

Abstract
Kainic acid (KA) induces status epilepticus in both adult and young rats but with different consequences on pathology and gene expression. In adults, GluR2(B) AMPA subunit expression is markedly reduced in CA3 neurons before neurodegeneration. In pups, the GluR2(B) subunit is sustained, possibly contributing to neuronal survival. Mechanisms underlying the reduced vulnerability of developing neurons to seizures was investigated by examining the effects of unilateral microinfusions of GluR2(B) antisense oligodeoxynucleotides (AS-ODNs) into the hippocampus of young rats in the presence or absence of a subconvulsive dose of KA. GluR2(B) AS-ODN infusions resulted in spontaneous seizure-like behavior, high stimulus intensity population spikes in the absence of long-term potentiation, and neurodegeneration of CA3 neurons lateral to the infusion site. Electroencephalography revealed paroxysmal activity and high-frequency high-amplitude discharges associated with vigorous and continuous scratching, wild running, or bilateral jerking movements. Pups lacking phenotypic behavior exhibited high-rhythmic oscillations and status epilepticus by the dose of KA used. Radiolabeled AS-ODNs accumulated throughout the ipsilateral dorsal hippocampus. GluR2(B) but not GluR1(A) receptor protein was markedly reduced after GluR2(B) knockdown. In contrast, GluR1(A) knockdown reduced GluR1(A) but not GluR2(B) protein without change in behavior or morphology. Therefore, unilateral downregulation of hippocampal GluR2(B) but not GluR1(A) protein reduces the seizure threshold and survival of CA3 neurons in the immature hippocampus, possibly providing a novel partial seizure model in the developing rat.
Keywords: epilepsy, development, GluR2(B), knockdown, hippocampus, neurodegeneration
Human temporal lobe epilepsy is associated with marked neurodegeneration of the hippocampus (Babb and Brown, 1987; Bruton, 1988). Sustained Ca2+influx through glutamate receptor channels may influence delayed seizure-induced damage (Siesjo and Bengtsson, 1989; Choi, 1990;Dingledine et al., 1990). AMPA-type glutamate receptors lacking the GluR2(B) subunit are highly Ca2+permeable, whereas heteromeric assemblies containing the GluR2(B) subunit are relatively impermeable to Ca2+(Hollman et al., 1991; Verdoorn et al., 1991; Burnashev et al., 1992) and may be involved in the seizure-induced damage and glutamate toxicity (the “GluR2(B) hypothesis”) (Pollard et al., 1993;Friedman et al., 1994; 1997; Friedman, 1998).
In adult rats, kainic acid (KA)-induced status epilepticus results in a marked but nonuniform reduction of GluR2(B) mRNA (Pollard et al., 1993;Friedman et al., 1994) and protein (Friedman, 1998) within vulnerable CA3 neurons at times preceding significant cell loss. Similar downregulations are observed in limbic structures after amygdala kindling (Prince et al., 1995) or in resected CA3 (Lynd-Balta et al., 1996; Mathern et al., 1998) and neocortex (DeFelipe et al., 1994) of human patients with temporal lobe epilepsy. In young rats, which are highly prone to seizures but resistant to KA seizure-induced brain damage (Cherubini et al., 1983; Albala et al., 1984; Nitecka et al., 1984; Holmes and Thompson, 1988; Wonziak et al., 1991; Friedman et al., 1997a,b), GluR2(B) hippocampal mRNA and protein expression remain constant and may contribute to neuronal survival (Friedman et al., 1997a). The functional role of Ca2+ influx through AMPA receptors at the physiological level has been unclear, but it is now known that different levels of GluR2(B) gene expression can result in a number of neurological deficits and age-dependent CA3 vulnerability (Brusa et al., 1995; Feldmeyer et al., 1999). In keeping with this, we found that hippocampal GluR2(B) antisense knockdown results in seizure-like behavior and CA3 neurosclerosis during a specific postnatal period (third week) (Friedman and Velísková, 1998). In contrast, adult GluR2(B) knockout mutants show no obvious alterations in hippocampal morphology or electroencephalography (EEG) activity, although during the third postnatal week they display reduced body weight, increased mortality, and enhancement of long-term potentiation (LTP) (Jia et al., 1996). Together, GluR2(B) antisense and genetic findings demonstrate that expression of the edited GluR2(B) subunit is essential during a particular stage in development to prevent occurrence of limbic seizures, maintain hippocampal plasticity, and contribute to CA3 neuronal survival.
Regulation of the GluR2(B) subunit with phosphodiesterized antisense oligodeoxynucleotides (AS-ODNs) allows one to examine possible roles of AMPA receptor subunits within a specific brain region and age. It is possible that regulation of the GluR2(B) gene after severe seizures may influence transcriptional factors or result in post-translational modifications that may trigger cell death genes (Schreiber and Baudry, 1995; Charriaut-Marlangue et al., 1996). The present study identifies molecular, cellular, and physiological consequences of modifying AMPA-mediated Ca2+ permeability during the third postnatal week to directly test the GluR2(B) hypothesis. Developmental aspects are emphasized because both antisense and genetic approaches reveal age-specific epileptogenesis and selective CA3 hippocampal neurodegeneration at a time when the brain has a high propensity toward seizures but is relatively resistant to seizure-induced damage.
MATERIALS AND METHODS
Unilateral intracranial cannula/bipolar electrode implantation and ODN administration. Sprague Dawley male albino pups [postnatal day 13 (P13), 25–30 gm; n = 74] were kept on a 12 hr light/dark cycle at room temperature (55% humidity). Rat pups were anesthetized intraperitoneally with a mixture of 70 mg/kg ketamine and 6 mg/kg of xylazine and then stereotaxically implanted with either chronic guide (25-G) cannulae or cannula/bipolar electrode assemblies into the right hippocampus to administer ODNs and obtain ipsilateral hippocampal EEG recordings (coordinates in millimeters with respect to bregma of P13 pups: anteroposterior, −3.2; lateral, 2.4; dorsal, −2.4; incisor bar at −3.5) (Paxinos and Watson, 1986). The cannulae were angled at 0° from the vertical sagittal plane. After surgery, young rats were kept in a box under a lamp and then returned to their lactating mother. Pups recovered from anesthesia and became active 1–3 hr after the surgery. Animals were infused with GluR2(B) AS-ODNs to selectively decrease the hippocampal synthesis of the GluR2(B) subunit as previously described (Friedman and Velísková, 1998). The infusion cannula was force-fitted onto Teflon tubing attached to a Hamilton syringe. Infusions of Tris buffer, antisense (AS), sense (S), or missense (NS, scrambled) ODNs [GluR2(B) AS-ODN, n = 32; GluR2(B) S-ODN,n = 16; NS-ODN, n = 9; Tris buffer,n = 2; GluR1(A) AS-ODN, n = 8; GluR1(A) S-ODN, n = 4] were initiated ∼3–4 hr after recovery from the anesthesia twice daily (9:00–10:00 A.M. and 5:00–6:00 P.M.). A phosphorothiolated 21 mer was designed from base pairs 30–50, near the initiation codon start site to selectively decrease the hippocampal synthesis of the GluR1(A) subunit: 5′-CGGTTTTCTAGGTGCGGTTGT-3′. This sequence was effective and not identical with other mammalian sequences as determined by a search of the GeneBank/European Molecular Biology Laboratory database. The sequence of the GluR1(A) S-ODN was (5′-ACAACCGCACCTAGAAAACCG-3′). Rat pups were killed 24 hr after 4 d of treatment. The injection volume (0.25 μl of 4 μg/μl dissolved in 0.1 mm Tris, pH 7.4) was delivered over 2 min to reduce spread of the ODNs. In three rats,33P-radiolabeled GluR2(B) AS-ODNs were also infused to determine the extent of ODN spread and accumulation. Behavior was monitored daily between 10:00–3:00 P.M. and 4:00–8:00 P.M. for behavioral changes.
33P autoradiography. To determine that the ipsilateral dorsal hippocampus was selectively labeled by the infusion paradigm used, the GluR2(B) AS-ODNs were 5′ end-labeled with [33P] γ-ATP for 30 min at 37°C, using a DuPont/NEN oligonucleotide 5′ end-labeling system. Labeled probes were purified on NENSORB20 nucleic acid purification cartridges, lyophilized, and resuspended in a small volume (10 μl, 1–10 ng/μl) for microinfusion into the hippocampus. Approximately 1 μCi of33P-labeled GluR2(B) AS-ODNs was microinfused twice daily into the hippocampus, and33P autoradiography was performed after 4 d of exposure to the radiolabeled GluR2(B) AS-ODNs. Brains were removed and quick-frozen, and serial coronal sections (20 μm) were cut in a cryostat at −18°C. Sections were mounted and air-dried onto double-gelatinized slides, then dried overnight in a vacuum desiccator in the presence of silica crystals. Slides were apposed to x-ray film for 4 d at −70°C to generate the33P autoradiographs. Films were scanned with a CCD camera attached to a Macintosh computer (Quadra 840V). Optical density measurements of hippocampal accumulations of33P-labeled ODNs, detected throughout the dorsal hippocampus, were analyzed with NIH image software to estimate the extent of ODN spread and accumulation. Optical density readings were not statistically analyzed because of the small number of pups injected with radioactivity (n = 3).
EEG recordings and statistical analysis of duration and number of scratching events. To determine whether behavioral manifestations corresponded to changes in electrographic activity, EEG recordings were obtained from a subset of rat pups that received the GluR2(B) AS-ODN sequence [GluR2(B) AS-ODNs, n = 9; S-ODNs,n = 5; and NS-ODNs, n = 5] [GluR2(B) AS-ODNs, n = 4] for at least 30–60 min twice daily. EEG recordings were also obtained from a separate group of rat pups that received a low, subconvulsive dose of KA acid (2.0 mg/kg) administered intraperitoneally (n = 5), after cannula implantation and 4 d of S-ODN infusions to compare chemoconvulsant-induced automatisms (e.g., scratching) with spontaneous automatisms arising in pups treated only with GluR2(B) AS-ODNs. Duration and number of scratching events were quantified from animals in the presence and absence of KA after antisense and control sequence treatments. Other manifestations have been reported (Friedman and Velísková, 1998). The data were compared by one-way ANOVA with post hoc Fischer protected least significant difference test (ANOVA). Duration of scratching events was measured only from EEG recording traces, for accuracy. In another subgroup of GluR2(B) AS-ODN-treated rat pups (n= 6), the progression of scratching was assessed and evaluated by the Student's paired t test. The level of significance was preset to p < 0.05.
Ex vivo hippocampal slice physiology of deprived GluR2(B) circuitry. For CA1 hippocampal slice recordings, 13-d-old Wistar rats were raised in our local departmental animal facility in Rehovot. Experiments were performed in three types of control animals: (1) littermate no treatment, (2) S-ODN treatment, or (3) slices prepared from the contralateral hemisphere. After 4 d of intrahippocampal GluR2(B) AS-ODN infusions, brains were rapidly removed and placed into cold (2–4°C) artificial CSF (ACSF), pH 7.4, containing (in mm): 124 NaCl, 2.0 KCl, 1.24 KH2PO4, 2.0 MgSO4, 2.5 CaCl2, 26 NaHCO3, and 10 d-glucose) saturated with 95% O2/5% CO2 gas mixture (flow rate 0.4 l/min) and adjusted with sucrose to 320 mOsm osmolarity as described previously (Auerbach and Segal, 1997; Koudinov et al., 1998). Hippocampal slices (400 μm) were prepared with a McIlwain tissue slicer. After incubation in a recreation chamber at room temperature for 1.5 hr in ACSF, slices were immediately transferred to a recording chamber held at constant temperature (32°C). Slices were submerged and superfused with ACSF at a flow rate of 1.8–2.0 ml/min. GluR2(B) knockdown and control slices were tested sequentially in random order. One extracellular electrode (∼4 MΩ, 0.75 mm NaCl) was guided centrally into stratum radiatum of CA1 (200 μm deep) near the infusion site with a Nikon binocular system (4× objective) to obtain two channel measurements of field EPSP (fEPSP) recordings. Two stimulating electrodes (bipolar stainless steel) were placed into CA1 stratum radiatum, either near the infusion site or distally toward the CA2 region, and then stimulated from either of the two sites. Stimulations were delivered every 30 sec for each channel at 50 μsec pulse duration with a 15 sec time interval between the channels. Stimulus intensity was adjusted so that initial baseline evoked responses (∼0.8 mV) were approximately one-half of the maximum possible (∼1.6 mV), and 2–6 mA stimulation intensities were used for input/output curve generation. After stable baseline recordings were established, tetanic LTP (tLTP) was induced by delivering a 100 Hz, 1 sec train through the stimulation electrode at the test baseline stimulus intensity (Koudinov et al., 1998). Data were collected, stored, and analyzed on an IBM-compatible computer using Asyst 3.1 data acquisition and analysis software. fEPSPs recorded from a single site in response to alternately delivered stimuli to two separate inputs in control versus experimental slices were averaged from both pathways (three to five hippocampal slices per animal). Values from control slices were similar whether recorded from contralateral hippocampus, sham, or sense-infused pups and therefore averaged. Values from slices obtained from pups with seizure behavior were also similar in both pathways and therefore averaged. In slices (n = 4) from an animal that did not exhibit seizures, there were differences in the two stimulation pathways; therefore these data were averaged separately. All data were normalized with respect to the steady baseline values and expressed as mean ± SEM. Nonparametric unpaired Mann–Whitney test was used for determining significant differences between potentiation levels of control and GluR2(B) knockdown slices at the indicated times. A probability ofp < 0.05 or less was accepted as statistically significant.
Histology. To elucidate direct effects of the cannulae versus GluR2(B) knockdown-induced injury in the hippocampus, Nissl (thionin) or chromatin (hematoxylin/eosin) histological stains were used on serial sections (20–30 μm), cut from a cryostat, to examine cell loss and alterations in morphology. Approximately 30 slides were prepared from animals that were not processed for immunocytochemistry. Three or four sections were mounted per slide from an anterior to posterior direction so that each slide contained sections from several hippocampal levels. Every fourth or fifth slide was stained with thionin to evaluate cell loss at anterior and posterior levels, and hematoxylin/eosin was used on remaining sections to detect subtle histopathology (e.g., eosinophilia) throughout the hippocampus. Approximately six alternate sections were collected for histology from pups processed for immunolabeling studies. Cannula placement or disruption of hippocampal tissue produced by the S/NS-ODN control sequence administration was measured and distinguished under light microscopy, from the distant neurodegeneration of the CA3a region after appearance of seizure behavior by the GluR2(B) AS-ODNs.
In situ hybridization. In situ hybridization for AMPA and NR1 mRNA detection was used as described (Friedman et al., 1994) to determine whether GluR2(B) mRNA levels were stable in response to the AS-ODN treatment (at 24 hr after the last infusion): GluR2(B) AS-ODNs, n = 5; GluR2(B) S-/NS-ODNs, n= 5). GluR2(B) and NR1 cDNAs were transcribed with [35S]UTP-labeled RNA and T7 RNA polymerase. Coronal sections (20 μm, −16°C) from antisense and S/NS-ODN-treated animals were cut and thaw-mounted on gelatin/poly-ml-lysine-coated slides. Sections were post-fixed with 4% (w/v) paraformaldehyde, acetylated with acetic anhydride (0.25%), and stored in 95% ethanol at 4°C. On the next day, prehybridization solution (100 μl) was added directly to sections and incubated for 2 hr at 50°C [50% (w/v) formamide/2.5 × Denhardt's solution/0.6 mNaCl/10 mmTris.HCl/1 mmEDTA/0.05% SDS/herring sperm DNA (150 μg/ml)/total yeast RNA (50 μg/ml)]. Hybridization solution (100 μl) containing the35S-labeled riboprobe (106 cpm/section, 1 ng/μl), 10% (w/v) dextran sulfate, and 10 mm dithiothreitol was added overnight (50°C). Adjacent sections from the same rats were labeled with GluR1(A), GluR2(B), and NR1 probes. The next day, sections were washed, treated with RNase A (20 μg/ml), washed for 2 hr in 0.2 × SSC at 50°C, and dehydrated in graded ethanols. For autoradiography, slides were apposed (1–7 d) to Kodak XR5 film. For cellular resolution, sections were dipped in Kodak NTB-2 emulsion, developed after 1–2 weeks, counterstained with hematoxylin/eosin, and examined under a light microscope with phase contrast (Nikon Eclipse 800).
Specificity of mRNA labeling and quantification. Conditions were of sufficiently high stringency as to rule out cross-hybridization among GluR1(A), GluR2(B), and GluR3 (Friedman et al., 1994) and more distantly related glutamate receptor subunits (GluR5–GluR7, KA1, KA2). GluR4 expression is prominent only in the cerebellum (Keinänen et al., 1990). GluR1(A), GluR2(B), and GluR3 are “pan” probes (Sommer et al., 1990) in that they label both “flip” and “flop” splice variants. The NR1 probe shares <20% sequence identity with NR2A-C and does not cross-react with other NMDA receptor subtypes (Nakanishi et al., 1992). Autoradiographs and emulsion-dipped sections were scanned with a CCD camera, and densitometric measurements were obtained and analyzed with NIH image software. Individual grains overlying CA3 neurons were also counted manually with NIH tools. Student's pairedt test was used for statistical analysis, and significance was set at p < 0.05.
Immunohistochemistry. To examine whether GluR2(B) or GluR1(A) proteins were selectively downregulated by the GluR2(B) or GluR1(A) AS-ODN treatment, respectively, immunocytochemistry was performed with AMPA subunit-specific antibodies in vibratome (40 μm) sections prepared from AS-ODN-, S-ODN-, or NS-ODN-treated rat pups at the level of the hippocampus as described (Vissavajjhala et al., 1996;Friedman, 1998; Friedman and Velísková, 1998). Twenty-four hours after 4 d of GluR2(B) or GluR1(A) AS-ODN infusions, pups were killed by transcardial perfusion. The anti-GluR1(A) (0.6 μg/ml) and anti-GluR2(B) (0.6 μg/ml) primary antibodies were used. Free-floating sections were washed in PBS (two times), then incubated with 0.5% H202 for 15 min to remove endogenous peroxides. Sections were then washed (four times for 10 min each). For the GluR2(B)-specific antibody (6C4), sections were blocked for 30 min in 5% horse serum/0.5% BSA–PBS. For the polyclonal antibody, GluR1(A), sections were blocked in 5% goat serum/0.5% BSA–PBS. Monoclonal and polyclonal primary antibodies were incubated for 48 hr at 4°C. Tissue sections were washed (three times for 10 min each) in PBS to remove primary antibodies. Secondary biotinylated goat anti-mouse or anti-rabbit IgG (H + L, diluted 1:200) were added for 2 hr at 25°C. After three washes, ABC solution was added for 1 hr. For visualization, the sections were reacted with 3′, 3-diaminobenzidine tetrahydrochloride (10 mg/20 ml and 3 μl hydrogen peroxide). Sections were then washed, mounted, dehydrated, cleared, and coverslipped.
Controls. To assess immunocytochemical signal specificity, additional sections were incubated with (1) PBS followed by goat anti-rabbit or horse anti-mouse secondary antibody, (2) preimmune mouse or rabbit serum at the same dilution as the primary antibody, or (3) primary antibody, which had been preadsorbed with the peptide antigen, followed by secondary antibody. All three control conditions gave no specific labeling.
Drugs and chemicals. Oligodeoxynucleotides were synthesized by Genosys Biotechnologies (The Woodlands, Texas). Surgical supplies were obtained from Plastic One (Roanoke, VA). Chemicals were bought from commercial sources. GluR1(A) and GluR2(B) cDNAs were generously provided by Dr. P. Seeburg (University of Heidelberg, Germany). The specific monoclonal antibodies (mAbs) GluR2(B) (6C4) and GluR2(4) (3A11) were generous gifts from Dr. J. Morrison (Mount Sinai Medical Center, NY), and polyclonal GluR1(A) and GluR2(B) antibodies were purchased commercially from Chemicon International (Temecula, CA).
RESULTS
Anatomical localization of GluR2(B) AS-ODNs by33P autoradiography
To determine that the ipsilateral dorsal hippocampus was selectively labeled by the infusion paradigm used, the GluR2(B) AS-ODNs were 5′ end-labeled and processed for 33P autoradiography. Stereotaxic coordinates of the intrahippocampal infusion and recording site are illustrated (Fig.1A). After 4 d of knockdown treatment, autoradiographs of full coronal sections revealed that accumulation and spread of the microinfused ODNs were highly restricted to the dorsal hippocampus of the ipsilateral hemisphere (Fig. 1B, a–c). All major subfields were labeled, whereas other structures, except for small portions of the dorsal thalamus, were absent in signal or similar to film background (entire section is illustrated by a schematic outline of the center section) (Fig. 1B, b). The GluR2(B) AS-ODNs also did not accumulate in the ventral hippocampus. The measured distance of the intrahippocampal GluR2(B) AS-ODN spread and regional accumulation extending from the medial septum to the posterior region of the dorsal hippocampus was ∼2.4–2.8 mm3 (Fig. 1B,a–c). Optical density measurements (NIH image software) were averaged from medial and lateral portions of the hippocampus around the infusion site at anterior, medial, and posterior levels. GluR2(B) AS-ODN radioactivity formed a mediolateral concentration gradient showing that the highest accumulations occurred in or around the cannula infusion site within the medial portion of the hippocampus (Fig. 1B, b, Table1). The concentration gradient diminished in posterior sections distant from the point of injection (Fig.1B, c).
Fig. 1.

A, Cannula/bipolar electrode assembly location is indicated by arrow.B, Nine entire coronal sections labeled by33P GluR2(B) AS-ODN autoradiography are on display (as indicated by outline of center section) from the most anterior portion of the dorsal hippocampus to the ventral posterior region. Distance between sections on display is 600–800 μm (a–c) so that the infused GluR2(B) AS-ODN spread or regional accumulation was approximately 2.4–2.8 mm3within the ipsilateral dorsal but not ventral hippocampus. Mediolateral gradients diminished in posterior sections (B,c), and ODNs did not spread to the contralateral hippocampus or to other brain regions (not visible or seen aswhite background).
Table 1.
Optical density measurements of 33P GluR2(B) AS-ODN autoradiography
| Level | Medial | Lateral |
|---|---|---|
| Anterior | 104 ± 8 | 143 ± 10 |
| Medial | 122 ± 5 | 194 ± 10 |
| Posterior | 100 ± 3 | 113 ± 2 |
| Infusion site | 197 ± 3 | nd |
P13 pups were infused with 33P-radiolabeled GluR2(B) AS-ODNs for 4 d into the right hippocampus as described in Materials and Methods. nd, Not done.
GluR2(B) knockdown-induced automatisms and seizure-like behavior
Control rat pups treated with equal volumes of sense (S) or missense (NS, scrambled) ODN sequences exhibited normal behavior similar to pups that did not undergo surgery or that were infused with Tris-buffer. Behavior included free movement around the recording chamber with sniffing and washing movements, occasional scratching or wet dog shakes, or rearing. In control animals infused with vehicle or S/NS-ODNs, scratching movements were generally short lasting (Fig.2A, Table2). In contrast, GluR2(B) knockdown resulted in altered motor behavior in 80% of the pups receiving GluR2(B) AS-ODNs; approximately one-half of the animals exhibited automatisms by 48 hr. Automatisms were characterized by long episodes of vigorous and continuous scratching (Table 2). This scratching resembled behavior that occurs in rat pups administered with either low or high doses of KA at similar ages (Cherubini et al., 1983; Albala et al., 1984; Tremblay et al., 1984). An apparent difference was that the number of scratching events progressed with time (day 2: 1.03 ± 0.8 vs day 3: 1.68 ± 0.8, n = 6,p < 0.05) in the GluR2(B) AS-ODN intrahippocampal-infused pups compared with KA control injected pups. The greatest number of scratching events appeared on day 4. In 40% of rat pups infused with GluR2(B) AS-ODNs (13 of 32), seizure-like symptoms progressed. There were jerking and stumbling movements, ball curling, head nods and shaking, wet dog shakes, or quadrilateral forelimb jerking movements, with backward jumping and loss of postural control. Ipsilateral followed by contralateral tremor with wild running and immobility with forelimb extension occurred in 25% of the GluR2(B) AS-ODN treated pups. Spontaneous seizure-like behavior often continued for 30–60 min, stopped, and then reappeared at different times. These seizure-type behaviors have been quantified and recently reported by us (Friedman and Velísková, 1998). Despite recurring seizure-like behaviors, GluR2(B) AS-ODN pups fed regularly, so that weight and mortality were not significantly affected. No seizure-like behavior was observed in control untreated or S/NS-ODN-treated groups. Moreover, a separate group of pups was infused unilaterally with AS-ODNs targeted to the GluR1 subunit. GluR1(A) hippocampal knockdown did not alter behavior in any of these animals (n = 8).
Fig. 2.
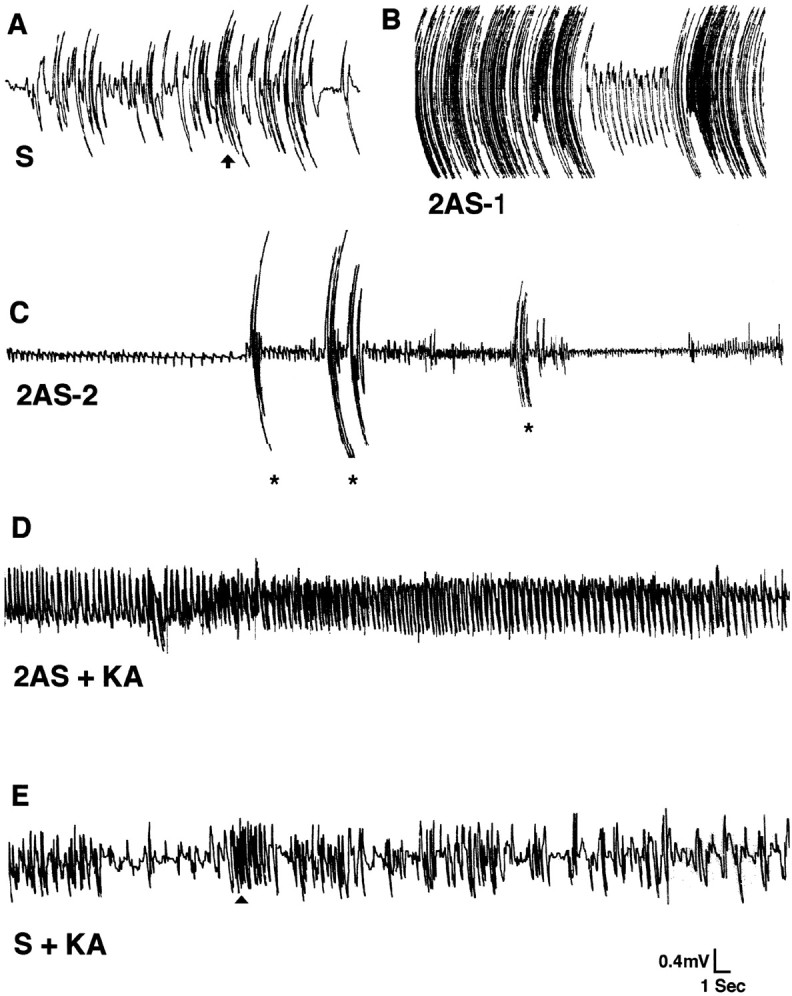
EEG recordings after GluR2(B) knockdown from ipsilateral pup hippocampus in a freely moving recording chamber.A, S-ODN (S) baseline EEG with normal movement artifacts showing a typical short-lasting (<1 sec) scratching event (vertical arrow) (left portion). B, GluR2(B) AS-ODN-treated pup (2AS-1) exhibited vigorous and continuous scratching that produced high-frequency, high-amplitude movement artifacts; occasional spikes occurred between scratching events (right portion). C, In another GluR2(B) AS-ODN-treated pup (2AS-2), convulsive seizure-like behavior including body tonus with forelimb extension and laying on one side occurred and was associated with high-frequency low-amplitude rhythmical activity of increasing size. Large-movement artifacts shown represent intermittent bilateral jerking movements (asterisks). D, In GluR2(B) AS-ODN-treated pups lacking the phenotypic behavior (2AS + KA), a low concentration of KA produced high-frequency rhythmical oscillations associated with tonic-clonic seizure behavior.E, In an S-ODN-treated pup (S + KA), a subconvulsive KA dose produced classic automatisms (e.g., wet dog shakes, arrowhead) but not the high-frequency rhythmical oscillations.
Table 2.
Quantification of scratching manifestations after GluR2(B) hippocampal knockdown
| ODN | Events per minute | n | Duration | n |
|---|---|---|---|---|
| S | 0.08 ± 0.30 | 11 | 0.97 ± 0.78 | 5 |
| NS | 0.10 ± 0.04 | 9 | 0.89 ± 0.48 | 5 |
| AS | 2.64 ± 0.53* | 25 | 5.81 ± 0.81* | 9 |
| AS + KA | 2.17 ± 0.59* | 6 | 4.16 ± 0.60* | 6 |
| S + KA | 1.38 ± 0.23* | 5 | 2.44 ± 57* | 3 |
P13 pups were infused with GluR2(B) AS-, S-, and NS-ODNs for 4 d into the right hippocampus. The number of scratching events and duration were calculated as described in Materials and Methods. Values expressed represent the mean ± SEM of control and experimental groups. *p < 0.0001, ANOVA.
EEG activity after GluR2(B) knockdown in the presence and absence of KA
Electrographic baseline recordings from control untreated, S-ODN-, or NS-ODN-treated pups showed normal hippocampal activity and movement artifacts (Fig. 2A). Low amplitude spikes were associated with either sleeping or immobility with eyes open (data not shown). Walking, roaming about the recording chamber, climbing, or normal short-lasting scratching events produced movement artifacts of high amplitude and low frequency (Fig. 2A, and quantification in Table 2). The EEG baseline from GluR2(B) AS-ODN-treated pups before onset of behavioral manifestations was indistinguishable from that of S/NS-ODN-treated pups. After onset of seizure-like behavior, the EEG revealed persistent high-frequency bursts accompanying the long-lasting vigorous scratching episodes. These produced highly pronounced movement artifacts with occasional rhythmical spikes that resembled epileptiform activity (Fig.2B). Wild running followed by forelimb extension and lying on one side were associated with high-frequency spikes of increasing amplitude (Fig. 2C, left portion). There were also high-frequency low-amplitude spikes that correlated with backward jumping, jerking movements involving all limbs, followed by loss of postural control and immobility (Fig. 2C). In other GluR2(B) AS-ODN-infused pups lacking phenotypic expression of seizure-like behavior, a low, subconvulsant dose of KA (2 mg/kg) produced high-frequency rhythmical oscillations and behavioral status epilepticus. These do not occur in rat pups of similar ages treated with similar doses of KA (Cherubini et al., 1983; Albala et al., 1984;Tremblay et al., 1984) or in S-ODN-, N-ODN-, or Tris buffer-infused pups (Fig. 2D). Similarly, control rat pups pretreated with S-ODNs and followed by injection with KA showed increased automatisms (i.e., scratching and wet dog shakes) in the absence of high-frequency rhythmical oscillations (Fig.2E).
GluR2(B) knockdown produces CA3 cell loss in pups with seizure behavior
Histology was performed with hematoxylin/eosin or thionin on serial air-dried coronal sections from GluR2(B) AS-ODN-treated and control ODN-treated animals to monitor eosinophilia and/or cell loss. In rat pups infused with GluR2(B) AS-ODNs, Nissl-stained sections showed specific neurodegeneration that was particularly notable in the CA3a subregion and distant and lateral to the more medially placed cannula infusion site (Fig.3A–C). Cell loss and changes in morphology of CA3 pyramidal neurons were detectable throughout the rostrocaudal plane of the dorsal hippocampus. Subtle histopathological changes were observed within CA3a neurons in more distant brain sections posterior to the cannula infusion site. Hematoxylin/eosin staining of nuclei revealed eosinophilic pyramidal CA3 neurons with irregular shape, particularly around the CA3a bend subregion (Fig.3C, F). Many surviving cells around the lesion site appeared either basophilic and swollen or eosinophilic and shrunken (see Fig. 8F). Small cells, probably glia, surrounded the lesion, suggesting an acute reactive gliosis (Fig.3F). In rat pups lacking phenotype, similar to control S-ODN- or NS-ODN-infused pups, there was no obvious change in morphology (see Fig. 9E). CA3b/c neurons near the cannula infusion site were intact, with normal cytoarchitecture (Fig.3D,E). CA3a lesions were apparent only in pups that exhibited continuous seizure-like behavior. Within the contralateral hippocampus in all but one GluR2(B) AS-ODN-treated pup, morphology of pyramidal and granule cell types was indistinguishable from control untreated sections. A subconvulsive dose of KA was injected intraperitoneally into animals pretreated with GluR2(B) AS-ODNs but lacking the phenotypic behavior. This resulted in CA3a cell loss that was more extensive and obvious compared with GluR2(B) AS-ODN pups exhibiting spontaneous behaviors, but CA1, hilus, and DG remained spared (data not shown).
Fig. 3.

Photomicrographs depicting GluR2(B) knockdown-induced cell loss and morphological alterations in CA3 hippocampal neurons after 4 d of infusions. A, Thionin-stained section from GluR2(B) AS-ODN-treated pup at low magnification shows marked loss of CA3a pyramidal neurons (betweensmall arrows) just after the bend and posterior to the level of infusion site (large arrow). B, Loss of CA3a neurons by thionin is shown at higher magnification.C, F, Hematoxylin/eosin-stained section 360 μm posterior to section displayed in B at low (C) and high (F) magnifications reveal eosinophilic neurons (large arrows) and apparent glial cell proliferation (short arrows) in posterior sections around the distant lesion.D, NS-ODN control section at level of infusion site and at low and high (E) magnifications shows that CA3 neurons distant from cannula infusion site are intact with normal cytoarchitecture. Scale bar, 50 μm. i, Infusion site;AS, antisense; NS, nonsense.
Fig. 8.
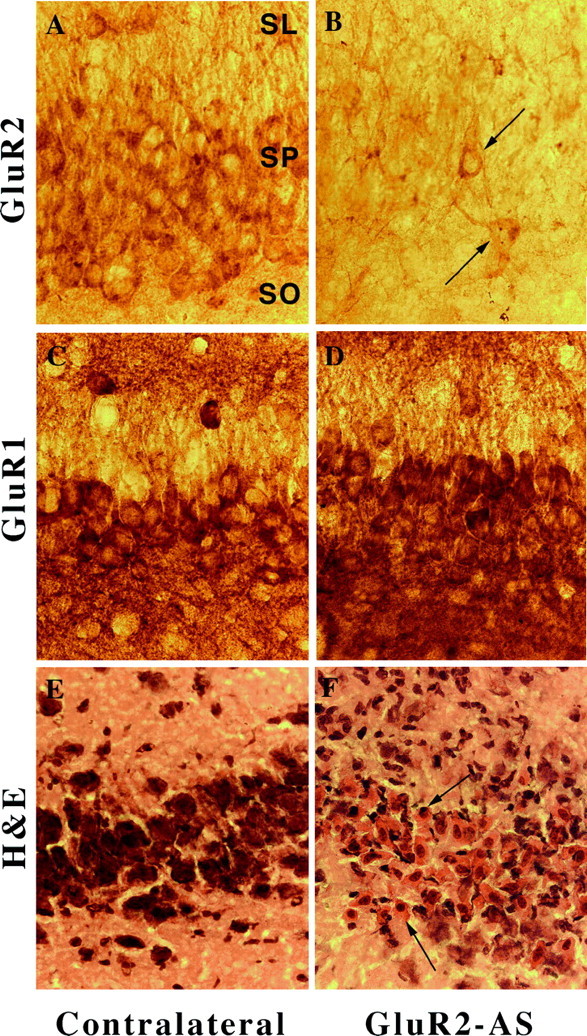
Photomicrographs of GluR2(B) and GluR1(A) immunolabeling of CA3 neurons after GluR2(B) knockdown at high magnification (400×). A, In control GluR2(B) sections, robust labeling of soma and proximal dendrites was observed. B, CA3 neurons were particularly decreased in GluR2(B) immunoreactivity; a few neurons were immunopositive near the lesion (arrows). C, GluR1(A) control immunoreactivity was dense and continuous with apical and basilar dendrites. D, GluR1(A) immunoreactivity near the lesion was unchanged. E, Control hematoxylin/eosin stain of CA3 just before the bend from contralateral hippocampus. F, Shrunken nuclei and eosinophilia of CA3a neurons were detected by hematoxylin/eosin stain (arrows). SR, Stratum radiatum; SP, stratum pyramidale;SO, stratum oriens.
Fig. 9.
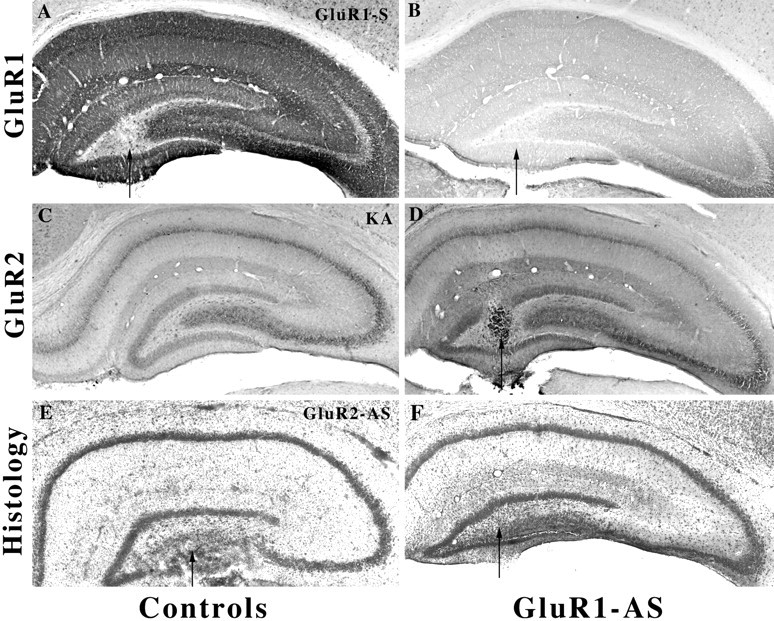
GluR1(A) knockdown decreases GluR1(A) but not GluR2(B) immunoprotein without change in histology. A, GluR1(A) S-ODN control microinfused pup (arrow).B, After GluR1(A) hippocampal knockdown (arrow), GluR1(A) immunoreactivity was markedly reduced throughout the immature hippocampus. C, After kainate-induced status epilepticus GluR2(B), immunoreactivity was intense and unaltered throughout the hippocampus at this young age.D, GluR2(B) immunoreactivity was unchanged, suggesting that a decrease in the GluR1(A)/GluR2(B) protein was achieved.E, No seizure control; thionin-stained section after GluR2(B) knockdown without phenotypic behavior showed damage to the ventral blade of the DG (n = 4).F, GluR1(A) knockdown demonstrates intact morphology and cytoarchitecture in adjacent Nissl-stained sections from the same rat shown in B and D.
Normal fEPSP activity but loss of LTP in CA1 hippocampal slices after GluR2(B) knockdown
After GluR2(B) knockdown, changes in synaptic function were studied from the hippocampal CA1 subfield in slices prepared from GluR2(B) AS-ODN-treated pups versus control animals including (1) no treatment, (2) S-ODN treatment, or (3) slices prepared from the contralateral hemisphere. Although high-frequency rhythmical activity was found in the EEG, epileptiform activity was not detectable in field recordings in the presence or absence of seizure behavior, similar to seizure phenotype GluR2(B) mutants with partial Q/R editing site incompetence (Feldmeyer et al., 1999). There were no significant differences between the antisense and control input/output characteristics of the slices, although knockdown slices tended to have larger responses (measured as a slope of the fEPSP) for the same stimulation intensity (Fig.4B). Similar fEPSP half-maximal baseline responses were evoked in GluR2(B) knockdown slices and in the various types of control slices used (0.7667 ± 0.04975 mV, n = 8 and 0.7750 ± 0.0366 mV fEPSP maximum, n = 12, for knockdown and control slices, respectively; both values were not significantly different) (Fig.4C,D). All slices also exhibited similar maximal fEPSP (1.6442 ± 0.1064 mV, n = 12 and 1.635 ± 0.07962 mV, n = 8, respectively).
Fig. 4.

fEPSPs recorded from a single site in response to alternately delivered stimuli to two separate inputs from control versus experimental slices after GluR2(B) hippocampal knockdown.A, Cross section showing stimulating (stim 1 and stim 2) and recording electrode (rec) arrangements in respect to the infusion site (asterisk). B, Averaged input–stimulus/output–response relationship (input/output curve) in GluR2(B) AS-ODN (circles) and control (squares) slices. C, Field synaptic responses in sense and antisense knockdown slices obtained before (1), immediately after (2), and 13 min after (3) the high-frequency train of stimuli. Note that the immediate post-tetanic trace from knockdown slices reveals a population spike over the fEPSP. D, Normalized fEPSP slope changes in GluR2(B) AS-ODN (●) and control (■) slices show impairment of tetanic LTP in CA1 averaged from both sites; arrow indicates time of tetanus.E, fEPSP recorded from a representative animal that did not exhibit seizure behavior. First and second tetanic stimulations (arrow) were delivered to inputs near (▪) and distal (▵) to the infusion site.
In contrast, in GluR2(B) knockdown slices prepared from pups that exhibited seizure behavior, there was a population spike over the maximal fEPSP in both stimulation pathways, suggesting that electrical properties of the preparation were modified (data not shown). Identical population spike activity over the fEPSP in GluR2(B) knockdown slices occurred in the first post-tetanic trace after induction of tLTP with a 100 Hz, 1 sec tetanus (Fig. 4C). The amount of initial post-tetanic potentiation in CA1 field recordings of knockdown and control slices, averaged from both pathways, also did not reach statistical significance (99.2 ± 15.9%, n = 12, and 113.7 ± 4.2%, n = 8, p = 0.08, respectively) (Fig. 4C,D). Interestingly, slice preparations from animals with seizure behavior did not maintain LTP whether stimulated near or distant from the infusion site, suggesting that GluR2(B) knockdown increased excitability in medial and distal regions of the hippocampus. Within 3 min potentiation, levels dropped to near baseline values (7.8 ± 2.4%), whereas control slices maintained LTP throughout the 30 min post-tetanic recording time (60.9 ± 6.1%) (Fig. 4D). Differences in two-channel recordings were observed in slices prepared from an animal that did not exhibit seizure behavior (Fig. 4E). Within 5 min potentiation, tLTP was markedly reduced when stimulations were delivered close to the infusion site (13.6 ± 6.3%;n = 4 slices) but maintained at near control levels when stimulated from the distal site (56.7 ± 8.4%;n = 4 slices).
Specificity of labeling by in situ hybridization and immunohistochemistry
Because AS-ODNs can decrease expression of their target mRNA attributable to binding of the oligo probes to the same sequence of the endogenous mRNA, GluR2(B) and a more distantly related glutamate receptor, NR1, mRNA expression were assessed by in situhybridization autoradiography (Fig. 5) and emulsion-dipping (Fig. 6). In control sections, GluR2(B) and NR1 mRNA expression were prominent and uniformly expressed throughout pyramidal and granule cell layers as described previously (Nakanishi et al., 1992; Friedman et al., 1994,1997a,b; Standley et al., 1995). After intrahippocampal infusion of GluR2(B) AS-ODNs, in situ hybridization autoradiographs showed that GluR2(B) and NR1 mRNAs were unaltered throughout the surviving regions of the CA1, CA3, and DG hippocampal subfields in both control and GluR2(B) AS-ODN-treated groups (CA1: 98 ± 5%; CA3: 91 ± 7%; DG: 97 ± 8%; vs control regions,n = 3). In contrast, densitometric measurements (NIH image) from the CA3 area of cell degeneration revealed an apparent reduction in the expression of both GluR2(B) and NR1 mRNAs (to 49.2 ± 12%, n = 3, p < 0.05 vs controls and 50 ± 2%, n = 2, respectively) (Fig.5). However, light microscopy of emulsion-dipped sections counterstained with hematoxylin/eosin confirmed that the decreases were attributable to cell loss and that individual CA3a neurons expressed control numbers of GluR2(B) and NR1 mRNA in situhybridization grains (GluR2 mRNA: 365 ± 16 vs 355 ± 12; control vs GluR2(B) knockdown; and NR1 mRNA: 51 ± 3 versus 50 ± 4; control versus GluR2(B) knockdown). Therefore, hippocampal mRNA levels within surviving CA3 neurons were not altered by our AS-ODN infusion procedure.
Fig. 5.
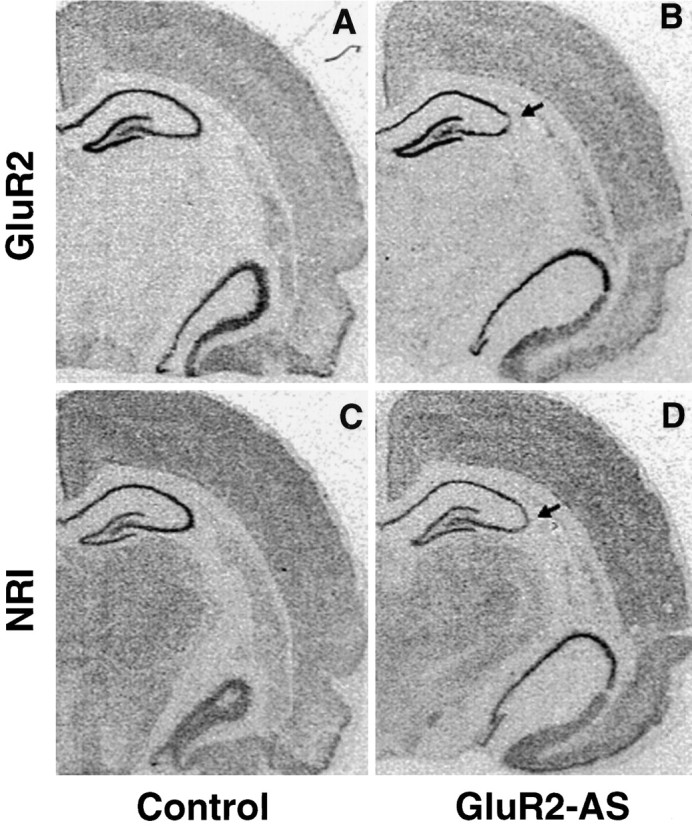
In situ hybridization showed that GluR2(B) knockdown had no effect on the overall level of GluR2(B) or NR1 mRNA expression. Loss of GluR2(B) expression was reduced only in the area of lesion for GluR2(B) (A, B) and for NR1 (C, D).
Fig. 6.
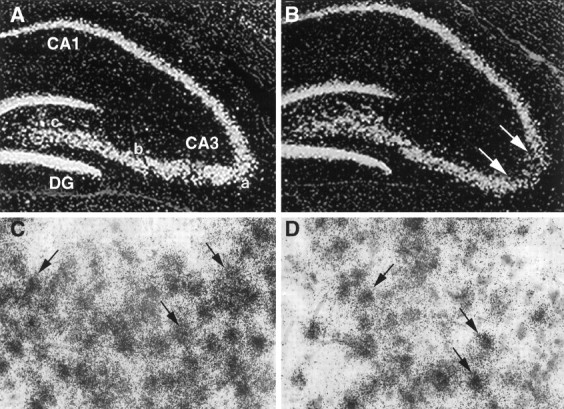
Emulsion-dipped sections showed control levels of GluR2(B) mRNA in individual neurons within the region of cell loss.A, Control GluR2(B) mRNA (40×). B, GluR2(B) mRNA after GluR2(B) knockdown. C, Control GluR2(B) hybridization grains were dense and uniform, overlying individual neurons (100×). D, At the single-cell level, GluR2(B) mRNA in situ hybridization grains in GluR2(B) knockdown sections were indistinguishable from control sections.
In contrast, AMPA receptor immunocytochemistry demonstrated specificity for both oligonucleotide sequences used for selective antisense-induced downregulation of GluR2(B) or GluR1(A) hippocampal proteins. In the contralateral hippocampus and in control sections after S-ODN or NS-ODN infusions, the pattern of cytoplasmic label of principal neurons for GluR1(A) and GluR2(B) was dense and evenly distributed throughout the hippocampus, as described previously (Rogers et al., 1991; Wenthold et al., 1992; Vissavajjhala et al., 1996; Friedman et al., 1997a,b;Friedman, 1998) (Figs. 7-9). After GluR2(B) knockdown, the use of several available GluR2(B)-specific antibodies (mAb 3A11 GluR2(4) and mAb 6C4 provided by Dr. John Morrison and the polyclonal mAb from Chemicon) and the polyclonal GluR1(A) antibody (provided by Dr. Scott Rogers or purchased from Chemicon) repeatedly showed that GluR2(B) but not GluR1(A) protein was downregulated by the GluR2(B) AS-ODN infusion paradigm similar to our previously reported Western blot data generated from adult rats (Friedman and Velísková 1998) (Figs. 7-8). Within the stratum lucidum, pyramidal cell dendrites lacked their typical diffuse and punctate immunolabeling patterns. Distal dendrites of many CA3 neurons were barely detectable near the lesion, although a few lightly stained neurons could be seen (Fig.8A,B). The anatomical advantage of immunocytochemistry performed herein showed not only that GluR2(B) immunolabel selectively declined throughout the ipsilateral hippocampus relative to the contralateral side or control ODN-infused pups, but also that the greatest reductions were highly apparent within CA2–CA3a–c subfields (Fig. 7B). However, intense downregulation of GluR2(B) protein was found regardless of presence or location of hippocampal degeneration, indicating that loss of the GluR2(B) subunit alone is insufficient to kill neurons. Neither antibodies labeled areas containing many eosinophilic neurons (Fig.8E,F), but GluR1(A) immunoreactivity was abundant all around the lesioned area (Fig.8C,D) after the GluR2(B) AS-ODN intrahippocampal infusions. Therefore, a significant unilateral shift in the GluR1(A)/GluR2(B) protein ratio appeared to be achieved by the method used. Moreover, kainate-induced status epilepticus in age-matched pups did not cause immunohistochemical downregulation of the GluR2 subunit or damage (Fig.9B), as reported previously by us (Friedman et al., 1997a,b), so that seizure activity alone is also not responsible for cell death. Similarly, some animals (n = 4) that did not exhibit seizure behavior after GluR2(B) knockdown also did not have CA3a lesions, possibly because of destruction of ventral blade granule cells by placement of the cannula (Fig. 9E). Other pups infused with GluR2(B) AS-ODNs that did not have spontaneous seizure behavior showed no obvious differences in cannula placement or damage produced by the infusions. In contrast, rat pups that exhibited both spontaneous seizures and GluR2(B) downregulation had CA3a neurodegeneration after GluR2(B) knockdown (Fig. 3), suggesting that both seizures and loss of the GluR2(B) subunit are involved in the selective cell death.
Fig. 7.

A, Photomicrograph showing that GluR2(B) immunolabeling (6C4) of CA1–CA3 stratum pyramidale was dense and uniform; GluR2(B) antibodies predominantly label soma, whereas GluR1(A) antisera label soma and dendrites. A, In sense-treated animal sections, GluR2(B) immunolabel was dense and evenly distributed throughout the cytoplasm of CA3 neurons.B, After GluR2(B) hippocampal knockdown (bold arrowhead), GluR2(B) immunoreactivity was markedly reduced throughout the CA2, CA3a–b, and part of CA3c subregions (betweenarrows); CA1 and DG were downregulated but to a lesser extent. C, GluR1(A) immunoreactivity after S-ODNs was intense throughout the hippocampus. D, GluR1(A) immunoreactivity was unchanged, suggesting that an increase in the GluR1(A)/GluR2(B) protein was achieved by the GluR2(B) knockdown.
GluR1(A) hippocampal knockdown produced the opposite effect such that GluR1(A) immunolabeling was markedly decreased and GluR2(B) immunolabeling in adjacent sections was unchanged (Fig. 9). GluR1(A) immunoreactivity was more evenly reduced throughout all hippocampal subfields of the ipsilateral hemisphere, with a slightly higher expression observed in CA3a neurons. There was no change in behavior or hippocampal morphology (Fig. 9F), similar to a recent report on genetically manipulated GluR1(A) knockout mice (Zamanillo et al., 1999). Thus, it appears that selective decreases in the GluR2(B) but not GluR1(A) subunit have functional and morphological consequences in the developing hippocampus. Several pups (n = 3) coinjected with GluR1(A) and GluR2(B) AS-ODNS also did not exhibit seizure behavior or damage, but because of a <50% success rate, many more animals would need to be injected to determine whether loss of the GluR1(A) subunit would prevent deleterious effects resulting from the GluR2(B) AS-ODNs.
DISCUSSION
Our previous molecular studies show that KA-induced status epilepticus has age-related consequences on gene expression, whereby AMPA and GABAA α1 subunits are decreased in adult CA3 neurons destined to die (Friedman et al., 1994; Friedman, 1998) and are relatively unaltered in immature neurons resistant to damage (Friedman et al., 1997a). This article demonstrates that a certain balance of GluR1(A) and GluR2(B) subunit expression within a single hippocampus is needed to prevent epileptogenesis and neurodegeneration in the developing brain.
Unexpectedly, we also recently reported that unilateral GluR2(B) hippocampal knockdown does not acutely affect EEG activity in adult rats or neonates during the first postnatal week (Friedman and Velísková, 1998). Accordingly, reducing GluR2(B) (Brusa et al., 1995; Feldmeyer et al., 1999) but not GluR1(A) (Zamanillo et al., 1999) receptor expression and function by genetic strategies results in age-dependent seizures. This sensitive period coincides with a stage in development when constituent GluR1(A) and GluR2(B) mRNA levels are high (Moyner et al., 1991;Standley et al., 1995) and may be a molecular mechanism to prevent epileptogenesis at a certain age. Lowering extracellular Ca2+ levels in young animals also can induce spontaneous paroxysmal activity (Albrecht and Heinemann, 1989). Our data suggest that intracellular Ca2+levels increase because of a shift in the GluR1(A)/GluR2(B) protein ratio induced by the GluR2(B) knockdown procedure and may partially lead to the observed alterations in behavior, EEG, and LTP patterns. We have supportive evidence that GluR2(B) AS-ODN exposure to dissociated hippocampal neurons also increases the GluR1(A)/GluR2(B) protein ratio and leads to pharmacologically selective increases in AMPA-mediated Ca2+ influx as determined by dye imaging with fura-2 AM (Friedman et al., 1997b). However, it is also possible that reduction of AMPA receptor GluR2(B) subunit levels may increase single-channel conductance of AMPA receptors, which in turn could lead to hyperexcitability within hippocampal circuits and be responsible for the population spike observed after tetanic stimulation. Changes in AMPA-mediated Ca2+permeability or single-channel conductance would occur presumably via increased formation of GluR1(A) heteromeric assemblies with GluR3(C) or GluR4(D) subunits or by GluR1(A) homomers. In contrast, selective downregulation of the hippocampal GluR1(A) subunit did not affect behavior or morphology, further supporting the possibility that the presence of the GluR1(A) subunit is needed to mediate the deleterious effects produced by the GluR2(B) knockdown procedure.
Specificity of the antisense infusions by in situhybridization analyses showed that GluR2(B) mRNA was not significantly decreased as predicted by the procedure, although antisense-induced decreases in mRNA have been reported (for review, see Loose-Mitchell, 1988; Uhlmann and Peyman, 1990). Control expression of GluR2(B) mRNA after GluR2(B) knockdown in our study is a result of detecting the endogenous mRNA with full-length cRNA probes that bind to a much longer sequence than the AS-ODNs used. Consistent with previous histochemical findings (Friedman and Velísková, 1998), there was pronounced cell loss in the distant CA3a subregion, with discrete damage along the rostrocaudal plane and little cytotoxicity near the site of infusion. However, here and previously (Friedman and Velísková, 1998) we have shown compelling data in adult rats that downregulation alone of the GluR2(B) subunit is insufficient to induce neurodegeneration in the knockdown model, which is also in agreement with several surviving GluR2(B) editing-deficient phenotypes (Kask et al., 1998; Feldmeyer et al., 1999), GluR2(B) knockout mice (Jia et al., 1996), and ischemia-resistant interneurons (Catani et al., 1995). 33P autoradiography demonstrated that the diffusion of ODNs to all regions of the hippocampus, therefore sparing of the CA1 and DG regions, was not caused by lack of uptake. Radiolabeled accumulations of the GluR2(B) ODNs were highest in the CA3 and may explain why depletion of GluR2(B) protein and damage were greatest in this region. Moreover, GluR1(A) knockdown did not alter behavior or cell survival, further supporting the possibility that the level of the GluR2(B) subunit within a single hippocampus may be an important limiting factor in age-dependent epileptogenesis.
Rat pups exhibiting no seizure behavior or only scratching automatisms did not display CA3 damage, possibly because of individual differences in (1) cannula placement, (2) amount of ODN delivered and accumulated, or (3) extent of GluR2(B) downregulation. It should be noted, however, that there were differences in the pattern of CA3 cell loss in pups after GluR2(B) knockdown when compared with the classical damage described after KA-induced status epilepticus in adults. Hippocampal neurodegeneration in our knockdown pups was mostly restricted to the CA3a subregion in or after the bend, despite the large decreases in GluR2(B) protein observed in the injected hippocampus that caused qualitative decreases in immunoreactivity throughout all areas of the CA3a–c. The dramatic loss of GluR2(B) immunolabeling within dendrites near the associated lesion and sustained GluR1(A) immunolabel is unlikely to be attributable to an artifact of the particular antibody used because three specific GluR2(B) antibodies revealed the same effects, suggesting that dendritic turnover of the GluR2(B) subunit may be less efficient relative to the soma. Similar alterations in the GluR1(A)/GluR2(B) ratio were observed in cultured single hippocampal neurons exposed to the GluR2(B) AS-ODNs (our unpublished observations). Hilar interneurons appeared mostly unaffected by the antisense-induced downregulation of hippocampal GluR2(B) protein, whereas these neurons are highly vulnerable to seizure-induced damage in adults in a number of seizure models (Nadler, 1981; Ben-Ari, 1985; Sperk 1994). A reduction in seizure threshold is supported by the following observations: (1) behavioral manifestations in young rats produced by the GluR2(B) AS-ODNs correspond to high-frequency paroxysmal activity in the EEG; (2) a subconvulsive dose of KA produced high-rhythmical oscillations and behavioral status epilepticus in GluR2(B) AS-ODN-pretreated pups lacking the phenotypic behavior; (3) high-rhythmical activity was not detected in control infused animals; (4) field recordings in ex vivo slices showed rapid onset of population spikes to high-intensity stimulation only in animals that were recipient to the GluR2(B) hippocampal knockdown treatment; and (5) GluR1(A) unilateral hippocampal knockdown did not result in seizures or damage. Combined data suggest that certain levels of glutamate, overexpression of Ca2+-permeable AMPA receptor channels, changes in single-channel conductance properties, or postsynaptic activity of mossy fibers together may be necessary to induce the selective CA3 cell loss. It appears that a particular amount of GluR2(B) expression must be lost in both antisense and genetic models before sufficient changes in synaptic activity occur within the hippocampal network and that neuronal populations affected within the deprived GluR2(B) circuit to regulate the activity may arise from the CA3. Recent studies of Feldmeyer et al. (1999) prove that induction of an age-dependent seizure-prone mutant phenotype is negatively correlated with the level of editing-efficient GluR2(B) gene expression.
Conventional tetanus-induced LTP in CA1 (a long-lasting increase in synaptic transmission efficacy) is dependent on the activation of NMDA receptors and a rise in intracellular calcium concentration (Bliss and Lomo 1973; Auerbach and Segal, 1997a). LTP is complex in that it can also be induced by means other than tetanic stimulation and has been shown to be dependent on or independent of NMDA receptor activation (Auerbach and Segal, 1994, 1997b; Cavus and Teyler, 1996). After in vivo GluR2(B) knockdown, induction of LTP was unaltered, as was NR1 expression, suggesting that hippocampal NMDA receptors were operational after the antisense infusions. However, LTP responses dropped off rapidly in GluR2(B) knockdown slices from animals with seizure behavior, suggesting that the stimulus train induced a transient increase in the fEPSPs, similar to the short-term potentiation (STP) observed when subthreshold tetani are applied (Malenka and Nicoll, 1993; Aniksztejn and Ben-Ari, 1995). This STP was associated with population spikes highly similar to our observations, suggesting increased excitability of the dendrites possibly by rises in Ca2+ that appear to be supported by the higher CA1 responses recorded from GluR2(B) knockdown slices. In an animal without altered behavior, tLTP was impaired near the infusion site, but normal tLTP was recorded when stimulation was delivered to the distal site, suggesting that changes in the electrical properties of the circuit did not spread to distal regions. A deprived circuit of the GluR2(B) subunit in both medial and lateral regions of the immature hippocampus may be responsible for epileptogenesis, possibly by causing an increased transfer of currents from the fEPSP generated in dendrites to increase their excitability and the spike-generating mechanisms of the cell body region.
In contrast to our observations, slices prepared from GluR2(B) knockout mice without seizure phenotype had enhanced LTP (Jia et al., 1996) and partial editing-incompetent GluR2(B) mutants with seizure phenotype-expressed LTP similar to wild type when NMDA receptor antagonist APV was not applied (Feldmeyer et al., 1999). It is likely that differences in developmental compensatory mechanisms exist in the various GluR2(B) mutant genotypes compared with our acute model of GluR2(B) knockdown to explain the different effects on tLTP. However, we also expect that differences exist because of the various tetanic stimulation characteristics used. For example, induction of tLTP in GluR2(B) editing-incompetent mutants (Feldmeyer et al., 1999) was achieved by using a different stimulation paradigm than ours (20–90 μsec, 0.2 Hz vs 50 μsec, 0.033 Hz stimulations, respectively) and required a higher stimulus intensity. In addition, our antisense model is not only less compensated because of its acute nature but also because reductions in GluR2(B) were restricted to a single hippocampus rather than the entire brain. Consistent with our observations, slices prepared from seizure-phenotype GluR2(B) mutants (1) did not exhibit spontaneous epileptiform activity, and (2) certain genotypes presented increased number of failures to induce LTP or they required 30% higher stimulus intensity compared with other genotypes. An alternative explanation as to why we lose LTP in CA1 of our GluR2(B) knockdown slices may be an induced overexcitability attributable to an acute saturation of intracellular Ca2+levels within the hippocampus, particularly in CA3, altered Na+ conductance, and lack of GluR2(B) assembly with the GluR1(A) subunit at mossy fiber and Schaffer collateral synapses. GluR1(A) gene knockouts, completely lacking LTP, exhibit marked reduction in translocation of GluR2(B) subunits into dendrites, probably because of their inability to assemble with GluR(A) subunits (Zamanillo et al., 1999). In fact, the presence of the GluR2(B) subunit facilitates assembly of AMPA receptors (Wenthold et al., 1996). Further pharmacological analysis is in progress to define the role of glutamate receptor types in our knockdown model by antagonist application and the effects of reduced GluR2(B) expression on LTD.
In conclusion, unilateral hippocampal GluR2(B) knockdown may provide a powerful strategy for studying the roles of certain receptor subunits in young rat pups that are highly prone to seizures but relatively resistant to damage. It appears that the GluR2(B) subunit is needed for normal development of hippocampal plasticity and CA3 neuronal survival at a time when the brain is undergoing major changes in CNS inhibitory and excitatory amino acid function. Our study supports the importance of examining various AS-ODNs and AMPA receptor Ca2+ blockers or antagonists as neuroprotective agents in epilepsy at several ages. Although why CA3a pyramidal neurons are particularly vulnerable to cell death after GluR2(B) knockdown remains to be elucidated, chronic unilateral downregulation of the GluR2(B) hippocampal subunit by in vivo infusion of GluR2(B) AS-ODNs may provide a novel partial seizure model in the developing rat.
Footnotes
Received June 16,1999; revised Aug. 4, 1999; accepted Aug. 23, 1999.
This work was supported by March of Dimes and National Institutes of Health Grant NS-38069 (L.K.F.), and a Sir Charles Clore Fellowship (A.R.K.). We are indebted to Dr. Jana Velísková, Dr. Solomon L. Moshé, Dr. Menachem Segal, and Dr. Yehezkel Ben-Ari for their invaluable comments and for providing necessary space to carry out experiments. We thank Bonaventure Magrys for technical assistance with GluR1(A) immunocytochemical studies and Mr. Howard Rubin and the Graphic Arts Center at Albert Einstein College of Medicine for expert photography.
Correspondence should be addressed to Dr. Linda K. Friedman, Department of Neuroscience, Seton Hall University, 400 South Orange Avenue, South Orange, NJ 07079. E-mail: lfriedma@aol.com.
REFERENCES
- 1.Albala BJ, Moshé SL, Okada R. Kainic-acid-induced seizures: a developmental study. Dev Brain Res. 1984;13:139–148. doi: 10.1016/0165-3806(84)90085-3. [DOI] [PubMed] [Google Scholar]
- 2.Aniksztejn L, Ben-Ari Y. Expression of LTP by AMPA and/or NMDA receptors is determined by the extent of NMDA receptors activation during the tetanus. J Neurophysiol. 1995;74:2349–2357. doi: 10.1152/jn.1995.74.6.2349. [DOI] [PubMed] [Google Scholar]
- 3.Auerbach J, Segal M. A novel cholinergic induction of long term potentiation in rat hippocampus. J Neurophysiol. 1994;72:2034–2040. doi: 10.1152/jn.1994.72.4.2034. [DOI] [PubMed] [Google Scholar]
- 4.Auerbach J, Segal M. Peroxide modulation of slow onset potentiation in rat hippocampus. J Neurosci. 1997a;17:8695–8701. doi: 10.1523/JNEUROSCI.17-22-08695.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Auerbach J, Segal M. Muscarinic receptors involved in hippocampal plasticity. Life Sci. 1997b;60:1085–1091. doi: 10.1016/s0024-3205(97)00051-9. [DOI] [PubMed] [Google Scholar]
- 6.Albrecht D, Heinemann U. Low calcium-induced epileptiform activity in hippocampus slices from infant rats. Dev Brain Res. 1989;48:316–320. doi: 10.1016/0165-3806(89)90085-0. [DOI] [PubMed] [Google Scholar]
- 7.Babb TL, Brown WJ. Pathological findings in epilepsy. In: Engel J Jr, editor. Surgical treatment of the epilepsies. Raven; New York: 1987. pp. 511–540. [Google Scholar]
- 8.Ben-Ari Y. Limbic seizure and brain damage produced by kainic acid: mechanisms and relevance to human temporal lobe epilepsy. Neuroscience. 1985;14:375–403. doi: 10.1016/0306-4522(85)90299-4. [DOI] [PubMed] [Google Scholar]
- 9.Bliss T, Lomo T. Long-lasting potentiation of synaptic transmission in the dentate area of the anaesthetized rabbit following stimulation of the perforant path. J Physiol (Lond) 1973;232:331–356. doi: 10.1113/jphysiol.1973.sp010273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Brusa RF, Zimmermann, Koh D-S, Feldmeyer D, Gass P, Seeburg PH, Sprengel R. Early onset epilepsy and postnatal lethality associated with an editing-deficient GluR-B allele in mice. Science. 1995;270:1677–1680. doi: 10.1126/science.270.5242.1677. [DOI] [PubMed] [Google Scholar]
- 11.Bruton CJ. The neuropathology of human temporal lobe epilepsy. Oxford UP; New York: 1988. [Google Scholar]
- 12.Burnashev N, Monyer H, Seeburg PH, Sakmann B. Divalent ion permeability of AMPA receptor channels is dominated by the edited form of a single subunit. Neuron. 1992;8:189–198. doi: 10.1016/0896-6273(92)90120-3. [DOI] [PubMed] [Google Scholar]
- 13.Catani MV, Tolle TR, Monyer H. Differential expression of AMPA receptor subunits in NOS-positive neurons of cortex, striatum, and hippocampus. J Neurosci. 1995;15:7046–7061. doi: 10.1523/JNEUROSCI.15-11-07046.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cavus I, Teyler T. Two forms of long-term potentiation in area CA1 activate different signal transduction cascades. J Neurophysiol. 1996;76:3038–3047. doi: 10.1152/jn.1996.76.5.3038. [DOI] [PubMed] [Google Scholar]
- 15.Charriaut-Marlangue C, Aggoun-Zouaoui D, Repressa A, Ben-Ari Y. Apoptotic features of selective neuronal death in ischemia, epilepsy, and gp 120 toxicity. Trends Neurosci. 1996;19:109–114. doi: 10.1016/s0166-2236(96)80039-7. [DOI] [PubMed] [Google Scholar]
- 16.Cherubini E, De Feo MR, Mecarelli O, Ricci GF. Behavioral and electrographic patterns induced by systemic administration of kainic acid in developing rats. Dev Brain Res. 1983;9:69–77. doi: 10.1016/0165-3806(83)90110-4. [DOI] [PubMed] [Google Scholar]
- 17.Choi D. Glutamate neurotoxicity and diseases of the nervous system. Neuron. 1990;1:623–634. doi: 10.1016/0896-6273(88)90162-6. [DOI] [PubMed] [Google Scholar]
- 18.DeFelipe J, Huntly GW, del Rio MR, Sola RG. Microzonal decreases in the immunostaining for non-NMDA ionotropic excitatory amino acid receptor subunits GluR2(B)/3 and GluR5/6/7 in the human epileptogenic neocortex. Brain Res. 1994;657:150–158. doi: 10.1016/0006-8993(94)90962-8. [DOI] [PubMed] [Google Scholar]
- 19.Dingledine R, McBain CJ, McNamara JO. Excitatory amino acid receptors in epilepsy. Trends Pharmacol. 1990;11:334–338. doi: 10.1016/0165-6147(90)90238-4. [DOI] [PubMed] [Google Scholar]
- 20.Feldmeyer D, Kask K, Brusa R, Kornau HC, Kolhekar R, Burnashev N, Jensen V, Hvalby O, Sprengel R, Seeburg PH. Neurological dysfunctions in mice expressing different levels of the Q/R site-unedited AMPAR subunit GluR-B. Nat Neurosci. 1999;2:57–64. doi: 10.1038/4561. [DOI] [PubMed] [Google Scholar]
- 21.Friedman LK. Selective reduction of GluR2(B) protein in adult hippocampal CA3 neurons following status epilepticus but prior to cell loss. Hippocampus. 1998;8:511–525. doi: 10.1002/(SICI)1098-1063(1998)8:5<511::AID-HIPO9>3.0.CO;2-W. [DOI] [PubMed] [Google Scholar]
- 22.Friedman LK, Velísková J. GluR2(B) hippocampal knockdown reveals developmental regulation of epileptogenicity and neurodegeneration. Mol Brain Res. 1998;61:224–231. doi: 10.1016/s0169-328x(98)00213-7. [DOI] [PubMed] [Google Scholar]
- 23.Friedman LK, Pellegrini-Giampietro DE, Sperber EF, Bennett MVL, Moshé SL, Zukin RS. Kainate-induced status epilepticus alters glutamate and GABAA receptor gene expression in adult rat hippocampus: an in situ hybridization study. J Neurosci. 1994;14:2697–2707. doi: 10.1523/JNEUROSCI.14-05-02697.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Friedman LK, Sperber EF, Moshé SL, Bennett MVL, Zukin RS. Developmental regulation of glutamate and GABAA receptor gene expression in rat hippocampus following kainate-induced status epilepticus. Dev Neurosci. 1997a;19:529–542. doi: 10.1159/000111257. [DOI] [PubMed] [Google Scholar]
- 25.Friedman LK, Velísková J, Spray DC, Segal M. GluR2 hippocampal knockdown enhances and accelerates kainate seizure-induced damage in adult rats and increases Ca2+ permeability in primary culture. ISN/ASN Satellite Symposium and Aspet Colloqium on Excitatory Amino Acids. 1997b.
- 26.Hollman M, Hartley M, Heinemann S. Ca2+ permeability of KA-AMPA-gated glutamate receptor channels depends on subunit composition. Science. 1991;253:1028–1031. doi: 10.1126/science.1709304. [DOI] [PubMed] [Google Scholar]
- 27.Holmes GL, Thompson JL. Effects of kainic acid on seizure susceptibility in the developing brain. Dev Brain Res. 1988;39:51–59. doi: 10.1016/0165-3806(88)90066-1. [DOI] [PubMed] [Google Scholar]
- 28.Jia Z, Nadia A, Miu P, Xiong Z, Henderson J, Geriai R, Taverna FA, Velumian A, Mac Donald J, Carlen P, Abrominov-Newerly W, Roder J. Enhanced LTP in mice deficient in the AMPA receptor GluR2(B). Neuron. 1996;17:945–956. doi: 10.1016/s0896-6273(00)80225-1. [DOI] [PubMed] [Google Scholar]
- 29.Kask K, Zamanillo D, Burnashev N, Sprengel R, Seeburg PH. The AMPA receptor subunit GluR-B in its Q/R site-unedited form is not essential for brain development and function. Proc Natl Acad Sci USA. 1998;95:13777–13782. doi: 10.1073/pnas.95.23.13777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Keinänen K, Wisden W, Sommer B, Werner P, Herb A, Verdoorn TA, Sakmann B, Seeburg P. A family of AMPA-selective glutamate receptors. Science. 1990;249:556–560. doi: 10.1126/science.2166337. [DOI] [PubMed] [Google Scholar]
- 31.Koudinov AR, Groner Y, Segal M. Cu/Zn-SOD transgenic mice are impaired in slow onset, long term potentiation. Neurosci Lett. 1998;51:S23. [Google Scholar]
- 32.Loose-Mitchell DS. Antisense nucleic acids as a potential class of pharmaceutical agents. Trends Pharmacol. 1988;9:45–47. doi: 10.1016/0165-6147(88)90112-5. [DOI] [PubMed] [Google Scholar]
- 33.Lynd-Balta E, Pilcher WH, Joseph SA. Distribution of AMPA receptor subunits in the hippocampal formation of temporal lobe epilepsy patients. Neuroscience. 1996;72:15–29. doi: 10.1016/0306-4522(95)00554-4. [DOI] [PubMed] [Google Scholar]
- 34.Malenka RC, Nicoll RA. NMDA-receptor-dependent synaptic plasticity: multiple forms and mechanisms. Trends Neurosci. 1993;16:521–527. doi: 10.1016/0166-2236(93)90197-t. [DOI] [PubMed] [Google Scholar]
- 35.Mathern GW, Pretorious JK, Kornblum HI, Mendoza D, Lozada A, Leite MF, Adelson D, Peacock WJ. Human hippocampal AMPA and NMDA mRNA levels in temporal lobe epilepsy patients. Brain. 1998;120:101–105. doi: 10.1093/brain/120.11.1937. [DOI] [PubMed] [Google Scholar]
- 36.Monyer H, Seeburg PH, Wisden W. Glutamate-operated channels: developmentally early and mature forms arise by alternative splicing. Neuron. 1991;6:799–810. doi: 10.1016/0896-6273(91)90176-z. [DOI] [PubMed] [Google Scholar]
- 37.Nadler JV. Kainic acid as a tool for the study of temporal lobe epilepsy. Life Sci. 1981;29:31–42. doi: 10.1016/0024-3205(81)90659-7. [DOI] [PubMed] [Google Scholar]
- 38.Nakanishi N, Axel R, Schneider NA. Alternative splicing generates functionally distinct N-methyl-d-aspartate receptors. Proc Natl Acad Sci USA. 1992;89:8552–8556. doi: 10.1073/pnas.89.18.8552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Nitecka LE, Tremblay G, Charton Bouillot JP, Berger ML, Ben-Ari Y. Maturation of kainic acid seizure brain damage syndrome in the rat. II. Histopathological sequelae. Neuroscience. 1984;13:1073–1094. doi: 10.1016/0306-4522(84)90289-6. [DOI] [PubMed] [Google Scholar]
- 40.Paxinos G, Watson C. The rat brain in stereotaxic coordinates, Ed 2. Academic; Sydney: 1986. [DOI] [PubMed] [Google Scholar]
- 41.Pollard H, Moreau HJ, Ben-Ari Y, Khrestchatisky M. Alterations of the GluR-B AMPA receptor subunit flip/flop expression in kainate-induced epilepsy and ischemia. Neuroscience. 1993;57:545–554. doi: 10.1016/0306-4522(93)90004-y. [DOI] [PubMed] [Google Scholar]
- 42.Prince HK, Conn PJ, Blackstone CD, Huganir RL, Levey AI. Down-regulation of AMPA receptor subunit GluR2(B) in amygdaloid kindling. J Neurochem. 1995;64:462–465. doi: 10.1046/j.1471-4159.1995.64010462.x. [DOI] [PubMed] [Google Scholar]
- 43.Rogers SW, Hughes TE, Hollmann M, Gasic GP, Deneris ES, Heinemann S. The characterization and localization of the glutamate receptor subunit GluR1(A) in the rat brain. J Neurosci. 1991;11:2713–2724. doi: 10.1523/JNEUROSCI.11-09-02713.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Schreiber SS, Baudry M. Selective neuronal vulnerability in the hippocampus: a role for gene expression? Trends Neurosci. 1995;18:446–450. doi: 10.1016/0166-2236(95)94495-q. [DOI] [PubMed] [Google Scholar]
- 45.Siesjo BK, Bengtsson F. Ca2+ fluxes, Ca2+ antagonists, and Ca2+-mediated pathology in brain ischemia, hypoglycemia, and spreading depression. J Cereb Blood Flow Metab. 1989;9:127–140. doi: 10.1038/jcbfm.1989.20. [DOI] [PubMed] [Google Scholar]
- 46.Sommer B, Keinänen K, Verdoon TA, Wisden W, Burnashev N, Herb A, Kohler M, Takagi T, Sakmann B, Seeburg PH. Flip and flop: a cell specific functional switch in operated channels of the CNS. Science. 1990;249:1580–1585. doi: 10.1126/science.1699275. [DOI] [PubMed] [Google Scholar]
- 47.Sperk G. Kainic acid seizures in the rat. Prog Neurobiol. 1994;42:1–32. doi: 10.1016/0301-0082(94)90019-1. [DOI] [PubMed] [Google Scholar]
- 48.Standley S, Tocco G, Tourigny MF, Massicotte G, Thompson RF, Baudry M. Developmental changes in α-amino-3-hydroxy-5-methyl-4-isoxazole propionate receptor properties and expression in the rat hippocampal formation. Neuroscience. 1995;67:881–892. doi: 10.1016/0306-4522(95)00075-t. [DOI] [PubMed] [Google Scholar]
- 49.Tremblay E, Nitecka L, Berger ML, Ben-Ari Y. Maturation of kainic acid seizure-brain damage syndrome in the rat: clinical, electrographic and metabolic observations. Neuroscience. 1984;13:1051–1072. doi: 10.1016/0306-4522(84)90288-4. [DOI] [PubMed] [Google Scholar]
- 50.Uhlmann E, Peyman A. Antisense oligonucleotides: a new therapeutic principle. Chem Rev. 1990;90:544–584. [Google Scholar]
- 51.Verdoorn TA, Burnashev N, Monyer H, Seeburg PH, Sakmann B. Structural determinants of ion flow through recombinant glutamate receptor channels. Science. 1991;252:1715–1718. doi: 10.1126/science.1710829. [DOI] [PubMed] [Google Scholar]
- 52.Vissavajjhala P, Janssen WGM, Hu Y, Gazzaley AH, Moran T, Hof PR, Morrison JH. Synaptic distribution of the AMPA-GluR2(B) subunit and its colocalization with calcium-binding proteins in rat and cerebral cortex: an immunohistochemical study using a GluR2-specific monoclonal antibody. Exp Neurol. 1996;142:295–311. doi: 10.1006/exnr.1996.0199. [DOI] [PubMed] [Google Scholar]
- 53.Wenthold RJ, Yokotani N, Doi K, Wada K. Immunochemical characterization of the non-NMDA glutamate receptor using subunit specific antibodies: evidence for a hetero-oligomeric structure in rat brain. J Biol Chem. 1992;267:501–507. [PubMed] [Google Scholar]
- 54.Wenthold RJ, Petralia RS, Blahos J, II, Niedzielski AS. Evidence for multiple AMPA receptor complexes in hippocampal CA1/CA2 neurons. J Neurosci. 1996;16:1982–1989. doi: 10.1523/JNEUROSCI.16-06-01982.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Wonziak DF, Stewart GR, Miller P, Olney JW. Age-related sensitivity to kainate neurotoxicity. Exp Neurol. 1991;114:250–253. doi: 10.1016/0014-4886(91)90042-b. [DOI] [PubMed] [Google Scholar]
- 56.Zamanillo D, Sprengel R, Hvalby Ø, Jensen V, Burnashev N, Rozov A, Kaiser KM, Käster HJ, Borchardt T, Worley P, Lÿbke J, Frotscher M, Kelly PH, Sommer B, Anderson P, Seeburg PH, Sakmann B. Importance of AMPA receptors for hippocampal synaptic plasticity but not for spatial learning. Science. 1999;284:1805–1810. doi: 10.1126/science.284.5421.1805. [DOI] [PubMed] [Google Scholar]