

Abstract
Forced overexpression of wild-type Alzheimer amyloid precursor protein (APP) causes postmitotic neurons to degenerate. Caspase-3 (CPP32) is a principal cell death protease involved in neuronal apoptosis during physiological development and under pathological conditions. Here, we investigated whether APP overexpression activates caspase-3 in human postmitotic neurons using adenovirus-mediated gene transfer. When a recombinant adenovirus vector expressing human wild-type APP695 was infected in vitro into neurally differentiated embryonal carcinoma NT2 cells, only postmitotic neurons underwent severe degeneration. Before neurodegeneration, full-length APP- and Aβ-immunoreactive peptides were accumulated in infected neurons, and caspase-3-like protease activity was markedly elevated. Western blot analysis revealed that activated caspase-3 subunits were generated in APP-accumulating neurons. Such neuronal caspase-3 activation was undetectable in NT2 neurons infected with β-galactosidase-expressing adenovirus. Addition of the caspase-3 inhibitor acetyl-Asp-Glu-Val-Asp-aldehyde to the culture medium significantly reduced the severity of degeneration exhibited by APP-overexpressing neurons. Immunocytochemical analyses revealed that some APP-accumulating neurons contained activated caspase-3 subunits and exhibited the characteristics of apoptosis, such as chromatin condensation and DNA fragmentation. Activation of caspase-3 was also observed in vivo in rat hippocampal neurons infected with the APP-expressing adenovirus. These results suggest that wild-type APP is an intrinsic activator of caspase-3-mediated death machinery in postmitotic neurons.
Keywords: amyloid precursor protein, caspase-3, apoptosis, adenovirus vector, postmitotic neurons, Alzheimer’s disease
Alzheimer’s disease (AD) is a neurodegenerative disease characterized by massive amounts of neuronal death accompanied by extracellular deposition of amyloid fibrils. The principal component of the amyloid fibrils is Aβ, which is generated by aberrant processing of the amyloid precursor protein (APP). APP and Aβ have been regarded as key molecules involved in the pathogenesis of this disease (Selkoe, 1994; Yankner, 1996). Although detailed mechanisms underlying neuronal death seen in the brain affected by AD remain unknown, earlier studies have suggested that Aβ peptides and APP mutants induce neurodegeneration characteristic of apoptosis (Loo et al., 1993; Moechars et al., 1996; Yamatsuji et al., 1996; Zhao et al., 1997). However, little is known about neuropathological roles of wild-type APP, which is expressed in the brain of all AD patients, except familial AD cases that bear mutant APP genes.
We have reported previously that overexpression of wild-type APP induces degeneration in vitro of postmitotic neurons derived from embryonal carcinoma P19 cells (Yoshikawa et al., 1992). Recently, we found that adenovirus-mediated overexpression of wild-type APP695 in the rat hippocampus in vivo causes severe neurodegeneration and that some APP-accumulating neurons show apoptosis-like features, such as severe membrane blebbing and DNA fragmentation (Nishimura et al., 1998). More recently, gene transfer experiments using a herpes simplex virus vector have demonstrated that overexpression of wild-type APP in primary cortical neurons showed a significant increase in the number of apoptotic cells and an increase in DNA fragmentation (Bursztajn et al., 1998). These findings raise the possibility that overexpression of wild-type APP induces degeneration of postmitotic neurons by apoptotic pathway.
Apoptosis is a type of cell death that requires specialized cellular machinery, including a family of cysteine proteases termed caspases (for review, see Thornberry and Lazebnik, 1998). Among the identified caspases, caspase-3 (CPP32) (Nicholson et al., 1995) is a potent effector of neuronal death during nervous system development and under certain pathological conditions. For example, caspases have a regulatory role in programmed death of chick spinal motoneurons (Milligan et al., 1995). Recently, the APP gene has been identified as one of the upregulated genes in dying motoneurons deprived of trophic support, and APP serves as a substrate for caspase-3 (Barnes et al., 1998), suggesting a link between APP and caspase-3 in this type of neuronal death. In cerebral neurons in vivo, ischemia causes both caspase-3 activation (Chen et al., 1998; Namura et al., 1998) and elevation of endogenous APP levels (Stephenson et al., 1992; Saido et al., 1994). These findings prompted us to investigate the effects of overexpression of wild-type APP on neuronal caspase-3. Here, we demonstrate, using an adenovirus vector and human postmitotic neurons in combination, that intracellular accumulation of wild-type APP markedly activates neuronal caspase-3 in a cleavage-dependent manner. The present findings may provide insights into the mechanisms whereby intracellular accumulation of wild-type APP induces neuronal death.
MATERIALS AND METHODS
Cosmid construction. Recombinant adenovirus expressing APP695 was constructed as described previously (Nishimura et al., 1998). Briefly, full-length cDNA of human APP695 (Kang et al., 1987; Yoshikawa et al., 1992) was blunt-ended and inserted in theSwaI site of pAxCAwt cosmid (Miyake et al., 1996). Cosmid DNA was cotransfected with the EcoT221-digested DNA-terminal protein complex of Ad5-dlX into 293 cells to generate the recombinant virus AxCAYAP by homologous recombination. Adenovirus expressing β galactosidase (AxCALacZ) was provided by Dr. I. Saito (University of Tokyo, Tokyo, Japan). The recombinant viruses were propagated in 293 cells. After the third propagation, virions were extracted from 293 cells, purified by double cesium step-gradient purification (Kanegae et al., 1994), dialyzed against a vehicle solution containing 10% glycerol in PBS, pH 7.4, and stored at −80°C. The titers of recombinant viruses were determined by the modified end-point cytopathic effect assay on 293 cells (Kanegae et al., 1994) and expressed in plaque-forming units. Positive expression of the inserted gene product was confirmed by immunohistochemical detection using COS-1 cells or NIH3T3 cells. Experiments using recombinant adenoviruses were approved by the Recombinant DNA Committee of the Osaka University and performed according to the institutional guidelines.
Adenovirus infection into NT2 cells. Human embryonal carcinoma cells of NTera2/cl.D1 (NT2) (Andrews, 1984) cell line (Stratagene, La Jolla, CA) were cultured and neurally differentiated as reported previously (Pleasure et al., 1992). NT2 cells were treated with 10 μm all-trans retinoic acid (Sigma, St. Louis, MO) for 35 d and subcultured at 3.5 × 105 cells per 35 mm dish in the medium Opti-MEM (Life Technologies, Grand Island, NY) supplemented with 10% fetal calf serum containing 1 μm cytosine arabinoside (Sigma). Neurally differentiated cells (mixed cell populations of neurons and non-neuronal cells) were exposed to AxCAYAP or AxCALacZ at 1 × 107 pfu/ml culture medium for 12 hr (multiplicity of infection 10–30) and then incubated in a virus-free fresh medium up to 120 hr. Infected cells were photographed with a phase-contrast microscope (Diaphot TMD; Nikon, Tokyo, Japan). β-Galactosidase was histochemically stained by immersion in 5 mmK3Fe(CN)6, 5 mmK4Fe(CN)6, 2 mm MgCl2, and 1 mg/ml 5-bromo-4-chloro-3-indolyl-β-galactoside in PBS at 37°C overnight (Schöler et al., 1989). Under these conditions, 50–80% of postmitotic neurons were infected as determined by infection with AxCALacZ. For cell viability analysis, cells were incubated with 10 μm ethidium homodimer (EthD-1; Molecular Probes, Eugene, OR) in PBS at room temperature for 40 min and observed with a fluorescence microscope (BX 50–34-FLAD1; Olympus Optical, Tokyo, Japan). For caspase-3 inhibitor protection analysis, AxCAYAP-infected cells were incubated for 96 hr in the absence and presence of 100 μmacetyl-Asp-Glu-Val-Asp-H(aldehyde) (Ac-DEVD-CHO) (Peptide Institute, Osaka, Japan) (Nicholson et al., 1995) in the medium. The caspase-3 inhibitor was added freshly to the medium that was replaced at 24 hr intervals.
Immunocytochemistry. Neurally differentiated NT2 cells grown on collagen-coated coverslips were infected with adenoviruses and fixed with 4% formaldehyde at 4°C for 10 min and methanol-acetone (1:1) at −20°C for 15 min. For immunodetection of APP and microtubule-associated protein 2 (MAP2), fixed cells were incubated with rabbit polyclonal antibody AC1 raised against the C-terminal 25 amino acid residues of APP (amino acids 671–695; 1:1000) (Yoshikawa et al., 1992) and mouse monoclonal anti-MAP2 antibody (1:250) (Sigma). For immunodetection of APP and activated caspase-3 subunits, fixed cells were incubated with mouse monoclonal antibody P2–1 against APP N terminus (Van Nostrand et al., 1989) (1:1000) and anti-caspase-3 antibody (anti-p20/17; 1:200) (Kouroku et al., 1998). The immunolabeled cells were incubated with fluorescein isothiocyanate (FITC)-conjugated anti-rabbit immunoglobulin (IgG) (Cappel, Aurora, OH) and rhodamine B-conjugated anti-mouse IgG and visualized by fluorescence microscopy. Chromosomal DNA was stained with 5 μm Hoechst 33342 (Sigma) for 15 min at room temperature. Immunoreactive cells were photographed with the fluorescence microscope, and images of APP and caspase-3 (p20/17) were superimposed with those of Hoechst staining using Adobe Photoshop 5.0 software (Adobe Systems, San Jose, CA). Nuclear DNA fragmentation was analyzed by terminal deoxynucleotidyl transferase-mediated dUTP-biotin nick-end labeling (TUNEL) method (Gavrieli et al., 1992) combined with immunostaining for APP with AC1. TUNEL and APP were visualized with Texas Red and FITC, respectively, by confocal laser scanning fluorescence microscopy (μRadiance; Bio-Rad, Hercules, CA).
Western blot analysis. NT2 neurons infected with adenovirus were dislodged by pipetting the medium up and down with a Pasteur pipette several times. Most of the non-neuronal cells remained attached after this treatment. Detached neurons were then collected by centrifugation at 150 × g for 5 min. Enriched neurons were lysed in PBS containing 0.5% Nonidet P-40, 0.1% SDS, and 100 μm phenylmethanesulfonyl fluoride. Proteins (5 μg/lane) were separated by 10% SDS-PAGE, transferred to polyvinylidene difluoride (PVDF) membrane (Millipore, Bedford, MA), and immunoblotted with antibodies AC1 and P2–1. For detection of Aβ peptides, total cell lysates (10 μg of protein) were separated by 16% Tris-Tricine SDS-PAGE, transferred to PVDF membrane, boiled in PBS for 5 min (Ida et al., 1996), and blotted with mouse monoclonal antibody recognizing Aβ (amino acids 17–24) (4G8; Wako, Tokyo, Japan). For Western blot analysis of caspase-3 protein, infected neurons were lysed in 10 mmTris-HCl, pH 7.5, 1 mm EDTA, and 100 mm phenylmethanesulfonyl fluoride and centrifuged at 15,000 × g for 20 min. Proteins (20 μg/lane) were separated by 12% SDS-PAGE, transferred to PVDF membrane, and blotted with rabbit polyclonal anti-caspase-3-antibody (65906E; PharMingen, San Diego, CA), which recognizes pro-caspase-3 and active subunits. The membrane was incubated with peroxidase-labeled anti-rabbit or anti-mouse IgG, and the signals were detected with chemiluminescence reagents (Renaissance; NEN, Boston, MA).
Measurement of caspase-like protease activities.Adenovirus-infected cells were collected and incubated at 37°C for 10 min in 50 mm Tris-HCl, pH7.5, 1 mm EDTA, 10 mm EGTA, and 10 μm digitonin (Kanuka et al., 1999). Lysates were centrifuged at 15,000 × g for 10 min at 4°C, and the supernatant (1 μg of protein) was used for caspase-3-like protease assay. Caspase-3-like activity was measured by cleavage of the substrate carbobenzoxy-Asp-Glu-Val-Asp-7-amino-4-trifluoromethyl coumarin (Z-DEVD-AFC) using a kit (FluorAce Apopain kit; Bio-Rad) and a spectrofluorometer (VersaFluor; Bio-Rad). Caspase-1-like enzyme activity in neuronal extracts used for caspase-3-like enzyme assay was measured using Acetyl-Tyr-Val-Ala-Asp-(4-methyl-coumaryl-7-amide) (Ac-YVAD-MCA) (Peptide Institute, Osaka, Japan) as a fluorogenic substrate. One unit was defined as the amount of enzyme required to cleave 1 pmol AFC (for caspase-3) or 7-amino-4-methyl-coumarin (for caspase-1) per 3 min incubation at 37°C (Kanuka et al., 1999).
Adenovirus injection in vivo and immunohistochemistry. Adenovirus microinjection and immunohistochemical detection were performed according to the methods descried previously (Nishimura et al., 1998). Briefly, AxCAYAP (2.4 × 107 pfu) suspended in 5 μl of 1 m mannitol solution was stereotactically injected into the dorsal hippocampus of rats, which were anesthetized with sodium pentobarbital (25 mg/kg). After 72 hr, rats were deeply anesthetized with sodium pentobarbital (50 mg/kg) and fixed by intracardiac perfusion with 200 ml of 4% paraformaldehyde in 0.1m phosphate buffer, pH 7.4. The brain tissues, including the hilus of the dentate gyrus, were removed, post-fixed with the same fixative overnight, and frozen. Cryosections were made and incubated with antibodies P2–1 and anti-p20/17 and then with rhodamine B-conjugated anti-mouse IgG and FITC-conjugated anti-rabbit IgG. APP and active caspase-3 subunits were visualized by fluorescence microscopy.
RESULTS
Degeneration of human postmitotic neurons by intracellular accumulation of wild-type APP695
We infected a recombinant adenovirus (AxCAYAP) that expresses human wild-type APP695 (Nishimura et al., 1998), an APP isoform expressed preferentially in neurons, into neurally differentiated NT2 embryonal carcinoma cells (Pleasure et al., 1992). NT2-derived postmitotic neurons, which bear scant cytoplasm and extended neurites, were easily distinguishable from large, flat non-neuronal cells by phase-contrast micrography (Fig.1A). These neurons showed intact morphology until 48 hr (Fig. 1B) and started to degenerate at 72 hr (Fig. 1C). A considerable number of neurons showed severe degeneration 96 hr after infection, whereas non-neuronal cells remained intact (Fig. 1D). On the other hand, neurons infected with a β-galactosidase-expressing adenovirus (AxCALacZ), a negative control, had intact morphologies even at 120 hr after infection (Fig. 1E,arrowheads). Many of the AxCAYAP-infected cells were dead at 96 hr because they retained ethidium homodimer, a fluorescent dye that is excluded by viable cells (Fig. 1F).
Fig. 1.

Degeneration of NT2-derived neurons infected with an APP695-expressing adenovirus. Neurally differentiated NT2 cells were infected with AxCAYAP.A–E, Phase-contrast micrographs at 0 (A), 48 (B), 72 (C), and 96 (D) hr.E, β-Galactosidase staining of AxCALacZ-infected cells at 96 hr. β-Galactosidase activity was histochemically stained and observed by phase-contrast micrography. Arrowheads, Aggregations of postmitotic neurons. F, Ethidium homodimer staining of AxCAYAP-infected cells at 120 hr.Arrowheads, Positive dead cells; arrows, negative viable cells. Scale bar (in A):A–E, 200 μm; F, 100 μm.
We identified AxCAYAP-infected cells by double immunostaining for APP and MAP2, a neuronal marker (Fig.2A). APP-accumulating neurons (APP+/MAP2+) showed intense degenerative changes, such as severe membrane blebbing and complete neurite retraction, whereas APP-accumulating non-neuronal cells (APP+/MAP2−) showed no degeneration. Uninfected neurons (APP−/MAP2+), which were weakly stained for endogenous APP, were morphologically intact. We quantified degenerated neurons among APP-accumulating neurons (APP+/MAP2+) (Fig.2B). APP-accumulating neurons remained intact until 48 hr, whereas 39 and 88% of APP-accumulating neurons showed severe degenerative changes at 72 and 96 hr, respectively. Virtually all of the APP-accumulating neurons completely degenerated at 120 hr after AxCAYAP infection, and many degenerated neurons were detached from culture plates. These results indicate that only the APP-accumulating postmitotic neurons undergo degeneration.
Fig. 2.

Degeneration of AxCAYAP-infected neurons. A, Double immunostaining for APP (left) and MAP2 (right) in AxCAYAP-infected cells at 96 hr. Neurally differentiated NT2 cells were infected with AxCAYAP and fixed 96 hr later. Fixed cells were double stained with an antibody against APP-C terminus (AC1) and anti-MAP2 antibody and visualized by fluorescence microscopy.Arrowheads, APP-accumulating neurons (APP+/MAP2+);double arrowheads, APP-accumulating non-neuronal cells (APP+/MAP2−); arrows, neurons without APP accumulation (APP−/MAP2+). Scale bar, 50 μm. B, Quantification of neurodegeneration. Morphological changes of AxCAYAP-infected NT2 cells were examined at each time point by double immunostaining for APP and MAP2 as shown in A. Degenerated cells showing severe membrane blebbing and complete neurite retraction among 100 cells of each group were counted (mean ± SEM; n = 3).
Accumulations of APP and its Aβ-containing derivatives in infected neurons
To elucidate the relationship between neurodegeneration and intracellular accumulation of APP, we analyzed the levels of APP and its Aβ-containing derivatives in enriched neurons at each time point after AxCAYAP infection. Western blot analysis using an antibody against APP C terminus revealed that intense signals of ∼100 kDa APP695 were detected in AxCAYAP-infected cells at 24 hr or later (Fig.3A, left). The signals at ∼100 kDa were also detected with an antibody recognizing APP N terminus (Fig. 3A, right), suggesting that full-length APP695 is accumulated at 24–96 hr after AxCAYAP infection. On the other hand, intense signals of small 8–14.5 kDa APP-derivatives containing an Aβ epitope (amino acids 17–24) were detected at 24 hr, and smaller 6–11 kDa Aβ peptides were generated at 48 hr or later (Fig. 3B). These Aβ-immunoreactive peptides were undetectable in uninfected cells (0 hr). It is noteworthy that APP and Aβ peptides were accumulated in neurons at 24–48 hr after infection, a period in which no appreciable neurodegeneration was observed (see Fig. 2B).
Fig. 3.

Western blot analysis of APP and Aβ peptides in AxCAYAP-infected neurons. A, APP-immunoreactive proteins. Lysates were prepared from enriched neurons at indicated time points after AxCAYAP infection. Proteins (5 μg/lane) were separated by 10% SDS-PAGE and transferred to PVDF membrane. APP C terminus (APP-C, left) and N terminus (APP-N, right) were detected with antibodies AC1 and P2–1, respectively. B, Aβ-immunoreactive peptides (Aβ). Proteins in AxCAYAP-infected neurons (10 μg/lane) were separated by 16% Tris-Tricine SDS-PAGE and transferred to PVDF membrane. After the membrane was boiled in PBS for 5 min, Aβ-immunoreactive peptides were detected with an antibody against Aβ (amino acids 17–24) (4G8). Size markers (in kilodaltons) are on the left.DF, Dye front.
Extracellular Aβ peptides induce death of primary rat hippocampal neurons in vitro (Yankner et al., 1990). However, Aβ peptides in the medium of 96–120 hr cultures were undetected (<1 μm) by Western blot analysis, and synthetic Aβ (amino acids 1–40) added to the medium at 3 μm, which causes neurodegeneration of primary cultured neurons (Yankner et al., 1990), had no degenerative effects on NT2-derived neurons (data not shown). These results suggest that APP-induced neurodegeneration is not attributable to extracellular Aβ peptides secreted from the infected cells in this NT2 system.
Activation of caspase-3 in APP-accumulating neurons
We measured caspase-3-like protease activity in enriched neurons using the fluorogenic substrate Z-DEVD-AFC (Fig.4, left). In AxCAYAP-infected cultures, the activity markedly increased, reached a peak at 48 hr, and declined thereafter. Caspase-3-like activity was undetected in uninfected neurons during the incubation period. On the other hand, caspase-1-like activity was slightly elevated at 72 hr after AxCAYAP infection (Fig. 4, right). AxCALacZ infection caused no appreciable increase in caspase-3-like activity or caspase-1-like activity. The substrate used for the caspase-3 protease activity is also cleaved by several caspases other than caspase-3. Because pro-caspase-3 protein (32 kDa) is cleaved into 20, 19, or 17 kDa subunits (Fernandes-Alnemri et al., 1996), we analyzed these active subunits by Western blotting (Fig. 5). Major and minor bands corresponding to the 17 and 19 kDa subunits, respectively, were detected in enriched neurons at 48–96 hr after AxCAYAP infection (Fig. 5, left), whereas these subunits were undetectable in AxCALacZ-infected cells (Fig. 5,right). These results suggest that overexpressed APP695 induces a cleavage-dependent activation of caspase-3. Signal intensities of the activated caspase-3 subunits at 72–96 hr were greater than those expected from the data of caspase-3-like protease activity (Fig. 4A, left), suggesting the presence of endogenous inhibitor(s) (at 72–96 hr) or activator(s) (at 24–48 hr) of caspase-3 in AxCAYAP-infected neurons.
Fig. 4.

Activation of caspase-3 in AxCAYAP-infected NT2 cultures. Neurally differentiated NT2 cells were infected with AxCAYAP or AxCALacZ, and lysates of enriched neurons were prepared at indicated time points. Caspase-3-like protease activity (Caspase-3, left) and caspase-1-like protease activity (Caspase-1, right) were measured by cleavage of the fluorogenic substrates Z-DEVD-AFC and Ac-YVAD-MCA, respectively (mean ± SEM; n = 3).
Fig. 5.

Western blot analysis of caspase-3 protein. Enriched neurons infected with AxCAYAP (left) or AxCALacZ (right) were harvested and lysed at each time point indicated. Proteins (20 μg/lane) were separated by 12% SDS-PAGE and transferred to PVDF membrane. Caspase-3-immunoreactive bands were detected with an anti-caspase-3 antibody recognizing both pro-caspase-3 and active subunits. Arrows, Pro-caspase-3 (p32) and its active caspase-3 subunits (p19, p17). Size markers (in kilodaltons) are on the left.
Protection of APP-accumulating neurons from degeneration by a caspase-3 inhibitor
To further confirm the involvement of caspase-3 in APP-induced neurodegeneration, AxCAYAP-infected neurons were treated with the caspase-3 inhibitor Ac-DEVD-CHO, and morphological changes were examined at 96 hr (Fig. 6). In the absence of the inhibitor, most of the APP-accumulating neurons (APP+/MAP2+) showed severe membrane blebbing and complete neurite retraction (Fig. 6A, a, b). In the presence of the inhibitor, the number of degenerated neurons was reduced, and many APP-accumulating neurons showed either intact morphologies or mild-to-moderate degenerative changes, such as disintegration of neurites and swelling of cell bodies (Fig.6A, c, d). We quantified the protective effect of the caspase-3 inhibitor on APP-induced neurodegeneration by counting the neurons that were classified into three stages of degeneration (Fig. 6B). In the absence of the inhibitor, 87% of APP-positive neurons severely degenerated (stage III), whereas the inhibitor significantly reduced the number of these degenerated neurons to 30%. In the presence of the inhibitor, APP-accumulating neurons showed intact morphology (stage I, 16%) or mild-to-moderate degeneration (stage II, 54%). Only the neurons in stage III were dead as assessed by ethidium homodimer exclusion test (data not shown), indicating that inhibition of caspase-3-like activity prevented APP-accumulating neurons from death. These results also support the proposition that APP-induced neuronal death is mediated by caspase-3.
Fig. 6.
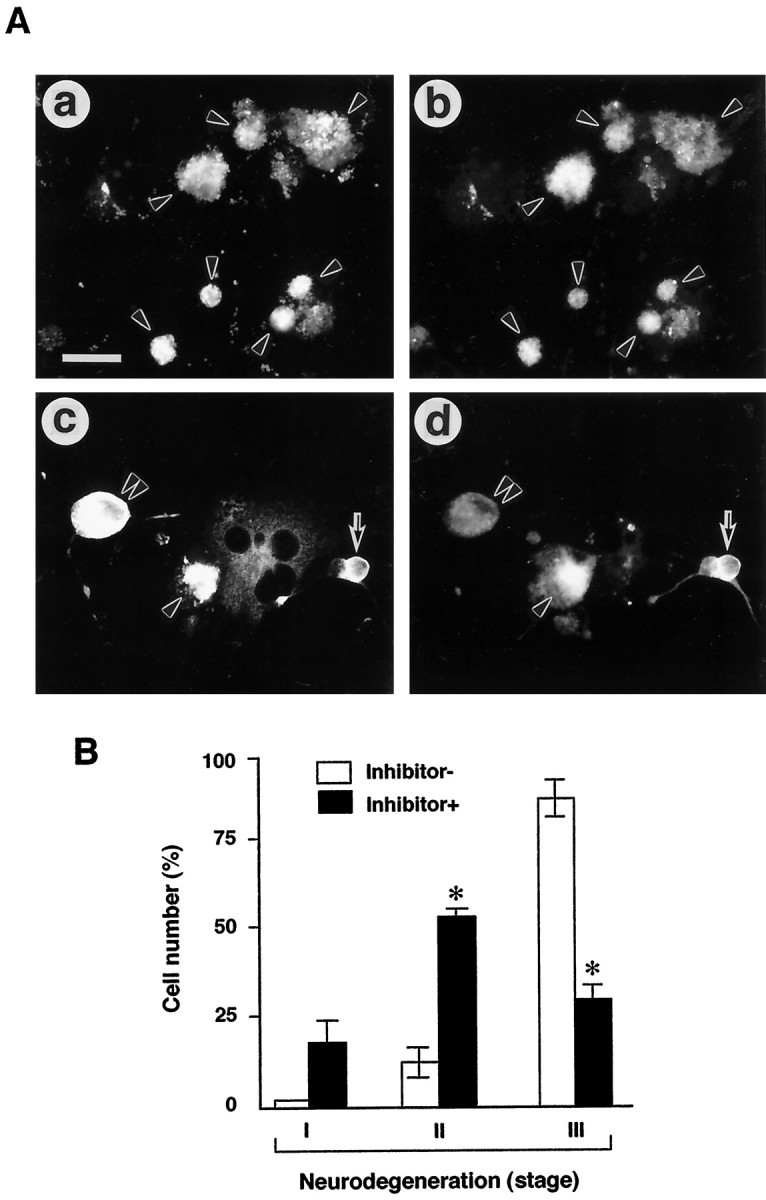
Protection of AxCAYAP-infected neurons from degeneration by caspase-3 inhibitor. AxCAYAP-infected cells were incubated for 96 hr in the absence (Inhibitor−) and presence (Inhibitor+) of 100 μmAc-DEVD-CHO. Cells were fixed and double immunostained for APP C terminus (a, c) and MAP2 (b, d). A, Morphological changes of AxCAYAP-infected neurons in the absence (a,b) and presence (c, d) of the inhibitor. The severity of degeneration displayed by APP-accumulating neurons was classified into three stages: stage I, intact morphology (arrows in c,d); stage II, mild membrane blebbing, disorganized neurites, or swelling of cell body (double arrowheads inc, d); and stage III, severe membrane blebbing and complete neurite retraction (arrowheads ina–d). Scale bar (in a),a–d, 50 μm. B, Quantification of the protective effect of caspase-3 inhibitor on neurodegeneration. AxCAYAP-infected neurons in each group (stages I–III) were counted after double staining for APP and MAP2 (mean ± SEM;n = 3; ≥200 cells per each group). *p < 0.05, significantly different from the Inhibitor− values by Student’s t test.
Generation of activated caspase-3 subunits in APP-accumulating neurons
We then immunocytochemically identified AxCAYAP-infected cells that contain activated caspase-3 (Fig.7). Some APP-immunopositive neurons showing moderate plasma membrane blebbing and disorganized neurites were intensely stained with an antibody that specifically recognizes active caspase-3 subunits (p20, p19, and p17) (Kouroku et al., 1998) (Fig. 7A,B, arrowheads), whereas APP-accumulating neurons showing mild degenerative changes, presumably at an early phase of degeneration, were weakly caspase-3-immunoreactive (Fig.7A,B, arrows). The population of intensely caspase-3-immunopositive neurons was ∼30% of total APP-accumulating neurons at 72 hr after infection. On the other hand, activated caspase-3 subunits were undetectable in APP-accumulating non-neuronal cells (Fig.7A,B, double arrowheads). The nuclei of APP-accumulating neurons that contained activated caspase-3 subunits showed strong chromatin condensation and nuclear fragmentation as assessed by staining with Hoechst 33342 (Fig.7C,D, arrowheads), suggesting that APP-accumulating neurons undergo caspase-3-dependent apoptosis. On the other hand, neither caspase-3 subunits nor chromatin condensation was observed in APP-accumulating non-neuronal cells (Fig.7C,D, arrows), which had flat, large cell bodies as observed by phase-contrast micrography (data not shown). TUNEL for DNA fragmentation, a most decisive marker for apoptosis, revealed that >90% of the shrunken nuclei in APP-accumulating neurons had fragmented DNA (Fig. 7E, arrowheads).
Fig. 7.
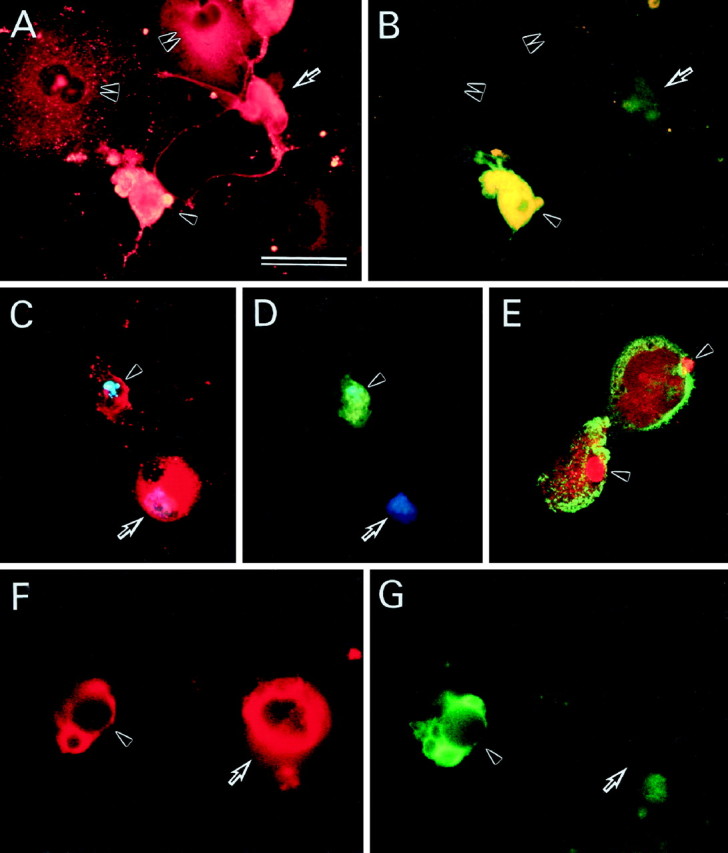
APP and caspase-3 immunoreactivities in AxCAYAP-infected neurons in vitro and in vivo. Neurally differentiated NT2 cells were fixed at 72 hr after AxCAYAP infection and stained for APP (A), activated caspase-3 (B), APP (red) and DNA (blue) (C), activated caspase-3 (green) and DNA (blue) (D), and APP (green) and TUNEL (red) (E). Rat hippocampal tissues were fixed in vivo at 72 hr after AxCAYAP injection and stained for APP (F) and activated caspase-3 (G). AxCAYAP-infected cells were double immunostained for APP N terminus and active caspase-3 subunits with antibodies P2–1 (rhodamine-B) and anti-p20/17 (FITC), respectively (A and B, C andD, and F and G, respectively). The image of chromosomal DNA was superimposed with those of APP and caspase-3 (p20/17) immunoreactivities (C, D). DNA fragmentation and APP were visualized by TUNEL (Texas Red) and immunostaining for APP C terminus with an antibody AC-1 (FITC) by confocal laser scanning fluorescence microscopy (E). See Results for indicated cells. Scale bar (in A), A–D, 50 μm;E–G, 35 μm.
We have reported previously that adenovirus-mediated overexpression of APP695 in the rat hippocampus in vivo causes neurons to degenerate and that some degenerating neurons are TUNEL-positive (Nishimura et al., 1998). Thus, we examined whether caspase-3 is also activated in vivo by APP overexpression in rat hippocampal neurons. Morphological changes of neurons in the hilus of the dentate gyrus were examined after microinjection with AxCAYAP suspended in 1m mannitol solution at the dorsal hippocampus. Under these conditions, only neurons are infected in the hilus (Nishimura et al., 1998). APP-accumulating degenerating neurons contained active caspase-3 subunits at 72 hr after AxCAYAP injection (Fig. 7F,G, arrowheads), whereas APP-accumulating neurons showing less degenerative changes had very weak caspase-3-immunoreactivity (Fig.7F,G, arrows). These results suggest that accumulation of APP induces neurodegeneration of rat hippocampal neurons in vivo through caspase-3 activation.
DISCUSSION
Previous gene transfer experiments using APP-expressing viral vectors have demonstrated that overexpression of wild-type APP in postmitotic neurons induces morphological changes characteristic of apoptosis (Bursztajn et al., 1998; Nishimura et al., 1998). However, it has been unclear whether APP-induced neurodegeneration occurs by the typical apoptotic pathway that involves caspases. We demonstrated here that infection of APP-expressing adenovirus increases the amounts of full-length APP and its degraded products at 24 hr or later (Fig. 3), activates caspase-3 maximally at 48 hr (Fig. 4), and induces neuronal degeneration at 72 hr or later (Fig. 2B). The time-dependent evolution of neurodegeneration suggests that APP accumulation induces caspase-3 activation, which subsequently causes neuronal death. Wild-type APP induced severe degeneration under the conditions in which β-galactosidase, a negative control, had no neurotoxic effects. We found that adenovirus-mediated overexpression of β-galactosidase also exerts degenerative effects on some types of neuronal cells (e.g., IMR32 neuroblastoma cells and P19 embryonal carcinoma cells) and primary cultured neurons (T. Uetsuki and K. Yoshikawa, unpublished observations). Therefore, NT2-derived postmitotic neurons are resistant to the accumulation of β-galactosidase but are vulnerable to that of wild-type APP. The specificity of wild-type APP has been supported by the previous findings that primary rat hippocampal neurons undergo apoptotic death by herpes virus-mediated overexpression of wild-type APP but not by that of presenilin-1 or its mutants (Bursztajn et al., 1998). We are now constructing mutant APPs that show different neurotoxic potencies by deleting specific domains or replacing them with other peptide sequences.
The fact that only postmitotic neurons in mixed cell populations undergo severe degeneration (Yoshikawa et al., 1992; Nishimura et al., 1998; this study) suggests the existence of neuron-specific mechanisms for APP-induced degeneration. We have reported that overexpression of full-length APP695 significantly enhances glutamate-induced elevation of intracellular calcium concentration in primary hippocampal neurons (Tominaga et al., 1997). The cytoplasmic domain of APP interacts with neuronal adapter proteins such as Fe65 and X11 (for review, see Russo et al., 1998). These findings suggest that full-length APP accumulated in neuronal membranes interacts, either directly or indirectly, with neuron-specific channels and receptors, resulting in the disintegration of intraneuronal settings essential for cell survival. Another possibility is that neurons are vulnerable to specific Aβ species that are generated in neurons. We detected small Aβ-immunoreactive peptides (6–11 kDa) in AxCAYAP-infected neurons before neuronal degeneration (Fig. 3B). These Aβ peptides are smaller than the APP C-terminal fragment consisting of 100 amino acid residues spanning the entire Aβ and cytoplasmic domains (APP-C100) (∼16 kDa) (Maruyama et al., 1990). Thus, the small Aβ-immunoreactive bands may correspond to peptides containing the entire ∼4 kDa Aβ domain with short flanking sequences. Thus, these Aβ-immunoreactive peptides, such as APP-C100 overexpressed in COS cells (Maruyama et al., 1990), may aggregate within neurons and exert neurotoxic effects.
Programmed death of chick spinal motoneurons has been extensively characterized as a typical apoptotic event occurring during physiological nervous system development (Oppenheim, 1991). The survival of spinal cord motoneurons depends on adequate supplies of trophic support from their target cells, and these neurons undergo caspase-mediated apoptosis as a result of trophic factor deprivation (Milligan et al., 1995). APP gene expression is upregulated in trophic support-deprived motoneurons in which large amounts of APP and Aβ are accumulated (Barnes et al., 1998). The present study demonstrated that APP overexpression induces caspase-3 activation, followed by neuronal death. Together, these findings raise the possibility that trophic factor-deprivation leads to the accumulation of endogenous APP, which contributes, at least in part, to the caspase-3-dependent apoptosis of spinal motoneurons. Several lines of evidence suggest that APP accumulation and caspase-3 activation are induced in neurons under similar pathological conditions. Cerebral ischemia causes intraneuronal accumulations of APP-immunoreactive materials (Stephenson et al., 1992;Saido et al., 1994) and activates caspase-3 (Chen et al., 1998;Namura et al., 1998). Furthermore, axotomy increases APP mRNA in dorsal root ganglion neurons (Scott et al., 1991) and APP C-terminal immunoreactivity in facial nucleus motoneurons (Palacios et al., 1992), and optic nerve transection induces activation of caspase-3 in axotomized retinal ganglion cells (Kermer et al., 1998). It is tempting to speculate that activation of the APP-caspase-3 system is a universal phenomenon seen in condemned neurons that are eliminated during nervous system development and in neurodegenerative situations.
Previous histopathological studies using various antibodies against different APP epitopes have revealed that neurons in AD brain contain abnormally dense APP-immunoreactive materials (Benowitz et al., 1989;Cole et al., 1991; Cummings et al., 1992). The areas containing these APP-accumulating neurons are consistent with those showing the most intense neuropathology in AD. Thus, neurons affected by AD are likely to accumulate APP as a result of loss of trophic support, gain of neurotoxic insult, or both. Aging-associated reduction of APP metabolism in neurons might facilitate the accumulation of endogenous APP. The prevailing idea is that neurodegeneration seen in AD is closely associated with, or caused by, extracellular amyloid depositions. In contrast, we have found previously that adenovirus-mediated overexpression of wild-type APP induces rapid neuronal degeneration in vivo in the absence of extracellular Aβ depositions and proposed the idea that intracellular APP accumulation per se causes a specific type of neuronal death independently of extracellular Aβ deposition (Nishimura et al., 1998). In the frontal cortex of AD brain, neurons displaying DNA fragmentation contain high levels of activated caspase-3 (p20 subunit), and no apparent amyloid depositions are noted in close proximity to caspase-3-immunopositive neurons (Masliah et al., 1998). We speculate that APP and active caspase-3 subunits are concurrently elevated in affected neurons without adjacent Aβ depositions in AD brain. After submitting this paper, we encountered a report on the involvement of caspases in proteolytic cleavage of APP and Aβ formation (Gervais et al., 1999). They found that acute excitotoxic or ischemic brain injury generates a caspase-3-cleaved APP fragment in hippocampal neuronsin vivo and that the cleaved APP fragment is colocalized with Aβ in senile plaques in AD brain. These data, together with the present findings, support the idea that neurotoxic insults induce APP elevation, followed by caspase-3 activation, which causes both apoptosis and the proteolytic processing of APP that leads to Aβ formation. Further studies on the APP-induced neuronal death may provide important clues to the pathogenesis of AD and to the development of the therapeutic strategies.
Footnotes
This work was partly supported by Health Sciences Research Grants (Research on Brain Science) from the Ministry of Health and Welfare of Japan (to K.Y.), by grants-in-aid for scientific research from the Ministry of Education, Science, Sports, and Culture of Japan (to T.U. and K.Y.), and by Research Fellowships of the Japan Society for the Promotion of Science for Young Scientists (to I.N.). We thank Dr. Izumu Saito for adenovirus vectors, Dr. William Van Nostrand for P2–1 antibody, and Dr. Takako Aizawa for advice of NT-2 cell culture.
Correspondence should be addressed to Dr. Kazuaki Yoshikawa, Division of Regulation of Macromolecular Functions, Institute for Protein Research, Osaka University, Yamadaoka 3–2, Suita, Osaka 565–0871, Japan.
REFERENCES
- 1.Andrews PW. Retinoic acid induces neuronal differentiation of a cloned human embryonal carcinoma cell line in vitro. Dev Biol. 1984;103:285–293. doi: 10.1016/0012-1606(84)90316-6. [DOI] [PubMed] [Google Scholar]
- 2.Barnes NY, Li L, Yoshikawa K, Schwartz LM, Oppenheim RW, Milligan CE. Increased production of amyloid precursor protein provides a substrate for caspase-3 in dying motoneurons. J Neurosci. 1998;18:5869–5880. doi: 10.1523/JNEUROSCI.18-15-05869.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Benowitz LI, Rodriguez W, Paskevich P, Mufson EJ, Schenk D, Neve RL. The amyloid precursor protein is concentrated in neuronal lysosomes in normal and Alzheimer disease subjects. Exp Neurol. 1989;106:237–250. doi: 10.1016/0014-4886(89)90156-8. [DOI] [PubMed] [Google Scholar]
- 4.Bursztajn S, DeSouza R, McPhie DL, Berman SA, Shioi J, Robakis NK, Neve RL. Overexpression in neurons of human presenilin-1 or a presenilin-1 familial Alzheimer’s disease mutant does not enhance apoptosis. J Neurosci. 1998;18:9790–9799. doi: 10.1523/JNEUROSCI.18-23-09790.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chen J, Nagayama T, Jin K, Stetler RA, Zhu RL, Graham SH, Simon RP. Induction of caspase-3-like protease may mediate delayed neuronal death in the hippocampus after transient cerebral ischemia. J Neurosci. 1998;18:4914–4928. doi: 10.1523/JNEUROSCI.18-13-04914.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cole GM, Masliah E, Shelton ER, Chan HW, Terry RD, Saitoh T. Accumulation of amyloid precursor fragment in Alzheimer plaques. Neurobiol Aging. 1991;12:85–91. doi: 10.1016/0197-4580(91)90046-m. [DOI] [PubMed] [Google Scholar]
- 7.Cummings BJ, Su JH, Geddes JW, Van Nostrand WE, Wagner SL, Cunningham DD, Cotman CW. Aggregation of the amyloid precursor protein within degenerating neurons and dystrophic neurites in Alzheimer’s disease. Neuroscience. 1992;48:763–777. doi: 10.1016/0306-4522(92)90265-4. [DOI] [PubMed] [Google Scholar]
- 8.Fernandes-Alnemri T, Armstrong RC, Krebs J, Srinivasula SM, Wang L, Bullrich F, Fritz LC, Trapani JA, Tomaselli KJ, Litwack G, Alnemri ES. In vitro activation of CPP32 and Mch3 by Mch4, a novel human apoptotic cysteine protease containing two FADD-like domains. Proc Natl Acad Sci USA. 1996;93:7464–7469. doi: 10.1073/pnas.93.15.7464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gavrieli Y, Sherman Y, Ben-Sasson SA. Identification of programmed cell death in situ via specific labeling of nuclear DNA fragmentation. J Cell Biol. 1992;119:493–501. doi: 10.1083/jcb.119.3.493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gervais FG, Xu D, Robertson GS, Vaillancourt JP, Zhu Y, Huang J, LeBlanc A, Smith D, Rigby M, Shearman MS, Clarke EE, Zheng H, Van Der Ploeg LH, Ruffolo SC, Thornberry NA, Xanthoudakis S, Zamboni RJ, Roy S, Nicholson DW. Involvement of caspases in proteolytic cleavage of Alzheimer’s amyloid-β precursor protein and amyloidogenic Aβ peptide formation. Cell. 1999;97:395–406. doi: 10.1016/s0092-8674(00)80748-5. [DOI] [PubMed] [Google Scholar]
- 11.Ida N, Hartmann T, Pantel J, Schroder J, Zerfass R, Forstl H, Sandbrink R, Masters CL, Beyreuther K. Analysis of heterogeneous A4 peptides in human cerebrospinal fluid and blood by a newly developed sensitive Western blot assay. J Biol Chem. 1996;271:22908–22914. doi: 10.1074/jbc.271.37.22908. [DOI] [PubMed] [Google Scholar]
- 12.Kanegae Y, Makimura M, Saito I. A simple and efficient method for purification of infectious recombinant adenovirus. Jpn J Med Sci Biol. 1994;47:157–166. doi: 10.7883/yoken1952.47.157. [DOI] [PubMed] [Google Scholar]
- 13.Kang J, Lemaire HG, Unterbeck A, Salbaum JM, Masters CL, Grzeschik KH, Multhaup G, Beyreuther K, Muller-Hill B. The precursor of Alzheimer’s disease amyloid A4 protein resembles a cell-surface receptor. Nature. 1987;325:733–736. doi: 10.1038/325733a0. [DOI] [PubMed] [Google Scholar]
- 14.Kanuka H, Hisahara S, Sawamoto K, Shoji S, Okano H, Miura M. Proapoptotic activity of Caenorhabditis elegans CED-4 protein in Drosophila: implicated mechanisms for caspase activation. Proc Natl Acad Sci USA. 1999;96:145–150. doi: 10.1073/pnas.96.1.145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kermer P, Klocker N, Labes M, Bahr M. Inhibition of CPP32-like proteases rescues axotomized retinal ganglion cells from secondary cell death in vivo. J Neurosci. 1998;18:4656–4662. doi: 10.1523/JNEUROSCI.18-12-04656.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kouroku Y, Urase K, Fujita E, Isahara K, Ohsawa Y, Uchiyama Y, Momoi MY, Momoi T. Detection of activated Caspase-3 by a cleavage site-directed antiserum during naturally occurring DRG neurons apoptosis. Biochem Biophys Res Commun. 1998;247:780–784. doi: 10.1006/bbrc.1998.8815. [DOI] [PubMed] [Google Scholar]
- 17.Loo DT, Copani A, Pike CJ, Whittemore ER, Walencewicz AJ, Cotman CW. Apoptosis is induced by β-amyloid in cultured central nervous system neurons. Proc Natl Acad Sci USA. 1993;90:7951–7955. doi: 10.1073/pnas.90.17.7951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Maruyama K, Terakado K, Usami M, Yoshikawa K. Formation of amyloid-like fibrils in COS cells overexpressing part of the Alzheimer amyloid protein precursor. Nature. 1990;347:566–569. doi: 10.1038/347566a0. [DOI] [PubMed] [Google Scholar]
- 19.Masliah E, Mallory M, Alford M, Tanaka S, Hansen LA. Caspase dependent DNA fragmentation might be associated with excitotoxicity in Alzheimer’s disease. J Neuropathol Exp Neurol. 1998;57:1041–1052. doi: 10.1097/00005072-199811000-00007. [DOI] [PubMed] [Google Scholar]
- 20.Milligan CE, Prevette D, Yaginuma H, Homma S, Cardwell C, Fritz LC, Tomaselli KJ, Oppenheim RW, Schwartz LM. Peptide inhibitors of the ICE protease family arrest programmed cell death of motoneurons in vivo and in vitro. Neuron. 1995;15:385–393. doi: 10.1016/0896-6273(95)90042-x. [DOI] [PubMed] [Google Scholar]
- 21.Miyake S, Makimura M, Kanegae Y, Harada S, Sato Y, Takamori K, Tokuda C, Saito I. Efficient generation of recombinant adenoviruses using adenovirus DNA-terminal protein complex and a cosmid bearing the full-length virus genome. Proc Natl Acad Sci USA. 1996;93:1320–1324. doi: 10.1073/pnas.93.3.1320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Moechars D, Lorent K, De Strooper B, Dewachter I, Van Leuven F. Expression in brain of amyloid precursor protein mutated in the α-secretase site causes disturbed behavior, neuronal degeneration and premature death in transgenic mice. EMBO J. 1996;15:1265–1274. [PMC free article] [PubMed] [Google Scholar]
- 23.Namura S, Zhu J, Fink K, Endres M, Srinivasan A, Tomaselli KJ, Yuan J, Moskowitz MA. Activation and cleavage of caspase-3 in apoptosis induced by experimental cerebral ischemia. J Neurosci. 1998;18:3659–3668. doi: 10.1523/JNEUROSCI.18-10-03659.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Nicholson DW, Ali A, Thornberry NA, Vaillancourt JP, Ding CK, Gallant M, Gareau Y, Griffin PR, Labelle M, Lazebnik YA, Munday NA, Raju SM, Smulson ME, Yamin T, Yu VL, Miller DK. Identification and inhibition of the ICE/CED-3 protease necessary for mammalian apoptosis. Nature. 1995;376:37–43. doi: 10.1038/376037a0. [DOI] [PubMed] [Google Scholar]
- 25.Nishimura I, Uetsuki T, Dani SU, Ohsawa Y, Saito I, Okamura H, Uchiyama Y, Yoshikawa K. Degeneration in vivo of rat hippocampal neurons by wild-type Alzheimer amyloid precursor protein overexpressed by adenovirus-mediated gene transfer. J Neurosci. 1998;18:2387–2398. doi: 10.1523/JNEUROSCI.18-07-02387.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Oppenheim RW. Cell death during development of the nervous system. Annu Rev Neurosci. 1991;14:453–501. doi: 10.1146/annurev.ne.14.030191.002321. [DOI] [PubMed] [Google Scholar]
- 27.Palacios G, Palacios JM, Mengod G, Frey P. β-Amyloid precursor protein localization in the Golgi apparatus in neurons and oligodendrocytes. An immunocytochemical structural and ultrastructural study in normal and axotomized neurons. Brain Res Mol Brain Res. 1992;15:195–206. doi: 10.1016/0169-328x(92)90109-o. [DOI] [PubMed] [Google Scholar]
- 28.Pleasure SJ, Page C, Lee VM. Pure, postmitotic, polarized human neurons derived from NTera 2 cells provide a system for expressing exogenous proteins in terminally differentiated neurons. J Neurosci. 1992;12:1802–1815. doi: 10.1523/JNEUROSCI.12-05-01802.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Russo T, Faraonio R, Minopoli G, De Candia P, De Renzis S, Zambrano N. Fe65 and the protein network centered around the cytosolic domain of the Alzheimer’s β-amyloid precursor protein. FEBS Lett. 1998;434:1–7. doi: 10.1016/s0014-5793(98)00941-7. [DOI] [PubMed] [Google Scholar]
- 30.Saido TC, Yokota M, Maruyama K, Yamao-Harigaya W, Tani E, Ihara Y, Kawashima S. Spatial resolution of the primary β-amyloidogenic process induced in postischemic hippocampus. J Biol Chem. 1994;269:15253–15257. [PubMed] [Google Scholar]
- 31.Schöler HR, Balling R, Hatzopoulos AK, Suzuki N, Gruss P. Octamer binding proteins confer transcriptional activity in early mouse embryogenesis. EMBO J. 1989;8:2551–2557. doi: 10.1002/j.1460-2075.1989.tb08393.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Scott JN, Parhad IM, Clark AW. β-Amyloid precursor protein gene is differentially expressed in axotomized sensory and motor systems. Brain Res Mol Brain Res. 1991;10:315–325. doi: 10.1016/0169-328x(91)90090-k. [DOI] [PubMed] [Google Scholar]
- 33.Selkoe DJ. Normal and abnormal biology of the β-amyloid precursor protein. Annu Rev Neurosci. 1994;17:489–517. doi: 10.1146/annurev.ne.17.030194.002421. [DOI] [PubMed] [Google Scholar]
- 34.Stephenson DT, Rash K, Clemens JA. Amyloid precursor protein accumulates in regions of neurodegeneration following focal cerebral ischemia in the rat. Brain Res. 1992;593:128–135. doi: 10.1016/0006-8993(92)91274-i. [DOI] [PubMed] [Google Scholar]
- 35.Thornberry NA, Lazebnik Y. Caspases: enemies within. Science. 1998;281:1312–1316. doi: 10.1126/science.281.5381.1312. [DOI] [PubMed] [Google Scholar]
- 36.Tominaga K, Uetsuki T, Ogura A, Yoshikawa K. Glutamate responsiveness enhanced in neurones expressing amyloid precursor protein. NeuroReport. 1997;8:2067–2072. doi: 10.1097/00001756-199705260-00051. [DOI] [PubMed] [Google Scholar]
- 37.Van Nostrand WE, Wagner SL, Suzuki M, Choi BH, Farrow JS, Geddes JW, Cotman CW, Cunningham DD. Protease nexin-II, a potent antichymotrypsin, shows identity to amyloid β-protein precursor. Nature. 1989;341:546–549. doi: 10.1038/341546a0. [DOI] [PubMed] [Google Scholar]
- 38.Yamatsuji T, Matsui T, Okamoto T, Komatsuzaki K, Takeda S, Fukumoto H, Iwatsubo T, Suzuki N, Asami-Odaka A, Ireland S, Kinane TB, Giambarella U, Nishimoto I. G protein-mediated neuronal DNA fragmentation induced by familial Alzheimer’s disease-associated mutants of APP. Science. 1996;272:1349–1352. doi: 10.1126/science.272.5266.1349. [DOI] [PubMed] [Google Scholar]
- 39.Yankner BA. Mechanisms of neuronal degeneration in Alzheimer’s disease. Neuron. 1996;16:921–932. doi: 10.1016/s0896-6273(00)80115-4. [DOI] [PubMed] [Google Scholar]
- 40.Yankner BA, Duffy LK, Kirschner DA. Neurotrophic and neurotoxic effects of amyloid β protein: reversal by tachykinin neuropeptides. Science. 1990;250:279–282. doi: 10.1126/science.2218531. [DOI] [PubMed] [Google Scholar]
- 41.Yoshikawa K, Aizawa T, Hayashi Y. Degeneration in vitro of post-mitotic neurons overexpressing the Alzheimer amyloid protein precursor. Nature. 1992;359:64–67. doi: 10.1038/359064a0. [DOI] [PubMed] [Google Scholar]
- 42.Zhao B, Chrest FJ, Horton WE, Sisodia SS, Jr, Kusiak JW. Expression of mutant amyloid precursor proteins induces apoptosis in PC12 cells. J Neurosci Res. 1997;47:253–263. [PubMed] [Google Scholar]