

Abstract
Taurine, brain derived neurotrophic factor (BDNF), and basic fibroblast growth factor (bFGF) are known to control the development of early postnatal cerebellar granule cells. This study attempted to investigate possible mechanisms of this control by determining neuronal survival, calcium homeostasis, and related calcium-mediated functions, as well as the site of action during glutamate-induced excitotoxicity in cultures of cerebellar granule cells. We report that stimulation of glutamate receptors induced a rapid increase in intracellular calcium concentrations ([Ca2+]i) and a decrease in mitochondrial energy metabolism. These effects of glutamate were time- and concentration-dependent and could be specifically blocked by glutamate receptor antagonists. Taurine and bFGF but not BDNF differently regulated [Ca2+]i, and preserved the mitochondrial energy metabolism in the presence of glutamate. The regulation of [Ca2+]i by bFGF and taurine required pretreatment of cells with these factors. Confocal microscope analysis of [Ca2+]i and45Ca2+ uptake studies showed that bFGF reduced the magnitude of glutamate-induced calcium uptake with no apparent regulation thereafter. Taurine, on the other hand, did not affect the level of calcium uptake induced by glutamate but rather the duration of the maximal response; this maximal response was transient and returned to basal levels ∼10 min after glutamate receptor stimulation. We conclude from these data that bFGF and taurine prevent glutamate excitotoxicity through regulation of [Ca2+]i and mitochondrial energy metabolism. Furthermore, the neuroprotective role of taurine and bFGF was enhanced by their collaboration.
Keywords: cerebellar granule cells, excitotoxicity, growth factors, taurine, glutamate, calcium, energy metabolism, BDNF, bFGF
The development and maintenance of the CNS are regulated by a balanced interaction of multiple factors. Slight alterations in the relative expression of any one signal or combination thereof can result in significant functional and structural neuronal changes and might, under certain conditions, lead to pathological states and cell death.
Glutamate is the major excitatory neurotransmitter in mammalian brain (Watkins and Evans, 1981; Olney et al., 1987). Activation of glutamate receptors causes extracellular calcium influx and mobilization of additional calcium from intracellular stores (Jaffe and Brown, 1994). Calcium serves physiologically important functions as second messenger (Lynch et al., 1983; Kater et al., 1988). However, excessive elevation of intracellular calcium levels results in structural damage to neurons (Beal et al., 1993; Mattson, 1994; Macaya et al., 1996; Stout et al., 1998). Thus, the control of intracellular calcium concentrations is a fundamental process in neuronal survival and function (Choi, 1990; Dykens et al., 1987; Siesjo, 1988; Mattson, 1992).
Recently, mechanisms have been described involving growth factors (GFs) that protect neurons from excitotoxicity (Pechan et al., 1992; Mattson and Cheng, 1993; El Idrissi et al., 1998). GFs regulate the development, maintenance, and survival of neurons (Barde, 1989) and are implicated in neuronal functions. These processes are generally mediated through autophosphorylation of cell surface receptors, leading to various transcriptional events (Chao, 1992; Barbacid, 1994; Greene and Kaplan, 1995; Bothwell, 1996).
We have shown that taurine (2-aminoethanesulfonic acid) plays a major role in modulating glutamate-induced signaling events (Trenkner, 1990;Trenkner et al., 1996; El Idrissi et al., 1998). Taurine prevented excitotoxicity in vitro (Trenkner and Dykens, 1986;Trenkner, 1990), suggesting that the neuroprotective effect of taurine, as well as GFs, may be mediated primarily through modulation of intracellular calcium homeostasis.
Energy metabolism is recognized as one of the fundamental processes necessary for the maintenance of neuronal structures and functions (Mattson et al., 1993; Beal, 1995). The activity of the mitochondrial electrochemical gradient (MtECG) and the amount of energy it produces are calcium-regulated (Hertz et al., 1988; White and Reynolds, 1995;Budd and Nicholls, 1996). Furthermore, mitochondria may be involved in glutamate toxicity (Schinder et al., 1996; White and Reynolds, 1996). Because the mitochondria have large capacity for calcium uptake (Nicholls and Akerman, 1982; Gunter et al., 1994), they might have a neuroprotective role by removing calcium from the cytoplasm (Budd and Nicholls, 1996; Stout et al., 1998). Mitochondria also have been found to be essential in controlling certain apoptotic pathways (Green and Reed, 1998) through the release of caspase activators, such as cytochrome c (Liu et al., 1996) and apoptosis-inducing factors (Susin et al., 1996). We and others have demonstrated that depletion of cellular energy levels increased the vulnerability toward excitotoxins, leading to cell death (Budd and Nicholls, 1995; Trenkner et al., 1996;Schinder et al., 1996).
This study shows that a sustained rise in intracellular calcium levels and a decrease in MtECG were primarily responsible for the degenerative actions of glutamate, which can be controlled through different mechanisms by taurine and basic fibroblast growth factor (bFGF).
MATERIALS AND METHODS
Brain derived neurotrophic factor (BDNF) and bFGF were obtained from Promega (Madison, WI). Minimum essential medium (MEM), horse serum (HS), fetal calf serum (FCS), penicillin, streptomycin, and N-2 supplement were purchased from Life Technologies (Grand Island, NY). Culture dishes were obtained from Falcon (Lincoln Park, NJ). Trypsin and DNase were purchased from Worthington (Freehold, NJ). NMDA, kainate, and MK-801 came from Tocris Cookson (Bristol, UK). Fluo-3, fluorescein diacetate (FDA), and propidium iodide (PI), were obtained from Molecular Probes (Eugene, OR). Percoll, Triton X-100, cytosine arabinoside (Ara C), poly-d-lysine (PDL), rhodamine 123, taurine, and glutamate were obtained from Sigma (St. Louis, MO). 45CaCl2was received from Amersham Pharmacia Biotech (Arlington Heights, IL). All other chemicals were of high-quality cell culture grade.
Cell preparation and culture conditions. The development of cerebellar granule neurons in vitro depends on growth conditions. To characterize the role of glial cells and exogenously added growth factors on the survival and function of granule neurons, we used four culture conditions, as follows. (1) Mixed cultures: Cerebellar granule cells were prepared from 7-d-old mice as described previously (Trenkner and Sidman, 1977; Trenkner, 1991). Briefly, the entire cerebellum was removed, and single cell suspensions were prepared by trypsinization and trituration in 1% trypsin in Ca2+–Mg2+-free isotonic phosphate buffer (CMF-PBS). Cells were washed in CMF-PBS and resuspended in culture medium (MEM), supplemented with 0.25% glucose, 2 mm glutamine, 10% HS, 5% FCS, and 25 U/ml both penicillin and streptomycin. Cells were seeded into PDL-coated dishes and incubated at 37°C in a moist chamber under 5% CO2. (2) Enriched neuronal cultures: Mixed cultures were prepared as described above, but after 24 hr in vitro the medium was replaced with serum-free medium containing 15% N-2 supplement (Bottenstein et al., 1980), consisting of 100 μg/ml transferrin, 20 μg/ml putrescine, 12.8 ng/ml progesterone, 10.4 ng/ml selenium, 25 ng/ml insulin, and 0.8 ng/ml thyroxine. The mitotic inhibitor Ara C was added during medium exchange (2 μm); this curtailed the number of astrocytes that develop in cultures. The cultures were maintained in a humidified 5% CO2–95% air atmosphere at 37°C in slightly modified MEM with elevated glucose (30 mm) and reduced glutamine (0.8 mm) concentration (Peng et al., 1991). (3) Purified neurons in serum-containing medium: Cells were obtained from 3- or 4-d-old mice, and single-cell suspensions were prepared as described above. Granule neurons were purified in a Percoll gradient (35–65%) based on cellular size and selective adhesiveness of neurons and glial cells to plastic (for review, see Trenkner, 1991). (4) Purified neurons in serum-free medium: Cultures were prepared as above and seeded in serum-containing medium for 24 hr, and medium was replaced by serum-free medium consisting of MEM supplemented with 15% N-2. Although these cell preparations were almost free of non-neuronal cells, the number of cerebellar granule cells after gradient separation was insufficient for most of the experiments described. Therefore, these four culture conditions were used only to confirm the role of glial cells in vitro. Most of the experiments were conducted in enriched neuronal cultures (condition 2) in which the number of non-neuronal cells was significantly reduced.
Assessment of cell survival. We have used both morphological and biochemical markers to determine cell viability. Morphological assessment was as follows. When growing in monolayer, the integrity of presumptive neurons, glia, and fibroblasts was appraised on the basis of morphology and phase-refraction characteristics using phase-contrast microscopy (Trenkner and Sidman, 1977; Hatten, 1985). Neurons were scored as viable if they had neurites that were smooth in appearance and cell bodies that were smooth and round-to-oval in shape. In degenerating neurons, neurites were fragmented and beaded, and the soma was rough, swollen, vacuolated, and irregular in shape. The percentages of live neurons represent counts of all presumptive granule neurons within two randomly chosen rectangular fields encompassing almost 60% of the total well area.
Intravital staining of cultured neurons was performed using the method of Faravon et al. (1988). Living cells were labeled with FDA (15 μg/ml), whereas the nuclei of dead cells were labeled with PI (4.5 μg/ml), which interacts with DNA to yield a bright red fluorescent complex. Cells were immediately observed with a standard epi-illumination fluorescent microscope (450 excitation., 520 barrier). Three to four fields containing ∼200 cells were examined.
Activity of the mitochondrial electrochemical gradient.Quantitative determination of rhodamine 123 uptake was as follows. As described by Chen (1989), a stock solution (1 mg/ml) of rhodamine 123 was prepared in DMSO and stored at 4°C. After treatment, medium was removed, and rhodamine 123 was added to a final concentration of 10 μg/ml. After 30 min at 37°C, the cultures were washed three times in the growth medium. To release accumulated rhodamine from cells, 2 ml of butanol was added to each culture dish and incubated at room temperature for 1 hr. Cellular accumulation of rhodamine 123 was determined using a spectrofluorometer with the excitation wavelength set at 508 nm and emission wavelength set at 536 nm and was normalized to total protein concentration.
45Ca2+accumulation. Cells were washed twice with Locke's solution containing (in mm): 154 NaCl, 5.6 KCl, 3.6 NaHCO3, 1.3 CaCl2, 5.6 glucose, and 5 mm HEPES, pH 7.4. Additions were made to a final volume of 0.25 ml including 2 × 105 cpm of45CaCl2, which was added 10 sec before the addition of the agonist. After 20 min (or as indicated) at room temperature, the cells were rapidly washed three times in 0.5 ml of Locke's solution containing 2 μm MK-801. High concentration of MK-801 was used in the45Ca2+ uptake assay to ensure fast channel block. Finally, the total amount of45Ca2+ was determined in the lysate after the cells were dissolved in 0.5 ml of 0.1 m NaOH. All45Ca2+ uptake determinations were measured in triplicate.
Calcium imaging. Quantitative measurements of intracellular calcium concentration ([Ca2+]i) were performed in morphologically identified cerebellar granule cells grown on PDL-coated coverslips for 4 d. Stock solution (1 mm) of the acetoxymethylester fluo-3 was prepared in anhydrous DMSO and stored desiccated at −20°C. Cells were loaded with fluo-3 (5 μm) for 30 min at 37°C in 1 ml of magnesium-free Locke's solution. Subsequently, cells were rinsed with Locke's solution, and coverslips were mounted into the recording chamber. To achieve rapid and even distribution, 0.5 ml of the compounds to be tested were added in double concentration to 0.5 ml of Locke's solution in the recording chamber.
Confocal images of cellular fluorescence were obtained using a Nikon (Tokyo, Japan) inverted epifluorescence microscope equipped with an oil immersion 60×, 1.4 NA objective. The excitation wavelength used was 488 nm, and the emission wavelength was 505 ± 30 nm. In all experiments, fluorescence was measured in cell somata rather than neurites. All recordings were performed at room temperature (22–25°C). Background fluorescence values, determined in cell-free regions of each coverslip, were subtracted from all values.
At the end of each experiments, fluorescence intensities were calibrated to determine [Ca2+]i. Maximal fluorescence value (Fmax) was determined by permeabilizing cells with 1% Triton X-100 to attain the saturation level of binding with the trapped fluo-3. Subsequently, the minimum fluorescence value (Fmin) was measured in the presence of 10 mm EGTA to chelate all the free calcium. The free cytosolic calcium concentration was then calculated using the following equation: [Ca2+]i =Kd (F −Fmin)/(Fmax− F) (Grynkiewicz et al., 1985;Kao, 1994; Veliçelebi et al., 1998; Lin et al., 1999; Mestdagh and Wülfert, 1999), where F is the observed fluorescence value in intact cells. An equilibrium dissociation constant (Kd) of 390 nm was used for fluo-3. The calibration procedure described here is based on the assumption that the fluorescence properties (Fmin andFmax) and theKd of the indicator are the same in cells as in vitro.
Protein determination. Protein concentrations were determined by the method of Bradford (1976) using bovine serum albumin as standard protein (Bio-Rad, Hercules, CA).
Statistical analysis. Multifactorial ANOVA and ANCOVA were used to identify overall condition effects. Significant changes were determined by post hoc comparisons of means using Tukey's honest significant difference test. Significance was set at a confidence level of 95%. Data are presented as mean ± SEM.
RESULTS
bFGF and taurine attenuate glutamate-induced excitotoxicity
The neuroprotective effects of bFGF and taurine against glutamate excitotoxicity were assessed under four different culture conditions (see Materials and Methods) (Fig. 1). We confirmed that neuronal survival was dependent on astroglial cells (Trenkner and Sidman, 1977; Hatten et al., 1988; Trenkner, 1991;Rosenberg, 1991), because the highest degree of neuronal survival was reached in serum-supplemented medium containing neurons and glial cells (Fig. 1A). Under these culture conditions, bFGF, with or without taurine, had little effect on neuronal survival.
Fig. 1.
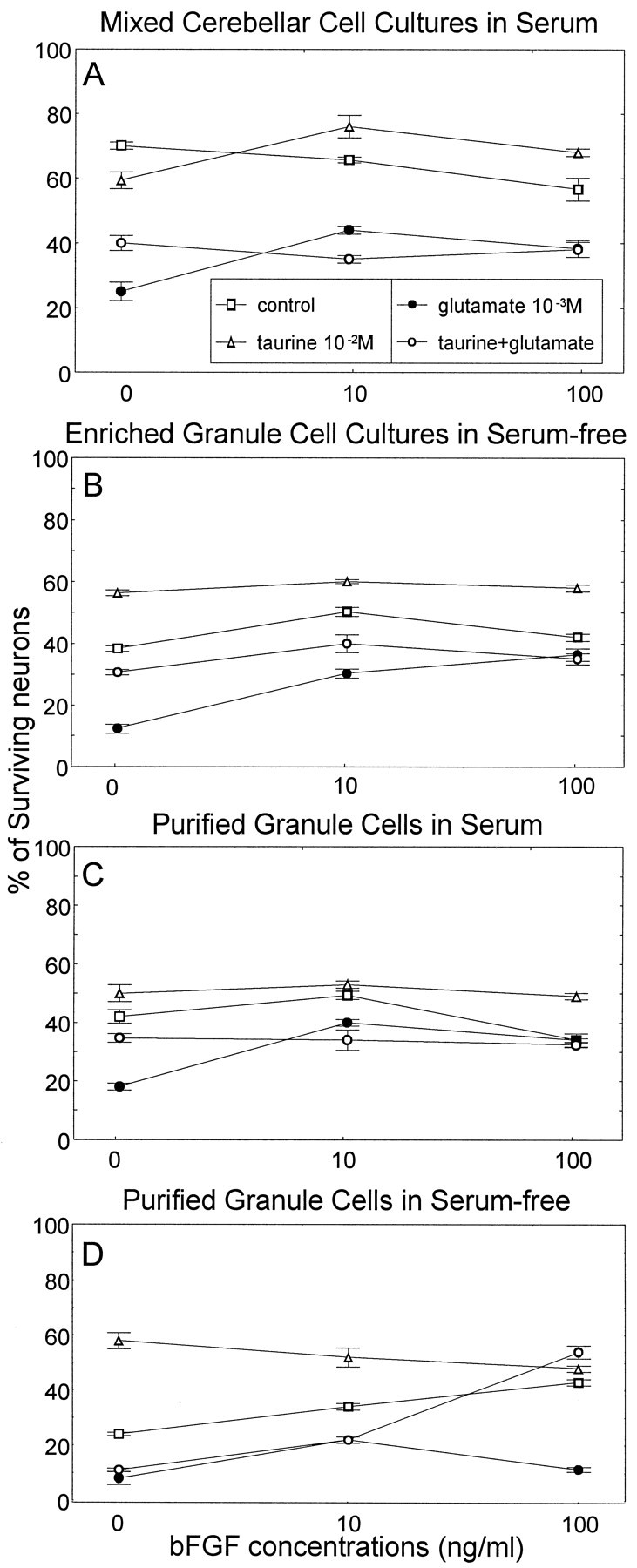
Effects of bFGF and taurine on glutamate-induced excitotoxicity. The survival of cerebellar granule neurons, isolated form early postnatal C57Bl/J mice (postnatal days 6–8), was determinedin vitro as a function of bFGF (0.1–100 ng/ml), taurine (10 mm), and glutamate (1nm). Four conditions were compared: A, mixed cerebellar cells in MEM containing 10% HS and 5% FCS; B, enriched granule cells (5% glia and 95% neurons) in serum-free medium (MEM plus 15% N-2 supplement); C, purified cerebellar neurons maintained in MEM with serum (10% HS and 5% FCS); D, purified cerebellar neurons maintained in serum-free medium (MEM plus 15% N-2 supplement). Cells were initially plated in serum-containing medium (MEM plus 10% HS plus 5% FCS). After 24 hr, the culture medium was replaced with the growth medium as indicated. Cells were preincubated with bFGF and/or taurine for 24 hr before glutamate. The number of living cells was determined 6–10 hr after glutamate addition. A three-way ANOVA showed a statistically significant interaction between NAAs × bFGF concentrations × culture condition (F(18,96) = 19.24;p < 0.0001). There was a significant main effect of NAAs (F(3,96) = 612.45;p < 0.0001), bFGF concentrations (F(2,96) = 81.08; p< 0.0001), and culture conditions (F(3,96) = 208.97;p < 0.0001). Post hoc tests showed that cell survival of controls in mixed cultures was significantly higher than controls in all other cultures (p < 0.001). Cell survival of controls of purified neurons in serum-free medium was significantly lower than that of controls from all other cultures (p < 0.001). Glutamate reduced cell viability in all culture conditions compared with the corresponding controls (p < 0.001). Under all four culture conditions (except in D), taurine alone or combined with different concentrations of bFGF significantly increased cell viability in glutamate-treated cultures (p < 0.005).
One millimolar glutamate significantly reduced the number of surviving granule neurons under every culture condition examined (Fig.1A–D). This excitotoxic effect of glutamate was more pronounced in cultures of purified or enriched cerebellar neurons than in mixed cultures, suggesting a protective role of astroglia during glutamate excitotoxicity (Rosenberg and Li, 1995). bFGF and/or taurine had little effect on neuronal survival in mixed cerebellar cultures but improved neuronal survival of purified granule cells significantly in numbers similar to controls (Fig. 1C), suggesting that these factors can replace the role of astrocytes. In serum-free medium, on the other hand, the highest neuroprotection against glutamate excitotoxicity was observed only when taurine and high concentrations of bFGF were combined, although both factors alone could not prevent glutamate-induced cell death (Fig. 1D). Thus, taurine can support bFGF function under certain conditions.
Taurine and bFGF did not act synergistically nor additively in the presence of glutamate, but rather both factors were capable of providing optimal cell survival. However, under serum-free conditions, neuroprotection was obtained only through collaboration of taurine and bFGF. Because both factors act through different mechanisms, a variety of alternative mechanisms might be activated to control cell survival.
Intracellular calcium accumulation as a function of glutamate receptor activation
Because increases in intracellular calcium have been implicated in excitotoxic cell death in several different paradigms (Dykens et al., 1987; Choi, 1988; Mattson et al., 1988; Siesjo, 1988; Mattson, 1992; Stout et al., 1998) and because elevation in intracellular calcium is known to mediate the glutamate-induced excitotoxicity, we performed experiments to determine the intracellular accumulation of45Ca2+ in the presence of increasing concentrations of glutamate receptor agonists. Figure 2 shows that45Ca2+accumulation in enriched granule cell cultures under serum-free conditions depended on concentrations of extracellular glutamate or NMDA but not on kainate concentrations. Depolarization with 1 mm glutamate or NMDA for 15 min induced a significant45Ca2+accumulation in these cells, whereas activation of kainate receptors, in the presence of MK-801, did not induce any. The antagonists of the NMDA or kainate subtype of glutamate receptors, MK-801 and DNQX, respectively, eliminated the glutamate-induced45Ca2+accumulation, indicating that both of these receptor subtypes are important for glutamate neuroexcitation. However, we found that kainate-induced45Ca2+accumulation in the presence of MK-801, an NMDA receptor antagonist, was not significantly different from baseline. Thus, kainate receptors do not directly mediate calcium influx in this system but rather indirectly through membrane depolarization and removal of the magnesium block from the NMDA receptors (Mayer et al., 1984; Nowak et al., 1984), therefore activating these receptors. The finding that NMDA induced less 45Ca2+accumulation than glutamate suggests calcium entry through other calcium channels than NMDA receptor-mediated channels. Stimulation of glutamate receptors also activates the voltage-dependent calcium channels through membrane depolarization. This was shown by adding 5 μm of ω-conotoxin GVIA, a blocker of the N-type calcium channels, which significantly reduced the glutamate-induced45Ca2+accumulation (Fig. 2). From these findings, we have concluded that the NMDA receptors are the main route of calcium entry into cerebellar granule cells and that the non-NMDA and voltage-dependent calcium channels are equally important during glutamate stimulation.
Fig. 2.

Calcium accumulation as a function of glutamate receptor activation. Cells were depolarized with various concentrations of glutamate, NMDA plus glycine (10 μm), and kainate plus MK-801 (10 μm) as indicated.45Ca2+ accumulation was determined 15 min after depolarization. Each data point represents mean ± SEM of three experiments. ANOVA showed significant main effect of agonists (F(2,30) = 85.30; p< 0.0001) and concentrations (F(4,30)= 121.91; p < 0.0001). The interaction was also significant (F(8,30) = 25.32;p < 0.0001). Post hoc tests indicated that 10, 100, and 1000 μm glutamate caused a significant increase in 45Ca2+accumulation above control levels (p < 0.001), whereas 100 and 1000 μm NMDA were significantly different from controls (p < 0.001). Kainate in the presence of 10 μm MK-801 did not induce a significant increase above baseline. Ten micromolar the kainate receptor antagonist DNQX or 10 μm the NMDA receptor antagonist MK-801 completely blocked glutamate-induced45Ca2+ accumulation.
Taurine and heterologous serum downregulate glutamate-induced calcium accumulation
It has been established in recent years that intracellular calcium concentrations determine, in part, neuronal cell survival and function. Therefore, the mechanism by which taurine prevents excitotoxic cell death may function through regulating calcium influx into neuronal cells during glutamate receptor-mediated depolarization. As shown in Figure 3, glutamate induced a significant increase in45Ca2+accumulation into cerebellar granule cells grown in serum-free medium. Such an increase was significantly reduced when cells were pretreated for 24 hr with 10 mm taurine. Taurine alone had no effect on calcium accumulation. Similarly, glutamate-induced45Ca2+accumulation was significantly reduced when 2% HS was added to the culture medium (plus 13% N-2 supplement), and it was completely abolished when the culture medium was supplemented with 10% HS and 5% N-2. These data further indicate that the composition of the growth medium significantly controls the cellular response to external stimuli and further suggest that horse serum contains a factor(s) that suppresses the glutamate-induced calcium accumulation.
Fig. 3.

Taurine and horse serum downregulate glutamate-induced calcium accumulation. Taurine modulation of glutamate-induced 45Ca2+ accumulation was analyzed in early postnatal cerebellar granule cells cultured in serum-free medium and supplemented with different concentrations of HS (0, 2, and 10%, respectively). Cells were initially plated under serum conditions (10% HS plus 5% FCS). After 24 hr in vitro, cultures were switched to serum-free medium (15% N-2 supplement). At 3 days in vitro, 0.25 ml of culture medium was removed and replaced with 0.25 ml of fresh medium supplemented with HS to give the desired final concentration (0, 2, or 10%). Ten millimolar taurine was added at this time. Twenty-four hours later,45Ca2+ accumulation was measured after depolarization with 1 mm glutamate for 30 min. Data represent mean ± SEM from at least three separate experiments. A two-way ANOVA showed a statistically significant interaction between HS concentrations and NAA treatment (F(6,24) = 65.82; p< 0.0001). There was a significant main effect of HS concentrations (F(2,24) = 200; p< 0.0001) and NAA treatment (F(3,24) = 200; p < 0.0001). Post hoc tests showed that glutamate-induced 45Ca2+accumulation was significantly higher than control (p < 0.001) under 0 and 2%, but not 10%, HS.
Taurine modulation of glutamate-induced calcium accumulation is time-dependent
Time course studies of glutamate-induced45Ca2+accumulation showed that depolarization with 1 mm glutamate caused a significant increase in intracellular45Ca2+ after a 2 min stimulation. The linear increase in45Ca2+accumulation continued over time up to 30 min (Fig.4A,B).
Fig. 4.

Time course accumulation of calcium under glutamate depolarization. A, Taurine was added simultaneously with glutamate. B, Cultures were pretreated with taurine (10 mm) for 24 hr before addition glutamate (1 mm). Each data point represents the mean ± SEM of three separate experiments. A two-way ANOVA showed a statistically significant main effect of taurine treatment (F(2,48) = 33.88; p< 0.0001) and time of depolarization (F(7,48) = 103.92;p < 0.0001). The interaction between taurine and time was also significant (F(14,48) = 5.53; p < 0.0001). Post hoc tests indicated that glutamate caused a significant (p < 0.05) increase in45Ca2+ accumulation after 5 min and thereafter (p < 0.0001). A, Only at 30 min did taurine significantly (p< 0.001) reduce 45Ca2+ accumulation. InB, taurine induced a significant (p < 0.05) reduction 15 min after depolarization.
To determine the effectiveness of taurine, we measured45Ca2+accumulation in cultures treated with taurine and glutamate simultaneously (Fig. 4A) and in cultures pretreated with taurine for 24 hr before the addition of glutamate (Fig.4B). At each time point examined, taurine pretreatment significantly reduced glutamate-induced45Ca2+accumulation starting 2 min after glutamate stimulation (Fig.4B). However, when taurine and glutamate were added simultaneously to the cultures, taurine did not affect the glutamate-induced45Ca2+accumulation for 20 min (Fig. 4A). After 20 min, taurine did reduce intracellular45Ca2+content significantly (Fig. 4A). These results indicate that the modulation of45Ca2+accumulation by taurine occurs in the cytoplasm and not at receptor levels. Because a minimum of 20 min were required for taurine to act (Fig. 4A), this would be consistent with a mechanism of transporter-mediated taurine uptake rather then an interaction with cell surface receptors.
bFGF and taurine protect against excitotoxicity through modulation of intracellular calcium
bFGF, similar to taurine, modulated calcium influx in response to glutamate. As shown in Figure 5, bFGF at 10 ng/ml significantly reduced the glutamate-induced45Ca2+accumulation, suggesting that bFGF, possibly through regulation of calcium uptake, protects neurons from glutamate excitotoxicity. On the other hand, BDNF at 10 ng/ml had no significant effect on glutamate-induced45Ca2+accumulation (Fig. 5), although BDNF was neuroprotective (data not shown). This suggests that the neuroprotection of BDNF may be mediated through mechanisms different from those suggested for taurine and bFGF. The collaboration between BDNF and bFGF (10 ng/ml) also significantly reduced the glutamate-induced45Ca2+accumulation to levels ranging between those of BDNF and bFGF (Fig. 5), indicating that BDNF can affect bFGF activity.
Fig. 5.

Interaction of GFs and taurine during calcium uptake modulation. Cells were pretreated with bFGF and BDNF (10 ng/ml) and with taurine (10 mm) or a combination thereof for 24 hr. Glutamate was added for 30 min, and45Ca2+ and accumulation were measured as described. Data represent mean ± SEM from three separate experiments. The two-way ANOVA showed a statistically significant interaction between growth factors and NAAs (F(9,32) = 25.10; p< 0.0001). There was also a significant main effect of growth factors (F(3,32) = 32.09; p< 0.0001) and NAAs (F(3,32) = 304.52;p < 0.0001). Post hoc tests indicated that glutamate significantly increased intracellular45Ca2+ under all conditions examined (p < 0.005). bFGF or bFGF and BDNF caused a significant reduction in glutamate-induced45Ca2+ accumulation (p < 0.001). Taurine alone or combined with GFs significantly reduced 45Ca2+accumulation (p < 0.0005) in the presence of glutamate.
To characterize the role of taurine in the presence of BDNF and bFGF, glutamate-induced45Ca2+accumulation was measured in cultures pretreated with these factors for 24 hr. As expected,45Ca2+accumulation was significantly reduced when cultures were pretreated with taurine. The levels obtained were comparable with those obtained with bFGF (Fig. 5). Furthermore, taurine was more efficient in reducing the glutamate-induced45Ca2+accumulation when cells were pretreated with either BDNF or bFGF (10 ng/ml). When cells were pretreated with all three factors, glutamate-induced45Ca2+accumulation significantly exceeded the levels of mediation seen with taurine alone or in combination with either growth factor. However, the combination of BDNF and bFGF did not reduce45Ca2+accumulation further in the presence of taurine, as could have been expected in view of BDNF and bFGF interactions (Fig. 5). Under nondepolarizing conditions,45Ca2+accumulation in the presence of GFs and taurine was similar to that of untreated controls.
Intracellular calcium is regulated differently by taurine and bFGF
[Ca2+]i was measured by fluorescence microscopy in cerebellar neurons with granule cell-like morphology after loading the cells with fluo-3. Approximately 95% of these cells responded to glutamate (1 mm) with a rapid increase in [Ca2+]i (Fig.6A). Basal [Ca2+]i in these neurons was ∼200 nm but was increased sixfold after glutamate was added to the medium to reach levels of ∼1.2 μm (Fig. 6A). In the presence of glutamate, most neurons showed a sustained increase in [Ca2+]i with no recovery to the basal levels over a period of 20 min (Fig.6A). However, in cultures pretreated for 24 hr with 10 mm taurine, glutamate induced a rapid increase of [Ca2+]i (Fig.6B), which returned to basal levels ∼10 min later. In contrast, pretreatment with bFGF (10 ng/ml) resulted in significantly reduced but constant [Ca2+]i after adding glutamate (less than twofold increase), with no recovery to basal levels over 23 min (Fig. 6C). As shown in Figure6D, photobleaching of intracellular fluo-3 signals did not interfere with the [Ca2+]idetermination. [Ca2+]i in cells did not change significantly over a period of 20 min under the same image-acquisition settings.
Fig. 6.
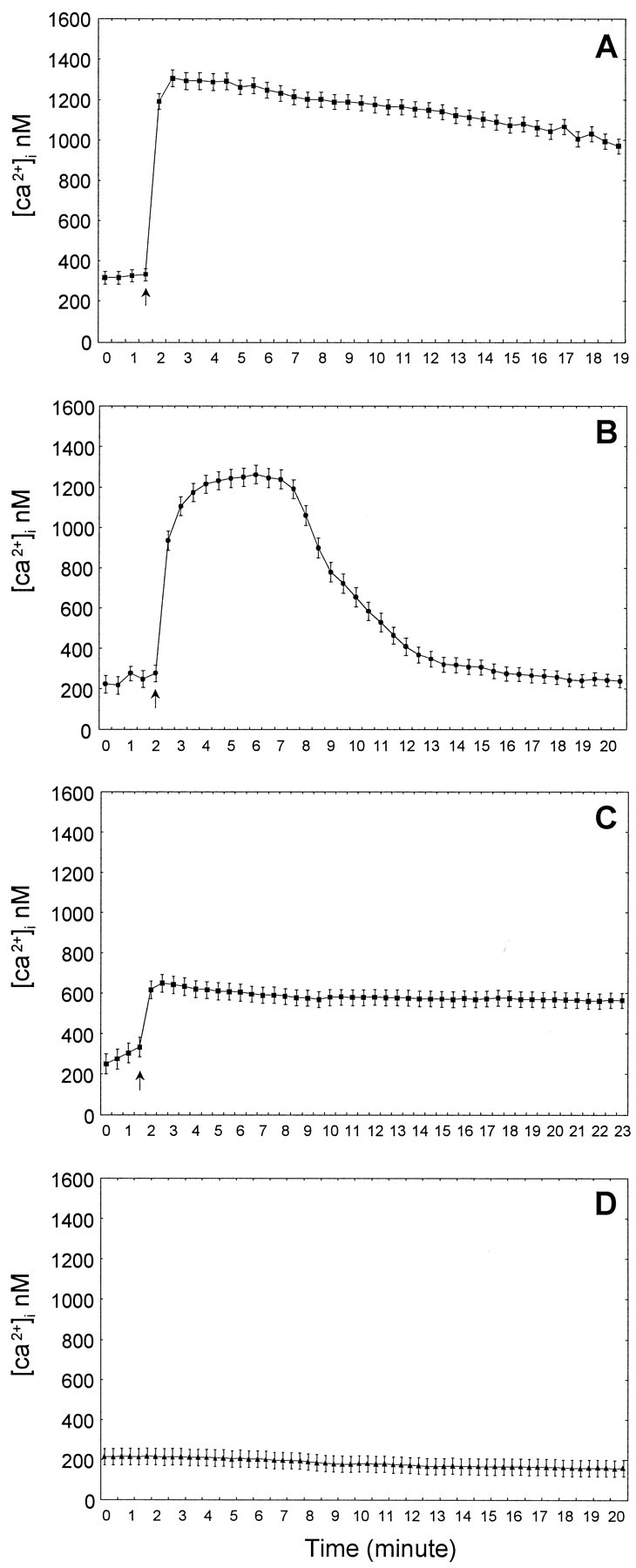
Regulation of [Ca2+]i by taurine and bFGF. [Ca2+]i was determined by confocal microscopy in cells attached to coverslips and loaded with 5 μm fluo-3. Images were acquired at 30 sec intervals.A–D show one set of data of one representative experiment. Each determination was repeated at least twice or more. Although the measurements vary from one experiment to another, the outcome was identical. Each point represents the mean ± SEM of [Ca2+]i determined as indicated in Materials and Methods. A, Glutamate (1 mm) was applied directly to the recording chamber as indicated by thearrow and was present throughout the recording period (n = 73 cells). B, Cells were pretreated with 10 mm taurine for 24 hr before glutamate addition (n = 82). Taurine was also present during the recording. C, Cultures were pretreated for 24 hr with 10 ng/ml bFGF (n = 65). D, The extent of photobleaching of the signal intensities. The software and microscope settings were maintained the same as forA–C, but images were acquired at 20 sec intervals (n = 57).
Growth factors and taurine enhance the mitochondrial function
Neuronal development and maintenance of function depend on cellular energy. Energy metabolism is recognized as one of the fundamental factors that control the required balance between maintenance of neuronal structures and their function during development and throughout adult life (Beal, 1995; Hoyer, 1993).We and others recently demonstrated that depleted cellular energy levels caused by malfunction of the MtECG increased the vulnerability of cultured neuronal cells toward excitotoxins and neurotoxins, leading to neuronal malfunction and subsequently to cell death (Dykens, 1994; Budd and Nicholls, 1996; Trenkner et al., 1996; El Idrissi et al., 1998). In this study, we examined the potential protective effects of growth factors and taurine on the mitochondrial function under excitotoxic conditions.
Treatment of cerebellar granule cells with BDNF or bFGF significantly increased the MtECG activity compared with controls (Fig.7). Similarly, the addition of taurine (10 mm) alone or in combination with BDNF and bFGF resulted in a significant increase of rhodamine uptake over untreated controls. Thus, taurine, BDNF, and bFGF may provide trophic support to cerebellar granule cells, as indicated by enhanced MtECG activity. Whereas taurine and GFs increased the mitochondrial activity, glutamate had opposite effects (Fig. 7). However, pretreatment with taurine and bFGF together before glutamate depolarization restored the MtECG to levels comparable with controls (Fig. 7). This increase in mitochondrial function with both taurine and bFGF under depolarizing condition may well be the result of their ability to regulate the intracellular calcium homeostasis. Therefore, different mechanisms and combinations of those analyzed here could lead to trophic support through modulating the activity of MtECG.
Fig. 7.

Combination of taurine and bFGF counteracted the glutamate-induced decrease in MtECG. Cells were treated with BDNF (10 ng/ml), bFGF (10 ng/ml), or taurine (10 mm) as indicated. Twenty-four hours later, cells were depolarized with 100 μm glutamate for 30 min. Data represent mean ± SEM of three experiments. A two-way ANOVA involving the interactions between NAAs and GFs was significant (F(6,24) = 2.65; p< 0.05). The main effect of NAAs was highly significant (F(3,24) = 155.9; p< 0.0001). The effect of GFs was also significant (F(2,24) = 30.35; p< 0.0001). Post hoc tests indicated that BDNF and bFGF induced significantly (p < 0.005) increased rhodamine uptake. Glutamate significantly (p< 0.01) reduced rhodamine uptake but not in cultures pretreated with taurine and bFGF compared with untreated controls.
Taurine specificity
The taurine analogs β-alanine and guanidinoethan sulfonate (GES) have been used as taurine-specific uptake blockers or competitors (Quesada et al., 1984; Moran and Pasantes-Morales, 1991). We have used both analogs in most of the experiments described here to demonstrate taurine-specific involvement. However, unexpectedly, β-alanine in equal concentrations to taurine (10 mm) had no effect on45Ca2+accumulation (Fig. 8), nor did it block the role of taurine as calcium modulator during prevention of excitotoxic cell death. Thus, β-alanine, although similar in structure to taurine, does not inhibit the modulatory functions of taurine. Similar results were obtained with GES, a taurine analog. These data suggest that taurine plays an active role in these regulatory mechanisms and that β-alanine and GES may not be used as specific taurine uptake blockers under these experimental conditions.
Fig. 8.

Effects of taurine and β-alanine on glutamate-induced calcium accumulation. Cells were pretreated with taurine, β-alanine, or a combination of the two at 10 mm.45Ca2+ accumulation was measured 30 min after depolarization with glutamate. Data represent mean ± SEM from three separate experiments. A two-way ANOVA showed a statistically significant interaction between glutamate and treatment with taurine and β-alanine (F(3,16) = 10.48;p < 0.0005). There was a significant main effect of glutamate (F(1,16) = 221.88;p < 0.0001) and taurine and/or β-alanine (F(3,16) = 14.16; p< 0.0001). Post hoc tests indicated that glutamate induced a significant (p < 0.001) increase in 45Ca2+ accumulation, which was significantly (p < 0.001) reduced by taurine and by taurine and β-alanine (p < 0.001). β-Alanine had no significant effect on glutamate-induced45Ca2+accumulation.
DISCUSSION
Because one of the essential mechanisms in neuronal development is the regulation and maintenance of calcium homeostasis (Mattson, 1988,1992; Mattson and Cheng, 1993), we attempted to determine how and whether neuroactive amino acids (NAAs) and GFs, known to mediate survival and function of cerebellar granule cells, interact in the regulation of calcium homeostasis during crucial steps in early postnatal cerebellar development and during excitotoxic conditions. Calcium ions are ubiquitous intracellular second messengers and act as key regulators of numerous cellular functions. Therefore, neurons must tightly regulate the concentrations of free cytoplasmic calcium to survive and function. Deregulation of intracellular calcium homeostasis causes numerous brain pathologies, including glutamate excitotoxicity (Dykens et al., 1987; Choi, 1987, 1988; Mattson and Cheng, 1993;Mattson et al., 1993; Eimerl and Schramm, 1995).
We have used taurine and glutamate as representatives of neuroactive amino acids, and bFGF and BDNF as examples of two distinct growth factor families. Because all of these molecules are present at a given time in the intracellular and extracellular environment, we have tried to characterize their interaction and questioned whether these molecules induce the same cellular regulatory systems and could substitute for each other, whether the regulation of calcium influx is synergistic or additive, and whether these mechanisms are complementary depending on their concentration and availability.
It is well established that the viability of neurons is greatly affected by their extracellular environment; e.g., more neurons survived in the presence of serum and astroglial cells (Fig.1A). Additions of factors with known neurotrophic support on cerebellar granule cells (bFGF and taurine) did not further improve cell survival beyond untreated controls, suggesting that optimal conditions were reached. In particular, both horse serum and astroglial cells had a significant influence on cell survival, as was shown by others as well as in this study (Trenkner et al., 1984; Hatten et al., 1988; Trenkner, 1990; Casper and Blum, 1995; Wood et al., 1997). When glial cells were removed from these cultures, the viability of purified cerebellar granule neurons was significantly reduced, providing evidence that serum and astroglial cells have trophic and protective effects.
Figure 1 demonstrates clearly the protective effects of the particular media under excitotoxic stress. Serum-containing cultures with astrocytes provided the most protection, possibly because trophic factors in serum and released glial factors protected to a maximum. Alternatively, in mixed cultures, glutamate may be removed by high-affinity uptake transporters (Fonnum, 1984; Rosenberg and Aizenman, 1989; Rosenberg, 1991), as Casper and Blum (1995) have shown. They determined that, in mixed cultures of dopaminergic neurons, glutamate uptake was significantly increased by bFGF through its mitogenic effect on astrocytes, reducing therefore its extracellular concentration and hence the excitotoxicity.
Our results suggest that neuroprotection is controlled by other mechanisms in addition to those involving transport and elimination of glutamate from the medium. These include regulation of intracellular calcium and neuronal energy metabolism. Support for this alternative mechanism comes from experiments in which the glutamate-induced calcium uptake was assessed in the presence of growth factors (Mattson et al., 1993; Cheng et al., 1995), showing quantitative correlations between excitotoxicity and total glutamate receptor-mediated calcium accumulation in neurons (Choi, 1988; Hartley et al., 1993; Eimerl and Schramm, 1994). Furthermore, we showed that glutamate-induced intracellular45Ca2+accumulation was time- and dose-dependent (Figs. 2, 4).
Although both taurine and bFGF regulated glutamate-induced45Ca2+accumulation (Fig. 5), they seem to act through different mechanisms (Fig. 6B,C). These differences became obvious through the two ways we determined [Ca2+]i. Although the content of45Ca2+ in cells determines the amount of45Ca2+ that has accumulated in these cells over time, this method does not differentiate between the total concentration, additional calcium uptake, extrusion, sequestration, or buffering. Confocal microscopy, on the other hand, allowed us to measure the total amount, as well as the increase of cytoplasmic free calcium over time (Fig.6B,C). We used fluo-3 as calcium indicator and not fura-2 or other calcium binding dyes because fuo-3 did not bleach over the period of observation (30 min). The importance of this fact was investigated by others (Becker and Fay, 1987), as well as in this study (Fig. 6D), and resulted in a semiquantitative determination of [Ca2+]i. Furthermore, the combination of these two methods provided a better understanding of calcium kinetics after glutamate depolarization and led us to postulate two regulatory mechanisms mediated by either taurine or bFGF.
As shown by Cheng et al. (1995), bFGF increases the expression levels of the GluR1 subunit of the AMPA receptor and suppresses the expression of a 71 kDa NMDA receptor protein in hippocampal (Mattson et al., 1993) and cerebellar neurons (Brandoli et al., 1998). If this is the case, suppression of the number of NMDA receptors would explain our results that bFGF reduces [Ca2+]i in the presence of glutamate, thus reducing neuronal vulnerability to calcium-mediated glutamate excitotoxicity.
Taurine, on the other hand, did not affect the magnitude of [Ca2+]i after glutamate stimulation but rather downregulated [Ca2+]i to basal levels ∼10 min after glutamate addition. These increases were transient, and [Ca2+]i recovered to basal levels in minutes (Fig. 6B), an effect not seen with glutamate alone (Fig. 6A) or in the presence of bFGF (Fig. 6C). The full extent of how taurine restores the [Ca2+]i after glutamate stimulation is not yet known. However, our results suggest that taurine does not affect the glutamate receptor-mediated calcium uptake directly but rather activates mechanisms inside the cell that reduce or redistribute cytoplasmic free calcium into different pools by extrusion, sequestration, or even buffering. We are presently addressing these questions.
Several mechanisms have been proposed to explain the neuroprotective role of taurine. Taurine is present in neurons and astroglial cells in multiple areas of the brain (Palkovits et al., 1986; Huxtable, 1992;Sturman, 1993). Moreover, it was shown that taurine is freely exchanged between these cells. We and others have suggested that taurine balances glutamate activity, particularly under excitotoxic conditions (Trenkner, 1990; Trenkner et al., 1996). The release and immediate uptake of taurine from neurons and glial cells has been associated with this neuroprotective effect (Trenkner, 1990; Bianchi et al., 1996;Trenkner et al., 1996; Katoh et al., 1997; Saransaari and Oja, 1997;Segovia et al., 1997). Furthermore, increasing the concentration of extracellular glutamate by infusion of selective inhibitors of the high-affinity glutamate uptake correlated with increasing concentrations of extracellular taurine. This increase was blocked by NMDA and AMPA–kainate receptor antagonists (Segovia et al., 1997). Taking these results together, the glutamate-induced taurine release might be neuroprotective. Yet another mechanism has been proposed involving taurine as an inhibitor of depolarization through increasing membrane chloride conductance via the activation of GABA and glycine receptors (Quinn and Miller, 1992). We have examined the possibility as to whether the ability of taurine to interact with GABA or glycine receptors had functional consequences (El Idrissi et al., 1998;Trenkner et al., 1998), but we were unable to detect any effect of GABA or glycine on the inhibition of45Ca2+accumulation by taurine. However, we found that low concentrations of glycine (10 μm) enhanced NMDA receptor activities, as described by Johnson and Asher (1987), in the absence of additional taurine. Recent electrophysiological studies of neocortical cultures have shown that taurine activates glycine receptors directly (Flint et al., 1998). In our culture system, however, taurine did not interact with either the glycine receptors as demonstrated by insensitivity to strychnine, a glycine receptor antagonist, or the glycine binding site of the NMDA receptor, as shown by the fact that taurine actions required pretreatment of the cells with taurine; however, glycine acted immediately (data not shown).
MtECG plays a pivotal role in controlling the maintenance of neuronal structures and their function throughout life (Mattson et al., 1993;Trenkner et al., 1994; Beal, 1995; Budd et al., 1997); the depletion of cellular energy, caused by malfunction of the MtECG, increased the vulnerability toward excitotoxins leading to neuronal cell death (Budd and Nicholls, 1995, 1996; El Idrissi et al., 1996; Trenkner et al., 1996). We have shown here that taurine (10 mm) in vitro alone or in combination with bFGF or BDNF facilitated an increase in mitochondrial activity as measured by an increase in rhodamine 123 uptake (Fig. 7). Furthermore, the selective collaboration of taurine and bFGF restored the MtECG after glutamate stimulation, possibly as a result of calcium regulation.
Although the mechanism(s) of the action of taurine is not fully understood, taurine plays a significant role in the regulation of calcium-mediated glutamate-induced signaling events. In particular, taurine has opposing effects to those of glutamate, modulated by the interaction with growth factors.
In conclusion, GFs and NAAs interact through modulation of calcium homeostasis after glutamate depolarization. Some of these factors induce the same regulatory mechanisms and appear to support each other. These findings further suggest that neuronal cells use alternative pathways to regulate and maintain their function. These pathways are accessible to cells depending on their developmental stages, the availability of these factors, and neuronal energy levels. We conclude that, under conditions in which more than one factor is present, as is likely in vivo, these factors differentially regulate the functions of each other to promote an optimal environment for cell survival and function.
Footnotes
This study was supported in part by funds from the New York State Office of Mental Retardation and Developmental Disabilities and the Center of Developmental Neuroscience and Developmental Disabilities. We are grateful to Drs A. Rabe, T. Lidsky, and M. Quinn for their time to critically read and discuss this manuscript with us and for their suggestions to make it readable.
Correspondence should be addressed to Dr. Ekkehart Trenkner, Institute for Basic Research in Developmental Disabilities, 1050 Forest Hill Road, Staten Island, NY 10314. E-mail: trenkner@postbox.csi.cuny.eduand trenkner@hotmail.com.
REFERENCES
- 1.Barbacid M. The trk family of neurotrophin receptors. J Neurobiol. 1994;25:1386–1403. doi: 10.1002/neu.480251107. [DOI] [PubMed] [Google Scholar]
- 2.Barde Y-A. Trophic factors and neuronal survival. Neuron. 1989;2:1525–1534. doi: 10.1016/0896-6273(89)90040-8. [DOI] [PubMed] [Google Scholar]
- 3.Beal MF. Aging, energy, and oxidative stress in neurodegenerative diseases. Ann Neurol. 1995;18:357–366. doi: 10.1002/ana.410380304. [DOI] [PubMed] [Google Scholar]
- 4.Beal MF, Hyman BT, Koroshetz W. Do defects in mitochondrial energy metabolism underlie the pathology of neurodegenerative diseases ? Trends Neurosci. 1993;16:125–131. doi: 10.1016/0166-2236(93)90117-5. [DOI] [PubMed] [Google Scholar]
- 5.Becker PL, Fay FS. Photobleaching of fura-2 and its effect on determination of calcium concentrations. Am J Physiol. 1987;253:C613–C618. doi: 10.1152/ajpcell.1987.253.4.C613. [DOI] [PubMed] [Google Scholar]
- 6.Bianchi LM, Conover JC, Fritzsch B, DeChiara T, Lindsay RM, Yancopoulos GD. Degeneration of vestibular neurons in late embryogenesis of both heterozygous BDNF null mutant mice. Development. 1996;122:1965–1973. doi: 10.1242/dev.122.6.1965. [DOI] [PubMed] [Google Scholar]
- 7.Bothwell M. p75NTR: a receptor after all. Science. 1996;272:506–507. doi: 10.1126/science.272.5261.506. [DOI] [PubMed] [Google Scholar]
- 8.Bottenstein JE, Skaper SD, Varon SS, Sato GH. Selective survival of neurons from chick embryo sensory ganglionic dissociates utilizing serum-free supplemented medium. Exp Cell Res. 1980;125:183–190. doi: 10.1016/0014-4827(80)90202-5. [DOI] [PubMed] [Google Scholar]
- 9.Bradford MM. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem. 1976;72:248–254. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- 10.Brandoli C, Sanna A, DeBernardi MA, Follesa P, Brooker G, Mocchetti I. Brain-derived neurotrophic factor and basic fibroblast growth factor down regulate NMDA receptor function in cerebellar granule cells. J Neurosci. 1998;18:7953–7961. doi: 10.1523/JNEUROSCI.18-19-07953.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Budd SL, Nicholls DG. A reevaluation of the role of mitochondria in neuronal Ca2+ homeostasis. J Neurochem. 1995;66:403–411. doi: 10.1046/j.1471-4159.1996.66010403.x. [DOI] [PubMed] [Google Scholar]
- 12.Budd SL, Nicholls DG. Mitochondria, calcium regulation, and acute glutamate excitotoxicity in cultured cerebellar granule cells. J Neurochem. 1996;67:2282–2291. doi: 10.1046/j.1471-4159.1996.67062282.x. [DOI] [PubMed] [Google Scholar]
- 13.Budd SL, Castilho RF, Nicholls DG. Mitochondrial membrane potential and hydroethidine-monitored superoxide generation in cultured cerebellar granule cells. FEBS Lett. 1997;415:21–24. doi: 10.1016/s0014-5793(97)01088-0. [DOI] [PubMed] [Google Scholar]
- 14.Casper D, Blum M. Epidermal growth factor and basic fibroblast growth factor protect dopaminergic neurons from glutamate toxicity in culture. J Neurochem. 1995;65:1016–1026. doi: 10.1046/j.1471-4159.1995.65031016.x. [DOI] [PubMed] [Google Scholar]
- 15.Chao MV. Neurotrophin receptors: a window into neuronal differentiation. Neuron. 1992;9:583–593. doi: 10.1016/0896-6273(92)90023-7. [DOI] [PubMed] [Google Scholar]
- 16.Chen LB. Fluorescent labeling of mitochondria. Methods Cell Biol. 1989;29:103–123. doi: 10.1016/s0091-679x(08)60190-9. [DOI] [PubMed] [Google Scholar]
- 17.Cheng B, Furukawa K, O'Keefe JA, Goodman Y, Kihiko M, Fabian T, Mattson MP. Basic fibroblast growth factor selectively increases AMPA-receptor subunit GluR1 protein level and differentially modulates Ca2+ responses to AMPA and NMDA in hippocampal neurons. Neurochemistry. 1995;65:2525–2536. doi: 10.1046/j.1471-4159.1995.65062525.x. [DOI] [PubMed] [Google Scholar]
- 18.Choi DW. Ionic dependence of glutamate neurotoxicity. J Neurosci. 1987;7:369–379. doi: 10.1523/JNEUROSCI.07-02-00369.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Choi DW. Glutamate neurotoxicity and diseases of the nervous system. Neuron. 1988;1:623–634. doi: 10.1016/0896-6273(88)90162-6. [DOI] [PubMed] [Google Scholar]
- 20.Choi DW. The role of glutamate neurotoxicity in hypoxic-ischemic neuronal death. Annu Rev Neurosci. 1990;13:171–182. doi: 10.1146/annurev.ne.13.030190.001131. [DOI] [PubMed] [Google Scholar]
- 21.Dykens JA. Isolated cerebral and cerebellar mitochondria produce free radicals when exposed to elevated Ca2+ and Na+: implication for neurodegeneration. J Neurochem. 1994;63:584–591. doi: 10.1046/j.1471-4159.1994.63020584.x. [DOI] [PubMed] [Google Scholar]
- 22.Dykens JA, Stern A, Trenkner E. Mechanism of kainate toxicity to cerebellar neurons in vitro is analogous to reperfusion tissue injury. J Neurochem. 1987;49:1222–1228. doi: 10.1111/j.1471-4159.1987.tb10014.x. [DOI] [PubMed] [Google Scholar]
- 23.Eimerl S, Schramm M. The quantity of calcium that appears to induce neuronal death. J Neurochem. 1994;62:1223–1226. doi: 10.1046/j.1471-4159.1994.62031223.x. [DOI] [PubMed] [Google Scholar]
- 24.Eimerl S, Schramm M. Resuscitation of brain neurons in the presence of Ca2+after toxic NMDA-receptor activity. J Neurochem. 1995;65:739–744. doi: 10.1046/j.1471-4159.1995.65020739.x. [DOI] [PubMed] [Google Scholar]
- 25.El Idrissi A, Harris A, Trenkner E. Neurotrophins, neuro-active amino acids and the mitochondrial respiratory chain together maintain neuronal survival and function. Soc Neurosci Abstr. 1996;22:770. [Google Scholar]
- 26.El Idrissi A, Harris C, Trenkner E. Taurine modulates glutamate- and growth factors-mediated signaling mechanisms. Adv Exp Med Biol. 1998;442:385–396. doi: 10.1007/978-1-4899-0117-0_48. [DOI] [PubMed] [Google Scholar]
- 27.Favaron M, Manev H, Alho H, Bertolino M, Ferret B, Guidotti A, Costa E. Gangliosides prevent glutamate and kainate neurotoxicity in primary neuronal cultures of neonatal rat cerebellum and cortex. Proc Natl Acad Sci USA. 1988;85:7351–7355. doi: 10.1073/pnas.85.19.7351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Flint AC, Liu X, Kriegstein AR. Nonsynaptic glycine receptor activation during early neocortical development. Neuron. 1998;20:43–53. doi: 10.1016/s0896-6273(00)80433-x. [DOI] [PubMed] [Google Scholar]
- 29.Fonnum F. Glutamate: a neurotransmitter in mammalian brain. J Neurochem. 1984;42:1–11. doi: 10.1111/j.1471-4159.1984.tb09689.x. [DOI] [PubMed] [Google Scholar]
- 30.Green DR, Reed JC. Mitochondria and apoptosis. Science. 1998;281:1309–1312. doi: 10.1126/science.281.5381.1309. [DOI] [PubMed] [Google Scholar]
- 31.Greene LA, Kaplan DR. Early events in neurotrophin signaling via Trk and p75 receptors. Curr Top Neurobiol. 1995;5:579–587. doi: 10.1016/0959-4388(95)80062-x. [DOI] [PubMed] [Google Scholar]
- 32.Grynkiewicz G, Peonie M, Tsien RY. A new generation of calcium indicators with greatly improved fluorescence properties. J Biol Chem. 1985;260:3440–3450. [PubMed] [Google Scholar]
- 33.Gunter TE, Gunter KK, Sheu SS, Gavin CE. Mitochondrial calcium transport: physiological and pathological relevance. Am J Physiol. 1994;267:313–339. doi: 10.1152/ajpcell.1994.267.2.C313. [DOI] [PubMed] [Google Scholar]
- 34.Hartley DM, Kurth MC, Bjerkness L, Weiss JH, Choi DW. Glutamate receptor-induced 45Ca2+ accumulation in cortical cell culture correlates with subsequent neuronal degeneration. J Neurosci. 1993;13:1993–2000. doi: 10.1523/JNEUROSCI.13-05-01993.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hatten ME. Neuronal regulation of astroglial morphology and proliferation in vitro. J Cell Biol. 1985;100:384–396. doi: 10.1083/jcb.100.2.384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hatten ME, Lynch M, Rydel RE, Sanchez J, Joseph-Silverstein J, Moscatelli D, Rifkin DB. In vitro neurite extension by granule neurons is dependent upon astroglial-derived fibroblast growth factor. Dev Biol. 1988;125:280–289. doi: 10.1016/0012-1606(88)90211-4. [DOI] [PubMed] [Google Scholar]
- 37.Hertz L, Drejer J, Schousboe A. Energy metabolism in glutamatergic neurons, GABAergic neurons and astrocytes in primary cultures. Neurochem Res. 1988;13:605–610. doi: 10.1007/BF00973275. [DOI] [PubMed] [Google Scholar]
- 38.Hoyer S. Brain oxidative energy and related metabolism, neuronal stress, and Alzheimer's disease: a speculative synthesis. J Geriatr Psychiatry Neurol. 1993;1:3–13. doi: 10.1177/002383099300600101. [DOI] [PubMed] [Google Scholar]
- 39.Huxtable RJ. The physiological actions of taurine. Physiol Rev. 1992;72:101–163. doi: 10.1152/physrev.1992.72.1.101. [DOI] [PubMed] [Google Scholar]
- 40.Jaffe DB, Brown TH. Metabotropic glutamate receptor activation induces calcium waves within hippocampal dendrites. J Neurophysiol. 1994;72:471–474. doi: 10.1152/jn.1994.72.1.471. [DOI] [PubMed] [Google Scholar]
- 41.Johnson JW, Ascher P. Glycine potentiates the NMDA response in cultured mouse brain neurons. Nature. 1987;325:529–531. doi: 10.1038/325529a0. [DOI] [PubMed] [Google Scholar]
- 42.Kao JP. Practical aspects of measuring [Ca2+] with fluorescent indicators. Methods Cell Biol. 1994;40:155–181. doi: 10.1016/s0091-679x(08)61114-0. [DOI] [PubMed] [Google Scholar]
- 43.Kater SB, Mattson MP, Cohan C, Connor J. Calcium regulation of the neuronal growth cone. Trends Neurosci. 1988;11:315–321. doi: 10.1016/0166-2236(88)90094-x. [DOI] [PubMed] [Google Scholar]
- 44.Katoh H, Sima K, Nawashiro H, Wada K, Chigasaki H. The effect of MK-801 on extracellular neuroactive amino acids in hippocampus after closed head injury followed by hypoxia in rats. Brain Res. 1997;758:153–162. doi: 10.1016/s0006-8993(97)00213-8. [DOI] [PubMed] [Google Scholar]
- 45.Lin K, Sadée W, Quillan JM. Rapid measurements of intracellular calcium using a fluorescence plate reader. Biotechniques. 1999;26:318–326. doi: 10.2144/99262rr02. [DOI] [PubMed] [Google Scholar]
- 46.Liu X, Kim CN, Yang J, Jemmerson R, Wang X. Induction of apoptotic program in cell-free extracts: requirements of dATP and cytochrome c. Cell. 1996;86:147–157. doi: 10.1016/s0092-8674(00)80085-9. [DOI] [PubMed] [Google Scholar]
- 47.Lynch G, Larson J, Kelso S, Barrionuevo G, Schottler F. Intracellular injections of EGTA block induction of hippocampal long-term potentiation. Nature. 1983;305:719–721. doi: 10.1038/305719a0. [DOI] [PubMed] [Google Scholar]
- 48.Macaya A, Munell F, Reventos J, Ferrer I. Molecular factors of cerebral hypoxia-ischemia. Rev Neurol. 1996;24:855–864. [PubMed] [Google Scholar]
- 49.Mattson MP. Neurotransmitters in the regulation of cytoarchitecture. Brain Res Rev. 1988;13:179–212. doi: 10.1016/0165-0173(88)90020-3. [DOI] [PubMed] [Google Scholar]
- 50.Mattson MP. Calcium as sculptor and destroyer of neural circuitry. Exp Gerontol. 1992;27:29–49. doi: 10.1016/0531-5565(92)90027-w. [DOI] [PubMed] [Google Scholar]
- 51.Mattson MP. Calcium and neuronal injury in Alzheimer's disease. Contributions of beta-amyloid precursor protein mismetabolism, free radicals, and metabolic compromise. Ann NY Acad Sci. 1994;15:50–76. [PubMed] [Google Scholar]
- 52.Mattson MP, Cheng B. Growth factors protect neurons against excitotoxic/ischemic damage by stabilizing calcium homeostasis. Stroke. 1993;24:1136–1140. [PubMed] [Google Scholar]
- 53.Mattson MP, Guthrie PB, Kater SB. Intracellular messengers in the generation and degeneration of hippocampal neuroarchitecture. J Neurosci Res. 1988;21:447–464. doi: 10.1002/jnr.490210236. [DOI] [PubMed] [Google Scholar]
- 54.Mattson MP, Zhang Y, Bose S. Growth factors prevent mitochondrial dysfunction, loss of calcium homeostasis, and cell injury, but not ATP depletion in hippocampal neurons deprived of glucose. Exp Neurol. 1993;121:1–13. doi: 10.1006/exnr.1993.1066. [DOI] [PubMed] [Google Scholar]
- 55.Mayer ML, Westbrook GL, Guthrie PB. Voltage-dependent block by Mg2+ of NMDA responses in spinal cord neurons. Nature. 1984;309:261–263. doi: 10.1038/309261a0. [DOI] [PubMed] [Google Scholar]
- 56.Mestadagh N, Wülfert E. Bicuculine increases Ca2+ transients in rat cerebellar granule cells through non-GABAB receptor associated mechanisms. Neurosci Lett. 1999;265:95–98. doi: 10.1016/s0304-3940(99)00213-x. [DOI] [PubMed] [Google Scholar]
- 57.Moran J, Pasantes-Morales H. Taurine-deficient cultured cerebellar astrocytes and granule neurons obtained by treatment with guanidinoethane sulfonate. J Neurosci Res. 1991;29:533–537. doi: 10.1002/jnr.490290414. [DOI] [PubMed] [Google Scholar]
- 58.Nicolls DG, Akerman KEO. Mitochondrial calcium transport. Biochim Biophys Acta. 1982;683:57–88. doi: 10.1016/0304-4173(82)90013-1. [DOI] [PubMed] [Google Scholar]
- 59.Nowak L, Bregestovski P, Ascher P, Herbet A, Prochiantz A. Magnesium gates glutamate-activated channels in mouse central neurones. Nature. 1984;307:462–465. doi: 10.1038/307462a0. [DOI] [PubMed] [Google Scholar]
- 60.Olney JW, Price MT, Salles KS, Labruyere J, Ryerson R, Mahan K, Frierdich G, Samson L. l-Homocysteic acid: an endogenous excitotoxic ligand of the NMDA receptor. Brain Res Bull. 1987;19:597–602. doi: 10.1016/0361-9230(87)90077-3. [DOI] [PubMed] [Google Scholar]
- 61.Palkovits M, Elekes I, Lang T, Patthy A. Taurine levels in discreet brain nuclei of rats. J Neurochem. 1986;47:1333–1335. doi: 10.1111/j.1471-4159.1986.tb00761.x. [DOI] [PubMed] [Google Scholar]
- 62.Pechan PA, Chowdhury K, Seifert W. Free radicals induce gene expression of NGF and bFGF in rat astrocyte culture. NeuroReport. 1992;3:469–472. doi: 10.1097/00001756-199206000-00003. [DOI] [PubMed] [Google Scholar]
- 63.Peng LA, Juurlink BH, Hertz L. Differences in transmitter release, morphology, and ischemia-induced cell injury between cerebellar granule cell cultures developing in the presence and in the absence of a depolarizing potassium concentration. Dev Brain Res. 1991;63:1–12. doi: 10.1016/0165-3806(91)90061-m. [DOI] [PubMed] [Google Scholar]
- 64.Quesada O, Huxtable RJ, Pasantes-Morales H. Effect of guanidinoethane sulfonate on taurine uptake by rat retina. J Neurosci Res. 1984;11:179–186. doi: 10.1002/jnr.490110207. [DOI] [PubMed] [Google Scholar]
- 65.Quinn MR, Miller CL. Taurine allosterically modulates flunitrazepam binding to synaptic membranes. J Neurosci Res. 1992;33:136–141. doi: 10.1002/jnr.490330117. [DOI] [PubMed] [Google Scholar]
- 66.Rosenberg PA. Accumulation of extracellular glutamate and neuronal death in astrocyte-poor cortical cultures exposed to glutamine. Glia. 1991;4:91–100. doi: 10.1002/glia.440040111. [DOI] [PubMed] [Google Scholar]
- 67.Rosenberg PA, Aizenman E. Hundred-fold increase in neuronal vulnerability to glutamate toxicity in astrocyte-poor cultures of rat cerebral cortex. Neurosci Lett. 1989;103:162–168. doi: 10.1016/0304-3940(89)90569-7. [DOI] [PubMed] [Google Scholar]
- 68.Rosenberg PA, Li Y. Adenylyl cyclase activation underlies intracellular cyclic AMP accumulation, cyclic AMP transport, and extracellular adenosine accumulation evoked by beta-adrenergic receptor stimulation in mixed cultures of neurons and astrocytes derived from rat cerebral cortex. Brain Res. 1995;692:227–232. doi: 10.1016/0006-8993(95)00668-g. [DOI] [PubMed] [Google Scholar]
- 69.Saransaari P, Oja SS. Enhanced taurine release in cell damaging conditions in the developing and ageing mouse hippocampus. Neuroscience. 1997;79:847–854. doi: 10.1016/s0306-4522(97)00038-9. [DOI] [PubMed] [Google Scholar]
- 70.Schinder AF, Olson EC, Spitzer NC, Montal M. Mitochondrial dysfunction is a primary event in glutamate neurotoxicity. J Neurosci. 1996;16:6125–6133. doi: 10.1523/JNEUROSCI.16-19-06125.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Segovia G, Del Acro A, Mora F. Endogenous glutamate increases extracellular concentrations of dopamine, GABA and taurine through NMDA and AMPA/kainate receptors in striatum of the freely moving rat: a microdialysis study. J Neurochem. 1997;69:1476–1483. doi: 10.1046/j.1471-4159.1997.69041476.x. [DOI] [PubMed] [Google Scholar]
- 72.Siesjo BK. Historical overview. Calcium, ischemia, and death of brain cells. Ann NY Acad Sci. 1988;522:638–661. doi: 10.1111/j.1749-6632.1988.tb33410.x. [DOI] [PubMed] [Google Scholar]
- 73.Stout AK, Raphael HM, Kanterewicz BI, Klann E, Reynolds IJ. Glutamate-induced neuron death requires mitochondrial calcium uptake. Nat Neurosci. 1998;1:366–373. doi: 10.1038/1577. [DOI] [PubMed] [Google Scholar]
- 74.Sturman JA. Taurine in development. Physiol Rev. 1993;73:119–147. doi: 10.1152/physrev.1993.73.1.119. [DOI] [PubMed] [Google Scholar]
- 75.Susin SA, Zamzami N, Castedo M, Hirsch T, Marchetti P, Macho A, Daugas E, Geuskens M, Kroemer G. Bcl-2 inhibits the mitochondrial release of an apoptogenic protease. J Exp Med. 1996;184:1331–1341. doi: 10.1084/jem.184.4.1331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Trenkner E. Possible role of glutamate with taurine in neuron-glia interaction during cerebellar development. Prog Clin Biol Res. 1990;351:133–140. [PubMed] [Google Scholar]
- 77.Trenkner E. Cerebellar cells in culture. In: Banker G, Goslin K, editors. Culturing nerve cells. MIT; Cambridge, MA: 1991. pp. 283–307. [Google Scholar]
- 78.Trenkner E, Dykens JA. Taurine moderates kynurenine excitotoxicity in vitro. Soc Neurosci Abstr. 1986;16:29. [Google Scholar]
- 79.Trenkner E, Sidman RL. Histogenesis of mouse cerebellum in microwell cultures: cell reaggregation and migration, fiber and synapse formation. J Cell Biol. 1977;75:915–940. doi: 10.1083/jcb.75.3.915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Trenkner E, Smith D, Segil N. Is cerebellar granule cell migration regulated by an internal clock? J Neurosci. 1984;4:2850–2855. doi: 10.1523/JNEUROSCI.04-11-02850.1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Trenkner E, Liu D, Harris C, Sturman J. Regulation of protein kinase C activity by taurine and beta-alanine during excitotoxicity in cat and mouse cerebellar cultures. Adv Exp Med Biol. 1994;359:309–316. doi: 10.1007/978-1-4899-1471-2_31. [DOI] [PubMed] [Google Scholar]
- 82.Trenkner E, El Idrissi A, Harris C. Balanced interaction of growth factors and taurine regulate energy metabolism, neuronal survival and function of mouse cerebellar granule cells under depolarizing conditions. Adv Exp Med Biol. 1996;403:507–517. doi: 10.1007/978-1-4899-0182-8_55. [DOI] [PubMed] [Google Scholar]
- 83.Trenkner E, El Idrissi A, Dumas R, Rabe A. Functional consequences of calcium uptake modulation by taurine in vivo and in vitro. Adv Exp Med Biol. 1998;442:277–284. doi: 10.1007/978-1-4899-0117-0_35. [DOI] [PubMed] [Google Scholar]
- 84.Valiçelebi G, Stauderman K, Varney MA, Akong M, Hess SD, Johnson EC. Fluorescence techniques for measuring channel activity. Methods Enzymol. 1998;294:20–47. doi: 10.1016/s0076-6879(99)94005-3. [DOI] [PubMed] [Google Scholar]
- 85.Watkins JC, Evans RH. Excitatory amino acid transmitter. Annu Rev Pharmacol Toxicol. 1981;21:165–204. doi: 10.1146/annurev.pa.21.040181.001121. [DOI] [PubMed] [Google Scholar]
- 86.White RJ, Reynods IJ. Mitochondria and Na+/Ca2+ exchange buffer glutamate-induced calcium loads in cultured cortical neurons. J Neurosci. 1995;15:1318–1328. doi: 10.1523/JNEUROSCI.15-02-01318.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.White RJ, Reynods IJ. Mitochondrial depolarization-in glutamate stimulated neurons: an early signal specific to excitotoxin exposure. J Neurosci. 1996;16:5688–5697. doi: 10.1523/JNEUROSCI.16-18-05688.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Wood AM, Tiwari P, Bristow DR. Media composition modulates excitatory amino acid-induced death of rat cerebellar granule cells. Hum Exp Toxicol. 1997;16:350–355. doi: 10.1177/096032719701600702. [DOI] [PubMed] [Google Scholar]