

Abstract
The main olfactory bulb is a critical relay step between the olfactory epithelium and the olfactory cortex. A marked feature of the bulb is its massive innervation by cholinergic inputs from the basal forebrain. In this study, we addressed the functional interaction between cholinergic inputs and intrinsic bulbar circuitry. Determining the roles of acetylcholine (ACh) requires the characterization of cholinergic effects on both neural excitability and synaptic transmission. For this purpose, we used electrophysiological techniques to localize and characterize the diverse roles of ACh in mouse olfactory bulb slices. We found that cholinergic inputs have a surprising number of target receptor populations that are expressed on three different neuronal types in the bulb. Specifically, nicotinic acetylcholine receptors excite both the output neurons of the bulb, i.e., the mitral cells, as well as interneurons located in the periglomerular regions. These nicotine-induced responses in interneurons are short lasting, whereas responses in mitral cells are long lasting. In contrast, muscarinic receptors have an inhibitory effect on the firing rate of interneurons from a deeper layer, granule cells, while at the same time they increase the degree of activity-independent transmitter release from these cells onto mitral cells.
Cholinergic signaling thus was found to have multiple and opposing roles in the olfactory bulb. These dual cholinergic effects on mitral cells and interneurons may be important in modulating olfactory bulb output to central structures required for driven behaviors and may be relevant to understanding mechanisms underlying the perturbations of cholinergic inputs to cortex that occur in Alzheimer's disease.
Keywords: mitral cells, inhibitory interneurons, basal forebrain, GABA, nicotinic receptors, muscarinic receptors, olfaction
Considerable evidence exists for an extrinsic cholinergic influence in the maturation and function of the main olfactory bulb (Halász and Shepherd, 1983; Le Jeune et al., 1996). It is also well established that a number of neurodegenerative disorders related to cholinergic systems are accompanied by olfactory dysfunctions (Serby et al., 1985; Kovacs et al., 1998) (for review, seeDoty, 1991; Quirion, 1993; Kasa et al., 1997). Indeed, the presence of cholinergic centrifugal fibers originating from the magnocellular basal forebrain nuclei [the substantia innominata and the medial part of the horizontal limb of the diagonal band of Broca (HDB)] has been revealed in the olfactory bulb (Shute and Lewis, 1975; Macrides et al., 1981). In vertebrates, the olfactory bulb is the first relay for olfactory processing, receiving information from the olfactory epithelium and conveying it to higher brain structures via its principal neurons, the mitral and tufted cells. These neurons interact with two classes of local inhibitory interneurons: (1) periglomerular cells that make synapses onto the primary dendrites of mitral/tufted cells, and (2) granule cells that release GABA onto mitral cell secondary dendrites (Ribak et al., 1977; Nowycky et al., 1981; Jahr and Nicoll, 1982). Cholinergic inputs were once thought to terminate predominantly onto granule spines, leading to the conclusion that the main role of acetylcholine (ACh) is to modulate granule cell inhibition of mitral cells (for review, see Halász and Shepherd, 1983; Macrides and Davis, 1983). However, more recent anatomical observations have reported significant cholinergic innervation of several layers of the olfactory bulb (Kasa et al., 1995), which led us to hypothesize far more varied roles for ACh.
Although behavioral studies have stressed the importance of cholinergic inputs in olfactory memory (Ravel et al., 1994; Lévy et al., 1995), the sites and mechanisms of cholinergic action have not been clearly defined. Electrophysiological experiments have yielded contradictory results. For example, electrical stimulation of the HDBin vivo was reported either to depress (Nickell and Shipley, 1988b) or to increase (Kunze et al., 1991) mitral cell firing through cholinergic modulation of GABAergic inhibition. In addition, application of ACh in vivo either decreased or increased mitral cell firing activity (Ravel et al., 1990; Elaagouby and Gervais, 1992).
The effects of ACh in the CNS were long thought to be mediated only by muscarinic ACh receptors (mAChRs), because their pharmacological blockade results in serious disruption of multiple brain functions (Ghonheim and Mewaldt, 1975) (for review, see Hagan and Moris, 1989). However, nicotinic AChRs (nAChRs) have been shown in recent years to play major roles in the CNS as well (Levin, 1992; Changeux et al., 1998). In this study, we took advantage of the slice preparation to explore the effects of the activation of both nicotinic and muscarinic ACh receptors on cell excitability and synaptic transmission in the olfactory bulb network. We report opposite effects of cholinergic modulation of excitability of mitral and granule cells. We have also found opposite modulatory action of ACh on GABAergic synaptic inputs onto granule and mitral cells; the latter may be critical for olfactory processing and learning (Shepherd and Greer, 1998). All of these cholinergic actions in conjunction with correlated anatomical findings suggest specific roles of the cholinergic system in odor processing as well as in olfactory learning.
MATERIALS AND METHODS
Slice preparation. Experiments were performed on olfactory bulb slices obtained from 2- to 5-week-old C57/Bl6 mice. Bulbs were rapidly removed and immediately placed in 4°C oxygenated artificial CSF (ACSF) containing (in mm): 124 NaCl, 3 KCl, 2 CaCl2, 1.3 MgCl2, 25 NaHCO3, 1.25 NaH2PO4, 10d-glucose, pH 7.3, when bubbled with 95% O2/5% CO2 (310 mOsm). Horizontal slices (300 μm) were cut with a vibrating microslicer (Vibratome 1000; TPI, St. Louis, MO) and kept in oxygenated ACSF at 32°C for ∼30 min. Subsequently, the slices were stored at room temperature (20–22°C) for 4–6 hr. Individual slices were then transferred to a submerged-slice recording chamber where they were superfused with oxygenated ACSF (same composition as above) at a rate of 2–3 ml/min. The horizontal plane was chosen because this orientation preserves the primary and secondary dendrites of mitral cells.
Electrophysiological recordings. Recordings were performed using the patch-clamp technique in the cell-attached and whole-cell configurations with a RK300 patch-clamp amplifier (Biological Science Instruments, Claix, France). Cells were recorded under visual control with an upright Zeiss Axioskop microscope (40× water immersion objective lens; 0.75 numerical aperture) and infrared differential interference contrast videomicroscopy (IR-DIC; filter, 850 nm). Images of neurons were captured and processed with an image processor (PXC200 Precision Color, Imagination). Mitral cells, periglomerular interneurons, and granule cells were easily discriminated based on morphology and location (Shepherd and Greer, 1998). To confirm visual identification of recorded neurons, a subset of cells was filled with Lucifer yellow (lithium salt from Sigma, St. Louis, MO) in the whole-cell recording configuration. The morphology of the patched cells was then sketched after recordings. All experiments were performed at room temperature (22–24°C). Microelectrodes with access resistance of ∼6 and 12 MΩ were used to record mitral and granule cells, respectively. An internal solution of the following composition was used (in mm): 123 Cs-gluconate, 10 CsCl, 8 NaCl, 1 CaCl2, 10 Cs-EGTA, 10 Na-HEPES, 10d-glucose, 0.3 GTP, 2 Mg-ATP, 0.2 AMPc, pH 7.3 (280 mOsm). Unless indicated otherwise, IPSCs were recorded either in the presence of the AMPA receptor antagonist 1,2,3,4-tetrahydro-6-nitro-2,3-dioxobenzo[f]quinoxaline-7-sulfonamide (NBQX; 10–20 μm), with the NMDA receptor antagonistd,l-2-amino-5-phosphonopentanoic acid (d,l-APV; 100 μm), or in the presence of a nonspecific ionotropic glutamate receptor antagonist, kynurenic acid (5 mm).
Data analysis. Synaptic responses were filtered at 1–5 kHz with an eight-pole Bessel filter, digitized at 4 kHz on a TL-1 interface (Axon Instruments, Foster City, CA), and collected on an IBM-compatible computer. On- and off-line data analyses were performed with Acquis1 (Gérard Sadoc, Centre National de la Recherche Scientifique-Agence Nationale pour la Valorisation de la Recherche, Paris, France). The frequency and amplitude of IPSCs were determined off-line, and our detection threshold was set at a di/dt of 10 pA/msec with minimum and maximum rise times set at 0.1 and 4 msec, respectively. The minimal acceptable amplitude for a miniature event was 6 pA. Statistical significance of differences between means was assessed using Student's ttest. Kolmogorov–Smirnov statistics were used to determine whether there were statistically significant changes in the frequency and amplitude of IPSCs. The level of significance was set atp < 0.05 unless indicated otherwise.
To investigate cholinergic effects in the cell-attached mode, a stable frequency of firing was recorded for at least 100 sec to establish a baseline expressed as mean firing rate (defined as 100%). During drug applications, effects were then quantified as a maximum firing frequency relative to baseline for mitral cells and mean for frequency relative to baseline for interneurons. For whole-cell recordings, the reported membrane potentials were corrected for junction potentials, which were measured to be 11 mV, for the solutions used. During all experiments, the access resistance (Ra) and the input membrane resistance (Rm) were constantly monitored, and acquisition was terminated when these parameters changed more than 15%. Capacitance compensation was used to decrease the charging time of the pipette. Because of the small voltage error from theRa, no series resistance compensation was used. Group measurements were expressed as mean ± SEM.
Solution and drugs. Drug solutions were bath-applied using a gravity-driven perfusion system, and maximally effective concentrations of the drugs were achieved within 30 sec (see Fig.1B for time course of wash-in). NBQX,d,l-APV and oxotremorine were from Tocris; all other drugs and salts were purchased from Sigma.
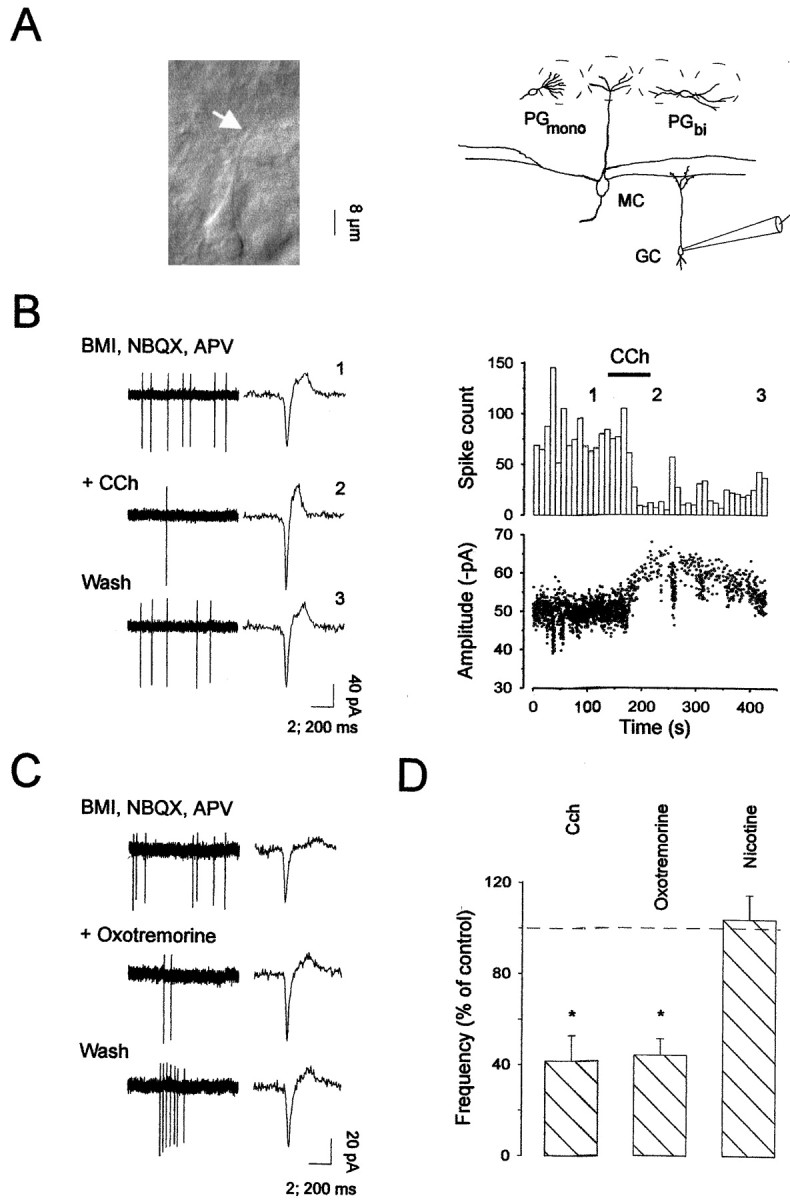
Fig. 1.

Activation of nicotinic receptors enhances mitral cell firing frequency in the main olfactory bulb. A(left), Infrared image of a mitral cell visualized in cell-attached mode under IR-DIC microscopy (arrowindicates the location of the patch electrode) and schema (right) illustrating the location of the recording pipette and the major cell types analyzed in the present study.MC, Mitral cell; GC, granule cell;PGmono, monopolar periglomerular cell; PGbi, bipolar periglomerular cell.B (left), Extracellular recording in cell-attached mode from a mitral cell, in normal conditions (STD), during carbachol (CCh, 50 μm) application, and after drug washout (Wash). Single action potentials are shown on theright side of each trace at an expanded time scale (note the different time calibration bar). B(right), The time courses for changes in action potential frequency (bin size: 10 sec) and amplitude after bath application of CCh are shown (numbersindicate the times of traces chosen for illustration in the left panel). C, Mitral cell firing activity in the presence of BMI (20 μm), NBQX (10 μm), and d,l-APV (100 μm) was increased after bath application of 30 μm nicotine. This nicotine-induced effect was reversible on washout of the drug (Wash).D, Average of the firing frequency changes induced by cholinergic drug applications in different treatments and normalized to the control responses. Changes in frequency were compared between control conditions and drug applications using a pairedt test. Ago: 50 μmcarbachol or 30 μm nicotine (*p< 0.05; **p < 0.01).
RESULTS
During the course of this study, whole-cell or cell-attached patch-clamp recordings were performed on 142 mitral cells (principal neurons) and different subtypes of interneurons: 9 bipolar periglomerular cells, 6 monopolar periglomerular cells, and 75 granule cells. Successfully patched mitral cells and interneurons were held for periods ranging from 15 min to >1 hr. Membrane resistances ranged from 89 to 325 MΩ (mean of 153 ± 10 MΩ) for mitral cells and were two- to eightfold higher in interneurons (mean of 703 ± 46 MΩ). The location and morphometric features used here to characterize each interneuron were consistent with previous morphological classifications (Schneider and Macrides, 1978) (for review, see Mori, 1987; Shepherd and Greer, 1998).
Activation of nicotinic receptors increases mitral cell firing frequency
To determine whether cholinergic receptor activation modulates neuronal excitability in the main olfactory bulb, we first assessed the firing activity of mitral cells in the cell-attached mode, and the effects of different cholinergic agonists were evaluated (Fig.1A; see the recording arrangement). All recordings from the somata of visually identified mitral cells showed spontaneous firing activity under our standard conditions (Fig.1B). The firing rates for mitral cells ranged from 1 to 11 Hz, with a mean of 5.5 ± 1.3 Hz (n = 7), in good agreement with previous in vivo reports (Pager, 1983;Motokizawa, 1996). The application of 10 μmNBQX, 100 μmd,l-APV, and 20 μm bicuculline methiodide (BMI), to block AMPA, NMDA, and GABAA receptors, respectively, only slightly reduced the basal firing rate of mitral cells, indicating that mitral cells were able to fire action potentials spontaneously even in the absence of fast glutamatergic and GABAergic synaptic inputs (n = 28). The broad-spectrum cholinergic receptor agonist carbachol was bath-applied to olfactory bulb slices at concentrations ranging from 10 to 100 μm. As illustrated in Figure 1B,D, application of 50 μm carbachol caused a dramatic increase in the frequency of spontaneous action potential discharge of all neurons recorded in standard conditions (a mean rate of 5.5 Hz, range 1–11 Hz in control conditions and a mean of 28 Hz, range 9–54 Hz in the presence of cholinergic agonist;p < 0.015; n = 8). This carbachol-induced response was fully reversible with washout of the drug (Fig. 1B). Previous studies have shown that cholinergic receptors can cause the release of neurotransmitters through the activation of ACh receptors on the presynaptic terminals (Wonnacott, 1997). We sought to determine whether the carbachol responses we observed were caused by the direct activation of postsynaptic receptors or indirectly through the release of neurotransmitter onto the mitral cells. Blocking ionotropic glutamate receptors (NBQX, 10 μm andd,l-APV, 100 μm) and GABAA receptors (BMI, 20 μm) did not inhibit carbachol responses (n = 5; p > 0.05), suggesting a direct effect onto mitral cells (Fig. 1D). In the presence of these synaptic blockers, application of 30 μm nicotine (Fig. 1C) mimicked the carbachol effect (n = 8). The mean frequency was increased from 6 Hz (range 0.9–10 Hz) to 58 Hz, (range 19–95 Hz;n = 8) after nicotinic application (Fig.1D). Because there are many subtypes of neuronal nicotinic receptors in the CNS (for review, see Wonnacott, 1997;Changeux et al., 1998), we used the nicotinic receptor antagonist mecamylamine at a concentration unlikely to distinguish between these different subtypes. We found that 50 μmmecamylamine completely blocked the enhancing actions of carbachol or nicotine on mitral cell firing frequency (n = 4) (Fig.1D). Moreover, oxotremorine (50 μm), used as a specific mAChR agonist, did not change the cell firing rate (n = 4; data not shown).
The spike amplitude reduction observed after nAChR activation (Fig.1B, right) strongly suggested that the firing activity enhancement was caused by a membrane potential depolarization. In agreement with this interpretation, bath application of nicotine at concentrations ranging from 10 to 100 μm always elicited an inward current accompanied by an increase in noise (Fig.2) (n = 42). In standard medium, the mean maximum amplitude of the response induced by nicotine application (30 μm) was −645 ± 138 pA (range from −238 to −1382 pA; n = 11) at −80 mV. As illustrated in Figure 2A, these responses were dose dependent and specifically blocked by mecamylamine (50 μm; n = 4). Rundown of the nicotine-induced current was not observed during the course of these experiments using our pipette medium (Fig. 2A1). This nicotine-induced current was also mimicked by 50 μm carbachol application (data not shown) and was revealed to be a direct response of mitral cells. Hence, nicotine (30 μm) still evoked a significant inward current (range from −113 to −570 pA; n = 17) in the presence of BMI and NBQX–APV (Fig. 2B1), as well as in the presence of blockers of voltage-dependent sodium channels (TTX, 1 μm) and voltage-dependent calcium channels (200 μm Cd2+ plus a mixture of external high Mg2+/0 Ca2+ ratio) (mean current: −244 ± 41 pA; n = 15) (Fig. 2B2), arguing for a direct effect on mitral cells. Altogether, these findings support the existence of functional nicotinic receptors on mitral cells that enhance their firing activity by directly depolarizing the membrane potential.
Fig. 2.

Characterization of nicotine-induced inward currents in mitral cells. A, Repeated applications of nicotine consistently elicited an inward current. Two consecutive bath applications of 100 μm nicotine showed no evidence of rundown (1). This current showed dose dependency (2) and was blocked by 50 μmmecamylamine (3). Sample recordings were taken from three different cells bathed in standard medium. B, The nicotine-induced current was also observed in the presence of a cocktail that included 20 μm BMI, 10 μmNBQX, and 100 μmd,l-APV (1), or on a different cell bathed with extracellular Ca2+ substituted with Mg2+ and containing 1 μm TTX (2). C, Long applications of nicotine revealed a slow decay. Inward currents were obtained from two different mitral cells (1 and 2) and showed identical decay kinetics when scaled (1 + 2). For all cells shown in A–C,Vh = −80 mV. D, Subtraction of currents elicited during a voltage ramp without and with nicotine application reveals the voltage dependence of nicotine-induced current. The recording was performed in the BMI–NBQX–APV mixture supplemented with TTX (1 μm) and Cd2+(200 μm) (top trace). The corresponding current–voltage relationship (bottom) shows inward rectification.
As illustrated in Figure 2C, a long (up to 400 sec) application of nicotine (30 μm) showed a slow decay, with a mean half-decay time ranging between 160 and 285 sec (247 ± 18 sec; n = 6). This time constant was relatively independent of the peak current amplitude (Fig.2C, bottom traces). To further characterize this slowly decaying current, voltage-ramp commands were applied to evaluate its relationship with voltage (Fig. 2D). Neuronal nAChRs have previously been shown to have inwardly rectifying current–voltage relationships, passing little outward current and having zero current between 0 and +20 mV (for review, see Role, 1992). Consistent with previous findings, voltage ramps given in the absence and presence of nicotine (30 μm) revealed a reversal potential (Erev) ranging from −5 to + 10 mV (a mean value of 3.2 ± 2.7 mV; n = 6) and an inward current, which strongly rectified such that outward current was not observed for positive membrane potential values, up to +50 mV (Fig. 2D). Thus, the current–voltage relationship of the slowly decaying nicotinic response is consistent with the activation of neuronal nAChRs.
Activation of muscarinic receptors decreases granule cell firing frequency
Because granule cells constitute the largest neuronal population in the main olfactory bulb, these interneurons are extremely influential in the control of olfactory bulb circuitry (for review, seeMori, 1987). We thus investigated whether they were subjected to cholinergic modulation. When recorded in the cell-attached configuration with BMI and NBQX–APV, some granule cells were also found to be spontaneously active (Fig.3B,C), with a mean firing rate of 6.0 ± 1.3 Hz (n = 7). Bath application of carbachol (50–100 μm) reduced spike discharge of granule cells (a mean reduction of 58 ± 11%;n = 4) (Fig. 3B,D). A dose-dependent effect was found within the range of 10–100 μm of carbachol (n = 5), and the reduction of granule cell firing activity was slowly reversible on washout of the agonist (Fig. 3B). The muscarinic nature of this carbachol effect on granule cell firing was revealed by oxotremorine (50 μm) (Fig. 3C,D) (n = 4), whereas nicotine (up to 50 μm) treatment did not affect their firing frequency (Fig. 3D) (n = 4). The reduction of spike discharge induced by muscarinic receptor activation may result from a membrane potential hyperpolarization because the amplitude of currents generated by action potentials was increased (Fig. 3B, right).
Fig. 3.
Muscarinic receptor activation decreases granule cell firing frequency. A, A granule cell visualized under IR-DIC microscopy with the arrow indicating the cell dendrite (left); right, schema of a recorded cell filled with Lucifer yellow. B(left), Extracellular recording in cell-attached mode from a granule cell recorded before, during, and afterCCh (50 μm) (Wash). Single action potentials are shown on the right (note different time calibration bars). B (right), The time course for changes in action potential frequency (bin size: 10 sec) and amplitude during CCh treatment is shown (numbers indicate when traces were extracted for illustration in the left panel).C, Oxotremorine (50 μm) decreased the firing frequency; this effect was reversible on washout (Wash). All granule cells were recorded in a mixture that included BMI (20 μm), NBQX (10 μm), and d,l-APV (100 μm). D, Average of the firing frequency changes induced by cholinergic drug applications in different treatments and normalized to the control responses. Changes in frequency were compared between control medium and drug applications using paired t test (*p < 0.05).
Altogether, these findings demonstrate that ACh modulates, in opposite directions, the spontaneous firing of mitral and granule cells through the activation of nicotinic and muscarinic receptors, respectively. Moreover, because these dual effects were also seen after blockade of ionotropic glutamatergic and GABAergic synaptic transmission, these two cholinergic actions probably result from direct membrane potential changes. It is noteworthy that our findings do not support a direct nicotinic effect on granule cells, whereas others have reported nicotine-induced responses in olfactory bulb interneurons (Alkondon and Albuquerque, 1994; Alkondon et al., 1996). We therefore decided to further investigate the nicotinic actions on other olfactory bulb interneuron subtypes.
Whole-cell nicotinic currents in interneurons
To examine a direct effect of nicotinic receptor activation on olfactory bulb interneurons (i.e., monopolar periglomerular cells, bipolar periglomerular cells, and granule cells), whole-cell recordings were performed with patch electrodes containing Lucifer yellow. This dye was used to determine whether the effects induced by cholinergic receptor activation could be correlated with distinct cell morphology and location. For all interneuron subtypes recorded, only bipolar periglomerular cells were sensitive to nicotine (30 μm). In the presence of BMI and NBQX–APV, a nicotine-induced current was observed from four of four recorded cells (−113 ± 84 pA) (Fig.4A,B), but up to 100 μm nicotine application failed to induce a response from all tested monopolar periglomerular (n= 6) and granule cells (n = 4) (Fig.4A). Bipolar periglomerular cell firing activity was also dramatically enhanced during nicotine treatment in the presence of BMI and NBQX-APV (data not shown). Figure 4 also shows that the response to nicotine (30 μm) progressively decays during prolonged application of the agonist; as reported for mitral cells, the decay time was independent of the amplitude (Fig.4B). This amplitude independence allowed us to compare the rate of decay across cell types. As depicted in Figure4C, the decay was found to be faster in interneurons than in mitral cells (mean times to half-decay of 94 ± 7 and 247 ± 18 sec for four bipolar periglomerular cells and six mitral cells, respectively; p < 0.001). This discrepancy could result from different compositions of nAChR subunits. Indeed, similarly fast decay time constants have been reported for nicotinic responses recorded in cultured olfactory bulb interneurons expressing the α7-subunit (Alkondon and Albuquerque, 1994); it is this α7 subunit that has been found to be highly expressed in the glomerular layer (Le Jeune et al., 1995). With the finding that functional nAChRs and mAChRs are present in the olfactory bulb network and that activation of these receptors strongly modulates neuronal excitability, we then analyzed in detail the ability of cholinergic receptors to control GABA release from interneurons.
Fig. 4.
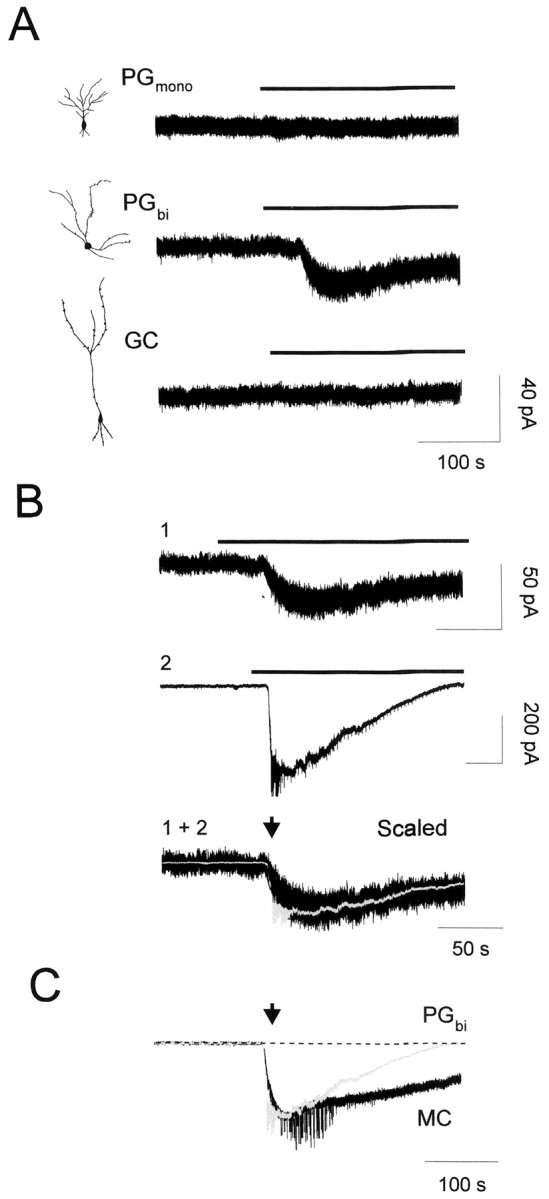
Selective responses of nicotine on olfactory bulb interneurons. The effect of bath application of nicotine was examined for three interneuron subtypes: monopolar periglomerular cell (PGmono), bipolar periglomerular cell (PGbi), and granule cell (GC). These cells were identified by their location, morphology, and pattern of electrophysiological activity.A, Membrane current traces of individual interneurons showed that only bipolar periglomerular cells respond to nicotine application. Insets, Camera lucida drawings of the Lucifer yellow-filled interneurons performed after whole-cell recordings. B, Inward currents obtained from two different periglomerular interneurons (1 and2) showed identical decay kinetics when scaled (1 + 2). C, Long applications of nicotine revealed a faster decay for a periglomerular cell (PG) than for a mitral cell (MC) shown in Fig.2C2. The holding potential was −80 mV for all recordings.
Spontaneous and glutamate-evoked GABA release onto mitral cells
Spontaneous synaptic inputs onto mitral cells were measured in the whole-cell recording configuration. Excitatory and inhibitory synaptic activities were distinguished under control conditions by using a low-chloride internal solution to separate the reversal potentials of excitatory and inhibitory synaptic currents. Under these conditions, spontaneous IPSCs were seen as inward at −80 mV and outward at 0 mV (the reversal potential for excitatory synaptic events) (Fig.5A). As expected, these spontaneous events reversed at the chloride equilibrium potential (ECl of −48 mV) and were completely and reversibly blocked by bicuculline (20 μm;n = 6), indicating that they were mediated by activation of GABAA receptors (Fig.5A). It is noteworthy that during the course of these recordings, GABAergic events remained stable even after long periods of recording in control conditions. This lack of rundown contrasts with the receptors observed in hippocampal cells (Stelzer et al., 1988).
Fig. 5.

GABAergic synaptic events recorded in mitral cells. A, GABAergic spontaneous synaptic currents were recorded at membrane potentials indicated at the rightof the traces. Outward spontaneous currents recorded at 0 mV (middle trace) correspond to pure GABAergic IPSCs because they were completely blocked by 20 μm bicuculline methiodide (+ BMI). B, The distributions of interevent intervals were constructed from 60 sec of continuous data measured at −80 mV (thin line, 784 events) or 0 mV (thick line, 2826 events).C, Spontaneous IPSCs recorded before (STD) and during application of 10 μm NBQX with 100 μmd,l-APV. Subsequent application of BMI (20 μm) completely abolished synaptic activity (bottom traces) (Vh = 0 mV). D, Corresponding cumulative distributions of spontaneous IPSC intervals (left) and amplitudes (right). Plots were constructed from 100 sec of continuous recording. Both frequency and amplitude distributions changed significantly (Kolmogorov–Smirnov test, p< 0.001) in the presence of NBQX–APV (thick line, 305 events; thin line, 1788 events). For this cell, the mean spontaneous IPSC frequency was 17.4 and 3.1 Hz, and mean amplitude was 55 ± 32 pA (SD) and 28 ± 16 pA in STD andNBQX, APV, respectively.
GABA release from granule cells onto mitral cells is located in a structure called the dendrodendritic reciprocal synapse, consisting of an excitatory synapse directly adjacent to an inhibitory granule-to-mitral cell synapse (Price and Powell, 1970). It has been demonstrated that evoked GABA release is mediated by glutamate excitation (Nowycky et al., 1981; Jahr and Nicoll, 1982; Isaacson and Strowbridge, 1998; Schoppa et al., 1998). However, little is known about spontaneous granule-to-mitral cell GABA release in the absence of excitation. To quantify spontaneous GABA release, cells were recorded in the presence of NBQX–APV (Fig. 5C,D) or the broad-spectrum ionotropic glutamatergic antagonist kynurenate (5 mm). Glutamatergic receptor blockers dramatically decreased spontaneous IPSC frequency (a mean reduction of 81 ± 4%, with p < 0.0005; n = 9) as well as the number of large spontaneous IPSCs (Fig. 5D). In sum, mitral cells receive spontaneous IPSCs driven by glutamate receptor activation that are characterized by large amplitudes and high frequency as well as spontaneous IPSCs independent of glutamate receptor activation characterized by smaller amplitudes and a lower frequency. Having described these two types of GABAergic events onto mitral cells, we then investigated their potential sensitivity to cholinergic modulation.
Activation of muscarinic receptors modulates GABAergic synaptic inputs onto mitral cells
As shown in Figure6A, bath application of carbachol in standard external medium markedly increased the frequency of spontaneous IPSCs; the relative interevent interval curve was shifted to the left, indicating an increase of the frequency of GABAergic events (Fig. 6C, left graph). The mean baseline frequency of these spontaneous events (5.0 ± 1.1 Hz) was increased during carbachol treatment by an average of threefold (15.4 ± 3.2 Hz) in seven of seven mitral cells. In most of these neurons, the increase in frequency was attributable to an increase in the number of small amplitude events, which therefore resulted in an apparent decrease of mean IPSC amplitude (Fig. 6B). As illustrated in Figure 6C (right graph), the cumulative amplitude plot indicates a reduction in the number of large amplitude GABAergic events. However, when spontaneous IPSCs were averaged and superimposed to compare kinetics, rise times and decay rates were unchanged (Fig. 6B). On average, no changes in cumulative rise time distributions of spontaneous IPSCs before and during carbachol treatment were seen (data not shown;n = 5; p > 0.05). The carbachol effect on spontaneous IPSC frequency reached a steady state within 20–40 sec and showed no evidence of desensitization over an application period of up to 3 min (n = 6).
Fig. 6.

Carbachol increases the frequency of both spontaneous IPSCs (A–C) and miniature IPSCs (D–F) from mitral cells. A, Sample recordings of spontaneous IPSCs are depicted in standard medium (Std) and after 50 μm carbachol application (CCh). B, Spontaneous IPSCs were averaged and scaled before (Std, 220 events) and after carbachol application (CCh, 191 events). Note that carbachol caused no change in the time course of these events.C, Cumulative probability plots of event frequency (left graph) and amplitude (right graph) were constructed from 125 sec of continuous recording (394 and 967 events in Std and CCh, respectively).CCh enhanced the frequency of spontaneous IPSCs (a mean interevent interval of 0.32 ± 0.4 (SD) sec and 0.13 ± 0.15 sec before and after bath application of carbachol, respectively). The mean spontaneous IPSC amplitude in Std was 49 ± 46 pA (SD) versus 38 ± 31 pA in CCh. Both changes were statistically significant (Kolmogorov–Smirnov test,p < 0.001). D, Sample recordings of miniature IPSCs from a different cell before (Ctrl) and during application of 50 μm carbachol (CCh) in the presence of 1 μm TTX. E, Averaged miniature IPSCs before (Ctrl, 230 events) and during carbachol treatment (CCh, 206 events). As in B, traces are superimposed and scaled on the right (Scaled) to illustrate that carbachol had no effect on the time course of miniature IPSCs. F, Cumulative probability plots of synaptic current frequency and amplitude were constructed from 80 sec of continuous recording (305 and 617 events in Ctrl andCCh, respectively). CCh enhanced the frequency of miniature IPSCs [mean interevent interval of 0.26 ± 0.25 sec (SD) and 0.13 ± 0.12 sec before and after bath application of carbachol, respectively]. The mean miniature IPSC amplitude in controls was 55 ± 54 pA versus 39 ± 29 pA in CCh. Both changes were statistically significant (Kolmogorov–Smirnov test,p < 0.001). D–F, Cells in this figure were recorded in the presence of 10 μm NBQX and 100 μmd,l-APV and voltage-clamped at 0 mV.
This carbachol-induced enhancement of spontaneous IPSC frequency could be caused by an increase in mitral cell firing or a direct increase in the dendrodendritic synaptic transmission, or both. To test this possibility, slices were bathed with glutamatergic receptor antagonists, which should prevent this effect. Carbachol still increased the frequency of GABAergic events in all recorded neurons (n = 4). We thus decided to explore more precisely the mechanism by which carbachol could directly increase GABA release.
To evaluate the contribution of action potential-independent transmitter release, synaptic events were measured in the presence of both tetrodotoxin (TTX; 0.5–1 μm, perfused for a minimum of 5 min) to block action potentials, and glutamatergic antagonists, to isolate miniature IPSCs reflecting quantal release by granule cells (n = 39 experiments) (Fig.6D–F). The baseline frequency of miniature IPSCs (2.0 ± 0.5 Hz; n = 12) was markedly increased in all tested cells by carbachol treatment (4.8 ± 1 Hz;n = 12; p = 0.004), and cumulative amplitude distributions of miniature IPSCs demonstrated significant changes in 9 of 12 cells (p < 0.001 with Kolmogorov–Smirnov test) (Fig.6D,F, left panel). In Figure 6, only the small amplitude (<40 pA) synaptic events seemed to be sensitive to carbachol. In other cells, only large synaptic currents were affected by carbachol (data not shown). However, in any case, a correlation with a change in the rise time could be observed, which argues against a distinct localization of the population of carbachol-sensitive GABAergic synapses that impinge on mitral cells. Thus, carbachol enhanced GABA release through a mechanism independent of the spontaneous firing of granule cells or glutamatergic excitatory drive, indicating that carbachol directly modulates quantal release of GABA from presynaptic terminals.
The carbachol-induced enhancement of miniature GABAergic events was not reproduced by 10–30 μm nicotine (Fig.7A) (n = 6) but could be mimicked by 50 μm oxotremorine (a mean increase of 318 ± 124% of control frequency; n = 4; p < 0.001), indicating that this enhancement was mediated through activation of mAChRs (Fig. 7B). Activation of mAChRs could increase miniature IPSC frequency either by a direct effect on the exocytotic machinery or by increasing calcium influx into presynaptic terminals, or both. We therefore examined whether carbachol affected the frequency of miniature IPSCs in the presence of the Ca2+ channel blocker Cd2+ (100–200 μm) added in the external medium. A change in the frequency distribution was still clearly observed (Fig. 7C), with a mean increase of 319 ± 220% (n = 4). We thus conclude that mAChRs facilitate GABA release onto mitral cells through a direct presynaptic effect that excludes modulation of Ca2+ influx through voltage-gated Ca2+ channels.
Fig. 7.

Activation of muscarinic receptors enhances the frequency of miniature IPSCs recorded in mitral cells.A, Miniature IPSCs recorded before and during bath application of 30 μm nicotine (left traces), and corresponding cumulative interval distributions are shown on the right. Nicotine (thick line) had no effect on the miniature IPSC frequency (Kolmogorov–Smirnov test, p > 0.05).B, Recordings from a different cell before and after 50 μm oxotremorine application (left traces). The corresponding cumulative display of interval distributions on theright shows that oxotremorine increased miniature IPSC frequency (Kolmogorov–Smirnov test, p < 0.001). C, Carbachol (+CCh) still increased miniature IPSC frequency in the presence of 100 μm Cd2+(Kolmogorov–Smirnov test, p < 0.001). Cells were voltage-clamped at 0 mV and recorded in normal ACSF with 10 μm NBQX, 100 μmd,l-APV, and 1 μm TTX (Ctrl).
Evidence for two distinct GABAergic synaptic events in the olfactory bulb
During cell-attached recordings of granule cells, the spontaneous firing frequency was increased when bicuculline was bath-applied (data not shown). This increase may reflect a tonic GABAergic inhibition that could protect granule cells from long-lasting depolarization. Whether the GABA involved in this modulation is released from intrinsic interneurons or from centrifugal fibers is unknown. First, to characterize spontaneous inhibitory synaptic inputs to granule cells, synaptic currents were recorded in the presence of NBQX–APV (as control condition) at +30 mV. Similar to the mitral cell experiments, inhibitory postsynaptic events were identified as GABAA receptor-mediated events by (1) complete blockade by bicuculline (20 μm) (n = 11) and (2) similar Erev reported for IPSCs detected in mitral cells. In all granule cells (n= 43), spontaneous GABAergic inputs were observed with a mean rate of ∼1 Hz (n = 21). For comparison, the mean rate of spontaneous IPSCs recorded in mitral cells under similar conditions was higher (1.88 ± 0.31 Hz; p < 0.05;n = 9) (Fig.8A, top traces). Because all granule cells showed spontaneous IPSCs, the source of GABA onto granule cells was probably neighboring interneurons rather than centrifugal fibers [also see Wellis and Kauer (1994)].
Fig. 8.

Two distinct inhibitory synaptic responses in olfactory bulb neurons. A, Spontaneous GABAergic IPSC activity from a granule (top) and a mitral cell (bottom) recorded before and during application of TTX (1 μm). Corresponding cumulative interval distributions are depicted before (thin lines) and in the presence of TTX (thick lines) (mitral cell: 220 events before and 177 events during TTX; granule cell: 664 events before and 147 events during TTX). Note that TTX strongly reduced the frequency of IPSCs from the granule cell. Neurons were recorded in the presence of NBQX and APV and voltage-clamped at 0 mV. B, Cumulative probability distribution of the reduction of IPSC frequency induced by TTX, measured in mitral cells (MC; n = 8) and granule cells (GC; n = 7) (mean reductions of 30 ± 9 and 60 ± 10%, for mitral cells and granule cells, respectively). C, Comparison of spontaneous IPSCs (no TTX included) recorded from a granule cell (GC) and a mitral cell (MC) is shown (top and middle traces). The averaged IPSCs (180 and 250 events, respectively, for granule and mitral cells) are superimposed and scaled (granule-IPSC and mitral-IPSC decay time constants: 40 msec and 10 msec, respectively). Both cells were voltage-clamped at 0 mV.
To assess the contribution of action potentials to GABA release onto mitral and granule cells, synaptic inputs were measured before and after addition of TTX (1 μm) in the presence of NBQX–APV (Fig. 8A,B). A dramatic change in the distribution of interevent intervals was observed in the cumulative distributions from a granule cell shown in Figure 8A (top panel) but not from a mitral cell (Fig.8A, bottom panel). Hence, whereas addition of TTX to the bathing medium decreased the frequency of spontaneous IPSCs detected in granule cells by 60 ± 10% (n = 8), it only decreased spontaneous IPSC frequency from mitral cells by 30 ± 9% (n = 8) (Fig.8B). This observation indicates that most of the GABAergic inputs onto granule cells depend on cell firing, whereas the large majority of spontaneous inhibitory events onto mitral cells depend on ionotropic glutamate receptor activation (Fig.5C,D). We also have found that spontaneous IPSCs, as well as miniature IPSCs in granule cells, last longer than those inhibitory events recorded from mitral cells. For example, spontaneous IPSCs in granule cells (n = 11) showed a mean half-decay time of 38.2 ± 3.6 msec (Fig. 8C, top traces) when compared with those from mitral cells, with fourfold faster decay rates (10.9 ± 1.1 msec; p < 0.0001; n= 9) (Fig. 8C, middle traces). Altogether, these results demonstrate that spontaneous GABAA-mediated IPSCs recorded from granule cells were distinct from those detected in mitral cells by (1) their low spontaneous frequency, (2) their dependence on firing activity, and (3) their slow time course of decay.
To test whether these inhibitory synapses could also be distinguished by their sensitivity to cholinergic modulation, spontaneous inhibitory events were recorded from granule cells in the presence of cholinergic receptor agonists. Figure9A,Bshows that 50 μm oxotremorine dramatically decreased the frequency of spontaneous IPSCs recorded in control conditions (a mean reduction of 38.8 ± 3.6%; n = 5), whereas nicotine application had no effect (n = 5) (Fig. 9C). This reduction could result from the decrease in granule cell firing activity reported above. To test this possibility, cells were first bathed with 1 μm TTX before application of oxotremorine. As shown in Figure 9C, in the absence of action potentials oxotremorine has no more effect, indicating that muscarinic modulation of GABA release onto granule cells requires firing activity. Furthermore, these results strongly suggest that GABAergic inputs onto granule cells originate from neighboring granule cells. Together, these data illustrate a dual modulation mediated by muscarinic receptors: on the one hand they increase the level of tonic GABAergic inhibition onto mitral cells while also relieving inhibition onto granule cells.
Fig. 9.

Activation of muscarinic receptors decreased the frequency of spontaneous IPSCs recorded in granule cells.A, Spontaneous IPSCs recorded in NBQX–APV before (Std) and during application of 50 μmoxotremorine while voltage-clamping the cell at 0 mV. B,Corresponding interval (top) and amplitude (bottom) cumulative distribution plots were constructed from 150 sec of continuous data before (442 events) and after (261 events) carbachol. The amplitude distribution remained unchanged (Kolmogorov–Smirnov test, p > 0.05; mean amplitude in control, 28.4 vs 28.7 pA in carbachol), whereas the interval distributions were significantly different (Kolmogorov–Smirnov test, p < 0.001).C, Bar graph summarizing the effect of 50 μm oxotremorine (Oxo) and 50 μm nicotine (Nic) on spontaneous IPSC (sIPSC) or miniature IPSC (mIPSC) frequency.
DISCUSSION
This study demonstrates the presence of at least four different targets for cholinergic inputs into the main olfactory bulb. We found that ACh induces a prominent increase in the firing activity of both mitral cells and bipolar periglomerular neurons through activation of distinct nAChR populations. In addition, activation of mAChRs both increases GABA release from granule cells onto mitral cells and inhibits granule cell firing. We propose that through varied mechanisms, including extrasynaptic ACh diffusion, receptor subunit composition differences, and subcellular compartmentalization, cholinergic inputs have multiple points of control over the olfactory bulb network.
Anatomical localization of cholinergic receptors in the olfactory bulb
The olfactory bulb, like other forebrain cortical structures, receives strong cholinergic inputs from the basal forebrain (Wenk et al., 1977; Macrides and Davis, 1983; Zaborsky et al., 1986). The bulb itself is virtually devoid of intrinsic cholinergic neurons (Woolf et al., 1984; Nickell and Shipley, 1988a; Senut et al., 1989). Autoradiographic and binding studies have revealed two important characteristics of cholinergic receptors in the olfactory bulb: both nAChRs and mAChRs are present at high densities, and their distribution within the olfactory bulb is well segregated with a low degree of overlap. This segregation of muscarinic and nicotinic receptors may reflect a dual organization of the olfactory bulb cholinergic system: the glomerular and mitral cell layers are more susceptible to modulation by nAChRs, whereas deeper layers (plexiform and granule cell layers) may be modulated primarily by mAChRs.
Fine structural observations have shown that centrifugal cholinergic projections terminate primarily onto interneurons (Nickell and Shipley, 1988a; Le Jeune and Jourdan, 1994; Kasa et al., 1995). For example, when choline acetyltransferase was used to label cholinergic axons, ultrastructural analysis revealed that cholinergic synapses were selectively targeted onto the dendrites of periglomerular or granule cells but never on mitral cells (Le Jeune and Jourdan, 1993). Because no cholinergic synapses have been observed on mitral cells, it has been thought that neuromodulatory effects of ACh in the bulb are mediated only through cholinergic synapses onto interneurons (Kasa et al., 1995). During the course of this study, however, a remarkable direct effect on mitral cells was consistently found after nAChR activation, suggesting an action of extrasynaptic diffusion of ACh (see below).
Two distinct nicotinic currents in the main olfactory bulb
Nicotinic AChRs have been found to be restricted to glomerular and mitral cell layers (Hunt and Schmidt, 1978; Hill et al., 1993; Le Jeune et al., 1995, 1996). This specific distribution supports the selective sensitivity to nicotinic agonists that we observed for periglomerular and mitral cells. Our data also agree with previous reports showing that ACh was able to increase the firing of interneurons from the glomerular layer (Pager, 1983) and induce inward currents in cultured interneurons (Alkondon and Albuquerque, 1994; Alkondon et al., 1996). These reports did not identify the cell types from which cholinergic responses were induced. Moreover, recordings of cultured neurons have reported only one type of cholinoceptive neuron (Alkondon and Albuquerque, 1994; Alkondon et al., 1996). This discrepancy may result from the absence of mitral cells in cultures prepared from neonatal animals (Trombley and Westbrook, 1990).
Despite the fact that cholinergic projections have not been found to form synapses onto mitral cells, these cells still respond to cholinergic activation with fast inward currents. The apparent lack of cholinergic synapses onto mitral cells suggests that ACh reaches postsynaptic nAChRs by long-range diffusion (Fig.10A). Extrasynaptic release of ACh has been proposed to occur in several systems in the CNS and may participate in a volume transmission phenomenon (Agnati et al., 1995) consistent with the diffuse anatomical organization of cholinergic fibers (Woolf, 1991).
Fig. 10.

Schematic representation of cholinergic targets in the main olfactory bulb. This schema summarizes findings from the present study and takes into account previous anatomical observations.A, Cholinergic fibers activate postsynaptic nicotinic receptors located on mitral cells (MC) where ACh may be delivered through a nonsynaptic relationship to act as a neuromodulator and also on bipolar periglomerular cells (PGbi) by a synaptic relationship where ACh rather acts as a neurotransmitter. Through this dual effect, ACh may facilitate the sensory transfer toward upper cortical centers.B, Cholinergic inputs into plexiform layers and granule cell layer allow activation of muscarinic receptors that contribute to a tonic inhibition of output neurons and a reduction of inhibition (disinhibition) onto granule cells. MC, Mitral cell;GC, granule cell; PGbi, bipolar periglomerular cell; O.N., olfactory nerve;HDB, the horizontal limb of the diagonal band of Broca.
Numerous nAChR subunits have been described that, in various combinations, form receptors with different functional properties (Marks et al., 1986; Happe et al., 1994). For instance, α7-containing nAChRs rapidly desensitize (Couturier et al., 1990; Gerzanich et al., 1994; Zhang et al., 1994), whereas α4β2-containing nAChRs show slower desensitization rates (Lindstrom, 1996; Léna and Changeux, 1997; Changeux et al., 1998). Immunohistochemical studies have found mitral cell bodies to be exclusively and intensely stained with β2-subunit antisera (Hill et al., 1993), whereas the α7 subunit has been found mainly in the periglomerular layer (Le Jeune et al., 1996). Thus, the different decay times of nicotinic currents that we observed could reflect the presence of putative nAChR containing α4β2 subunits (hereafter referred to as α4β2 nAChR, although the exact subunit composition remains to be determined) located on mitral cells, and α7 subunit located on periglomerular cells. In general, α7-containing nAChRs are found clustered in synaptic locations, whereas β2-containing receptors have been localized perisynaptically (for review, see Clarke, 1993; Role and Berg, 1996). It is noteworthy that α4β2-containing nAChRs show a high-affinity ACh binding site and therefore are more suitable to activation through paracrine ACh action. In addition, the different time courses of decay of nicotinic responses found in mitral and bipolar periglomerular cells suggest that nAChRs are best suited for mediating a tonic action in mitral cells and a phasic effect in interneurons, as recently suggested for different hippocampal interneuron subtypes (Alkondon et al., 1999; McQuiston and Madison, 1999).
By increasing firing rates of both mitral cells and periglomerular neurons, nAChRs can exert opposing effects on the final output firing rate of mitral cells (Fig. 10A). Diffusion of ACh from cholinergic terminals to mitral cells would activate the slowly desensitizing nicotinic receptors to bring the resting membrane potential closer to the threshold for initiation of action potentials. At the same time, in glomeruli, ACh could indirectly inhibit olfactory nerve inputs onto mitral cell dendrites through activation of periglomerular cells. Indeed, it has been proposed that the periglomerular neurons sensitive to ACh are dopaminergic interneurons (Nickell and Shipley, 1988a), and we have recently reported that dopamine receptor activation reduces the strength of olfactory nerve inputs (Hsia et al., 1999).
Dual forms of modulation by muscarinic receptor activation on granule cells
The selective effect of muscarinic receptor activation on granule cells described here is consistent with previous anatomical reports, which find binding for both M1- and M2-like receptors localized to the plexiform and granule cell layers, with relatively little muscarinic binding in the glomerular layer (Hunt and Schmidt, 1978). We found that mAChR activation modulates the output of granule cells in opposing directions, depending on the subcellular localization of the mAChR population: somatically, by reducing excitability of granule cells, and presynaptically, by enhancing GABA release onto mitral cells. At the level of the network, it should also be noted that the muscarinic inhibition of granule cell firing would lead to a disinhibition of neighboring granule cells, because GABAergic inputs onto these interneurons are strongly dependent on action potentials (Fig.8A,B).
The reduction of granule cell firing by mAChR activation may result from a membrane potential hyperpolarization caused by activation of a potassium conductance, as shown in other brain structures (Egan and North, 1986; McCormick and Pape, 1988) (for review, see Brown et al., 1997). As to how mAChRs could stimulate neurotransmitter release, most studies have instead reported a reduction of synaptic transmission mediated by mAChRs (for review, see Nicoll et al., 1990). Because the enhancement of GABA release onto mitral cells was still observed in the presence of TTX and Cd2+, we propose that mAChR activation increases GABA release either by recruiting Ca2+ release from intracellular pools or by directly modulating exocytotic machinery. Muscarinic receptors are therefore likely to be located near the transmitter release site on GABAergic terminals.
Integration of cholinergic modulation in the main olfactory bulb
A consideration of the entire network of connections in the bulb can lead to more complex interpretations. First, as mentioned above, an indirect effect of decreasing granule cell body excitability would be a decrease in the degree of inhibition—a disinhibition—of neighboring granule cells. Second, a consideration of the ensemble of nicotinic and muscarinic AChR effects would be necessary to understand a potential role for cholinergic inputs in complex temporal and spatial patterns of circuit activity. For example, the cholinergic projection to the hippocampus has been proposed to serve as the pacemaker for the hippocampal theta rhythm. Indeed, this rhythm is present in the olfactory bulb as well and has been shown to be phase-locked with both the sniff cycle and the hippocampal theta during odor learning tasks (Macrides et al., 1982). Temporal coordination of circuit activity may be critical to the olfactory system (Stopfer et al., 1997), which lacks the point-to-point spatial organization of other sensory systems.
Our characterization of four distinct targets for ACh in the olfactory bulb elucidates the critical role of cholinergic inputs in modulating various elements of this network. Because of the widespread and opposing effects of ACh receptor activation, changes in any of these four points of control will change the state of activity and the output of the network. Further investigation of how these sites of cholinergic modulation interact and change during olfactory processing and learning should deepen our understanding of this complex circuit.
Footnotes
This study was supported by the Centre National de la Recherche Scientifique, the Institut Universitaire de France, a grant from Evaluation-Orientation de la Coopération Scientifique (ECOS-Uruguay), and a grant from the French Ministère de la Recherche et de l'Enseignement (A.C.). We are grateful to G. Sadoc for help with the analysis and acquisition software, to R. Gervais, F. Jourdan, and G. Shepherd for reading this manuscript, and to A. Hsia for critical comments.
P.E.C. and A.C. contributed equally to this work.
Correspondence should be addressed to Dr. Pierre-Marie Lledo, Centre National de la Recherche Scientifique, Institut Alfred Fessard, Avenue de la Terrasse, 91198 Gif-sur-Yvette Cedex, France. E-mail:Pierre-Marie.Lledo@iaf.cnrs-gif.fr.
REFERENCES
- 1.Agnati LF, Zoli M, Strömberg I, Fuxe K. Intercellular communication in the brain: wiring versus volume transmission. Neuroscience. 1995;69:711–726. doi: 10.1016/0306-4522(95)00308-6. [DOI] [PubMed] [Google Scholar]
- 2.Alkondon M, Albuquerque EX. Presence of α-bungarotoxin-sensitive nicotinic ACh receptors in rat olfactory bulb neurons. Neurosci Lett. 1994;176:152–156. doi: 10.1016/0304-3940(94)90070-1. [DOI] [PubMed] [Google Scholar]
- 3.Alkondon M, Rocha ES, Maelicke A, Albuquerque EX. Diversity of nicotinic ACh receptors in rat brain. V. α-bungarotoxin-sensitive nicotinic receptors in olfactory bulb neurons and presynaptic modulation of glutamate release. J Pharmacol Exp Ther. 1996;278:1460–1471. [PubMed] [Google Scholar]
- 4.Alkondon M, Pereira EFR, Eisenberg HM, Albuquerque EX. Choline and selective antagonists identify two subtypes of nicotinic acetylcholine receptors that modulate GABA release from CA1 interneurons in rat hippocampal slices. J Neurosci. 1999;19:2693–2705. doi: 10.1523/JNEUROSCI.19-07-02693.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Brown DA, Abogadie FC, Allen TG, Buckley NJ, Caulfield MP, Delmas P, Haley JE, Lamas JA, Selyanko AA. Muscarinic mechanisms in nerve cells. Life Sci. 1997;60:1137–1144. doi: 10.1016/s0024-3205(97)00058-1. [DOI] [PubMed] [Google Scholar]
- 6.Changeux JP, Bertrand D, Corringer PJ, Dehaene S, Edelstein S, Léna C, Le Novëre N, Marubio L, Picciotto M, Zoli M. Brain nicotinic receptors: structure and regulation, role in learning and reinforcement. Brain Res Rev. 1998;26:198–216. doi: 10.1016/s0165-0173(97)00040-4. [DOI] [PubMed] [Google Scholar]
- 7.Clarke PB. Nicotinic receptors in mammalian brain: localization and relation to cholinergic innervation. Prog Brain Res. 1993;98:77–83. doi: 10.1016/s0079-6123(08)62383-3. [DOI] [PubMed] [Google Scholar]
- 8.Couturier S, Bertrand D, Matter J-M, Hernandez M-C, Bertrand S, Millar N, Valera S, Barkas T, Ballivet M. A neuronal nicotinic ACh receptor subunit (α7) is developmentally regulated and forms a homo-oligomeric channel blocker by α-BTX. Neuron. 1990;5:847–856. doi: 10.1016/0896-6273(90)90344-f. [DOI] [PubMed] [Google Scholar]
- 9.Doty RL. Olfactory dysfunction in neurodegenerative disorders. In: Getchell TV, Doty RL, Bartoshuk LM, Snow JB, editors. Smell and taste in health and disease. Raven; New York: 1991. pp. 735–751. [Google Scholar]
- 10.Egan TM, North RA. Acetylcholine hyperpolarizes central neurones by acting on an M2 muscarinic receptor. Nature. 1986;319:405–407. doi: 10.1038/319405a0. [DOI] [PubMed] [Google Scholar]
- 11.Elaagouby A, Gervais R. Acetylcholine-induced long-lasting enhancement in excitability of output cells of the rat olfactory bulb. NeuroReport. 1992;3:10–12. doi: 10.1097/00001756-199201000-00002. [DOI] [PubMed] [Google Scholar]
- 12.Gerzanich V, Anand R, Lindstrom JM. Homomers of α8 and α7 subunits of nicotinic receptors exhibit similar channel but contrasting binding site properties. Mol Pharmacol. 1994;45:212–220. [PubMed] [Google Scholar]
- 13.Ghonheim MM, Mewaldt SP. The effect of diazepam and fentanyl on mental, psychomotor and electroencephalographic functions and their rate of recovery. Psychopharmacologia. 1975;44:257–262. doi: 10.1007/BF00421185. [DOI] [PubMed] [Google Scholar]
- 14.Hagan JJ, Moris RG. M. In: Iversen LL, Iversen SD, Snyder SJ, editors. Psychopharmacology of the aging nervous system. Plenum; New York: 1989. pp. 237–323. [Google Scholar]
- 15.Halász N, Shepherd GM. Neurochemistry of the vertebrate olfactory bulb. Neuroscience. 1983;10:579–619. doi: 10.1016/0306-4522(83)90206-3. [DOI] [PubMed] [Google Scholar]
- 16.Happe HK, Peters JL, Bergman DA, Murrin LC. Localization of nicotinic cholinergic receptors in rat brain: autoradiographic studies with [3H]cytisine. Neuroscience. 1994;62:929–944. doi: 10.1016/0306-4522(94)90484-7. [DOI] [PubMed] [Google Scholar]
- 17.Hill JA, Zoli M, Bourgeois JP, Changeux JP. Immunocytochemical localization of a neuronal nAChR: the β2-subunit. J Neurosci. 1993;13:1551–1568. doi: 10.1523/JNEUROSCI.13-04-01551.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hsia AY, Vincent J-D, Lledo P-M (1999) Dopamine depresses synaptic inputs into the olfactory bulb. J Neurophysiol, in press. [DOI] [PubMed]
- 19.Hunt S, Schmidt J. Some observations on the binding patterns of α-bungarotoxin in the central nervous system of the rat. Brain Res. 1978;157:213–232. doi: 10.1016/0006-8993(78)90025-2. [DOI] [PubMed] [Google Scholar]
- 20.Isaacson JS, Strowbridge BW. Olfactory reciprocal synapses: dendritic signaling in the CNS. Neuron. 1998;20:749–761. doi: 10.1016/s0896-6273(00)81013-2. [DOI] [PubMed] [Google Scholar]
- 21.Jahr CE, Nicoll RA. An intracellular analysis of dendrodendritic inhibition in the turtle in vitro olfactory bulb. J Physiol (Lond) 1982;326:213–234. doi: 10.1113/jphysiol.1982.sp014187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kasa P, Hlavati I, Dobo E, Wolff A, Joo F, Wolff JR. Synaptic and non-synaptic cholinergic innervation of the various types of neurons in the main olfactory bulb of adult rat: immunocytochemistry of choline acetyltransferase. Neuroscience. 1995;67:667–677. doi: 10.1016/0306-4522(95)00031-d. [DOI] [PubMed] [Google Scholar]
- 23.Kasa P, Rakonczay Z, Gulya K. The cholinergic system in Alzheimer's disease. Prog Neurobiol. 1997;52:511–535. doi: 10.1016/s0301-0082(97)00028-2. [DOI] [PubMed] [Google Scholar]
- 24.Kovacs I, Torok I, Zombori J, Kasa P. Cholinergic structures and neuropathologic alterations in the olfactory bulb of Alzheimer's disease brain samples. Brain Res. 1998;789:167–170. doi: 10.1016/s0006-8993(98)00097-3. [DOI] [PubMed] [Google Scholar]
- 25.Kunze WAA, Shafton AD, Kemm RE, McKenzie JS. Effect of stimulating the nucleus of the horizontal limb of the diagonal band on single unit activity in the olfactory bulb. Neuroscience. 1991;40:21–27. doi: 10.1016/0306-4522(91)90171-j. [DOI] [PubMed] [Google Scholar]
- 26.Le Jeune H, Jourdan F. Cholinergic innervation of olfactory glomeruli in the rat: an ultrastructural immunocytochemical study. J Comp Neurol. 1993;336:279–292. doi: 10.1002/cne.903360209. [DOI] [PubMed] [Google Scholar]
- 27.Le Jeune H, Jourdan F. Acetylcholinesterase-containing intrinsic neurons in the rat main olfactory bulb: cytological and neurochemical features. Eur J Neurosci. 1994;6:1432–1444. doi: 10.1111/j.1460-9568.1994.tb01005.x. [DOI] [PubMed] [Google Scholar]
- 28.Le Jeune H, Aubert I, Jourdan F, Quirion R. Comparative laminar distribution of various autoradiographic cholinergic markers in adult rat main olfactory bulb. J Chem Neuroanat. 1995;9:99–112. doi: 10.1016/0891-0618(95)00070-n. [DOI] [PubMed] [Google Scholar]
- 29.Le Jeune H, Aubert I, Jourdan F, Quirion R. Developmental profiles of various cholinergic markers in the rat main olfactory bulb using quantitative autoradiography. J Comp Neurol. 1996;373:433–450. doi: 10.1002/(SICI)1096-9861(19960923)373:3<433::AID-CNE8>3.0.CO;2-3. [DOI] [PubMed] [Google Scholar]
- 30.Léna C, Changeux J-P. Pathological mutations of nicotinic receptors and nicotine-based therapies for brain disorders. Curr Opin Neurobiol. 1997;7:674–682. doi: 10.1016/s0959-4388(97)80088-8. [DOI] [PubMed] [Google Scholar]
- 31.Levin ED. Nicotinic systems and cognitive function. Psychopharmacology. 1992;108:417–431. doi: 10.1007/BF02247415. [DOI] [PubMed] [Google Scholar]
- 32.Lévy F, Kendrick KM, Goode JA, Guevara-Guzman R, Keverne EB. Oxytocin and vasopressin release in the olfactory bulb of parturient ewes: changes with maternal experience and effects on acetylcholine, γ-aminobutyric acid, glutamate and noradrenaline release. Brain Res. 1995;669:197–206. doi: 10.1016/0006-8993(94)01236-b. [DOI] [PubMed] [Google Scholar]
- 33.Lindstrom J. Neuronal nicotinic ACh receptors. Ion Channels. 1996;4:377–450. doi: 10.1007/978-1-4899-1775-1_10. [DOI] [PubMed] [Google Scholar]
- 34.Macrides F, Davis BJ. The olfactory bulb. In: Emson PC, editor. Chemical neuroanatomy. Raven; New York: 1983. pp. 391–426. [Google Scholar]
- 35.Macrides F, Davis BJ, Youngs WM, Nadis NS, Margolis FL. Cholinergic and catecholaminergic afferents to the olfactory bulb in the hamster: a neuroanatomical, biochemical and histochemical investigation. J Comp Neurol. 1981;203:497–516. doi: 10.1002/cne.902030311. [DOI] [PubMed] [Google Scholar]
- 36.Macrides F, Eichenbaum HB, Forbes WB. Temporal relationship between sniffing and the limbic rhythm during odor discrimination reversal learning. J Neurosci. 1982;2:1705–1717. doi: 10.1523/JNEUROSCI.02-12-01705.1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Marks MJ, Stitzel JA, Romn E, Wehner JM, Collins AC. Nicotinic binding sites in rat and mouse brain: comparison of acetylcholine, nicotine and α-bungarotoxin. Mol Pharmacol. 1986;30:427–436. [PubMed] [Google Scholar]
- 38.McCormick DA, Pape HS. Acetylcholine inhibits identified interneurons in the cat lateral geniculate nucleus. Nature. 1988;334:246–248. doi: 10.1038/334246a0. [DOI] [PubMed] [Google Scholar]
- 39.McQuiston AR, Madison DV. Nicotinic receptor activation excites distinct subtypes of interneurons in the rat hippocampus. J Neurosci. 1999;19:2887–2896. doi: 10.1523/JNEUROSCI.19-08-02887.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Mori K. Membrane and synaptic properties of identified neurons in the olfactory bulb. Prog Neurobiol. 1987;29:275–320. doi: 10.1016/0301-0082(87)90024-4. [DOI] [PubMed] [Google Scholar]
- 41.Motokizawa F. Odor representation and discrimination in mitral/tufted cells of the rat olfactory bulb. Exp Brain Res. 1996;112:24–34. doi: 10.1007/BF00227174. [DOI] [PubMed] [Google Scholar]
- 42.Nickell WT, Shipley MT. Two anatomically specific classes of candidate cholinoceptive neurons in the rat olfactory bulb. J Neurosci. 1988a;8:4482–4491. doi: 10.1523/JNEUROSCI.08-12-04482.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Nickell WT, Shipley MT. Neurophysiology of magnocellular forebrain inputs to the olfactory bulb in the rat: frequency potentiation of field potentials and inhibition of output neurons. J Neurosci. 1988b;8:4492–4502. doi: 10.1523/JNEUROSCI.08-12-04492.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Nicoll RA, Malenka RC, Kauer JA. Functional comparison of neurotransmitter receptor subtypes in mammalian central nervous system. Physiol Rev. 1990;70:513–565. doi: 10.1152/physrev.1990.70.2.513. [DOI] [PubMed] [Google Scholar]
- 45.Nowycky MC, Mori K, Shepherd GM. GABAergic mechanisms of dendrodendritic synapses in isolated turtle olfactory bulb. J Neurophysiol. 1981;46:639–648. doi: 10.1152/jn.1981.46.3.639. [DOI] [PubMed] [Google Scholar]
- 46.Pager J. Unit responses changing with behavioral outcome in the olfactory bulb of unrestrained rats. Brain Res. 1983;289:87–98. doi: 10.1016/0006-8993(83)90009-4. [DOI] [PubMed] [Google Scholar]
- 47.Price JL, Powell TPS. The morphology of granule cells within the olfactory bulb. J Cell Sci. 1970;7:91–123. doi: 10.1242/jcs.7.1.91. [DOI] [PubMed] [Google Scholar]
- 48.Quirion R. Cholinergic markers in Alzheimer disease and the autoregulation of ACh release. J Psychiatry Neurosci. 1993;18:226–243. [PMC free article] [PubMed] [Google Scholar]
- 49.Ravel N, Akaoka H, Gervais R, Chouvet G. The effect of ACh on rat olfactory bulb unit activity. Brain Res Bull. 1990;24:151–155. doi: 10.1016/0361-9230(90)90199-a. [DOI] [PubMed] [Google Scholar]
- 50.Ravel N, Elaagouby A, Gervais R. Scopolamine injection into the olfactory bulb impairs short-term olfactory memory in rats. Behav Neurosci. 1994;108:317–324. doi: 10.1037//0735-7044.108.2.317. [DOI] [PubMed] [Google Scholar]
- 51.Ribak CE, Vaughn JE, Saito K, Barber R, Roberts E. Glutamate decarboxylase localization in neurons of the olfactory bulb. Brain Res. 1977;126:1–18. doi: 10.1016/0006-8993(77)90211-6. [DOI] [PubMed] [Google Scholar]
- 52.Role LW. Diversity in primary structure and function of neuronal nicotinic acetylcholine receptor channels. Curr Opin Neurobiol. 1992;2:254–262. doi: 10.1016/0959-4388(92)90112-x. [DOI] [PubMed] [Google Scholar]
- 53.Role LW, Berg DK. Nicotinic receptors in the development and modulation of CNS synapses. Neuron. 1996;16:1077–1085. doi: 10.1016/s0896-6273(00)80134-8. [DOI] [PubMed] [Google Scholar]
- 54.Schneider SP, Macrides F. Laminar distribution of interneurons in the main olfactory bulb of the adult hamster. Brain Res Bull. 1978;3:73–82. doi: 10.1016/0361-9230(78)90063-1. [DOI] [PubMed] [Google Scholar]
- 55.Schoppa NE, Kinzie JM, Sahara Y, Segerson TP, Westbrook GL. Dendrodendritic inhibition in the olfactory bulb is driven by NMDA receptors. J Neurosci. 1998;18:6790–6802. doi: 10.1523/JNEUROSCI.18-17-06790.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Senut MC, Menetrey D, Lamour Y. Cholinergic and peptide projections from the medial septum and the nucleus of the diagonal band of Broca to dorsal hippocampus, cingulate cortex, and olfactory bulb: a combined wheatgerm agglutinin-apohorseradish peroxidase-gold immunohistochemical study. Neuroscience. 1989;30:385–403. doi: 10.1016/0306-4522(89)90260-1. [DOI] [PubMed] [Google Scholar]
- 57.Serby M, Corwin J, Conrad P, Rotrosen J. Olfactory dysfunction in Alzheimer's disease and Parkinson's disease. Am J Psychiatry. 1985;142:781–782. doi: 10.1176/ajp.142.6.aj1426781. [DOI] [PubMed] [Google Scholar]
- 58.Shepherd GM, Greer CA. Olfactory bulb. In: Shepherd GM, editor. The synaptic organization of the brain, Ed 4. Oxford UP; New York: 1998. pp. 159–203. [Google Scholar]
- 59.Shute CCD, Lewis PR. Cholinergic pathways. Pharmacol Ther. 1975;1B:79–87. doi: 10.1016/0306-039x(75)90017-3. [DOI] [PubMed] [Google Scholar]
- 60.Stelzer A, Kay AR, Wong RK. GABAA-receptor function in hippocampal cells is maintained by phosphorylation factors. Science. 1988;241:339–341. doi: 10.1126/science.2455347. [DOI] [PubMed] [Google Scholar]
- 61.Stopfer M, Bhagavan S, Smith BH, Laurent G. Impaired odour discrimination on desynchronization of odour-encoding neural assemblies. Nature. 1997;390:70–74. doi: 10.1038/36335. [DOI] [PubMed] [Google Scholar]
- 62.Trombley PQ, Westbrook GL. Excitatory synaptic transmission in cultures of rat olfactory bulb. J Neurophysiol. 1990;64:598–606. doi: 10.1152/jn.1990.64.2.598. [DOI] [PubMed] [Google Scholar]
- 63.Wellis DP, Kauer JS. GABAergic and glutamatergic synaptic input to identified granule cells in salamander olfactory bulb. J Physiol (Lond) 1994;475:419–431. doi: 10.1113/jphysiol.1994.sp020082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Wenk HU, Meye U, Bigl V. Centrifugal cholinergic connections in the olfactory system of rats. Neuroscience. 1977;2:797–800. doi: 10.1016/0306-4522(77)90033-1. [DOI] [PubMed] [Google Scholar]
- 65.Wonnacott S. Presynaptic nicotinic ACh receptors. Trends Neurosci. 1997;20:92–98. doi: 10.1016/s0166-2236(96)10073-4. [DOI] [PubMed] [Google Scholar]
- 66.Woolf NC. Cholinergic systems in mammalian brain and spinal cord. Prog Neurobiol. 1991;37:475–524. doi: 10.1016/0301-0082(91)90006-m. [DOI] [PubMed] [Google Scholar]
- 67.Woolf NC, Eckenstein F, Butcher LL. Cholinergic systems in the rat brain: I. projections to the limbic telencephalon. Brain Res Bull. 1984;13:751–784. doi: 10.1016/0361-9230(84)90236-3. [DOI] [PubMed] [Google Scholar]
- 68.Zaborsky L, Carlsen HR, Brashear HR, Heimer L. Cholinergic and GABAergic afferents to the olfactory bulb in the rat with special emphasis on the projection from the horizontal limb of the diagonal band. J Comp Neurol. 1986;243:488–509. doi: 10.1002/cne.902430405. [DOI] [PubMed] [Google Scholar]
- 69.Zhang Z-W, Vijayaraghavan S, Berg DK. Neuronal ACh receptors that bind α-bungarotoxin with high affinity function as ligand-gated channels. Neuron. 1994;12:167–177. doi: 10.1016/0896-6273(94)90161-9. [DOI] [PubMed] [Google Scholar]