

Abstract
Extracellular adenosine critically modulates ischemic brain injury, at least in part through activation of the A1 adenosine receptor. However, the role played by the A2A receptor has been obscured by intrinsic limitations of A2A adenosinergic agents. To overcome these pharmacological limitations, we explored the consequences of deleting the A2A adenosine receptor on brain damage after transient focal ischemia. Cerebral morphology, as well as vascular and physiological measures (before, during, and after ischemia) did not differ between A2A receptor knock-out and wild-type littermates. The volume of cerebral infarction, as well as the associated neurological deficit induced by transient filament occlusion of the middle cerebral artery, were significantly attenuated in A2A receptor knock-out mice. This neuroprotective phenotype of A2A receptor-deficient mice was observed in different genetic backgrounds, confirming A2A receptor disruption as its cause. Together with complimentary pharmacological studies, these data suggest that A2A receptors play a prominent role in the development of ischemic injury within brain and demonstrate the potential for anatomical and functional neuroprotection against stroke by A2A receptor antagonists.
Keywords: A2A adenosine receptor, ischemia, stroke, purine receptor, knock-out, neuroprotection
The ubiquitous metabolic intermediary and nucleoside adenosine also serves as a neuromodulator under physiological conditions (Fredholm et al., 1994). Growing evidence supports an important role for adenosine in modulating ischemic neuronal injury as well (Rudolphi et al., 1992; Deckert and Gleiter, 1994; Phillis, 1997; von Lubitz, 1997). First, adenosine levels markedly increase in response to cerebral ischemia and hypoxia as ATP breakdown dramatically increases the formation of adenosine. The extracellular levels of adenosine often rise faster (within minutes), higher (by more than 50-fold), and after smaller reductions in cerebral blood flow (CBF) compared with the levels of neurotransmitters such as glutamate (Hagberg et al., 1987; Matsumoto et al., 1992). Second, elevating extracellular adenosine levels by inhibiting adenosine degradation or uptake reduces ischemia-induced brain damage (Rudolphi et al., 1992). Third, adenosine analogs can attenuate hypoxic–ischemic neuronal injury, whereas certain adenosine antagonists exacerbate it (Rudolphi et al., 1992; Phillis, 1997; von Lubitz, 1997).
Although early studies suggested that adenosine acts predominantly as a neuroprotectant during cerebral ischemia (Deckert and Gleiter, 1994;Rudolphi et al., 1992), the complexity of the role of adenosine has been increasingly appreciated with the identification of four major adenosine receptor subtypes (A1, A2A, A2B, and A3), each having a unique distribution among brain regions and their neuronal, glial, and vascular elements (Fredholm et al., 1994). Furthermore, these receptors are differentially coupled through G-protein receptors to second messengers, including cAMP and calcium (Fredholm et al., 1994, 1997). Nevertheless, the neuroprotective effects of adenosine can be attributed at least in part to A1 receptor stimulation, as A1-specific agonists and antagonists consistently attenuate and potentiate ischemic brain injury, respectively (Rudolphi et al., 1992). A1receptor-mediated neuroprotection may be a result of the inhibitory action of A1 receptors on the release of excitatory amino acids such as glutamate (Rudolphi et al., 1992).
Much less is known about the role of A2Areceptors in ischemic damage (Phillis, 1997; von Lubitz, 1997). A2A receptors are expressed at high levels in the striatum (Shiffmann et al., 1991; Fink et al., 1992; Svenningsson et al., 1998) but are also present in other brain regions, such as the cortex and hippocampus (Johansson et al., 1993; Weaver, 1993; Rosin et al., 1998), and on the endothelial and smooth muscle cells of the cerebral vasculature (Kalaria and Hank, 1986). Efforts to clarify the role of A2A receptors in ischemic injury have produced mixed results. The relatively specific A2A agonist CGS 21680 reduces ischemic or excitotoxic hippocampal damage (Scheardown and Knutsen, 1996;Jones et al., 1998). A2A receptor-mediated vasodilation (Phillis, 1989; Ibayashi et al., 1991), inhibition of platelet aggregation (Sandoli et al., 1994; Ledent et al., 1997), and suppression of neutrophil superoxide generation (Cronstein, 1994;Jordan et al., 1997) may account for A2Areceptor-mediated protection. These beneficial vascular effects of A2A receptor activation have been suggested as a partial explanation for the exacerbation of ischemic brain damage induced by the nonselective adenosine antagonists theophylline and caffeine (Rudolphi et al., 1992). On the other hand, several relatively specific A2A antagonists have been found to reduce ischemic damage in animal models of global or permanent ischemia, as well as in excitotoxic neuronal damage (Gao et al., 1994;Phillis, 1995; von Lubitz et al., 1995; Jones et al., 1998; Monopoli et al., 1998). A2A receptor-mediated facilitation of glutamate release observed in ischemic cortex (O'Regan et al., 1992;Simpson et al., 1992) and striatum (Popoli et al., 1995; Corsi et al., 1997) may explain a protective effect of A2Aantagonists. Thus, the contradictory data on A2Areceptors in cerebral ischemia may reflect their potential to produce opposing effects through different (vascular and neuronal) mechanisms.
In addition, our understanding of how A2Areceptors influence ischemic injury has been confounded by the poor specificity and solubility of adenosine drugs. Almost all A2A adenosine receptor agonists and antagonists also have some effects on A1 or A3 receptors (Jacobson et al., 1992; Ongini and Fredholm, 1996; Ongini et al., 1999). To help clarify the role that A2A receptors play in neurological disorders such as stroke, we generated A2A receptor knock-out (A2A KO) mouse strains (which are distinct from a previously reported A2a KO strain) (Ledent et al., 1997) and examined the susceptibility of these mice to ischemic brain injury. We demonstrate that A2A receptor inactivation attenuates brain damage and preserves neurological function after transient middle cerebral arterial (MCA) occlusion. These results strongly support the prospect that A2A receptor blockade may offer neuroprotection against brain damage induced by transient focal ischemia.
MATERIALS AND METHODS
Generation of A2A KO mice. Three independent genomic clones (∼20 kb genomic DNA fragment) encoding a putative A2A receptor gene from a mouse 129-Steel genomic library were isolated using the rat A2A receptor cDNA as a probe. Characterization of the mouse A2A receptor gene revealed an additional (previously unknown) exon in the 5′ untranslated region (Chen and Fink, 1996) (Fig.1A). Based on this A2A receptor genomic map, a standard replacement-type vector was constructed to inactivate the A2A receptor gene. It consists of 5 and 4.5 kb of A2A receptor genomic fragments (as theleft and right arms of the inset, respectively) flanking a positive selection marker (PGK-Neocassette). This target vector disrupts the A2Areceptor gene by replacing the 3′ end of exon 2 (12 bp from the splice junction site) and the adjacent intron sequences (0.9 kb from 5′ end of the splice junction site) with the PGK-Neo cassette. A dysfunctional mutant gene product was expected because the deleted 3′ end of exon 2 in the mouse A2A receptor gene corresponds to a highly conserved region of the mouse A2A receptor between the third and fourth transmembrane domains (Peterfreund et al., 1996).
Fig. 1.

Generation of A2A KO mice with target inactivation of the A2A receptor.A, Schematic diagram of the A2A receptor targeting vector; a standard replacement-type vector was constructed with 5 and 4.5 kb A2A receptor genomic fragments split by a positive selection marker (Neo cassette), which replaced the 3′ end of exon 2 (E2) and the adjacent 5′ splice junction and intron sequences. Digestion of wild-type and mutant A2A receptor genes with BamHI (at sites labeled B) generates 7.5 and 5.0 kb fragments, respectively, that can be distinguished using a nonoverlapping 3′ probe (as in B). B, Genomic Southern analysis of WT (+/+), heterozygous (+/−), and homozygous (−/−) mice with respect to the A2A receptor gene was performed as described in Materials and Methods, using the 3′ nonoverlapping probe illustrated in A. WT mice displayed a single 7.5 kb band, whereas homozygous A2A KO mice showed a single 5.0 kb band corresponding to the restriction fragments for WT and mutant alleles, respectively. Heterozygous mice showed both 7.5 and 5.0 kb bands.C, Homozygous A2A receptor KO mice are defi-cient in A2A receptors detected by receptor autoradiography; A2A receptor binding was determined using3H-CGS 21680 as a ligand. A representative coronal brain section from a WT mouse shows specific labeling of A2Areceptors in striatum (caudate putamen, CP; nucleus accumbens, NA) and olfactory tubercle (OT), whereas that from a homozygous mouse shows no 3H-CGS 21680 binding. D, Behavioral responses to the A2A agonist CGS 21680 in WT and A2A KO mice; ambulation was measured in WT and A2A KO mice (n = 14–16) before and after challenge with CGS 21680 (0.2 mg/kg, i.p.) by recording contiguous photobeam interruptions (ambulation) for 60 min. Error bars represent the mean ± SEM. *p < 0.05 (Student's t test) when compared with ambulation in the WT mice before treatment.
Embryonic stem (ES) cells [129/SvJae, “Steel substrain” (Simpson et al., 1997)] were obtained from Dr. E. Li (Li et al., 1992) in the Knockout Core Facility at Massachusetts General Hospital. The A2A receptor targeting vector was transferred into ES cells by electroporation. Targeted ES clones were selected and expanded in medium containing the aminoglycoside antibiotic G418. Mutant clones with the desired recombinant allele were identified by Southern blotting using a nonoverlapping 3′ probe after digestion withBamHI (at sites designated by B in Fig.1A). One of the ES cell clones (#50) containing the recombinant allele was injected into blastocysts and transferred to a host in the Knockout Core Facility. Viable chimeric mice were maintained until weaning, and these chimeric mice (F0) were bred to C57BL/6 (Taconic, Germantown, NY) or 129/SvEvTac mice [Steel substrain (Simpson et al., 1997), Taconic]. The germ line-transmitting mice with the A2A receptor mutation were identified by Southern blot (F1). Heterozygous female and male mice from different founder mice were interbred to generate homozygous, heterozygous, and wild-type (WT) littermates mice, which were delivered at gestation day 21 in a normal mendelian distribution of A2A receptor genotypes. The F2–F4 generations of A2A homozygous, heterozygous, and wild-type littermates were used here. The hybrid (C57BL/6 × 129-Steel) mice were used for anatomical, immunohistochemical, and behavioral characterization of A2A KO mice. Ischemic injury studies (including hemodynamic and other physiological measurements) were performed in both the hybrid (C57BL/6 × 129-Steel) and pure 129-Steel strains.
Receptor autoradiography and immunohistochemistry. Receptor autoradiography for detecting A2A and NMDA receptors using the specific ligands3H-CGS 21680 (46.0 Ci/mmol; NEN, Boston, MA) and 3H-MK-801 (22.5 Ci/mmol; NEN), respectively, was performed as described previously (Johansson et al., 1993). For A2A receptor binding, coronal brain sections were preincubated at room temperate with 50 mm Tris-HCl buffer, pH 7.7, and 1 U of adenosine deaminase for 20 min and then incubated with the Tris buffer containing 2.5 nm3H-CGS 21680 for 60 min. For NMDA receptor binding, the slides were preincubated in 50 mmTris-acetate buffer twice at 4°C for 15 min each time and were then incubated with 5.0 nm3H-MK-801 in the presence of 30 μm glutamate and 10 μmglycine. To define nonspecific binding for the A2A and NMDA receptors, 20 μm of 2-chloroadenosine or 5.0 μm MK-801, respectively, was coincubated in adjacent sections.
For immunohistochemistry, mice were anesthetized with Avertin (2% tribromoethanol, 1% tertiary amylalcohol) and fixed by transcardial perfusion with 4% paraformaldehyde in 0.1 msodium cacodylate buffer, pH 7.4. The brains were post-fixed in the same solution for 2 hr and then cryoprotected in 20% glycerol. Brains were cut coronally in 25 μm sections with a sliding microtome. Immunostaining was performed in free-floating sections following standard avidin–biotin procedures described previously (Moratalla et al., 1996).
Hemodynamic and other physiological measurements. All procedures, measurements, and analyses were performed in a manner blinded to A2A receptor genotype. Adult littermate mice (male and female, weighing 18–25 gm) were housed in the Massachusetts General Hospital Knockout Core facility under conditions of diurnal light cycling with access to food and waterad libitum. Anesthesia was induced by 2% halothane and maintained with 1% halothane in 70% N2O and 30% O2 using a Fluotec 3 vaporizer (Colonial Medical). In randomly selected mice (n = 6 for each group), the right femoral artery was cannulated with PE-10 polyethylene tubes for arterial blood pressure and heart rate measurement (ETH 400 transducer and MacLab/8 data acquisition system; AD Instruments) and blood gas determination using a pH/Blood Gas Analyzer (Corning 178; Ciba Corning Diagnostics, Medfield, MA). Core temperature was measured using a BAT-12 thermometer (Physitemp, Clifton, NJ). Core temperature was maintained at ∼36.5–37.0°C with a thermostat (Frederick Haer Company, Bowdoinham, ME). Because hypothermia is a well known complication of prolonged ischemia, mice were kept in an incubator (ThermoCare Systems) at 32°C and 45% humidity for 6 hr after ischemia.
Focal transient ischemia (MCA occlusion) model. Focal cerebral ischemia was induced by occlusion of the left MCA with an 8–0 nylon monofilament (Ethicon, New Brunswick, NJ) coated with a mixture of silicone resin (Xantopren, Osaka, Japan) and a hardener (Elastomer Activator; Bayer, Etobicoke, Ontario, Canada) as described previously (Huang et al., 1994; Hara et al., 1996; Bonventre et al., 1997). This coated filament was introduced into the internal carotid artery through the external carotid artery, up to the origin of the anterior cerebral artery to occlude the MCA and anterior cerebral artery for 2 hr. For filament withdrawal, mice were briefly reanesthetized with halothane. In randomly selected mice (n = 6 for each group), cortical CBF was determined by a PF2B laser–Doppler flowmetry (Perimed, Stockholm, Sweden) and recorded on a MacLab/8 data acquisition system (AD Instruments). The tip of the probe was fixed 2 mm posterior and 6 mm lateral to bregma on the ipsilateral hemisphere. These coordinates identified the site on the convex brain surface within the vascular territory supplied by proximal segments of the MCA, and they corresponded to brain ischemic core area (Huang et al., 1994). Steady-state baseline values were recorded before MCA occlusion. Cortical CBF was recorded continuously before, during, and after ischemia and reperfusion and was expressed as percentage relative to the baseline value.
Measurement of neurological deficits and locomotion. For scoring neurological deficits, mice were ranked as described previously (Hara et al., 1996): 0, no observable neurological deficit (normal); 1, failure to extend right forepaw (mild); 2, circling to the contralateral side (moderate); and 3, loss of walking or righting reflex (severe). The animals were rated by an observer blinded to the genotypes.
Horizontal locomotor activity was assessed in polypropylene cages that were placed into adjustable frames equipped with seven infrared photocell beams, recorded, and analyzed on a computer (San Diego Instruments, San Diego, CA). Ambulation was quantified as the number of sequential breaks in adjacent beams. Mice were habituated in the test cages for at least 120 min before recording basal locomotion for 60 min. In assessing the motor-depressant effect of CGS 21680, locomotor activity was measured during the dark phase of the light cycle to obtain high basal locomotion. CGS 21680 was administered intraperitoneally (0.1 ml/10 gm), and locomotion was recorded for an additional 60 min.
Infarct volume measurement. Twenty-two hours after reperfusion, animals were decapitated under deep halothane anesthesia, and the brains were removed. For 2,3,5-triphenyltetrazolium chloride (TTC)-stained sections, brains were sectioned coronally into five 2 mm slices in a mouse brain matrix (RBM-2000C; Activational Systems). Slices were stained with 2% TTC (Sigma, St. Louis, MO) in PBS, followed by 10% formalin overnight. For hematoxylin–eosin stained cryostat sections, the brains were first immediately frozen in 2-methylbutane on dry ice and then sectioned coronally into 10 20-μm-thick slices (from +2.80 to −4.84 mm relative to bregma) in a microtome. Coronal sections were stained with hematoxylin and eosin. The infarct area (in square millimeters) of each TTC-stained section or hematoxylin–eosin-stained cryostat section was measured using an image analysis system (M4; Imaging Research, St. Catharine's, Ontario, Canada) on the posterior surface of each section. The total infarct volume was calculated by summing the volumes of the sections as described previously (Huang et al., 1994).
RESULTS
Targeted inactivation of the A2A receptor in homozygous mutant mice
To generate mice lacking the A2A receptor, a gene targeting vector was constructed with 10 kb of the murine A2A receptor gene disrupted by a positive selection marker, Neo (Fig. 1A). The replacement of a critical stretch of nucleotides at the junction of exon 2 and its 3′ intron with the Neo cassette was designed to ensure that the resulting mutant gene does not encode a functional A2A receptor. The A2Areceptor genotypes of mice generated with this vector were determined by Southern blot analysis, yielding the expected 7.5 and 5.0 kb labeled restriction fragments for wild-type and mutant alleles, respectively (Fig. 1A,B).
The absence of functional A2A receptors in A2A KO mice was demonstrated by receptor autoradiography with the A2A receptor agonist3H-CGS 21680 (Fig. 1C). WT mice show specific labeling of A2A receptors in the striatum and olfactory bulb, whereas homozygous A2A KO mice show no3H-CGS 21680 binding in these regions. Finally, we examined the behavioral response to the A2A receptor agonist CGS 21680 in A2A KO mice to confirm the functional inactivation of A2A receptors in the CNS. CGS 21680 (0.2 mg/kg) significantly decreased locomotor activity (p < 0.05) in WT mice. Although spontaneous locomotion in A2A KO mice was lower than in WT mice, it was not decreased further by treatment with CGS 21680 (Fig.1D). Similarly, the A2Aantagonist 8-(3-chlorostyryl)caffeine (CSC) induced motor stimulation in WT mice but not in A2A KO mice (data not shown). Together, these genetic, neurochemical, and behavioral data demonstrate the functional disruption of A2A receptors in homozygous mutant mice.
Development of striatum, cortex, and cerebral vasculature in the absence of A2A receptors
A2A KO mice appeared healthy and displayed no gross anatomical or behavioral abnormalities. The average body weight of WT and A2A KO mice between postnatal days 76 and 90 did not differ (30.3 ± 1.0 and 30.1 ± 1.0 gm, respectively, for male mice; 25.7 ± 1.2 and 27.3 ± 0.8 gm, respectively, for female mice; n = 23–24).
The neuropeptide enkephalin is highly colocalized with A2A receptors in the striatopallidal projection neurons (Shiffmann et al., 1991; Fink et al., 1992; Svenningsson et al., 1998), and thus its pattern of expression may be most sensitive to the absence of these receptors during development. The extent and distribution of enkephalin immunoreactivity appeared normal in KO mice (Fig. 2). Enkephalin immunostaining is concentrated in the matrix compartment of the striatum, and this pattern is preserved in A2A KO mice. Conversely, dynorphin is a relatively specific marker for the striosomal compartment (Graybiel, 1990). Characteristic clusters of dynorphin-immunoreactive neurons in A2A KO striatum were indistinguishable from those in WT mice (Fig. 2). In addition, cortical lamination assessed by Nissl staining was indistinguishable between A2A WT and KO mice (data not shown). These results indicate that striatal and cortical architecture appears to have developed normally in the absence of A2A receptors.
Fig. 2.
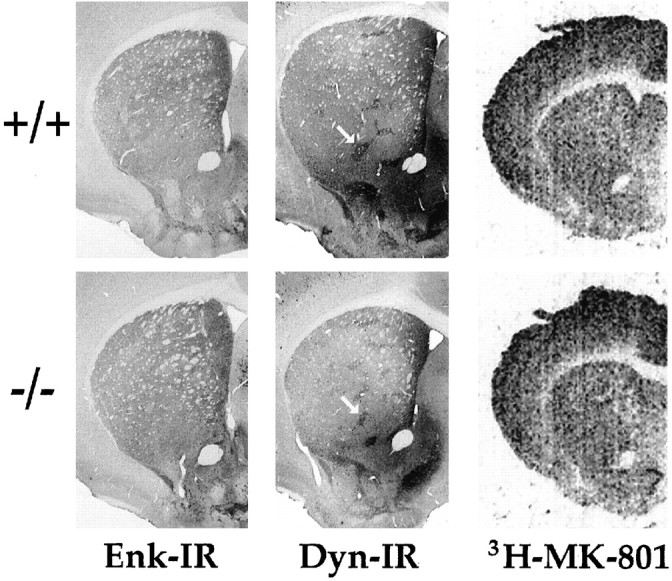
Neurochemical markers of striatal and cortical development in the absence of A2A receptor. Representative coronal sections through corresponding levels of cortex and striatum are shown for A2A WT (+/+, top) and KO (−/−, bottom) adult mice. Immunohistochemistry for enkephalin (Enk-IR) or dynorphin (Dyn-IR) and receptor autoradiography for NMDA receptor (3H-MK-801) in striatum and cortex were performed on brain sections as described in Materials and Methods. The characteristic striosomal pattern of dynorphin in striatum was indistinguishable between A2A WT and KO mice (arrows).
The possibility of neurochemical adaptations to the absence of A2A receptors in the KO mice was also considered. Because NMDA receptors play a critical role in focal ischemia-induced neuronal cell death, we measured their binding density in cortex and striatum by receptor autoradiography using3H-MK-801 (dizocilpine). The levels for MK-801 binding sites were relatively high in cortex and moderate in striatum, and there was no significant difference between A2A KO mice and their WT littermate (n = 5–6). The MK-801 binding densities in cortex were 856 ± 84 and 747 ± 37 fmol/mg tissue, and in striatum were 840 ± 89 and 777 ± 59 fmol/mg tissue, for A2A KO and WT mice, respectively (Fig. 2).
Cerebrovascular and systemic physiology are indistinguishable between A2A KO and WT mice in the MCA occlusion ischemia model
In the A2A KO and WT mice with hybrid genetic background (C57BL/6 × 129-Steel; n = 6) (Table 1), there were no significant differences in basal mean arterial blood pressure (MABP) and heart rate. MABP in unanesthetized free-moving mice (pure 129-steel strain) also did not differ between the two groups (115 ± 5 and 110 ± 3.7 mmHg for A2A WT and KO mice, respectively). Furthermore, preliminary study showed no significant difference in absolute blood flow between the two groups (data not shown). To exclude the potential contribution of strain-specific genes to the phenotypes, we also measured these physiological parameters in anesthetized A2A KO and WT mice of a pure 129-Steel substrain (Simpson et al., 1997). Although we detected higher basal MABP in the pure 129-Steel mice (96 ± 12 and 98 ± 3 mmHg for A2A KO and WT mice, respectively) compared with the hybrid C57BL/6 × 129-Steel mice (77 ± 4 and 74 ± 5 mmHg for A2A KO and WT mice, respectively), there was no significant difference in MABP between A2A KO and WT mice in either genetic background. Of note, Ledent et al. (1997) reported a hypertensive phenotype of A2A KO mice, in contrast to the normal MABP we observed. The discrepancy in MABP may caused by the different strains of A2A KO mice generated by Ledent et al. (1997)(hybrid CD-1 × 129/Sv strain) and our group (a hybrid C57BL/6 × 129-Steel strain, as well as a pure 129-Steel substrain).
Table 1.
Cerebrovascular and systemic physiology before, during, and after MCA occlusion-induced ischemia in A2A wild-type and knock-out mice
| Mean arterial BP (mmHg) | Cortical blood flow (%) | Heart rate (beats/min) | ||||
| Hemodynamics | A2A+/+ | A2a −/− | A2A+/+ | A2a −/− | A2A+/+ | A2a −/− |
| Before ischemia | ||||||
| 5′ | 74 ± 5 | 77 ± 4 | 100 ± 0 | 100 ± 0 | 486 ± 45 | 466 ± 38 |
| During ischemia | ||||||
| 5′ | 80 ± 10 | 78 ± 13 | 20 ± 7 | 18 ± 3 | 494 ± 101 | 483 ± 39 |
| 10′ | 79 ± 10 | 76 ± 13 | 22 ± 8 | 19 ± 4 | 529 ± 70 | 495 ± 46 |
| 20′ | 80 ± 11 | 74 ± 8 | 21 ± 6 | 19 ± 4 | 542 ± 51 | 504 ± 63 |
| 30′ | 82 ± 10 | 75 ± 8 | 22 ± 5 | 20 ± 3 | 557 ± 75 | 506 ± 64 |
| Before reperfusion | ||||||
| 5′ | 81 ± 7 | 84 ± 5 | 22 ± 7 | 20 ± 3 | 480 ± 21 | 471 ± 79 |
| During reperfusion | ||||||
| 5′ | 81 ± 5 | 83 ± 7 | 66 ± 32 | 63 ± 21 | 474 ± 24 | 473 ± 70 |
| 10′ | 86 ± 9 | 83 ± 8 | 86 ± 19 | 82 ± 20 | 476 ± 23 | 461 ± 62 |
| 20′ | 85 ± 11 | 87 ± 12 | 104 ± 20 | 96 ± 21 | 498 ± 62 | 473 ± 108 |
| 30′ | 87 ± 9 | 85 ± 13 | 110 ± 11 | 108 ± 20 | 485 ± 80 | 475 ± 108 |
| pH | PaCO2 (mmHg) | PaO2 (mmHg) | ||||
| Blood gas analysis | A2A+/+ | A2a −/− | A2A+/+ | A2a −/− | A2A+/+ | A2a −/− |
| Before ischemia | 7.34 ± 0.08 | 7.25 ± 0.05 | 40 ± 7 | 48 ± 4 | 140 ± 25 | 133 ± 26 |
| After ischemia | 7.30 ± 0.05 | 7.28 ± 0.05 | 46 ± 6 | 50 ± 7 | 166 ± 31 | 163 ± 52 |
| Body temperature (°C) | A2A+/+ | A2a −/− | ||||
| Before ischemia | 36.9 ± 0.31 | 36.9 ± 0.23 | ||||
| After ischemia | ||||||
| 1 hr | 36.9 ± 0.39 | 36.8 ± 0.30 | ||||
| 3 hr | 36.6 ± 0.32 | 36.7 ± 0.28 | ||||
| 6 hr | 36.5 ± 0.26 | 36.6 ± 0.16 | ||||
| 20 hr | 36.4 ± 0.20 | 36.4 ± 0.15 | ||||
Other physiological parameters that may influence ischemic injury did not differ between the two groups at any time point during the experiment. Immediately after MCA occlusion, cortical CBF decreased to ∼20% of baseline and remained at this level during the 2 hr of ischemia (n = 6) (Table 1). After reperfusion, cortical CBF increased to 98–100% in both groups within 5 min. There were no significant differences in cortical CBF before, during, and after MCA occlusion between A2A WT and KO mice with hybrid genetic background (C57BL/6 × 129-Steel) (Table 1) or with pure 129-Steel genetic background (data not shown). Finally, before and after ischemia, there was also no difference in core body temperature, arterial pH, or blood gas (PaO2 and PaCO2) values of mice with a hybrid C57BL/6 × 129-Steel strain (Table 1) or a pure 129-Steel substrain (data not shown) between A2A KO and WT littermates.
A2A receptor inactivation attenuates transient MCA occlusion-induced cerebral infarction
Twenty-two hours after reperfusion (i.e., 24 hr after onset of ischemia), total and regional (cortical and striatal) infarction was assessed by hematoxylin and eosin staining with volumetric analysis. Total infarct volume was reduced by 26% in A2AKO (C57BL/6 × 129-Steel) mice compared with their WT littermates (61.2 ± 9.1 compared with 87.2 ± 3.2 mm3, respectively; n = 6;p < 0.05) (Fig.3A). Similarly, in pure 129-Steel mice, infarct volume was reduced by 30% in A2A KO mice (51.0 ± 4.9 mm3) compared with that of their WT littermates (72.8 ± 3.5 mm3;p < 0.05). In a separate set of experiments, ischemic lesion volume was also determined by staining with TTC, a marker of intact cellular metabolism. A2A KO mice (C57BL/6 × 129-Steel) showed an even more pronounced (77%) reduction of total lesion volume compared with their WT littermate control (18.0 ± 4.7 compared with 77.6 ± 13.4 mm3, respectively; n = 8–9; p < 0.05). Lesion volumes appeared intermediate in size for A2A heterozygous mice (56.1 ± 20.2 mm3; n = 6;p > 0.05 compared with WT, and p < 0.05 compared KO mice).
Fig. 3.
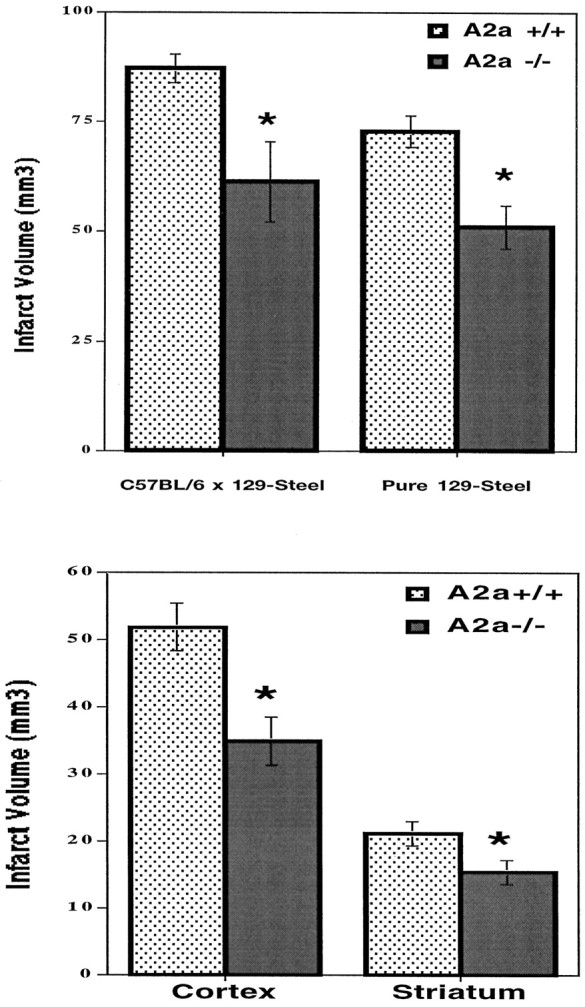
Inactivation of A2A receptors attenuated MCA occlusion-induced infarction. A, Twenty-two hours after reperfusion, infarct volumes were determined using hematoxylin and eosin staining as described in Materials and Methods for A2A KO and WT mice with hybrid C57BL/6 × 129-Steel genetic background (n = 6) as well as with pure 129-Steel genetic background (n = 11–12). B, Regional infarct volume was analyzed with respect to cerebral cortex and striatum of pure 129-Steel substrain mice (n = 11–12). *p < 0.05 when comparing infarct volumes of A2A KO mice with those of WT littermates (Student's t test).
Analysis of discrete infarct areas shows significant reductions in both cerebral cortex (33%) and striatum (27%) of A2AKO mice (pure 129-Steel) when compared with the WT littermates (Fig.3B). Cortical infarct volumes were 51.9 ± 3.1 and 35.0 ± 3.6 mm3 for A2A WT and KO mice, respectively (n = 6; p < 0.05). Striatal infarct volumes were 21.2 ± 1.3 and 15.5 ± 1.8 mm3 for A2AWT and KO mice, respectively (p < 0.05). Furthermore, there was no significant induction of A2A receptors in cortex or striatum 24 hr after ischemia in WT mice. In fact, receptor autoradiography showed that focal ischemia significantly reduced binding density in the ipsilateral striatum (data not shown).
A2A receptor inactivation preserves behavioral function after ischemic injury
We also evaluated functional outcome after MCA occlusion. A2A KO mice displayed significantly fewer signs of neurological deficit compared with their WT littermates 24 hr after ischemia. Neurological deficit scores, assigned by an observer blinded to genotype, were reduced by 50–60% in A2A KO compared with WT mice, in both hybrid C57BL/6 × 129-Steel (n = 6; p < 0.05) and pure 129-Steel (n = 11–12; p < 0.05) genetic backgrounds (Fig. 4).
Fig. 4.
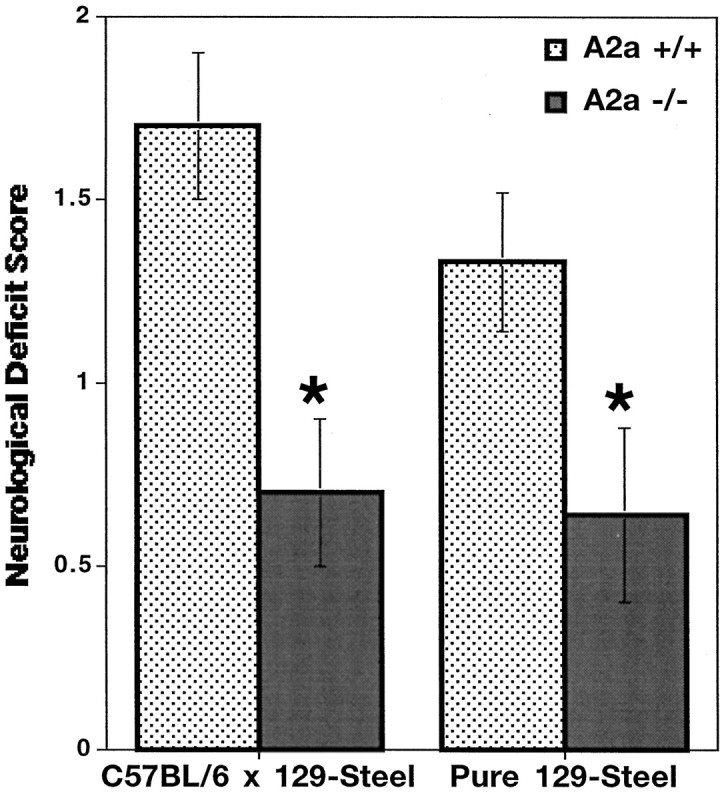
Inactivation of A2A receptors enhances neurological function after MCA occlusion. Neurological deficit behavioral scores were assessed by a trained observer in a blinded manner as described in Materials and Methods. Neurological deficits were determined for A2A KO and WT mice with the hybrid C57BL/6 × 129-Steel strain (n = 6) and with the pure 129-Steel strain (n = 11–12). *p < 0.05 when comparing neurological deficit scores of A2A KO mice with those of their WT littermates (Mann–Whitney U test).
DISCUSSION
The present study using an A2A KO model clearly demonstrates that inactivation of the A2Areceptor protects the brain from transient focal ischemia. MCA occlusion followed by reperfusion produces significantly smaller infarct volumes and fewer neurological deficits in mice lacking the A2A receptor. These data establish an important role for the A2A receptor in neuroprotec- tion against ischemic injury and advance the prospects for A2A receptor blockade as a pharmacological strategy to reduce ischemic brain injury.
Transgenic inactivation of A2A receptors attenuates ischemic injury
Pharmacological analyses of A2A receptor involvement in cerebral ischemia have produced conflicting results. Nonselective antagonists caffeine and theophylline have been shown to either potentiate or attenuate ischemia-induced brain damage depending on treatment paradigm (Rudolphi et al., 1992; Jacobson et al., 1996). Furthermore, both agonists (Scheardown and Knutsen, 1996; Jones et al., 1998) and antagonists (Phillis, 1995; Jones et al., 1998; Monopoli et al., 1998) with relative selectivity for A2Areceptors have been shown to protect against brain damage in animal models of ischemic and excitotoxic neuronal injury. These mixed results may reflect complex actions of A2A receptor activation during ischemia, as well as the intrinsic pharmacokinetic limitations of A2A adenosine agents. For example, CGS 21680, one of the most selective and widely used A2A receptor agonists, displays only a 140-fold selectivity for A2A over A1receptors (Jacobson et al., 1992; Ongini and Fredholm, 1996). However, the effective concentrations of CGS 21680 used in different studies to modify neuronal death have differed by as much as 5000-fold (Scheardown and Knutsen, 1996; Jones et al., 1998). Similarly, CSC, an A2A receptor antagonist frequently used to explore the effects of A2A receptor blockade on ischemic damage, possesses high A2A over A1 receptor selectivity but displays poor solubility and CNS permeability, and it rapidly photoisomerizes to an inactive form (Ongini and Fredholm, 1996). These pharmacological limitations are overcome by genetic deletion of the A2A receptor, leading to complete and selective inactivation of the A2A receptor in A2A KO mice. Thus, attenuation of MCA occlusion-induced cerebral infarction and neurological disability in A2A KO mice provides the strongest evidence to date that blockade of the A2A receptor reduces ischemic damage. Together with previous demonstrations of A2A antagonist-induced protection from global or permanent ischemia, the neuroprotection observed in a transient focal ischemia model in A2A KO mice advances the prospects for pharmacological intervention in ischemic stroke by blocking A2A receptors.
Despite their important advantages, transgenic models raise unique considerations that must be addressed to properly interpret the data they generate (Silva et al., 1997). Most critically, a potential contribution of genetic background to a KO phenotype must be ruled out before the phenotype can be definitely attributed to the disruption of the “knocked-out” gene (Banbury Conference on Genetic Background in Mice, 1997). Transgenic studies of neuroprotection may be particularly susceptible to misinterpretation because of differences in genetic background (Schauwecker and Steward, 1997). In the present study, the attenuation of ischemic brain damage observed in A2A KO mice on a standard hybrid genetic background (C57BL/6 × 129-Steel) was confirmed in separately derived A2A KO mice of a pure 129-Steel substrain. This result verifies A2A receptor inactivation as the basis for neuroprotection from cerebral ischemia in A2A KO mice.
Also in contrast to pharmacological approaches, an A2A receptor KO model does not readily distinguish between developmental, chronic, and acute effects of receptor inactivation. The detection of A2Areceptor mRNA in the CNS as early as embryonic day 15 in rats (Weaver, 1993) raises the possibility that brain development could be altered in A2A KO mice such that their predisposition to ischemic injury is reduced. Similarly, chronic A2A receptor antagonism may lead to upregulation of other receptors such as the A1receptor, which can itself attenuate ischemic injury. Indeed, neuroprotection offered by chronic treatment with the nonselective adenosine antagonist caffeine has been attributed to upregulation of A1 receptors (Jacobson et al., 1996).
We found no evidence for contributing developmental or chronic changes in relevant anatomical and neurochemical systems. Vascular and parenchymal brain structures that we assessed were indistinguishable between A2A KO and WT mice. Nissl staining and neuropeptide immunohistochemistry demonstrate normal laminar and compartmental patterns of cortical and striatal organization, respectively. Focal ischemic injury can be reduced by the NMDA receptor antagonist MK-801 (Pulsinelli et al., 1993) and the A1 receptor agonist CHA (Rudolphi et al., 1992; von Lubitz, 1997). However, we demonstrated normal tritiated MK-801 binding density (Fig. 2C) and found no evidence for upregulation of A1 receptor binding site density using the tritiated A1 receptor ligands N6 cyclohexyladenosine (CHA) and 1,3-dipropyl-8-cyclopentylxanthine (J.-F. Chen and M. A. Schwarzschild, unpublished observations). Thus, neurochemical assessment of these receptor binding sites showed no alteration in A2A KO brains to account for their resistance to ischemia.
Alternatively, neuroprotection from cerebral ischemia in A2A KO mice may reflect an effect of A2A receptor inactivation during ischemic injury. Recent studies with a new generation of more specific A2A receptor antagonists have suggested that blocking A2A receptors can directly contribute to neuroprotection in a model of kainate-induced hippocampal damage (Jones et al., 1998), neonatal hypoxia–ischemia (Bona et al., 1997), and cerebral ischemia (Monopoli et al., 1998). Monopoli et al. (1998), for example, found that low doses of the A2A receptor antagonist SCH 58261 (which binds A2A receptors with 500-fold greater affinity than A1 receptors) significantly reduces cortical infarction volume, even when administered 10 min after MCA occlusion. These emerging data are consistent with an acute effect of A2A receptor deficiency in the neuroprotection observed in A2A KO mice.
Multiple mechanisms may underlie the neuroprotection offered by A2A KO mice
The apparent contradictions of published data on the A2A receptor in neuroprotection point not only to the shortcomings of A2A receptor pharmacology but also to the complexity of A2A receptor biology. Indeed, multiple mechanisms, involving neuronal, vascular, and microglial elements, may underlie protection from cerebral ischemia offered by A2A receptor deficiency in A2A KO mice.
A neuronal basis for A2A receptor modulation of ischemic injury has been suggested by studies showing adenosinergic regulation of glutamate and aspartate release. A massive release of these excitatory amino acids during brain ischemia plays a critical role in subsequent neuronal death. A1 receptor stimulation attenuates this release and in this way likely attenuates ischemic damage (Rudolphi et al., 1992; von Lubitz, 1997). Conversely, A2A receptor agonists enhance the release of glutamate under ischemic and nonischemic conditions (O'Regan et al., 1992; Simpson et al., 1992; Popoli et al., 1995), as well as the release of other neurotransmitters such as acetylcholine (Sebastiao and Ribeiro, 1996; Dunwiddie and Fredholm, 1997). To the extent that the inhibition of release by the A1 receptor can be attributed to its negative coupling to calcium influx and/or cAMP production (Fredholm et al., 1994, 1997), the enhancement of neurotransmitter release by the A2A receptor may be caused by its positive coupling to these second messenger systems (Gubitz et al., 1996; Fredholm et al., 1997). Thus, a pharmacological blockade or transgenic deficiency of the A2Areceptor may afford neuroprotection after ischemia because of reduced glutamate release and excitotoxicity.
Interestingly, the prominence of cortical as well as subcortical (striatal) protection from ischemia in A2A KO mice belies the intense localization of brain A2Areceptors to the striatum (Fig. 1C). This apparent mismatch of regional protection and receptor density may be explained by the ability of the relatively sparse but well documented cortical A2A receptors to markedly enhance glutamate release (Johansson et al., 1993; Sebastiao and Ribeiro, 1996; Dunwiddie and Fredholm, 1997). Evidence that these relatively low levels of A2A receptor are in fact sufficient for potentiating neurotransmitter release is provided by demonstrations of A2A receptors agonist-induced release in cortical (synaptosomal and slice) preparations (Sebastiao and Ribeiro, 1996;Dunwiddie and Fredholm, 1997). Monopoli et al. (1998) also noted the discrepancy between cortical protection by A2Areceptor inactivation and the dearth of cortical A2A receptors. Although they raise the interesting possibility that ischemia may induce A2A receptors in cortical glia, we found no autoradiographic evidence for cortical A2Areceptor induction 24 hr after transient ischemia. Alternatively, A2A receptors may act trans-synaptically (at a neuronal network level) to modify cortical ischemic damage. For example, the extensive feedback projection from striatum to cortex via glutamatergic thalamocortical neurons potentially links striatal A2A receptors with excitatory nerve terminals in cortex.
A vascular basis for the anti-ischemic phenotype of A2A KO mice might also explain the widespread cerebral protection (i.e., far beyond the high density of neuronal A2A receptors in striatum). A2A adenosine agents are well known for their vasoactive properties (Phillis, 1989; Ibayashi et al., 1991), which result from the functions of A2A receptors located on cerebral, as well as systemic, vasculature (Kalaria and Hank, 1986). However, activation of A2Areceptors on cerebral vascular smooth muscle and endothelial cells produces vasodilatation and thus may increase cerebral blood flow (Phillis, 1989; Ibayashi et al., 1991). Indeed, under hypoxic conditions, cortical blood flow is enhanced by the A2A receptor agonist CGS 21680 and is reduced by the specific A2A receptor antagonist ZM 241385 (Coney and Marshall, 1988). Hence, the pharmacological data would not predict a vascular mechanism of attenuated ischemic damage in A2A KO mice. Moreover, direct comparison of cerebral blood flow and systemic cardiovascular parameters showed no difference between A2A KO and WT mice before, during, or after MCA occlusion.
A2A receptor activation also regulates the aggregation of platelets and the generation of reactive oxygen species, which may participate in the development of ischemic injury. Again however, A2A receptor pharmacology would suggest that these functions do not contribute to the cerebroprotective phenotype of A2A KO mice. A2A receptor agonists have been shown to inhibit platelet aggregation (Sandoli et al., 1994; Ledent et al., 1997) and free radical generation by neutrophils (Cronstein, 1994; Jordan et al., 1997). Thus, A2A receptor inactivation may be expected to enhance platelet initiation of vascular occlusion and neutrophil-triggered oxidative damage, neither of which would account for the observed reduction in infarct size in mice lacking the A2A receptor. Furthermore, in our study, all other physiological parameters assessed for their potential contribution to ischemic injury (body temperature and blood pH, oxygenation and CO2 content) were indistinguishable between KO and WT mice, both before and after ischemia. Together with previously published A2Areceptor pharmacology, the physiological and anatomical data reported here argue against a vascular mechanism underlying the neuroprotection seen in A2A KO mice. Because of offsetting vascular actions of A2A receptors, the potential for neuroprotection by A2A receptor inactivation may be significantly underestimated. Selective blockade of neuronal A2A receptors may therefore provide further protection against transient focal ischemia in brain than was observed in A2A KO mice.
In conclusion, the A2A KO model presented here demonstrates that A2A receptor deficiency attenuates cerebral damage and dysfunction induced by transient focal ischemia and suggests that A2A receptor stimulation may normally exacerbate cerebral infarction. Together with the well established protective effect of A1receptor stimulation, our data support a more refined view of adenosine signaling in ischemic brain injury. The high levels of extracellular adenosine in ischemic brain tissues may trigger offsetting A1 and A2A receptor effects on neurotoxicity, possibly through opposing influences on glutamate release. The marked preservation of neurological function associated with attenuated cerebral infarction in A2A KO mice highlights the potential benefit of A2Areceptor antagonists in the treatment of ischemic stroke. Moreover, the proposed model encourages the rational development of neuroprotective strategies involving potentially additive or synergistic effects of A2A receptor blockade combined with A1 receptor stimulation.
Footnotes
This work was supported by National Institutes of Health Grants DA07496, 5P50 NS10828, NS01729, and NS31579 and grants from the National Alliance for Research on Schizophrenia and Depression, the Scottish-Rite, and the National Parkinson Foundation. We thank Dr. E. Li for assistance in the generation of the A2A knock-out mice, and Yuehang Xu and Mark Beilstein for excellent technical assistance.
Correspondence should be addressed to Dr. Jiang-Fan Chen, Molecular Neurobiology Laboratory, 149 Massachusetts General Hospital East, 13th Street, Charlestown, MA 02129. E-mail: chenjf@helix.mgh.harvard.edu.
REFERENCES
- 1.Banbury conference on genetic background in mice. Mutant mice and neuroscience: recommendations concerning genetic background. Neuron. 1997;19:755–759. doi: 10.1016/s0896-6273(00)80958-7. [DOI] [PubMed] [Google Scholar]
- 2.Bona E, Aden U, Gilland E, Fredholm BB, Hagberg H. Neonatal cerebral hypoxia ischemia: the effect of adenosine receptor antagonists. Neuropharmacology. 1997;36:1327–1338. doi: 10.1016/s0028-3908(97)00139-1. [DOI] [PubMed] [Google Scholar]
- 3.Bonventre JV, Huang Z, Taheri MR, O'Leary E, Li E, Moskowitz MA, Sapirstein A. Reduced fertility and postischemic brain injury in mice deficient in cytosolic phospholipase A2. Nature. 1997;390:622–625. doi: 10.1038/37635. [DOI] [PubMed] [Google Scholar]
- 4.Chen J-F, Fink JS. Characterization of the genomic structure of mouse A2aR adenosine receptor gene. Soc Neurosci Abstr. 1996;22:1569. [Google Scholar]
- 5.Coney AM, Marshall JM. Role of adenosine and its receptors in the vasodilation induced in the cerebral cortex of the rat by systemic hypoxia. J Physiol (Lond) 1988;509:507–518. doi: 10.1111/j.1469-7793.1998.507bn.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Corsi C, Pazzagli M, Bianchi L, Della Corte L, Pepeu G, Pedata F. In vivo amino acid release from the striatum of aging rats: adenosine modulation. Neurobiol Aging. 1997;18:243–250. doi: 10.1016/s0197-4580(97)00002-x. [DOI] [PubMed] [Google Scholar]
- 7.Cronstein BN. Adenosine, an endogenous anti-inflammatory agent. J Appl Physiol. 1994;76:5–13. doi: 10.1152/jappl.1994.76.1.5. [DOI] [PubMed] [Google Scholar]
- 8.Deckert J, Gleiter CH. Adenosine-an endogenous neuroprotective metabolite and neuromodulator. J Neural Transm. 1994;43:23–31. [PubMed] [Google Scholar]
- 9.Dunwiddie TV, Fredholm BB. Adenosine neuromodulation. In: Jacobson KA, Jarvis MF, editors. Purinergic approaches in experimental therapeutics. Wiley-Liss; New York: 1997. pp. 359–382. [Google Scholar]
- 10.Fink JS, Weaver DR, Rivkees SA, Peterfreund RA, Pollack AE, Adler EM, Reppert SM. Molecular cloning of the rat A2 adenosine receptor: selective coexpression with D2 dopamine receptors in rat striatum. Mol Brain Res. 1992;14:186–195. doi: 10.1016/0169-328x(92)90173-9. [DOI] [PubMed] [Google Scholar]
- 11.Fredholm BB, Abbracchio MP, Burnstock G, Daly JW, Harden TK, Jacobson KA, Leff P, Williams M. Nomenclature and classification of purinoceptors. Pharmacol Rev. 1994;46:143–156. [PMC free article] [PubMed] [Google Scholar]
- 12.Fredholm BB, Arslan G, Kull B, Kontny E, Svenningsson P. Adenosine (P1) receptor signalling. Drug Dev Res. 1997;39:262–268. [Google Scholar]
- 13.Gao Y, Phillis JW. CGS 15943, an adenosine A2 receptor antagonist, reduces cerebral ischaemic injury in the Mongolian gerbil. Life Sci. 1994;55:PL61–PL65. doi: 10.1016/0024-3205(94)00889-2. [DOI] [PubMed] [Google Scholar]
- 14.Graybiel AM. Neurotransmitters and neuromodulators in the basal ganglia. Trends Neurosci. 1990;13:244–254. doi: 10.1016/0166-2236(90)90104-i. [DOI] [PubMed] [Google Scholar]
- 15.Gubitz AK, Widdowson L, Kurokawa M, Kirkpatrick KA, Richardson PJ. Dual signalling by the adenosine A2A receptor involves activation of both N- and P-type calcium channels by different G proteins and protein kinases in the same striatal nerve terminals. J Neurochem. 1996;67:374–381. doi: 10.1046/j.1471-4159.1996.67010374.x. [DOI] [PubMed] [Google Scholar]
- 16.Hagberg H, Andersson P, Lacarewicz J, Jacobson I, Butcher S, Sandberg M. Extracellular adenosine, inosine, hypoxanthine, and xanthine in relation to tissue nucleotides and purines in rat striatum during transient ischemia. J Neurochem. 1987;49:227–231. doi: 10.1111/j.1471-4159.1987.tb03419.x. [DOI] [PubMed] [Google Scholar]
- 17.Hara H, Huang PL, Panahian N, Fishman MC, Moskowitz MA. Reduced brain edema and infarction volume in mice lacking the neuronal isoform of nitric oxide synthase after transient MCA occlusion. J Cereb Blood Flow Metab. 1996;16:605–611. doi: 10.1097/00004647-199607000-00010. [DOI] [PubMed] [Google Scholar]
- 18.Huang Z, Huang PL, Panahian N, Dalkara T, Fishman MC, Moskowitz MA. Effects of cerebral ischemia in mice deficient in neuronal nitric oxide synthase. Science. 1994;265:1883–1885. doi: 10.1126/science.7522345. [DOI] [PubMed] [Google Scholar]
- 19.Ibayashi S, Ngai AC, Meno JR, Winn HR. Effects of topical adenosine analogs and forskolin on rat pial arterioles in vivo. J Cereb Blood Flow Metab. 1991;11:72–76. doi: 10.1038/jcbfm.1991.8. [DOI] [PubMed] [Google Scholar]
- 20.Jacobson KA, van Galen PJ, Williams M. Adenosine receptors –pharmacology, structure activity relationships, and therapeutic potential. J Med Chem. 1992;35:407–422. doi: 10.1021/jm00081a001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jacobson KA, von Lubitz DK, Daly JW, Fredholm BB. Adenosine receptor ligands: differences with acute versus chronic treatment. Trend Pharmacol Sci. 1996;17:108–113. doi: 10.1016/0165-6147(96)10002-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Johansson B, Georgiev V, Parkinson FE, Fredholm BB. The binding of the adenosine A2A receptor selective agonist [3H]CGS 21680 to rat cortex differs from its binding to rat striatum. Eur J Pharmacol. 1993;247:103–110. doi: 10.1016/0922-4106(93)90066-i. [DOI] [PubMed] [Google Scholar]
- 23.Jones PA, Smith RA, Stone TW. Protection against kainate-induced excitotoxicity by adenosine A2A receptor agonists and antagonists. Neuroscience. 1998;85:229–237. doi: 10.1016/s0306-4522(97)00613-1. [DOI] [PubMed] [Google Scholar]
- 24.Jordan JE, Zhao Z-Q, Sato H, Taft S, Vinten-Johansen J. Adenosine A2receptor activation attenuates reperfusion injury by inhibiting neutrophil accumulation, superoxide generation and coronary endothelial adherence. J Pharmacol Exp Ther. 1997;280:301–309. [PubMed] [Google Scholar]
- 25.Kalaria RN, Harik SI. Adenosine receptors of cerebral microvessels and choroid plexus. J Cereb Blood Flow Metab. 1986;6:463–470. doi: 10.1038/jcbfm.1986.80. [DOI] [PubMed] [Google Scholar]
- 26.Ledent C, Vaugeois J-M, Schiffmann SN, Pedrazzini T, Yacoubi M, Vanderhaeghen J-J, Costentin J, Heath JK, Vassart G, Parmentier M. Aggressiveness, hypoalgesia and high blood pressure in mice lacking the adenosine A2A receptor. Nature. 1997;388:674–678. doi: 10.1038/41771. [DOI] [PubMed] [Google Scholar]
- 27.Li E, Bestor TH, Jaenisch R. Targeted mutation of the DNA methyltransferase gene results in embryonic lethality. Cell. 1992;69:915–926. doi: 10.1016/0092-8674(92)90611-f. [DOI] [PubMed] [Google Scholar]
- 28.Matsumoto K, Graf R, Rosner G, Shimada N, Heiss W-D. Flow thresholds for extracellular purine catabolite elevation in cat focal ischemia. Brain Res. 1992;579:309–314. doi: 10.1016/0006-8993(92)90066-i. [DOI] [PubMed] [Google Scholar]
- 29.Monopoli A, Lozza G, Forlani A, Mattavelli A, Ongini E. Blockade of A2A adenosine receptors by SCH 58261 results in neuroprotective effects in cerebral ischaemia in rats. NeuroReport. 1998;9:3955–3959. doi: 10.1097/00001756-199812010-00034. [DOI] [PubMed] [Google Scholar]
- 30.Moratalla R, Elibol B, Vallejo M, Graybiel AM. Network-level changes in inducible Fos-Jun proteins in the striatum during chronic cocaine treatment and withdrawal. Neuron. 1996;17:147–156. doi: 10.1016/s0896-6273(00)80288-3. [DOI] [PubMed] [Google Scholar]
- 31.Ongini E, Fredholm BB. Pharmacology of adenosine A2A receptors. Trend Pharmacol Sci. 1996;17:364–372. [PubMed] [Google Scholar]
- 32.Ongini E, Dionisotti S, Gessi S, Irenius E, Fredholm BB. Comparison of CGS 15943, ZM 241385 and SCH 58261 as antagonists at human adenosine receptors. Naunyn Schmiedebergs Arch Pharmacol. 1999;359:7–10. doi: 10.1007/pl00005326. [DOI] [PubMed] [Google Scholar]
- 33.O'Regan MH, Simpson RE, Perkins LM, Phillis JW. The selective A2 agonist CGS 21680 enhances excitatory transmitter amino acid release from the ischemic rat cerebral cortex. Neurosci Lett. 1992;138:169–172. doi: 10.1016/0304-3940(92)90498-v. [DOI] [PubMed] [Google Scholar]
- 34.Peterfreund RA, MacCollin M, Gusella J, Fink JS. Characterization and expression of the human A2aR adenosine receptor gene. J Neurochem. 1996;66:362–368. doi: 10.1046/j.1471-4159.1996.66010362.x. [DOI] [PubMed] [Google Scholar]
- 35.Phillis JW. Adenosine in the control of the cerebral circulation. Cerebrovasc Brain Metab Rev. 1989;1:26–54. [PubMed] [Google Scholar]
- 36.Phillis JW. The effects of selective A1 and A2a adenosine receptor antagonists on cerebral ischemic injury in the gerbil. Brain Res. 1995;705:79–84. doi: 10.1016/0006-8993(95)01153-6. [DOI] [PubMed] [Google Scholar]
- 37.Phillis JW. Adenosine agonists and antagonists. In: Welch KMA, Caplan LR, Reis DJ, Siesjo BK, Weir B, editors. Primer on cerebrovascular disease. Academic; San Diego: 1997. pp. 250–253. [Google Scholar]
- 38.Popoli P, Betto P, Reggio R, Ricciarello G. Adenosine A2A receptor stimulation enhances striatal extracellular glutamate levels in rats. Eur J Pharmacol. 1995;287:215–217. doi: 10.1016/0014-2999(95)00679-6. [DOI] [PubMed] [Google Scholar]
- 39.Pulsinelli W, Sarokin A, Buchan A. Antagonism of the NMDA and non-NMDA receptors in global versus focal brain ischemia. Prog Brain Res. 1993;96:125–135. doi: 10.1016/s0079-6123(08)63262-8. [DOI] [PubMed] [Google Scholar]
- 40.Rosin DL, Robeva A, Woodard RL, Guyenet PG, Linden J. Immunohistochemical localization of adenosine A2A receptors in the rat central nervous system. J Comp Neurol. 1998;401:163–186. [PubMed] [Google Scholar]
- 41.Rudolphi K, Schubert P, Parkinson FE, Fredholm BB. Neuroprotective role of adenosine in cerebral ischaemia. Trends Pharmacol Sci. 1992;13:439–445. doi: 10.1016/0165-6147(92)90141-r. [DOI] [PubMed] [Google Scholar]
- 42.Sandoli D, Chiu PJS, Chintala M, Dionisotti S, Ongini E. In vivo and ex vivo effects of adenosine A1 and A2 receptor agonists on platelet aggregation in the rabbit. Eur J Pharmacol. 1994;259:43–49. doi: 10.1016/0014-2999(94)90155-4. [DOI] [PubMed] [Google Scholar]
- 43.Schauwecker PE, Steward O. Genetic determinants of susceptibility to excitotoxic cell death: Implications for gene targeting approaches. Proc Natl Acad Sci USA. 1997;94:4103–4108. doi: 10.1073/pnas.94.8.4103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Scheardown MJ, Knutsen LJS. Unexpected neuroprotection ob- served with the adenosine A2A receptor agonist CGS 21680. Drug Dev Res. 1996;39:108–114. [Google Scholar]
- 45.Schiffmann SN, Jacobs O, Vanderhaeghen J-J. Striatal restricted adenosine A2 receptor (RDC8) is expressed by enkephalin but not by substance P neurons: an in situ hybridization histochemistry study. J Neurochem. 1991;57:1062–1067. doi: 10.1111/j.1471-4159.1991.tb08257.x. [DOI] [PubMed] [Google Scholar]
- 46.Sebastiao AM, Ribeiro JA. Adenosine A2 receptor-mediated excitatory actions on the nervous system. Prog Neurobiol. 1996;48:167–189. doi: 10.1016/0301-0082(95)00035-6. [DOI] [PubMed] [Google Scholar]
- 47.Silva AJ, Smith AM, Gieses KP. Gene targeting and the biology of learning and memory. Annu Rev Genet. 1997;31:527–546. doi: 10.1146/annurev.genet.31.1.527. [DOI] [PubMed] [Google Scholar]
- 48.Simpson EM, Linder CC, Sargent EE, Davisson MT, Mobraaten LE, Sharp JJ. Genetic variation among 129 substrains and its importance for targeted mutagenesis in mice. Nat Genet. 1997;16:19–27. doi: 10.1038/ng0597-19. [DOI] [PubMed] [Google Scholar]
- 49.Simpson RE, O'Regan MH, Perkins LM, Phillis JW. Excitatory transmitter amino acid release from the ischemic rat cerebral cortex: effects of adenosine receptor agonists and antagonists. J Neurochem. 1992;58:1683–1690. doi: 10.1111/j.1471-4159.1992.tb10041.x. [DOI] [PubMed] [Google Scholar]
- 50.Svenningsson P, Le Moine C, Aubert I, Burbaud P, Fredholm BB, Bloch B. Cellular distribution of adenosine A2A receptor mRNA in the primate striatum. J Comp Neurol. 1998;399:229–240. doi: 10.1002/(sici)1096-9861(19980921)399:2<229::aid-cne6>3.0.co;2-2. [DOI] [PubMed] [Google Scholar]
- 51.von Lubitz DJKE. Adenosine and acute treatment of cerebral ischemia and stroke—“put out more flags.”. In: Jacobson KA, Jarvis MF, editors. Purinergic approaches in experimental therapeutics. New York. Wiley-Liss; 1997. pp. 449–470. [Google Scholar]
- 52.von Lubitz DJKE, Lin RCS, Jacobson KA. Cerebral ischemia in gerbils: effects of acute and chronic treatment with adenosine A2A receptor agonist and antagonist. Eur J Pharmacol. 1995;287:295–302. doi: 10.1016/0014-2999(95)00498-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Weaver DR. A2A adenosine receptor gene expression in developing rat brain. Brain Res Mol Brain Res. 1993;20:313–327. doi: 10.1016/0169-328x(93)90058-w. [DOI] [PubMed] [Google Scholar]