

Abstract
High-frequency stimulation (HFS) applied to stratum radiatum of a rat hippocampal slice in the presence of ionotropic glutamate receptor antagonists evokes a biphasic GABAAreceptor-dependent response in CA1 pyramidal neurons, with a brief hyperpolarizing IPSP (hIPSP) followed by a long-lasting depolarization. We show now that it is possible to pharmacologically separate the hIPSP and late depolarization from one another. In neurons intracellularly perfused for 1–2 hr with F− as the major anion and no ATP, the hIPSP (and the corresponding current, hIPSC) evoked by HFS was blocked, whereas neither the late depolarization nor its underlying current was attenuated. In contrast, internal perfusion with a high concentration (5 mm) of the impermeant lidocaine derivative QX-314 selectively abolished the depolarizing component of the biphasic response and also strongly reduced depolarizations evoked by extracellular microinjection of K+. Bath application of quinine (0.2–0.5 mm) or quinidine (0.1 mm) resulted in a pronounced inhibition of the HFS-induced extracellular K+ concentration ([K+]o) transient but not of the bicarbonate-dependent alkaline shift in extracellular pH. The attenuation of the [K+]o transient was closely paralleled by a suppression of the HFS-evoked depolarization but not of the hIPSP. Quini(di)ne did not affect depolarizations induced by exogenous K+ either.
These data provide direct pharmacological evidence for the view that the HFS-induced biphasic response of the pyramidal neuron is composed of mechanistically distinct components: a direct GABAA receptor-mediated phase, which is followed by a slow, nonsynaptic [K+]o-mediated depolarization. The bicarbonate-dependent, activity-induced [K+]o transient can be blocked by quini(di)ne, whereas its depolarizing action in the pyramidal neuron is inhibited by internal QX-314. The presence of fundamentally distinct components in GABAA receptor-mediated actions evoked by HFS calls for further investigations of their functional role(s) in standard experimental maneuvers, such as those used in studies of synaptic plasticity and induction of γoscillations.
Keywords: GABA-mediated depolarization, nonsynaptic transmission, interstitial potassium, quinine, quinidine, QX-314
In the presence of blockers of ionotropic glutamate receptors (iGluRs) and GABABreceptors, single stimuli applied to the CA1 stratum radiatum evoke hyperpolarizing fast IPSPs (fIPSPs) in pyramidal neurons, which are mediated by GABAA receptors (GABAARs) (Davies et al., 1990; Davies and Collingridge, 1993). With high-frequency stimulation (HFS), a more complex biphasic postsynaptic response is seen (Davies and Collingridge, 1993; Grover et al., 1993; Staley et al., 1995; Kaila et al., 1997). With 40 pulses applied at 100 Hz, the biphasic response consists of a fused sequence of fIPSPs yielding an initial 0.2–0.3 sec hyperpolarizing response [the hyperpolarizing IPSP (hIPSP)] that is followed by a long-lasting depolarization with a duration of up to a few seconds.
Despite the fact that biphasic GABAAreceptor-mediated responses have been subject to extensive studies under various experimental conditions (see references above; Avoli et al., 1996; Perkins and Wong, 1996; Burg et al., 1998), their ionic and biophysical bases have remained controversial (Kaila, 1994;Lambert and Grover, 1995; Perkins, 1999). The mechanisms proposed to account for them have included, e.g., postulations of GABAARs with distinct permeabilities to Cl− and HCO3− (Perkins and Wong, 1996; Perkins, 1999) (also see Alger and Nicoll, 1982) and anionic redistribution (Staley et al., 1995) analogous to that demonstrated earlier with direct recordings of the relevant ions in crayfish muscle fibers (Kaila and Voipio, 1987; Kaila et al., 1989) and neurons (Voipio et al., 1991).
That bicarbonate has a direct depolarizing contribution to fast postsynaptic GABAAR-mediated currents in mammalian neurons was originally demonstrated in experiments on neocortical preparations (Kaila et al., 1993). However, in a recent study (Kaila et al., 1997), we provided evidence for two different mechanisms being responsible for the HFS-induced GABA-mediated biphasic behavior of the postsynaptic membrane potential: the hIPSP is mediated by the GABAA receptors of the target cell, whereas the major part of long-lasting depolarization is caused by a bicarbonate-dependent, activity-induced extracellular [K+]o([K+]o) transient. In our previous work (Kaila et al., 1997; Taira et al., 1997; Autere et al., 1999), the long-lasting depolarization was called, the GABA-mediated depolarizing postsynaptic potential (GDPSP). In light of the present verification of its nonsynaptic nature, we will change this acronym to the GABA-mediated depolarizing nonsynaptic potential (GDNSP).
Our earlier key observations, which indicated that the GDNSP is a [K+]o-evoked response (Kaila et al., 1997), were that (1) activation of the target neuron GABAAR-mediated inputs by a second HFS train applied at the peak of the GDNSP induced a prompt hyperpolarization; (2) the I–V relationships of the hIPSC and the GDNSC (the current underlying the GDNSP) were qualitatively distinct, the hIPSC showing outward rectification typical for GABAAR currents, whereas the GDNSC exhibited inward rectification at positive voltages; (3), very often no clear reversal of the GDNSC was observed even at voltages slightly positive to 0 mV; and (4) the GDNSP was paralleled by, and showed a sub-Nernstian relationship to, the HFS-induced [K+]otransient.
In this study, we present direct pharmacological evidence favoring the dual-mechanism hypothesis. Specifically, our mechanistic scheme (cf.Kaila et al., 1997, their figure 12) predicts that (1) blocking GABAA receptors from the inside within the target cell should abolish the fIPSP and hIPSP, leaving the GDPSP unaffected; (2) using an internal blocker to inhibit those K+ channels of the target cell that are involved in GDPSP generation should block the GDPSP, leaving the fIPSP and hIPSP unaffected; and (3) inhibition of the activity-induced, bicarbonate-dependent [K+]o transient should inhibit the GDPSP, leaving the fIPSP and hIPSP intact. The present findings fully confirm these predictions. They are also of importance in understanding the functions of the inhibitory circuitry and nonsynaptic transmission in a number of standard experimental designs, including those in which HFS is used to induce changes in synaptic efficacy (Bliss and Collingridge, 1993) and neuronal oscillatory activity (Whittington et al., 1997; Bracci et al., 1999).
Some preliminary results have been published in abstract form (Smirnov and Kaila, 1997).
MATERIALS AND METHODS
The methods used in the present study were similar to those before (Kaila et al., 1997). Hippocampal slices (400 μm) were prepared from brains of 30- to 45-d-old male Wistar rats (100–120 gm), which were decapitated under deep pentobarbital anesthesia. The standard physiological solution contained (in mm): NaCl 124, KCl 3.0, CaCl2 2.0, NaHCO3 25, NaH2PO4 1.1, MgSO4 2.0, and d-glucose 10. It was continuously gassed with 95% O2 and 5% CO2, pH 7.4 at the experimental temperature of 32°C.
Neuronal responses were evoked by stimuli (15–30 V, 100 μsec) delivered via a bipolar electrode placed in stratum radiatum at a distance of ≤500 μm from the recording site. Single pulses were given at a rate of ≤0.05 Hz; trains of stimuli (40 pulses/100 Hz) were delivered once per 3–5 min.
All experiments were done in the continuous presence of 10 μm 6-nitro-7-sulfamoylbenzo(f)-quinoxaline-2,3-dione (NBQX), an antagonist of AMPA and kainate receptors; 40 μmd-2-amino-5-phosphonopentoate (AP-5), and 50 μm ketamine, a competitive and a noncompetative antagonist of NMDA receptors. GABAB receptors were blocked with bath-applied 100–200 μm CGP 35348 or 5 μm CGP 55845A (kindly donated by Dr. M. Pozza, Ciba-Geigy, Basel, Switzerland). Quinine (200–500 μm), quinidine (100 μm), and picrotoxin (100 μm) were applied in the bath. One millimolar Cs+, 50 μm UL-FS 49 (a kind donation from Prof. H.-C. Pape, University of Magdeburg), 100 μmBa2+, and 10 mmtetraethylammonium (TEA+) were directly dissolved in the saline (Ba2+ application was done in the absence of sulfate and phosphate). The lidocainen-ethyl bromide quaternary salt QX-314 bromide (see below) was a gift from Astra (Sodertalje, Sweden). NBQX and AP-5 were from Tocris Cookson (Bristol, UK), and all other commercially available chemicals were from Sigma (St. Louis, MO).
Intracellular recordings from CA1 hippocampal pyramidal neurons were obtained with an Axoclamp 2B amplifier (Axon Instruments, Foster City, CA) either in bridge or in continuous voltage-clamp mode. Sharp microelectrodes were filled with 1.5 m potassium acetate plus 1.5 m potassium methyl sulfate and 5 mmKCl (resistance, 80–120 MΩ). Cells with a stable resting membrane potential of −67 to −75 mV 20 min after impalement and an input impedance of 30–60 MΩ were used in the measurements. Whole-cell recordings (Blanton et al., 1989) were obtained using micropipettes with a resistance of 5–7 MΩ when filled with the standard intracellular solution containing (in mm): K-gluconate 120, MgCl2 1.5, Mg-ATP 2, HEPES 10, CaCl2 0.5, K-bis(2-aminophenoxy)ethane-N,N,N′,N′-tetraacetate (K-BAPTA) or K-EGTA 5, d-glucose 20, and NaOH 4.8, pH 7.1, with KOH. Cells with a stable resting membrane potential of −50 mV or more negative in whole-cell configuration and with an input impedance of 100–200 MΩ were accepted for further measurements. The access resistance was 8–12 MΩ. The solution used for intracellular inhibition of GABAA receptors (Nelson et al., 1994; Khazipov et al., 1997) contained (in mm): KF 135, MgCl2 1.5, HEPES 10, CaCl2 0.5, and K-BAPTA or K-EGTA 5.0, pH 7.1 with NaOH. We also made several attempts to block the GABAA receptors from within using pipette solutions containing (in mm):4,4′-diisothiocyanatostilbene-2,2′-disulfonic acid 1, 4,4′-dinitrostilbene-2,2′-disulfonic acid up to 1, and picrotoxin 1 (cf. Nelson et al., 1994), but none of these drugs was effective in our hands.
In preliminary experiments with internal QX-314, 2–3 mm of the tip of the pipette were prefilled with the standard intracellular solution, and thereafter the pipette was back-filled with the QX-314-containing solution. The bulk of experiments with QX-314 were performed using a perfused pipette (Alford et al., 1993). A thin plastic capillary (outer tip diameter, 200 μm) was inserted into the pipette with a 1–1.5 mm distance between the tips of the capillary and the patch pipette. Because Br− has an effect on the Ag/AgCl wire, the standard intracellular solution was supplemented with 5 mm NaBr, which after the control recording period was replaced by 5 mm QX-314 bromide. The change in pipette solution was done with a syringe connected to the capillary. The efficacy of the perfusion (achieved with 5–10 μl) was tested using a pipette perfusion solution with a K+concentration 10 times higher than that of the standard filling solution. Measurements of the local potassium concentration just outside the mouth of the patch pipette with a K+-selective microelectrode showed that a complete solution change was achieved within 20–30 sec.
Potassium transients based on exogenous K+were generated within the tissue by delivering short pulses of high pressure (20–100 msec, three or four bars) into glass micropipettes (tip diameter, 2–5 μm) filled with 150 mm KCl. The stability of the applications remained constant for several hours (much longer than what was required for the experiments) as verified in control recordings of the associated extracellular K+ transients using K+-selective microelectrodes.
The double-barreled K+-selective and H+-selective microelectrodes with a short column of the liquid membrane solution (60398 or 95293, respectively; Fluka, Buchs, Switzerland) were made from borosilicate glass pipettes (Voipio et al., 1994).
The data are presented as mean ± SEM or as the range of measured values, where n refers to the number of slices.
RESULTS
Effects of internal fluoride
To examine the role of the target cell GABAAreceptors in the generation of the biphasic hIPSP/GDNSP voltage response and the corresponding currents (hIPSC/GDNSC), we made recordings using whole-cell patch pipettes devoid of ATP and glucose and with F− (Bormann et al., 1987; Akaike et al., 1989) as the prevalent anion (Nelson et al., 1994; Khazipov et al., 1997). Shortly after the rupture of the membrane, the resting membrane potential of the cell underwent a slight transient positive shift (5–7 mV), thereafter recovering to its initial value. The input resistance increased by 38 ± 13%.
As shown in Figure 1, 5–15 min after the establishment of the whole-cell configuration, the fIPSP/C and hIPSP/C were identical to what was typically seen under control conditions (Kaila et al., 1997). However, prolonged perfusion with the F−/0 ATP solution for 1–2 hr resulted in a block of the single-pulse-evoked fIPSP and in the corresponding current (fIPSC) in all six experiments of this kind (Fig.1A). The small remaining conductance (3–5% of the control) was associated with a slight positive shift in the GABAA reversal potential, which is fully attributable to the current carried by F−in fluoride-loaded neurons (Akaike et al., 1989). In light of the aims of the present study, the most significant observation was that the abolishment of the fIPSP/C was tightly paralleled by a block of the HFS-induced hIPSP, as well as the hIPSC (Fig. 1B). All the recordings in Figure 1 demonstrate a near-complete abolishment of synaptically mediated GABAA inputs into the target cell after F− perfusion.
Fig. 1.

Intracellular block of GABAA receptors of the pyramidal neuron does not suppress the GDNSP. Internal perfusion with fluoride and no ATP for >1 hr blocks fIPSCs measured at different holding voltages (−50 and −70 mV; A) as well as hIPSP (B, top traces) and associated hIPSCs measured at −50 mV (B, bottom traces) without attenuating the GDNSP and GDNSC. Voltage recordings inB (top traces) were taken at resting membrane potential (−54 mV). Values given above the traces represent time after rupture of the membrane. Here and in illustrations below,horizontal bars indicate stimulation trains (100 Hz, 0.4 sec, 15–20 V).
In sharp contrast to its blocking action on fIPSP/C and hIPSP/C, internal perfusion with the fluoride solution did not result in any attenuation of the GDNSP/C (Fig. 1B). In most cases (four of six), a clear increase in GDNSP amplitude and in the slope of its rising phase was observed, as would be expected if the hIPSP and GDNSP were to show some temporal overlap.
Effects of internal QX-314
In our previous study (Kaila et al., 1997), we routinely used 50 μm QX-314 in the patch pipette filling solution to block spiking. During the course of these experiments we found, quite fortuitously, that a high concentration of QX-314 had a selective blocking action on the GDNSP (Kaila et al., 1995). To rule out the possibility that irreversible extracellular contamination by this lidocaine derivative during cell approach was the primary reason for its blocking effect on the GDNSP (cf. Connors and Prince, 1982; Hara et al., 1995), we either prefilled the tip of the patch pipette with the control solution or used a perfused pipette (see Materials and Methods). In all neurons perfused with 5 mm QX-314, spiking, if observed under control condition, was abolished shortly after perfusion, which was followed by a more delayed suppression of the GDNSP.
Figure 2A represents a typical experiment of eight with internal perfusion of 5 mm QX-314. In five of eight cells, a near-complete block (≥95%) of the GDNSP was seen after this kind of internal perfusion, and on average, the peak amplitude of the GDNSP was decreased by 89 ± 5%.
Fig. 2.

Intracellular perfusion with 5 mmQX-314 selectively blocks GDNSP (A) as well as the depolarization in response to pressure-injected K+ (B). In the voltage-clamp recordings shown below each main trace, the cell was held at its resting membrane potential (−62 mV in A, −57 mV inB), and the voltage steps were 0, −10, −20, −30, and −40 mV. Note block of Iq and increase in steady-state input resistance after QX-314 perfusion.Arrows below the traces in B represent exogenous K+ injections.
As reported by Perkins and Wong (1995), internal application of 5 mm QX-314 also resulted in a block of an inward current (Iq) evoked at hyperpolarized potentials. The suppression of Iq(Fig. 2, insets) was used as an independent verification of the efficacy of the intracellular perfusion with QX-314, but it should be noted here that Iq plays no role in the generation of the GDNSP/C (see below).
In view of the aims of the present work, the most important aspect of the action of 5 mm internal QX-314 was that, despite the abolishment of the GDNSP/C, it did not attenuate the fIPSP/C or the hIPSP/C. In fact, we often observed a slight increase in hIPSP, which indicates that the peak of the hyperpolarizing phase shows a temporal overlap with the mechanisms that generate the GDNSP (see above, Fig. 1;Kaila et al., 1997).
The block of Iq by intracellular QX-314 did not play a role in blocking the GDNSP in the target cell, because application of 1 mmCs+ or 50 μm UL-FS 49 (Pape, 1996) did not have any inhibitory action of the GDNSP/C amplitude (data not shown). However, the I–Vrecordings (Fig. 2, insets) showed a significant increase in the steady-state input resistance of the neuron, which agrees with previous findings demonstrating that internal application of lidocaine derivatives such as QX-314 and QX-572 block various kinds of K+ channels (Segal, 1988; Nathan et al., 1990). However, we found that the membrane chord conductance measured using positive voltage pulses was much less sensitive to intracellular QX-314 than responses to negative voltage steps. This QX-314-induced inhibition of steady-state inward rectification (Perkins and Wong, 1995) is consistent with the observations that intracellular application of the drug had only a slight depolarizing action on resting neurons (+3 ± 0.5 mV; n = 12) but had a strong inhibitory action on membrane depolarization in response to a local injection of K+ (see below).
The experiments depicted in Figures 1 and 2A support the conclusion that the GDNSP is not a consequence of GABAAR activation in the target neuron. The involvement of an extracellular K+transient in the generation of this long-lasting depolarization (Kaila et al., 1997) prompted us to examine the effects of internal QX-314 on the voltage responses of the neurons to extracellular microinjections of K+. As shown in Figure2B, the depolarizing responses evoked in the presence of iGluR and GABA antagonists by exogenous potassium transients were strongly suppressed after internal application of QX-314. A pronounced inhibition (by 65–77%) of the K+-induced depolarization was seen in all four experiments of this kind.
Effects of bath-applied quinine and quinidine
So far, we have demonstrated that a selective blockade of either the HFS-induced hIPSP or the subsequent GDNSP can be achieved by intraneuronal manipulation with F− or QX-314. To gain further evidence for the involvement of a [K+]o transient in the generation of the GDNSP, we examined the actions of bath-applied drugs to see whether we could find a useful one capable of blocking the HFS-induced GABAergic [K+]o transient. We made pilot experiments with some conventional potassium channel antagonists, including TEA+ and Ba2+ (Hille, 1992), but a major problem with these drugs was a large increase in gross excitability, which made them futile in the present context (data not shown).
In contrast to the agents above, quinine, a K+ channel antagonist (Fishman and Spector, 1981; Bokvist et al., 1990a,b; Doi et al., 1995) (also seeSeki et al., 1993) that has been shown to inhibit the increase in [K+]o, which takes place in the rat brain in vivo after a short period of hypoxia (Zetterström et al., 1995), turned out to be a most effective tool in terms of specificity of action. The effects of quinidine, a closely related compound (Fishman and Spector, 1981; Doi et al., 1995), were also examined in parallel experiments.
Figure 3, A and B, shows simultaneous measurements of pyramidal cell membrane potential and of [K+]o in stratum pyramidale. In experiments of this kind, the distance between the two recording sites was ≤50 μm. We did not perform systematic concentration–effect studies, but quinine was found to have a clear inhibitory action on the [K+]o transients and neuronal depolarizations at a concentration of 50 μm, with a marked block of both responses at 0.2 mm. Within ∼20–30 min after the onset of 0.2 mm quinine application, the peak values of both the GDNSP and [K+]o transient were inhibited (by 75–94 and 65–78%, respectively) with strikingly similar time courses (half-time of inhibition was 10–12 min;n = 5). We found quinidine to have a similar effect on these responses (n = 4), but it turned out to be about two times more potent than quinine.
Fig. 3.
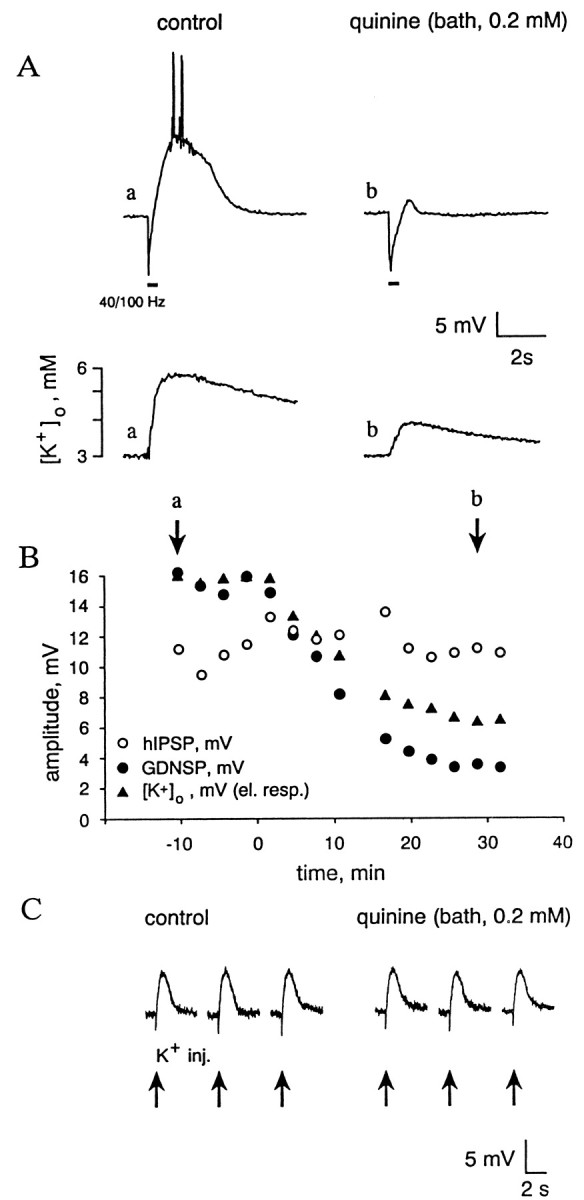
Bath-applied quinine produces a parallel inhibition of the [K+]o transient and the GDNSP without changing the K+ sensitivity of the cell membrane. A, Simultaneous specimen recordings of membrane potential and [K+]o(a, b; membrane potential is recorded with a sharp microelectrode). Voltage calibration applies for both membrane potential (top traces) and differential signal measured by the K+ electrode (bottom traces). A calibration in terms of [K+]o is also given on the left of the K+recordings. B, Time course of the quinine-induced decrease in peak amplitude of the GDNSP (filled circles), hIPSP (open circles), and [K+]o transient (filled triangles); a and b refer to the specimen recordings in A. Quinine (0.2 mm) application starts at the time point indicated as 0. C, Depolarizing responses evoked by K+ pressure injections (arrows below the traces) before and after (35–45 min) quinine application (sharp microelectrode recording).
The well known capability of quini(di)ne to block certain kinds of K+ channels (Fishman and Spector, 1981;Bokvist et al., 1990a,b; Doi et al., 1995) might suggest that the inhibition of the GDNSP is a result of a decrease in the [K+]o sensitivity of the target neuron, as was the case with intracellular perfusion of QX-314 (see Fig. 2). However, in striking contrast to this action of QX-314, quinine (n = 4) and quinidine (n = 4) had no effect on the depolarizing response evoked by exogenous K+ (Fig.3C).
Measurements of fIPSP/Cs and hIPSP/Cs showed that quinine had no effect on the immediate GABAA responses mediated by the receptor channels of the target neuron (Fig.4). The fact that the GABAA reversal potential (−81 ± 2 mV;n = 4) remained unaffected as measured using fIPSP/Cs (Fig. 4A) indicates that 0.2–0.5 mm quinine did not have a significant inhibitory action on the ability of the pyramidal cell K+–Cl−cotransporter (Rivera et al., 1999) to maintain a constant resting Cl− equilibrium potential (ECl−) under the present experimental conditions (Ellory et al., 1982; Adragna and Lauf, 1994). As was clearly seen in experiments of the kind illustrated in Figure 4,B and C, quini(di)ne exerted a selective blocking effect on the GDNSC, in agreement with its selective action on the GDNSP and on the [K+]o transient (see above, Fig. 3).
Fig. 4.
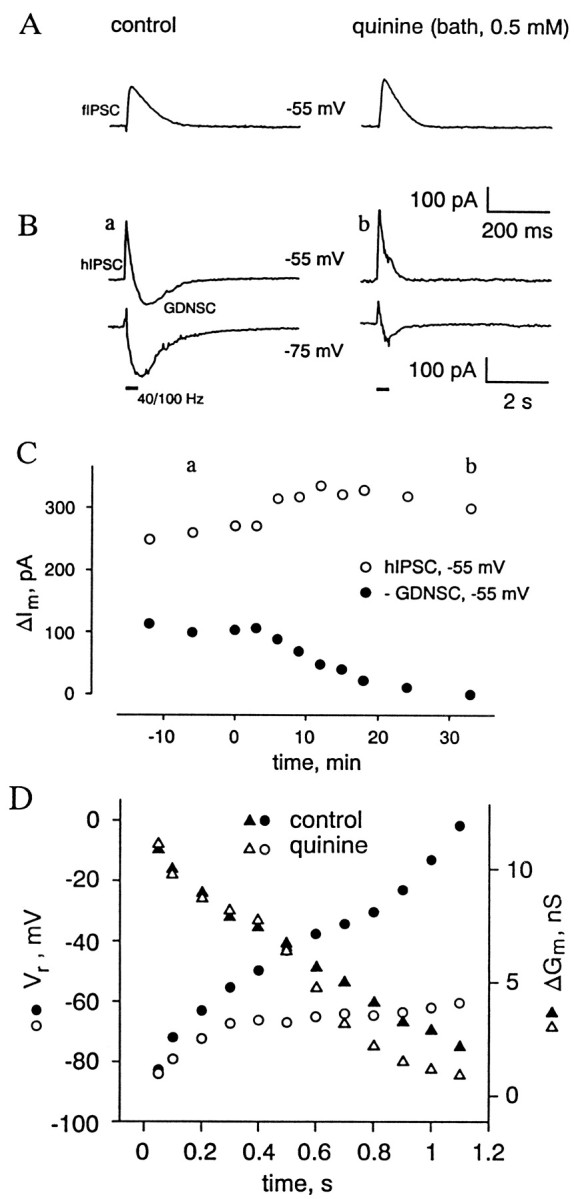
Bath-applied quinine blocks GDNSC but leaves the fIPSC and hIPSC intact. The cell was held using whole-cell clamp at its resting membrane potential (−55 mV). A, fIPSCs recorded at −55 mV in the absence and presence of 0.5 mm quinine.B, Sample recordings (a,b) of hIPSCs and GDNSCs (at −55 and −75 mV).C, Peak amplitudes (ΔIm) of the GDNSC (filled circles) and hIPSC (open circles) as a function of time after exposure to quinine (0.5 mm; the onset of the application is at 0 min).D, Temporal behavior of the hIPSC–GDNSC reversal potential (Vr, circles) and associated increase in conductance (ΔGm, triangles) as examined during an HFS-induced response in the absence (filled symbols) and presence of quinine (open symbols). Time point 0 represents the onset of HFS.
In our previous work (Kaila et al., 1997), one of the key findings leading to the idea that the HFS-induced GDNSC is largely caused by an activity-induced increase in [K+]o resulted from experiments in which the temporal characteristics of the GDNSC reversal potential were examined during an HFS-induced response. In that study, measurements of the current sequence hIPSC–GDNSC reversal at successive time points with 100 msec intervals revealed a progressive buildup of pronounced inward rectification (i.e., qualitatively opposite to GABAAR-mediated currents; Kaila, 1994), with a simultaneous reduction of the HFS-induced conductance increase and a frequent absence of a clear reversal for the late phase of the GDNSC even at potentials more positive than 0 mV. This kind of behavior is depicted in the control values shown in Figure 4D. In striking contrast to this, the GDNSCs measured in the presence of quini(di)ne had a reversal potential consistently more negative than −50 mV (n = 6) throughout the current response (Fig. 4D).
Finally, we addressed the question of whether the selective inhibition of the GDNSP by quini(di)ne could be attributable to a block of the depolarizing HCO3− component of GABAA receptor-mediated currents involved in GDNSP/C generation (Kaila et al., 1997; Lamsa and Kaila, 1997), which can be monitored in measurements of extracellular pH (pHo) transients (Voipio et al., 1995). An inhibition of the bicarbonate current might result from a number of reasons, including a drug-induced collapse in the poststsynaptic pH–HCO3− gradient. As shown in Figure5A, quinine and quinidine suppressed the GABAAR-mediated alkaline pHo shifts (Kaila et al., 1989, 1992) associated with the GDNSPs only by 25 ± 8% (n = 4) and 27 ± 9% (n = 4), respectively. This inhibition most likely reflects the decrease in network excitability after the drug-induced block of the [K+]o transient (Avoli et al., 1996; Lamsa and Kaila, 1997). Nevertheless, increasing the stimulation intensity to reestablish the peak amplitude of the alkalosis did not lead to a parallel recovery of the GDNSP (Fig.5A). That there was no quini(di)ne-induced change in the ionic basis of the alkaline transient was confirmed in experiments with benzolamide (an inhibitor of extracellular carbonic anhydrase; Kaila et al., 1989, 1992; Chen and Chesler, 1992; Voipio et al., 1995) in which the alkaloses were fully blocked (Fig. 5B), indicating that they were caused by a GABAA receptor-mediated CO2–HCO3− shuttle.
Fig. 5.

Block of GDNSP by bath-applied quinine is not associated with a significant inhibition of HFS-induced GABAAR-mediated interstitial alkaline transient.A, Quinine (0.2 mm) abolished the GDNSP, which was linked to a small suppression of the HFS-induced extracellular alkalosis. The original amplitude of the alkaline transient was fully reestablished by increasing the stimulation intensity (from 15 to 20 V) with little recovery of the GDNSP. The twotop traces are simultaneous measurements shown at an identical time scale (as indicated by the thin vetical lines); the bottom traces depict the full duration of the same pHo responses. B, Benzolamide (BA) blocks the HFS-induced alkaline pHo transient evoked in the presence of quinine.
DISCUSSION
In the present study, the various approaches for pharmacological dissection of the hIPSP and GDNSP using internal F−/0 ATP, internal QX-314 (5 mm), and bath-applied quini(di)ne are in good agreement with our previous conclusion (Kaila et al., 1997) that the biphasic voltage response mediated by GABAA receptors in pyramidal neurons during high-frequency stimulation is caused by a dual mechanism: a direct GABAA-mediated transient hyperpolarization (hIPSP), which fades rapidly (cf. Thompson and Gähwiler, 1989; Davies et al., 1990), is followed by a slow depolarization evoked by an activity-induced increase in [K+]o. In particular, the experiments described here and previously (Kaila et al., 1997) clearly demonstrate that a bicarbonate-dependent accumulation of Cl− in the target neuron (Staley et al., 1995), an ionic redistribution mechanism originally examined in crayfish preparations (Kaila and Voipio, 1987; Kaila et al., 1989; Voipio et al., 1991), is not responsible for the generation of the HFS-induced long-lasting GDNSP. The data obtained in the presence of quini(di)ne (Fig. 4D) indicated that the anionic redistribution that takes place in the target cell in the absence of an extracellular K+ transient does not induce a shift in the reversal potential of the HFS-induced GABAAR-mediated current to values more positive than −50 mV.
In line with our dual-mechanism scheme, blocking the GABAA receptors of the target cell from within using F− and depletion of ATP (Bormann et al., 1987; Akaike et al., 1989; Nelson et al., 1994; Khazipov et al., 1997) resulted in a selective block of fIPSP/C and of the hIPSP/C, leaving the GDNSP/C unaffected. On the other hand, internal application of 5 mm QX-314 had the opposite effect, preserving the fIPSP/C and hIPSP/C but inhibiting the GDNSP/C.
An important finding made presently was that internal QX-314 strongly suppressed the sensitivity of the target neuron to the depolarizing action of exogenous K+. It is relevant to note here that intracellular application of QX-314 blocks various kinds of potassium-permeable channels, including voltage-insensitive (Bräu et al., 1995) (also see Segal, 1988), Ca2+-activated (Oda et al., 1992), as well as hyperpolarization-activated cation channels, which mediate theIq (Perkins and Wong, 1995). Nevertheless, Iq seems not to be involved in the generation of the GDNSP, because it was not affected by bath-applied Cs+ (1 mm) or UL-FS 49 (Perkins and Wong, 1995; Pape, 1996), both of which abolished Iq in the present experiments.
The blocking action of QX-314 on the GDNSP may account for the discrepancy between our results concerning the generation of the GDNSP (Kaila et al., 1997) and those on inward GABAA-mediated currents obtained by Perkins and Wong (1996) in the presence of 4-aminopyridine, because the latter study was carried out using whole-cell electrodes with 10 mm QX-314 in their filling solution. In particular, the rather negative reversal potential and brief time to peak of the GABA-mediated depolarization measured by Perkins and Wong (1996) are consistent with a QX-induced block of the late [K+]o-mediated component in their generation (cf. Lamsa and Kaila, 1997).
Application of extracellular quini(di)ne mimicked the effect of QX-314 in that it selectively blocked the GDNSP. A major finding in the present study was that the block of the GDNSP/C by quini(di)ne was faithfully paralleled by an inhibition of the activity-induced [K+]otransient.
Quin(di)ine is known to have distinct concentration-dependent actions on hippocampal neurons (Yoshida et al., 1986). However, at the concentrations and exposure durations (≤30 min) relevant for this study we did not detect any “nonspecific” actions that might have affected the present conclusions.
The possibility that quini(di)ne inhibits K+ channels, which are involved in the generation of the GDNSP in the target cell, was clearly eliminated by the observation that, unlike internal QX-314, quini(di)ne had no blocking effect on depolarizations evoked by exogenous K+. Taken together with the observations made with QX-314, the actions of quini(di)ne are fully consistent with the idea that the activity-induced [K+]o transient is the immediate cause of the GDNSP/C.
When applied at relatively high concentrations, quini(di)ne is known to have an inhibitory action on Cl−extrusion by K+–Cl−cotransport (Ellory et al., 1982; Adragna and Lauf, 1994). However, it is hard to see how an effect of this kind would lead to a selective suppression of the GDNSP, especially because in the present work quini(di)ne had no effect on the reversal potential of single pulse-mediated fIPSPs. A further important finding was that quini(di)ne had no significant effect on the HFS-induced GABAAR-mediated net efflux of bicarbonate (Kaila et al., 1993) as measured on the basis of the benzolamide-sensitive alkaline pHo transient associated with the GDNSP (Voipio et al., 1995; Kaila and Chesler, 1998).
The present work demonstrates that it is possible to use pharmacological agents to dissect the HFS-evoked GABAergic biphasic response of pyramidal neurons into its two underlying components, the direct GABAA receptor-mediated hIPSP and the nonsynaptically generated, slow depolarizing GDNSP. Although [K+]o-mediated responses that are phenomenologically similar to conventional postsynaptic potentials have been demonstrated in invertebrate preparations (Yarom et al., 1982), the present work is, to the best of our knowledge, the first one to directly demonstrate stimulation-induced fast neuron-to-neuron signaling mediated by potassium in mammalian brain tissue.
We think that it is now timely and significant to make a change in the previously used abbreviation GDPSP (which would imply a postsynaptic potential only) to GDNSP, thereby underscoring the nonsynaptic chain of events triggered during HFS. This is because the HFS-induced bicarbonate-dependent increase in [K+]o does not affect only those neurons that are postsynaptic with regard to the stimulated ones, but the K+ transient will have a depolarizing influence on all nearby neurons, glial cells, as well as presynaptic terminals (for the spatial properties of the HFS-induced potassium transient, see Kaila et al., 1997, their Fig. 9).
A point worth noting here is that the time courses of the extracellular K+ transients and GDNSPs often show a temporal deviation during the repolarization of the neuron, which is substantially faster than the potassium transient (Fig. 3A; also see Kaila et al., 1997). This deviation can be explained on the basis that first, the resting membrane potential is not identical to the K+ equilibrium potential and shows a non-Nernstian dependence on [K+]o (Jensen et al., 1993) and second, various intrinsic conductances (Fricker et al., 1999) are modified during the GDNSP. An intriguing possibility here is that the potassium-activated voltage-sensitive K+ conductance demonstrated by Filippov and Krishtal (1999) participates in the generation of the GDNSP.
The main findings and conclusions of this work are summarized in the scheme depicted in Figure 6. In agreement with our previous conclusions (Kaila et al., 1997), the bicarbonate-dependent, quini(di)ne-sensitive increase in [K+]o exerts its main contribution to GDNSP generation via a direct depolarizing action mediated by K+-selective channels that are open at and around the resting voltage level and are sensitive to high concentrations of internal QX-314.
Fig. 6.

Pharmacological isolation of the two components (hIPSP and GDNSP) of the HFS-induced GABAergic response. The synaptically generated, hyperpolarizing hIPSP is abolished by internal F−, whereas the depolarizing, nonsynaptic GDNSP can be blocked either by quini(di)ne, which inhibits the activity-dependent increase in [K]o, or by a high internal concentration (5 mm) of QX-314, which suppresses the sensitivity of the neuronal resting membrane potential to the [K]o transient.
It has been suggested that activity-induced [K+]o shifts play an important role in long-term potentiation (LTP) in the hippocampus (Collingridge, 1992). Hence, an interesting question that emerges here is the role of HFS-induced GABAergic [K+]o shifts and their nonsynaptic actions in LTP and in other manifestations of neuronal plasticity, including pathophysiological phenomena (Heinemann et al., 1990; Jefferys, 1995; also see Lopantsev and Avoli, 1998), as well as in the generation of tetanus-induced neuronal oscillations (e.g., Whittington et al., 1997; Bracci et al., 1999; Traub et al., 1999). Another phenomenon to be thoroughly examined in future work is the mechanism of the biphasic voltage response evoked byexogenous GABAAR agonists (Barker and Ransom, 1978; Alger and Nicoll, 1982), because we have recently obtained evidence (Voipio et al., 1999) that despite a gross similarity (cf. Grover et al., 1993; Staley et al., 1995) its depolarizing phase is not generated in a manner identical to the GDNSP.
Footnotes
This study was supported by grants from the Academy of Finland and from the Sigrid Jusélius Foundation. We thank Dr. Katherine Perkins for helpful discussions.
Correspondence should be addressed to Kai Kaila, Department of Biosciences, Division of Animal Physiology, University of Helsinki, Helsinki FIN-00014, Finland. E-mail: kai.kaila@helsinki.fi.
REFERENCES
- 1.Adragna NC, Lauf PK. Quinine and quinidine inhibit and reveal heterogeneity of K-Cl cotransport in low K sheep erythrocytes. J Membr Biol. 1994;142:195–207. doi: 10.1007/BF00234941. [DOI] [PubMed] [Google Scholar]
- 2.Akaike N, Inomata N, Yakushiji T. Differential effects of extra- and intracellular anions on GABA-activated currents in bullfrog sensory neurons. J Neurophysiol. 1989;62:1388–1399. doi: 10.1152/jn.1989.62.6.1388. [DOI] [PubMed] [Google Scholar]
- 3.Alford S, Frenguelli BG, Schofield JG, Collingridge GL. Characterization of Ca2+ signals induced in hippocampal CA1 neurones by the synaptic activation of NMDA receptors. J Physiol (Lond) 1993;469:693–716. doi: 10.1113/jphysiol.1993.sp019838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Alger BE, Nicoll RA. Pharmacological evidence for two kinds of GABA receptor on rat hippocampal pyramidal cells studied in vitro. J Physiol (Lond) 1982;328:125–141. doi: 10.1113/jphysiol.1982.sp014256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Autere A-M, Lamsa K, Kaila K, Taira T. Synaptic activation of GABAA receptors induces neuronal uptake of Ca2+ in adult rat hippocampal slices. J Neurophysiol. 1999;81:811–816. doi: 10.1152/jn.1999.81.2.811. [DOI] [PubMed] [Google Scholar]
- 6.Avoli M, Louvel J, Kurcewicz I, Pumain R, Barbarosie M. Extracellular free potassium and calcium during synchronous activity induced by 4-aminopyridine in the juvenile rat hippocampus. J Physiol (Lond) 1996;493:707–717. doi: 10.1113/jphysiol.1996.sp021416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Barker JL, Ransom BR. Amino acid pharmacology of mammalian central neurones grown in tissue culture. J Physiol (Lond) 1978;280:331–354. doi: 10.1113/jphysiol.1978.sp012387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Blanton MG, Loturco JJ, Kriegstein AR. Whole cell recording from neurons in slices of reptilian and mammalian cerebral cortex. J Neurosci Methods. 1989;30:203–210. doi: 10.1016/0165-0270(89)90131-3. [DOI] [PubMed] [Google Scholar]
- 9.Bliss TVP, Collingridge GL. A synaptic model of memory: long-term potentiation in the hippocampus. Nature. 1993;361:31–39. doi: 10.1038/361031a0. [DOI] [PubMed] [Google Scholar]
- 10.Bokvist K, Rorsman P, Smith PA. Effects of external tetraethylammonium ions and quinine on delayed rectifying K+ channels in mouse pancreatic beta-cells. J Physiol (Lond) 1990a;423:311–325. doi: 10.1113/jphysiol.1990.sp018024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bokvist K, Rorsman P, Smith PA. Block of ATP-regulated and Ca2+-activated K+ channels in mouse pancreatic beta-cells by external tetraethylammonium and quinine. J Physiol (Lond) 1990b;423:327–342. doi: 10.1113/jphysiol.1990.sp018025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bormann J, Hamill OP, Sakmann B. Mechanism of anion permeation through channels gated by glycine and γ-aminobutyric acid in mouse cultured spinal neurones. J Physiol (Lond) 1987;385:243–286. doi: 10.1113/jphysiol.1987.sp016493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bracci E, Vreugdenhil M, Hack SP, Jefferys JGR. On the synchronizing mechanisms of tetanically induced hippocampal oscillations. J Neurosci. 1999;19:8104–8113. doi: 10.1523/JNEUROSCI.19-18-08104.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bräu ME, Nau C, Hempelmann G, Vogel W. Local anesthetics potently block a potential insensitive potassium channel in myelinated nerve. J Gen Physiol. 1995;105:485–505. doi: 10.1085/jgp.105.4.485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Burg M, Heinemann U, Schmitz D. Neuroactive steroids induce GABAA receptor-mediated depolarizing postsynaptic potentials in hippocampal CA1 pyramidal cells of the rat. Eur J Neurosci. 1998;10:2880–2886. doi: 10.1111/j.1460-9568.1998.00297.x. [DOI] [PubMed] [Google Scholar]
- 16.Chen JC, Chesler M. pH transients evoked by excitatory synaptic transmission are increased by inhibition of extracellular carbonic anhydrase. Proc Natl Acad Sci USA. 1992;89:7786–7790. doi: 10.1073/pnas.89.16.7786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Collingridge GL. The mechanism of induction of NMDA receptor-dependent long-term potentiation in the hippocampus. Exp Physiol. 1992;77:771–797. doi: 10.1113/expphysiol.1992.sp003645. [DOI] [PubMed] [Google Scholar]
- 18.Connors BW, Prince DA. Effects of local anesthetic QX-314 on the membrane properties of hippocampal pyramidal neurons. J Pharmacol Exp Ther. 1982;220:476–481. [PubMed] [Google Scholar]
- 19.Davies CH, Collingridge GL. The physiological regulation of synaptic inhibition by GABAB autoreceptors in rat hippocampus. J Physiol (Lond) 1993;472:245–265. doi: 10.1113/jphysiol.1993.sp019945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Davies CH, Davies SN, Collingridge GL. Paired-pulse depression of monosynaptic GABA-mediated inhibitory postsynaptic responses in rat hippocampus. J Physiol (Lond) 1990;424:513–531. doi: 10.1113/jphysiol.1990.sp018080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Doi T, Fakler B, Schultz JH, Ehmke H, Brändle U, Zenner H-P, Süßbrich H, Lang F, Ruppersberg JP, Busch AE. Subunit-specific inhibition of inward-rectifier K+ channels by quinidine. FEBS Lett. 1995;375:193–196. doi: 10.1016/0014-5793(95)01182-e. [DOI] [PubMed] [Google Scholar]
- 22.Ellory JC, Dunham PB, Logue PJ, Stewart GW. Anion-dependent cation transport in erythrocytes. Philos Trans R Soc Lond B Biol Sci. 1982;299:483–495. doi: 10.1098/rstb.1982.0146. [DOI] [PubMed] [Google Scholar]
- 23.Filippov V, Krishtal O. The mechanism gated by external potassium and sodium controls the resting conductance in hippocampal and cortical neurons. Neuroscience. 1999;92:1231–1242. doi: 10.1016/s0306-4522(99)00081-0. [DOI] [PubMed] [Google Scholar]
- 24.Fishman MC, Spector I. Potassium current suppression by quinidine reveals additional calcium currents in neuroblastoma cells. Proc Natl Acad Sci USA. 1981;78:5245–5249. doi: 10.1073/pnas.78.8.5245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Fricker D, Verheugen JA, Miles R. Cell-attached measurements of the firing threshold of rat hippocampal neurones. J Physiol (Lond) 1999;517:791–804. doi: 10.1111/j.1469-7793.1999.0791s.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Grover LM, Lambert NA, Schwartzkroin PA, Teyler TJ. Role of HCO3--ions in depolarizing GABAA receptor-mediated responses in pyramidal cells of rat hippocampus. J Neurophysiol. 1993;69:1541–1555. doi: 10.1152/jn.1993.69.5.1541. [DOI] [PubMed] [Google Scholar]
- 27.Hara M, Kai Y, Ikemoto Y. Local anesthetics reduce the inhibitory neurotransmitter-induced current in dissociated hippocampal neurons of the rat. Eur J Pharmacol. 1995;283:83–89. doi: 10.1016/0014-2999(95)00293-t. [DOI] [PubMed] [Google Scholar]
- 28.Heinemann U, Stabel J, Rausche G. Activity-dependent ionic changes and neuronal plasticity in rat hippocampus. Prog Brain Res. 1990;83:197–214. doi: 10.1016/s0079-6123(08)61250-9. [DOI] [PubMed] [Google Scholar]
- 29.Hille B. Ionic currents of excitable membranes, Ed 2. Sinauer; Sunderland, MA: 1992. [Google Scholar]
- 30.Jefferys JGR. Nonsynaptic modulation of neuronal activity in the brain: electric currents and extracellular ions. Physiol Rev. 1995;75:689–723. doi: 10.1152/physrev.1995.75.4.689. [DOI] [PubMed] [Google Scholar]
- 31.Jensen MS, Cherubini E, Yaari Y. Opponent effects of potassium on GABAA-mediated postsynaptic inhibition in the rat hippocampus. J Neurophysiol. 1993;69:764–771. doi: 10.1152/jn.1993.69.3.764. [DOI] [PubMed] [Google Scholar]
- 32.Kaila K. Ionic basis of GABAA receptor channel function in the nervous system. Prog Neurobiol. 1994;42:489–537. doi: 10.1016/0301-0082(94)90049-3. [DOI] [PubMed] [Google Scholar]
- 33.Kaila K, Chesler M. Activity-evoked changes in extracellular pH. In: Kaila K, Ransom BR, editors. pH and brain function. Wiley; New York: 1998. pp. 309–337. [Google Scholar]
- 34.Kaila K, Voipio J. Postsynaptic fall in intracellular pH induced by GABA-activated bicarbonate conductance. Nature. 1987;330:163–165. doi: 10.1038/330163a0. [DOI] [PubMed] [Google Scholar]
- 35.Kaila K, Pasternack M, Saarikoski J, Voipio J. Influence of GABA-gated bicarbonate conductance on membrane potential, current and intracellular chloride in crayfish muscle fibres. J Physiol (Lond) 1989;416:161–181. doi: 10.1113/jphysiol.1989.sp017755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kaila K, Paalasmaa P, Taira T, Voipio J. pH transients due to monosynaptic activation of GABAA receptors in rat hippocampal slices. NeuroReport. 1992;3:105–108. doi: 10.1097/00001756-199201000-00028. [DOI] [PubMed] [Google Scholar]
- 37.Kaila K, Voipio J, Paalasmaa P, Pasternack M, Deisz RA. The role of bicarbonate in GABAA receptor-mediated IPSPs in rat neocortical neurons. J Physiol (Lond) 1993;464:273–289. doi: 10.1113/jphysiol.1993.sp019634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kaila K, Smirnov S, Taira T, Voipio J. Both anionic and cationic mechanisms contribute to GABAA receptor-mediated depolarizing synaptic responses in CA1 pyramidal neurons. Soc Neurosci Abstr. 1995;21:244.4. [Google Scholar]
- 39.Kaila K, Lamsa K, Smirnov S, Taira T, Voipio J. Long-lasting GABA-mediated depolarization evoked by high-frequency stimulation in pyramidal neurons of rat hippocampal slice is attributable to a network-driven, bicarbonate-dependent K+ transient. J Neurosci. 1997;17:7662–7672. doi: 10.1523/JNEUROSCI.17-20-07662.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Khazipov R, Leinekugel X, Khalilov I, Gaiarsa JL, Ben-Ari Y. Synchronization of GABAergic interneuronal network in CA3 subfield of neonatal rat hippocampal slices. J Physiol (Lond) 1997;498:763–772. doi: 10.1113/jphysiol.1997.sp021900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lambert N, Grover L. The mechanism of biphasic GABA responses. Science. 1995;269:928–929. doi: 10.1126/science.7638614. [DOI] [PubMed] [Google Scholar]
- 42.Lamsa K, Kaila K. Ionic mechanisms of spontaneous GABAergic events in rat hippocampal slices exposed to 4-aminopyridine. J Neurophysiol. 1997;78:2582–2591. doi: 10.1152/jn.1997.78.5.2582. [DOI] [PubMed] [Google Scholar]
- 43.Lopantsev V, Avoli M. Participation of GABAA-mediated inhibition in ictallike discharges in the rat entorhinal cortex. J Neurophysiol. 1998;79:352–360. doi: 10.1152/jn.1998.79.1.352. [DOI] [PubMed] [Google Scholar]
- 44.Nathan T, Jensen MS, Lambert JDC. The slow inhibitory postsynaptic potential in rat hippocampal neurons is blocked by intracellular injection of QX-314. Neurosci Lett. 1990;110:309–313. doi: 10.1016/0304-3940(90)90865-7. [DOI] [PubMed] [Google Scholar]
- 45.Nelson S, Toth L, Sheth B, Sur M. Orientation selectivity of cortical neurons during intracellular blockade of inhibition. Science. 1994;265:774–777. doi: 10.1126/science.8047882. [DOI] [PubMed] [Google Scholar]
- 46.Oda M, Yoshida A, Ikemoto Y. Blockade by local anaesthetics of the single Ca2+-activated K+ channel in rat hippocampal neurones. Br J Pharmacol. 1992;105:63–70. doi: 10.1111/j.1476-5381.1992.tb14211.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Pape H-C. Queer current and pacemaker: the hyperpolarization-activated cation current in neurons. Annu Rev Physiol. 1996;58:299–327. doi: 10.1146/annurev.ph.58.030196.001503. [DOI] [PubMed] [Google Scholar]
- 48.Perkins KL. Cl− accumulation does not account for the depolarizing phase of the synaptic GABA response in hippocampal pyramidal cells. J Neurophysiol. 1999;82:768–777. doi: 10.1152/jn.1999.82.2.768. [DOI] [PubMed] [Google Scholar]
- 49.Perkins KL, Wong RKS. Intracellular QX-314 blocks the hyperpolarization-activated inward current Iq in hippocampal CA1 pyramidal cells. J Neurophysiol. 1995;73:911–915. doi: 10.1152/jn.1995.73.2.911. [DOI] [PubMed] [Google Scholar]
- 50.Perkins KL, Wong RKS. Ionic basis of the postsynaptic depolarizing GABA response in hippocampal pyramidal cells. J Neurophysiol. 1996;76:3886–3894. doi: 10.1152/jn.1996.76.6.3886. [DOI] [PubMed] [Google Scholar]
- 51.Rivera C, Voipio J, Payne JA, Ruusuvuori E, Lahtinen H, Lamsa K, Pirvola U, Saarma M, Kaila K. The K+/Cl− co-transporter KCC2 renders GABA hyperpolarizing during neuronal maturation. Nature. 1999;397:251–255. doi: 10.1038/16697. [DOI] [PubMed] [Google Scholar]
- 52.Segal M. Effects of a lidocaine derivative QX-572 on CA1 neuronal responses to electrical and chemical stimuli in a hippocampal slice. Neuroscience. 1988;27:905–909. doi: 10.1016/0306-4522(88)90193-5. [DOI] [PubMed] [Google Scholar]
- 53.Seki G, Taniguchi S, Uwatoko S, Suzuki K, Kurokawa K. Evidence for conductive Cl− pathway in the basolateral membrane of rabbit renal proximal tubule S3 segment. J Clin Invest. 1993;92:1229–1235. doi: 10.1172/JCI116694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Smirnov S, Kaila K. Selective pharmacological inhibition of the hyperpolarizing and depolarizing components of the biphasic postsynaptic response evoked in hippocampal pyramidal neurons by high-frequency stimulation of GABAergic interneurons. Soc Neurosci Abstr. 1997;23:892.11. [Google Scholar]
- 55.Staley KJ, Soldo BL, Proctor WR. Ionic mechanisms of neuronal excitation by inhibitory GABAA receptors. Science. 1995;269:977–981. doi: 10.1126/science.7638623. [DOI] [PubMed] [Google Scholar]
- 56.Taira T, Lamsa K, Kaila K. Post-tetanic excitation mediated by GABAA receptors in rat CA1 pyramidal neurons. J Neurophysiol. 1997;77:2213–2218. doi: 10.1152/jn.1997.77.4.2213. [DOI] [PubMed] [Google Scholar]
- 57.Thompson SM, Gähwiler BH. Activity-dependent disinhibition. I. Repetitive stimulation reduces IPSP driving force and conductance in the hippocampus in vitro. J Neurophysiol. 1989;61:501–511. doi: 10.1152/jn.1989.61.3.501. [DOI] [PubMed] [Google Scholar]
- 58.Traub RD, Whittington MA, Buhl EH, Jefferys JG, Faulkner HJ. On the mechanism of the gamma → beta frequency shift in neuronal oscillations induced in rat hippocampal slices by tetanic stimulation. J Neurosci. 1999;19:1088–1105. doi: 10.1523/JNEUROSCI.19-03-01088.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Voipio J, Pasternack M, Rydqvist B, Kaila K. Effect of γ-aminobutyric acid on intracellular pH in the crayfish stretch-receptor neurone. J Exp Biol. 1991;156:349–361. doi: 10.1242/jeb.156.1.349. [DOI] [PubMed] [Google Scholar]
- 60.Voipio J, Pasternack M, MacLeod K. Ion-sensitive microelectrodes. In: Ogden D, editor. Microelectrode techniques, the Plymouth Workshop handbook, Ed 2. Company of Biologists; Cambridge, UK: 1994. pp. 275–316. [Google Scholar]
- 61.Voipio J, Paalasmaa P, Taira T, Kaila K. Pharmacological characterization of extracellular pH transients evoked by selective synaptic and exogenous activation of AMPA, NMDA, and GABAA receptors in the rat hippocampal slice. J Neurophysiol. 1995;74:633–642. doi: 10.1152/jn.1995.74.2.633. [DOI] [PubMed] [Google Scholar]
- 62.Voipio J, Ruusuvuori E, Kaila K. GABAA receptor channel-mediated anionic load and the consequent increase in interstitial K+: a developmental study sheds light on the molecular mechanisms involved. Soc Neurosci Abstr. 1999;25:710.18. [Google Scholar]
- 63.Whittington MA, Stanford IM, Colling SB, Jefferys JG, Traub RD. Spatiotemporal patterns of gamma frequency oscillations tetanically induced in the rat hippocampal slice. J Physiol (Lond) 1997;502:591–607. doi: 10.1111/j.1469-7793.1997.591bj.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Yarom Y, Grossman Y, Gutnick MJ, Spira ME. Transient extracellular potassium accumulation produced prolonged depolarizations during synchronized bursts in picrotoxin-treated cockroach CNS. J Neurophysiol. 1982;48:1089–1097. doi: 10.1152/jn.1982.48.5.1089. [DOI] [PubMed] [Google Scholar]
- 65.Yoshida S, Fujimura K, Matsuda Y. Effects of quinidine and quinine on the excitability of pyramidal neurons in guinea-pig hippocampal slices. Pflugers Arch. 1986;406:544–546. doi: 10.1007/BF00583380. [DOI] [PubMed] [Google Scholar]
- 66.Zetterström TS, Vaughan-Jones RD, Grahame-Smith DG. A short period of hypoxia produces a rapid and transient rise in [K+]e in rat hippocampus in vivo which is inhibited by certain K+-channel blocking agents. Neuroscience. 1995;67:815–821. doi: 10.1016/0306-4522(95)00107-t. [DOI] [PubMed] [Google Scholar]