

Abstract
Considerable evidence suggests that Ca2+-permeable AMPA receptors are critical mediators of the delayed, selective neuronal death associated with transient global ischemia and sustained seizures. Global ischemia suppresses mRNA and protein expression of the glutamate receptor subunit GluR2 and increases AMPA receptor-mediated Ca2+ influx into vulnerable neurons of the hippocampal CA1 before the onset of neurodegeneration. Status epilepticus suppresses GluR2 mRNA and protein in CA3 before neurodegeneration in this region. To examine whether acute downregulation of the GluR2 subunit, even in the absence of a neurological insult, can cause neuronal cell death, we performed GluR2 “knockdown” experiments. Intracerebral injection of antisense oligodeoxynucleotides targeted to GluR2 mRNA induced delayed death of pyramidal neurons in CA1 and CA3. Antisense-induced neurodegeneration was preceded by a reduction in GluR2 mRNA, as indicated by in situ hybridization, and in GluR2 protein, as indicated by Western blot analysis. GluR2 antisense suppressed GluR2 mRNA in the dentate gyrus but did not cause cell death. The AMPA receptor antagonist 6-cyano-7-nitroquinoxiline-2,3-dione (CNQX) and the Ca2+-permeable AMPA receptor channel blocker 1-naphthyl acetyl spermine protected against antisense-induced cell death. This result indicates that antisense-induced cell death is mediated by Ca2+-permeable AMPA receptors. GluR2 antisense and brief sublethal global ischemia acted synergistically to cause degeneration of pyramidal neurons, consistent with action by a common mechanism. These findings demonstrate that downregulation of GluR2 is sufficient to induce delayed death of specific neuronal populations.
Keywords: antisense oligonucleotides, neurodegeneration, AMPA receptors, gene expression, hippocampus, excitotoxicity, cerebral ischemia, status epilepticus
Transient severe global ischemia, occurring in patients during cardiorespiratory arrest or induced experimentally in animals, causes selective and delayed neuronal cell death, particularly of the hippocampal CA1 (Schmidt-Kastner and Freund, 1991; Hsu and Buzsaki, 1993). The delayed cell death after ischemia requires an initial translocation of Zn2+(Koh et al., 1996) and is associated with a late influx of toxic amounts of Ca2+ into vulnerable neurons, which may be mediated by Ca2+-permeable AMPA receptors (Pellegrini-Giampietro et al., 1997). During the ischemic episode, cells depolarize, exhibit a rise in intracellular Ca2+ (Silver and Erecinska, 1992), and become inexcitable. After reperfusion, cells appear morphologically normal, exhibit normal intracellular Ca2+(Silver and Erecinska, 1992), and are again able to generate action potentials for 24–72 hr after the ischemic insult (Gorter et al., 1997). Ultimately, intracellular Ca2+again rises in vulnerable neurons, and cell death ensues, exhibiting a number of the hallmarks of apoptosis (Kirino, 1982; Takei and Endo, 1994; Choi, 1995; Chen et al., 1998; but see Colbourne et al., 1999). Delayed neurodegeneration is also seen after kainate-induced status epilepticus, but in this paradigm, it is the pyramidal neurons of CA3 that are selectively vulnerable (Friedman et al., 1994).
AMPA receptors containing an edited form of the glutamate receptor subunit GluR2 (or GluR-B) are relatively impermeable to divalent cations; receptors lacking this subunit are much more permeable to Ca2+ (and Zn2+) (Hollmann et al., 1991; Verdoorn et al., 1991; Burnashev et al., 1992; Bowie and Mayer, 1995;Washburn et al., 1997). Most principal neurons of adult hippocampus express heteromeric AMPA receptors that contain edited GluR2 and have low permeability to Ca2+ (Bochet et al., 1994; Jonas et al., 1994; Geiger et al., 1995). In these cells, an acute reduction in GluR2 expression could act as a “molecular switch,” leading to increased Ca2+permeability of AMPA receptors and enhanced toxicity of endogenous glutamate.
A number of studies provide evidence that Ca2+-permeable AMPA receptors are mediators of the delayed neurodegeneration associated with global ischemia and status epilepticus. AMPA receptor antagonists, but not NMDA antagonists, protect against ischemia-induced neuronal death, even when administered many hours after a brief ischemic insult (Pulsinelli et al., 1993; Sheardown et al., 1993). The relevance of this finding, however, is unclear, because protection may be caused by antagonist-induced hypothermia rather than by block of Ca2+-permeable AMPA receptors in vulnerable neurons (Nurse and Corbett, 1996). Global ischemia induces downregulation of GluR2 mRNA and protein expression in vulnerable neurons before cell death (for review, see Bennett et al., 1996; Pellegrini-Giampietro et al., 1997). In slices from postischemic animals, CA1 neurons with robust action potentials exhibit greatly enhanced AMPA-elicited rises in intracellular Ca2+ compared with those in cells from control animals (Gorter et al., 1997). EPSCs in postischemic CA1 neurons exhibit an enhanced Ca2+-dependent component that appears to be mediated by Ca2+-permeable AMPA receptors (Tsubokawa et al., 1994, 1995). These studies provide evidence of Ca2+ influx through AMPA receptors in neurons destined to die. Kainate-induced status epilepticus also leads to downregulation of GluR2 mRNA and protein expression before the onset of delayed neuronal death in CA3 (Friedman et al., 1994, 1997).
To examine whether acute reduction in the level of GluR2 is sufficient to cause neurodegeneration, even in the absence of the other changes caused by global ischemia or status epilepticus, we performed “knockdown” experiments with GluR2 antisense oligonucleotides targeted to GluR2 mRNA. Knockdown of receptor subunits has proven valuable in investigation of receptor function in vivo andin vitro (for review, see Wahlestedt, 1994; Weiss et al., 1997). Intracerebroventricular injection of GluR2 antisense induced delayed degeneration of pyramidal neurons in CA1 and CA3, regions vulnerable to global ischemia and status epilepticus, respectively. Degeneration in these regions was preceded by a significant reduction in the levels of GluR2 mRNA and protein. Blockers of AMPA receptors protected against antisense-induced death. Moreover, GluR2 antisense acted synergistically with brief, sublethal ischemia to cause degeneration of pyramidal neurons, consistent with the mediation of postischemic death by downregulation of GluR2.
MATERIALS AND METHODS
Antisense oligodeoxynucleotides.Oligodeoxynucleotides (21 bases in length) corresponding to specific sequences in the 5′-coding region of the GluR2 cDNA were synthesized in the antisense orientation (Genosys Biotechnologies, The Woodlands, TX). The specific antisense oligonucleotide designated AS1 (5′-CTGTATGCTGTTAGAAGAGAC-3′), complementary to positions 64–84 of rat GluR2 cDNA (GenBank M36419), was designed to target the 5′-coding region of GluR2 mRNA just after the signal sequence. The specific antisense designated AS2 (5′-AACCATTTTATCCACTTCACT-3′), complementary to positions 1168–1188 of the GluR2 cDNA, was designed to target the region of GluR2 mRNA encoding a unique sequence in the N-terminal domain recognized by polyclonal antibody GluR2–23 (Wenthold et al., 1992). SC1 or “scrambled” antisense (5′-CTAACCTCCAATCTTATTCTA-3′), in which the bases of the 21-mer AS2 were randomized, was synthesized for use as a control. A search of GenBank revealed no rodent sequence, other than that of the GluR2 gene, that was identical to any of the three oligonucleotides for a length of 15 or more base pairs. Identity over shorter sequences was not considered, because the predicted Tm of 15 mers would be ∼15°C or lower, well below rodent body temperature. Specificity of the oligos can also be inferred from their effects described in the Results. To increase stability of the injected oligonucleotides, phosphorothioate bonds were incorporated between three terminal nucleotides at the 5′ and 3′ ends. For stereotaxic injections, oligonucleotides were dissolved in saline (0.9% NaCl). Each single injection was 10 nmol in 5 μl for rats or 2 nmol in 3 μl for gerbils.
Intracerebroventricular administration of GluR2 antisense in rats. For administration of antisense oligonucleotides, adult male Sprague Dawley rats (Charles River Laboratories, Wilmington, MA), weighing 260–280 gm, were anesthetized with 5% halothane, after which they were transferred to a surgical chamber and maintained in 2% halothane in a mixture of N2O/O2 (70:30) by means of a Vapomatic anesthetic vaporizer (CWE, Ardmore, PA). Body temperature and anesthesia level were monitored continuously during surgery. Animals were positioned in a stereotaxic frame, and a cannula (28 gauge; stainless steel; inner diameter, 0.18 mm; outer diameter, 0.36 mm) was lowered stereotaxically into the right cerebral ventricle to a position defined by the following coordinates: −0.92 mm posterior from bregma, −1.2 mm lateral to bregma, and −3.6 mm below the skull surface. Specific GluR2 antisense AS1, antisense AS2, or randomized (scrambled) antisense SC1 (10 nmol in 5 μl of saline) or saline (5 μl) was delivered via the cannula at a flow rate of 1 μl/min. A single injection of antisense or saline was given, or four injections were given at 12 hr intervals. Both specific and scrambled antisense caused some reduction in activity of the animals for several hours after each injection; animals were never observed to exhibit seizures.
Administration of antisense oligonucleotides and sublethal global ischemia in gerbils. To examine the combined effects of GluR2 antisense oligonucleotide administration and transient, forebrain ischemia, we anesthetized adult male Mongolian gerbils (Charles River Laboratories), weighing 60–80 gm, with 4% halothane and then maintained them in 2% halothane in a mixture of N2O/O2 (70:30) as described above. For intracerebroventricular injection, animals were held in a stereotaxic frame, and the injection cannula was inserted to a position defined by the following stereotaxic coordinates: −0.4 mm posterior from bregma, −1.2 mm lateral to bregma, and −2.2 mm below the skull surface. Specific GluR2 antisense AS2 or randomized (scrambled) antisense SC1 (2 nmol in 3 μl of saline) or saline (3 μl) was delivered via the cannula at a flow rate of 1 μl/min. A single injection of antisense or saline was given, or four injections were given at 12 hr intervals. One-half of the animals received no further treatment, and one-half were subjected to sublethal (2 min) forebrain ischemia immediately after the last injection. The ischemia was produced by temporary bilateral occlusion of the carotid arteries using a modification of our previously described method (Oguro et al., 1995). The common carotid arteries were exposed by means of a ventral midline neck incision and occluded for 2 min with nontraumatic aneurism clips. At the end of the occlusion period, the clamps were released, allowing restoration of carotid blood flow, and the incision was sutured. Halothane administration was discontinued immediately after carotid occlusion. Body temperature was monitored and maintained close to 37.5 ± 0.5°C with a rectal thermistor and heat lamp until the animal had fully recovered from anesthesia.
Animal care. Animals were maintained in a temperature- and light-controlled environment with a 14:10 hr light/dark cycle and were treated in accordance with the principles and procedures of theNational Institutes of Health Guidelines for the Care and Use of Laboratory Animals.
Histological analysis. In rats, neuronal damage was monitored by histological examination of brain sections at the level of the hippocampus of animals killed at 1, 2, and 7 d after specific antisense treatments and at 7 d after control treatments (saline or scrambled antisense). In gerbils, histological analysis was of animals killed at 7 d after experimental and control treatments. Animals were fixed by transcardiac perfusion with 4% paraformaldehyde in 10 mm PBS (200 ml/30 min) under deep chloroform anesthesia, and their brains were removed and immersed in fixative at 4°C overnight. Coronal sections (30 μm) were cut with a vibratome and stained with toluidine blue. Hippocampal injury was assessed quantitatively by counting the number of pyramidal cells appearing histologically normal at a position distant to the site of antisense infusion (rats, −3.3 mm from bregma; gerbils, −1.5 mm from bregma). Cell counts from a minimum of four microscopic sections for each animal were statistically analyzed by means of the Student's unpairedt test.
In situ hybridization. An [35S]UTP-labeled RNA probe directed against GluR2 was transcribed by incubation (1 hr at 37°C) of the rat GluR2 cDNA with T7 polymerase in the presence of labeled and unlabeled nucleotides using a Stratagene transcription kit (La Jolla, CA). Radiolabeled probe was purified by phenol and chloroform extraction.
For quantitation of GluR2 mRNA expression, antisense-injected rats were anesthetized with chloroform and decapitated 6, 12, 24, 48, and 72 hr after the last injection. Saline-injected rats (four injections at 12 hr intervals) were killed 6 hr after the last injection. GluR2 mRNA expression was assessed by in situ hybridization on sections of brain at the level of the hippocampus (Pellegrini-Giampietro et al., 1992a). In brief, brains were rapidly removed, frozen by immersion in 2-methylbutane at −35°C, and stored at −70°C until sectioned. Coronal sections (18 μm) were cut on a cryotome and thaw-mounted on gelatin- or poly-l-lysine-coated slides. After fixation with 4% paraformaldehyde in 10 mmPBS with 5 mm MgCl2for 15 min, sections were rinsed in PBS, dehydrated in graded ethanols, and stored in 95% ethanol (4°C) until use. Before application of riboprobes, slides were subjected to acetylation and incubated for 2 hr at 50°C with prehybridization solution. For hybridization, slides were incubated overnight at 50°C with35S-labeled RNA probe (106 cpm/section; 1 ng/ml). Sections were washed, treated (30 min at room temperature) with RNase A (20 μg/ml), and again dehydrated in graded ethanols. Slides were apposed to Kodak XAR 5 film (Eastman Kodak, Rochester, NY) for 48 hr.
Signal specificity was assessed by competition experiments with excess unlabeled oligonucleotides (Pellegrini-Giampietro et al., 1992a). These resulted in virtually blank autoradiograms. In separate control studies, application of sense RNA probes to sections pretreated with RNase A (100 μg/ml) showed no detectable labeling. The GluR2 probe is a “pan” probe in that it labels both flip and flop splice variants.
For quantitation of GluR2 mRNA levels, autoradiograms were analyzed with a Scan Jet 4-C computing densitometer using NIH Image 1.61 image-processing and analysis software. Films were scanned at 600 dots per inch resolution, and images of each section (∼1 × 106 pixels) were created. Mean optical densities in regions of maximal labeling of individual hippocampal subfields were averaged from a minimum of two sections from each animal, and film background was subtracted. Optical density values were expressed as grand means (± SDs) of individual means from three rats for each time point and were normalized to optical density values for the corresponding regions of control brains. To enable comparisons between groups for any time points, sections were cut from brains of control and experimental rats in the same session, incubated with the same solutions of RNA probes, and apposed to the same sheet of film. Mean optical density values were statistically analyzed by the Student's unpaired t test. The rationale of the quantitative analysis was based on the following considerations: (1) optical density readings taken from the region of interest varied little among sections from a single animal, (2) the concentration of RNA probe (106 cpm/section) used in the experiment produced saturating levels of hybridization and a maximal signal-to-noise ratio, and (3) density readings of [35S]UTP-labeled standards made of brain paste indicated that exposure times were in the linear response range of the film (Pellegrini-Giampietro et al., 1992a).
Western blotting. For quantitation of protein expression levels, rats were anesthetized with chloroform and killed by decapitation 6, 12, 24, 48, and 72 hr after the last (fourth) injection of GluR2 antisense or 6, 12, and 24 hr after a single injection of antisense AS2. Control rats receiving a single injection or four injections of saline at 12 hr intervals were killed 6 hr after the last injection. Hippocampi were quickly dissected out and placed in ice-cold PBS, and thick slices of dorsal hippocampus (1 mm) were cut on a Mcllwain tissue chopper. CA1 and the bend region of CA3 were rapidly separated from the slices by microdissection and stored at −70°C before use. Tissue was placed in a sonicator in 200 μl of 1 mm NaHNO3 buffer, pH 6.8, containing the protease inhibitor PMSF (phenylmethylsulfonyl fluoride, 1 mm; Sigma, St. Louis, MO) and sonicated until the solution was uniform. The tissue sample was lysed overnight at 4°C in Laemmli sample buffer (0.025 m Tris-HCl, 5% glycerol, 1% SDS, 0.05% bromphenol blue, 0.1 m dithiothreitol, 2.5% β-mercaptoethanol, 1 mm PMSF, and 0.5 mmNaHNO3 buffer, pH 6.8). Protein concentration was measured using the bicinchoninic acid protein assay kit (Pierce, Rockford, IL), and protein samples (10 μg) were loaded on 10% polyacrylamide minigels (Bio-Rad, Richmond, CA) and separated by gel electrophoresis. Protein bands were transferred from the gel to nitrocellulose (Bio-Rad) in blotting buffer containing 0.192m glycine and 20% methanol. After blocking for 30 min with 25 mm Tris-HCl buffer, pH 8.0, 125 mm NaCl, 0.1% Tween 20, and 4% skim milk, the membrane was incubated for 1 hr at room temperature with the primary antibody diluted 1:1000 in PBS. Two primary antibodies were used: a mouse monoclonal antibody directed to a sequence within the N-terminal domain of the GluR2 subunit (Vissavajjhala et al., 1996) (gift of John H. Morrison, Mt. Sinai, NY) and a rabbit polyclonal antibody directed to a sequence within the C-terminal domain of the GluR1 subunit (Wenthold et al., 1992) (PharMingen, San Diego, CA). The membrane was then incubated for 1 hr at room temperature with secondary antibody diluted 1:1000 in PBS and enhanced chemiluminescence reagents to visualize the antibody reaction (ECL; Amersham, Arlington Heights, IL) and finally exposed to XAR 5 x-ray film (Eastman Kodak). Secondary antibodies used were, for GluR2, a horseradish peroxidase (HRP)-conjugated anti-mouse IgG and, for GluR1, an HRP-conjugated anti-rabbit IgG (Vector Laboratories, Burlingame, CA).
For quantitation of GluR1 and GluR2 protein levels, Western blots were analyzed with a Scan Jet 4-C computing densitometer using NIH Image 1.61 image-processing and analysis software. Mean optical densities of bands for two samples per animal were determined, and the film background was subtracted. Optical density values were expressed as grand means (± SDs) of individual means from a minimum of three rats. Changes in optical density for antisense-injected rats were normalized to optical density values for the corresponding brain region of control rats to enable comparisons of band densities of immunoblots apposed to different films. Mean band densities were statistically analyzed with the Student's unpaired t test to determine significance.
Administration of glutamate receptor antagonists. For pharmacological studies of neuroprotection by glutamate antagonists, antagonist (10 mm in 5 μl of saline) or saline was injected into the right lateral ventricle at the time of the second or fourth antisense injection or 24 hr after the fourth (last) antisense injection. Antagonists used were as follows: 1-naphthyl acetyl spermine (Naspm), channel blocker of Ca2+-permeable AMPA receptors (Research Biochemicals, Natick, MA); 6-cyano-7-nitroquinoxiline-2,3-dione disodium (CNQX), AMPA receptor antagonist (Research Biochemicals); andd-2-amino-5-phosphonovalerate (AP-5), NMDA receptor antagonist (Tocris, Ballwin, MO).
RESULTS
GluR2 antisense induces neuronal cell death in the hippocampal CA1 and CA3
To examine the effect of GluR2 knockdown on the morphology and viability of the principal neurons of the hippocampus, we administered GluR2 antisense oligonucleotides AS1 and AS2 into the right ventricle of rats, either as a single injection or as four injections at 12 hr intervals. Control rats were administered four injections of scrambled antisense SC1 or saline at 12 hr intervals. Experimental rats were killed at 1, 2, and 7 d after the last injection, control rats were killed at 7 d after the last injection, and brain sections were examined histologically.
The specific GluR2 antisense AS1 induced selective, delayed neurodegeneration in the pyramidal cell layer of the hippocampus (Figs.1, 2). At 1 d after the last of four antisense injections, cell counts of toluidine blue-stained brain sections did not yet reveal any indication of cell death in the pyramidal cell layers in CA1 or CA3 (histology not shown; cell counts in Fig. 2; n = 5). At 2 d after the last antisense injection, the number of normal-appearing cells was reduced by one-third in both CA1 and CA3 on both right and left sides compared with that in saline- or scrambled antisense-injected controls (Figs. 1E,F, 2; n = 5;p < 0.01 for each region, right and left CA1 and CA3; saline injected, n = 5; SC1 injected, n= 5; control animals killed at 7 d). At this time the deteriorating cells showed nuclear pyknosis and shrinkage of cytoplasm. At 7 d after the last AS1 injection, the entire pyramidal cell layer of the hippocampus was reduced in thickness, and loss of ∼50% of cells was evident in CA1 and CA3 (Figs. 1H,I, 2;n = 5; p < 0.01 for each region). Cell loss was not associated with inflammation. There was no indication of degeneration or inflammation in the controls, and cell counts were not significantly different from those in untreated animals (data not shown). Although there was a slight trend toward greater cell death on the injected (right) side versus the contralateral side and in CA1 versus CA3, the extent of neuronal damage did not differ significantly on the two sides or in the two regions at any time examined (Fig.2A,B). There was no histologically detectable degeneration in the granule cell layer of the dentate gyrus at any time examined.
Fig. 1.

GluR2 antisense induces selective, delayed neurodegeneration in hippocampal CA1 and CA3. Toluidine blue staining of coronal brain sections shows the dorsal hippocampus.A–C, Control brain from an animal killed at 7 d after the last of four injections of saline is shown.D–F, By 2 d after the last of four injections of GluR2 antisense AS1, the pyramidal cell layer of CA1 and CA3 exhibited loss of neurons and degenerative changes. G–I, By 7 d after the last antisense injection, there was further cell loss in CA1 and CA3. Scale bars: A, D, G, 100 μm;B, C, E, F, H, I, 50 μm.
Fig. 2.

Quantitation of antisense-induced cell loss. Cell number per millimeter on the right (rt), injected side, and on the left (lt) side of CA1 (A), CA3 (B), and the dentate gyrus (DG; C) is shown. Eachfilledcircle represents a single animal. Control animals received four injections of saline at 12 hr intervals or four injections of scrambled antisense SC1 at 12 hr intervals and were killed at 7 d. Controls showed no sign of neurodegeneration. GluR2 antisense AS1 was injected four times at 12 hr intervals, and animals were killed at 1, 2, and 7 d after the last injection. GluR2 antisense caused degeneration in the pyramidal cell layer in CA1 and CA3 on both sides of the brain. There was no degeneration in either DG. Cell loss relative to controls was observed after 2 d and was greater after 7 d. The number of pyramidal cells in CA1 and CA3 was reduced to ∼70% of the control value at 2 d and 50% of the control value at 7 d (**p < 0.01).
The specific GluR2 antisense AS2 (four injections at 12 hr intervals) induced a similar pattern of selective neurodegeneration in the pyramidal cell layer measured at 7 d (n = 5; data not shown). However, a single injection of either AS1 or AS2 did not produce any detectable neurodegeneration at 7 d in any region examined (for AS1, n = 5; for AS2, n = 4; data not shown).
None of the animals exhibited seizure activity, and postictal processes are unlikely to have contributed to the observed degeneration.
GluR2 antisense decreases GluR2 mRNA levels in CA1 and CA3 pyramidal layers
To investigate whether GluR2 antisense depresses GluR2 mRNA levels, we performed in situ hybridization on brain sections from AS2- and saline-injected rats with a riboprobe specific for GluR2 mRNA. In saline-injected control animals (four injections; killed 6 hr after the last injection), autoradiograms of coronal sections at the level of the dorsal hippocampus showed high levels of GluR2 mRNA throughout the pyramidal cell layers of CA1 and CA3 and in the granule cell layer of the dentate gyrus (Fig. 3,top left; right hippocampus, side of injection), as reported previously in untreated animals (see Pellegrini-Giampietro et al., 1992a). At 6 and 12 hr after the fourth antisense injection, GluR2 mRNA expression was reduced throughout the hippocampus, including the dentate gyrus (DG), but recovered to control values at 24–48 hr. Quantitation of film densities of autoradiograms showed reduction of GluR2 mRNA in the right CA1 to 70 ± 3.9% of control at 6 hr (n = 3; p < 0.01) and to 74 ± 1.4% of control at 12 hr (n = 3; p < 0.01; Fig. 3, bottom).
Fig. 3.

Decreases in the GluR2 mRNA level in the hippocampus induced by injection of GluR2 antisense.Top, Film autoradiograms of in situhybridization of coronal sections of the dorsal hippocampus of a control rat (4 saline injections at 12 hr intervals) and GluR2 antisense AS2-injected rats (4 injections at 12 hr intervals) at 6, 12, 24, 48, and 72 hr after the last of four injections are shown. The autoradiograms indicate GluR2 knockdown in the entire right hippocampus (injected side) at 6 and 12 hr and recovery by 48 hr.Bottom, Mean densities in CA1 normalized to the mean value for control rats (data from the right, injected side) show that the relative abundance of mRNA declined to a minimum of 70 ± 3.9% of control at 6 hr after the last injection and largely recovered by 24 hr (n = 3 for each time point; **p < 0.01). C, Control.
There was a trend toward greater reduction in the GluR2 mRNA level on the injected (right) side compared with that on the contralateral side, but the difference was not statistically significant (data not shown). GluR2 antisense did not induce any detectable alteration in GluR2 mRNA levels in the piriform cortex or in the hypothalamus (measured in the same sections used for Fig. 3).
GluR2 antisense reduces the GluR2 protein level in CA1 and CA3
Antisense-induced effects in GluR1 and GluR2 protein levels in CA1 and the bend region of CA3 were examined by quantitative Western blot analysis with subunit-specific antibodies (Wenthold et al., 1992;Vissavajjhala et al., 1996). Antisense treatment produced a substantial reduction in the level of GluR2 protein in CA1 relative to that in control animals (representative blots for four injections are shown in Fig. 4A, top; the control received four injections of saline and was killed at 6 hr after the last injection). A single antisense injection produced little reduction in GluR2 protein at 6 hr and maximal reduction at 12 hr (to 68 ± 7.0% of the saline-injected control value;p < 0.01; n = 3 for each time point and control) with partial recovery by 24 hr (Fig. 4A, bottom left). Four injections of antisense produced reductions in the GluR2 protein in CA1 at 12 and 24 hr after the last injection that did not differ significantly from those after a single injection (Fig.4A,bottom right; n = 3), indicating that the effects of successive injections were not cumulative. GluR2 protein at 6 hr after the last of four injections was lower than that at 6 hr after a single injection but higher than that at 12 hr after either regimen. This observation suggests that there is partial recovery of GluR2 protein between successive injections. Because GluR2 mRNA recovers somewhat by 12 hr after the last of four injections (Fig. 3), it would be expected to recover between injections at 12 hr intervals with a small time lag for uptake and action of each additional antisense injection. GluR2 protein should also recover, but with an additional time lag for protein synthesis. By 48 and 72 hr after the last of four injections, GluR2 protein had largely recovered. The small (but not statistically significant) residual depression may have been caused by cell morbidity. The reduction in GluR2 protein was bilateral and did not differ significantly in ipsilateral versus contralateral CA1 (data not shown). Similar changes were observed in CA3 bend region (data not shown).
Fig. 4.
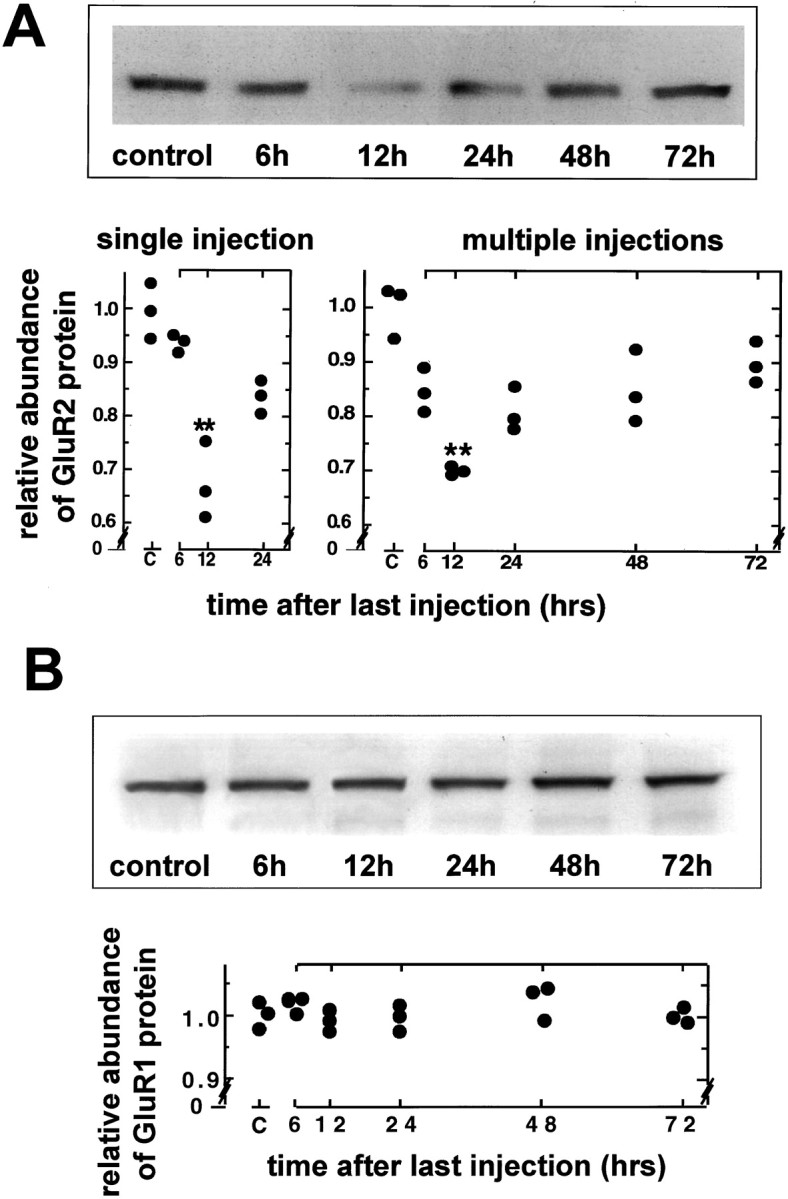
Injection of GluR2 antisense decreases GluR2 protein levels in CA1 but does not affect GluR1 protein levels.A, Film autoradiograms of Western blotting for GluR2 protein in the right CA1 (injected side) of control (4 saline injections at 12 hr intervals; rat killed at 6 hr after the last injection) and antisense AS2-injected rats at 6, 12, 24, 48, and 72 hr after the last of four injections (top) and relative band densities after a single saline or antisense injection (bottomleft; saline control killed at 6 hr) or after the multiple injections of saline or antisense (bottomright) are shown. Band densities are normalized to that of the saline-injected controls. The GluR2 protein level of antisense-injected rats was significantly decreased at 12 hr after either a single injection or the last of four injections (**p < 0.01) and then recovered toward control levels (n = 3 for each time point).B, Film autoradiograms of Western blotting for GluR1 protein expression (top) and density measurements (bottom) showed no significant alteration in GluR1 protein expression in the right CA1 after four GluR2 antisense injections (same animals used in A).
The GluR2 antisense treatment did not alter the level of GluR1 protein in CA1 (Fig. 4B) or in CA3 (data not shown) at any time examined. This finding indicates that the effects of GluR2 antisense were specific and not simply caused by toxicity.
Naspm and CNQX protect against GluR2 antisense-induced neuronal death
To examine whether cell death induced by multiple injections of GluR2 antisense was mediated by Ca2+-permeable AMPA receptors, we administered glutamate receptor antagonists at the time of the second or fourth injection or at 24 hr after the fourth injection (Fig.5, bottom). As a control for the injection of antagonists, saline was injected at 12 hr into a group of antisense-treated animals. Cell counts were performed on animals killed at 7 d after the last injection and normalized to counts in untreated animals defined as 100%. Naspm, an open channel blocker selective for Ca2+-permeable, GluR2-lacking AMPA receptors (Koike et al., 1997), greatly reduced antisense-induced neurodegeneration in CA1, i.e., increased cell survival, when given at the time of the second or fourth injection of antisense (Fig. 5). Rather than the ∼50% cell loss observed in antisense-injected animals (saline plus antisense in Fig. 5; see Fig. 2for antisense alone), there was only ∼10% cell loss in the protected animals (p < 0.05 for each of the two times of injection compared with antisense plus saline, i.e., significant protection, or with untreated, i.e., still significant residual cell loss; n = 5 for each group). CNQX, a competitive blocker of AMPA/kainate receptors injected at the time of the second or the fourth antisense injection, also restricted cell loss in CA1 to only ∼10% (Fig. 5; p < 0.05 for each time of injection compared with the antisense plus saline control andp < 0.01 compared with untreated; n = 5 for each group). These data suggest that GluR2 antisense-induced cell death is mediated by Ca2+-permeable, GluR2-lacking AMPA receptors. Naspm and CNQX injected 24 hr after the last antisense injection did not have a significant protective effect, although analysis of a larger sample might establish a small degree of protection. The failure of protection at 24 hr suggests that by this time many neurons had had sufficient Ca2+influx through GluR2-lacking receptors to initiate the processes of dying, although there were no obvious morphological changes at this time after antisense injection alone (compare Fig. 2). The role of AMPA receptors was confirmed by the observation that AP-5, a competitive NMDA receptor antagonist, administered at any of the three times during or after antisense administration, failed to afford neuroprotection against antisense-induced cell death at any of the times examined. Similar effects of the glutamate antagonists were observed in CA3 and on the left side contralateral to the injection site, but these results were not quantified.
Fig. 5.
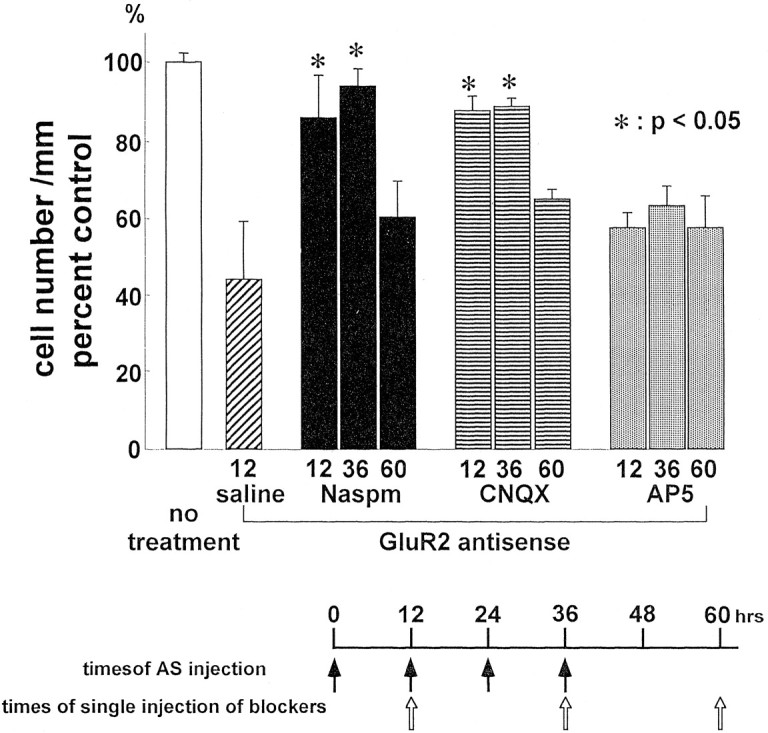
AMPA antagonists reduce antisense-induced neurodegeneration. Naspm and CNQX were injected into the right cerebral ventricle concomitantly with either the second or fourth of four injections at 12 hr intervals of GluR2 antisense AS2 (i.e., at 12 or 36 hr after the first injection) or 24 hr after the last injection (at 60 hr after the first injection; see bottom for timing). Animals were killed at 7 d after the last antisense injection. Cell counts are normalized to the value for untreated rats defined as 100% (n = 5 for each treatment and time point). Naspm and CNQX largely prevented antisense-induced delayed neurodegeneration in CA1 (p < 0.05;n = 5 for each antagonist) compared with saline injection at the time of the second antisense injection, which had no protective effect (compare Fig. 2). Protection was not complete at 12 or 36 hr (∼10% residual loss; p < 0.05 for Naspm; p < 0.01 for CNQX). Naspm or CNQX injected at 60 hr after the first AS injection afforded no significant neuroprotection. The NMDA receptor antagonist AP-5 administered at any of the three time points afforded no significant neuroprotection against GluR2 antisense-induced neurodegeneration. *p < 0.05 for antisense plus antagonist versus antisense plus saline-injected animals (labeledsaline).
GluR2 antisense injection and sublethal ischemia synergistically induce neuronal death in CA1 and CA3
To investigate the possibility that GluR2 antisense treatment and brief, sublethal ischemia summate to produce greater neuronal cell death than is observed for either treatment alone, we administered GluR2 antisense AS2 to gerbils and subjected them to a sublethal episode of forebrain ischemia by a 2 min occlusion of the carotid arteries. A single injection of antisense AS2 induced little or no neurodegeneration at 7 d after injection, and four injections produced a modest degree of degeneration of 25% or fewer of the pyramidal cells in CA1 and CA3 (data not shown). The lesser effect of multiple antisense alone injections in the gerbil compared with the rat, in spite of the higher quantity given per unit body weight (∼30 vs ∼20 nmol/kg), may reflect differences in the gerbil versus rat sequence or other species differences. In separate control experiments, a single injection or four injections of saline with or without 2 min of ischemia and four injections of scrambled antisense with or without 2 min of ischemia (n = 3 for each group) produced no neuronal death in the hippocampal CA1 or CA3 at 7 d. However, a single injection of GluR2 antisense immediately followed by a 2 min ischemic period induced pyramidal cell loss in CA1 (∼50%;p < 0.01; n = 3) but not CA3 (Figs.6D–F,7). Four antisense injections followed by 2 min of ischemia caused much greater cell loss in CA1 and CA3 (to ∼5 and ∼50%, respectively, of the values of controls injected with saline and subjected to ischemia; Figs. 6G–I, 7;p < 0.01; n = 3 for each group). Cell loss was much greater than the sum of the losses caused by each treatment alone, indicating synergistic action of the two treatments.
Fig. 7.

Quantitation of cell loss after antisense injection and sublethal ischemia. Cell counts are as described in Figure 2 in the right CA1 and CA3 of sections represented in Figure 6. Four injections of saline (salinex4) or scrambled antisense SC1 (Scx4) followed by 2 min of ischemia caused no cell loss in CA1 or CA3. A single antisense AS2 injection (ASx1) followed by 2 min of ischemia caused ∼50% cell loss in CA1 (**p < 0.01 compared with saline-injected plus ischemia; n = 3 for each group) and little if any loss in CA3. Four antisense injections (ASx4) followed by 2 min of ischemia caused nearly complete cell loss in CA1 and ∼50% loss in CA3 (**p < 0.01 for both CA1 and CA3;n = 3 for each group).
Fig. 6.

GluR2 antisense and brief, sublethal ischemia induce cell death in the hippocampus of gerbils. Toluidine blue labeling of coronal brain sections at the level of the dorsal hippocampus from gerbils at 7 d after 2 min of ischemia associated with saline (A–C) or GluR2 antisense AS2 injection (D–I) are shown. Top row, Low magnification view of the right hippocampus is shown.Middle, bottom rows, Higher magnification views of CA1 and CA3, respectively, are shown. A–C, Two minutes of ischemia immediately after four injections of saline produced no neuronal death in any hippocampal region. D–F, A single GluR2 antisense injection immediately followed by 2 min of ischemia produced partial cell loss in the pyramidal cell layers of CA1 and CA3.G–I, Four injections of antisense at 12 hr intervals immediately followed by 2 min of ischemia produced virtually complete loss of CA1 pyramidal cells and partial loss of CA3 pyramidal cells. Scale bars: A, D, G, 100 μm; B, C, E, F, H, I, 50 μm.
DISCUSSION
Antisense-induced neuronal death
Considerable evidence indicates that Ca2+-permeable AMPA receptors are critical mediators of ischemia-induced delayed neuronal death (Bennett et al., 1996; Pellegrini-Giampietro et al., 1997). Transient forebrain ischemia induces downregulation of GluR2 mRNA and protein expression and enhanced AMPA-elicited Ca2+ influx into vulnerable CA1 neurons before the onset of neurodegeneration. AMPA antagonists protect the neurons but do not prevent downregulation of GluR2. Suppression of GluR2 expression would be expected to lead to increased formation of Ca2+-permeable AMPA receptors and excessive entry of Ca2+(and/or Zn2+) in response to endogenous glutamate (the GluR2 hypothesis of delayed neurodegeneration). Ca2+-permeable AMPA receptors are also implicated in the delayed neurodegeneration associated with kainate-induced status epilepticus (Friedman et al., 1994, 1997). In that experimental model, CA3 pyramidal neurons are more vulnerable to degeneration, and GluR2 mRNA and protein expression are selectively suppressed in them before they die.
The present study examined the consequences of intraventricular administration of antisense oligonucleotides targeting GluR2 mRNA for neuronal survival in the intact rat and gerbil. We sought to determine whether a reduction in the level of GluR2 is in itself sufficient to cause neuronal death in the absence of other changes caused by neurological insults such as global ischemia and status epilepticus. We demonstrated that GluR2 knockdown causes degeneration of pyramidal cells in CA1 of the hippocampus (as occurs in global ischemia) and in CA3 (as occurs in status epilepticus). We do not exclude that the other changes in gene expression after global ischemia and status epilepticus modulate the cell death caused by downregulation of GluR2; some of these changes may be neuroprotective, e.g., upregulation of a Zn2+ transporter (Tsuda et al., 1997), whereas others may potentiate death.
To analyze molecular mechanisms underlying neurodegeneration caused by GluR2 antisense, we examined the effects of GluR2 knockdown on the levels of GluR2 mRNA by in situ hybridization and of GluR2 protein by Western blotting. Antisense treatment reduced GluR2 mRNA in the hippocampal CA1 and CA3, regions that undergo degeneration, and in the dentate gyrus, a region resistant to cell death (see below). GluR2 mRNA levels in the hypothalamus and piriform cortex were unaffected, presumably because antisense oligonucleotide administered into the right ventricle failed to reach these structures. Naspm and CNQX protected against antisense-induced death. Thus, GluR2 knockdown in vulnerable neurons is the most likely proximate cause of antisense-induced death.
The degree and time course of GluR2 mRNA and protein suppression
The level and duration of knockdown required for neuronal death are issues that will require further investigation. In the present study, GluR2 knockdown caused ∼30% reduction in GluR2 mRNA and protein in CA1, whereas global ischemia led to ∼70% reduction in CA1 (Pellegrini-Giampietro et al., 1992a; Gorter et al., 1997), and status epilepticus caused ∼40% reduction in CA3 (Friedman et al., 1994). Maximal cell loss was ∼50% after antisense treatment and close to 100% after global ischemia. After status epilepticus, cells were lost entirely in some regions and survived in neighboring regions where GluR2 recovered.
As would be expected, a reduction in GluR2 mRNA caused by antisense preceded a reduction in the level of GluR2 protein. GluR2 mRNA was maximally reduced at 6 hr after the last of four antisense injections. The GluR2 protein level was maximally reduced 6 hr later or 12 hr after the last injection, but the reduction was not greater than that at 12 hr after a single injection. Neuronal death was not produced by a single injection and was first observed 48 hr after the last of the four injections. Thus, a period of 36–48 hr of GluR2 protein suppression is required to trigger neuronal cell death, consistent with the time course in rats of delayed ischemia-induced neuronal death in CA1 (Pellegrini-Giampietro et al., 1992a) and of status epilepticus-induced cell death in CA3 (Friedman et al., 1994). In both paradigms, histopathological changes are detected 48–72 hr after the insult (Nadler, 1981; Pulsinelli et al., 1982; Ben-Ari, 1985) or 36–48 hr after downregulation of GluR2 mRNA (Pellegrini-Giampietro et al., 1992a; Friedman et al., 1994).
The synergistic action of GluR2 antisense and sublethal global ischemia
In the present study we also showed that GluR2 knockdown and sublethal global ischemia act synergistically to cause degeneration of CA1 and CA3 pyramidal neurons. This observation is consistent with a mechanism by which depression of GluR2 levels by antisense and by sublethal ischemia summate to cause neuronal death. Although brief (10 min) global ischemia in rats does not cause neuronal death in CA3, it does suppress GluR2 mRNA expression in this region to a lesser degree than in CA1 (Pellegrini-Giampietro et al., 1992a). Sublethal ischemia would be also expected to cause some suppression of GluR2 expression. Thus, the synergistic action of antisense and sublethal ischemia may reflect a nonlinear response to GluR2 suppression by the two treatments.
Gerbils subjected to sublethal transient global ischemia exhibit reduced hippocampal CA1 neuronal death after a more severe ischemic insult 24–48 hr later, a phenomenon termed “ischemic tolerance” (Heurteaux et al., 1995). Although molecular mechanisms underlying ischemic tolerance are unclear, sublethal global ischemia is known to prevent GluR2 downregulation after a subsequent more severe ischemic insult (Heurteaux et al., 1995). This finding suggests that the protection afforded by ischemic preconditioning involves a step in the cascade of events “upstream” from GluR2 gene expression. If so, one would predict that ischemic preconditioning would not protect against subsequent GluR2 antisense-induced neuronal death. Similarly, a single antisense injection would be unlikely to affect upstream processes and therefore would not protect against ischemia-induced damage.
Neuronal survival and antisense and transgenic methods to suppress GluR2
In recent years, antisense treatment has been used to study the role of specific receptor subtypes both in vivo and in vitro (for review, see Wahlestedt, 1994; Weiss et al., 1997). In intact animals, antisense has been used to knockdown the μ opioid receptor, the 5-HT-3 receptor, and the NMDA receptor (Wahlestedt et al., 1993; Weiss et al., 1993). In the present study, GluR2 antisense was used to bypass the mechanisms underlying downregulation of GluR2 induced by ischemia or status epilepticus and to ask whether GluR2 downregulation is sufficient to kill neurons. Acute knockdown of GluR2 expression by antisense administration offers an important advantage over targeted disruption or gene knockout in that it permits examination of acute reduction in GluR2 levels in an animal that has developed under normal conditions. The GluR2(−/−) mouse has a normal complement of pyramidal neurons in CA1 and CA3, although the Ca2+ permeability of AMPA receptors is high because of the absence of GluR2 (Jia et al., 1996). This mouse is viable and does not exhibit seizures. Development of the brain in the absence of GluR2 (or of its correctly edited form) may induce compensatory changes, such as enhanced expression of Ca2+-binding proteins and/or transporters. The viability of hippocampal neurons in the GluR2 knockout mouse and of GABAergic interneurons, which normally express GluR2-lacking receptors (Bochet et al., 1994; Jonas et al., 1994; Geiger et al., 1995), indicates that a rapid decrease in the level of GluR2 and a rapid increase in AMPA receptor permeability to Ca2+ may be necessary to induce death of pyramidal cells. Other neurons, such as the granule cells of the DG, are resistant to acute (but partial) GluR2 knockdown, and levels of GluR2 protein may be maintained. Moreover, the Ca2+-binding protein calbindin is found in granule cells and may contribute to their lesser sensitivity to GluR2 knockdown (Freund et al., 1990).
Mice with editing-deficient GluR2 alleles show defects depending on the expression level of the allele (Brusa et al., 1995; Feldmeyer et al., 1999). Homozygotes with an allele having a low level of expression are comparable with the GluR2(−/−) animal, whereas heterozygotes carrying an allele with a high level of expression exhibit epileptic seizures and premature death. Higher levels of editing-deficient GluR2 lead to larger AMPA currents, in addition to the increased Ca2+ permeability of these receptors. Editing-deficient alleles may also be more deleterious than knockout because of the role of the GluR2 subunit in channel assembly and targeting (Song et al., 1998; Sheng and Pak, 1999).
Technical problems inherent in the antisense approach
Several considerations support the specificity of targeting and the lack of toxicity of the GluR2 antisense oligonucleotides used in the present study: (1) The two specific oligonucleotides would not be expected to bind to any other known rodent sequence. (2) These oligonucleotides caused delayed degeneration of pyramidal neurons in CA1 and CA3 but not of granule cells in the dentate gyrus. (3) Western blot analyses indicated that the one oligonucleotide tested suppressed GluR2, but not GluR1, protein before degeneration. (4) A scrambled version of one oligonucleotide had no effect on cell viability.
Oligonucleotides in their unmodified form are phosphodiesters and are subject to rapid degradation by nucleases in blood and CSF. To enhance stability and minimize toxicity of the GluR2 antisense oligonucleotides used in this study, we introduced phosphorothioate groups at three positions at both the 5′ and 3′ ends (Campbell et al., 1990; Agrawal, 1996). In situ hybridization and Western studies indicated the effectiveness of our GluR2 antisense oligonucleotides in that GluR2 mRNA was suppressed for 24 hr and GluR2 protein for somewhat longer. Diffusion was extensive, because antisense actions extended to CA3 and were bilateral. Others have reported penetration of phosphothiorated oligonucleotides (but not of unmodified oligonucleotides) deep into the brain parenchyma after intraventricular injection (Yaida and Nowak, 1995; Gyurko et al., 1997).
Role of Ca2+-permeable AMPA receptors in delayed neurodegeneration
Downregulation of GluR2 leading to formation of Ca2+-permeable AMPA receptors has been implicated in the pathogenesis associated with delayed selective neuronal death after global ischemia and kainate-induced status epilepticus (Bennett et al., 1996; Pellegrini-Giampietro et al., 1997). The mechanisms underlying downregulation of GluR2 expression in these paradigms remain to be determined; presumably, they are part of the physiological controls under normal physiological conditions. GluR2 levels are low in early postnatal development (Pellegrini-Giampietro et al., 1992b), and GluR2 downregulation may represent a return to an earlier developmental state. Perhaps such a change would be advantageous in promoting neuronal regrowth but is clearly deleterious after global ischemia or sustained seizures.
The present study indicates that a reduction in the level of GluR2 is sufficient to cause neuronal death and provide new evidence that Ca2+-permeable AMPA receptors are critical mediators of delayed neurodegeneration.
Footnotes
This work was supported by National Institutes of Health Grants NS 20752 and NS 31282 to R.S.Z. and NS 007412 to M.V.L.B. M.V.L.B. is the Sylvia and Robert S. Olnick Professor of Neuroscience. We thank Dr. Thoralf Opitz for his helpful comments.
Correspondence should be addressed to Dr. Michael V. L. Bennett, Department of Neuroscience, Albert Einstein College of Medicine, 1300 Morris Park Avenue, Bronx, NY 10461. E-mail: mbennett@aecom.yu.edu.
REFERENCES
- 1.Agrawal S. Antisense oligonucleotides: towards clinical trials. Trends Biotechnol. 1996;14:376–387. doi: 10.1016/0167-7799(96)10053-6. [DOI] [PubMed] [Google Scholar]
- 2.Ben-Ari Y. Limbic seizure and brain damage produced by kainic acid: mechanisms and relevance to human temporal lobe epilepsy. Neuroscience. 1985;14:375–403. doi: 10.1016/0306-4522(85)90299-4. [DOI] [PubMed] [Google Scholar]
- 3.Bennett MV, Pellegrini-Giampietro DE, Gorter JA, Aronica E, Connor JA, Zukin RS. The GluR2 hypothesis: Ca(++)-permeable AMPA receptors in delayed neurodegeneration. Cold Spring Harb Symp Quant Biol. 1996;61:373–384. [PubMed] [Google Scholar]
- 4.Bochet P, Audinat E, Lambolez B, Crepel F, Rossier J, Iino M, Tsuzuki K, Ozawa S. Subunit composition at the single-cell level explains functional properties of a glutamate-gated channel. Neuron. 1994;12:383–388. doi: 10.1016/0896-6273(94)90279-8. [DOI] [PubMed] [Google Scholar]
- 5.Bowie D, Mayer ML. Inward rectification of both AMPA and kainate subtype glutamate receptors generated by polyamine-mediated ion channel block. Neuron. 1995;15:453–462. doi: 10.1016/0896-6273(95)90049-7. [DOI] [PubMed] [Google Scholar]
- 6.Brusa R, Zimmermann F, Koh DS, Feldmeyer D, Gass P, Seeburg PH, Sprengel R. Early-onset epilepsy and postnatal lethality associated with an editing-deficient GluR-B allele in mice. Science. 1995;270:1677–1680. doi: 10.1126/science.270.5242.1677. [DOI] [PubMed] [Google Scholar]
- 7.Burnashev N, Monyer H, Seeburg PH, Sakmann B. Divalent ion permeability of AMPA receptor channels is dominated by the edited form of a single subunit. Neuron. 1992;8:189–198. doi: 10.1016/0896-6273(92)90120-3. [DOI] [PubMed] [Google Scholar]
- 8.Campbell JM, Bacon TA, Wickstrom E. Oligodeoxynucleoside phosphorothioate stability in subcellular extracts, culture media, sera and cerebrospinal fluid. J Biochem Biophys Methods. 1990;20:259–267. doi: 10.1016/0165-022x(90)90084-p. [DOI] [PubMed] [Google Scholar]
- 9.Chen J, Nagayama T, Jin K, Stetler RA, Zhu RL, Graham SH, Simon RP. Induction of caspase-3-like protease may mediate delayed neuronal death in the hippocampus after transient cerebral ischemia. J Neurosci. 1998;18:4914–4928. doi: 10.1523/JNEUROSCI.18-13-04914.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Choi DW. Calcium: still center-stage in hypoxic-ischemic neuronal death. Trends Neurosci. 1995;18:58–60. [PubMed] [Google Scholar]
- 11.Colbourne F, Sutherland GR, Auer RN. Electron microscopic evidence against apoptosis as the mechanism of neuronal death in global ischemia. J Neurosci. 1999;19:4200–4210. doi: 10.1523/JNEUROSCI.19-11-04200.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Feldmeyer D, Kask K, Brusa R, Kornau HC, Kolhekar R, Rozov A, Burnashev N, Jensen V, Hvalby O, Sprengel R, Seeburg PH. Neurological dysfunctions in mice expressing different levels of the Q/R site-unedited AMPAR subunit GluR-B. Nat Neurosci. 1999;2:57–64. doi: 10.1038/4561. [DOI] [PubMed] [Google Scholar]
- 13.Freund TF, Buzsaki G, Leon A, Baimbridge KG, Somogyi P. Relationship of neuronal vulnerability and calcium binding protein immunoreactivity in ischemia. Exp Brain Res. 1990;83:55–66. doi: 10.1007/BF00232193. [DOI] [PubMed] [Google Scholar]
- 14.Friedman LK, Pellegrini-Giampietro DE, Sperber EF, Bennett MV, Moshe SL, Zukin RS. Kainate-induced status epilepticus alters glutamate and GABAA receptor gene expression in adult rat hippocampus: an in situ hybridization study. J Neurosci. 1994;14:2697–2707. doi: 10.1523/JNEUROSCI.14-05-02697.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Friedman LK, Sperber EF, Moshe SL, Bennett MV, Zukin RS. Developmental regulation of glutamate and GABA(A) receptor gene expression in rat hippocampus following kainate-induced status epilepticus. Dev Neurosci. 1997;19:529–542. doi: 10.1159/000111257. [DOI] [PubMed] [Google Scholar]
- 16.Geiger JR, Melcher T, Koh DS, Sakmann B, Seeburg PH, Jonas P, Monyer H. Relative abundance of subunit mRNAs determines gating and Ca2+ permeability of AMPA receptors in principal neurons and interneurons in rat CNS. Neuron. 1995;15:193–204. doi: 10.1016/0896-6273(95)90076-4. [DOI] [PubMed] [Google Scholar]
- 17.Gorter JA, Petrozzino JJ, Aronica EM, Rosenbaum DM, Opitz T, Bennett MV, Connor JA, Zukin RS. Global ischemia induces downregulation of GluR2 mRNA and increases AMPA receptor-mediated Ca2+ influx in hippocampal CA1 neurons of gerbil. J Neurosci. 1997;17:6179–6188. doi: 10.1523/JNEUROSCI.17-16-06179.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gyurko R, Tran D, Phillips MI. Time course of inhibition of hypertension by antisense oligonucleotides targeted to AT1 angiotensin receptor mRNA in spontaneously hypertensive rats. Am J Hypertens. 1997;10:56S–62S. [PubMed] [Google Scholar]
- 19.Heurteaux C, Lauritzen I, Widmann C, Lazdunski M. Essential role of adenosine, adenosine A1 receptors, and ATP-sensitive K+ channels in cerebral ischemic preconditioning. Proc Natl Acad Sci USA. 1995;92:4666–4670. doi: 10.1073/pnas.92.10.4666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hollmann M, Hartley M, Heinemann S. Ca2+ permeability of KA-AMPA–gated glutamate receptor channels depends on subunit composition. Science. 1991;252:851–853. doi: 10.1126/science.1709304. [DOI] [PubMed] [Google Scholar]
- 21.Hsu M, Buzsaki G. Vulnerability of mossy fiber targets in the rat hippocampus to forebrain ischemia. J Neurosci. 1993;13:3964–3979. doi: 10.1523/JNEUROSCI.13-09-03964.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jia Z, Agopyan N, Miu P, Xiong Z, Henderson J, Gerlai R, Taverna FA, Velumian A, MacDonald J, Carlen P, Abramow-Newerly W, Roder J. Enhanced LTP in mice deficient in the AMPA receptor GluR2. Neuron. 1996;17:945–956. doi: 10.1016/s0896-6273(00)80225-1. [DOI] [PubMed] [Google Scholar]
- 23.Jonas P, Racca C, Sakmann B, Seeburg PH, Monyer H. Differences in Ca2+ permeability of AMPA-type glutamate receptor channels in neocortical neurons caused by differential GluR-B subunit expression. Neuron. 1994;12:1281–1289. doi: 10.1016/0896-6273(94)90444-8. [DOI] [PubMed] [Google Scholar]
- 24.Kirino T. Delayed neuronal death in the gerbil hippocampus following ischemia. Brain Res. 1982;239:57–69. doi: 10.1016/0006-8993(82)90833-2. [DOI] [PubMed] [Google Scholar]
- 25.Koh JY, Suh SW, Gwag BJ, He YY, Hsu CY, Choi DW. The role of zinc in selective neuronal death after transient global cerebral ischemia. Science. 1996;272:1013–1016. doi: 10.1126/science.272.5264.1013. [DOI] [PubMed] [Google Scholar]
- 26.Koike M, Iino M, Ozawa S. Blocking effect of 1-naphthyl acetyl spermine on Ca(2+)-permeable AMPA receptors in cultured rat hippocampal neurons. Neurosci Res. 1997;29:27–36. doi: 10.1016/s0168-0102(97)00067-9. [DOI] [PubMed] [Google Scholar]
- 27.Nadler JV. Minireview. Kainic acid as a tool for the study of temporal lobe epilepsy. Life Sci. 1981;29:2031–2042. doi: 10.1016/0024-3205(81)90659-7. [DOI] [PubMed] [Google Scholar]
- 28.Nurse S, Corbett D. Neuroprotection after several days of mild, drug-induced hypothermia. J Cereb Blood Flow Metab. 1996;16:474–480. doi: 10.1097/00004647-199605000-00014. [DOI] [PubMed] [Google Scholar]
- 29.Oguro K, Nakamura M, Masuzawa T. Histochemical study of Ca(2+)-ATPase activity in ischemic CA1 pyramidal neurons in the gerbil hippocampus. Acta Neuropathol (Berl) 1995;90:448–453. doi: 10.1007/BF00294804. [DOI] [PubMed] [Google Scholar]
- 30. Pellegrini-Giampietro DE, Zukin RS, Bennett MV, Cho S, Pulsinelli WA. Switch in glutamate receptor subunit gene expression in CA1 subfield of hippocampus following global ischemia in rats. Proc Natl Acad Sci USA 89 1992a. 10499 10503[Erratum(1993)90:780]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Pellegrini-Giampietro DE, Bennett MV, Zukin RS. Are Ca(2+)-permeable kainate/AMPA receptors more abundant in immature brain? Neurosci Lett. 1992b;144:65–69. doi: 10.1016/0304-3940(92)90717-l. [DOI] [PubMed] [Google Scholar]
- 32.Pellegrini-Giampietro DE, Gorter JA, Bennett MV, Zukin RS. The GluR2 (GluR-B) hypothesis: Ca(2+)-permeable AMPA receptors in neurological disorders. Trends Neurosci. 1997;20:464–470. doi: 10.1016/s0166-2236(97)01100-4. [DOI] [PubMed] [Google Scholar]
- 33.Pulsinelli W, Sarokin A, Buchan A. Antagonism of the NMDA and non-NMDA receptors in global versus focal brain ischemia. Prog Brain Res. 1993;96:125–135. doi: 10.1016/s0079-6123(08)63262-8. [DOI] [PubMed] [Google Scholar]
- 34.Pulsinelli WA, Brierley JB, Plum F. Temporal profile of neuronal damage in a model of transient forebrain ischemia. Ann Neurol. 1982;11:491–498. doi: 10.1002/ana.410110509. [DOI] [PubMed] [Google Scholar]
- 35.Schmidt-Kastner R, Freund TF. Selective vulnerability of the hippocampus in brain ischemia. Neuroscience. 1991;40:599–636. doi: 10.1016/0306-4522(91)90001-5. [DOI] [PubMed] [Google Scholar]
- 36.Sheardown MJ, Suzdak PD, Nordholm L. AMPA, but not NMDA, receptor antagonism is neuroprotective in gerbil global ischaemia, even when delayed 24 h. Eur J Pharmacol. 1993;236:347–353. doi: 10.1016/0014-2999(93)90470-3. [DOI] [PubMed] [Google Scholar]
- 37.Sheng M, Pak DT. Glutamate receptor anchoring proteins and the molecular organization of excitatory synapses. Ann NY Acad Sci. 1999;868:483–493. doi: 10.1111/j.1749-6632.1999.tb11317.x. [DOI] [PubMed] [Google Scholar]
- 38.Silver IA, Erecinska M. Ion homeostasis in rat brain in vivo: intra- and extracellular [Ca2+] and [H+] in the hippocampus during recovery from short-term, transient ischemia. J Cereb Blood Flow Metab. 1992;12:759–772. doi: 10.1038/jcbfm.1992.107. [DOI] [PubMed] [Google Scholar]
- 39.Song I, Kamboj S, Xia J, Dong H, Liao D, Huganir RL. Interaction of the N-ethylmaleimide-sensitive factor with AMPA receptors. Neuron. 1998;21:393–400. doi: 10.1016/s0896-6273(00)80548-6. [DOI] [PubMed] [Google Scholar]
- 40.Takei N, Endo Y. Ca2+ ionophore-induced apoptosis on cultured embryonic rat cortical neurons. Brain Res. 1994;652:65–70. doi: 10.1016/0006-8993(94)90317-4. [DOI] [PubMed] [Google Scholar]
- 41.Tsubokawa H, Oguro K, Masuzawa T, Kawai N. Ca(2+)-dependent non-NMDA receptor-mediated synaptic currents in ischemic CA1 hippocampal neurons. J Neurophysiol. 1994;71:1190–1196. doi: 10.1152/jn.1994.71.3.1190. [DOI] [PubMed] [Google Scholar]
- 42.Tsubokawa H, Oguro K, Masuzawa T, Nakaima T, Kawai N. Effects of a spider toxin and its analogue on glutamate-activated currents in the hippocampal CA1 neuron after ischemia. J Neurophysiol. 1995;74:218–225. doi: 10.1152/jn.1995.74.1.218. [DOI] [PubMed] [Google Scholar]
- 43.Tsuda M, Imaizumi K, Katayama T, Kitagawa K, Wanaka A, Tohyama M, Takagi T. Expression of zinc transporter gene, ZnT-1, is induced after transient forebrain ischemia in the gerbil. J Neurosci. 1997;17:6678–6684. doi: 10.1523/JNEUROSCI.17-17-06678.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Verdoorn TA, Burnashev N, Monyer H, Seeburg PH, Sakmann B. Structural determinants of ion flow through recombinant glutamate receptor channels. Science. 1991;252:1715–1718. doi: 10.1126/science.1710829. [DOI] [PubMed] [Google Scholar]
- 45.Vissavajjhala P, Janssen WG, Hu Y, Gazzaley AH, Moran T, Hof PR, Morrison JH. Synaptic distribution of the AMPA-GluR2 subunit and its colocalization with calcium-binding proteins in rat cerebral cortex: an immunohistochemical study using a GluR2-specific monoclonal antibody. Exp Neurol. 1996;142:296–312. doi: 10.1006/exnr.1996.0199. [DOI] [PubMed] [Google Scholar]
- 46.Wahlestedt C. Antisense oligonucleotide strategies in neuropharmacology. Trends Pharmacol Sci. 1994;15:42–46. doi: 10.1016/0165-6147(94)90107-4. [DOI] [PubMed] [Google Scholar]
- 47.Wahlestedt C, Golanov E, Yamamoto S, Yee F, Ericson H, Yoo H, Inturrisi CE, Reis DJ. Antisense oligodeoxynucleotides to NMDA-R1 receptor channel protect cortical neurons from excitotoxicity and reduce focal ischaemic infarctions. Nature. 1993;363:260–263. doi: 10.1038/363260a0. [DOI] [PubMed] [Google Scholar]
- 48.Washburn MS, Numberger M, Zhang S, Dingledine R. Differential dependence on GluR2 expression of three characteristic features of AMPA receptors. J Neurosci. 1997;17:9393–9406. doi: 10.1523/JNEUROSCI.17-24-09393.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Weiss B, Zhou LW, Zhang SP, Qin ZH. Antisense oligodeoxynucleotide inhibits D2 dopamine receptor-mediated behavior and D2 messenger RNA. Neuroscience. 1993;55:607–612. doi: 10.1016/0306-4522(93)90426-g. [DOI] [PubMed] [Google Scholar]
- 50.Weiss B, Davidkova G, Zhang SP. Antisense strategies in neurobiology. Neurochem Int. 1997;31:321–348. doi: 10.1016/s0197-0186(96)00105-2. [DOI] [PubMed] [Google Scholar]
- 51.Wenthold RJ, Yokotani N, Doi K, Wada K. Immunochemical characterization of the non-NMDA glutamate receptor using subunit-specific antibodies. Evidence for a hetero-oligomeric structure in rat brain. J Biol Chem. 1992;267:501–507. [PubMed] [Google Scholar]
- 52.Yaida Y, Nowak TS., Jr Distribution of phosphodiester and phosphorothioate oligonucleotides in rat brain after intraventricular and intrahippocampal administration determined by in situ hybridization. Regul Pept. 1995;59:193–199. doi: 10.1016/0167-0115(95)00100-p. [DOI] [PubMed] [Google Scholar]