGE. Full-length apoE was cut from the gel and electroeluted using the S&S Elutrap device (Schleicher & Schuell, Keene, NH). Mouse IgG (Pierce) was treated the same way and used as a control.
Preparation of recombinant RAP. Recombinant RAP was prepared on the basis of human placental RAP cDNA (Strickland et al., 1991) as described previously (Williams et al., 1992). Briefly, RAP cDNA was cloned into a pGEX2T vector (Pharmacia) designed to produce a protein fusion of the insert encoded protein and glutathioneS-transferase (GST) from Schistosoma japonicum. The construct also contains a thrombin cleavage site that permits the release of RAP. The expression vector was subsequently transformed into the DH5αF′ strain of E. coli. The fusion protein was purified from other proteins contained in the bacterial lysate on a GST affinity column (Herz et al., 1991). After purification, the GST was removed by digestion with thrombin (Calbiochem) then passing the digestion mix once again over the GST affinity column.
Fura-2 AM measurements of intracellular free calcium levels. Fluorescence ratio imaging of the calcium indicator dye fura-2 AM was used to quantify [Ca2+]i using previously described procedures (Mattson et al., 1989; Mark et al., 1995; Mattson et al., 1995). Briefly, the cells were loaded at 37°C for 20–30 min with 10 μm fura-2 AM (Molecular Probes, Eugene, OR). Immediately before imaging, the cells were washed twice with Locke’s solution containing (in mm): 154 NaCl, 20 glucose, 5.6 KCl, 2.3 CaCl2, 1 MgCl2, 3.6 NaHCO3, 5 HEPES, and 0.002% gentamycin, pH 7.2, with or without calcium chloride, as indicated, and then imaged using a Zeiss Axiovert microscope linked to a Zeiss/Attofluor imaging system to acquire and process the images. The [Ca2+]i was determined in the neuronal cell bodies from the ratio of the fluorescence emission using two different excitation wavelengths (340 and 380 nm) after subtraction of background fluorescence.
Quantification of neuronal survival. For toxicity studies performed with dissociated chick sympathetic neurons, the percentage of living cells remaining after overnight incubation with the experimental treatments was assessed using a vital dye (5-carboxyfluorescein diacetate, acetoxymethyl ester; Molecular Probes) as described previously (Tolar et al., 1997). The number of living neurons was determined from images that were captured from four wells per treatment. Each data point is based on the average number from these quadruplicate determinations.
Survival of rat hippocampal neurons was quantified using methods described in previous studies (Mattson et al., 1995). Undamaged neurons in the same 10× microscope fields (phase-contrast optics) were counted immediately before, and at appropriate time points after, exposure to experimental treatments. Neurons were considered undamaged if they had neurites that were uniform in diameter and smooth in appearance and somata that were smooth and round to oval in shape. Degenerating neurons with fragmented and beaded neurites and a swollen and vacuolated soma with an irregular shape were considered nonviable. The quantitation was performed by an observer without knowledge of the experimental condition. Statistical comparisons were made using paired and unpaired Student’s t test (two-tailed) or one-way ANOVA and Scheffe’s post hoc test where appropriate.
RESULTS
Protease inhibitors reduce the production of truncated apoE and attenuate neurotoxicity of full-length apoE
The apoE peptide, E(141–149)2 (consisting of a duplicated sequence of apoE amino acids 141 through 149), truncated apoE, and full-length apoE4 exhibited significant neurotoxicity when tested against dissociated chick sympathetic neurons. The toxic effect of the apoE peptide and of apoE4 are shown in Figure 1. These results are consistent with previous reports. Also, as found previously, exposure of neurons to full-length apoE resulted in the appearance of lower molecular weight fragments of apoE, including a band running with an approximate molecular weight of 22 kDa (Fig. 1), which most likely represents the major N-terminal fragment of apoE (truncated apoE). The other bands presumably represent other proteolytic fragments of apoE. The addition of protease inhibitors at the time of treatment reduced both the toxicity of apoE and the generation of truncated apoE (Fig. 1), suggesting that metabolism of the full-length protein is involved in its toxicity. On the other hand, the protease inhibitors did not attenuate the toxicity produced by the apoE peptide (Fig. 1) or truncated apoE (results not shown). This suggests that the mechanism of toxicity associated with the shorter molecules does not involve proteolysis.
Fig. 1.

ApoE neurotoxicity may involve proteolysis. Dissociated chick sympathetic neurons were exposed to the apoE peptide (ApoEp) or full-length apoE4 (apoE4) in the presence or absence of protease inhibitors (PI). Controls included cells not receiving any experimental treatment (Control), receiving protease inhibitors alone (PI), or treated with an antibody against mouse IgG (IgG) that had been subjected to the same purification procedure as that used for apoE4. Significant toxicity was obtained with the apoE peptide and apoE4 but not with the control treatments. Protease inhibitors prevented the toxic effects of apoE4 but not of the apoE peptide. Medium from the cultures treated with apoE4 with or without PI was subjected to Western blotting using an antibody to apoE. The results are shown below the corresponding treatments in the bar graph. PI treatment reduced the number and intensity of bands representing low molecular weight apoE fragments in the medium. The approximate location of the molecular weight standards is indicated to the left of the Western blot.
ApoE peptide and truncated apoE4 cause death of rat hippocampal neurons
The toxicity of apoE peptides has previously been demonstrated for lymphocytes, chick sympathetic and cortical neurons, and rat hippocampal neurons (Crutcher et al., 1994; Clay et al., 1995; Tolar et al., 1997; Moulder et al., 1999). Exposure of hippocampal cultures to 2 μm apoE peptide resulted in a time-dependent decrease in neuronal survival such that 20, 50, and 80% of the neurons were degenerated at 6, 12, and 24 hr after exposure to the peptide (Fig.2A). Neuronal degeneration was induced in a concentration-dependent manner by the peptide at concentrations ranging from 0.05 to 2.5 μm (Fig. 2B). Neurons exposed for up to 24 hr to different concentrations of the peptide exhibited progressive degeneration (Fig. 2C). The degeneration of neurons started with both swelling of the cell bodies (Fig.2C, arrows) and beading of the neurites (Fig.2C, A2, arrowheads), followed by neurite fragmentation (Fig. 2C, A3, arrowheads) and cell lysis.
Fig. 2.
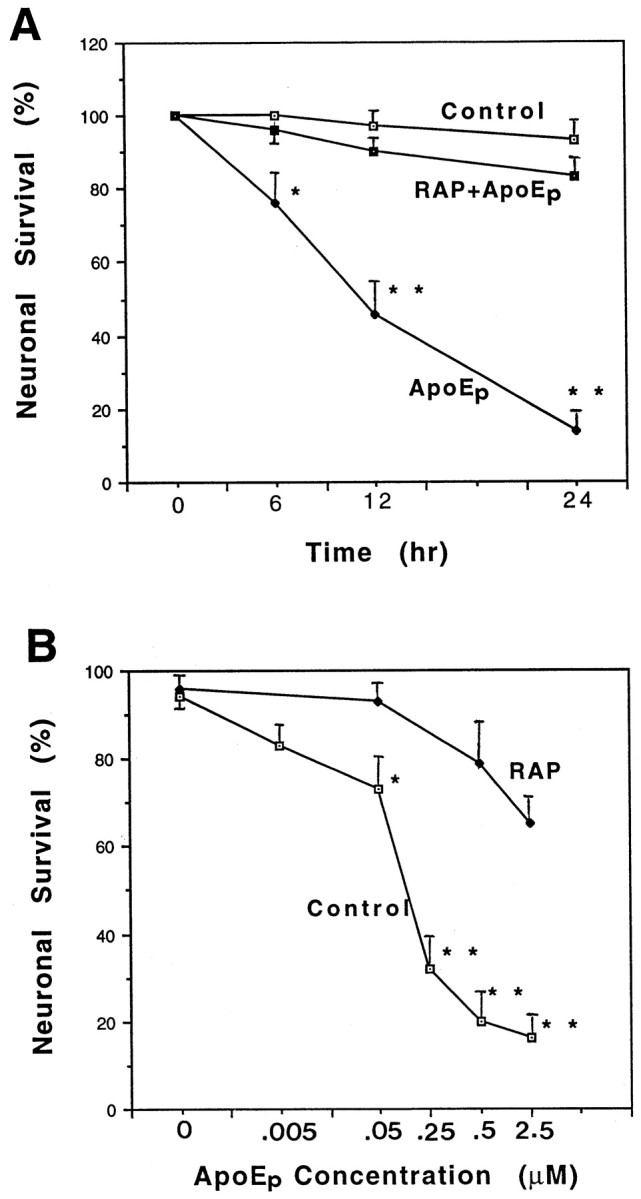
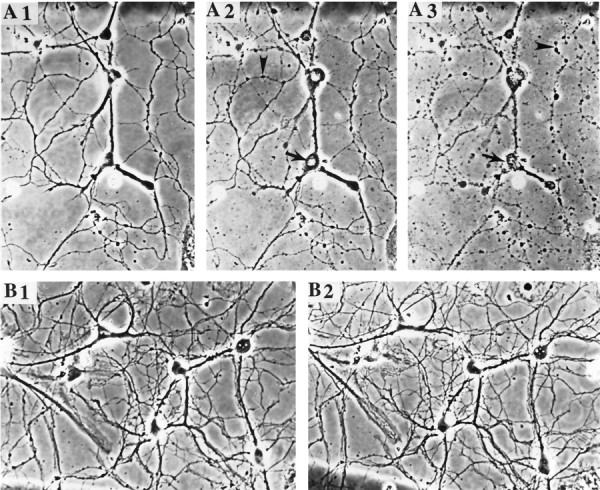
ApoE peptide is neurotoxic in hippocampal cell cultures. A, Cultures were exposed for the indicated times to saline (Control), 2 μmapoE peptide (ApoEp), or 4 μm RAP plus 2 μm apoE peptide (RAP+ApoEp). The percentage of neurons surviving at each time point was quantified; values are the mean and SEM of determinations made in four separate cultures. *p < 0.05, **p < 0.01 compared with corresponding values in control and RAP + apoE cultures (ANOVA with Scheffe’s post hoc test). B, Cultures were exposed for 24 hr to the indicated concentrations of apoE peptide (ApoEp) in the absence (Control) or presence of 4 μmRAP. The percentage of neurons was quantified, and values are the mean and SEM of determinations made in four separate cultures. *p < 0.05, **p < 0.01 compared with corresponding values in RAP-treated cultures (ANOVA with Scheffe’s post hoc test). C, Phase-contrast micrographs of hippocampal neurons in a culture before treatment (A1), and 6 hr (A2) and 24 hr (A3) after exposure to 0.5 μm apoE peptide. B1 and B2 are micrographs of neurons in a culture before and 24 hr after exposure to saline, respectively. Note the degeneration of neuronal cell bodies (arrow) and neurites (arrowhead) in the culture exposed to the apoE peptide. (Figure 2continues.)
To determine whether the apoE peptide-mediated neurotoxicity might be mediated by receptors for apoE, the toxicity of the peptide was tested in the presence of recombinant human RAP, a protein that blocks the interaction of all known ligands for LRP. Hippocampal neurons were preincubated with RAP for 1 hr followed by exposure to peptide E(141–149)2. RAP (4 μm) significantly protected the neurons against toxicity of the peptide (Fig. 2A,B).
Although the quantity of truncated apoE4 that was available for these experiments was more limited, similar neurotoxicity was obtained after exposure of hippocampal neurons to increasing concentrations of this protein (Fig. 3A). When added at 75 or 125 μg/ml (∼3 or 6 μm), hippocampal neuronal survival was reduced by 30 and 60%, respectively.
Fig. 3.
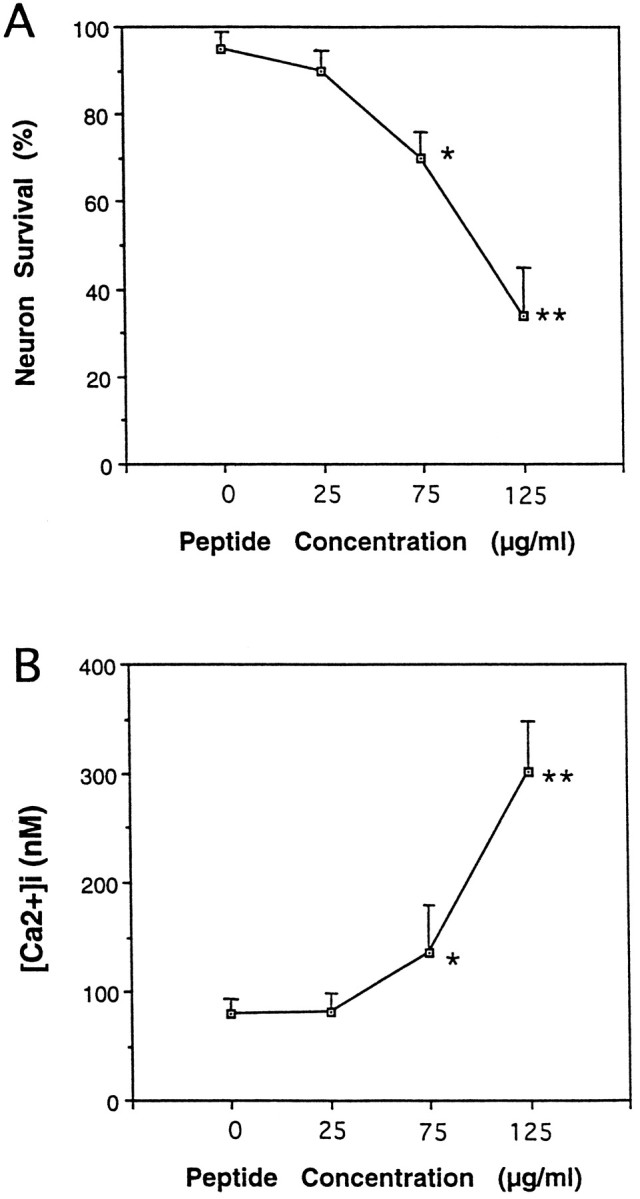
Truncated apoE4 is neurotoxic and increases [Ca2+]i in cultured rat hippocampal neurons. A, Cultures were exposed to the indicated concentrations of truncated apoE4 for 24 hr, and neuronal survival was quantified. Values are the mean and SEM of determinations made in three separate cultures. B, Cultures were exposed to the indicated concentrations of truncated apoE4 for 2 hr, and the [Ca2+]i was measured in 12–17 neurons/culture. Values are the mean and SEM of determinations made in three separate cultures.
ApoE peptide and truncated apoE4 also stimulate an influx of Ca2+
The similarity of the neurite-degenerative effects of the apoE peptide and truncated apoE4 to the neurotoxic effects of glutamate suggested that calcium influx may also occur. Fluorescence ratio imaging of the calcium indicator dye fura-2 AM was used to assess changes in [Ca2+]iin neuronal cell bodies and neurites. A significant increase in intracellular calcium was observed in hippocampal neurons 2 hr after exposure to either 75 or 125 μg/ml of truncated apoE4 (Fig.3B). Additional studies of this effect on intracellular calcium were performed with the apoE peptide. Treatment of rat hippocampal or chick sympathetic neurons with the apoE peptide resulted in a rapid and sustained increase in [Ca2+]i (Figs.4,5A). The magnitude of the rise in [Ca2+]i was dependent on the concentration of the peptide. Lower concentrations resulted in less of an increase of [Ca2+]i (data not shown). The magnitude and onset of the increase in calcium was similar in both types of neurons. On average, a twofold increase in calcium within 1–2 min after addition of the peptide was observed; however, not all cells showed the same response. Some exhibited only minor increases, whereas others showed much larger (four- to fivefold) increases.
Fig. 4.

ApoE peptide induces a rapid and sustained elevation of [Ca2+]i in chick sympathetic neurons that is inhibited by MK-801. Pretreatment of chick sympathetic neurons with 200 μm MK-801 for 30 min attenuates the peptide-induced increase in [Ca2+]i. MK-801 pretreatment also resulted in an increase in the resting [Ca2+]i. The [Ca2+]i was measured by imaging the fluorescent calcium indicator dye fura-2 AM in neurons before and after exposure to 8 μm of the peptide at 80 sec (AEP, arrow). Values are the mean of determinations made in 70–80 cells in 10–13 separate cultures in either Neurobasal or F-12 medium.
Fig. 5.


ApoE peptide induces rapid and sustained elevations of [Ca2+]i in rat hippocampal neurons. The [Ca2+]i was monitored by fluorescence ratio imaging of the calcium indicator dye fura-2 AM in neuronal somata before and after exposure to 2 μm apoE peptide (arrow denotes time of addition of the apoE peptide) in the absence (A) or presence (B) of 1 μm RAP. Values represent mean [Ca2+]i in 10–15 neurons and are representative of results obtained in at least six separate experiments. C, Images of intracellular free calcium levels in hippocampal neurons obtained by imaging of the calcium indicator dye fura-2 AM. The [Ca2+]i is represented on acolor scale with values in nanomoles as indicated on the scale bar. Images were taken immediately before treatment and 1.5, 3, and 10 min after exposure to 2 μm apoE peptide. Note the rapid and sustained elevation of [Ca2+]i in neuronal somata and in neurites, as well as swelling of the neuronal cell bodies and fragmentation of neurites (arrowheads). (Figure 5continues.)
More detailed studies of the calcium response were performed with cultured rat hippocampal neurons, where the regulation of calcium has been studied extensively . An increase in [Ca2+]i was also found within the neurites and was accompanied by profound changes in neuronal morphology. Neurite beading and fragmentation appeared within 10 min of exposure to the peptide (Fig. 5C, arrowheads) and was accompanied by swelling of the cell body.
ApoE peptide-induced elevation of [Ca2+]i is caused primarily by extracellular calcium and attenuated by RAP and MK-801
The contribution of extracellular calcium to the intracellular increase elicited by the apoE peptide was examined by eliminating extracellular calcium through the use of calcium-free medium and chelators. The [Ca2+]i rise was significantly decreased and of shorter duration when cells were incubated in Locke’s medium lacking Ca2+and containing 1 mm EGTA (Fig.6B), suggesting that extracellular sources of free calcium participate in calcium elevation. Because incubation of the cells in calcium-free medium did not completely eliminate the rise in [Ca2+]i, some release of calcium from intracellular stores after exposure to the neurotoxic peptide was also suggested. This conclusion was also supported by the fact that depletion of intracellular calcium stores with thapsigargin attenuated the peak [Ca2+]i rise (data not shown).
Fig. 6.
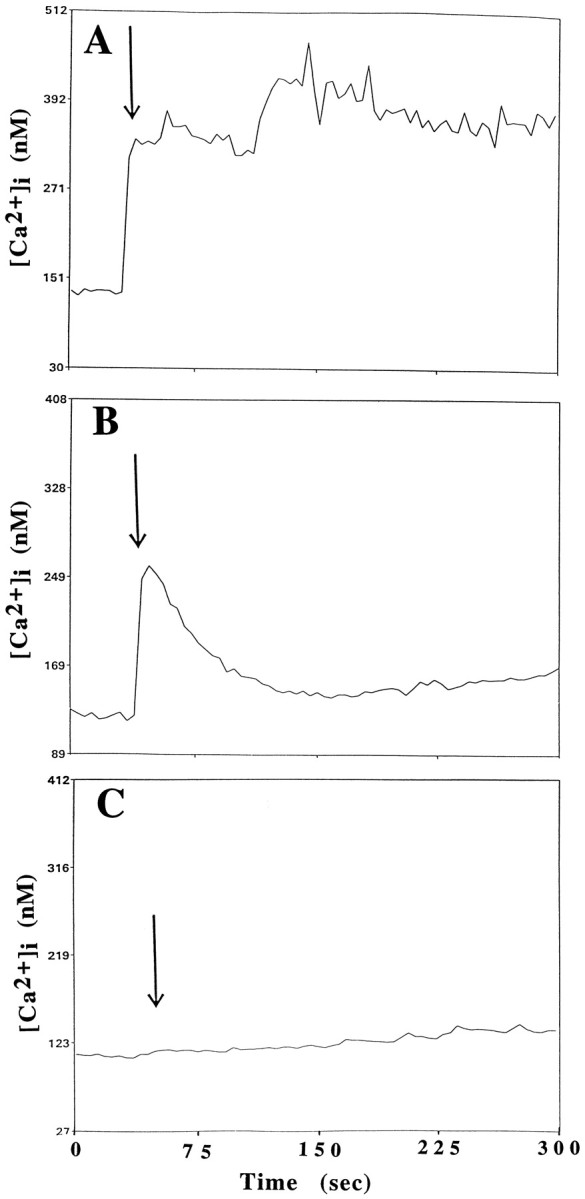
ApoE-induced elevation of [Ca2+]i involves calcium release from intracellular stores and is attenuated by MK-801. The [Ca2+]i was monitored before and after exposure of rat hippocampal neurons to 2 μm apoE peptide (added at the time point indicated by arrow) in a control culture (A), a culture incubated in medium lacking Ca2+ and containing 0.5 mm EGTA (B), and a culture pretreated with 200 μm MK-801 (C). Values represent the mean [Ca2+]i in at least 10 neurons and are representative of results obtained in at least six separate experiments.
In previous studies, RAP, a ligand for LRP and related receptors, was found to attenuate the toxicity of the apoE peptide (Tolar et al., 1997). The extent to which RAP might also affect the calcium response was examined by incubating hippocampal neuronal cultures with 1 μm human recombinant RAP before addition of the peptide. As shown in Figure 6C, human recombinant RAP was able to significantly attenuate the increase in calcium caused by the apoE peptide.
In hippocampal neurons, the influx of extracellular calcium is tightly regulated by ligand-gated and voltage-dependent calcium channels. For example, glutamate, which exerts neurotoxic effects when applied to these neurons, causes an increase in intracellular calcium that is mediated by NMDA-type receptors (Mattson et al., 1993b; Cheng et al., 1995). Because the apoE peptide also causes neurotoxicity and an influx of calcium, we tested specific blockers of calcium channels to investigate their potential contribution to the calcium response elicited by the apoE peptide. A 30 min preincubation with MK-801, a noncompetitive NMDA receptor antagonist, prevented the apoE peptide-induced influx of calcium in both chick sympathetic and rat hippocampal neurons (Figs. 4, 6C). In contrast, a 5 min preincubation of hippocampal neurons with 200 μm of CNQX, an antagonist of the AMPA/kainate subclass of glutamate receptors, was ineffective (data not shown). Nifedipine (100 μm), a specific blocker of voltage-sensitive calcium channels, also did not provide any protection against the rise in intracellular calcium in hippocampal neurons (data not shown).
Contribution of calcium influx and MK-801 sites to the neurotoxic action of the apoE peptide
Because NMDA receptor activation and subsequent calcium influx have been shown to precede the death of CNS neurons in culture (Ankarcrona et al., 1995; DeLorenzo and Limbrick, 1996; Orrenius and Ankarcrona, 1996), the contribution of NMDA receptor activation and calcium influx to the neurotoxic action of the apoE peptide was tested. Rat hippocampal cultures were exposed for 24 hr to 2 μmapoE peptide in the presence and absence of extracellular calcium. The percentage of neurons surviving in each treatment group was quantified (Fig. 7). The absence of extracellular calcium significantly, but only partially, enhanced neuronal survival after exposure to the apoE peptide. Also, the effect of 200 μm MK-801 (MK8) on the toxicity of 2 μmapoE peptide was tested. MK-801 was found to significantly, although not completely, prevent the toxicity of the apoE peptide, suggesting a role for NMDA-type glutamate receptors.
Fig. 7.

The neurotoxic action of the apoE peptide is attenuated by MK-801 or removal of extracellular calcium. Hippocampal cultures were exposed for 24 hr to saline (Cont), 2 μm apoE peptide (ApoEp), 200 μm MK-801 (MK8) plus 2 μm apoE peptide, or medium lacking calcium plus 2 μm apoE peptide. The percentage of neurons surviving was quantified, and values are the mean and SEM of determinations made in four separate cultures. *p < 0.05 compared with values for cultures exposed to apoE peptide alone. **p < 0.01 compared with values in control cultures and cultures exposed to MK-801, and MK-801 + apoE peptide (ANOVA with Scheffe’s post hoc test).
Experiments were also performed to determine whether MK-801 would protect chick sympathetic neurons from the toxic effects of the peptide. Cultures were treated with the peptide in the presence or absence of MK-801 (ranging from 100 to 800 μm), and the extent of survival was assessed at 4 or 20 hr after treatment. There was no protection provided by MK-801 at either time point or at any concentration (data not shown). To determine whether these neurons are sensitive to glutamate, additional cultures were treated with 100 μm glutamate. No toxicity was observed (data not shown).
DISCUSSION
Proteolysis may play a role in apoE neurotoxicity
As found previously (Crutcher et al., 1994; Marques et al., 1996,1997), a synthetic apoE-related peptide, the N-terminal portion of apoE4 (truncated apoE4), and full-length apoE4 exhibited neurotoxic activity. Protease inhibitors reduced both the toxicity of full-length apoE4 and the appearance of truncated apoE fragments, suggesting that proteolysis of apoE may mediate its toxic effects. If so, this might help explain the variability that exists in the literature. In some studies, apoE causes neurotoxic effects (Marques et al., 1997;Jordán et al., 1998; DeMattos et al., 1999), but in other studies, no toxicity has been found (Bellosta et al., 1995; Nathan et al., 1995; DeMattos et al., 1998). In one study, cytosolic expression of apoE in neuroblastoma cells was found to be toxic (DeMattos et al., 1999), but no information was provided on whether apoE was degraded under these conditions. The present results suggest that conditions may be required in which truncated apoE is generated to observe toxic effects of apoE. We have also observed that serum blocks apoE peptide toxicity (M. Marques and K. Crutcher, unpublished observations), indicating that composition of the medium may be critical.
The contribution of apoE proteolysis to neurotoxicity may also have some bearing on understanding the role of apoE in vivo. Fragments of apoE are present in both the brain and CSF, and the most abundant fragment, with an apparent molecular weight of 22 kDa, likely represents the N-terminal thrombin cleavage fragment of apoE (Marques et al., 1996).
Calcium influx caused by apoE-related molecules
Both truncated apoE and the apoE peptide were found to elicit sustained elevation of [Ca2+]i in cultured rat hippocampal neurons. The apoE peptide was also found to have a similar effect on chick sympathetic neurons. Other investigators have reported effects of full-length apoE on neuronal calcium (Hartmann et al., 1994; Müller et al., 1998). The effect of the apoE peptide on [Ca2+]iis also consistent with studies in which the same peptide was found to cause an increase in [Ca2+]i that was blocked by removal of extracellular calcium but not blocked by inhibitors of voltage-gated calcium channels (Wang and Gruenstein, 1997). In contrast, we observed significant attenuation of the increase in [Ca2+]i by RAP and MK-801.
ApoE peptide effects may involve apoE receptors
Although the RAP used in our experiments provided significant protection against both the calcium influx and neurotoxicity of the apoE peptide, other groups have found RAP not to be effective in blocking the neurotoxicity of either full-length apoE (Jordán et al., 1998) or the apoE peptide (Moulder et al., 1999), nor did RAP inhibit the calcium influx elicited by the peptide on rat cortical neurons (Wang and Gruenstein, 1997). These discrepancies may be caused by different experimental conditions or the RAP preparation. For example, we have tested the RAP preparation used by Jordán et al. (1998) and Molder et al. (1999) (recombinant rat RAP, kindly provided by Dr. G. Bu, Washington University) and found that it did not protect chick sympathetic neurons under the same conditions in which recombinant human RAP was protective (tested at concentrations ranging from 0.5 to 4 μm under the same conditions used for previous toxicity studies). It is possible that there is some species-specific difference in the RAP or that other biochemical differences contribute to the differing results. Wang and Gruenstein (1997) did not provide details on their RAP experiments, but in surprising contrast to their results with RAP, they reported that an anti-LRP antibody virtually abolished the calcium response to the apoE peptide, consistent with results demonstrating inhibition of toxicity with the same antibody (Tolar et al., 1997).
ApoE binding and internalization, which can be mediated by several structurally related cell surface receptors of the LDL receptor family, including the LDL receptor and LRP (Yamamoto et al., 1984; Herz et al., 1988; Beisiegel et al., 1989; Raychowdhury et al., 1989; Takahasi et al., 1992; Kim et al., 1996; Novak et al., 1996), are not thought to involve signaling pathways. Nevertheless, findings from studies of non-neuronal cells suggest that exposure of lipoproteins to various cells can stimulate second messenger pathways with alterations in intracellular calcium (Haller et al., 1994; Tasaki et al., 1994; Hjalm et al., 1996). More recently, LRP has been found to interact with a heterotrimeric G-protein with downstream activation of cAMP-dependent protein kinase (Goretzki and Mueller, 1998).
Is LRP involved in the toxic effects of the apoE peptide?
LRP, also known as the α 2-macroglobulin (α2 m)receptor, in conjunction with heparan sulfate proteoglycan may participate in the toxicity of the apoE peptide (Tolar et al., 1997). The role of this LDL-related receptor in cell function is enigmatic. It has been demonstrated to mediate binding and internalization of various ligands, including apoE (Strickland et al., 1990). Binding of apoE to its receptor is usually assumed to require lipid, but the present experiments were carried out without exogenously added lipid. Interestingly, a recent study reported that lipid-free apoE can be catabolized via LRP (Yu et al., 1998). If so, it is likely that both truncated apoE and the apoE peptide could be taken up in the same manner. Also of potential relevance is the fact that LRP can mediate the uptake and internalization of various cytotoxic compounds, including Pseudomonas exotoxin (Mucci et al., 1995; Gu et al., 1996), certain proteases (Maeda et al., 1987, 1989), and ribosome-inactivating proteins such as saporin (Cavallaro et al., 1995).
The fact that mutations in both LRP (Kang et al., 1997; Hollenbach et al., 1998; Lambert et al., 1998) and α2 m (Blacker et al., 1998) genes have been linked to the risk of AD, along with the well established association with amyloid precursor protein (APP) mutations, draws attention to this pathway in disease pathogenesis. The hypothetical scenario is commonly cast in the light of the role that the amyloid β peptide plays. However, to the extent that apoE may be a source of neurotoxicity, an alternative possibility implicates modifications of the interactions of apoE with LRP or with LRP ligands (including APP and α2 m).
ApoE peptide effects may involve MK801-sensitive sites
The morphological manifestations of apoE peptide toxicity resemble those that occur during rapid excitotoxic degeneration triggered by glutamate and NMDA (Choi 1988; Mattson and Kater, 1989). The large increase of [Ca2+]i and cytotoxicity in hippocampal neurons may be explained by their expression of glutamate receptors, because pharmacological blockade of NMDA receptors with MK-801 prevented cell death. The AMPA/kainate receptor antagonist CNQX was ineffective, suggesting that non-NMDA glutamate receptors are not involved.
The precise site of action of the apoE peptide in these cultures is unknown. One possibility is that an LRP-like receptor triggers a second messenger cascade(s) that promotes activation of NMDA receptors through an intracellular pathway. Such a pathway might involve altered phosphorylation of NMDA receptors secondary to modulation of kinase or phosphatase activities (Lieberman and Mody, 1994; Wang and Salter, 1994; Wang et al., 1994). Alternatively, apoE peptide binding might induce membrane depolarization resulting in release of Mg2+ from the NMDA receptor channel, thereby permitting responses to glutamate (Mayer and Westbrook, 1987). It is also possible that the peptide stimulates release of glutamate from terminals.
The difficulty in identifying the precise nature of the interaction of MK-801 with the apoE peptide effect is underscored by the fact that MK-801 does not protect against the toxic effects of the peptide on chick sympathetic neurons, nor was glutamate found to exert neurotoxic effects in these cultures. Yet MK-801 does attenuate the acute effects of the apoE peptide on calcium influx, suggesting that either the effects on calcium are not directly related to subsequent toxicity or the effects of MK-801 are more complex than simply interference with NMDA receptors, or both.
A direct comparison between the neurotoxic and calcium effects of the apoE peptide is difficult to make because of the differences in experimental conditions (culture medium, time-interval, etc.). Although a correlation between the loss of calcium homeostasis and subsequent neuronal cell death has been shown in various paradigms of excitotoxic and metabolic injury (Mattson and Mark, 1996; Tymianski 1996), influx of calcium is unlikely, by itself, to account for toxicity. Depolarization of neurons with KCl, which results in calcium influx, actually protects against the toxic effects of the apoE peptide (Moulder et al., 1999; Marques and Crutcher, unpublished observations). This indicates that calcium loading per se does not predict subsequent toxicity, consistent with recent data suggesting that the route of calcium entry, not calcium load, determines neurotoxicity (Sattler et al., 1998).
ApoE, calcium, and neurodegeneration in AD
Does the activity observed in these experiments reflect a potential effect of apoE-derived molecules in neurodegeneration? If so, this would be consistent with evidence suggesting that neuronal calcium homeostasis is disrupted and that an excitotoxic mechanism of neuronal degeneration occurs in AD (Mattson et al., 1993a). The present results extend the documentation of neurotoxic effects of apoE-related peptides, indicating that proteolysis of apoE may play a role and that neurotoxicity is preceded by changes in calcium homeostasis. If these effects reflect endogenous activity of apoE or of apoE fragments, the rapid and potent increase in intracellular calcium may indicate a role for apoE in signal transduction. Perhaps more importantly, these results strengthen the possibility that the isoform-specific association of apoE with pathological processes, including Alzheimer’s disease, may indicate a negative role for this protein, rather than the absence of a positive role.
Footnotes
This work was supported by the Alzheimer’s Association (K.A.C.) and by National Institutes of Health Grants NS31410 (K.A.C.), NS29001 (M.P.M.), and NS30583 (M.P.M.). Human recombinant RAP protein was provided by Dr. J. A. K. Harmony (Department of Pharmacology and Cell Biophysics, University of Cincinnati). Escherichia coli expressing human truncated apoE were a generous gift from Dr. K. Weisgraber (Gladstone Institute), and HEK cells transfected with human apoE were kindly provided by Dr. M. J. LaDu (University of Chicago). The technical assistance of Alison Koch is gratefully acknowledged.
Correspondence should be addressed to Dr. Keith A. Crutcher, Department of Neurosurgery, University of Cincinnati, ML 515, Cincinnati, OH 45267-0515.
Dr. Tolar’s present address: Boston Medical Center, Boston University School of Medicine, C-314, Department of Neurology, 715 Albany Street, Boston, MA 02118.
REFERENCES
- 1.Ankarcrona M, Dypbukt JM, Bonfoco E, Zhivotovsky B, Orrenius S, Lipton SA, Nicotera P. Glutamate-induced neuronal death: a succession of necrosis or apoptosis depending on mitochondrial function. Neuron. 1995;15:961–973. doi: 10.1016/0896-6273(95)90186-8. [DOI] [PubMed] [Google Scholar]
- 2.Beisiegel U, Weber W, Ihrke G, Herz J, Stanley KK. The LDL-receptor-related protein, LRP, is an apolipoprotein E-binding protein. Nature. 1989;341:162–164. doi: 10.1038/341162a0. [DOI] [PubMed] [Google Scholar]
- 3.Bellosta S, Nathan BP, Orth M, Dong L-M, Mahley RW, Pitas RE. Stable expression and secretion of apolipoproteins E3 and E4 in mouse neuroblastoma cells produces differential effects on neurite outgrowth. J Biol Chem. 1995;270:27063–27071. doi: 10.1074/jbc.270.45.27063. [DOI] [PubMed] [Google Scholar]
- 4.Blacker D, Wilcox M, Laird N, Rodes L, Horvath SM, Go RCP, Perry R, Watson B, Jr, Bassett SS, McInnis MG, Albert MS, Hyman BT, Tanzi RE. Alpha-2 macroglobulin is genetically associated with Alzheimer disease. Nat Genet. 1998;19:357–360. doi: 10.1038/1243. [DOI] [PubMed] [Google Scholar]
- 5.Cavallaro U, Nykjaer A, Nielsen M, Soria MR. Alpha 2-macroglobulin receptor mediates binding and cytotoxicity of plant ribosome-inactivating proteins. Eur J Biochem. 1995;232:165–171. doi: 10.1111/j.1432-1033.1995.tb20795.x. [DOI] [PubMed] [Google Scholar]
- 6.Cheng B, Furukawa K, O’Keefe JA, Goodman Y, Kihiko M, Fabian T, Mattson MP. Basic fibroblast growth factor selectively increases AMPA-receptor subunit GluR1 protein level and differentially modulates Ca2+ responses to AMPA and NMDA in hippocampal neurons. J Neurochem. 1995;65:2525–2536. doi: 10.1046/j.1471-4159.1995.65062525.x. [DOI] [PubMed] [Google Scholar]
- 7.Choi DW. Glutamate neurotoxicity in cortical cell culture is calcium dependent. Neurosci Lett. 1985;58:293–297. doi: 10.1016/0304-3940(85)90069-2. [DOI] [PubMed] [Google Scholar]
- 8.Choi DW. Glutamate neurotoxicity and the diseases of the nervous system. Neuron. 1988;1:623–634. doi: 10.1016/0896-6273(88)90162-6. [DOI] [PubMed] [Google Scholar]
- 9.Clay MA, Anantharamaiah GM, Mistry MJ, Balasubramaniam A, Harmony JAK. Localization of a domain in apolipoprotein E with both cytostatic and cytotoxic activity. Biochemistry. 1995;34:11142–11151. doi: 10.1021/bi00035a020. [DOI] [PubMed] [Google Scholar]
- 10.Corder EH, Saunders AM, Strittmatter WJ, Schmechel DE, Gaskell PC, Small GW, Roses AD, Haines JL, Pericak-Vance MA. Gene dose of apolipoprotein E type 4 allele and the risk of Alzheimer’s disease in late onset families. Science. 1993;261:921–923. doi: 10.1126/science.8346443. [DOI] [PubMed] [Google Scholar]
- 11.Crutcher KA, Clay MA, Scott SA, Tian X, Tolar M, Harmony JAK. Neurite degeneration elicited by apolipoprotein E peptides. Exp Neurol. 1994;130:120–126. doi: 10.1006/exnr.1994.1191. [DOI] [PubMed] [Google Scholar]
- 12.Crutcher KA, Tolar M, Harmony JAK, Marques MA. A new hypothesis for the role of apolipoprotein E in Alzheimer’s disease pathology. In: Iqbal K, Winblad B, Nishimura T, Takeda M, Wisniewski HM, editors. Alzheimer’s disease: biology, diagnosis and therapeutics. Wiley; Chichester: 1997. pp. 545–554. [Google Scholar]
- 13.DeLorenzo RJ, Limbrick DD. Effects of glutamate on calcium influx and sequestration/extrusion mechanism in hippocampal neurons. Adv Neurol. 1996;71:37–46. [PubMed] [Google Scholar]
- 14.DeMattos RB, Curtiss LK, Williams DL. A minimally lipidated form of cell-derived apolipoprotein E exhibits isoform-specific stimulation of neurite outgrowth in the absence of exogenous lipids or lipoproteins. J Biol Chem. 1998;273:4206–4212. doi: 10.1074/jbc.273.7.4206. [DOI] [PubMed] [Google Scholar]
- 15.DeMattos RB, Thorngate FE, Williams DL. A test of the cytosolic apolipoprotein E hypothesis fails to detect the escape of apolipoprotein E from the endocytic pathway into the cytosol and shows that direct expression of apolipoprotein E in the cytosol is cytotoxic. J Neurosci. 1999;19:2464–2473. doi: 10.1523/JNEUROSCI.19-07-02464.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Goretzki L, Mueller BM. Low-density-lipoprotein-receptor-related protein (LRP) interacts with a GTP-binding protein. Biochem J. 1998;336:381–386. doi: 10.1042/bj3360381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gu M, Gordon VM, Fitzgerald DJ, Leppla SH. Furin regulates both the activation of pseudomonas exotoxin A and the quantity of the toxin receptor expressed on target cells. Infect Immun. 1996;64:524–527. doi: 10.1128/iai.64.2.524-527.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Haller H, Rieger M, Lindschau C, Kuhlmann M, Philipp S, Luft FC. LDL increases Ca2+ in human endothelial cells and augments thrombin-induced cell signalling. J Lab Clin Med. 1994;124:708–714. [PubMed] [Google Scholar]
- 19.Hartmann H, Eckert A, Muller WE. Apolipoprotein E and cholesterol affect neuronal calcium signalling: the possible relationship to β-amyloid neurotoxicity. Biochem Biophys Res Commun. 1994;200:1185–1192. doi: 10.1006/bbrc.1994.1576. [DOI] [PubMed] [Google Scholar]
- 20.Herz J, Hamann U, Rogne S, Myklebost O, Gausepohl H, Stanley KK. Surface location and high affinity for calcium of a 500-kd liver membrane protein closely related to the LDL-receptor suggest a physiological role as lipoprotein receptor. EMBO J. 1988;7:4119–4127. doi: 10.1002/j.1460-2075.1988.tb03306.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Herz J, Goldstein JL, Strickland DK, Ho YK, Brown MS. 39-kDa protein modulates binding of ligands to low density lipoprotein receptor-related protein/α2-macroglobulin receptor. J Biol Chem. 1991;266:1–7. [PubMed] [Google Scholar]
- 22.Hjalm G, Murray E, Crumley G, Harazim W, Lundgren S, Onyango I, Ek B, Larsson M, Juhlin C, Hellman P. Cloning and sequencing of human gp330, a Ca2+-binding receptor with potential intracellular signaling properties. Eur J Biochem. 1996;239:132–137. doi: 10.1111/j.1432-1033.1996.0132u.x. [DOI] [PubMed] [Google Scholar]
- 23.Hollenbach E, Ackermann S, Hyman B, Rebeck GW. Confirmation of an association between a polymorphism in exon 3 of the low-density lipoprotein receptor-related protein gene and Alzheimer’s disease. Neurology. 1998;50:1905–1907. doi: 10.1212/wnl.50.6.1905. [DOI] [PubMed] [Google Scholar]
- 24.Innerarity TL, Friedlander EJ, Rall SJ, Weisgraber KH, Mahley RW. The receptor-binding domain of human apolipoprotein E. Binding of apolipoprotein E fragments. J Biol Chem. 1983;258:12341–12347. [PubMed] [Google Scholar]
- 25.Jancso G, Karcsu S, Kiraly E, Szebeni A, Toth L, Bacsy E, Joo F, Parducz A. Neurotoxin induced nerve cell degeneration: possible involvement of calcium. Brain Res. 1984;295:211–216. doi: 10.1016/0006-8993(84)90969-7. [DOI] [PubMed] [Google Scholar]
- 26.Jordán J, Galindo MF, Miller RJ, Reardon CA, Getz GS, LaDu MJ. Isoform-specific effect of apolipoprotein E on cell survival and β-amyloid-induced toxicity in rat hippocampal pyramidal neuronal cultures. J Neurosci. 1998;18:195–204. doi: 10.1523/JNEUROSCI.18-01-00195.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kang DE, Saitoh T, Chen X, Xia Y, Masliah E, Hansen LA, Thomas RG, Thal LJ, Katzman R. Genetic association of the low-density lipoprotein receptor-related protein gene (LRP), an apolipoprotein E receptor, with late-onset Alzheimer’s disease. Neurology. 1997;49:56–61. doi: 10.1212/wnl.49.1.56. [DOI] [PubMed] [Google Scholar]
- 28.Kim D-H, Iijima H, Goto K, Sakai J, Ishii H, Kim H-J, Suzuki H, Kondo H, Saeki S, Yamamoto T. Human apolipoprotein E receptor 2. J Biol Chem. 1996;271:8373–8380. doi: 10.1074/jbc.271.14.8373. [DOI] [PubMed] [Google Scholar]
- 29.Lalazar A, Weisgraber KH, Rall SC, Jr, Giladi H, Innerarity TL, Levanon AZ, Boyles JK, Amit B, Gorecki M, Mahley RW, Vogel T. Site-specific mutagenesis of human apolipoprotein E: receptor binding activity of variants with single amino acid substitutions. J Biol Chem. 1988;263:3542–3545. [PubMed] [Google Scholar]
- 30.Lambert J-C, Wavrant-De Vrieze F, Amouyel P, Chartier-Harlin M-C. Association at LRP gene locus with sporadic late-onset Alzheimer’s disease. Lancet. 1998;351:1787–1788. doi: 10.1016/s0140-6736(05)78749-3. [DOI] [PubMed] [Google Scholar]
- 31.Laskowitz DT, Roses AD. Apolipoprotein E: an expanding role in the neurobiology of disease. Alzheimer’s Reports. 1998;1:5–12. [Google Scholar]
- 32.Lieberman DN, Mody I. Regulation of NMDA channel function by endogenous Ca2+-dependent phosphatase. Nature. 1994;369:235–239. doi: 10.1038/369235a0. [DOI] [PubMed] [Google Scholar]
- 33.Lucas DR, Newhouse JP. The toxic effects of sodium L-glutamate on the inner layers of the retina. Arch Ophthalmol. 1957;58:193–201. doi: 10.1001/archopht.1957.00940010205006. [DOI] [PubMed] [Google Scholar]
- 34.Maeda H, Molla A, Oda T, Katsuki T. Internalization of serratial protease into cells as an enzyme-inhibitor complex with alpha 2-macroglobulin and regeneration of protease activity and cytotoxicity. J Biol Chem. 1987;262:10946–10950. [PubMed] [Google Scholar]
- 35.Maeda H, Molla A, Sakamoto K, Murakami A, Matsumura Y. Cytotoxicity of bacterial proteases in various tumor cells mediated through alpha 2-macroglobulin receptor. Cancer Res. 1989;49:660–664. [PubMed] [Google Scholar]
- 36.Mahley RW. Apolipoprotein E: cholesterol transport protein with expanding role in cell biology. Science. 1988;240:622–630. doi: 10.1126/science.3283935. [DOI] [PubMed] [Google Scholar]
- 37.Mark RJ, Hensley K, Butterfield DA, Mattson MP. Amyloid beta-peptide impairs ion-motive ATPase activities: evidence for a role in loss of neuronal Ca2+ homeostasis and cell death. J Neurosci. 1995;15:6239–6249. doi: 10.1523/JNEUROSCI.15-09-06239.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Marques MA, Tolar M, Harmony JAK, Crutcher KA. A thrombin cleavage fragment of apolipoprotein E exhibits isoform-specific neurotoxicity. NeuroReport. 1996;7:2529–2532. doi: 10.1097/00001756-199611040-00025. [DOI] [PubMed] [Google Scholar]
- 39.Marques MA, Tolar M, Crutcher KA. Apolipoprotein E exhibits isoform-specific neurotoxicity. Alzheimer’s Res. 1997;3:1–6. doi: 10.1097/00001756-199611040-00025. [DOI] [PubMed] [Google Scholar]
- 40.Mattson MP, Kater SB. Excitatory and inhibitory neurotransmitters in the generation and degeneration of hippocampal neuroarchitecture. Brain Res. 1989;478:337–348. doi: 10.1016/0006-8993(89)91514-x. [DOI] [PubMed] [Google Scholar]
- 41.Mattson MP, Mark RJ. Excitotoxicity and excitoprotection in vitro. Adv Neurol. 1996;71:1–30. [PubMed] [Google Scholar]
- 42.Mattson MP, Murrain M, Guthrie PB, Kater SB. Fibroblast growth factor and glutamate: opposing roles in the generation and degeneration of hippocampal cytoarchitecture. J Neurosci. 1989;9:3728–3740. doi: 10.1523/JNEUROSCI.09-11-03728.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Mattson MP, Barger SW, Cheng B, Lieberburg I, Smith-Swintosky VL, Rydel RE. β-amyloid precursor protein metabolites and loss of neuronal Ca2+ homeostasis in Alzheimer’s disease. Trends Neurosci. 1993a;16:409–14. doi: 10.1016/0166-2236(93)90009-b. [DOI] [PubMed] [Google Scholar]
- 44.Mattson MP, Kumar K, Cheng B, Wang H, Michaelis EK. Basic FGF regulates the expression of a functional 71 kDa NMDA receptor protein that mediates calcium influx and neurotoxicity in cultured hippocampal neurons. J Neurosci. 1993b;13:4575–4588. doi: 10.1523/JNEUROSCI.13-11-04575.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Mattson MP, Kumar KN, Wang H, Cheng B, Michaelis EK. Basic FGF regulates the expression of a functional 71 kDa NMDA receptor protein that mediates calcium influx and neurotoxicity in hippocampal neurons. J Neurosci. 1993c;13:4575–4588. doi: 10.1523/JNEUROSCI.13-11-04575.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Mattson MP, Barger SW, Begley JG, Mark RJ. Calcium, free radicals, and excitotoxic neuronal death in primary cell culture. Methods Cell Biol. 1995;46:187–216. doi: 10.1016/s0091-679x(08)61930-5. [DOI] [PubMed] [Google Scholar]
- 47.Mayer ML, Westbrook GL. The physiology of excitatory amino acids in the vertebrate central nervous system. Prog Neurobiol. 1987;28:197–276. doi: 10.1016/0301-0082(87)90011-6. [DOI] [PubMed] [Google Scholar]
- 48.Moulder KL, Narita M, Chang LK, Bu G, Johnson EM. Analysis of a novel mechanism of neuronal toxicity produced by an apolipoprotein E-derived peptide. J Neurochem. 1999;72:1069–1080. doi: 10.1046/j.1471-4159.1999.0721069.x. [DOI] [PubMed] [Google Scholar]
- 49.Mucci D, Forristal J, Strickland D, Morris R, Fitzgerald D, Saelinger CB. Level of receptor-associated protein moderates cellular susceptibility to pseudomonas exotoxin A. Infect Immun. 1995;63:2912–2918. doi: 10.1128/iai.63.8.2912-2918.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Müller W, Meske V, Berlin K, Scharnagl H, März W, Ohm TG. Apolipoprotein E isoforms increase intracellular Ca2+ differentially through a ω-agatoxin IVa-sensitive Ca2+-channel. Brain Pathol. 1998;8:641–653. doi: 10.1111/j.1750-3639.1998.tb00190.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Namba Y, Tomonaga M, Kawasaki H, Otomo E, Ikeda K. Apolipoprotein E immunoreactivity in cerebral amyloid deposits and neurofibrillary tangles in Alzheimer’s disease and kuru plaque amyloid in Creutzfeld-Jacob disease. Brain Res. 1991;541:163–166. doi: 10.1016/0006-8993(91)91092-f. [DOI] [PubMed] [Google Scholar]
- 52.Nathan BP, Chang K-C, Bellosta S, Brisch E, Ge N, Mahley RW, Pitas RE. The inhibitory effect of apolipoprotein E4 on neurite outgrowth is associated with microtubule depolymerization. J Biol Chem. 1995;270:19791–19799. doi: 10.1074/jbc.270.34.19791. [DOI] [PubMed] [Google Scholar]
- 53.Novak S, Hiesberger T, Schneider WJ, Nimpf J. A new low density lipoprotein receptor homologue with 8 ligand binding repeats in brain of chicken and mouse. J Biol Chem. 1996;271:11732–11736. doi: 10.1074/jbc.271.20.11732. [DOI] [PubMed] [Google Scholar]
- 54.Orrenius S, Ankarcona M, Nicotera P. Mechanisms of calcium-related cell death. Adv Neurol. 1996;71:137–149. [PubMed] [Google Scholar]
- 55.Raychowdhury RJ, Niles L, McCluskey RT, Smith JA. Autoimmune target in Heymann nephritis is a glycoprotein with homology to the LDL receptor. Science. 1989;244:1163–1165. doi: 10.1126/science.2786251. [DOI] [PubMed] [Google Scholar]
- 56.Sattler R, Charlton MP, Hafner M, Tymianski M. Distinct influx pathways, not calcium load, determine neuronal vulnerability to calcium neurotoxicity. J Neurochem. 1998;71:2349–2364. doi: 10.1046/j.1471-4159.1998.71062349.x. [DOI] [PubMed] [Google Scholar]
- 57.Saunders AM, Schmader K, Breitner JCS, Benson MD, Brown WT, Goldfarb L, Goldgaber D, Manwaring MG, Szymanski MH, McCown N, Dole KC, Schmechel DE, Strittmatter WJ, Pericak-Vance MA, Roses AD. Apolipoprotein E ε4 allele distributions in late-onset Alzheimer’s disease and in other amyloid-forming diseases. Lancet. 1993a;342:710–711. doi: 10.1016/0140-6736(93)91709-u. [DOI] [PubMed] [Google Scholar]
- 58.Saunders AM, Strittmatter WJ, Schmechel D, George-Hyslop PH, Pericak-Vance MA, Joo SH, Rosi BL, Gusella JF, Crapper-McLachlin DR, Alberts MJ, Hulette C, Crain B, Goldgaber D, Roses AD. Association of apolipoprotein E allele ε4 with late-onset familial and sporadic Alzheimer’s disease. Neurology. 1993b;43:1467–1472. doi: 10.1212/wnl.43.8.1467. [DOI] [PubMed] [Google Scholar]
- 59.Strickland DK, Ashcom JD, Williams S, Burgess WH, Migliorini M, Argraves WS. Sequence identity between α2-macroglobulin receptor and low density lipoprotein receptor-related protein suggests that this molecule is a multifunctional receptor. J Biol Chem. 1990;265:17401–17404. [PubMed] [Google Scholar]
- 60.Strickland DK, Ashcom JD, Williams S, Battey F, Behre E, McTigue K, Battey JF, Argraves WS. Primary structure of α2-macroglobulin receptor-associated protein. J Biol Chem. 1991;266:13364–13369. [PubMed] [Google Scholar]
- 61.Strittmatter WJ, Saunders AM, Schmechel D, Pericak-Vance M, Enghild J, Salvesen GS, Roses AD. Apolipoprotein E: high avidity binding to β-amyloid and increased frequency of type-4 allele in late-onset familial Alzheimer disease. Proc Natl Acad Sci USA. 1993;90:1977–1981. doi: 10.1073/pnas.90.5.1977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Takahasi S, Kawarabayasi Y, Nakai T, Sakai J, Yamamoto T. Rabbit very low density lipoprotein receptor: a low density lipoprotein receptor-like protein with distinct ligand specificity. Proc Natl Acad Sci USA. 1992;89:9252–9256. doi: 10.1073/pnas.89.19.9252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Tasaki H, Yamashita K, Nakashima Y, Kuroiwa A, Tulenko TN. Increase in intracellular calcium ion in smooth muscle cells induced by low-density lipoprotein. Gerontology. 1994;40:23–28. doi: 10.1159/000213624. [DOI] [PubMed] [Google Scholar]
- 64.Tolar M, Marques MA, Harmony JAK, Crutcher KA. Neurotoxicity of the 22 kDa thrombin cleavage fragment of apolipoprotein E and related synthetic peptides is receptor-mediated. J Neurosci. 1997;17:5678–5686. doi: 10.1523/JNEUROSCI.17-15-05678.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Tymianski M. Cytosolic calcium concentrations and cell death in vitro. Adv Neurol. 1996;71:85–105. [PubMed] [Google Scholar]
- 66.Wang LY, Orser BA, Brautigan DL, MacDonald JF. Regulation of NMDA receptors in cultured hippocampal neurons by protein phosphatases 1 and 2A. Nature. 1994;369:230–232. doi: 10.1038/369230a0. [DOI] [PubMed] [Google Scholar]
- 67.Wang X-S, Gruenstein E. Rapid elevation of neuronal cytoplasmic calcium by apolipoprotein E peptide. J Cell Physiol. 1997;173:73–83. doi: 10.1002/(SICI)1097-4652(199710)173:1<73::AID-JCP9>3.0.CO;2-G. [DOI] [PubMed] [Google Scholar]
- 68.Wang YT, Salter MW. Regulation of NMDA receptors by tyrosine kinases and phosphatases. Nature. 1994;369:233–235. doi: 10.1038/369233a0. [DOI] [PubMed] [Google Scholar]
- 69.Weisgraber KH. Apolipoprotein E: structure-function relationships. Adv Prot Chem. 1994;45:249–302. doi: 10.1016/s0065-3233(08)60642-7. [DOI] [PubMed] [Google Scholar]
- 70.Weisgraber KH, Innerarity TL, Harder KJ, Mahley RW, Milne RW, Marcel YL, Sparrow JT. The receptor-binding domain of human apolipoprotein E: monoclonal antibody inhibition of binding. J Biol Chem. 1983;258:12348–12354. [PubMed] [Google Scholar]
- 71.Weisgraber KH, Rall SC, Mahley RW, Milne RW, Marcel YL, Sparrow JT. Human apolipoprotein E: determination of the heparin binding sites of apolipoprotein E. J Biol Chem. 1986;261:2068–2076. [PubMed] [Google Scholar]
- 72.Williams SE, Ashcom JD, Argraves WS, Strickland DK. A novel mechanism for controlling the activity of α2-macroglobulin receptor/low density lipoprotein receptor-related protein. J Biol Chem. 1992;267:9035–9040. [PubMed] [Google Scholar]
- 73.Yamamoto T, Davis GC, Brown MS, Schneider WJ, Casey ML, Goldstein JL, Russel DW. The human LDL receptor: a cysteine-rich protein with multiple Alu sequences in its mRNA. Cell. 1984;39:27–38. doi: 10.1016/0092-8674(84)90188-0. [DOI] [PubMed] [Google Scholar]
- 74.Yu L, Narita M, Bu G, Schwartz A, Holtzman D. Lipid free apoE3 and apoE4 are differentially degraded via cellular lrp. Soc Neurosci Abstr. 1998;24:1712. [Google Scholar]