

Abstract
From the elegant studies of Ramon y Cajal (1909) to the current advances in molecular cloning (e.g., Farber and Danciger, 1997), the retina has served as an ideal model for the entire CNS. We have taken advantage of the well described anatomy, physiology, and molecular biology of the retina to begin to examine the role of the laminins, one component of the extracellular matrix, on the processes of neuronal differentiation and synapse formation in the CNS. We have examined the effect of the deletion of one laminin chain, the β2 chain, on retinal development. The gross development of retinas from laminin β2 chain-deficient animals appears normal, and photoreceptors are formed. However, these retinas exhibit several pathologies: laminin β2 chain-deficient mice display abnormal outer segment elongation, abnormal electroretinograms, and abnormal rod photoreceptor synapses. Morphologically, the outer segments are reduced by 50% in length; the outer plexiform layer of mutant animals is disrupted specifically, because only 7% of observed rod invaginating synapses appear normal, whereas the inner plexiform layer is undisturbed; finally, the rate of apoptosis in the mutant photoreceptor layer is twice that of control mice. Physiologically, the electroretinogram is altered; the amplitude of the b-wave and the slope of the b-wave intensity–response function are both decreased, consistent with synaptic disruption in the outer retina. Together, these results emphasize the prominence of the extracellular matrix and, in particular, the laminins in the development and maintenance of synaptic function and morphogenesis in the CNS.
Keywords: synapse development, laminin, photoreceptor, ERG, apoptosis, extracellular matrix
The development and maintenance of functional connections within the CNS is dependent on a wide variety of processes. These include, but are not limited to, the coordinated production of different cell types, the establishment of proper connections among different cell types, and the subsequent maintenance of these connections. Many environmental factors have been suggested to play roles during these three processes (for review, see Chiba and Keshishian, 1996; Pearlman and Sheppard, 1996; Higgins et al., 1997;Hunter and Brunken, 1997). Because it is well described and continues to mature postnatally, the retina is an excellent model system in which to dissect the role of environmental factors in these three processes.
We have begun to analyze the role of one element of the extracellular matrix, the laminins, in CNS development. The laminins are a complex family of extracellular glycoproteins, each composed of three independent gene products: an α, a β, and a γ chain. Eleven laminin chain isoforms (five α, three β, and three γ) are known (Timpl, 1996; Koch et al., 1999), and at least 14 potential heterotrimers have been postulated (Timpl, 1996; Miner et al., 1997;Koch et al., 1999) with more, no doubt, to be identified.
Mutations in several of the laminin chains are correlated with human disease. These include mutations in the α3, β3, and γ2 chains in the blistering disorders known as junctional epidermolysis bullosas (for review, see Ryan et al., 1996) and mutations in the α2 chain in certain types of muscular dystrophy (for review, see Arahata et al., 1995). The potential pathologies in the CNS have not been extensively studied; however, because α2 is expressed in several parts of the CNS (Morissette and Carbonetto, 1995; Hagg et al., 1997; Raabe et al., 1997; Tian et al., 1997), and muscular dystrophies frequently are associated with CNS dysfunction, it is reasonable to postulate that at least the α2 chain is important in maintenance of CNS function (Arahata et al., 1995).
β2-containing laminins are expressed on the apical surface of the retina (Hunter et al., 1992; Libby et al., 1996, 1997a,b) and in the outer plexiform layer (OPL; R. T. Libby, Y. Xu, E. P. Gibbons, M. F. Champliaud, M. Koch, R. E. Burgeson, D. D. Hunter, and W. J. Brunken, unpublished data) from early in development through adulthood and appear to be the product of the retinal Müller glial cell (Libby et al., 1997). We have shown that β2-containing laminins are important in retinal development (Hunter et al., 1992; Libby et al., 1996, 1997a; Hunter and Brunken, 1997). In the peripheral nervous system, β2-containing laminins have been shown to be important for the development and maintenance of synaptic contacts: mice that are deficient in production of the laminin β2 chain (Noakes et al., 1995a) exhibit abnormalities in their neuromuscular junctions. However, the effects of disruption of laminin β2 chain production in the CNS have not, as yet, been examined. Using histological and physiological techniques, we characterize the retinas of laminin β2 chain-deficient mice. Our data demonstrate that photoreceptor morphogenesis and synaptogenesis in the OPL are disrupted in these animals.
MATERIALS AND METHODS
Animals
All procedures involving animals were approved by the Boston College and Tufts University Animal Care Committees and were in accordance with the National Institutes of Health Guide for the Care and Use of Animals. Mice heterozygous for a null mutation in the laminin β2 chain gene (Noakes et al., 1995a) were a gift from Joshua Sanes (Washington University, Saint Louis, MO). These mice were created by a homologous recombination that targeted the second coding exon of the mouse laminin β2 chain gene. The mutation was made by inserting a neo cassette into exon 3 (the second coding exon) of the Lamb2 gene resulting in a gene that is 1.5 kb longer than the wild type (Noakes et al., 1995a). DNA transfer blot analysis has confirmed the presence of a disrupted laminin β2 chain gene (Noakes et al., 1995a). Stop codons were placed in all three frames of the inserted cassette, and skipping by alternate splicing of exon 3 would result in a frame shift and a stop in translation. Thus, no full-length protein should be produced in these animals. Protein transfer blot analysis has confirmed the absence of laminin β2 chain protein in homozygous nulls (Noakes et al., 1995a), which was confirmed in the kidney (Noakes et al., 1995b). However, parts of exons 2 and 3 could be translated, resulting in the production of the most N-terminal domains (V and VI) of the β2 chain. There are no reagents available to test for the production of this protein fragment. Nevertheless, if the short N terminus were made, it is highly unlikely that it would be exported, because only fully assembled trimers with intact long arms are exported (Yurchenco et al., 1997). Heterozygous animals, maintained in a 12 hr day/night cycle, were bred in our colony at Boston College. The day of birth was defined as postnatal day 0 (P0). Genotypes of offspring of heterozygous matings were determined as described previously (Noakes et al., 1995a) with minor modifications; to identify the mutated gene we used the following primer pairs: 5′-ccgggcgcccctgcgctgacagc-3′ and 5′-cgaattcgaacacgcagatgcag-3′; these primers yield a 250 bp PCR product.
In all respects, heterozygous animals appear indistinguishable from homozygous normal mice (data not shown); this observation is consistent with previous reports that characterized animals carrying the laminin β2 chain-null mutation. Thus, as in those previous reports, heterozygous (+/−) and homozygous (+/+) animals, both of which are phenotypically wild type, were used as controls.
Histology
Tissue preparation. Unfixed tissue was prepared, embedded, and frozen as described previously (Libby et al., 1996). For preparation of paraformaldehyde-fixed tissue, eyes were removed, and a hole was made at the ora serrata using a hypodermic needle. The tissue was then placed in 4% paraformaldehyde (Sigma, St. Louis, MO) in PBS (137 mm NaCl, 2.68 mm KCl, 10 mm Na2HPO4, 1.76 mmKH2PO4, pH 7.4) at 0°C. After ∼30 min, the anterior chamber and lens were removed, and the resultant eyecup was fixed for 2 hr or overnight. The tissue was then placed into an ascending series of sucrose solutions (5, 10, 15, and 20% in PBS), incubated overnight at 4°C in a mixture of 20% sucrose and 80% OCT (Miles, Elkhart, IN), and then changed to a fresh solution of 20% sucrose and 80% OCT. The tissue was then frozen and cut at 10 μm, as described previously (Libby et al., 1996). For some histological examinations, the tissue was fixed in 3% glutaraldehyde (Polysciences, Warrington, PA) and 2% formaldehyde in PBS and then treated as described above.
For electron microscopy, eyes were prepared as above but fixed in 3% glutaraldehyde and 2% formaldehyde in 0.1 m sodium cacodylate, pH 7.4, for 1 hr. Eyecups were sectioned into quadrants, washed in 0.1 m sodium cacodylate, pH 7.4, fixed in 2% glutaraldehyde and 1% osmium tetroxide for 1 hr, and then dehydrated in a graded series of ethanols, embedded in plastic, and sectioned. Semithin sections were taken to assure the orientation of the tissue, and then ultrathin sections were made and viewed and photographed on a Philips (Eindhoven, The Netherlands) CM-10 transmission electron microscope. Synaptic morphology was scored and counted from working prints. Synaptic ribbons were identified and the number of postsynaptic elements counted; synapses were grouped into four categories: triads, requiring the presence of three postsynaptic processes; dyads, in which either one or two distinct postsynaptic processes are present; floating ribbons, in which a ribbon but no apposed postsynaptic process is present; and two on one, in which two ribbons are apposed to one postsynaptic element.
Immunohistochemistry on unfixed and paraformaldehyde-fixed tissue was performed as described previously (Libby et al., 1996, 1997a).
Antibodies and lectins. A mouse monoclonal antibody that recognizes rhodopsin (Ret-P1; Fekete and Barnstable, 1983) was a gift from C. Barnstable (Yale University, New Haven, CT). Anti-laminin antibodies were a polyclonal anti-laminin α4 chain from J. R. Sanes; a monoclonal anti-laminin β2 chain (Hunter et al., 1989); and a polyclonal anti-laminin γ3 chain from R. E. Burgeson (Harvard University, Charlestown, MA). A mouse monoclonal antibody that recognizes synaptic ribbons was raised in our laboratories (B16;Balkema, 1991; Balkema and Rizkalla, 1996). A mouse monoclonal anti-synaptophysin antibody was obtained commercially (Boehringer Mannheim, Indianapolis, IN). Secondary antibodies were obtained from Sigma and Incstar (Stillwater, MN). Texas Red-labeled peanut agglutinin was obtained from Boehringer Mannheim.
Assay for apoptotic cell death
For consistency, sections from P15 and P20 animals were chosen that included the optic nerve or were close to it. Apoptotically dying cells in these sections were detected using an in situ cell death detection kit (Boehringer Mannheim). This kit detects dying cells by the terminal deoxynucleotidyl transferase-mediated dUTP nick end-labeling method (Gavrieli et al., 1992), which labels the ends of fragmented DNA with labeled UTP (here, the UTP is coupled to fluorescein), using the enzyme terminal deoxynucleotidyl transferase. Paraformaldehyde-fixed sections (10 μm; see above) were prepared for labeling by fixation onto slides with 4% paraformaldehyde in PBS for 20 min at room temperature and then were permeabilized and labeled according to the manufacturer's instructions. After labeling, sections were mounted in a glycerol-based solution containing paraphenylenediamine (1 mg/ml) to reduce photobleaching.
Electroretinography
Electroretinograms (ERGs) were performed on animals between postnatal days 20 and 26. All preparative procedures were performed in normal room light. Animals were anesthetized with Taractan (chlorporthixene; Roche, Nutley, NJ; ∼12.5 mg/kg), followed by Nembutal (∼65 mg/kg), and then placed into a stereotactic holder. A reference electrode, made from silver wire, was placed between the skin and the skull near bregma and secured with cynoacrylate. After suturing the eye open, a drop of atropine (0.54 mg/ml) was placed onto the eye for ∼1 min. The animal was then placed into a light-tight Faraday cage. After aligning the animal with the light source, a cotton wick electrode coupled to a silver-silver chloride half-cell was placed onto the animal's cornea in a position that did not attenuate the light flashes. A test flash was presented to the animal to check for proper electrode placement. After a 21 min dark adaptation, animals were presented 10 separate 50 msec flashes that were separated by 2 sec (PS22 Photopic stimulator; Grass Instruments, Quincy, MA); the responses were amplified using a Dam 50 differential amplifier (World Precision Instruments, Sarasota, FL) and recorded and averaged using MacLab 4S (AD Instruments, Mountain View, CA). For each animal, at least one set of recordings was made over a 4.2 log range of intensities, starting with least bright. A 21 min period of dark adaptation preceded any subsequent recording sessions.
RESULTS
Mice that are homozygous for the laminin β2-null mutation (here referred to as laminin β2 chain-deficient) have been previously reported to have disrupted neuromuscular junctions and to suffer from proteinuria (Noakes et al., 1995a,b). Laminin β2 chain-deficient mice stop gaining weight during the second postnatal week and normally live only into the fourth postnatal week. Here, we have characterized disruptions in one portion of the CNS of these laminin β2 chain-deficient animals through the fourth postnatal week. The animals lactate normally but are lethargic and move about with difficulty. No extensive behavioral tests were performed on these animals.
Histology
During the first 10 or 11 postnatal days, retinas from laminin β2 chain-deficient and control mice were not grossly different (results not shown). By P13, however, the first differences between the laminin β2 chain-deficient and control mice become evident; in particular, the thickness of the retinas in laminin β2 chain-deficient mice is reduced (results not shown). This difference in thickness is easily detectable by P15 (Fig.1). This reduction is attributable almost entirely to a decrease in photoreceptor length: although nuclear and plexiform (synaptic) layers in laminin β2 chain-deficient retinas appear largely normal (as judged by width), the photoreceptor outer segments, and possibly inner segments, are clearly shorter (Fig. 1). The period during which this difference becomes apparent is the period when outer segments are undergoing their maximal lengthening.
Fig. 1.
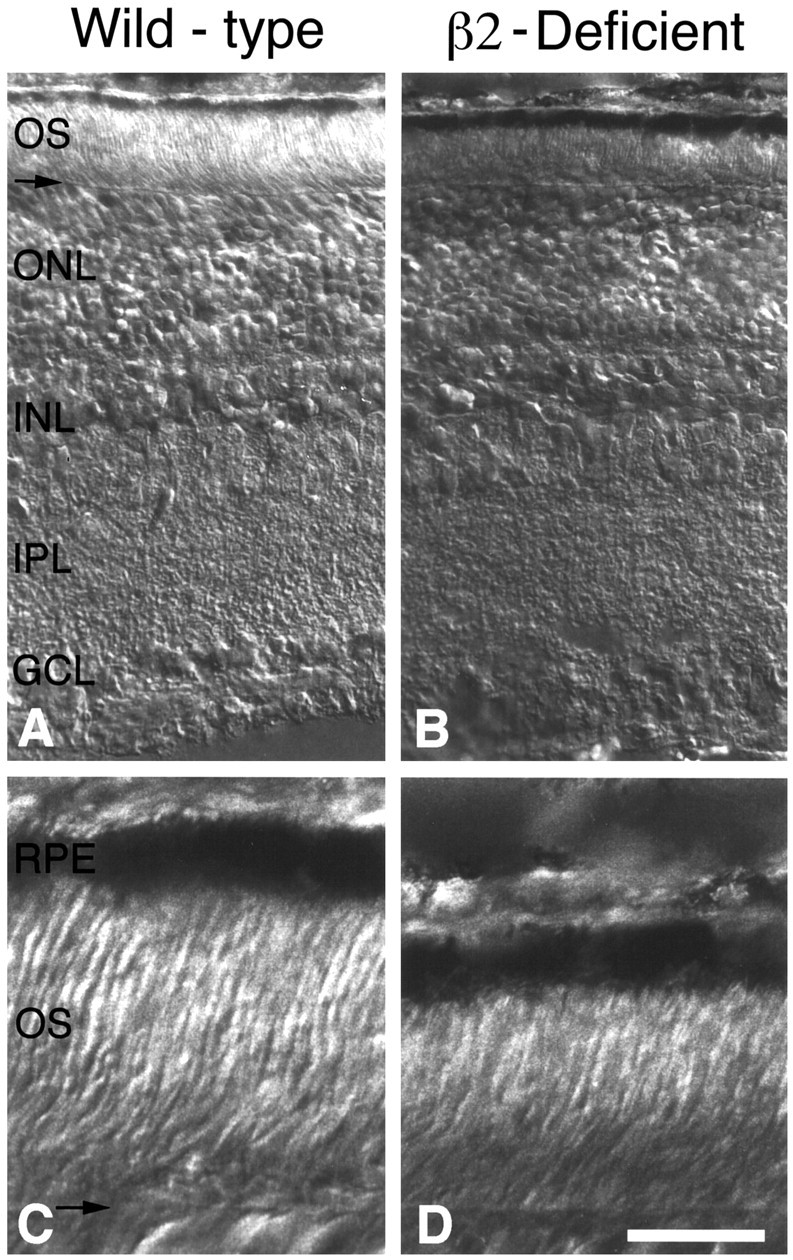
Comparison of wild-type and laminin β2 chain-deficient retinas at postnatal day 15. Sections of wild-type (A, C) and laminin β2 chain-deficient littermate (B, D) retinas from areas adjacent to the optic nerve head are shown. There are no gross effects of laminin β2 chain deficiency on the outer nuclear layer (ONL), inner nuclear layer (INL), IPL, or ganglion cell layer (GCL). However, the outer segments (OS) and inner segments of the laminin β2 chain-deficient animal are considerably smaller than those of the wild-type animal, as judged by the distance from the external limiting membrane (arrow) to the retinal pigmented epithelium (RPE). C, D, Enlargements of A and B for clarity. Scale bar: A, B, 50 μm; C, D, 12.5 μm.
In control mice, i.e., phenotypically wild type and genotypically either +/− or +/+, outer and inner segments reach their maximum length over the subsequent 2 postnatal weeks (weeks 3 and 4 after birth). In contrast, in laminin β2 chain-deficient mice, outer and inner segments fail to increase dramatically over this same period. At P25 in control mice, photoreceptor outer and inner segment lengths (∼32 and 16 μm, respectively) have reached the normal range for adult mice (LaVail, 1973); in contrast, outer and inner segments in retinas from laminin β2 chain-deficient mice are ∼50% shorter (Fig.2). Thus, by the fourth postnatal week (that is, at their maximal survival time), laminin β2 chain-deficient mice have severely retarded development of outer and inner segments or a disposition to shorter outer and inner segments, presumably as a consequence of the laminin β2 chain deficiency.
Fig. 2.
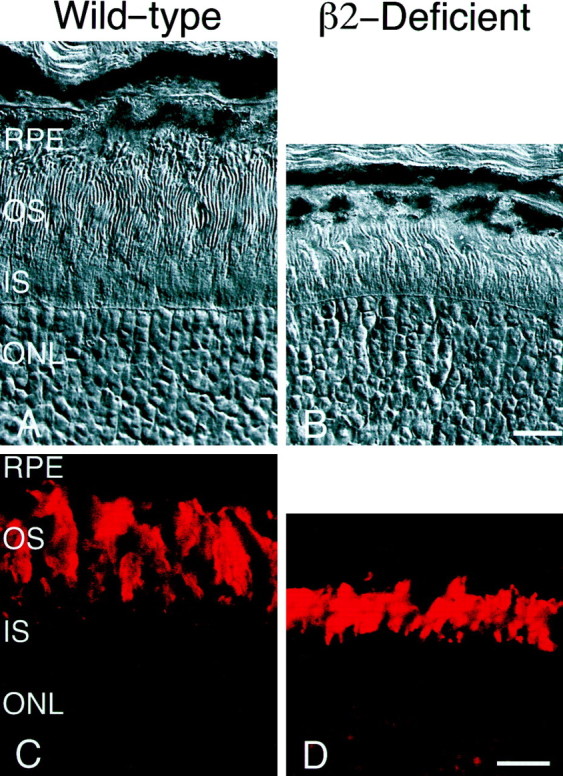
Comparison of wild-type and laminin β2 chain-deficient retinas at postnatal day 25. Sections of wild-type (A, C) and laminin β2 chain-deficient littermate (B, D) retinas, from similar regions, were examined histologically (A, B) or were reacted with an antibody specific for rhodopsin (Ret-P1; C, D). Inner segments (IS) and outer segments (OS) of the wild-type mouse have reached their adult length by postnatal day 25 (A); both inner and outer segments of the laminin β2 chain-deficient mouse are shorter than those of its littermate (B). Wild-type and laminin β2 chain-deficient retinas express rhodopsin and localize it to their outer segments (C, D). However, comparison of rhodopsin expression emphasizes the fact that wild-type OS (C) are significantly longer than those in laminin β2 chain-deficient retinas (D). RPE, Retinal pigmented epithelium; ONL, outer nuclear layer. Scale bars:A, B, 10 μm; C, D, 15 μm.
Although the morphology of the photoreceptors is clearly altered in laminin β2 chain-deficient mice, these mice still express the photopigment rhodopsin in their outer segments. An antibody to rhodopsin reveals rhodopsin in laminin β2 chain-deficient mice that is properly targeted to the outer segments (Fig. 2). Although immunohistochemistry cannot determine whether laminin β2 chain-deficient mice produce control amounts of rhodopsin, we predict that, judging by the production and location of photopigment, these photoreceptors would be capable of responding to light.
Ultrastructurally, the outer segments of the β2 chain-deficient animals are normal (Fig. 3). The membranous disks are well formed and appear normal (Fig. 3, compareA, B). We did note that the disks were more loosely packed in some β2 chain-deficient mice than in others, but we cannot rule out fixation artifacts as the source of this observation (data not shown). The outer segments of the β2 chain-deficient animals arise from basal bodies (Fig. 3C, arrows) in the inner segment, and the ciliary stalk appears normal. Another abnormality in the outer retina of the β2-deficient animals was the presence of aberrant processes in the interphotoreceptor matrix (IPM; Fig. 3D, boxed region shown in higher power inE). These processes appear to arise from deep in the retina and extend up to the retinal pigmented epithelium (Fig. 3D), where they appear to make tight junctions with the overlying retinal pigemented epithelial cells (Fig. 3E). Although we have not as yet identified the source of these fibers, we believe that they arise from Müller cells, which have been shown to sprout in response to retinal injury (Lewis et al., 1999). We did not observe any remarkable differences between control and laminin β2 chain-deficient mice in the ultrastructure of the retinal pigmented epithelium in the electron microscope, nor did we see any change in their attachment to their basement membrane, Bruch's membrane (data not shown).
Fig. 3.
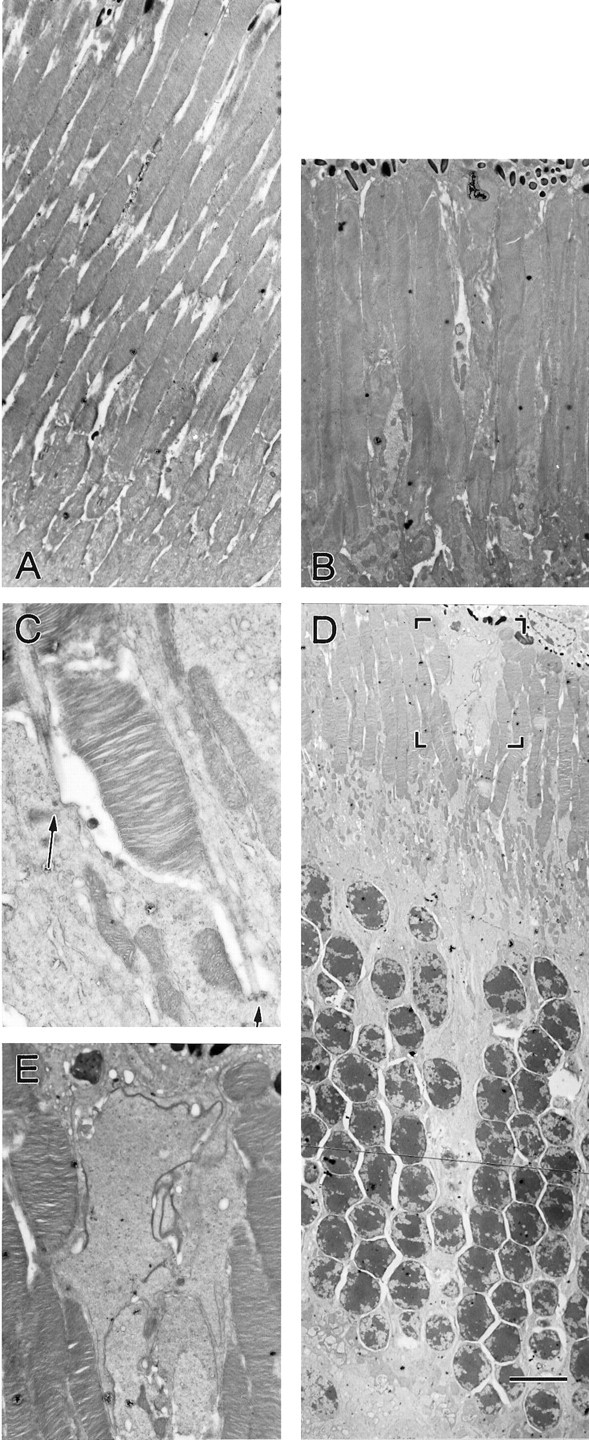
Ultrastructural comparison of wild-type and laminin β2 chain-deficient outer retinas at postnatal day 21. Sections of wild-type (A) and laminin β2 chain-deficient (B–E) outer retinas were viewed with transmission electron microscopy. Although photoreceptor inner and outer segments are shorter in the laminin β2 chain-deficient retinas (B), they appear structurally normal, including at the basal bodies (C, arrows). Aberrant processes were occasionally observed in the laminin β2 chain-deficient retinas (D), which extended through the outer nuclear layer and to the retinal pigmented epithelium. The boxed area in Dis shown at higher power in E. Scale bar: A, B, 4.4 μm; C, 1.1 μm; D, 8.5 μm; E, 2.5 μm.
In a screen of laminin β2 chain-deficient retinas, we did not detect any marked changes in the numbers or morphology at the light-microscopic level of several other cell types, including bipolar cells (using an antibody against protein kinase C; Greferath et al., 1990), horizontal cells (using an antibody against calbindin;Massey and Mills, 1996), and Müller cells (using an antibody to vimentin; Dräger et al., 1984) (data not shown). These data, when coupled with the apparent normal anatomy of the inner retinas from laminin β2 chain-deficient mice (see above), suggest that mainly photoreceptor development is altered by the removal of the laminin β2 chain.
Apoptosis
Programmed cell death, i.e., apoptosis, is a normal part of retinal morphogenesis, occurring during specific periods of retinal development. In the mouse retina, the number of cells undergoing cell death generally follows a Gaussian distribution, beginning before birth, peaking at ∼P9, and ceasing at ∼P25 (Young, 1984). Also, within this distribution, inner retinal cells (i.e., ganglion cells and amacrine cells) die earlier than outer cells (i.e., photoreceptors and bipolar cells). To test whether the laminin β2 chain-deficient mice were undergoing these normal developmental processes, we assayed for programmed cell death on two different critical days, P15 and P20.
At P15 in our control wild-type mice, the type (primarily rod photoreceptors, based on location) and number (16.5 ± 1.5 cells per section, mean ± SEM) of cells undergoing programmed cell death is similar to that previously reported (Young, 1984). Laminin β2 chain-deficient mice had approximately twice the amount of programmed cell death (35.7 ± 6.1 cells per section).
In normal mouse development, by the 20th postnatal day the number of cells undergoing cell death declines rapidly and is limited mainly to photoreceptors. At P20, wild-type retinas contained fewer apoptotic cells (3.6 ± 1.2 cells per section) than at P15; laminin β2 chain-deficient mice also have fewer apoptotic cells at P20 (9.3 ± 2.2 cells per section) than at P15.
Therefore, laminin β2 chain-deficient mice do have elevated programmed cell death; however, they are still following the basic developmental trend: a decrease in dying cells with age. Moreover, the rate of decrease in cell death is parallel to that of the rate in the littermate controls, suggesting that cell death in the laminin β2 chain-deficient mice is slowing as it should. In addition, the increase in dying cells in the laminin β2 chain-deficient mice is relatively small in number when compared with the total number of retinal cells (many thousands per retinal section), so that there is probably little or no effect on retinal function merely because of a paucity of cells. It should also be noted that at both ages examined there is no apparent clumping of apoptotic cells that would suggest necrosis (Vaux, 1993); apoptotic cells are generally surrounded by viable cells. This suggests that the retina of the laminin β2 chain-deficient mouse is not significantly affected by the other pathologies (e.g., kidney disturbances) occurring in the animal.
Laminins in the IPM of laminin β2 chain-deficient mice
Our early work on retinal laminins focused on the apical expression of the laminin β2 chain, i.e., within the interphotoreceptor matrix (Hunter et al., 1992, Libby et al., 1996, 1997). More recently, we have shown that the laminin β2 chain is co-expressed with the laminin α3, α4, and γ3 chains on the apical surface of the retina but also in the outer plexiform layer; the inner plexiform layer appears to contain other laminins, because of the known laminin chains only α4 is expressed there (Libby et al., 1997b; Libby, Xu, Gibbons, Champliaud, Koch, Burgeson, Hunter, and Brunken, unpublished data).
We have proposed that the retina contains two novel laminin heterotrimers, which we have termed laminin 13 (α3β2γ3) and laminin 14 (α4β2γ3) (Libby, Xu, Gibbons, Champliaud, Koch, Burgeson, Hunter, and Brunken, unpublished data). Does the lack of the laminin β2 chain result in a decreased synthesis or deposition of its potential partners in the retina? For example, disruptions in the gene encoding the β3 chain of laminin 5 (α3β3γ2) result in a failure of the other chain partners to be assembled and secreted (Matsui et al., 1995, 1998).
We have examined the retinas of wild-type and laminin β2 chain-deficient animals for the presence of the other components of laminins 13 (α3β2γ3) and 14 (α4β2γ3), i.e., the laminin α3, α4, and γ3 chains. We examined the expression of RNAs encoding these three chains (by in situ hybridization). We have not detected any changes in expression of RNA encoding these chains (results not shown), suggesting that there are no gross changes in transcription of these chains as a result of the removal of the laminin β2 chain.
We have only been able to examine the protein expression of two of the chain partners of β2, because there is currently no mouse-reactive anti-α3 chain reagent available. As we have shown elsewhere for normal rat and human retinas (Libby et al., 1997b; Libby, Xu, Gibbons, Champliaud, Koch, Burgeson, Hunter, and Brunken, unpublished data), the laminin α4 and γ3 chains are present in the retinas of wild-type mice (Fig.4A,C). In all three species, these laminin chains are present in three distinct locations: (1) the interphotoreceptor matrix, (2) the outer plexiform layer, and (3) the inner plexiform layer, where only the α4 chain is expressed. Although full-length laminin β2 chain protein is lacking in laminin β2 chain-deficient mouse, we cannot detect any gross disturbances in the distribution of its presumed partners, the laminin α4 and γ3 chains, in the laminin β2 chain-deficient retinas in any of these locations (Fig.4B,D). These data are consistent with the apparent lack of change in expression of RNA encoding these chains and suggest that the loss of the laminin β2 chain protein does not result in a gross change in the deposition of the laminin α4 and γ3 chains.
Fig. 4.

Other laminin chains are not disrupted by laminin β2 chain deficiency. Unfixed, transverse sections of wild-type and laminin β2 chain-deficient (β2−) littermate retinas were reacted with several antibodies that recognize other laminin chains. The expression pattern of the laminin α4 (A, B) and γ3 (C, D) chains, potential laminin trimer partners of the laminin β2 chain in the neuronal retina, appear to be identical in wild-type (A, C) and laminin β2 chain-deficient (B, D) mice: the laminin α4 and γ3 chains are still present in the subretinal space around inner segments (arrowheads) and in the outer plexiform layer (arrows). The laminin α4 chain (A, B) is also present in both mice throughout the inner retina, through the inner nuclear layer (INL) and the ganglion cell layer (GCL). ONL, Outer nuclear layer. Scale bar, 25 μm.
Laminins are assembled intracellularly, first as βγ dimers, to which α chains are added, allowing export (Matsui et al., 1995;Yurchenco et al., 1997). Notably, in the absence of the laminin β2 chain, the laminin α4 and γ3 chains continue to be expressed in the IPM, wherein they are almost certainly extracellular (Hunter et al., 1992; Libby et al., 1997b). These data suggest that there is a compensatory expression of another, perhaps unknown, laminin β chain in trimers containing the α4 and γ3 chains. Such compensatory mechanisms have been suggested to be present in the kidney of laminin β2 chain-deficient animals (Noakes et al., 1995b).
In the kidney of laminin β2 chain-deficient mice, the laminin β1 chain has been shown to be ectopically expressed in what would normally be a laminin β2 chain pattern. Therefore, we examined the distribution of the laminin β1 chain in the retina of these animals to ascertain whether it could substitute for the laminin β2 chain in retinal laminin trimers containing the α4 and γ3 chains. Using a polyclonal antiserum that reacts with all three chains of laminin-1 (α1, β1, and γ1), we can only detect the laminin chains that are associated with the retinal vasculature; we found no ectopic laminin β1 chain (or laminin α1 and γ1 chains) in the laminin β2 chain-deficient retinas (results not shown). We have no mouse-reactive anti-laminin β3 chain antibodies, leaving open the possibility that the laminin β3 chain, which is not normally associated with the adult neuronal retina (Libby et al., 1997b), may substitute for the laminin β2 chain in the laminin β2 chain-deficient retinas. However,in situ hybridizations using probes that detect RNA encoding the laminin β3 chain suggest that there are no alterations in expression of the RNA encoding this chain. It is, therefore, plausible that a novel laminin β chain may substitute for the laminin β2 chain in the interphotoreceptor matrix and outer plexiform layer of laminin β2 chain-deficient mice.
Light responses
An ERG documents the summed electrical activity of the retina, thereby yielding information about its overall physiology. In particular, an ERG can describe independently the response of photoreceptors and outer retinal interneurons to light (Dowling, 1960;Brown and Wiesel, 1961): photoreceptor responses are present as an initial downward deflection, known as the a-wave, whereas the transmission to second-order retinal interneurons, dominated by the contribution of depolarizing bipolar cells, is present as a subsequent upward deflection, known as the b-wave (Fig.5). The amplitude of these waves, their shape, and the time it takes to reach a peak voltage (“time to peak” or “implicit time”) can be used as diagnostic tools to determine the physiological state of the retina.
Fig. 5.

Electroretinography of wild-type and laminin β2 chain-deficient retinas. A, Top, The ERGs of a P21, wild-type mouse (left) and a P20, laminin β2 chain-deficient mouse (right) are shown at maximum light intensity. The laminin β2 chain-deficient mouse exhibits a normal a-wave; however, its b-wave is markedly attenuated.Bottom, Intensity–response series (over 4.2 log units, after 21 min of dark adaptation) of a P18, wild-type mouse (left) and a P20, laminin β2 chain-deficient mouse (right). The a- and b-waves of the wild-type mouse increase with increasing levels of light. The laminin β2 chain-deficient mouse shows a similar intensity–response profile, but the b-wave is clearly altered. Calibration: 20 μV (vertical); 100 msec (horizontal). B, The average a-wave amplitudes (triangles) and b-wave amplitudes (squares) for wild-type mice (open symbols) and laminin β2 chain-deficient mice (filled symbols) are shown over a wide range of light intensities. The a-waves of the wild-type mice and the laminin β2 chain-deficient mice are similar; in contrast, the b-waves are attenuated in laminin β2 chain-deficient mice over the entire range.
We performed ERGs on phenotypically wild-type (both +/+ and +/−) and laminin β2 chain-deficient (−/−) mice. At maximum stimulus intensity, the ERG of a control, wild-type mouse (Fig. 5A, top left trace) is typical, with a photoreceptor-driven a-wave overtaken at ∼50 msec by the field potential arising from the second-order interneurons, the b-wave. The b-wave reaches its peak at ∼100 msec, and then the ERG falls quickly back to baseline.
On the other hand, the ERGs obtained from the laminin β2 chain-deficient mouse are abnormal (Fig. 5A, top right record). The a-wave appears largely normal in terms of its amplitude at maximal light intensity; although the example shown in Figure 5A has a slightly increased amplitude, this is not representative of the population as a whole (see below). The mean a-wave amplitude for β2 chain-deficient mice (n = 7) was 69.9 ± 7.7 μV (mean ± SEM) compared with 85.3 ± 18.9 μV in control mice (n = 9). The time to peak (implicit time) was also unaltered (44.5 ± 1.4 msec in the β2 chain-deficient mice compared with 49.6 ± 2.4 msec in the wild-type mice). This suggests that, although the morphology of the photoreceptor outer segments is clearly affected by the lack of the laminin β2 chain, the overall ability of the photoreceptors to respond to light is relatively unaffected (see below).
In contrast to the largely normal a-wave, there is a marked disruption of the b-wave of ERGs from laminin β2 chain-deficient animals (Fig.5A, top right record). Although the b-waves have normal implicit times (98.8 ± 4.2 msec in −/− mice compared with 100.2 ± 7.7 msec in wild-type mice), their mean amplitudes at maximal light intensity are significantly reduced (to 136 ± 13.4 μV in the −/− mice from 269 ± 51.8 μV in control mice;p < 0.05). Moreover, the ERG does not return to baseline as in the wild-type mouse; rather, it generally remains well above baseline for several seconds. This standing wave is frequently persistent and can be seen at the outset of subsequent stimulation (2 sec after the previous stimulation); this is most evident at particularly high light intensities.
Next we examined the intensity–response functions of the ERG in both wild-type and mutant animals. Wild-type (+/− and +/+) mice exhibit the normal response of the retina to increases in light intensity (Fig.5A, bottom left records, plotted in B, open symbols): (1) at low light levels, the b-wave is the only electrical activity observable in the ERG; as the light intensity increases, its amplitude increases and its implicit time decreases; and (2) a-waves are first detectable at brighter light levels than b-waves; like b-waves, the a-wave amplitude increases and implicit time decreases with increasing light intensity.
The response of laminin β2 chain-deficient retinas to increases in light intensity is fundamentally similar to that of wild-type mice: the amplitudes of both a- and b-waves increase with increasing light intensity (Fig. 5A, bottom right records, plotted inB, filled symbols). However, at maximal intensities, although the a-waves of the laminin β2 chain-deficient retinas are similar to those of wild-type controls, the b-waves are greatly attenuated. The response–intensity functions for both a- and b-waves show that, over the whole range of light intensities, the laminin β2 chain-deficient retinas exhibit near-normal a-waves (Fig.5B, filled triangles). There is some apparent reduction at the maximal intensity of light in the a-wave amplitude, but this reduction is not statistically significant. Such a reduction would be expected given the reduction in the volume of the photoreceptors outer segment.
On the other hand, the b-waves of the laminin β2 chain-deficient mice are attenuated at all light intensities tested (Fig. 5B). The ratio of the b- to a-wave amplitudes is a sensitive measure of the transfer function between photoreceptors and second-order neurons. The b- to a-wave ratios (taken at maximal light intensity) are significantly lower in the laminin β2 chain-deficient mice (1.99 ± 0.13 vs 3.33 ± 0.19; p < 0.01). Together, these data suggest that the laminin β2 chain-deficient retinas are capable of detecting light nearly normally, but that information transfer from photoreceptors to second-order cells (the retinal bipolar cells) is compromised. Further support for this suggestion comes from the shape of the b-wave intensity–response curve in laminin β2 chain-deficient retinas (Fig. 5B, filled squares,heavy line), which is considerably different from that in control animals; the b-wave in β2 chain-deficient mice is flatter and approaches a linear function, rather than the exponential shape in the control (Fig. 5B, open squares). The exponential shape of the b-wave response–intensity function reflects the transfer properties of the synapse; the change from an exponential to a linear function in the mutant mouse suggests that the photoreceptor to bipolar transmission is not as efficacious in the mutant mice.
Synaptic integrity in laminin β2 chain-deficient retinas
The physiological data suggest that there is reduction in synaptic transmission in the outer retina, i.e., in the OPL. Therefore, we examined the OPL in detail for any defects in synaptic formation with both the light and electron microscopes. Our previous studies on laminin expression have focused on the apical surface of the retina (Hunter et al., 1992, Libby et al., 1996); however, more recently, we showed that the component chains of laminin 13 and 14 are expressed not only on the apical surface of the retina but also in the OPL of rat and human retinas (Libby et al., 1997b; Libby, Xu, Gibbons, Champliaud, Koch, Burgeson, Hunter, and Brunken, unpublished data). Unfortunately, we have no mouse-reactive anti-laminin β2 chain antibodies; however, in the rat OPL, laminin β2 chain immunoreactivity co-localizes with that of B16, a marker of the synaptic ribbon (Fig.6A–C). Because the α and γ chains of laminin 14 are present in the mouse OPL (see Fig. 4), we presume that the laminin β2 chain is expressed in the mouse OPL as it is the rat retina. Both reverse transcription PCR and in situ hybridizations (data not shown) have established that laminin β2 chain RNA is expressed in mouse retina, and, thus, we have no reason to think there any species difference among mouse, rat, and primate. Thus, laminins containing the β2 chain are localized to the photoreceptor synapse and ideally situated to mediate some of the processes of synaptic formation and stabilization.
Fig. 6.

The OPL is disrupted in laminin β2 chain-deficient mice. As a mouse-reactive laminin β2 chain antibody was not available, β2 chain expression is shown in rat OPL (A–C). B16 is an antibody that recognizes photoreceptor ribbon synapses (A) and co-localizes with the laminin β2 chain (B) in the OPL (images in A and B are merged inC). Note that the laminin β2 chain is also present in capillaries (*), whereas the B16 antigen is not. D–G, Transverse sections of postnatal day 25 wild-type (D, F) and laminin β2 chain-deficient littermate (E, G) retinas were reacted with a lectin (peanut agglutinin; D, E) that recognizes cone outer segments (OS) and cone photoreceptor terminals (arrows) or an antibody that recognizes all photoreceptor synaptic termini (anti-synaptophysin; F, G) within the OPL, located between the outer nuclear layer (ONL) and the inner nuclear layer (INL). Photoreceptor synapses in the laminin β2 chain-deficient retina appear disorganized. Scale bars, 25 μm.
Peanut lectin, which specifically labels cone photoreceptors, is normally expressed in discrete patches in the OPL, reflecting the regularly, and widely, spaced cone pedicles (Fig. 6D,arrows point to individual pedicles). In contrast, in the mutant mice, peanut lectin binding appears to be a nearly continuous band, as if adjacent cone pedicles have expanded their bases (Fig.6E). Synaptophysin, which is expressed at presynaptic sites, is confined to the terminals of wild-type mice (Fig.6F); in the β2 chain-deficient mouse, it is expressed along the axon of the photoreceptor as well as in the terminal, suggesting a disruption in the molecular anatomy of the terminals (Fig. 6G). These alterations, albeit minor, of the synapse suggested that ultrastructural investigations of the photoreceptor anatomy were warranted.
To describe further the synaptic phenotype, we examined retinas from seven animals (four mutants and three controls) at P21–P25, an age when the OPL is mature (Olney, 1968a,b; Blanks et al., 1974; McArdle et al., 1977). In wild-type mice, photoreceptor synapses usually are seen as a classic “triad,” in which the photoreceptor terminal, complete with its synaptic ribbon and underlying arciform density demarcating the active zone, are apposed to the dendrites of three postsynaptic cells: one central, bipolar element and two lateral, horizontal cell elements (Fig. 7A, *). This structure predominates in normal retinas (Table1). However, as a result of the complexity of the invaginations into the photoreceptor terminal, the central bipolar dendrite can be missed if the plane of section is not through the long axis of the terminal; under this circumstance, one only sees the horizontal processes (Table 1, dyads). Together, triads and dyads represent ∼95% of the synapses in the wild-type retina and confirm that OPL development is largely complete at this age.
Fig. 7.
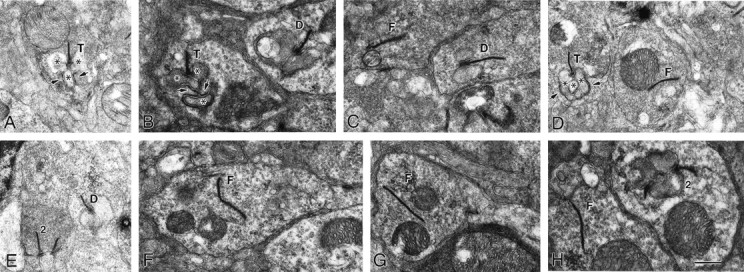
Ultrastructural comparison of synapses within the outer plexiform layer of wild-type and laminin β2 chain-deficient retinas at postnatal day 21. Sections of wild-type (A) and laminin β2 chain-deficient (B–H) retinas were viewed with transmission electron microscopy. In the normal retina, rod photoreceptors make synapses, called triads (T), with three postsynaptic elements (*); the ribbon is a presynaptic specialization that marks the active site for release; the three postsynaptic elements invaginate into the base of the photoreceptor such that two horizontal cell dendrites lie laterally, and one bipolar cell dendrite lies centrally. The photoreceptor wraps around the bipolar cell (arrows). In the β2 chain-deficient animal (B–H), several different types of synapses are present. Triads (T) like those in the wild type animal are rare; dyads, with only one or two horizontal cell processes apposed to the ribbon, are more common (D); dyads are seen in wild-type retinas as well (not illustrated). Also common in the β2 chain-deficient retinas are floating synapses (F), wherein a fully formed ribbon, often with vesicles associated, is seen without any postsynaptic element apposed. This type of synapse is seen extremely rarely in the wild-type retina (see Table 1). Finally, occasionally, two ribbons from the same photoreceptor will be apposed to a single postsynaptic element (2); this was also rarely observed in the wild-type animal. Scale bar: A, D, E, 850 nm; B, C, F–H, 630 nm.
Table 1.
Morphology of photoreceptor synapses in wild-type and laminin β2 chain-deficient retinas
| Wild-type (n = 95) | β2 Chain-deficient (n = 219) | |||
|---|---|---|---|---|
| n | % | n | % | |
| Triads | 50 | 52.63 | 16 | 7.31 |
| Dyads | 40 | 42.11 | 107 | 48.86 |
| Floating ribbons | 4 | 4.21 | 84 | 38.36 |
| 2 on 11-a | 1 | 1.05 | 12 | 5.48 |
Synapses from wild-type and laminin β2 chain-deficient retinas were observed in transmission electron micrographs and placed into one of four categories. Most wild-type synapses consisted of the typical triad arrangement (see Fig. 7A), whereas most laminin β2 chain-deficient synapses were abnormal.
Two ribbons apposed to a single postsynaptic element.
In marked contrast, the OPLs of laminin β2 chain-deficient mice rarely contain fully formed triads (Table 1); instead, a variety malformations are present (Fig. 7B–H). A few triads (T in Fig. 7B,D) are present; these represented <8% of all synapses. There were numerous examples of a ribbon complex apposed to one or two postsynaptic elements, which we could identify as horizontal cell dendrites by the presence of synaptic vesicles (D in Fig. 7B,C,E); these were scored together as dyads (Table 1) and accounted for nearly 50% of all synaptic complexes, which is not significantly different from the control retinas. Together, triads and dyads, relatively mature synapses, account for only ∼55% of the ribbon synapses in the mutant mice. We observed with low frequency multiple ribbons apposed on one postsynaptic element (2 in Fig. 7E,H). However, the most conspicuous malformations we saw were structures we call floating ribbons (Fig. 7C,D,F–H); these are fully assembled ribbon complexes with associated synaptic vesicles that are unapposed to any postsynaptic element; these structures account for 44% of all ribbon structures in the laminin β2 chain-deficient retinas (Table 1). This profound disruption in the anatomical arrangement of the photoreceptor output synapse is likely to account for the disruption in the b-wave in the mutant animals.
To ask whether the defect is a result of malformation or failure for bipolar cells to develop properly, we examined the inner plexiform layer (IPL). In the IPL, bipolar cells make output synapses with a ribbon structure onto amacrine and ganglion cells (Fig.8A,E, R). The IPL also contains conventional synapses between amacrine cells and from amacrine cells onto bipolar and ganglion cells (not illustrated for control). Examination of the IPL of the β2 chain-deficient mouse revealed no striking abnormalities; both well formed ribbon synapses (Fig. 8B,F, R) and conventional synapses (Fig. 8C,D,G, C) were present. Although not exhaustive both light (data not shown) and these electron microscopic studies suggest that the IPL is well developed, which is not unexpected, because the laminin β2 chain is not expressed in this region. Therefore, the apparent defect in the β2 chain-deficient mouse is in the outer retina, consistent with the expression pattern of this molecule, i.e., in the IPM and in the matrix of the OPL.
Fig. 8.
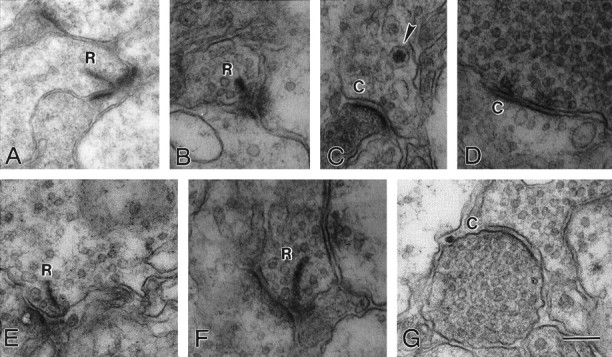
The ultrastructure of the inner plexiform layer of wild-type and β2 chain-deficient animals is similar. Bipolar cell output synapses are ribbon-type synapses (R), whereas amacrine cells make conventional synapses (C) onto other amacrine cells. Two examples of ribbon synapses in wild-type animals (A, E) are shown; apposing the ribbon are one or two postsynaptic processes of unknown origin. Synapses in β2 chain-deficient animals (B–D, F, G) illustrate that the IPL of the mutant animal has both normal ribbon synapses (B, F) and normal conventional synapses (C, D, G), one of which contains a dense-core vesicle (arrowhead). These data suggest that synaptogenesis has proceeded normally in the IPL. Scale bar:A, 340 nm; B–G, 250 nm.
DISCUSSION
Laminin β2 chain-deficient mice have two basic morphological abnormalities: (1) shortened inner and outer segments and (2) disrupted synaptic connections between photoreceptors and second order cells. These mice also have a basic functional abnormality: ineffective transmission of light stimuli from photoreceptors to retinal interneurons, presumably as a result of the disrupted photoreceptor synapses. We believe that these disruptions result from a block of normal photoreceptor differentiation in laminin β2 chain-deficient retinas. Synaptogenesis and outer segment morphogenesis occur in the first and second postnatal weeks and happen simultaneously (Fig.9). First, an apical process is elaborated as the growth cone of the axon makes contact with horizontal cell processes (P4); later, the outer segment arises, and horizontal cell dendrites invaginate into the terminal (P7); maturation is complete when the bipolar cell invades between flanking horizontal cells and the outer segments reach their adult length (P14). In the β2-deficient mouse, final stages of photoreceptor morphogenesis are halted.
Fig. 9.

Disruption of laminin β2 chain production leads to disruption of photoreceptor development. During normal development, photoreceptors begin to develop from an undifferentiated progenitor cell between P0 and P4, at which time they begin to form contacts with processes from interneurons within the outer plexiform layer. The normal developmental program begins to falter in laminin β2 chain-deficient retinas at this stage, with poor development of outer segments and synaptic contacts by P7 and little progress past this point. The net result is that, by P14, the morphogenesis of the photoreceptor is markedly disrupted. This is likely to contribute to the inadequate function that is readily apparent in laminin β2 chain-deficient retinas.
Differences in disruption of the β2 chain in vitroand in vivo
Using an in vitro system, we have suggested that the β2 chain, directly or indirectly, is at least partly responsible for the differentiation of rod photoreceptors (Hunter et al., 1992; Hunter and Brunken, 1997). This result is consistent with the in vivo data reported here, wherein a lack of the β2 chain results in poor differentiation of rods, i.e., their outer segments and synapses. Previously, we showed that the β2 chain modulates a choice between rod photoreceptor and rod bipolar cell fates in vitro (Hunter and Brunken, 1997), a choice that is not reflected in the data reported here.
Why are the results of our in vitro and in vivodisruptions of the β2 chain different? Perhaps the most unsatisfying, but most likely, explanation is that there are redundant developmental pathways in vivo that are absent in vitro. FGF-2 (Hicks and Courtois, 1992), taurine (Altshuler and Cepko, 1992), and retinoic acid (Kelly et al., 1994) have all been shown to promote rod photoreceptor development, whereas TGF-α (Lillien and Cepko, 1992), leukemia inhibitory factor (Neophytou et al., 1997), and CNTF (Ezzeddine et al., 1997; Neophytou et al., 1997) have all been shown to inhibit photoreceptor development. With so many different factors potentially influencing the development of photoreceptors, it is not surprising that the absence of any single factor may not be sufficient to prevent a progenitor cell from differentiating into a photoreceptor but may be sufficient to cause a disruption in the overall differentiation program.
Another possibility is that, although we have functionally removed the β2 chain in vitro and in vivo, the retinal laminins have been disrupted in different ways in the two systems. As noted above, several of the laminin chains that are thought to be chain partners with the β2 chain are present in the adult retina (Fig. 4); therefore, in vivo, a non-β2 chain may substitute in a laminin that may subserve some of the functions of β2 chain-containing laminins.
Is the health of the β2 chain-deficient mice contributing to the retinal phenotypes?
β2 chain-deficient mice stop growing during the second week of life and eventually die, probably as a result of proteinuria, during the fourth week of life. Because the animal is unhealthy during this period, it is possible that the alteration in phenotype that we have attributed to a direct effect of the lack of the β2 chain in the retina is, instead, secondary to extraretinal pathologies. However, it is unlikely that slow growth affects photoreceptor outer segment length: rodents that are malnourished and have growth curves similar to those of β2 chain-deficient mice do not have altered photoreceptor outer segment length during their third week of life (Sinning et al., 1984), the period when the β2 chain-deficient mice show considerably shorter outer and inner segments. Thus, simple nutritional deficiency is not sufficient to cause stunted outer and inner segments.
In situations of malnutrition, particularly vitamin A deficiency, ERG abnormalities are markedly different from those of β2 chain-deficient animals: the a-wave almost completely disappears before the b-wave begins to attenuate. In later stages of vitamin A deficiency, the photoreceptors begin to degenerate (Dowling, 1960). Similarly, in the photoreceptor degenerative disorders, generally, the a-wave is the first to be attenuated, followed by the b-wave. The normal a-wave in the ERG of the β2 chain-deficient mice suggests that the photoreceptors are healthy and, importantly, not undergoing degeneration.
Disruption of retinal morphology
The OPL is disrupted in retinas of β2 chain-deficient animals, whereas the IPL remains largely unaffected. There are at least two potential loci where the loss of the β2 chain could affect development of this layer. First, removal of the β2 chain from the ventricular surface of the retina could adversely affect not only outer segment formation but also overall photoreceptor development, such that all photoreceptor structures, including terminals, might be disrupted. Second, because β2 chain protein is expressed, along with its chain partners, in the matrix of the OPL, it is possible that the disruption of the OPL is a direct result of the loss of the β2 chain at or near the synapses.
It is novel, but not surprising, to find an effect of the β2 chain associated with a synaptic layer in the CNS. Laminins containing the β2 chain are found at a peripheral synapse, the neuromuscular junction (e.g., Hunter et al., 1989; Patton et al., 1997). Furthermore, the neuromuscular junction is similarly disorganized in the β2 chain-deficient mouse, suggesting that it is an important factor in differentiation and stabilization at this peripheral synapse. Thus, by analogy, the β2 chain could be important in the differentiation and function of particular central synapses, including the rod photoreceptor synapses within the OPL of the retina. The apparent disruption in the process of the Müller glial cell in the outer retina is also consistent with the finding at the neuromuscular junction in which the glial cell behavior is aberrant (Patton et al., 1998).
Disruption of retinal physiology
The ERG a-wave, a measure of photoreceptor activity (Dowling, 1960; Brown and Wiesel, 1961), has been related to the rhodopsin content of photoreceptors and, therefore, their size. Thus, the lack of an altered a-wave in the β2 chain-deficient mice, which have shorter outer segments, is counterintuitive. However, although rhodopsin concentration is likely an important determinant of the a-wave, during development in the rat a near-normal a-wave function can be achieved with only 50% of the normal content of rhodopsin (Fulton et al., 1995). Thus, it is possible that there is enough rhodopsin in the shortened outer segments of the β2 chain-deficient retinas to generate a nearly normal photoresponse.
The most striking alteration in the ERGs of β2 chain-deficient mice is in their severely attenuated b-wave, the wave generated by the activation of the second-order neurons. A likely explanation for the selective attenuation of the b-wave is the synaptic disturbance in the OPL itself. Because the β2 chain is associated with the OPL in mammals, a deficiency in the laminin β2 chain could result in an improperly formed or functioning OPL, which then could directly explain the attenuated b-wave. Moreover, the intensity–response relationship of the b-wave is markedly altered in the mutant mice (Fig.4B); in particular, the function becomes nearly linear with a marked attenuation at the higher intensities. These observations suggest that the photoreceptor output synapse is not able to function effectively at the higher rates of vesicular fusion seen at high light levels. These alterations in b-wave properties are analogous to the electrophysiological alterations observed at the β2 chain-deficient neuromuscular junction, where both a decrease in synaptic efficacy and rates of spontaneous release were seen (Noakes et al., 1995a).
The alterations in ERGs seen in the β2 chain-deficient retina are remarkably similar to those reported after disruptions in the protein dystrophin, as seen in humans (in Duchenne and Becker muscular dystrophy) and in mice (in the mdx andmdxCv3 mutants): a normal a-wave and an attenuated b-wave. Dystrophin is present in photoreceptor terminals in many mammals, including mouse and human, and the attenuated b-wave in dystrophin mutant mice is thought to be the result of disruptions at those terminals (Schmitz et al., 1993; Fitzgerald et al., 1994; Pillars et al., 1995; Ueda et al., 1997); however, disruptions of the synaptic terminals on the scale shown here are not reported in themdxCv3 mutant (Blank et al., 1999).
Dystrophin can interact with laminin via the dystroglycan complex (for review, see Sunada and Campbell, 1996); at least part of this complex is present at photoreceptor synapses. Because genetic disruptions in dystrophin and the laminin β2 chain cause similar attenuations of the b-wave, and the outer plexiform layer contains at least part of the complex that may link dystrophin and laminins, we speculate that these two components may act in concert in the differentiation and stabilization of photoreceptor synapses. This, again, would be analogous to the neuromuscular junction, where disruptions in both the laminin β2 chain and dystrophin result in neuromuscular pathologies. We further speculate that such interactions may be important at additional central synapses.
Footnotes
This work was supported by funds provided by Boston College (W.J.B., G.W.B.), Tufts University and the National Eye Institute (D.D.H.), and the Foundation Fighting Blindness and the E. Matilda Ziegler Foundation (W.J.B.). Some of this work was supported by the core facilities of the Cutaneous Biology Research Center (CBRC) under a Massachusetts General Hospital–Shiseido Co., Ltd. agreement. We thank Josh Sanes for the generous gift of the founder mice for our β2 knock-out colony, Colin Barnstable for the gift of the antibody against rhodopsin; Josh Sanes and Jeff Miner for the gift of the antibody against the laminin α4 chain, and Bob Burgeson and Marie-France Champliaud for the gift of the antibody against the laminin γ3 chain. W.J.B. thanks Bob Burgeson for encouragement and generosity during his leave of absence at the CBRC.
Correspondence should be addressed to Dr. William J. Brunken, Cutaneous Biology Research Center, Massachusetts General Hospital, Harvard Medical School, Building 149, 13th Street, Charlestown, MA 02129. E-mail: bill.brunken@cbrc2.mgh.harvard.edu.
REFERENCES
- 1.Altshuler D, Cepko C. A temporally regulated, diffusible activity is required for rod photoreceptor development in vitro. Development. 1992;114:942–957. doi: 10.1242/dev.114.4.947. [DOI] [PubMed] [Google Scholar]
- 2.Arahata K, Ishii H, Hayashi YK. Congenital muscular dystrophies. Curr Opin Neurol. 1995;8:385–390. doi: 10.1097/00019052-199510000-00011. [DOI] [PubMed] [Google Scholar]
- 3.Balkema GW. A synaptic antigen (B16) is localized in retinal synaptic ribbons. J Comp Neurol. 1991;312:573–583. doi: 10.1002/cne.903120408. [DOI] [PubMed] [Google Scholar]
- 4.Balkema GW, Rizkalla R. Ultrastructural localization of a synaptic ribbon protein recognized by antibody B16. J Neurocytol. 1996;25:565–571. doi: 10.1007/BF02284824. [DOI] [PubMed] [Google Scholar]
- 5.Blank M, Koulen P, Blake DJ, Kröger S. Dystrophin and beta-dystroglycan in photoreceptor terminals from normal and mdx3Cv mouse retinae. Eur J Neurosci. 1999;11:2121–2133. doi: 10.1046/j.1460-9568.1999.00636.x. [DOI] [PubMed] [Google Scholar]
- 6.Blanks JC, Adinolfi AM, Lolley RN. Synaptogenesis in the photoreceptor terminal of the mouse retina. J Comp Neurol. 1974;156:81–93. doi: 10.1002/cne.901560107. [DOI] [PubMed] [Google Scholar]
- 7.Brown KT, Wiesel TN. Localization of origins of electroretinogram components by intraretinal recording in the intact cat eye. J Physiol (Lond) 1961;158:257–280. doi: 10.1113/jphysiol.1961.sp006768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chiba A, Keshishian H. Neuronal pathfinding and recognition: roles of cell adhesion molecules. Dev Biol. 1996;180:424–432. doi: 10.1006/dbio.1996.0316. [DOI] [PubMed] [Google Scholar]
- 9.Dowling JE. Night blindness, dark adaptation and the electroretinogram. Am J Ophthalmol. 1960;50:875–889. doi: 10.1016/0002-9394(60)90340-8. [DOI] [PubMed] [Google Scholar]
- 10.Dräger UC, Edwards DL, Barnstable CJ. Antibodies against filamentous components in discrete cell types of the mouse retina. J Neurosci. 1984;4:2025–2042. doi: 10.1523/JNEUROSCI.04-08-02025.1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ezzeddine ZD, Yang X, DeChiara T, Yancopoulos G, Cepko CL. Postmitotic cells fated to become rod phoptoreceptors can be respecified by CNTF treatment of the retina. Development. 1997;124:1055–1067. doi: 10.1242/dev.124.5.1055. [DOI] [PubMed] [Google Scholar]
- 12.Fekete DM, Barnstable CJ. The subcellular localization of rat photoreceptor-specific antigens. J Neurocytol. 1983;12:785–803. doi: 10.1007/BF01258151. [DOI] [PubMed] [Google Scholar]
- 13.Fitzgerald KM, Cibis GW, Giambrone SA, Harris DJ. Retinal signal transmission in Duchenne muscular dystrophy: evidence for dysfunction in the photoreceptor/depolarizing bipolar cell pathway. J Clin Invest. 1994;93:2425–2430. doi: 10.1172/JCI117250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Fulton AB, Hansen RM, Findl O. The development of the rod photoresponse from dark-adapted rats. Invest Ophthalmol Vis Sci. 1995;36:1038–1045. [PubMed] [Google Scholar]
- 15.Gavrieli Y, Sherman Y, Ben-Sasson SA. Identification of programmed cell death in situ via specific labeling of nuclear DNA fragmentation. J Cell Biol. 1992;119:493–501. doi: 10.1083/jcb.119.3.493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Greferath U, Grunert U, Wässle H. Rod bipolar cells in mammalian retina show protein kinase C-like immunoreactivity. J Comp Neurol. 1990;301:433–442. doi: 10.1002/cne.903010308. [DOI] [PubMed] [Google Scholar]
- 17.Hagg T, Portera-Cailiau C, Jucker M, Engvall E. Laminins of the adult mammalian CNS: laminin-alpha2 (merosin M-) chain immunoreactivity is associated with neuronal processes. Brain Res. 1997;764:17–27. doi: 10.1016/s0006-8993(97)00419-8. [DOI] [PubMed] [Google Scholar]
- 18.Hicks D, Courtois Y. Fibroblast growth factor stimulates photoreceptor differentiation in vitro. J Neurosci. 1992;12:2022–2033. doi: 10.1523/JNEUROSCI.12-06-02022.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Higgins D, Burack M, Lein P, Banker G. Mechanisms of neuronal polarity. Curr Opin Neurobiol. 1997;7:599–604. doi: 10.1016/s0959-4388(97)80078-5. [DOI] [PubMed] [Google Scholar]
- 20.Hunter DD, Brunken WJ. β2 laminins modulate neuronal phenotype in the rat retina. Mol Cell Neurosci. 1997;10:7–15. doi: 10.1006/mcne.1997.0632. [DOI] [PubMed] [Google Scholar]
- 21.Hunter DD, Shah V, Merlie JP, Sanes JR. A laminin-like adhesive protein concentrated in the synaptic cleft of the neuromuscular junction. Nature. 1989;338:229–234. doi: 10.1038/338229a0. [DOI] [PubMed] [Google Scholar]
- 22.Hunter DD, Murphy MD, Olsson CV, Brunken WJ. S-laminin expression in adult and developing retinae: a potential cue for photoreceptor morphogenesis. Neuron. 1992;8:399–413. doi: 10.1016/0896-6273(92)90269-j. [DOI] [PubMed] [Google Scholar]
- 23.Kelly MW, Turner JK, Reh TA. Retinoic acid promotes differentiation of photoreceptors in vitro. Development. 1994;120:2091–2102. doi: 10.1242/dev.120.8.2091. [DOI] [PubMed] [Google Scholar]
- 24.Koch M, Olsen P, Albus A, Jin W, Hunter DD, Brunken WJ, Burgeson RE, Champlaiud M-F. Characterization and expression of the laminin γ3 chain: a novel, non-basement membrane, associated laminin chain. J Cell Biol. 1999;145:605–617. doi: 10.1083/jcb.145.3.605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.LaVail MM. Kinetics of rod outer segment renewal in the developing mouse retina. J Cell Biol. 1973;58:650–661. doi: 10.1083/jcb.58.3.650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Libby RT, Hunter DD, Brunken WJ. Developmental expression of laminin β2 in rat retina. Further support for a role in rod morphogenesis. Invest Ophthalmol Vis Sci. 1996;37:1651–1661. [PubMed] [Google Scholar]
- 27.Libby RT, Yin X, Selfors LM, Brunken WJ, Hunter DD. Identification of the cellular source of laminin β2 in adult and developing vertebrate retinae. J Comp Neurol. 1997a;389:355–367. doi: 10.1002/(sici)1096-9861(19971229)389:4<655::aid-cne8>3.0.co;2-#. [DOI] [PubMed] [Google Scholar]
- 28.Libby RT, Xu Y, Gibbons EP, Hunter DD, Brunken WJ. Identification and localization of laminin chains in human retina. Invest Ophthalmol Vis Sci. 1997b;38:S30. [Google Scholar]
- 29.Lillien L, Cepko C. Control of proliferation in the retina–temporal changes in responsiveness to FGF and TGFα. Development. 1992;115:253–266. doi: 10.1242/dev.115.1.253. [DOI] [PubMed] [Google Scholar]
- 30.Lewis GP, Valter K, Mervin K, Maslim J, Stone J, Fisher SK. Müller cell hyptertrophy following experimental retinal detachments association with cone photoreceptors. Invest Ophthalmol Vis Sci. 1999;40:S145. [Google Scholar]
- 31.Massey SC, Mills SL. A calbindin-immunoreactive cone bipolar cell type in the rabbit retina. J Comp Neurol. 1996;366:15–33. doi: 10.1002/(SICI)1096-9861(19960226)366:1<15::AID-CNE2>3.0.CO;2-N. [DOI] [PubMed] [Google Scholar]
- 32.Matsui C, Wang CK, Nelson CF, Bauer EA, Hoeffler K. The assembly of lamininin-5 subunits. J Biol Chem. 1995;270:23496–23503. doi: 10.1074/jbc.270.40.23496. [DOI] [PubMed] [Google Scholar]
- 33.Matsui C, Pereira P, Wang CK, Nelson CF, Kutzkey T, Lanigan C, Woodley D, Morohashi M, Welsh EA, Hoeffler K. Extent of laminin-5 assembly and secretion effect junctional epidermolysis bulbosa phenotype. J Exp Med. 1998;187:1273–1283. doi: 10.1084/jem.187.8.1273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.McArdle CB, Dowling JE, Masland RH. Development of outer segments and synapses in the rabbit retina. J Comp Neurol. 1977;175:253–274. doi: 10.1002/cne.901750302. [DOI] [PubMed] [Google Scholar]
- 35.Miner JH, Patton BL, Lentz SI, Gilbert DJ, Snider WD, Jenkins NA, Copeland NG, Sanes JR. The laminin alpha chains: expression, developmental transitions, and chromosomal locations of α1–5, identification of heterotrimeric laminins 8–11, and cloning of a novel α3 isoform. J Cell Biol. 1997;137:685–701. doi: 10.1083/jcb.137.3.685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Morissette N, Carbonetto S. Laminin α2 chain (M chain) is found within the pathway of avian and murine retinal projections. J Neurosci. 1995;15:8067–8082. doi: 10.1523/JNEUROSCI.15-12-08067.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Neophytou C, Vernallis AB, Smith A, Raff MC. Müller cell-derived leukaemia inhibitory factor arrests rod photoreceptor differentiation at a postmitotic pre-rod stage of development. Development. 1997;124:2345–2354. doi: 10.1242/dev.124.12.2345. [DOI] [PubMed] [Google Scholar]
- 38.Noakes PG, Gautam M, Mudd J, Sanes JR, Merlie JP. Aberrant differentiation of neuromuscular junctions in mice lacking s-laminin/laminin β2. Nature. 1995a;374:258–262. doi: 10.1038/374258a0. [DOI] [PubMed] [Google Scholar]
- 39.Noakes PG, Miner JH, Gautam M, Cunningham JM, Sanes JR, Merlie JP. The renal glomerulus of mice lacking s-laminin/laminin β2: nephrosis despite molecular compensation by laminin β1. Nat Genet. 1995b;10:400–406. doi: 10.1038/ng0895-400. [DOI] [PubMed] [Google Scholar]
- 40.Olney JW. An electron microscopic study of synapse formation, receptor outer segment development, and other aspects of developing mouse retina. Invest Ophthalmol. 1968a;7:250–268. [PubMed] [Google Scholar]
- 41.Olney JW. Centripetal sequence of appearance of receptor-bipolar synaptic structures in developing mouse retina. Nature. 1968b;218:281–282. doi: 10.1038/218281a0. [DOI] [PubMed] [Google Scholar]
- 42.Patton BL, Chiu AY, Sanes JR. Synaptic laminin prevents glial entry into the synaptic cleft. Nature. 1998;393:698–701. doi: 10.1038/31502. [DOI] [PubMed] [Google Scholar]
- 43.Patton BL, Miner JH, Chiu AY, Sanes JR. Distribution and function of laminins in the neuromuscular system of developing, adult, and mutant mice. J Cell Biol. 1997;139:1507–1521. doi: 10.1083/jcb.139.6.1507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Pearlman AL, Sheppard AM. Extracellular matrix in early cortical development. Prog Brain Res. 1996;108:117–134. [PubMed] [Google Scholar]
- 45.Pillers DM, Weleber RG, Woodward WR, Green DG, Chapman VM, Ray PN. MdxCv3 mouse is a model for electroretinography of Duchenne/Becker muscular dystrophy. Invest Ophthalmol Vis Sci. 1995;36:462–466. [PubMed] [Google Scholar]
- 46.Raabe EH, Yoshida K, Schwarting GA. Differential laminin isoform expression in the developing rat olfactory system. Dev Brain Res. 1997;101:187–196. doi: 10.1016/s0165-3806(97)00064-3. [DOI] [PubMed] [Google Scholar]
- 47.Ryan MC, Christiano AM, Engvall E, Wewer UM, Miner JH, Sanes JR, Burgeson RE. The functions of laminins: lessons from in vivo studies. Matrix Biol. 1996;15:369–381. doi: 10.1016/s0945-053x(96)90157-2. [DOI] [PubMed] [Google Scholar]
- 48.Schmitz F, Holbach M, Drenckhahn D. Colocalization of retinal dystrophin and actin in postsynaptic dendrites of rod and cone photoreceptor synapses. Histochemistry. 1993;100:473–479. doi: 10.1007/BF00267828. [DOI] [PubMed] [Google Scholar]
- 49.Sinning AR, Olson MD, Sandstead HH. The effects of zinc deficiency on developing photoreceptors in the rat retina: a scanning microscopic study. Scanning Electron Microsc 1984 (part II) 1984. pp. 867–873. [PubMed]
- 50.Sunada Y, Campbell KP. Dystroglycan: a novel laminin receptor and its involvement in the pathogenesis of muscular dystrophy. In: Ekblom P, Timpl R, editors. The laminins. Harwood; Amsterdam: 1996. pp. 291–316. [Google Scholar]
- 51.Tian M, Hagg T, Denisova N, Knusel B, Engvall E, Jucker M. Laminin-alpha2 chain-like antigens in CNS dendritic spines. Brain Res. 1997;764:28–38. doi: 10.1016/s0006-8993(97)00420-4. [DOI] [PubMed] [Google Scholar]
- 52.Timpl R. Macromolecular organization of basement membranes. Curr Opin Cell Biol. 1996;8:618–624. doi: 10.1016/s0955-0674(96)80102-5. [DOI] [PubMed] [Google Scholar]
- 53.Ueda J, Kato Y, Baba T, Terada N, Fujii Y, Tsukahara S, Ohno S. Immunocytochemical study of dystrophin localization in cone cells of mouse retinas. Invest Ophthalmol Vis Sci. 1997;38:1627–1630. [PubMed] [Google Scholar]
- 54.Vaux DL. Toward an understanding of the molecular mechanisms of physiological cell death. Proc Natl Acad Sci USA. 1993;90:786–789. doi: 10.1073/pnas.90.3.786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Young RW. Cell death during differentiation of the retina in the mouse. J Comp Neurol. 1984;229:362–373. doi: 10.1002/cne.902290307. [DOI] [PubMed] [Google Scholar]
- 56.Yurchenco PD, Quan Y, Colognato J, Mathus T, Harrison D, Yamada Y, Julian JJ. The α chain of laminin-1 is independently secreted and drives secretion of its β- and γ-chain partners. Proc Natl Acad Sci USA. 1997;94:10189–10194. doi: 10.1073/pnas.94.19.10189. [DOI] [PMC free article] [PubMed] [Google Scholar]