

Abstract
Synapse-specific expression of the nicotinic acetylcholine receptor (AChR) is believed to be mediated by neuregulin, an epidermal growth factor-like trophic factor released by somatic motoneurons at the neuromuscular junction (NMJ). Neuregulin stimulates ErbB2, ErbB3, and ErbB4, members of the ErbB family of receptor tyrosine kinases. SHP2 is a cytoplasmic protein tyrosine phosphatase containing two Src homology 2 domains near its N terminus, and has been shown to be a positive mediator of mitogenic responses to various growth factors. We found that SHP2 interacted with ErbB2 and ErbB3 after neuregulin stimulation of muscle cells. Expression of SHP2 in C2C12 mouse muscle cells attenuated the neuregulin-induced expression of an AChR ε-promoter reporter gene, whereas a catalytically inactive SHP2 mutant or a mutant lacking the N-terminal Src homology 2 (SH2) domain enhanced reporter expression, suggesting that SHP2 negatively regulates the neuregulin signaling pathway. In fibroblast cells that express a mutant SHP2 with a targeted deletion of the N-terminal SH2 domain, neuregulin-mediated activation of the Ras/Raf/extracellular signal-regulated kinase cascade was enhanced. Furthermore, we found that SHP2 immunoreactivity colocalized with the staining of α-bungarotoxin, a marker of the NMJ. These results demonstrate a negative role of SHP2 in the neuregulin signal that leads to AChR gene expression at the NMJ.
Keywords: neuregulin, ErbB, SHP2, AChR, neuromuscular junction, tyrosine phosphatase
The transcription of genes encoding AChR subunits (α, β, γ, or ε, and δ) is highest in nuclei localized to the synaptic regions of muscle, thereby contributing to the high density of AChRs at the postjunctional membrane. Such synapse-specific transcription is believed to be mediated by neuregulin, a trophic factor used by motoneurons to stimulate AChR synthesis (Jessell et al., 1979; Falls et al., 1993; Loeb and Fischbach, 1995). Neuregulin (also known as neu differentiation factor, glial growth factor, and acetylcholine receptor inducing activity) (Lemmon and Sclessinger, 1994) activates ErbB2, ErbB3, and ErbB4, members of the ErbB family of protein tyrosine kinases (Chu et al., 1995; Jo et al., 1995; Zhu et al., 1995; Si et al., 1996). Both neuregulin and ErbB proteins are localized to the adult neuromuscular junction (NMJ) (Altiok et al., 1995; Moscoso et al., 1995; Zhu et al., 1995), thus fulfilling a spatial requirement for synapse-specific signal transduction. Mice that are heterozygous for one neuregulin allele show deficiencies in neuromuscular transmission and have 50% fewer AChRs at the NMJ (Sandrock et al., 1997), although homozygous mice die before NMJ formation because of developmental defects of the heart (Meyer and Birchmeier, 1995). Current evidence therefore supports neuregulin as the best candidate for the primary signal of synapse-specific transcription.
The neuregulin signaling pathway that leads to AChR gene expression has become increasingly clear. Neuregulin stimulates ErbB tyrosine kinases and subsequently the Ras/Raf/MAP kinase signaling cascade (Marte et al., 1995; Si et al., 1996). Both Ras and Mek are required for neuregulin-stimulated induction of AChR genes (Si et al., 1996; Tansey et al., 1996; Altiok et al., 1997). Several studies have implicated Ets proteins, known targets of the MAP kinase pathway (Marais et al., 1993;Brunner et al., 1994; O'Hagan and Hassell, 1999), as components of the activated transcriptional machinery. In the δ-subunit promoter, a potential Ets-protein-binding site was identified as a neuregulin-response element (NRE) (Fromm and Burden, 1998), and GABPα, an Ets protein, and GABPβ, a dimerization partner, were implicated as NRE-binding proteins. Unlike the other AChR subunits, expression of the ε subunit is restricted to the synapse at all stages of development and is therefore the subject of particular attention. Ets-binding sites in the AChR ε 5′flanking region also appear to be necessary for synapse-specific expression (Duclert et al., 1996) and neuregulin responsiveness (Sapru et al., 1998). We have reported the requirement of a different non-Ets site for neuregulin induction of the mouse AChR ε gene in cultured muscle cells (Si et al., 1997).
In contrast to the extensive study of downstream mechanisms contributing to synapse-specific expression, less attention has focused on upstream mechanisms that may regulate neuregulin signaling, perhaps because of a presumed similarity to the epidermal growth factor (EGF) pathway. One signaling molecule known to modulate a large number of receptor-tyrosine kinase signaling pathways is SHP2. SHP2 (PTP1D, SHPTP2, or Syp) is a widely expressed cytoplasmic protein tyrosine phosphatase containing two Src homology 2 domains near its N terminus (Feng and Pawson, 1994). SHP2 associates with numerous receptor tyrosine kinases in a stimulation-dependent manner and acts as a positive regulator of mitogenic responses to EGF, insulin, and insulin growth factor (IGF)-1 (Feng et al., 1993; Xiao et al., 1994;Bennett et al., 1996). Although the precise mechanism by which SHP2 transduces positive signals is unknown, numerous studies have implicated the requirement of the catalytic activity of the protein and functional Src homology 2 (SH2) domains. Expression of an enzymatically inactive SHP2 mutant inhibits extracellular signal-regulated kinase (ERK) activation in response to insulin, IGF-1, and fibroblast growth factor (Noguchi et al., 1994; Tang et al., 1995; Yamauchi et al., 1995). Cells expressing a mutant SHP2 lacking a functional N-terminal SH2 domain show attenuated ERK activation after EGF, platelet-derived growth factor (PDGF), and IGF-1 stimulation (Shi et al., 1998). The role of SHP2 in the regulation of neuregulin signaling, however, is still unclear. We report here that SHP2 association with ErbB2 and ErbB3 and tyrosine phosphorylation are increased in neuregulin-stimulated cells. In addition, SHP2 was found to be localized to the NMJ in vivo. Our results from C2C12 mouse muscle cells and mutant fibroblasts suggest that SHP2 negatively regulates neuregulin signaling.
MATERIALS AND METHODS
Materials. Recombinant neuregulin (rHRG-β1177–244, a peptide of HRGβ1 residues 177–244) was generously provided by Mark Sliwkowski (Genentech, San Francisco, CA; Holmes et al., 1992). This peptide stimulates AChR gene expression in myotubes in primary culture (Altiok et al., 1995; Chu et al., 1995; Si et al., 1996, 1997). Cell culture medium and components were purchased from Life Technologies (Gaithersburg, MD). Polyclonal antibodies to ErbB2 (C-18), ErbB3 (C-17) EGF receptor (EGFR) (SC-O3), and monoclonal anti-SHP2 antibody (B-1) were from Santa Cruz Biotechnology (Santa Cruz, CA). The anti-phosphotyrosine antibody (RC20) was from Transduction Laboratories (Lexington, KY). The E10 monoclonal anti-phospho MAP Kinase antibody was purchased from New England BioLabs (Beverly, MA). The generation of rabbit anti-ERK has been described previously (Shi et al., 1998). Rat monoclonal Y13–259 anti-Ras antibody was obtained from culture medium of a hybridoma cell line (CRL-1742; American Type Culture Collection, Manassas, VA). Anti-Raf antibody, recombinant glutathioneS-transferase-MAP/ERK kinase (GST-MEK), and recombinant GST ERK1 were kindly provided by Dr. Z. Luo (Boston University). All other chemicals were from Sigma (St. Louis, MO).
Cell lines and culture conditions. The wild-type (Shp-2+/+) and mutant (Shp-2−/−) embryonic fibroblast cell lines have been described previously (Shi et al., 1998). C2C12 cells were obtained from Dr. E. S. Ralston (National Institute of Neurological Disorders and Stroke, National Institutes of Health, Bethesda, MD), which were a subclone of the C2 cells originally derived from mouse thigh muscle (Yaffe and Saxel, 1977). The C2C12 cells were maintained as undifferentiated myoblasts in DMEM with high glucose supplemented with 20% fetal bovine serum, 0.5% chicken embryo extract, 2 mml-glutamine, 100 U/ml penicillin, and 100 μg/ml streptomycin at 37° C in an atmosphere of 5% CO2 and 95% humidity (Si et al., 1996). Cells were split at ∼70% confluence using 0.05% trypsin and 0.02% EDTA in saline. Fusion of myoblasts into myotubes was induced by culturing myoblasts for 48 hr in differentiation medium (DM), DMEM supplemented with 5% horse serum and 2 mml-glutamine. DM was changed every 24 hr to keep myotubes healthy. Under our conditions, myotube formation was complete 48 hr after switching to DM. The C2C12 myotubes were stimulated with neuregulin at a final concentration of 2 nm at 37°C. Myotubes used for reporter gene expression assays were treated for 24 hr after complete fusion.
Transfection procedures. At ∼50–70% confluence, C2C12 myoblasts plated in six well cell culture plates were transiently transfected using the calcium phosphate method. (Ausubel et al., 1994;Si et al., 1996, 1997). Myoblasts were cotransfected with an experimental plasmid DNA (3 μg per well), plus the ε-subunit promoter-luciferase transgene (1 μg of DNA per well) and a control plasmid pCMVβ (0.1 μg of DNA per well) encoding β-galactosidase. The myoblasts were incubated with the calcium phosphate precipitate for 24 hr under normal growth conditions and then switched to DM to induce myotube formation.
Luciferase and β-galactosidase assays. The luciferase assay was performed using a kit from Promega (Madison, WI) following the manufacturer's instructions. Briefly, 100 μl of cell lysate was mixed in an equal volume of luciferase substrate solution containing 20 mm tricine, 1.07 mm(MgCO3)4 Mg (OH)2 · 5 H2O, 2.67 mm MgSO4, 0.1 mm EGTA, 33.3 mm DTT, 270 μm coenzyme A, 470 μm luciferin, and 530 μm ATP and placed in a microβ luminometer (Wallac, Turku, Finland) to measure light production for 10 sec. β-galactosidase activity was determined by hydrolysis ofo-nitrophenyl-β-d-galactopyranoside (ONPG) according to published methods (Sambrook et al., 1989; Si et al., 1996). Briefly, an aliquot of cell lysate was incubated with 67 mm sodium phosphate, pH 7.5, 1 mm MgCl2, 45 mm β-mercaptoethanol, and 0.88 mg/ml ONPG in a volume of 300 μl at 37°C until a faint yellow color developed. The absorbance at 420 nm was measured using a spectrophotometer. Luciferase activity of transgenes was normalized to β-galactosidase activity to correct for variations in transfection efficiency.
Immunoprecipitations. C2C12 myotubes or wild-type and mutant mouse fibroblasts were washed two times with cold PBS and then lysed in cold lysis buffer [50 mm Tris–HCl, pH 7.5, 150 mm NaCl, 2 mm EDTA, 2 mm sodium orthovanadate, 1% NP-40, 0.25% deoxycholate, 1 μmpepstatin, 1 μg/ml leupeptin, 2 μg/ml aprotinin, and 1 mm phenylmethylsulfonyl flouride (PMSF)]. Cell lysates were incubated on ice for 30 min and centrifuged at 13,000 ×g for 10 min at 4°C to remove cell debris, after which the protein concentration of the supernatant was determined by the method of Bradford using bovine serum albumin (BSA) as a standard. For immunoprecipitation, cleared lysates were incubated with specific antibodies for 1 hr at 4°C and were then incubated with 50 μl of 50% protein A-agarose beads overnight at 4°C on a rotating platform. After centrifugation, beads were washed four or five times with lysis buffer, and bound proteins were eluted with SDS sample.
Western blot analysis. Lysates or immunocomplexes after immunoprecipitation were subjected to SDS-PAGE and transferred to nitrocellulose membranes (Schleicher & Schuell, Keene, NH). Nitrocellulose blots were incubated for 1 hr in blocking buffer [5% milk in Tris-buffered saline with 0.1% Tween (TBS-T) or 3% BSA, for anti-phosphotyrosine antibodies] at room temperature. The blots were then incubated in 1% milk or BSA with the recommended concentrations of specific primary antibodies. After washing three times for 15 min with TBS-T, the blots were incubated with horseradish peroxidase-conjugated donkey anti-mouse or anti-rabbit IgG (Amersham Pharmacia Biotech, Piscataway, NJ) followed by washing. Immunoreactive bands were visualized with enhanced chemiluminescence substrate (Pierce, Rockford, IL).
ERK kinase assay. For kinase assay, ERK kinase was precipitated with antibody and protein A-agarose beads in HO buffer (50 mm HEPES, pH 7.5, 100 mm NaCl, 2 mm EDTA, 1% NP-40, 40 mmp-nitrophenylphosphate, 1 μmpepstatin, 1 μg/ml leupeptin, 0.2 mm sodium orthovanadate, and 2 μg/ml aqprotinin). The beads were washed twice in HO buffer and twice with kinase buffer (10 mmHEPES, pH 7.4, and 10 mm magnesium acetate). The assay was performed by mixing the beads with 1 mg/ml myelin basic protein (MBP), 75 μm ATP, and 2.5 μCi [γ-32P]ATP in kinase buffer and incubated for 15 min at 30°C. After centrifugation supernatants were either subjected to SDS-PAGE followed by autoradiography or spotted onto P81 Whatman paper followed by five washes in 180 mm phosphoric acid and one wash in 100% ethanol. Air-dried P81 paper or excised gel bands were counted in scintillation cocktail. Both methods gave similar results.
Raf kinase assay. Raf kinase was immunoprecipitated with polyclonal anti-Raf antibody and protein A-agarose beads in Raf lysis buffer (in mm: 50 Tris–HCl, pH 7.8, 50 β-glycerol phosphate, 1% Triton X-100, 1 sodium orthovanadate, 1 EDTA, 1 DTT, 1 PMSF, 1 μg/ml pepstatin, 1 μg/ml leupeptin, and 2 μg/ml aprotinin). Beads were washed twice in lysis buffer, two times in lysis buffer with 1 m NaCl, and two times in kinase the buffer (in mm: 50 Tris–HCl, pH 7.8, 1 MgCl2, and 1 DTT). Kinase assays were performed by mixing the beads with 4 μg/ml recombinant GST-MEK, 100 μm ATP, and 5 μCi [γ-32P] ATP, in the kinase buffer and incubated for 20 min at 30°C. After the addition of 20 μg/ml recombinant GST-ERK1, the samples were incubated for an additional 30 min and then subjected to SDS-PAGE.
Ras assay. Active GTP-bound Ras was precipitated by the minimal Ras-binding domain (RBD) (aa 51–131) of Raf1 (Rooij and Bos, 1997). Briefly, serum-starved CT and M2 cells were treated with neuregulin (5 nm) or EGF (50 ng/ml) for indicated times and lysed in Ras-binding buffer (15% glycerol, 50 mm Tris/HCl, pH 7.4, 1% NP-40, 200 mm NaCl, 10 mmMgCl2, 1 mm PMSF, 1 μg/ml pepstatin, 1 μg/ml leupeptin, and 2 μg/ml aprotinin). Cleared lysates were incubated for 1 hr with recombinant GST-RBD (15 μg/sample) that had been precoupled to glutathione–agarose beads. After four washes in Ras buffer, bead complexes were separated on a 12.5% SDS-PAGE and transferred to nitrocellulose membrane for Western blotting. Ras was detected using the rat monoclonal antibody Y13–259 followed by HRP-coupled goat anti-rat antiserum (Santa Cruz Biotechnology). Quantification was performed by image analysis of films by scanning the film with Personal Densitometer (Molecular Dynamics, Sunnyvale, CA), and the captured image was analyzed with ImageQuant software (Molecular Dynamics). Each gel included a series of lanes loaded with increasing amounts of a known quantity of sample lysate which was later used for calibration during densitometric analysis.
Protein concentration determination. Protein concentration was measured by the Bradford method using a Coomassie protein assay reagent (Pierce) and BSA as a standard (Bradford, 1976).
Immunofluorescence. Mouse diaphragm was isolated and frozen in isopentane. Cryosections (15 μm) were then thaw-mounted onto positively charged slides and incubated in blocking solution (2% normal goat serum in PBS) for 1 hr at room temperature. Sections were incubated for 1 hr with 1:1000 dilution of polyclonal anti-SHP2 in blocking solution at room temperature. After washing in PBS, sections were incubated with FITC-conjugated secondary antibody (Zymed Laboratories, San Francisco, CA) and rhodamine-conjugated α-bungarotoxin in blocking solution for 1 hr at room temperature. Indirect immunofluorescent images were captured on 35 mm film using an automatic exposure system. Exposure times for the FITC images (SHP2) were approximately twice that used for rhodamine images (α-bungarotoxin).
RESULTS
Stimulation-dependent interaction between SHP2 and ErbB proteins in C2C12 myotubes
SHP2 is expressed in C2C12 myotubes, a cell culture system routinely used to study NMJ formation. After stimulation of myotubes with neuregulin, both ErbB2 and ErbB3, the ErbBs that are expressed in this cell line, become tyrosine phosphorylated (Si et al., 1996; S. Won and L. Mei, unpublished observations). We determined whether SHP2 interacts with the tyrosine-phosphorylated ErbB proteins. C2C12 myotubes were stimulated with 2 nm neuregulin for various times, cells lysed, and SHP2-interacting proteins were coimmunoprecipitated by a monoclonal anti-SHP2 antibody. The immunocomplex was resolved on SDS-PAGE, transferred to nitrocellulose, and probed with an anti-phosphotyrosine antibody. As shown in Figure1, several proteins copurified with SHP2, among which were two bands migrating at ∼180 kDa. Reprobing of blots with specific antibodies revealed that they were ErbB2 (∼180 kDa) and ErbB3 (∼160 kDa) (data not shown), indicating that SHP2 interacts either directly, or indirectly with both proteins. Like the tyrosine phosphorylation of ErbB proteins, this interaction was increased by neuregulin stimulation (Fig. 1A). Moreover, neuregulin increased the tyrosine phosphorylation of SHP2 (Fig.1A). A protein migrating at ∼115 kDa also copurified with SHP2 in a stimulation-dependent manner (Fig.1A). This protein appeared to be SHPS-1/SIRP, which becomes associated with SHP2 after stimulation of cells with insulin or growth hormone (Baugh et al., 1991; Stofega et al., 1998).
Fig. 1.
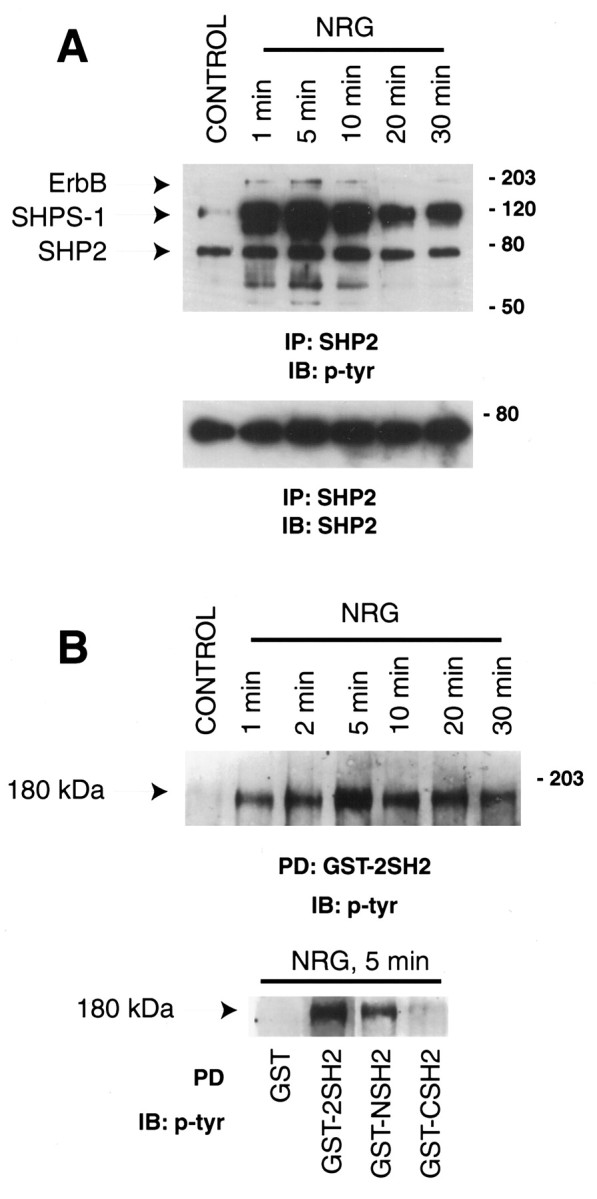
SHP2 interaction with ErbB proteins in neuregulin (NRG)-stimulated muscle cells.A, C2 myotubes were stimulated for the indicated times with 2 nm neuregulin. Cell lysates were subjected to immunoprecipitation with a SHP2 monoclonal antibody followed by SDS-PAGE and immunoblot using anti-phosphotyrosine antibody. A doublet was observed in the 180 kDa range, and subsequent reprobing demonstrated that the higher molecular weight band comigrated with erbB2, whereas the lower one comigrated with ErbB3. The 70 kDa protein that showed a stimulation-dependent increase in phosphotyrosine content was SHP2. Approximately equal amounts of SHP2 were immunoprecipitated for each time point, as indicated in the bottom panel.B, Interaction of SHP2 SH2 domains with 180 kDa proteins. C2 myotubes were stimulated with 2 nm neuregulin for 5 min and then incubated with agarose-bound GST or GST fusion proteins containing both SHP2 SH2 domains (2SH2), the N-terminal (NSH2), or the more C-terminal (CSH2) SH2 domain. Bound proteins were eluted in SDS-sample buffer followed by SDS-PAGE and immunoblot analysis using an anti-phosphotyrosine antibody. The 180 kDa proteins were determined to contain ErbB proteins based on parallel experiments with specific antibodies. No association occurred with GST alone.
SHP2 has two SH2 domains. To determine which SH2 domain interacts with ErbB proteins, we generated recombinant GST fusion proteins containing the N-terminal (GST-NSH2), the C-terminal (GST-CSH2), or both (GST-2SH2) SH2 domains. These proteins were immobilized on agarose beads and incubated with cell lysates. As shown in Figure1B, GST-2SH2 was able to pull down ∼180 kDa proteins in a neuregulin stimulation-dependent manner. The N-terminal SH2 appeared to have a higher affinity for these proteins than the C-terminal SH2 (Fig. 1B, bottom panel).
Wild-type SHP2 negatively regulates neuregulin-stimulated induction of an AChR reporter gene
SHP2 is believed to act as a positive mediator of signaling responses to various factors, including EGF. Moreover, a positive role of SHP2 has been shown for factor-induced gene expression by α/β interferon and prolactin (Ali et al., 1996; David et al., 1996). We were therefore interested in investigating the possible role of SHP2 in neuregulin-induced AChR expression in C2C12 myotubes. Cotransfection experiments were performed with various SHP2 constructs and ε416-Luc, an AChR ε-subunit 5′ flanking region that drives the expression of luciferase (Si et al., 1997). A β-galactosidase plasmid under the constitutive control of the cytomegalovirus (CMV) promoter was included in transfections to control for transfection efficiency and sample handling. Expression of the luciferase reporter increased to ∼2.5-fold after neuregulin stimulation of vector-transfected cells. As shown in Figure 2, overexpression of wild-type SHP2 reduced the level of induction to ∼70% of a vector control, although expression of a SHP2 construct coding for only the catalytic domain was without effect. Surprisingly, overexpression of a mutant SHP2 that lacks the N-terminal SH2 domain resulted in a significant increase in AChR reporter gene expression. These results suggested that SHP2 may negatively regulate neuregulin signaling. To further determine the negative role of SHP2 in the regulation of AChR gene expression, a phosphatase dead SHP2 mutant (SHP2Δ450–478) was introduced into C2C12 cells, and the effect on neuregulin-induced expression of the ε416-Luc transgene assessed. Previous studies have shown that a similar SHP2 mutant functions as a dominant negative mutant in the EGFR signaling pathway (Bennett et al., 1996). Expression of SHP2Δ450–478 also resulted in a higher fold of induction (Fig.2), as did a catalytically impaired SHP2 construct (SHP2Asp426Ala) containing an alanine substituted for a catalytically essential aspartic acid residue (data not shown) (Barford et al., 1994; Garton et al., 1996). Because the wild-type SHP2 was inhibitory, these results suggest that the N-terminal SH2 domain of SHP2 is required for the negative regulatory effect of SHP2 on neuregulin signaling.
Fig. 2.

Negative regulation of the NRG-induced expression of an AChR ε subunit reporter gene by SHP2. C2 cells were cotransfected with various SHP2 constructs and ε416-luc, which contains the ε-subunit 5′ flanking region driving the expression of luciferase. A β-galactosidase plasmid under the constitutive control of the CMV promoter was included to control for transfection efficiency and sample handling. Subsequent to myotube formation, transfectants were treated with or without 2 nm neuregulin for 24 hr. Luciferase activity was normalized to β-galactosidase activity. These data represent the mean ± SD of at least three independent experiments. Wild-type SHP2 reduced the neuregulin-induced expression to 72 ± 7% of vector control, whereas a catalytically inactive mutant (SHP2Δ450–478) and a mutant lacking the N-terminal SH2 domain (SHP2ΔNSH2) enhanced the reporter expression to 155+ 29% and 190 + 35%, respectively. Coexpression of the SHP2 catalytic domain (SHP2PTP) had a negligible effect. *p < 0.05.
Enhanced ERK activation in response to neuregulin in fibroblasts expressing a mutant SHP2
SHP2 mutant mice die early in embryonic development [embryonic day 9.5 (E9.5)] when it is almost impossible to isolate primary muscle cells for culture. To confirm the negative regulatory role of SHP2 in the neuregulin signaling pathway, we studied neuregulin and EGF signaling in mouse fibroblast cells that express a mutant SHP2 lacking the N-terminal SH2 domain (amino acids 46–110) (Saxton et al., 1997). This cell line is thought to represent a partial or nearly total loss of SHP2 function. Previous studies of these cells have shown a positive role of the SHP2 N-terminal SH2 domain in EGF signaling (Shi et al., 1998). Use of the mutant cells to study SHP2 function in neuregulin signaling precluded the necessity of overexpressing dominant negative mutants of SHP2 that might lead to nonspecific effects and, in addition, facilitated biochemical studies. The SHP2 mutant cells (M2) express a 57 kDa SHP2 protein at a reduced level compared to the full-length phosphatase expressed by the wild-type cells (CT) (Fig.3A) (Shi et al., 1998). The two cell lines express comparable amounts of ErbB2 and ErbB3 (Fig.3A). No ErbB4 was detected in either cell line. Both lines have previously been shown to express similar levels of EGFR, ERK1, and ERK2 (Shi et al., 1998).
Fig. 3.
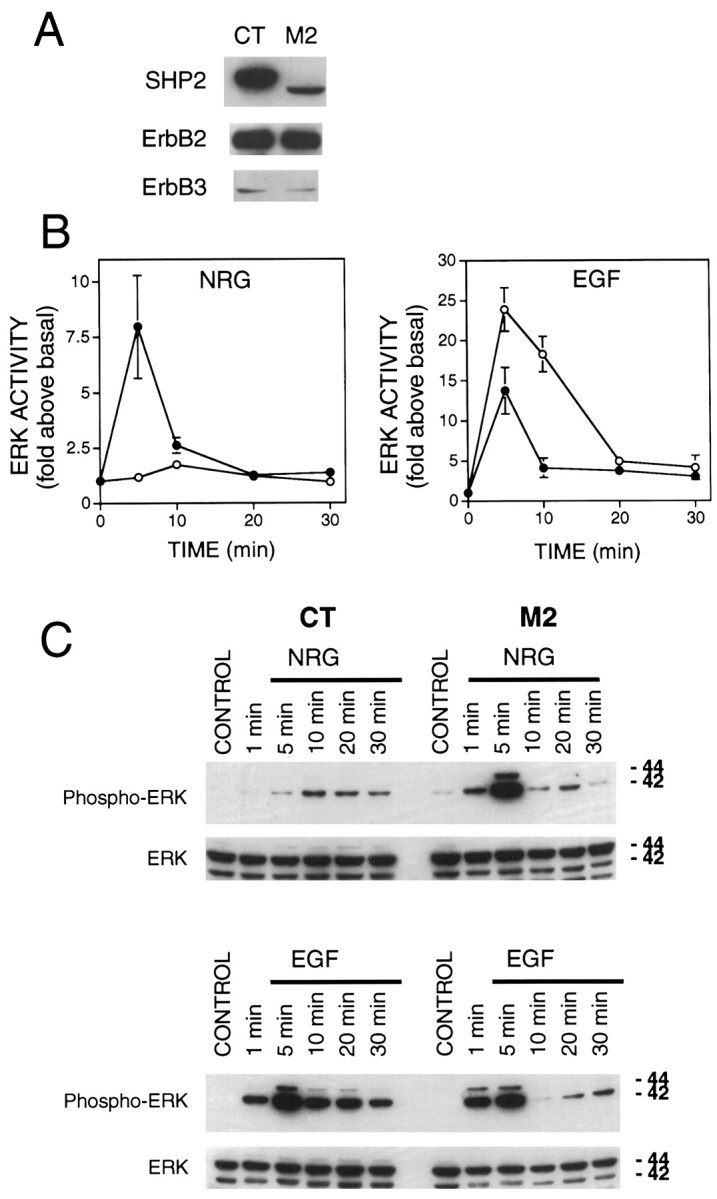
ERK activation in response to NRG and EGF in CT and M2 fibroblasts. A, Immunoblot analysis demonstrating the relative expression of ErbB2, ErbB3, and SHP2 in SHP2 mutant M2 cells, and wild-type CT cells. ErbB4 expression was not detected in either cell line. Notice the lower apparent molecular weight of the mutant SHP2, resulting from deletion of amino acids 46–110 of the N-terminal SH2 domain. B, ERK activation assayed using MBP as a substrate. Serum-starved CT (open circles) and M2 (closed circles) fibroblasts were stimulated with EGF or neuregulin for the indicated times. ERK1 was immunoprecipitated from cell lysates and subjected to an in vitro kinase assay using MBP as a substrate. Shown are the averages of ERK1 activity from four to six independent experiments. C, ERK activation assayed by anti-phospho-ERK antibodies. Lysates from cells treated as in B were subjected to SDS-PAGE and Western blot analysis with an antibody that specifically recognizes activated ERK1 and ERK2. The same membrane was stripped and reprobed with a different antibody that recognizes both phosphorylated and nonphosphorylated ERK1 and ERK2. Shown is a Western blot from a representative experiment that was repeated three times with similar results.
To study the role of SHP2 in neuregulin-mediated activation of the MAP kinase pathway, CT and M2 cells were treated with neuregulin and assayed for ERK activity using an in vitro kinase assay of immunoprecipitated ERK. The basal ERK kinase activity was at a low and similar level in either cell line. Parallel experiments demonstrated that EGF elicited ERK activation in both CT and M2 cells, however the activation seen in M2 cells was significantly attenuated, in agreement with previous results (Fig. 3B) (Shi et al., 1998). CT cells demonstrated only a modest activation of ERK in response to neuregulin, probably because of the relatively low expression level of ErbB3 (1.7 ± 0.2-fold above the basal; Fig. 3B). Strikingly, the mutant M2 cells displayed a fairly robust response (8.0 ± 2.3-fold) to neuregulin, despite the fact that they express ErbB2 and ErbB3 at similar levels as wild-type cells. Also worth noting is the difference in the time courses of neuregulin-stimulated ERK activation in CT and M2 cells, with the CT cells showing a relatively delayed response. Because the polyclonal ERK antibody used for immunoprecipitation in these assays preferentially precipitates ERK1 (p44 MAP kinase), we confirmed our results using an activation-specific antibody that recognizes both ERK1 and ERK2. Western blots of cell lysates from CT and M2 cells treated as for the in vitrokinase assays were probed with the E10 monoclonal antibody. This antibody is highly specific for ERK1 and ERK2 that are doubly phosphorylated on threonine and tyrosine residues corresponding to Thr202/Tyr204 of human p44 and p42 MAP kinases (Payne et al., 1991). Phosphorylation at these sites is essential for maximum activation of ERK1 and ERK2. As shown in Figure 3C, results from these experiments were consistent with the ERK activity data, providing further evidence of a negative role of SHP2 in neuregulin-stimulated ERK activation.
Enhanced activation by neuregulin of Raf and Ras in M2 mutant cells
In light of the opposing responses of M2 and CT cells to EGF and neuregulin, we were interested in characterizing points of divergence upstream of ERK activation. To this end, we characterized activation of Raf, an ERK kinase kinase, in response to neuregulin and EGF. Raf is a serine–threonine kinase that phosphorylates and activates the dual specificity kinase MEK (MAPKK) (Luo et al., 1996) which, in turn, phosphorylates and activates ERK. Raf was purified by immunoprecipitation with a polyclonal anti-Raf antibody, and its activity was assayed by a standard method with [32P]-labeled ERK1 as a readout (Luo et al., 1996). Raf activity in CT cells was increased within 5 min after treatment with EGF (Fig. 4). The Raf activation by EGF was less in M2 cells. Neuregulin activation of Raf was barely detectable in CT cells but was prominent in M2 cells. These experiments demonstrated that Raf activation followed a similar pattern to that of ERK activation in response to EGF and neuregulin, suggesting that the site of SHP2 differential regulation is upstream of Raf kinase.
Fig. 4.

EGF- and NRG-stimulated Raf kinase activity in CT and M2 fibroblasts. Serum-starved CT and M2 cells were treated with EGF or neuregulin for 5 and 10 min, lysed, and subjected to immunoprecipitation with an anti-Raf antibody. GST-ERK1 was added to assay Raf kinase activity 20 min after the addition of [γ-32P]ATP and GST-MEK. Shown is the autoradiograph of samples resolved on SDS-PAGE from a representative experiment that was repeated three times with similar results.
Ras acts proximally downstream to receptor activation through its conversion to the active, GTP-bound, state by the receptor-associated Grb2-SOS complex (Buday and Downward, 1993; Egan et al., 1993). Importantly, previous studies have indicated that the positive effects of wild-type SHP2 act upstream of, or parallel to, Ras (Noguchi et al., 1994; Yamauchi et al., 1995). Thus, Ras may represent an important convergence point for receptor-generated signals acting through SHP2. We used a method developed by Rooij and Bos (1997) that exploits the high affinity of the Raf1 RBD (aa 51–131) for GTP-bound Ras (Herrmann et al., 1995). The Raf RBD binds to GDP-bound Ras, however, with three orders of magnitude lower affinity. GST-Raf-RBD was used to purify active Ras from control and treated CT and M2 cells that was then analyzed by Western blot using Ras antibodies. Neuregulin stimulation of CT cells produced a barely detectable increase in active Ras, whereas the increase after EGF treatment was quite strong. In contrast, an increase in active Ras was readily apparent after both neuregulin and EGF treatment of M2 cells (Fig. 5). In addition, the time dependence of Ras activation was qualitatively similar to that seen of ERK activity. With each experiment, lysate equivalents were run in parallel to provide a standard for calibration. Using scanning densitometry, a semiquantitative analysis was performed, and the results are illustrated in bar graph form in Figure5B. The inverse relation of EGF and neuregulin-stimulated Ras activation is readily apparent. Thus, the overall pattern of Ras activation was consistent with downstream components of the ERK pathway, indicating that the primary divergence point was upstream of Ras.
Fig. 5.
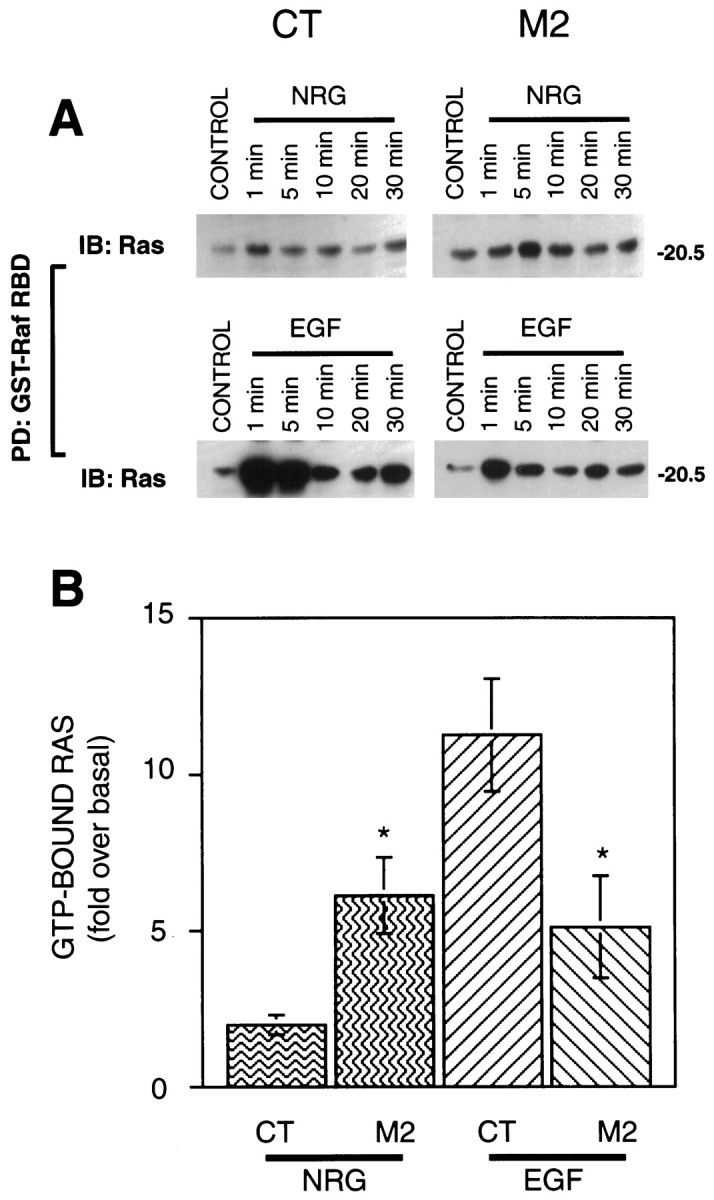
Neuregulin- and EGF-stimulated Ras activity in CT and M2 fibroblasts. A, CT and M2 cell lysates were incubated with Raf-RBD immobilized on agarose beads. Bound protein was eluted in SDS-sample buffer followed by SDS-PAGE and Western blot analysis with a rat monoclonal anti-Ras antibody. B,Comparison of Ras activation in CT and M2 cells in response to neuregulin and EGF (5 min). Each Western blot included varying amounts of lysate to calibrate each experiment for subsequent scanning densitometric analysis. Shown is the average of three independent experiments. *p < 0.05.
Hyperphosphorylation of ErbB proteins in M2 mutant cells
After ligand presentation the ErbB family of receptors become tyrosine-phosphorylated, thereby forming docking sites for SH2 domain-containing proteins such as the adaptor proteins Shc and Grb2 (Buday and Downward, 1993; Won and Mei, unpublished observations). Tyrosine phosphorylation is therefore considered a measure of receptor activation. To study the regulation of receptor activation by SHP2, we immunoprecipitated EGFR, ErbB2, and ErbB3 from CT and M2 cells stimulated with the appropriate growth factor and determined their tyrosine phosphorylation. EGFR demonstrated a dramatic increase in tyrosine phosphorylation in as little as 1 min, whereas there was a modest increase in tyrosine phosphorylation of ErbB2 and ErbB3 in wild-type cells (Fig. 6, left lanes). This increase in ErbB protein tyrosine-phosphorylation paralleled the activation of ERKs. Ligand-stimulated tyrosine phosphorylation of ErbB2 and ErbB3 was significantly higher in mutant cells than in wild-type cells, whereas phosphorylation of EGFR appeared to be modestly increased at later time points (Fig. 6, right lanes). Additionally, phosphorylation of ErbB3 occurred relatively later in wild-type cells. Probing of the membranes with a SHP2 monoclonal antibody revealed that SHP2 coprecipitated with ErbB2 and ErbB3 in both mutant and wild-type cells, suggesting the C-terminal SH2 domain is able to interact with those proteins. The lower amount of mutant SHP2 coprecipitating with ErbB2 and ErbB3 is likely a combination of its lower level of expression and a reduced affinity for the activated receptors. SHP2 coprecipitated with EGFR in wild-type cells; in contrast, no such interaction was seen in cells expressing the mutant SHP2. These results suggest that ErbB proteins are substrates of SHP2 and in addition, that the mutant SHP2 protein is incapable of interacting with the EGFR or forms a weaker association that does not survive immunoprecipitation.
Fig. 6.

Neuregulin (NRG)- and EGF-stimulated receptor tyrosine phosphorylation and association with SHP2. CT and M2 cells were stimulated with neuregulin or EGF for the indicated times. Cell lysates were subjected to immunoprecipitation with anti-ErbB2, anti-ErbB3, or anti-EGFR polyclonal antibodies. Immunocomplexes were resolved by SDS-PAGE, transferred to nitrocellulose, and probed with anti-phosphotyrosine and anti-SHP2 antibodies. Half of each immunocomplex was run on a parallel gel and probed with same antibody used for immunoprecipitation to demonstrate equal amount of immunoprecipitated protein.
SHP2 is localized at the neuromuscular junction
Neuregulin receptor ErbB proteins, as well as other tyrosine-phosphorylated proteins, are localized to the neuromuscular junction (Qu et al., 1990; Altiok et al., 1995; Moscoso et al., 1995;Zhu et al., 1995). We hypothesized that SHP2, containing two SH2 domains, binds to synaptic tyrosine-phosphorylated proteins and thus becomes concentrated at the NMJ. We therefore studied SHP2 localization using immunofluorescence techniques. Muscle sections were incubated with affinity-purified anti-SHP2 antibody and rhodamine-conjugated α-bungarotoxin, which labels the AChR. The SHP2 immunoreactivity was visualized with an FITC-conjugated secondary antibody. As shown in Figure 7, SHP2 showed a strikingly similar pattern of labeling (Fig. 7A) to AChRs labeled with α-bungarotoxin (Fig. 7A′). Merging of the two images indicated that SHP2 is localized in precise register with the AChR at the neuromuscular junction (data not shown). Specificity of the staining of SHP2 at the NMJ was demonstrated by the fact that the preimmune serum did not produce any staining above the background (Fig.7B,B′). Furthermore, the SHP2 staining was diminished by preabsorbing the antibody with the immunogen (Fig.7C,C′).
Fig. 7.

SHP2 localization at the NMJ. Mouse diaphragm sections were incubated with affinity-purified anti-SHP2 antibody (A) and rhodamine-conjugated α-bungarotoxin (α-BTX) (A′), which labels the AChR. The SHP2 immunoreactivity was visualized by FITC-conjugated secondary antibody.B shows staining by preimmune serum, andC shows anti-SHP2 antibodies preadsorbed with the immunogen. The exposure time for SHP2 immunoreactivity was approximately twice that of α-bungarotoxin labeling of the AChR, as determined by an automatic exposure system.
DISCUSSION
In this paper we report several lines of evidence implicating the involvement of SHP2 in neuregulin signaling at the NMJ. We demonstrate that association of SHP2 with both ErbB2 and ErbB3 is increased after neuregulin stimulation and that expression of mutant SHP2 proteins with defective phosphatase activity or deletion of the N-terminal SH2 domain result in increases in neuregulin-stimulated transcriptional activation in C2C12 myotubes. The negative regulatory role of SHP2 in the neuregulin signaling pathway was confirmed in fibroblasts expressing wild-type and mutant SHP2. Furthermore, our data suggests that the point of SHP2-negative regulation occurs at the receptor level, in contrast to signal-enhancing effects of SHP2 in the EGF signaling pathway, which appears to occur at the level of Ras activation. Finally, SHP2 was found to be localized to the neuromuscular junction, where neuregulin receptors are known to be concentrated and synapse-specific signal-transduction presumably occurs.
SHP2 at the neuromuscular junction
Tyrosine phosphorylation plays a paramount role in formation and stability of the neuromuscular synapse. This is first suggested by the immunohistochemical finding that tyrosine-phosphorylated proteins are concentrated at the NMJ. Both innervation of muscle during development and of myotubes cocultured with ganglionic neurons increases the phosphotyrosine immunostaining at synaptic sites (Qu et al., 1990). Several synapse-specific tyrosine kinases have been identified. The expression of ErbB2, ErbB3, and ErbB4 receptor tyrosine kinases is restricted to the adult NMJ (Altiok et al., 1995; Moscoso et al., 1995; Zhu et al., 1995), as is MuSK, a transmembrane tyrosine kinase whose activity is stimulated by agrin and that is both necessary and sufficient to cause clustering of the AChR (DeChiara et al., 1996;Jones et al., 1999). In addition, two cytoplasmic tyrosine kinases, Fyk and Fyn, bind via their SH2 domains to the AChR which itself is a substrate of tyrosine phosphorylation (Swope and Huganir, 1994). In contrast to the extensive study of tyrosine kinases that may be involved in NMJ formation, little is known of the relevant protein tyrosine phosphatases. We previously purified a protein tyrosine phosphatase from Torpedo electric organ that specifically dephosphorylates the AChR (Mei and Huganir, 1991). Moreover, the protein tryrosine phosphatase (PTP) activity in skeletal muscle appears to be regulated by motoneurons, because surgical denervation causes an increase in PTP activity in rat hindlimb muscles (Tanowitz and Mei, 1996). In C2C12 cells, we have previously demonstrated that inhibition of PTP activity by vanadate enhanced the basal and neuregulin-induced expression of an AChR reporter gene (Si et al., 1996). Here we have demonstrated that SHP2 negatively regulates neuregulin signaling, providing a possible explanation for this effect of vanadate treatment. This would also be consistent with the finding that overexpression of protein tyrosine phosphatases reduces expression of adult-type nicotinic receptors in primary muscle cultures (Sapru et al., 1994). Neuregulin is secreted by the somatic motoneuron and deposited in the synaptic basal lamina where it persists long after loss of the presynaptic neuron (Jo et al., 1995). Regulation of the neuregulin signal by changes in presynaptic deposition would appear to be an inefficient mechanism. The presence of SHP2 at the NMJ may reflect a role in fine tuning or adjusting the “gain” of the inductive effect of neuregulin.
If the concentration of SHP2 at the NMJ is a result of its association with ErbB receptors, synaptic ErbB proteins should exist in a more or less constitutively phosphorylated state. Although there is no direct evidence that ErbB proteins are constitutively phosphorylated at the NMJ, several lines of evidence support this hypothesis. Neuregulin is concentrated in the synaptic basal lamina (Goodearl et al., 1995; Jo et al., 1995), and ErbB proteins are concentrated at the NMJ (Moscoso et al., 1995; Zhu et al., 1995). Thus, it is a logical inference that synaptic ErbBs are constitutively phosphorylated and it is likely, therefore, that SHP2 localizes to the NMJ by an interaction with activated ErbB proteins. The concentration of SHP2 at the NMJ could indicate additional functions of SHP2, such as regulating the formation of cytoskeletal specializations that serve to anchor the AChR to the synapse. Huganir and his colleagues (Qu et al., 1990) have shown that tyrosine-phosphorylated proteins are concentrated at the NMJ. In addition to ErbB proteins, tyrosine-phosphorylated proteins at the NMJ include the AChR (Wallace et al., 1991) and MuSK (DeChiara et al., 1996). SHP2 may interact with and regulate the function of these proteins. Peng and his colleagues (Dai and Peng, 1998) recently proposed that a diffusible signal, which may be a cytoplasmic protein tyrosine phosphatase, is involved in the dispersal of spontaneous AChR clusters (“hot spots”). By virtue of its synaptic concentration, SHP2 may be an attractive candidate for this phosphatase. Within such a scenario, synaptically localized phosphoproteins concentrate SHP2, which in turn disrupts nearby spontaneous clustering and helps to sculpt the synaptic distribution of AChRs and/or other synaptic proteins.
Mechanisms of SHP2 regulation of neuregulin signaling
Importantly, we found that the mutant SHP2 protein lacking the N-terminal SH2 domain was capable of interacting with ErbB2 and ErbB3 and thus could, in theory, interfere with wild-type SHP2 function or substitute for it. If deletion of the N-terminal SH2 domain results in a SHP2 protein that can substitute for wild-type, overexpression of either protein would be expected to produce similar results. This was clearly not the case because overexpression of wild-type SHP2 exerted a negative effect on the AChR ε reporter in myotubes, yet overexpression of the N-terminal SH2 domain deletion mutant yielded a nearly 100% increase in neuregulin-stimulated transgene expression. Neither is it likely that the effect of mutant SHP2 is caused by a loss of proper SH2-mediated localization because expression of the PTP region alone had negligible effect. A reasonable interpretation is that the SHP2ΔSH2, retaining the C-terminal SH2 domain, was able to displace the endogenous protein and interfere with its negative regulatory effect. The increased receptor tyrosine phosphorylation in M2 cells indicates that the negative influence of wild-type SHP2 is mediated by dephosphorylation and inactivation of ErbB2 and/or ErbB3. The mutant protein is perhaps incapable of dephosphorylating the ErbB receptors because of improper alignment or targeting of the catalytic region with the relevant phosphotyrosine(s). Ideally, the mechanism of SHP2 regulation of AChR subunit gene expression should be examined in muscle cells isolated from SHP2 mutant mice. Unfortunately, early embryonic lethality prevents the isolation of primary muscle cells for culture. The fibroblasts used in the present study were a viable alternative because they express both ErbB2 and ErbB3 and exhibit neuregulin responses.
It is possible that SHP2 exerts both positive and negative influences after neuregulin signaling where the dominant, negative role requires the N-terminal SH2 domain, whereas the C-terminal SH2 domain is capable of mediating a positive effect. ErbB2 has a number of tyrosine residues that when phosphorylated may become either positive or negative regulatory sites for downstream signaling. Deletion of a negative site leads to increased Grb2 and Shc binding to the activated receptor (Dankort et al., 1997). Alternatively, the target of SHP2 catalytic activity could be one or more phosphotyrosines that regulate ErbB tyrosine kinase activity. The phosphorylation of residues in the activation motif, or so called “A-loop”, of the catalytic domain is central to the activation of many protein kinases (Johnson et al., 1996), including the insulin receptor (Hubbard et al., 1994).
Although the catalytic activity of SHP2 is clearly important for both positive and negative influences, a role for SHP2 tyrosine phosphorylation cannot be discounted. The N-terminal deletion mutant displayed increased tyrosine phosphorylation after growth factor treatment (data not shown). This was also reported of the same mutant following PDGF treatment of primary fibroblasts (Saxton et al., 1997). SHP2 may function as an adapter and has been shown to bind Grb2. Such an adaptor function of SHP2 has been proposed for PDGF signaling (Bennett et al., 1994), although a functional requirement of SHP2 tyrosine phosphorylation has never been demonstrated. Tyrosine phosphorylation of CORKSCREW (CSW), the Drosophila SHP2 homolog, occurs after stimulation of the TORSO receptor tyrosine kinase. Phosphorylated CSW may participate in linking DRK, theDrosophila Grb2 homolog, to TORSO (Cleghon et al., 1998). Whether SHP2 can function as an adapter in the neuregulin signaling pathway remains to be determined.
In summary, the protein tyrosine phosphatase SHP2 is concentrated at the NMJ, negatively regulates neuregulin-induced AChR expression, and may serve other functions at the synapse.
Footnotes
This work was supported by the National Institutes of Health Grants NS34062 to L.M. and GM53660 to G.S.F. and grants from the Muscular Dystrophy Association and the March of Dimes Birth Defects Foundation to L.M, a predoctoral fellowship from Pharmaceutical Research and Manufacturers of America Foundation to M.T., and a postdoctoral fellowship (2T32CA09109) to J.S. We thank Drs. M. Sliwkowski (Genentech) and Z. J. Luo (Boston University) for providing reagents, and members of the Mei lab for helpful discussion.
Correspondence should be addressed to Dr. Lin Mei, Department of Neurobiology, University of Alabama at Birmingham, LHRB531, 1530 Third Avenue South, Birmingham, AL 35294-0007. E-mail: lmei@nrc.uab.edu.
REFERENCES
- 1.Ali S, Chen Z, Lebrun J-J, Vogel W, Kharitonenkov A, Kelly PA, Ullrich A. PTP1D is a positive regulator of the prolactin signal leading to beta-casein promoter activation. EMBO J. 1996;15:135–142. [PMC free article] [PubMed] [Google Scholar]
- 2.Altiok N, Bessereau J-L, Changeux J-P. ErbB3 and ErbB2/neu mediate the effect of heregulin on acetylcholine receptor gene expression in muscle: differential expression at the endplate. EMBO J. 1995;14:4258–4266. doi: 10.1002/j.1460-2075.1995.tb00100.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Altiok N, Altiok S, Changeux JP. Heregulin-stimulated acetylcholine receptor gene expression in muscle: requirement for MAP kinase and evidence for a parallel inhibitory pathway independent of electrical activity. EMBO J. 1997;16:717–725. doi: 10.1093/emboj/16.4.717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ausubel FM, Brent P, Kingston RE, Moore DD, Seidman JA, Smith JA, Struhl K. Current protocols in molecular biology. Wiley; New York: 1994. [Google Scholar]
- 5.Barford D, Flint AJ, Tonks NK. Crystal structure of human protein tyrosine phosphatase 1B. Science. 1994;263:1397–1404. [PubMed] [Google Scholar]
- 6.Baugh LD, Allen EE, Liu RH, Langner JG, Fentress JC, Chadha SC, Cook LF, Walia AS. Evaluation of immunoassay methods for the screening of cocaine metabolites in urine. J Forensic Sci. 1991;36:79–85. [PubMed] [Google Scholar]
- 7.Bennett AM, Tang TL, Sugimoto S, Walsh CT, Neel BG. Protein-tyrosine-phosphatase SHPTP2 couples platelet-derived growth factor receptor beta to Ras. Proc Natl Acad Sci USA. 1994;91:7335–7339. doi: 10.1073/pnas.91.15.7335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bennett AM, Hausdorff SF, O'Reilly AM, Freeman RM, Neel BG. Multiple requirements for SHPTP2 in epidermal growth factor-mediated cell cycle progression. Mol Cell Biol. 1996;16:1189–1202. doi: 10.1128/mcb.16.3.1189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bradford MM. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem. 1976;72:248–254. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- 10.Brunner D, Ducker K, Oellers N, Hafen E, Scholz H, Klambt C. The ETS domain protein pointed-P2 is a target of MAP kinase in the sevenless signal transduction pathway. Nature. 1994;370:386–389. doi: 10.1038/370386a0. [DOI] [PubMed] [Google Scholar]
- 11.Buday L, Downward J. Epidermal growth factor regulates p21ras through formation of a complex of receptor, Grb2 adaptor protein, and Sos nucleotide exchange factor. Cell. 1993;73:611–620. doi: 10.1016/0092-8674(93)90146-h. [DOI] [PubMed] [Google Scholar]
- 12.Chu GC, Moscoso LM, Sliwkowski MX, Merlie JP. Regulation of the acetylcholine receptor e subunit gene by recombinant ARIA: an in vitro model for transynaptic gene regulation. Neuron. 1995;14:329–339. doi: 10.1016/0896-6273(95)90289-9. [DOI] [PubMed] [Google Scholar]
- 13.Cleghon V, Feldmann P, Ghiglione C, Copeland TD, Perrimon N, Hughes DA, Morrison DK. Opposing actions of CSW and RasGap modulate the strength of Torso RTK signaling in the Drosophila Terminal pathway. Mol Cell. 1998;2:719–727. doi: 10.1016/s1097-2765(00)80287-7. [DOI] [PubMed] [Google Scholar]
- 14.Dai Z, Peng B. A role of tyrosine phosphatase in acetylcholine receptor cluster dispersal and formation. J Cell Biol. 1998;141:1613–1624. doi: 10.1083/jcb.141.7.1613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dankort DL, Wang Z, Blackmore V, Moran MF, Muller WJ. Distinct tyrosine autophosphorylation sites negatively and positively modulate neu-mediated transformation. Mol Cell Biol. 1997;17:5410–5425. doi: 10.1128/mcb.17.9.5410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.David M, Zhou G, Pine R, Dixon J, Larner AC. J Biol Chem. 1996;271:15862–15865. doi: 10.1074/jbc.271.27.15862. [DOI] [PubMed] [Google Scholar]
- 17.DeChiara TM, Bowen DC, Valenzuela DM, Simmons MV, Poueymirou WT, Thomas S, Kinetz E, Compton DL, Rojas E, Park JS, Smith C, DiStefano PS, Glass DJ, Burden SJ, Yancopoulos GD. The receptor tyrosine kinase MuSK is required for neuromuscular junction formation in vivo. Cell. 1996;85:501–512. doi: 10.1016/s0092-8674(00)81251-9. [DOI] [PubMed] [Google Scholar]
- 18.Duclert A, Savatier N, Schaeffer L, Changeux J-P. Identification of an element crucial for the sub-synaptic expression of the acetylcholine receptor ε-subunit gene. J Biol Chem. 1996;271:17433–17438. doi: 10.1074/jbc.271.29.17433. [DOI] [PubMed] [Google Scholar]
- 19.Egan SE, Giddings BW, Brooks MW, Sizeland AM, Weinberg RA. Association of Sos Ras exchange protein with grb2 is implicated in tyrosine kinase signal transduction and transformation. Nature. 1993;363:45–51. doi: 10.1038/363045a0. [DOI] [PubMed] [Google Scholar]
- 20.Falls DL, Rosen KM, Corfas G, Lane WS, Fischbach GD. ARIA, a protein that stimulates acetylcholine receptor synthesis, is a member of the Neu ligand family. Cell. 1993;72:801–815. doi: 10.1016/0092-8674(93)90407-h. [DOI] [PubMed] [Google Scholar]
- 21.Feng G, Pawson T. Phosphotyrosine phosphatases with SH2 domains: regulators of signal transduction. Trends Genet. 1994;10:54–58. doi: 10.1016/0168-9525(94)90149-x. [DOI] [PubMed] [Google Scholar]
- 22.Feng G, Hui C, Pawson T. SH2-containing phosphotyrosine phosphatase as a target of protein-tyrosine kinases. Science. 1993;259:1607–1611. doi: 10.1126/science.8096088. [DOI] [PubMed] [Google Scholar]
- 23.Fromm L, Burden SJ. Synapse-specific and neuregulin-induced transcription require an Ets site that binds GABPa/GABPb. Genes Dev. 1998;12:3074–3083. doi: 10.1101/gad.12.19.3074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Garton AJ, Flint AJ, Tonks NK. Identification of p130cas as a substrate for the cytosolic protein tyrosine phosphatase PTP-PEST. Mol Cell Biol. 1996;16:6408–6418. doi: 10.1128/mcb.16.11.6408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Goodearl ADJ, Yee AG, Sandrock AW, Corfas G, Fischbach GD. ARIA is concentrated in the synaptic basal lamina of the developing chick neuromuscular junction. J Cell Biol. 1995;130:1423–1434. doi: 10.1083/jcb.130.6.1423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Herrmann C, Martin G, Wittinghofer A. Quantitative analysis of the complex between p21 and the Ras-binding domain of the human Raf-1 protein kinase. J Biol Chem. 1995;270:2901–2905. doi: 10.1074/jbc.270.7.2901. [DOI] [PubMed] [Google Scholar]
- 27.Holmes WE, Slikowski MX, Akita RW, Kenzel WJ, Lee J, Park JW, Yansura D, Abadi N, Raab H, Lewis GD, Shepard HM, Huang WJ, Wood WJ, Goeddel DV, Vandlen RL. Identification of heregulin, a specific activator of p185erbB2. Science. 1992;256:1205–1210. doi: 10.1126/science.256.5060.1205. [DOI] [PubMed] [Google Scholar]
- 28.Hubbard SR, Wei L, Ellis L, Hendrickson WA. Crystal structure of the tyrosine kinase domain of the human insulin receptor. Nature. 1994;372:746–754. doi: 10.1038/372746a0. [DOI] [PubMed] [Google Scholar]
- 29.Jessell TM, Siegel RE, Fischbach GD. Induction of acetylcholine receptors on cultured skeletal muscle by a factor extracted from brain and spinal cord. Proc Natl Acad Sci USA. 1979;76:5397–5401. doi: 10.1073/pnas.76.10.5397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Jo SA, Zhu X, Marchionni MA, Burden SJ. Neuregulins are concentrated at nerve-muscle synapses and activate ACh-receptor gene expression. Nature. 1995;373:158–161. doi: 10.1038/373158a0. [DOI] [PubMed] [Google Scholar]
- 31.Johnson LN, Noble MEM, Owen DJ. Active and inactive protein kinases: structural basis for regulation. Cell. 1996;85:149–158. doi: 10.1016/s0092-8674(00)81092-2. [DOI] [PubMed] [Google Scholar]
- 32.Jones G, Moore C, Hashemolhosseini S, Brenner HR. Constitutively active MuSK is clustered in the absence of agrin and induces ectopic postsynaptic-like membranes in skeletal muscle fibers. J Neurosci. 1999;19:3376–3383. doi: 10.1523/JNEUROSCI.19-09-03376.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lemmon MA, Sclessinger J. Regulation of signal transduction and signal diversity by receptor oligomerization. Trends Biochem Sci. 1994;19:459–463. doi: 10.1016/0968-0004(94)90130-9. [DOI] [PubMed] [Google Scholar]
- 34.Loeb JA, Fischbach GD. ARIA can be released from extracellular matrix through cleavage of a heparin-binding domain. J Cell Biol. 1995;130:127–135. doi: 10.1083/jcb.130.1.127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Luo Z, Tzivion G, Belshaw PJ, Vavvas D, Marshall M, Avruch J. Oligomerization activates c-Raf-1 through a Ras-dependent mechanism. Nature. 1996;383:181–185. doi: 10.1038/383181a0. [DOI] [PubMed] [Google Scholar]
- 36.Marais R, Wynne J, Treisman R. The SRF accessory protein Elk-1 contains a growth factor-regulated transcriptional activation domain. Cell. 1993;73:381–393. doi: 10.1016/0092-8674(93)90237-k. [DOI] [PubMed] [Google Scholar]
- 37.Marte BM, Graus-Porta D, Jeschke M, Fabbro D, Hynes NE, Taverna D. NDF/heregulin activates MAP kinase and p70/p85 S6 kinase during proliferation or differentiation of mammary epithelial cells. Oncogene. 1995;10:167–175. [PubMed] [Google Scholar]
- 38.Mei L, Huganir RL. Purification and characterization of a protein tyrosine phosphatase which dephosphorylates the nicotinic acetylcholine receptor. J Biol Chem. 1991;266:16063–16072. [PubMed] [Google Scholar]
- 39.Meyer D, Birchmeier C. Multiple essential functions of neuregulin in development. Nature. 1995;378:386–390. doi: 10.1038/378386a0. [DOI] [PubMed] [Google Scholar]
- 40.Milarski KL, Saltiel AR. Expression of catalytically inactive Syp phosphatase in 3T3 cells blocks stimulation of mitogen-activated protein kinase by insulin. J Biol Chem. 1994;269:21239–21243. [PubMed] [Google Scholar]
- 41.Moscoso LM, Chu GC, Gautam M, Noakes PG, Merlie JP, Sanes JR. Synapse-associated expression of an acetylcholine receptor-inducing protein, ARIA/heregulin, and its putative receptors, ErbB2 and ErbB3, in developing mammalian muscle. Dev Biol. 1995;172:158–169. doi: 10.1006/dbio.1995.0012. [DOI] [PubMed] [Google Scholar]
- 42.Noguchi T, Matozaki T, Horita K, Fujioka Y, Kasuga M. Role of SH-PTP2, a protein-tyrosine phosphatase with Src homology 2 domains, in insulin-stimulated Ras activation. Mol Cell Biol. 1994;14:6674–6682. doi: 10.1128/mcb.14.10.6674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.O'Hagan RC, Hassell JA. The PEA3 Ets transcription factor is a downstream target of the HER2/Neu receptor tyrosine kinase. Oncogene. 1999;16:301–310. doi: 10.1038/sj.onc.1201547. [DOI] [PubMed] [Google Scholar]
- 44.Payne DM, Rossomando AJ, Martino P, Erickson AK, Her JH, Shabanowitz J, Hunt DF, Weber MJ, Sturgill TW. Identification of the regulatory phosphorylation sites in pp42 mitogen-activated protein kinase (MAP kinase). EMBO J. 1991;10:885–892. doi: 10.1002/j.1460-2075.1991.tb08021.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Qu Z, Moritz E, Huganir RL. Regulation of tyrosine phosphorylation of the nicotinic acetylcholine receptor at the rat neuromuscular junction. Neuron. 1990;4:367–378. doi: 10.1016/0896-6273(90)90049-l. [DOI] [PubMed] [Google Scholar]
- 46.Rooij J, Bos JL. Minimal Ras-binding domain of Raf1 can be used as an activation-specific probe for Ras. Oncogene. 1997;14:623–625. doi: 10.1038/sj.onc.1201005. [DOI] [PubMed] [Google Scholar]
- 47.Sambrook J, Fritsch EF, Maniatis T. Molecular cloning: a laboratory manual. Cold Spring Harbor Laboratory; Cold Spring Harbor, NY: 1989. [Google Scholar]
- 48.Sandrock AW, Dryer SE, Rosen KM, Gozani SN, Kramer R, Theill LE, Fischbach GD. Maintenance of acetylcholine receptor number by neuregulins at the neuromuscular junction in vivo. Science. 1997;276:599–603. doi: 10.1126/science.276.5312.599. [DOI] [PubMed] [Google Scholar]
- 49.Sapru MK, Zhou G, Goldman D. Protein-tyrosine phosphatases specifically regulate muscle adult-type nicotinic acetylcholine receptor gene expression. J Biol Chem. 1994;269:27811–27814. [PubMed] [Google Scholar]
- 50.Sapru MK, Florance SK, Kirk C, Goldman D. Identification of a neuregulin and protein-tyrosine phosphatase response element in the nicotinic acetylcholine receptor e subunit gene: regulatory role of an ets transcription factor. Proc Natl Acad Sci USA. 1998;95:1289–1294. doi: 10.1073/pnas.95.3.1289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Saxton TM, Henkemeyer M, Gasca S, Shen R, Rossi DJ, Shalaby F, Feng GS, Pawson T. Abnormal mesoderm patterning in mouse embryos mutant for the SH2 tyrosine phosphatase Shp-2. EMBO J. 1997;16:2352–2364. doi: 10.1093/emboj/16.9.2352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Shi ZQ, Lu W, Feng GF. The Shp-2 tyrosine phosphatase has opposite effects in mediating the activation of extracellular signal-regulated and c-Jun NH2-terminal mitogen-activated protein kinases. J Biol Chem. 1998;273:4904–4908. doi: 10.1074/jbc.273.9.4904. [DOI] [PubMed] [Google Scholar]
- 53.Si J, Luo Z, Mei L. Induction of acetylcholine receptor gene expression by ARIA requires activation of mitogen-activated protein kinase. J Biol Chem. 1996;271:19752–19759. doi: 10.1074/jbc.271.33.19752. [DOI] [PubMed] [Google Scholar]
- 54.Si J, Miller DS, Mei L. Identification of an element required for acetylcholine receptor-inducing activity (ARIA)-induced expression of the acetylcholine receptor ε subunit gene. J Biol Chem. 1997;272:10367–10371. doi: 10.1074/jbc.272.16.10367. [DOI] [PubMed] [Google Scholar]
- 55.Stofega MR, Wang H, Ullrich A, Carter-Su C. J Biol Chem. 1998;273:7112–7117. doi: 10.1074/jbc.273.12.7112. [DOI] [PubMed] [Google Scholar]
- 56.Swope SL, Huganir RL. Binding of the nicotinic acetylcholine receptor to SH2 domains of Fyn and Fyk protein tyrosine kinases. J Biol Chem. 1994;269:29817–29824. [PubMed] [Google Scholar]
- 57.Tang TL, Freeman RM, Jr, O'Reilly AM, Neel BG, Sokol SY. The SH2-containing protein-tyrosine phosphatase SH-PTP2 is required upstream of MAP kinase for early Xenopus development. Cell. 1995;80:473–483. doi: 10.1016/0092-8674(95)90498-0. [DOI] [PubMed] [Google Scholar]
- 58.Tanowitz MB, Mei L. Surgical denervation increases protein tyrosine phosphatase activity in skeletal muscle. Brain Res. 1996;712:299–306. doi: 10.1016/0006-8993(95)01475-6. [DOI] [PubMed] [Google Scholar]
- 59.Tansey MG, Chu GC, Merlie JP. ARIA/HRG regulates AChR ε subunit gene expression at the neuromuscular synapse via activation of phosphatidylinositol 3-kinase and Ras/MAPK pathway. J Cell Biol. 1996;134:465–476. doi: 10.1083/jcb.134.2.465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Wallace BG, Qu Z, Huganir RL. Agrin induces phosphorylation of acetylcholine receptors. Neuron. 1991;6:869–878. doi: 10.1016/0896-6273(91)90227-q. [DOI] [PubMed] [Google Scholar]
- 61.Xiao S, Rose DW, Sasaoka HM, Burke TR, Roller PP, Shoelson SE, Olefsky JM. Syp (SH-PTP2) is a positive mediator of growth factor-stimulated mitogenic signal transduction. J Biol Chem. 1994;269:21244–21248. [PubMed] [Google Scholar]
- 62.Yaffe D, Saxel O. Serial passaging and differentiation of myogenic cells isolated from dystrophic mouse muscle. Nature. 1977;270:725–727. doi: 10.1038/270725a0. [DOI] [PubMed] [Google Scholar]
- 63.Yamauchi K, Milarski KL, Saltiel AR, Pessin JE. Protein-tyrosine-phosphatase SHPTP2 is a required positive effector for insulin downstream signaling. Proc Natl Acad Sci USA. 1995;92:664–668. doi: 10.1073/pnas.92.3.664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Zhu X, Lai C, Thomas S, Burden SJ. Neuregulin receptors, erbB3 and erbB4, are localized at neruomuscular synapses. EMBO J. 1995;23:5842–5848. doi: 10.1002/j.1460-2075.1995.tb00272.x. [DOI] [PMC free article] [PubMed] [Google Scholar]