

Abstract
Seizure causes neuronal cell loss in both animal models and human epilepsy. To determine the contribution of apoptotic mechanisms to seizure-induced neuronal cell death, rat brains were examined for the occurrence of terminal deoxynucleotidyl transferase-mediated UTP nick end labeling (TUNEL)-positive nuclei after pilocarpine-induced seizure. Numerous TUNEL-positive cells were observed throughout the postseizure hippocampus, piriform cortex, and entorhinal cortex. Combined TUNEL/NeuN immunocytochemistry demonstrated that the vast majority of TUNEL-positive cells were neurons. To identify components of the signal transduction cascade promoting postseizure apoptosis, the expression of the p75 neurotrophin receptor (p75NTR) was examined. Seizure-induced increases in p75NTR protein and mRNA were detected in hippocampus, piriform cortex, and entorhinal cortex. Immunohistochemical double labeling revealed almost complete correspondence between TUNEL-positive and p75NTR-expressing cells, suggesting that seizure-induced neuronal loss within the CNS occurs through apoptotic signaling cascades involving p75NTR.
Keywords: seizure, apoptosis, pilocarpine, p75NTR, piriform cortex, entorhinal cortex, hippocampus
The neurotrophins are a conserved family of proteins that play a critical role in the development and maintenance of the nervous system (for review, see Barde, 1989). Their cellular effects are mediated by two distinct classes of cell surface receptors. The trk receptors are a family of transmembrane receptor tyrosine kinases that selectively bind different members of the neurotrophin family, with trkA preferentially binding NGF, trkB preferring BDNF and neurotrophin (NT)-4/5, and trkC interacting with NT-3 (for review, see Kaplan and Miller, 1997). The second class of neurotrophin receptor contains a single family member, the p75 neurotrophin receptor (p75NTR), that binds all the neurotrophins (for review, see Barker, 1998; Casaccia-Bonnefil et al., 1998). p75NTR is a member of the tumor necrosis factor (TNF) receptor superfamily that includes CD27, CD30, CD40, 4–1BB, OX40, the fas antigen, and the tumor necrosis factor receptors TNFR1 and TNFR2 (Bazan, 1993). Two opposing functions have been proposed for p75NTR. When coexpressed with trkA, p75NTR enhances NGF-mediated survival by increasing the amount of NGF that binds the trkA receptor (Barker and Shooter, 1994; Mahadeo et al., 1994; Verdi et al., 1994). Conversely, in some systems, p75NTR appears to behave as a ligand-regulated proapoptotic receptor (Frade et al., 1996; Casaccia-Bonnefil et al., 1996; Majdan et al., 1997; Bamji et al., 1998; Frade and Barde, 1998). The signaling cascades that allow p75NTR to promote apoptosis remain unknown but may involve ceramide production through activation of sphingomyelinase (Dobrowsky et al., 1994, 1995), activation of c-Jun N-terminal kinase (JNK;Casaccia-Bonnefil et al., 1996; Yoon et al., 1998), and accumulation of p53 (Aloyz et al., 1998).
Neuronal cell death has been well documented in both human epilepsy and experimental seizure models (for review, see Represa et al., 1995;Morrison et al., 1996; Sloviter, 1996; Treiman, 1996). Although the specific contribution of cell death to the pathophysiology of epilepsy remains unclear, multiple studies suggest that damage produced by status epilepticus (SE) promotes the development of the recurrent spontaneous seizures characteristic of epilepsy (Aicardi and Chevrie, 1983; Cavalheiro et al., 1991; Priel et al., 1996). Pilocarpine-induced SE in the rat results in damage in multiple brain regions (Turski et al., 1983; Olney et al., 1986; Turski et al., 1987). Dystrophic neurons can be detected as early as 20 min after induction of SE, and much of this cell damage is likely necrotic (Fujikawa, 1996). Apoptotic cell death has been reported in some seizure models (Pollard et al., 1994;Morrison et al., 1996; Bengzon et al., 1997), but the specific contribution of apoptotic or necrotic death to seizure-induced neuronal loss is not clear, and the cellular mechanisms leading to the induction of apoptosis after seizure are unknown.
In addition to necrotic and apoptotic cell death, seizure also induces changes in gene expression, including marked alterations in neurotrophin and trk receptor expression (Gall et al., 1991a; for review, see Gall, 1993). Neurotrophins have been suggested to play a trophic role after seizure; however, the recent demonstration of a proapoptotic role for the p75 neurotrophin receptor suggests that increased neurotrophin expression after seizure could potentially promote cell death via p75NTR-dependent apoptotic mechanisms.
In this study, we demonstrate that pilocarpine-induced seizure results in marked and persistent apoptosis within hippocampal, piriform, and entorhinal cortical neurons. We show that this region-specific increase in neuronal apoptosis is accompanied by expression of p75NTR mRNA and protein. Furthermore, we demonstrate that neurons undergoing seizure-induced apoptosis invariably show strong induction of p75NTR, suggesting that upregulation of p75NTR expression and activation of p75NTR signaling cascades may facilitate neuronal apoptosis after seizure.
MATERIALS AND METHODS
Seizure induction. Adult male Sprague Dawley rats (200–300 gm; Charles River ) were used for all experiments and housed under environmentally controlled conditions. Status epilepticus (SE) was induced by the administration of pilocarpine (380 mg/kg, i.p.; ICN Biochemicals, Montréal, Québec, Canada). Thirty minutes before administering pilocarpine, animals received scopolamine methyl-bromide (1 mg/kg, i.p.; Sigma) to reduce the peripheral cholinergic effects of pilocarpine. During SE, the animals exhibited two to five stage 5 seizures, behaviorally similar to kindled stage 5 seizures (Racine, 1972). To reduce mortality caused by seizure, diazepam (10 mg/kg, i.p.; Hoffmann-La Roche) was injected 1 hr after the onset of SE. Control animals were treated identically to the experimental group, except that they received saline instead of pilocarpine. Animals were killed, and tissue was removed 1, 3, 7, or 14 d after pilocarpine injection.
Tissue preparation. For immunocytochemical and terminal deoxynucleotidyl transferase-mediated UTP nick end labeling (TUNEL) assays, animals were anesthetized by injection of sodium pentobarbital (50 mg/kg, i.p.; MTC Pharmaceuticals), and perfused intracardially with PBS plus heparin (5 μg/ml) followed by 4% paraformaldehyde, 15% picric acid, in PBS at pH 8 and 37°C. After perfusion, brains were removed, post-fixed for 3 d at room temperature (RT) and cryoprotected in 30% sucrose-containing fixative at RT for 48 hr before sectioning. Frozen 40 μm cryostat serial sections were stored in cryoprotectant at −20°C (30% sucrose and 30% ethylene glycol in PBS) and assayed within 3 months of sectioning. For immunoblot analysis, animals were euthanized with pentobarbital as above, the brain was removed, the cortex (combined neocortical and paleocortical tissue excluding hippocampus) and hippocampus were rapidly dissected, and total protein was extracted using Trizol (Life Technologies, Gaithersburg, MD), as suggested by the manufacturer.
Immunoblotting. p75NTR immunoreactivity was detected using anti-p75NTR-B1, a rabbit polyclonal antibody directed against a glutathione S-transferase (GST)-fusion protein containing amino acids 276–425 of the intracellular domain of rat p75NTR (Majdan et al., 1997). MC192 ascites fluid was produced as described previously (Barker and Shooter, 1994) and purified using an Immunopure column (Pierce, Rockford, IL). Protein content of brain tissue extracts was normalized using the BCA assay (Pierce). Twenty five micrograms of protein were then solubilized in sample buffer (Laemmli, 1970), separated on 10% SDS-PAGE and electroblotted to nitrocellulose. Blocking, primary antibody, and secondary antibody incubations for p75NTR immunoblots were all performed in 10 mmTris, pH 7.4, 150 mm NaCl, and 0.2% Tween 20 with 5% (w/v) dry skim milk powder using anti-p75NTR-B1 (1:2000). HRP-conjugated donkey anti-rabbit IgG (Jackson ImmunoResearch, West Grove, PA) was used at a dilution of 1:10,000. Immunoreactive bands were detected using enhanced chemiluminescence (ECL) according to the manufacturer’s instructions (DuPont). The immunoreactive band detected in brain homogenates comigrated with an immunoreactive band of the appropriate molecular weight present in cell homogenates derived from p75NTR-transfected 293 cells, and immunoreactivity could be blocked by a 6XHis-fusion protein corresponding to the intracellular domain of p75NTR (data not shown). Densitometry and quantification of the relative level of p75NTR protein was performed on scanned images of immunoblots (Epson ES 1200C) using NIH Image software (United States National Institutes of Health). The mean densitometric value corresponding to p75NTR expression was calculated for each time point (n = 3 per time point with the exception of the 1 d postseizure time point where n = 2), and the percent increase from controls determined by direct comparison with samples on the same immunoblot.
Immunocytochemistry. After cryostat sectioning, brain sections were washed briefly in PBS, and endogenous peroxidase activity was reduced by incubation in 75% methanol and 3% H2O2 for 30 min at RT. Blocking, primary, and secondary antibody incubations were performed in blocking solution (2% bovine serum albumin, 2% heat-inactivated normal goat serum, and 0.2% Triton X-100). Anti-p75NTR-B1 was used at a dilution of 1:500, and HRP-conjugated goat anti-rabbit IgG was used at a dilution of 1:1000 (Jackson ImmunoResearch). Immunocytochemistry for c-Jun expression was performed as described for p75NTR, using a monoclonal antibody against c-Jun at 1:2000 (Transduction Laboratories, Lexington, KY), and an HRP-conjugated goat anti-mouse IgG at 1:1000 (Jackson ImmunoResearch). Antibody complexes were detected with diaminobenzidine (DAB) and H2O2 as described (Vector Laboratories, Burlingame, CA). For p75NTR and TUNEL costaining, sections were directly blocked for 2 hr after the TUNEL reaction, followed by an overnight incubation with purified MC192 (3 μg/ml at 4°C), a monoclonal antibody that recognizes rat p75NTR (Chandler et al., 1984). Secondary antibody incubation was performed at RT for 2 hr using Cy3-conjugated goat anti-mouse IgG (Jackson ImmunoResearch) at a dilution of 1:1000. NeuN/TUNEL double labeling was performed as for p75NTR, using the NeuN mouse monoclonal antibody at a dilution of 1:25 (gift of Richard Mullen). During the washes, nuclei were stained using Hoechst 33258 as described (Molecular Probes, Eugene, OR). Bright-field images of p75NTR and c-Jun immunoreactivity and TUNEL were photographed using a Zeiss Axiophot microscope. Fluorescence was visualized using a Zeiss Axioscop microscope and photographed using a CCD camera and Northern Eclipse software (Empix Imaging).
In situ hybridization. After intracardial perfusion with 100 ml of 37°C saline with 5 μg/ml heparin, brains were immediately dissected, placed in ice-cold PBS, and frozen in isopentane (2-methyl butane) chilled in liquid nitrogen. Five micrometer cryostat sections were cut and fixed to slides (Superfrost/Plus; Fisher Scientific) with 4% paraformaldehyde and 15% picric acid in PBS.In situ hybridization was performed as described (Braissant and Wahli, 1998) using digoxigenin-labeled RNA probes, signal-amplified using the Tyramide Signal Amplification kit (NEN, Boston, MA), and peroxidase/DAB detection. Probes were sense and antisense transcripts of a 300 bp fragment corresponding to nucleotides 400–700 of rat p75NTR cDNA (Radeke et al., 1987).
In situ detection of DNA cleavage. TUNEL was performed using an in situ cell death detection kit as per the manufacturer’s instructions (Boehringer Mannheim). TUNEL-positive nuclei were detected using DAB and H2O2 with an HRP-conjugated anti-fluorescein antibody. For the colabeling studies described above, TUNEL-positive cells were identified directly using the FITC fluorescence of the incorporated dUTP. Positive cells in 12 fields sampled from layers II and III of the entorhinal cortex derived from two different rats (six fields each) at 3 d after seizure were scored for p75NTR immunoreactivity, TUNEL reactivity, and their colocalization. In separate experiments, coincidence of NeuN and TUNEL reactivity was similarly determined.
DNA extraction and agarose gel electrophoresis. Samples of hippocampi as well as frontal and temporal lobe were dissected from brains of control rats or from rats seized 1 or 3 d earlier and then immediately frozen in dry ice, and stored at −70°C. DNA was purified as described in Sankar et al. (1998) with some modifications. Tissues were homogenized using a Dounce homogenizer with a loose pestle in five volumes of a buffer containing 15 mm HEPES, pH 7.2, 0.25 m sucrose, 60 mm KCl, 10 mmNaCl, 1 mm EGTA, 5 mm EDTA, and 1 mm PMSF. Cells were then centrifuged at 2000 ×g for 10 min and incubated overnight in a buffer containing 50 mm NaCl, 10 mm Tris, pH 8.0, 25 mm EDTA, 0.5% SDS, 0.5 mg/ml proteinase K, and 0.1 mg/ml DNase-free RNase A at 55°C. The lysate was extracted twice using equal proportions of phenol: chloroform (1:1), and then the aqueous layer was incubated at 37°C with 0.1 mg/ml RNase A for 90 min. The phenol:chloroform extraction was repeated, and DNA was precipitated overnight with 2.5 vol of ethanol and 1/10 vol of sodium acetate at −20°C. Precipitated DNA was spun at 15,000 ×g for 30 min and washed three times with 70% ethanol. The DNA pellet was then air-dried and resuspended in 0.5–1 ml of 10 mm Tris and 1 mm EDTA. Spectrophotometry revealed A260/280 ratios of 1.6–1.9, indicating relatively pure DNA in concentrations of 0.8–1.3 mg/ml. Thirty micrograms of DNA was run on each lane of a 1% agarose gel containing 0.5% ethidium bromide at 5 V/cm gel length. The gel was viewed under UV transillumination and photographed using a Kohu CCD camera.
RESULTS
Pilocarpine-induced seizure leads to apoptosis in the adult rat brain
Brains of rats subjected to pilocarpine-induced seizure were examined for evidence of cell death using TUNEL, a method that detects apoptotic cells in situ (Gavrieli et al., 1992; Sgonc et al., 1994). Induction of SE by injection of pilocarpine caused severe generalized seizures (multiple class 5) that resulted in numerous TUNEL-positive nuclei in multiple brain regions. TUNEL-positive cells were clearly detected at both 1 and 3 d after seizure in piriform cortex, entorhinal cortex, and hippocampus, but TUNEL-positive cells were not observed in brain sections of control animals (Fig.1). The incidence of TUNEL-positive cells was greater 1 d after seizure in both the piriform and entorhinal cortices compared to 3 d. However, in the hippocampus, TUNEL staining was maximal at 3 d after seizure (n > 4 for each time point). Although the incidence of TUNEL-positive nuclei peaked at 1 d in entorhinal cortex after seizure, they were still detected in layers II and III of the entorhinal cortex 14 d after seizure (data not shown). In the hippocampus, most TUNEL-positive nuclei were concentrated in the CA1 region, with lower numbers in the granule cell layer of the dentate gyrus (Fig. 1I). Consistent with previous histochemical descriptions of the distribution of dystrophic cells produced by pilocarpine-induced seizure (Mello et al., 1993;Fujikawa, 1996), the amygdala, the perirhinal cortex, and the lateral posterior thalamic nucleus showed fewer but detectable TUNEL-positive nuclei after seizure (data not shown). Incidence of TUNEL-positive cells varied somewhat between animals, but the regional distribution of TUNEL-positive cells was consistent.
Fig. 1.
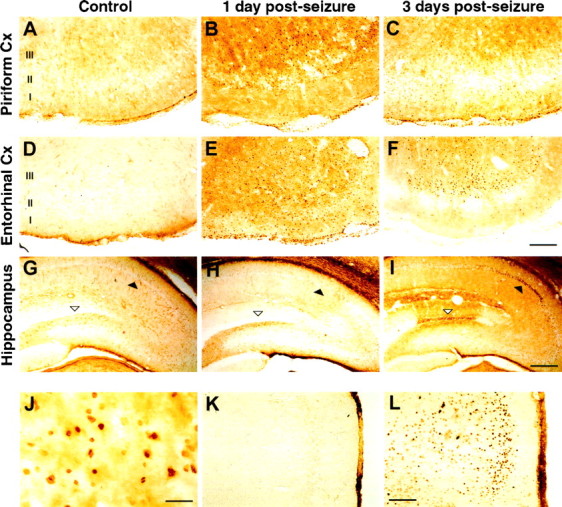
TUNEL staining and expression of c-Jun after pilocarpine-induced seizure in the rat brain. Cells positive for TUNEL reactivity (A–J) and for c-Jun expression (K, L) were visualized using peroxidase/DAB in brain sections from control rats (A,D, G, K), 1 d (B, E, H, L), or 3 d after seizure (C, F, I) in the piriform cortex (A–C), entorhinal cortex (D–F, K, L), and hippocampus (G–I). J is a higher magnification of the tissue in E showing TUNEL-positive nuclei. Filled arrowheads identify the hippocampal CA1 pyramidal cell layer. Open arrowheads identify the upper blade of the dentate granule cell layer. Cortical layers are indicated.Cx, Cortex. Scale bars: F, 200 μm;I, 400 μm; J, 20 μm;L, 160 μm.
To confirm that TUNEL staining within seized brain reflects seizure-induced apoptosis, genomic DNA was extracted from brains of control and seized rats and analyzed for oligosomal fragmentation, a biochemical hallmark of apoptosis. Figure2 shows that DNA fragmentation was undetectable in brains from control animals; however, DNA fragments with a periodicity of ∼180 bp were present in extracts of temporal cortex from seized animals at both 1 and 3 d after SE. DNA fragmentation was most prevalent in temporal cortex, consistent with the high level of TUNEL reactivity observed in the piriform and entorhinal cortices, but was also clearly detected in extracts of hippocampus and the frontal cortex. Sections costained with TUNEL and Hoechst 33258 demonstrate that a subset of the TUNEL-positive nuclei are condensed in a manner characteristic of apoptotic cell death (Fig.7). Together, these data demonstrate the presence of apoptotic cells within multiple CNS regions in the rat after pilocarpine-induced seizure.
Fig. 2.

DNA fragmentation after pilocarpine-induced seizure. DNA extracted from control, 1 d, and 3 d postseizure tissue was analyzed by agarose gel electrophoresis. DNA laddering is visible in extracts of rat brain 1 and 3 d after pilocarpine-induced seizure. DNA was extracted from dissected samples of frontal cortex (F), temporal cortex (T), and hippocampus (H).
Fig. 7.

p75NTR expression is induced after seizure in neurons undergoing apoptosis. Triple-immunofluorescence of p75NTR (A, D, H), TUNEL (B, E, I), and Hoechst 33258 (C, F, J) in layer III of the entorhinal cortex (between bregma −7.04 and −7.30;A–F) and within hippocampus (H–J), 3 d after pilocarpine-induced seizure. G is a composite of D–F, with p75NTR immunoreactivity visualized in red(Cy3-conjugated secondary antibody), TUNEL reaction ingreen (FITC), and Hoechst-stained nuclei inblue. In H–J, the dashed line indicates the boundary of the hippocampal CA1 layer, and the white bars indicate cells containing pyknotic nuclei. Scale bars: C, J, 50 μm;G, 10 μm.
Signaling pathways that result in c-Jun expression and phosphorylation are involved in neuronal apoptosis (for review, see Dragunow and Preston, 1995; Herdegen et al., 1997), and we therefore examined whether c-Jun protein expression was induced after pilocarpine-induced seizure. Consistent with previous results that showed induction of immediate-early response genes in other seizure models (Dragunow et al., 1993; Herdegen et al., 1997), prominent c-Jun expression was detected throughout the brain 1 d after pilocarpine-induced SE (Fig. 1L).
p75NTR protein expression increases after seizure
Neurotrophins promote neuronal survival by activating trk receptors, but recent studies also suggest that p75NTR may facilitate neuronal apoptosis. As an initial step toward identifying molecules that may promote neuronal apoptosis after seizure, levels of p75NTR protein were assayed after pilocarpine-induced seizure. Immunoblot analysis of tissue isolated from nonseized controls show that the level of p75NTR protein is low but detectable in protein extracted from hippocampal and cortical tissue. After seizure, p75NTR protein expression increased in a time-dependent manner (Fig.3). In the hippocampus (Fig.3B), p75NTR protein levels increased 1 d after seizure and then persisted as a fourfold to sixfold increase for at least 7 d. Two weeks after seizure, the level of p75NTR protein had returned to that of sham-treated controls. In cortical lysates (Fig.3A), the increase was delayed and more transient, with a fivefold increase in p75NTR expression 3 d after seizure but sharply reduced p75NTR levels by 7 d.
Fig. 3.

Increased p75NTR expression after seizure. Protein homogenates from the hippocampus and cortex (neocortical and paleocortical tissue) of control, 1, 3, 7, and 14 d after seizure. Representative immunoblots show the relative amount of p75NTR protein in cortex (A) and hippocampus (B). Each graph represents the normalized densitometric index of immunoblots from three animals per time point (± SD) except for the 1 d time point, at which two animals were analyzed.
To identify the cellular distribution of p75NTR protein, brain sections from seized and control rats were analyzed for p75NTR immunoreactivity 1 and 3 d after pilocarpine treatment. In control animals, p75NTR immunoreactivity was restricted to basal forebrain, as previously described (Kiss et al., 1988; Lee et al., 1998) (data not shown). After pilocarpine-induced seizure, p75NTR immunoreactivity was detected in the piriform cortex, entorhinal cortex, perirhinal cortex, and hippocampus (Fig. 4). Of these regions, entorhinal cortex showed the strongest p75NTR immunoreactivity, particularly 3 d after seizure. At 1 d after seizure, a diffuse increase in immunoreactivity was detected in entorhinal, piriform, and perirhinal cortices. By 3 d after seizure, strong p75NTR immunoreactivity was concentrated in cortical layers II and III and was clearly associated with cell bodies and processes. Immunoreactivity decreased gradually dorsal to perirhinal cortex. The immunoblotting studies did not reveal an increase in p75NTR protein in the cortex 1 d after seizure (Fig. 3), consistent with the restricted cortical expression observed by immunocytochemistry at this time point. In the hippocampus, increased p75NTR expression was observed in the dentate granule cell layer, dentate hilus, and CA1 pyramidal cell layer, and was most prominent 3 d after seizure.
Fig. 4.
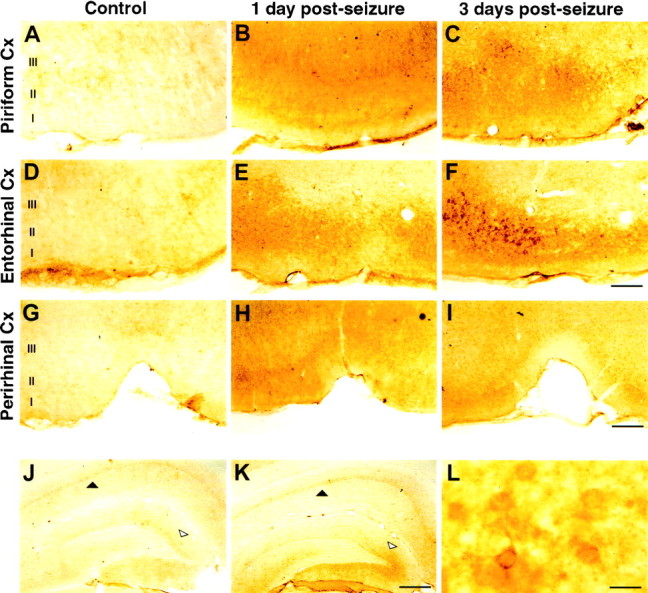
Cellular localization of p75NTR protein after seizure. p75NTR expression visualized with peroxidase/DAB in brain sections from control (A, D, G, J), 1 d (B, E, H), or 3 d after seizure (C, F, I, K). A–C show the piriform cortex,D–F the entorhinal cortex, G–I the perirhinal cortex, and J, K, the hippocampus. L is a higher magnification of the tissue in F showing specifically stained cell bodies and processes. Filled arrowheads identify the hippocampal CA1 pyramidal cell layer. Open arrowheads identify the upper blade of the dentate granule cell layer. Cortical layers are identified as indicated. Cx, Cortex. Scale bars:F, 160 μm; I, 250 μm;K, 400 μm; L, 20 μm.
p75NTR mRNA expression is increased in seized brain
To identify the cellular source of p75NTR expression after pilocarpine seizure, p75NTR mRNA distribution was determined byin situ hybridization in sections taken from pilocarpine-treated animals 1 and 3 d after seizure. Figure5A shows that p75NTR mRNA was readily detected in layers II and III of the entorhinal cortex using an antisense p75NTR cRNA. Within the hippocampus, p75NTR mRNA was detected primarily in the CA1 pyramidal cell layer and dentate granule cell layer, but lower levels were present in the hilus (Fig. 5D). No specific hybridization was observed when a control sense probe was used in the entorhinal cortex or the hippocampus (Fig.5B,E), and sense and antisense p75NTR probes produced no detectable signal in sections of entorhinal cortex and hippocampus from control, nonseized animals (data not shown).
Fig. 5.
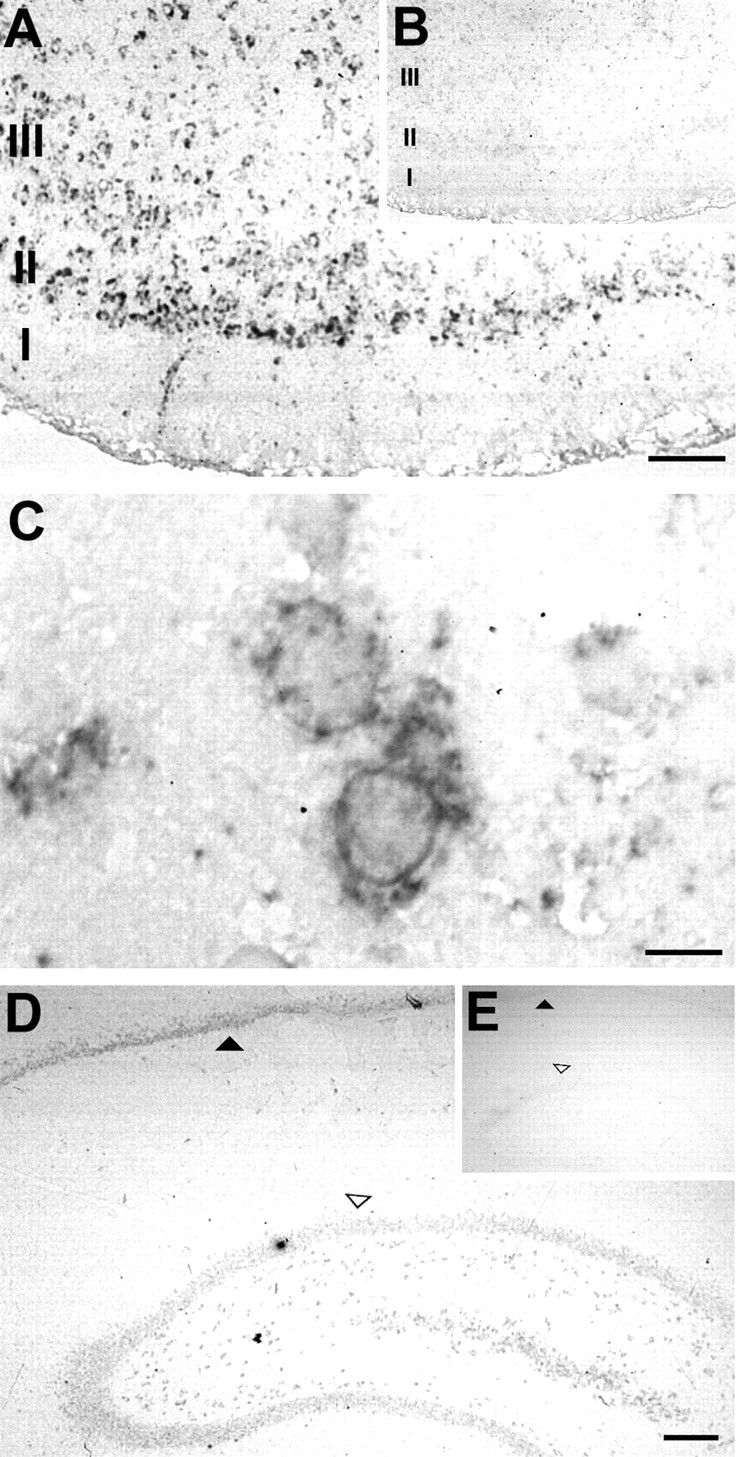
p75NTR mRNA expression after seizure. p75NTR mRNA was detected by in situ hybridization 1 d after seizure in the entorhinal cortex. A, p75NTR mRNA detected with an antisense probe in entorhinal cortex 1 d after seizure. B, No signal was detected using the corresponding sense probe on a adjacent section from the same brain.C, Higher magnification of the tissue inA, illustrating the cytoplasmic localization of the specific hybridization signal. D, p75NTR mRNA detected in the hippocampus 3 d after seizure using the antisense probe, and (E) no signal was detected using the sense probe on an adjacent section from the same brain. Filled arrowheads identify the hippocampal CA1 pyramidal cell layer.Open arrowheads identify the upper blade of the dentate granule cell layer. Cortical layers are indicated. Scale bars:A, 120 μm; C, 12 μm;D, 200 μm.
p75NTR expression is induced after seizure by neurons undergoing apoptosis
To determine if the p75NTR immunoreactive and TUNEL-positive cells were neurons, we first examined the coincidence of TUNEL and NeuN, a neuron-specific epitope (Mullen et al., 1992). Double label immunofluorescence showed that >90% of the TUNEL-positive cells in layers II and III of the entorhinal cortex 3 d after seizure were NeuN-positive, identifying them as neurons (Fig.6).
Fig. 6.

Cells undergoing apoptosis are neurons. Triple immunofluorescence demonstrating colocalization of Hoechst 33258 (A), NeuN (B), and TUNEL (C) in entorhinal cortex 3 d after pilocarpine-induced seizure. NeuN is neuron-specific, but not a pan-neuronal marker (Mullen et al., 1992), suggesting that some proportion of NeuN-negative cells may also be neuronal. Scale bar, 20 μm.
To determine if p75NTR expression correlated with TUNEL labeling of individual cells, colabeling was performed on sections of entorhinal cortex derived from animals 3 d after pilocarpine-induced SE. The mouse monoclonal antibody MC192 was used to detect p75NTR in these experiments because p75NTR-B1 antigenicity was incompatible with the TUNEL reaction. Control studies on serial sections demonstrated that MC192 and p75NTR-B1 produce identical patterns of immunoreactivity (data not shown). Colabeling within the entorhinal cortex demonstrated that most of the TUNEL-positive cells present in layers II and III 3 d after seizure were also p75NTR-immunoreactive (Fig.7A–G). An almost complete coincidence of TUNEL staining with p75NTR immunoreactivity was also observed in the CA1 region of the hippocampus (Fig.7H,I). Cell counts of layers II and III in the entorhinal cortex revealed a strong correlation between p75NTR immunoreactivity and TUNEL (n > 600; Table1): >83% of the cells that were positive for p75NTR were also TUNEL-positive, and >85% of TUNEL-positive cells were immunoreactive for p75NTR (Table 1). Therefore, there was an almost complete overlap between p75NTR expression and the presence of TUNEL-positive nuclei.
Table 1.
Proportion of TUNEL and p75NTR colabeled cells in the entorhinal cortex 3 d after seizure
| TUNEL-positive | p75NTR-positive | p75NTR-negative |
| 530 (100%) | 456 (85.8 ± 7.6%) | 74 (14.2 ± 2.4%) |
| p75NTR-positive | TUNEL-positive | TUNEL-negative |
| 545 (100%) | 456 (83.6 ± 7.4%) | 89 (16.4 ± 1.2%) |
Three days after seizure, brain sections were assayed for nicked DNA using TUNEL followed by p75NTR immunocytochemistry as shown in Figure 5. TUNEL-positive (FITC) and p75NTR-positive (Cy3) cells were counted, and the overlap between the two groups was determined. Values represent the percentage of each group compared to either the total p75NTR or total TUNEL-positive cells ± SE of 12 separate fields derived from two different animals.
DISCUSSION
In this study we demonstrate that pilocarpine-induced seizure produces a large increase in TUNEL-positive neurons in the hippocampal, entorhinal, and piriform cortices and a dramatic rise in cellular DNA cleavage, a hallmark of apoptosis. This regional damage is accompanied by a large increase in levels of p75NTR mRNA and protein in neurons within these same structures. The incidence of TUNEL within individual neurons correlates tightly with p75NTR expression, with >85% of the cells with TUNEL-positive nuclei showing induced p75NTR expression. TUNEL-positive cells are still observed many days after pilocarpine administration, indicating that apoptotic mechanisms, potentially mediated by p75NTR, may contribute to long-term cell loss after status epilepticus.
The relative proportions of necrotic and apoptotic cell death are not known in any seizure model, but both necrosis and apoptotic cell death occur after kainic acid-induced seizure and during kindling (Pollard et al., 1994; Morrison et al., 1996; Bengzon et al., 1997). Histochemical assays have shown that pilocarpine-induced seizure induces cell damage in numerous sites throughout the brain that include the hippocampal gyrus (CA1 and CA3 cell layers), the dentate gyrus (both granule cell and hilar layers), piriform cortex, and entorhinal cortex (Fujikawa, 1996). This pilocarpine-induced cell loss can be inhibited by NMDA antagonists (Rice and DeLorenzo, 1998), suggesting that much of it is triggered by excitotoxic mechanisms. We have found that pilocarpine-induced seizure results in a profound increase in TUNEL reactivity in neurons, particularly in entorhinal and piriform cortices but also within the hippocampus.
Necrosis and apoptosis are defined on the basis of morphological criteria but in mechanistic terms, apoptosis refers to active intracellular signaling that results in cellular suicide. In some systems, dying cells can show morphological features of both necrosis and apoptosis; for example, cells showing morphological hallmarks of necrotic death can also be TUNEL-positive (Charriaut-Marlangue and Ben-Ari, 1995). Pyknotic nuclei, which are characteristic of apoptosis, become numerous after pilocarpine-induced seizure paradigm (Fig. 7) but to confirm that intracellular apoptotic signaling cascades contribute to the cell death that occurs after pilocarpine-induced SE, DNA extracted from various areas of seized brains was examined for the DNA cleavage pattern characteristic of intracellular apoptotic mechanisms. Our demonstration that seizure induced by pilocarpine results in DNA fragmentation that correlates with the region-specific increase in TUNEL staining and pyknotic nuclei indicates that this seizure model results in widespread activation of intracellular apoptotic cascades. Kainate, which is widely used to induce seizure, results in damage primarily to the hippocampus, particularly within CA1, CA3, the hilus, and the subiculum (Morrison et al., 1996); our data suggest that pilocarpine-induced seizure results in a much more profound apoptotic response within the CNS, ultimately resulting in more widespread neuronal damage.
The expression of neurotrophins and trk receptors is regulated by kindling and after chemically-induced seizure. A transient increase in mRNA for NGF, BDNF, trkB, and trkC in the hippocampus and neocortex has been demonstrated during kindling (Ernfors et al., 1991; Bengzon et al., 1993; Merlio et al., 1993), and kainic acid or bicuculline-induced seizure results in increased expression of NGF, BDNF, and trkB mRNA levels (Zafra et al., 1990; Ballarin et al., 1991; Gall et al., 1991b;Isackson et al., 1991; Dugich-Djordjevic et al., 1992, 1995; Humpel et al., 1993; Wetmore et al., 1994). Studies examining NT-3 expression have suggested either no change (Ballarin et al., 1991; Ernfors et al., 1991; Merlio et al., 1993) or a decrease in expression by hippocampal neurons after seizure (Bengzon et al., 1992; Gall, 1992; Rocamora et al., 1992), suggesting that increases in neurotrophin expression are restricted to specific family members. In addition, trkA mRNA is unchanged by kindling or pilocarpine-induced seizure (for review, seeGall, 1993; Persson and Ibanez, 1993; Mudo et al., 1996). Traditionally, the injury-induced increase in neurotrophin expression has been thought to mediate cell survival or synaptic plasticity; however, the recently discovered apoptotic function of p75NTR may require re-evaluation of this hypothesis.
The p75NTR is widely expressed in the nervous system during development, but in the adult CNS, p75NTR expression is limited mainly to magnocellular neurons of the basal forebrain, cells within the caudate putamen, and cerebellar Purkinje cells (for review, see Barker, 1998). The expression profile of p75NTR after chemically-induced seizure has not been previously addressed, but p75NTR mRNA levels are unchanged by kindling (Merlio et al., 1993). p75NTR mRNA levels are increased in some forms of neuronal injury such as in motoneurons after sciatic nerve crush (Ernfors et al., 1989), in adult striatal cholinergic neurons after experimentally induced focal cerebral ischemia (Kokaia et al., 1998), and in Purkinje cells after axotomy (Armstrong et al., 1991; Dusart et al., 1994; Martinez-Murillo et al., 1998). The functional consequences of these changes in p75NTR expression after neuronal trauma are uncertain since p75NTR can, on the one hand, facilitate trkA activation and increase survival effects of the neurotrophins (Barker and Shooter, 1994; Verdi et al., 1994; Ryden et al., 1997) but also play a proapoptotic role (Rabizadeh et al., 1993; Barrett and Bartlett, 1994; Frade et al., 1996; Majdan et al., 1997; Bamji et al., 1998). This contrast in p75NTR function is illustrated by comparing wild-type and p75−/− sympathetic neurons under different experimental conditions. Sympathetic neurons derived from p75−/− mice require increased amounts of NGF to maintain survival (Lee et al., 1994; Ryden et al., 1997), indicating that p75NTR normally facilitates trkA activity, yet p75−/− neurons undergo apoptosis in response to neurotrophin withdrawal considerably more slowly than their wild-type counterparts (Bamji et al., 1998), indicating that p75NTR also normally facilitates apoptosis. It now seems very likely that there is considerable cell and developmental specificity to p75NTR function in vivo, with p75NTR enhancing survival in some circumstances and facilitating apoptosis in others.
We favor the hypothesis that p75NTR is induced after seizure through an activity-dependent mechanism and is then capable of activating apoptotic signaling cascades in response to bound neurotrophin. This model is consistent with findings that show that p75NTR expression is increased by potassium chloride treatment of cultured Purkinje cells (Cohen-Cory et al., 1993), with the action of other related apoptotic receptors, in which regulated receptor expression is necessary and sufficient for the initiation of an apoptotic cascade (Muller et al., 1998; Chan et al., 1999), and with findings that show that p75NTR activates apoptotic pathways in a ligand-dependent manner (Casaccia-Bonnefil et al., 1996; Frade et al., 1996; Majdan et al., 1997; Bamji et al., 1998; Frade and Barde, 1998, 1999).
Signaling pathways activated by p75NTR remain poorly characterized, but likely candidates that may be involved in this cascade include JNK and the p53 tumor suppressor. Mice lacking the gene for JNK3, an isoform of JNK enriched in the CNS, show reduced apoptosis in response to excitotoxic injury (Yang et al., 1997), and although p75NTR-mediated activation of JNK has not been reported in central neurons, JNK is induced after p75NTR activation in oligodendrocytes and sympathetic neurons (Casaccia-Bonnefil et al., 1996; Bamji et al., 1998; Yoon et al., 1998). Our results show that seizure resulted in increased c-Jun immunoreactivity, yet the distribution of both p75NTR and TUNEL staining was considerably more restricted than the increase in c-Jun expression. It is possible that production of the c-Jun protein may be a necessary prerequisite for seizure-induced neuronal apoptosis but is insufficient to mediate apoptosis on its own. Indeed, c-Jun may become phosphorylated by JNK (and therefore active) only in those cells expressing p75NTR. It is noteworthy that after brain ischemia, c-Jun is widely expressed yet c-jun phosphorylation occurs only in a proportion of piriform cortical cells undergoing apoptosis (Herdegen et al., 1998). Several findings also indicate that p53 may be involved in neuronal apoptosis after seizure. Morrison et al. (1996) have shown that hippocampal neurons that are normally lost after kainic acid-induced seizure are protected in mice lacking functional alleles for p53. Furthermore, p53 may be implicated in a p75NTR-dependent apoptotic pathway induced in sympathetic neurons withdrawn from NGF (Aloyz et al., 1998). Given the links between p75NTR, JNK activation, and p53, it will be interesting to test if p75NTR acts as an apoptotic receptor that mediates JNK and/or p53 activation in adult neurons and to determine if p75NTR expression constitutes the first step of a death process triggered by seizure.
The roles of p75NTR in enhancing trk activity and mediating apoptosis are complex, and signaling events evoked by p75NTR are not fully understood. There is a critical need for in vitro andin vivo models that will allow the elucidation of p75NTR function. Here, the concurrence of apoptosis and p75NTR expression observed in the CNS after pilocarpine-induced seizure indicates that analysis of the role of p75NTR in seizure-induced apoptosis will prove useful for identifying both p75NTR signaling mechanisms and the potential contribution of p75NTR to neuronal cell death in vivo.
Footnotes
This work was supported by grants from the Medical Research Council of Canada (MRC), the Neuroscience Network (Canada), and the Fond de la Recherche en Santé du Quebec. P.P.R. and M.A.C. were supported by a Jeanne Timmons Costello studentship and by a Savoy Foundation studentship, respectively. P.A.B. is a Killam Foundation Scholar and a Scholar of the MRC. T.E.K. is a Scholar of the MRC. We thank Drs. Dan McIntyre and Adriana Di Polo for critically reading this manuscript.
Correspondence should be addressed to Philip A. Barker or Timothy E. Kennedy, Centre for Neuronal Survival, Montreal Neurological Institute, McGill University, 3801 University Avenue, Montreal, Quebec, Canada H3A 2B4.
REFERENCES
- 1.Aicardi J, Chevrie JJ. Consequences of status epilepticus in infants and children. Adv Neurol. 1983;34:115–125. [PubMed] [Google Scholar]
- 2.Aloyz RS, Bamji SX, Pozniak CD, Toma JG, Atwal J, Kaplan DR, Miller FD. p53 is essential for developmental neuron death as regulated by the TrkA and p75 neurotrophin receptors. J Cell Biol. 1998;143:1691–1703. doi: 10.1083/jcb.143.6.1691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Armstrong DM, Brady R, Hersh LB, Hayes RC, Wiley RG. Expression of choline acetyltransferase and nerve growth factor receptor within hypoglossal motoneurons following nerve injury. J Comp Neurol. 1991;304:596–607. doi: 10.1002/cne.903040407. [DOI] [PubMed] [Google Scholar]
- 4.Ballarin M, Ernfors P, Lindefors N, Persson H. Hippocampal damage and kainic acid injection induce a rapid increase in mRNA for BDNF and NGF in the rat brain. Exp Neurol. 1991;114:35–43. doi: 10.1016/0014-4886(91)90082-n. [DOI] [PubMed] [Google Scholar]
- 5.Bamji SX, Majdan M, Pozniak CD, Belliveau DJ, Aloyz R, Kohn J, Causing CG, Miller FD. The p75 neurotrophin receptor mediates neuronal apoptosis and is essential for naturally occurring sympathetic neuron death. J Cell Biol. 1998;140:911–923. doi: 10.1083/jcb.140.4.911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Barde YA. Trophic factors and neuronal survival. Neuron. 1989;2:1525–1534. doi: 10.1016/0896-6273(89)90040-8. [DOI] [PubMed] [Google Scholar]
- 7.Barker PA. p75NTR: A study in contrasts. Cell Death Diff. 1998;5:346–356. doi: 10.1038/sj.cdd.4400375. [DOI] [PubMed] [Google Scholar]
- 8.Barker PA, Shooter EM. Disruption of NGF binding to the low affinity neurotrophin receptor, p75LNTR, reduces NGF binding to trkA on PC12 cells. Neuron. 1994;13:203–215. doi: 10.1016/0896-6273(94)90470-7. [DOI] [PubMed] [Google Scholar]
- 9.Barrett GL, Bartlett PF. The p75 nerve growth factor receptor mediates survival or death depending on the stage of sensory neuron development. Proc Natl Acad Sci USA. 1994;91:6501–6505. doi: 10.1073/pnas.91.14.6501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bazan JF. Emerging families of cytokines and receptors. Curr Biol. 1993;3:603–606. doi: 10.1016/0960-9822(93)90009-d. [DOI] [PubMed] [Google Scholar]
- 11.Bengzon J, Soderstrom S, Kokaia Z, Kokaia M, Ernfors P, Persson H, Ebendal T, Lindvall O. Widespread increase of nerve growth factor protein in the rat forebrain after kindling-induced seizures. Brain Res. 1992;587:338–342. doi: 10.1016/0006-8993(92)91016-8. [DOI] [PubMed] [Google Scholar]
- 12.Bengzon J, Kokaia Z, Ernfors P, Kokaia M, Leanza G, Nilsson OG, Persson H, Lindvall O. Regulation of neurotrophin and trkA, trkB and trkC tyrosine kinase receptor messenger RNA expression in kindling. Neuroscience. 1993;53:433–446. doi: 10.1016/0306-4522(93)90207-v. [DOI] [PubMed] [Google Scholar]
- 13.Bengzon J, Kokaia Z, Elmer E, Nanobashvili A, Kokaia M, Lindvall O. Apoptosis and proliferation of dentate gyrus neurons after single and intermittent limbic seizures. Proc Natl Acad Sci USA. 1997;94:10432–10437. doi: 10.1073/pnas.94.19.10432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Braissant O, Wahli W (1998) A simplified in situ hybridization protocol using non-radioactively labeled probes to detect abundant and rare mRNAs on tissue sections. Roche Molecular Biochemicals, Biochemica 1:1016.
- 15.Casaccia-Bonnefil P, Carter BD, Dobrowsky RT, Chao MV. Death of oligodendrocytes mediated by the interaction of nerve growth factor with its receptor p75. Nature. 1996;383:716–719. doi: 10.1038/383716a0. [DOI] [PubMed] [Google Scholar]
- 16.Casaccia-Bonnefil P, Kong H, Chao MV. Neurotrophins: the biological paradox of survival factors eliciting apoptosis. Cell Death Diff. 1998;5:357–364. doi: 10.1038/sj.cdd.4400377. [DOI] [PubMed] [Google Scholar]
- 17.Cavalheiro EA, Leite JP, Bortolotto ZA, Turski WA, Ikonomidou C, Turski L. Long-term effects of pilocarpine in rats: structural damage of the brain triggers kindling and spontaneous recurrent seizures. Epilepsia. 1991;32:778–782. doi: 10.1111/j.1528-1157.1991.tb05533.x. [DOI] [PubMed] [Google Scholar]
- 18.Chan H, Bartos DP, Owen-Schaub LB. Activation-dependent transcriptional regulation of the human Fas promoter requires NF-κ B p50–p65 recruitment. Mol Cell Biol. 1999;19:2098–2108. doi: 10.1128/mcb.19.3.2098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chandler CE, Parsons LM, Hosang M, Shooter EM. A monoclonal antibody modulates the interaction of nerve growth factor with PC12 cells. J Biol Chem. 1984;259:6882–6889. [PubMed] [Google Scholar]
- 20.Charriaut-Marlangue C, Ben-Ari Y. A cautionary note on the use of TUNEL stain to determine apoptosis. NeuroReport. 1995;7:61–64. [PubMed] [Google Scholar]
- 21.Cohen-Cory S, Elliott RC, Dreyfus CF, Black IB. Depolarizing influences increase low-affinity NGF receptor gene expression in cultured Purkinje neurons. Exp Neurol. 1993;119:165–175. doi: 10.1006/exnr.1993.1018. [DOI] [PubMed] [Google Scholar]
- 22.Dobrowsky RT, Werner MH, Castellino AM, Chao MV, Hannun YA. Activation of the sphingomyelin cycle through the low affinity neurotrophin receptor. Science. 1994;265:1596–1598. doi: 10.1126/science.8079174. [DOI] [PubMed] [Google Scholar]
- 23.Dobrowsky RT, Jenkins GM, Hannun YA. Neurotrophins induce sphingomyelin hydrolysis - modulation by co-expression of p75(ntr) with trk receptors. J Biol Chem. 1995;270:22135–22142. doi: 10.1074/jbc.270.38.22135. [DOI] [PubMed] [Google Scholar]
- 24.Dragunow M, Preston K. The role of inducible transcription factors in apoptotic nerve cell death. Brain Res Rev. 1995;21:1–28. doi: 10.1016/0165-0173(95)00003-l. [DOI] [PubMed] [Google Scholar]
- 25.Dragunow M, Young D, Hughes P, MacGibbon G, Lawlor P, Singleton K, Sirimanne E, Beilharz E, Gluckman P. Is c-Jun involved in nerve cell death following status epilepticus and hypoxic-ischaemic brain injury? Mol Brain Res. 1993;18:347–352. doi: 10.1016/0169-328x(93)90101-t. [DOI] [PubMed] [Google Scholar]
- 26.Dugich-Djordjevic MM, Tocco G, Willoughby DA, Najm I, Pasinetti G, Thompson RF, Baudry M, Lapchak PA, Hefti F. BDNF mRNA expression in the developing rat brain following kainic acid-induced seizure activity. Neuron. 1992;8:1127–1138. doi: 10.1016/0896-6273(92)90133-x. [DOI] [PubMed] [Google Scholar]
- 27.Dugich-Djordjevic MM, Ohsawa F, Okazaki T, Mori N, Day JR, Beck KD, Hefti F. Differential regulation of catalytic and non-catalytic trkB messenger RNAs in the rat hippocampus following seizures induced by systemic administration of kainate. Neuroscience. 1995;66:861–877. doi: 10.1016/0306-4522(94)00631-e. [DOI] [PubMed] [Google Scholar]
- 28.Dusart I, Morel MP, Sotelo C. Parasagittal compartmentation of adult rat Purkinje cells expressing the low-affinity nerve growth factor receptor: changes of pattern expression after a traumatic lesion. Neuroscience. 1994;63:351–356. doi: 10.1016/0306-4522(94)90534-7. [DOI] [PubMed] [Google Scholar]
- 29.Ernfors P, Henschen A, Olson L, Persson H. Expression of nerve growth factor receptor mRNA is developmentally regulated and increased after axotomy in rat spinal cord motoneurons. Neuron. 1989;2:1605–1613. doi: 10.1016/0896-6273(89)90049-4. [DOI] [PubMed] [Google Scholar]
- 30.Ernfors P, Bengzon J, Kokaia Z, Persson H, Lindvall O. Increased levels of messenger RNAs for neurotrophic factors in the brain during kindling epileptogenesis. Neuron. 1991;7:165–176. doi: 10.1016/0896-6273(91)90084-d. [DOI] [PubMed] [Google Scholar]
- 31.Frade JM, Barde YA. Nerve growth factor: two receptors, multiple functions. Bioessays. 1998;20:137–145. doi: 10.1002/(SICI)1521-1878(199802)20:2<137::AID-BIES6>3.0.CO;2-Q. [DOI] [PubMed] [Google Scholar]
- 32.Frade JM, Barde YA. Genetic evidence for cell death mediated by nerve growth factor and the neurotrophin receptor p75 in the developing mouse retina and spinal cord. Development. 1999;126:683–690. doi: 10.1242/dev.126.4.683. [DOI] [PubMed] [Google Scholar]
- 33.Frade JM, Rodriguez-Tebar A, Barde YA. Induction of cell death by endogenous nerve growth factor through its p75 receptor. Nature. 1996;383:166–168. doi: 10.1038/383166a0. [DOI] [PubMed] [Google Scholar]
- 34.Fujikawa DG. The temporal evolution of neuronal damage from pilocarpine-induced status epilepticus. Brain Res. 1996;725:11–22. doi: 10.1016/0006-8993(96)00203-x. [DOI] [PubMed] [Google Scholar]
- 35.Gall CM. Regulation of brain neurotrophin by physiological activity. Trends Pharmacol Sci. 1992;13:401–403. doi: 10.1016/0165-6147(92)90123-n. [DOI] [PubMed] [Google Scholar]
- 36.Gall CM. Seizure-induced changes in neurotrophin expression: implications for epilepsy. Exp Neurol. 1993;124:150–166. doi: 10.1006/exnr.1993.1186. [DOI] [PubMed] [Google Scholar]
- 37.Gall C, Lauterborn J, Bundman M, Murray K, Isackson P. Seizures and the regulation of neurotrophic factor and neuropeptide gene expression in brain. Epilepsy Res [Suppl] 1991a;4:225–245. [PubMed] [Google Scholar]
- 38.Gall C, Murray K, Isackson PJ. Kainic acid-induced seizures stimulate increased expression of nerve growth factor mRNA in rat hippocampus. Mol Brain Res. 1991b;9:113–123. doi: 10.1016/0169-328x(91)90136-l. [DOI] [PubMed] [Google Scholar]
- 39.Gavrieli Y, Sherman Y, Ben-Sasson SA. Identification of programmed cell death in situ via specific labeling of nuclear DNA fragmentation. J Cell Biol. 1992;119:493–501. doi: 10.1083/jcb.119.3.493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Herdegen T, Skene P, Bahr M. The c-Jun transcription factor–bipotential mediator of neuronal death, survival and regeneration. Trends Neurosci. 1997;20:227–231. doi: 10.1016/s0166-2236(96)01000-4. [DOI] [PubMed] [Google Scholar]
- 41.Herdegen T, Claret FX, Kallunki T, Martin-Villalba A, Winter C, Hunter T, Karin M. Lasting N-terminal phosphorylation of c-Jun and activation of c-Jun N-terminal kinases after neuronal injury. J Neurosci. 1998;18:5124–5135. doi: 10.1523/JNEUROSCI.18-14-05124.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Humpel C, Wetmore C, Olson L. Regulation of brain-derived neurotrophic factor messenger RNA and protein at the cellular level in pentylenetetrazol-induced epileptic seizures. Neuroscience. 1993;53:909–918. doi: 10.1016/0306-4522(93)90476-v. [DOI] [PubMed] [Google Scholar]
- 43.Isackson PJ, Huntsman MM, Murray KD, Gall CM. BDNF mRNA expression is increased in adult rat forebrain after limbic seizures: temporal patterns of induction distinct from NGF. Neuron. 1991;6:937–948. doi: 10.1016/0896-6273(91)90234-q. [DOI] [PubMed] [Google Scholar]
- 44.Kaplan DR, Miller FD. Signal transduction by the neurotrophin receptors. Curr Opin Cell Biol. 1997;9:213–221. doi: 10.1016/s0955-0674(97)80065-8. [DOI] [PubMed] [Google Scholar]
- 45.Kiss J, McGovern J, Patel AJ. Immunohistochemical localization of cells containing nerve growth factor receptors in the different regions of the adult rat forebrain. Neuroscience. 1988;27:731–748. doi: 10.1016/0306-4522(88)90179-0. [DOI] [PubMed] [Google Scholar]
- 46.Kokaia Z, Andsberg G, Martinez-Serrano A, Lindvall O. Focal cerebral ischemia in rats induces expression of p75 neurotrophin receptor in resistant striatal cholinergic neurons. Neuroscience. 1998;84:1113–1125. doi: 10.1016/s0306-4522(97)00579-4. [DOI] [PubMed] [Google Scholar]
- 47.Laemmli UK. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature. 1970;227:680–685. doi: 10.1038/227680a0. [DOI] [PubMed] [Google Scholar]
- 48.Lee KF, Davies AM, Jaenisch R. p75-deficient embryonic dorsal root sensory and neonatal sympathetic neurons display a decreased sensitivity to NGF. Development. 1994;120:1027–1033. doi: 10.1242/dev.120.4.1027. [DOI] [PubMed] [Google Scholar]
- 49.Lee TH, Kato H, Pan LH, Ryu JH, Kogure K, Itoyama Y. Localization of nerve growth factor, trkA and p75 immunoreactivity in the hippocampal formation and basal forebrain of adult rats. Neuroscience. 1998;83:335–349. doi: 10.1016/s0306-4522(97)00346-1. [DOI] [PubMed] [Google Scholar]
- 50.Mahadeo D, Kaplan L, Chao MV, Hempstead BL. High affinity nerve growth factor binding displays a faster rate of association than p140trk binding. Implications for multi-subunit polypeptide receptors. J Biol Chem. 1994;269:6884–6891. [PubMed] [Google Scholar]
- 51.Majdan M, Lachance C, Gloster A, Aloyz R, Zeindler C, Bamji S, Bhakar A, Belliveau D, Fawcett J, Miller FD, Barker PA. Transgenic mice expressing the intracellular domain of the p75 neurotrophin receptor undergo neuronal apoptosis. J Neurosci. 1997;17:6988–6998. doi: 10.1523/JNEUROSCI.17-18-06988.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Martinez-Murillo R, Fernandez AP, Bentura ML, Rodrigo J. Subcellular localization of low-affinity nerve growth factor receptor-immunoreactive protein in adult rat purkinje cells following traumatic injury. Exp Brain Res. 1998;119:47–57. doi: 10.1007/s002210050318. [DOI] [PubMed] [Google Scholar]
- 53.Mello LE, Cavalheiro EA, Tan AM, Kupfer WR, Pretorius JK, Babb TL, Finch DM. Circuit mechanisms of seizures in the pilocarpine model of chronic epilepsy: cell loss and mossy fiber sprouting. Epilepsia. 1993;34:985–995. doi: 10.1111/j.1528-1157.1993.tb02123.x. [DOI] [PubMed] [Google Scholar]
- 54.Merlio JP, Ernfors P, Kokaia Z, Middlemas DS, Bengzon J, Kokaia M, Smith ML, Siesjo BK, Hunter T, Lindvall O, Persson H. Increased production of the TrkB protein tyrosine kinase receptor after brain insults. Neuron. 1993;10:151–164. doi: 10.1016/0896-6273(93)90307-d. [DOI] [PubMed] [Google Scholar]
- 55.Morrison RS, Wenzel HJ, Kinoshita Y, Robbins CA, Donehower LA, Schwartzkroin PA. Loss of the p53 tumor suppressor gene protects neurons from kainate-induced cell death. J Neurosci. 1996;16:1337–1345. doi: 10.1523/JNEUROSCI.16-04-01337.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Mudo G, Jiang XH, Timmusk T, Bindoni M, Belluardo N. Change in neurotrophins and their receptor mRNAs in the rat forebrain after status epilepticus induced by pilocarpine. Epilepsia. 1996;37:198–207. doi: 10.1111/j.1528-1157.1996.tb00012.x. [DOI] [PubMed] [Google Scholar]
- 57.Mullen RJ, Buck CR, Smith AM. NeuN, a neuronal specific nuclear protein in vertebrates. Development. 1992;116:201–211. doi: 10.1242/dev.116.1.201. [DOI] [PubMed] [Google Scholar]
- 58.Muller M, Wilder S, Bannasch D, Israeli D, Lehlbach K, Li-Weber M, Friedman SL, Galle PR, Stremmel W, Oren M, Krammer PH. p53 activates the CD95 (APO-1/Fas) gene in response to DNA damage by anticancer drugs. J Exp Med. 1998;188:2033–2045. doi: 10.1084/jem.188.11.2033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Olney JW, Collins RC, Sloviter RS. Excitotoxic mechanisms of epileptic brain damage. Adv Neurol. 1986;44:857–877. [PubMed] [Google Scholar]
- 60.Persson H, Ibanez CF. Role and expression of neurotrophins and the trk family of tyrosine kinase receptors in neural growth and rescue after injury. Curr Opin Neurol Neurosurg. 1993;6:11–18. [PubMed] [Google Scholar]
- 61.Pollard H, Charriaut-Marlangue C, Cantagrel S, Represa A, Robain O, Moreau J, Ben-Ari Y. Kainate-induced apoptotic cell death in hippocampal neurons. Neuroscience. 1994;63:7–18. doi: 10.1016/0306-4522(94)90003-5. [DOI] [PubMed] [Google Scholar]
- 62.Priel MR, dos Santos NF, Cavalheiro EA. Developmental aspects of the pilocarpine model of epilepsy. Epilepsy Res. 1996;26:115–121. doi: 10.1016/s0920-1211(96)00047-2. [DOI] [PubMed] [Google Scholar]
- 63.Rabizadeh S, Oh J, Zhong LT, Yang J, Bitler CM, Butcher LL, Bredesen DE. Induction of apoptosis by the low-affinity NGF receptor. Science. 1993;261:345–248. doi: 10.1126/science.8332899. [DOI] [PubMed] [Google Scholar]
- 64.Racine RJ. Modification of seizure activity by electrical stimulation. II. Motor seizure. Electroencephalogr Clin Neurophysiol. 1972;32:281–294. doi: 10.1016/0013-4694(72)90177-0. [DOI] [PubMed] [Google Scholar]
- 65.Radeke MJ, Misko TP, Hsu C, Herzenberg LA, Shooter EM. Gene transfer and molecular cloning of the rat nerve growth factor receptor. Nature. 1987;325:593–597. doi: 10.1038/325593a0. [DOI] [PubMed] [Google Scholar]
- 66.Represa A, Niquet J, Pollard H, Ben-Ari Y. Cell death, gliosis, and synaptic remodeling in the hippocampus of epileptic rats. J Neurobiol. 1995;26:413–425. doi: 10.1002/neu.480260313. [DOI] [PubMed] [Google Scholar]
- 67.Rice AC, DeLorenzo RJ. NMDA receptor activation during status epilepticus is required for the development of epilepsy. Brain Res. 1998;782:240–247. doi: 10.1016/s0006-8993(97)01285-7. [DOI] [PubMed] [Google Scholar]
- 68.Rocamora N, Palacios JM, Mengod G. Limbic seizures induce a differential regulation of the expression of nerve growth factor, brain-derived neurotrophic factor and neurotrophin-3, in the rat hippocampus. Mol Brain Res. 1992;13:27–33. doi: 10.1016/0169-328x(92)90041-9. [DOI] [PubMed] [Google Scholar]
- 69.Ryden M, Hempstead B, Ibanez CF. Differential modulation of neuron survival during development by nerve growth factor binding to the p75 neurotrophin receptor. J Biol Chem. 1997;272:16322–16328. doi: 10.1074/jbc.272.26.16322. [DOI] [PubMed] [Google Scholar]
- 70.Sankar R, Shin DH, Liu H, Mazarati A, de Vasconcelos AP, Wasterlain CG. Patterns of status epilepticus-induced neuronal injury during development and long-term consequences. J Neurosci. 1998;18:8382–8393. doi: 10.1523/JNEUROSCI.18-20-08382.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Sgonc R, Boeck G, Dietrich H, Gruber J, Recheis H, Wick G. Simultaneous determination of cell surface antigens and apoptosis. Trends Genet. 1994;10:41–42. doi: 10.1016/0168-9525(94)90140-6. [DOI] [PubMed] [Google Scholar]
- 72.Sloviter RS. Hippocampal pathology and pathophysiology in temporal lobe epilepsy. Neurologia. 1996;11:29–32. [PubMed] [Google Scholar]
- 73.Treiman DM. Status epilepticus. Baillieres Clin Neurol. 1996;5:821–839. [PubMed] [Google Scholar]
- 74.Turski WA, Cavalheiro EA, Schwarz M, Czuczwar SJ, Kleinrok Z, Turski L. Limbic seizures produced by pilocarpine in rats: behavioural, electroencephalographic and neuropathological study. Behav Brain Res. 1983;9:315–335. doi: 10.1016/0166-4328(83)90136-5. [DOI] [PubMed] [Google Scholar]
- 75.Turski L, Meldrum BS, Cavalheiro EA, Calderazzo-Filho LS, Bortolotto ZA, Ikonomidou-Turski C, Turski WA. Paradoxical anticonvulsant activity of the excitatory amino acid N-methyl-d-aspartate in the rat caudate-putamen. Proc Natl Acad Sci USA. 1987;84:1689–1693. doi: 10.1073/pnas.84.6.1689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Verdi JM, Birren SJ, Ibanez CF, Persson H, Kaplan DR, Benedetti M, Chao MV, Anderson DJ. P75LNGFR regulates Trk signal transduction and NGF-induced neuronal differentiation in MAH cells. Neuron. 1994;12:733–745. doi: 10.1016/0896-6273(94)90327-1. [DOI] [PubMed] [Google Scholar]
- 77.Wetmore C, Olson L, Bean AJ. Regulation of brain-derived neurotrophic factor (BDNF) expression and release from hippocampal neurons is mediated by non-NMDA type glutamate receptors. J Neurosci. 1994;14:1688–1700. doi: 10.1523/JNEUROSCI.14-03-01688.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Yang DD, Kuan CY, Whitemarsh AJ, Rincon M, Zheng TS, Davis RJ, Rakic P, Flavell RA. Absence of excitotoxicity-induced apoptosis in the hippocampus of mice lacking the jnk3 gene. Nature. 1997;389:865–870. doi: 10.1038/39899. [DOI] [PubMed] [Google Scholar]
- 79.Yoon SO, Casaccia-Bonnefil P, Carter DB, Chao MV. Competitive signaling between TrkA and p75 nerve growth factor receptors determines cell survival. J Neurosci. 1998;18:3273–3281. doi: 10.1523/JNEUROSCI.18-09-03273.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Zafra F, Hengerer B, Leibrock J, Thoenen H, Lindholm D. Activity dependent regulation of BDNF and NGF mRNAs in the rat hippocampus is mediated by non-NMDA glutamate receptors. EMBO J. 1990;9:3545–3550. doi: 10.1002/j.1460-2075.1990.tb07564.x. [DOI] [PMC free article] [PubMed] [Google Scholar]