

Abstract
We have exploited a new monoclonal antibody against the tyrosine kinase A (TrkA) nerve growth factor (NGF) receptor to block the NGF–TrkA interaction in the rat basal forebrain. The monoclonal antibody MNAC13 is a potent antagonist that prevents the binding of NGF to TrkA in a variety of systems. This antibody was used to study the maintenance of the cholinergic phenotype in the rat basal forebrainin vivo, by the implant of antibody-secreting cells. Basal forebrain cholinergic neurons (BFCNs) are greatly affected by the antibody treatment, both in terms of cell number and of cell soma size. When antibody-secreting cells are implanted at postnatal day 2 (P2), the effects observed at P8 are as severe as those obtained with anti-NGF antibodies and, interestingly, are observed also if anti-TrkA cells are implanted at P8, when anti-NGF antibodies, delivered by the same route, are no longer effective (Molnar et al., 1998). The effects induced by anti-TrkA, as those induced by anti-NGF, are reversible, but the time required for recovery and the critical period in the sensitivity of BFCNs to the functional inactivation of TrkA is twice as long than that observed when NGF is intercepted. These results demonstrate that BFCNs are more sensitive to the block of TrkA activation than they are to the block of NGF. The cloning of MNAC13 variable regions and their assembly into a functional polypeptide of reduced size (single chain Fv fragment) will allow its use in gene transfer applications.
Keywords: TrkA, NGF receptors, antagonist monoclonal antibody, basal forebrain cholinergic neurons, recombinant antibody fragment, rat
Basal forebrain cholinergic neurons (BFCNs) represent the principal target of nerve growth factor (NGF) action in the CNS (Korsching, 1986). These actions are mediated by the p75 and tyrosine kinase A (TrkA) receptors (Radeke et al., 1987;Kaplan et al., 1991; Klein et al., 1991; Chao and Hempstead, 1995;Frade and Barde, 1998) that are expressed on BFCNs (Koh and Higgins, 1991; Holtzman et al., 1992, 1995). These neurons provide most of the cortical and hippocampal cholinergic innervation (Dutar et al., 1995) and exhibit selective uptake and retrograde transport of NGF (Sieler and Schwab, 1984; Di Stefano et al., 1992; Domenici et al., 1994a).
The administration of exogenous NGF into the brain (Hefti, 1986;Williams et al., 1986; Batchelor et al., 1989; Tuszynski et al., 1990;Kawaja et al., 1992) rescues cholinergic neurons from lesion-induced degeneration and increases the level of choline acetyltransferase (ChAT). The role of endogenous NGF in modulating the survival, differentiation, and function of BFCNs has been reassessed by implanting cells secreting a high-affinity, neurotrophin-specific, neutralizing anti-NGF antibody (Molnar et al., 1997, 1998; Avignone et al., 1998). These studies have shown that the effects of anti-NGF monoclonal antibodies on the developing basal forebrain are transient and reversible. The number and the size of ChAT-positive BFCNs is greatly reduced, if the delivery of anti-NGF antibodies is started at postnatal age 2 (P2). Antibodies delivered at P8 and onward do not affect the number and the size of BFCNs (Molnar et al., 1998). These results suggest that, as the development of the BF progresses, cholinergic neurons become less sensitive to NGF deprivation, possibly because other neurotrophic factors cooperate in the maintenance of their differentiated state. The role of TrkA receptor is still an open question, although it has been suggested that the p75 receptor mediates a developmental death in a subpopulation of BFCNs in the absence of TrkA (Van der Zee et al., 1996).
To clarify the role of the interaction between NGF and TrkA in the differentiation and maintenance of BFCNs phenotype, it is necessary to antagonize the NGF–TrkA interaction at the receptor level. Because no antagonistic anti-TrkA antibody has been isolated so far, in this study we describe the development and the characterization of a new monoclonal antibody (mAb) MNAC13, directed against the human TrkA neurotrophin receptor. This antibody inhibits the binding of NGF to cells expressing the human, or the rat, TrkA receptor. We show that this antibody is very effective in preventing the functional activation of TrkA by NGF in a variety of biological systems, both in vitro (PC12) and in vivo (rat BFCNs).
The study of the effects induced by the implant of anti-TrkA-secreting cells on rat BFCNs revealed that BFCNs are affected by the antibody treatment both at P2 and at P8, a time when anti-NGF antibodies, delivered by the same route, are no longer effective (Molnar et al., 1998). These results demonstrate that BFCNs are more sensitive to the block of TrkA activation than they are to the block of NGF.
MATERIALS AND METHODS
Immunization protocol. BALB/C 3T3-transfected cells, expressing 106 human TrkA molecules per cell, were kindly provided by Stefano Alema' [Consiglio Nazionale delle Ricerche (CNR) Institute of Cell Biology, Roma, Italy] (TrkA-BALB/C 3T3 cells). These cells were used in a congenic immunization protocol. Three groups of three female BALB/C mice were immunized with 105, 5 × 105, and 106living cells per mouse, respectively. After five injections at 2 week intervals, prefusion sera were tested for their ability to inhibit the binding of NGF to the TrkA receptor on TrkA-BALB/C 3T3 cells. The greatest inhibition of NGF-binding activity was found in the sera from the mice injected with 5 × 105 cells (binding inhibition still occurring at a 1/100 dilution).
Hybridoma production. Three days after a boost injection of TrkA-BALB/C 3T3 cells, mice were killed, the spleens were removed, and splenocytes were fused to NSO myeloma cells (10:1 ratio) with polyethylene glycol (PEG 1500), as described previously (Novak et al., 1991). Hybridoma growth and selection were performed according to standard methods (Galfre' and Milstein, 1981).
Inhibition of 125I-NGF binding to TrkA-BALB/C 3T3 cells. NGF (2.5 S) was purified from mouse submandibular glands (kind gift of Delio Mercanti at the CNR Institute of Neurobiology) and iodinated to a specific activity of 105 cpm/ng as described previously (Cattaneo et al., 1983). We plated 5 × 104 TrkA-BALB/C 3T3 cells in each well of flexible conical-bottomed 96 microwell plates in a volume of 50 ml culture medium [DMEM supplemented with 10% fetal calf serum (FCS)]. Aliquots of 50 ml of hybridoma supernatant were incubated for 1 hr with the cells, followed by the addition of the125I-NGF solution (5 × 104 cpm/well). Plates were processed as described (Cattaneo et al., 1988). Nonspecific binding was determined in parallel wells, in the presence of an excess (5 μg/ml) of unlabeled NGF. In parallel wells, binding was performed in the presence of a nonrelevant hybridoma supernatant (mAb Rab 50) or of the neutralizing anti-NGF antibody mAb αD11 (Cattaneo et al., 1988).
ELISA. Soluble TrkA and TrkB receptors were engineered as immunoadhesins (Chamow and Ashkenazi, 1996), and were produced by linking the extracellular domain of the human TrkA receptor to the Fc portion of camel IgG2, constituted of a long hinge (35 amino acid residues) followed by the CH2 and CH3 domains. The DNA sequences coding for the TrkA and TrkB immunoadhesins (TrkA-IgG and TrkB-IgG) were cloned into baculovirus [Autographa californica nuclear polyhedrosis virus (AcNPV)] transfer vectors for expression in insect cells (Baculogold transfection kit; PharMingen, San Diego, CA), and the proteins were purified by Protein A-Sepharose chromatography from serum-free culture medium of High Five insect cells. For ELISA, TrkA-IgG and TrkB-IgG were coated at 2 μg/ml, followed by incubation with 2 or 20 ng/ml of purified mAb MNAC13 and anti-mouse IgG, previously preabsorbed on camel Igs.
Fluorescence-activated cell sorter and immunofluorescence analysis. mAb MNAC13 was purified from hybridoma serum-free supernatants by Protein A Sepharose chromatography. TrkA-BALB/C 3T3 cells (5 × 104) were batch-incubated with purified mAb MNAC13 and analyzed on a Becton Dickinson (Franklin Lakes, NJ) fluorescence-activated cell sorter (FACS). For immunofluorescence, adherent cells were fixed for 10 min at room temperature with 3.7% paraformaldehyde in PBS and incubated with purified mAb MNAC13, followed by FITC-labeled anti-mouse IgG antibodies (Vector Laboratories, Burlingame, CA) and analyzed by confocal microscopy (Olympus, Hamburg, Germany).
NGF bioassay with PC12 cells. Rat PC12 pheochromocytoma cells (Greene and Tischler, 1976) were maintained in RPMI 1640 medium (Life Technologies, Milano, Italy), supplemented with 5% fetal calf serum and 10% heat-inactivated horse serum. For survival and differentiation assays, PC12 cells were washed with serum-free medium and plated in collagen-coated 35 mm Petri dishes at a density of 4 × 105 cells per dish, in 1% horse serum (1% HS). Cells were incubated with 20 ng/ml NGF for 4–6 d in the presence or absence of mAb MNAC13, mAb αD11, and mAb 9E10 (4 μg/ml; 30 min preincubation). PC12 cells were also incubated with the antibodies alone or with 20 ng/ml BDNF and 20 ng/ml NT-3 in the presence or absence of mAb MNAC13. PC12 cells treated with mAb MNAC13 and mAb αD11 were fixed, permeabilized, and stained with 4,6-diamidino-2-phenylindole (DAPI; Sigma, St. Louis, MO). Fluorescent staining was analyzed with a Zeiss Axiophot microscope (Zeiss, Oberkochen, Germany).
Alternatively, cells were primed with 50 ng/ml of NGF for 1 week and replated in the presence of 10 ng/ml NGF with or without the appropriate antibody, or recombinant single chain Fv. Neurite growth was scored 2 d later.
For immunostaining of PC12 with antibodies against activated MAP kinase, cells were fixed as above, permeabilized in ethanol absolute for 15 min at −20°C, and incubated with mouse monoclonal anti-activated MAP kinase (MAPK-YT; Sigma), followed by anti-mouse IgG alkaline phosphatase conjugate (diluted 1:100 in 10% fetal calf serum in PBST; Sigma).
Intraventricular hybridoma injections and immunohistochemistry. Intraventricular hybridoma injection and analysis of the cholinergic phenotype of basal forebrain neurons were performed essentially as described (Molnar et al., 1997, 1998). Briefly, MNAC13 hybridoma cells and control myeloma cells (P3X63Ag8) were suspended in HBBS at 2 × 105 cells/ml and injected into the right lateral ventricle of Wistar rats as described (Molnar et al., 1998). Care and handling of the animals were approved by the National Committee (Law on Animal Care number 116/1992, Italy). Injections were performed at P2, P8, or P15, and the animals were killed for analysis at P9, P16, or P22, as appropriate. After perfusion under deep anesthesia, brains were processed for ChAT immunohistochemistry as described (Molnar et al., 1997, 1998).
The level of MNAC13 antibodies in the CSF were determined by ELISA, using soluble TrkA receptors (TrkA IgG) as solid-phase antigens.
For immunochemistry with MNAC13 and anti-TrkC (Santa Cruz Biotechnology, Santa Cruz, CA) animals were anesthetized with ether and perfused with phosphate buffer (PB; 0.1 m, pH 7.4) followed by 4% paraformaldehyde–PB (pH 7.2 at 4°C). After dissection, brains were post-fixed in 4% paraformaldehyde–PB at 4°C for 2 hr and transferred to 25% sucrose–PBS. The next day, brains were frozen in isopentane at −20°C and sectioned on a cryostat. Coronal sections (14 μm) through the basal forebrain, the medial habenular, or the arcuate nuclei were collected onto gelatinized slides and stored at −20°C until processing. After blocking nonspecific binding in 10% FCS, 5% bovine serum albumin (BSA) in Tris–HCl (0.1 m, pH 7.4)–0.05% Triton X-100 (Buffer A) sections were incubated overnight at 4°C with anti-TrkA MNAC13 (6 μg/ml) or anti-TrkC (1:200) in 5% FCS–2% BSA in Buffer A. The next day, sections were incubated with a biotinylated anti-mouse or anti-rabbit IgG (Vector Laboratories) for 2 hr at room temperature and for 1 hr in ABC kit (Vector Laboratories). The reaction was developed in 3–3′ diaminobenzidine HCl (Sigma). After dehydration, sections were mounted in DPX.
Quantitative analysis. Cell counts were performed at 200× magnification using an ocular grid mounted on a Zeiss Axiophot microscope, as described (Molnar et al., 1998). ChAT-immunostained cells were counted at four representative levels indicated in the atlas by Paxinos and Watson (1986) through the rostrocaudal extention of the medial septum (MS) and diagonal band (DB) regions. MS was demarcated against DB by an horizontal line paralleling the anterior commissure. For each age, six controls, six anti-TrkA-, and three anti-NGF-treated animals were analyzed. To avoid aspecific slice shrinkage deriving from experimental variability that could affect the cell density, the analysis was performed on tissue from control and experimental animals processed in the same experiments (unless explicitly stated). To control for possible differential aspecific alterations in tissue volume, the distance between the external lateral margins of the anterior commissure was measured in control and experimental slices. The distributions obtained for the two groups were overlapping. For each group, the mean cell density was calculated (cells per square millimeter), and unpaired t test was performed to evaluate the statistical significance. Furthermore, in the same areas the cell body size was measured with the MicroComputer Imaging Device software (MCID; Imaging Research Incorporation, Ontario, Canada) and data were acquired and analyzed as described (Molnar et al., 1998).
Detection of apoptotic cells. The number of cells showing DNA fragmentation was evaluated using the TUNEL staining (ApopTagIn situ Apoptosis Detection Kit; Oncor, Gaithersburg, MD) on basal forebrain sections of P4 and P8 rats injected at P2 with MNAC13 or P3U cells. Animals were perfused as described above, and 40 μm brain sections were cut using a sliding microtome. Sections were digested with proteinase K for 5 min at room temperature. After washes, sections were processed according to manufacturer's instructions, then dehydrated in methanol and mounted as described above.
Cloning of the variable regions of mAb MNAC13. The cloning of the variable regions of the mAb MNAC13 was performed from hybridoma mRNA. The cloning was performed following closely the manual of the EMBO Course “Selecting from phage display libraries” (Bradbury et al., 1996). Variable region PCR was performed with the set of primers for mouse Igs previously described (Krebber et al., 1997). The amplified VH and VK variable regions were assembled into a ScFv format by PCR assembly and cloned into the phagemid vector pDAN (Bradbury et al., 1996). After fingerprinting analysis with BstNI restriction endonuclease, which confirmed a limited diversity of the resulting ScFvs, phage particles displaying ScFv fragments were subjected to phage ELISA, using TrkA–IgG as the solid-phase antigen. Phage ELISA was developed with secondary HRP-coupled anti-M13 antibodies. Positively identified phages were further assayed and finally used to produce soluble ScFv fragment in Escherichia coli. Bacterial supernatants were assayed by ELISA against TrkA–IgG, using a monoclonal antibody against the SV5 tag (Hanke et al., 1992) present in the ScFv fragment, followed by anti-mouse IgG conjugated with HRP.
RESULTS
Production and characterization of a monoclonal antibody that inhibits the binding of NGF to TrkA-expressing cells
To produce antibodies able to interfere with the neurotrophin-binding activity of the TrkA receptor, we exploited a congenic immunization protocol. BALB/C-3T3 cells expressing the human TrkA receptor (TrkA-BALB/C 3T3), produced by transfection of the human trk proto-oncogene, were used for immunization of BALB/C mice. The number of cells was found to be critical for the induction of serum antibodies neutralizing NGF-binding to target cells (see Materials and Methods).
Hybridoma supernatants were screened by a functional assay, namely their ability to inhibit the binding of125I-NGF to BALB/C 3T3-TrkA cells. Of 1266 wells in which hybridoma growth was occurring, only four supernatants showed NGF-neutralizing activity. The corresponding cells were subcloned, giving rise to clones MNAC13, MNAC30, MNAC191, and MNAC232. The ability of the antibodies produced by these clones to inhibit the binding of NGF to TrkA-BALB/C 3T3 cells is demonstrated in Figure1. These anti-TrkA antibodies inhibit the binding of NGF as efficiently as the neutralizing anti-NGF antibody αD11 (Fig. 1). Although the latter does so by binding to the active site of NGF, the former bind the TrkA receptor, most likely at or close to the NGF-docking site. These antibodies do not inhibit the binding of125I-NGF to C6 glioma cells, which express only the p75 receptors and not TrkA. The IgG antibody MNAC13 was selected for further studies.
Fig. 1.

Inhibition of binding of 125I-NGF to TrkA-BALB/C 3T3 cells. Hybridoma supernatants were preincubated with TrkA-BALB/C 3T3 cells before the addition of 125I-NGF. The histogram reports the inhibition of specific NGF binding to TrkA-BALB/C 3T3 cells by different antibodies. Specific binding was evaluated by subtracting from the total binding that obtained in the presence of an excess of unlabeled NGF. The values reported are the mean ± SEM of triplicates.
The inhibition of NGF binding is achieved by a direct interaction of the antibodies with the extracellular portion of the TrkA receptor, as demonstrated by a variety of binding studies. We first exploited a soluble form of the human TrkA receptor, engineered as an immunoadhesin (Chamow and Ashkenazi, 1996), in which the extracellular portion of the receptor is fused to the Fc domains of camel Igs (TrkA- and TrkB-IgG, see Materials and Methods). Figure2A shows that mAb MNAC13 binds to the TrkA immunoadhesin in an ELISA assay, although it fails to react with the related TrkB immunoadhesin. This confirms that mAb MNAC13 binds specifically to the extracellular portion of TrkA. This binding is unaffected if the solid-phase TrkA-IgG is previously saturated with NGF (Fig. 2B, left). Conversely, NGF can bind to solid-phase TrkA-IgG, regardless of previous incubation with saturating amounts of mAb MNAC13 (Fig.2B, right).
Fig. 2.
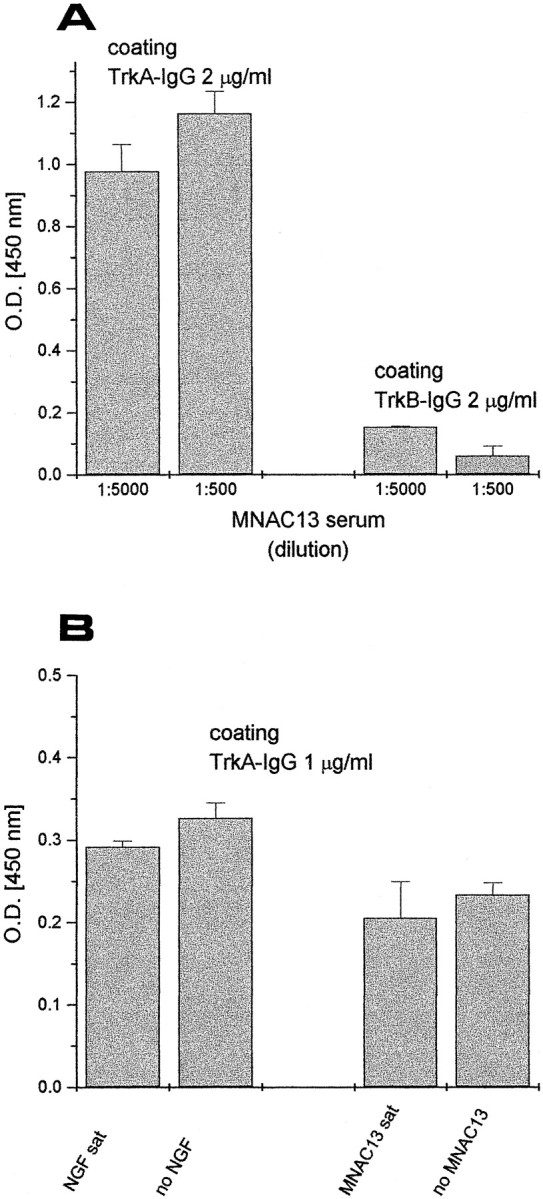
mAb MNAC13 recognizes the extracellular domain of TrkA receptor and does not inhibit NGF binding to soluble TrkA receptors. A, Soluble TrkA and TrkB receptors, engineered as immunoadhesins (TrkA-IgG and TrkB-IgG, see Materials and Methods) were used as solid-phase antigens for an ELISA assay and incubated with 2 or 20 ng/ml of purified mAb MNAC13. B,TrkA-IgG was used as solid-phase antigen for an ELISA assay, preincubated with saturating NGF (left) or saturating MNAC13 (right) and then incubated with MNAC13 (left) or NGF (right). MNC13 does not block NGF binding to TrkA-IgG and vice versa.
Figure 3 shows the result of a FACS analysis on TrkA-BALB/C 3T3 cells, demonstrating that mAb MNAC13 interacts with the human receptor expressed on the membrane of living cells and does not react to the parental 3T3 nontransfected cells. Immu- nofluorescence analysis confirms this conclusion (data not shown).
Fig. 3.

mAb MNAC13 recognizes the TrkA receptor on living cells. BALB/C 3T3 or TrkA-BALB/C 3T3 cells were incubated with purified mAb MNAC13 and subjected to FACS analysis.
The species specificity of mAb MNAC13 was tested by its ability to recognize TrkA receptors on rat neurons. Sections of rat brains were taken at the basal forebrain level (Fig.4A,B), which is rich in TrkA-positive neurons. The intense staining obtained in the basal forebrain with mAb MNAC13 (Fig. 4A) shows that this antibody, raised against the human TrkA receptor, also recognizes its rat counterpart. The antibody does not stain the medial habenular nuclei, known to be devoid of TrkA-positive neurons (Holtzman et al., 1995) (data not shown). Likewise, MNAC13 does not stain the TrkA-negative arcuate nucleus (Fig. 4C), which contains cells expressing the related receptor TrkC (Fig.4D).
Fig. 4.
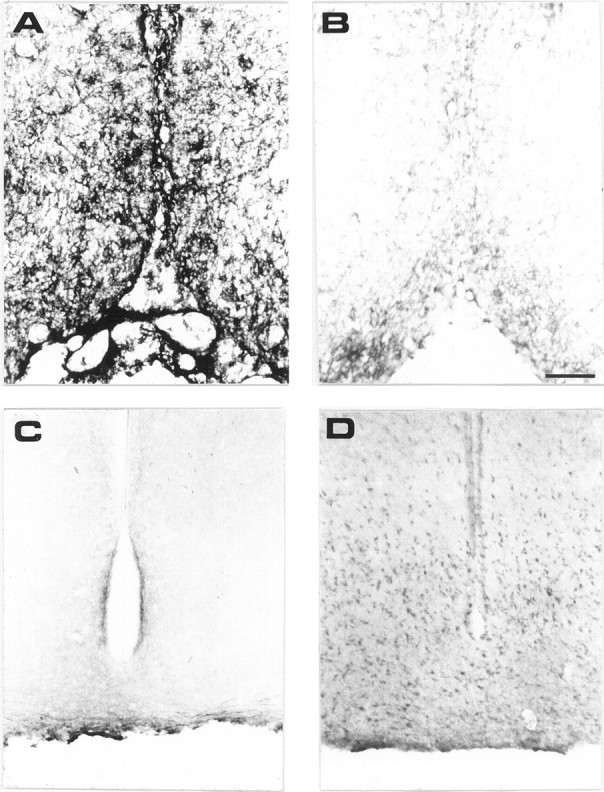
mAb MNAC13 labels the TrkA receptors on rat basal forebrain neurons and does not bind to TrkC. Coronal sections of P10 rat basal forebrain (A, B) were incubated in the presence (A) or in the absence (B) of mAb MNAC13. Coronal sections of P10 rat arcuate nucleus were incubated with mAb MNAC13 (C) or anti-TrkC (D). Scale bar, 98 μm.
The ability of mAb MNAC13 to inhibit the biological activation of the TrkA receptor by the NGF ligand was studied in rat PC12 cells (Fig.5).
Fig. 5.

mAb MNAC13 inhibits the NGF-induced differentiation of rat pheochromocytoma PC12 cells. PC12 cells were transferred to serum-free medium and incubated in the absence (A) or the presence (B–D) of 20 ng/ml NGF for 4 d. mAb MNAC13 (4 μg/ml) (C) completely inhibits NGF-induced survival and differentiation, whereas the control antibody 9E10 does not (D).
The survival and differentiation of PC12 cells by NGF (Fig.5B) was completely inhibited by incubation of the cultures with mAb MNAC13 (Fig. 5C) and with the anti-NGF neutralizing antibody αD11 (data not shown). NGF-induced differentiation occurred normally, in the presence of the nonrelevant 9E10 antibody (Fig.5D). PC12 cells treated with mAb MNAC13, in the absence of NGF or in the presence of BDNF or NT-3, did not show neurite outgrowth (data not shown).
The nuclear morphology of PC12 cells was observed after staining with DAPI under the different conditions of the assays. NGF-treated cells showed a normal nuclear morphology, whereas negative controls, BDNF-, BDNF plus mAb MNAC13-, NGF plus mAb MNAC13-, NGF plus mAbαD11-, mAb MNAC13-, and mAb αD11-treated cells showed a nuclear pyknotic morphology, a marker for the ongoing cell death. No difference was seen under the different conditions.
Activation of TrkA receptors by NGF leads to the downstream activation of MAP kinase, which can be visualized by immunostaining with antibodies against activated MAP kinase. Preincubation of PC12 cells with MNAC13 completely blocks the NGF-induced activation of MAP kinase (data not shown).
Altogether, these results show that MNAC13 neutralizes the binding of NGF to cellular TrkA.
Functional blockade of rat basal forebrain TrkA by MNAC13 hybridoma cell implant
Cholinergic neurons of the basal forebrain are a well known target for NGF action in the CNS (Korsching, 1986; Holtzman et al., 1992). Hybridoma cells secreting the MNAC13 antibody were implanted in the lateral ventricle of neonatal rats 2 d after birth (P2), and the cholinergic phenotype of the neurons was studied 1 week later (P9) by immunohistochemistry with antibodies against ChAT. This experimental approach has been used previously to study the effects of implanted cells secreting the anti-NGF monoclonal antibody αD11 (Berardi et al., 1994; Domenici et al., 1994b; Molnar et al., 1997, 1998). The levels of anti-TrkA antibodies found in the CSF were determined at different time points by ELISA (Table 1), and were found to be slightly lower than those observed when implanting the anti-NGF hybridoma αD11 (Molnar et al., 1998). One week after the implant at P2, the level of anti-TrkA antibodies was of 1.4 ng/μl (Table 1). The results show that the number of ChAT-positive cells is dramatically reduced in the brains implanted with the anti-TrkA antibody (Fig. 6B), with respect to controls (injected with a nonrelevant myeloma) (Fig.6A). The effect is as severe as that obtained in anti-NGF-treated animals (Fig. 6C). A quantitative evaluation of the number of ChAT-positive neurons (Table2) showed that this number is reduced by 73% in the medial septum and by 77% in the diagonal band of mAb MNAC13-implanted rats, with respect to controls, a greater effect than observed after anti-NGF treatment (Table 2). No sign whatsoever of DNA fragmentation, a diagnostic marker for cell death by apoptosis, was found in the basal forebrain, as assessed by the TUNEL method (data not shown). The remaining ChAT-positive neurons showed a marked shrinkage of the cell body, as shown in Figure7A. This demonstrates that mAb MNAC13 is very effective in blocking the activation of the TrkA receptor by endogenous NGF in the rat brain.
Table 1.
Levels of TrkA antibody in CSF measured by ELISA
| CSF (ng/μl) | |
|---|---|
| P9 inj. P2 | 1.40 ± 0.16 |
| P16 inj. P2 | 0.81 ± 0.01 |
| P22 inj. P2 | 0.30 ± 0.12 |
| P16 inj. P8 | 0.89 ± 0.07 |
| P22 inj. P8 | 0.70 ± 0.06 |
| P22 inj. P15 | 0.70 ± 0.15 |
Measures after 1, 2, and 3 weeks after injection (n = 6 for each age of brain analysis).
Fig. 6.
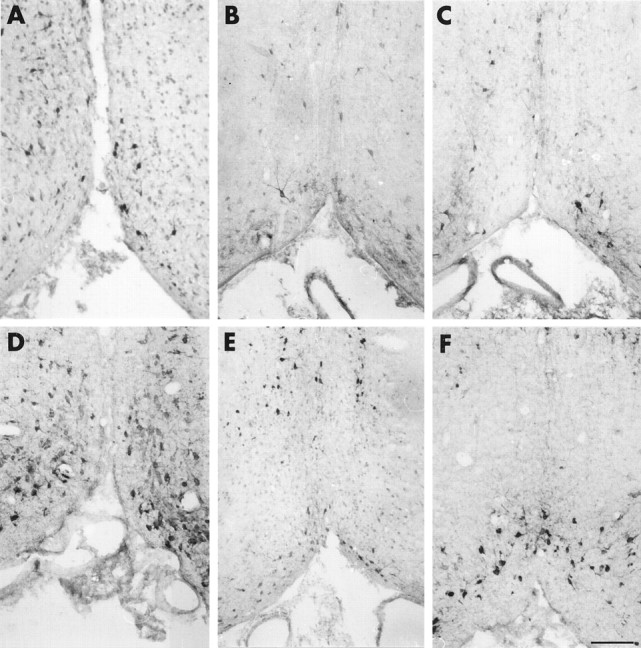
Implant of mAb MNAC13-secreting cells in the rat brain significantly reduces the number of cholinergic basal forebrain neurons. The cholinergic phenotype of P9 (A–C) and P16 (D–F) rat basal forebrain neurons was determined by immunoreactivity for ChAT after the intraventricular implant at P2 of cells secreting the anti-TrkA MNAC13 antibody (B,E), the anti-NGF αD11 antibody (C, F), or of control myeloma cells (A,D). Note the marked reduction of the number of ChAT-positive neurons in MNAC13-implanted rats (B). Scale bar, 75 μm.
Table 2.
Number of ChAT-positive neurons in the basal forebrain of rats injected with control myeloma cells (P3U) or with hybridoma cells (αTrkA or αD11)
| P3U control | αTrkA | αD11 | Variation αTrkA vs P3U | Variation αD11 vs P3U | ||
|---|---|---|---|---|---|---|
| P9 inj. P22-a | MS | 123 ± 8 | 36 ± 3 | 37 ± 2 | −73%* | −70%* |
| DB | 116 ± 11 | 27 ± 1 | 60 ± 7 | −77%* | −58%* | |
| P16 inj. P22-a | MS | 178 ± 10 | 30 ± 4 | 67 ± 5 | −62%* | −62%* |
| DB | 161 ± 11 | 70 ± 7 | 157 ± 13 | −57% | no diff. | |
| P22 inj. P22-b | MS | 171 ± 5 | 166 ± 5 | no diff. | ||
| DB | 165 ± 10 | 161 ± 6 | no diff. | |||
| MS | 195 ± 14 | 183 ± 44 | no diff. | |||
| DB | 259 ± 10 | 269 ± 16 | no diff. | |||
| P16 inj. P82-b | MS | 165 ± 4 | 37 ± 3 | −77%* | ||
| DB | 159 ± 4 | 64 ± 3 | −60%* | |||
| MS | 237 ± 30 | 196 ± 10 | no diff. | |||
| DB | 309 ± 25 | 302 ± 24 | no diff. | |||
| P22 inj. P82-a | MS | 168 ± 5 | 168 ± 5 | no diff. | ||
| DB | 165 ± 5 | 162 ± 6 | no diff. | |||
| P22 inj. P152-b | MS | 167 ± 3 | 167 ± 4 | no diff. | ||
| DB | 163 ± 4 | 166 ± 5 | no diff. | |||
| MS | 211 ± 19 | 207 ± 10 | no diff. | |||
| DB | 255 ± 40 | 265 ± 26 | no diff. |
MS, Medial septum; DB, diagonal band; no diff, not significantly different from P3U controls.
Measures taken from littermates injected with myeloma (P3U) or hybridoma cells (αTrkA or αD11) on the same day.
Measures from rats injected with αD11 hybridoma cells have been reported from Molnar et al. (1998).
Measures at P9, P15, or P22 injected at P2, P8, or P15 (n = 6; unpaired t test; *p< 0.01).
Fig. 7.
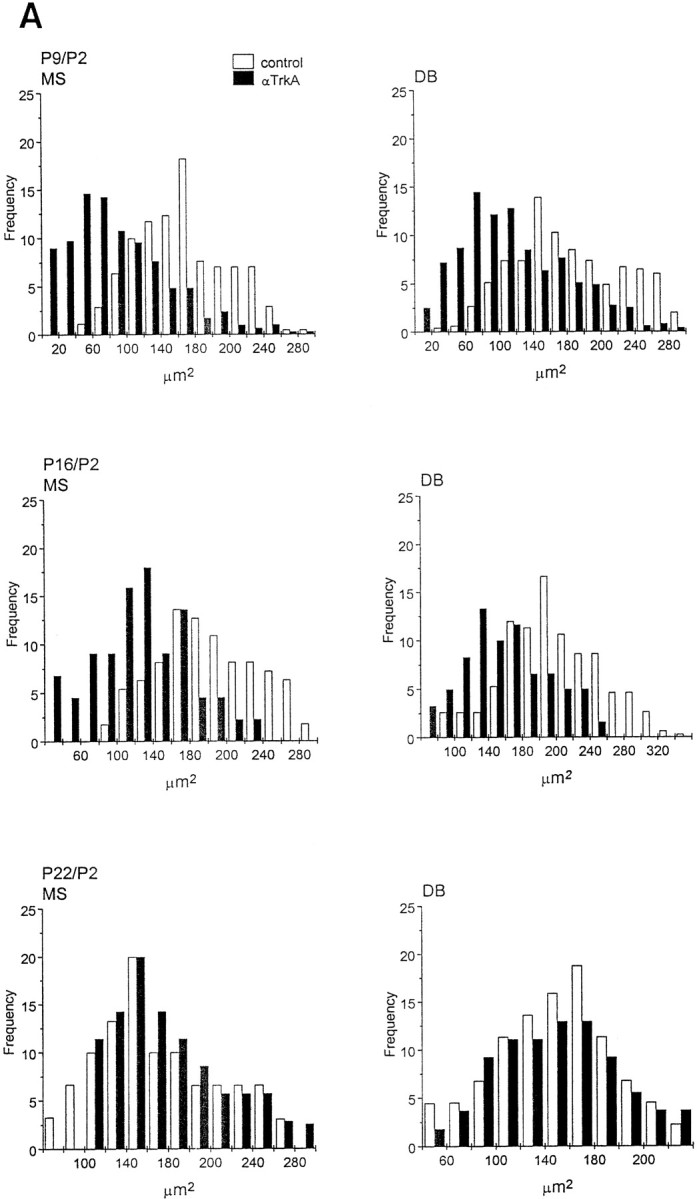
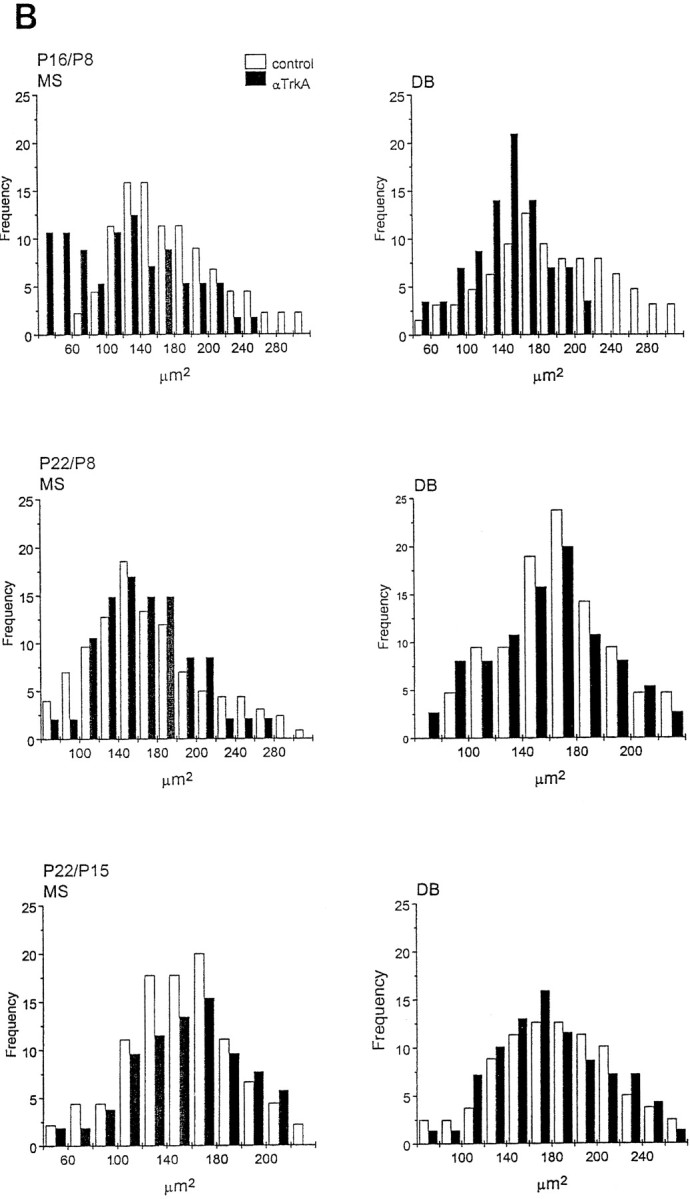
Soma size distribution of ChAT-immunopositive cells in the medial septum (MS) and diagonal band (DB) of animals implanted with mAb MNAC13-secreting cells. The soma size distribution of ChAT-immunopositive cells in the MS and DB of animals implanted with mAb MNAC13-secreting cells (black bars) or with myeloma cells (white bars). Animals were injected at P2 and killed at P9, P16, and P22 (A) or injected at P8 or P15 and killed at P16 or P22 (B) (Figure 7continues).
We have shown previously that in anti-NGF-treated rats, the effects observed on BFCNs are transient and reversible (Molnar et al., 1998). This was also observed in anti-TrkA-treated rats, but with a slower time scale of recovery. In fact, 2 weeks after the injection at P2, the number of ChAT-positive cells in the diagonal band is still greatly reduced in MNAC13-, but not in anti-NGF-treated animals (Table 2). Likewise, at this age the size of cell body is significantly reduced only after anti-TrkA treatment (Fig. 7A), but not after anti-NGF treatment (Molnar et al., 1998). Three weeks after the injection at P2, in both cases (anti-TrkA and anti-NGF treatment) the number of ChAT-positive neurons, and their sizes, are identical to those in controls.
If anti-TrkA-secreting cells are implanted at P8, a significant reduction in the number of ChAT-positive neurons (77% in the MS and 60% in the DB) (Table 2), as well as in the size of the remaining neurons (Fig. 7B), are observed. This is in marked contrast with what was observed with anti-NGF treatment in our previous study (Molnar et al., 1998), in which case BFCNs are not affected. A complete recovery is observed for this group at P22 (Table 2, Fig.7B).
Finally, also for anti-TrkA, as for anti-NGF, hybridoma implant at P15 does not affect the cholinergic phenotype (Table 2, Fig.7B).
In conclusion, the critical period in the sensitivity of BFCNs to the functional inactivation of TrkA is longer than that observed when NGF is intercepted. Thus, blocking the receptor is not the same as blocking the ligand.
Isolation of a recombinant functional form of mAb MNAC13 with a significantly reduced size
To expand the range of applications of the newly isolated anti-TrkA antibody, its variable regions were cloned and engineered into a recombinant antibody fragment of smaller size.
Cloning of the variable regions of monoclonal antibodies from the corresponding hybridoma can be complicated, leading to the cloning of artifactual unwanted variable regions (Ruberti et al., 1994). We therefore exploited phage display technology (Winter et al., 1994) to facilitate this task. The heavy (VH) and light chain (VL) variable regions of mAb MNAC13 were amplified by PCR from a cDNA derived from hybridoma mRNA, using mouse IgG-specific primers. The variable regions were assembled into the format of single-chain Fv (ScFv) polypeptides by PCR assembly and cloned into the phagemid vector pDAN, to allow the display of the cloned antibody fragments on the surface of filamentous phage. ScFv fragments represent a smaller form of the original antibody (Bird et al., 1988), consisting of the light and heavy variable regions joined by a linker peptide, linking the C terminus of the VL to the N terminus of the cognate VH. The PCR-assembled ScFv fragments were displayed on filamentous phage, as fusions to the phage protein p3. The minilibrary of phage particles was screened by phage-ELISA, using TrkA immunoadhesin as the solid-phase antigen. This led to the isolation of ELISA-positive phages, displaying the ScFv version of the parental mAb MNAC13 (ScFvMNAC13). The binding properties of this phage, as well as those of the soluble ScFv fragment derived from this phage, and expressed in E. coli, were characterized by a competition ELISA assay (Fig. 8). The TrkA immunoadhesin was coupled to solid phase and incubated with MNAC13 displaying phage particles (Fig. 8A), with soluble ScFvMNAC13 (Fig. 8B) or with parental mAb MNAC13 (Fig. 8C), in the presence of increasing amounts of competing soluble TrkA immunoadhesin. The binding reaction was revealed by secondary anti-M13 phage antibodies (Fig. 8A), by an antibody against the SV5 tag present at the C terminus of the ScFv fragment (Fig. 8B), or by anti-mouse IgG antibodies (Fig. 8C). The results confirm that the ScFv version of the parental monoclonal antibody, either displayed on phage or secreted from E. coli, binds TrkA as efficiently as the parental monoclonal antibody.
Fig. 8.

Recombinant forms of mAb MNAC13 bind TrkA. Phage-displayed ScFvMNAC13 (A), soluble ScFvMNAC13 (B), and the parental mAb MNAC13 (C) were used in a TrkA ELISA assay, using TrkA immunoadhesin as a solid-phase antigen in the presence of increasing concentrations of competing soluble TrkA immunoadhesin.Squares are 10-fold dilutions of the input antibody, with respect to triangles.
The biological activity of ScFvMNAC13 was tested on PC12 cells. Cells were primed for 1 week with 50 ng/ml of NGF, after which they were replated (to disrupt the neurite network), in the presence of NGF and ScFvMNAC13 antibody fragment. As shown in Figure9, ScFvMNAC13 totally inhibits the extension of neurites from primed PC12 cells, confirming that the recombinant single-chain Fv form of the MNAC13 antibody retains the neutralization properties of the parental antibody. The small size of this single polypeptide antibody form will allow to expand the range of applications of the anti-TrkA antibody characterized in this paper, facilitating its delivery, expression, and penetration within the nervous tissue.
Fig. 9.

Inhibition of NGF-induced neurite growth in PC12 cells by the recombinant ScFvMNAC13. Periplasmic fractions containing recombinant ScFvMNAC13 (B) or an irrelevant control ScFv fragment (anti-phox ScFv) were added, together with 10 ng/ml of NGF, to PC12 cells primed for 7 d with 50 ng/ml of mouse NGF, and replated at the beginning of the assay. The recombinant ScFvMNAC13 in B inhibits the regrowth of neurites in replated primed PC12 cells, mediated by the activation of TrkA by NGF.
DISCUSSION
The role of NGF in modulating the survival and the differentiation of BFCNs has been the object of many studies.
The rescue by NGF of BFCNs from lesion-induced cell death has led to the suggestion that NGF is a survival factor for these neurons in vivo (Hefti, 1986). Surprisingly, in NGF −/− knock-out mice (Crowley et al., 1994) ChAT-positive neurons were present and not grossly affected. On the other hand, atrophy and reduction of BFCNs were found in adult NGF +/− mice (Chen et al., 1997), which raises the intriguing possibility that exposure to suboptimal levels of NGF would induce a subsequent dependence of BFCNs from NGF, the so called “negative priming model” suggested in Molnar et al. (1998).
The role of TrkA and p75 receptors in the basal forebrain is still an open question that was not solved by the generation of TrkA knock-out mice (Smeyne et al., 1994; Fagan et al., 1997). It is possible that the gene knock-out has triggered some compensatory change, allowing BFCNs to overcome their presumed dependence on an effective interaction between NGF and TrkA.
Therefore, to clarify the mechanisms of differentiation and maintenance of the BFCNs cholinergic phenotype, the role of TrkA needs to be further investigated with new tools. Antibodies directed against the TrkA receptor, able to inhibit its activation by the natural ligand or ligands, would represent an essential reagent. One anti-TrkA antiserum with agonist properties (RTA) (Clary et al., 1994; Lucidi-Phillipi et al., 1996) and one agonist monoclonal antibody (mAb 5C3) (LeSauteur et al., 1996; Maliartchouk and Saragovi, 1997) have been described and used in vitro and in vivo. On the other hand, no antagonistic antibody against TrkA receptors of any species has been described and used for biological studies so far. The monoclonal antibody MNAC13, described in this paper, is the first anti-TrkA receptor with demonstrated antagonistic properties in biological systems. This antibody was raised against native human TrkA, but it also recognizes the rat TrkA receptor, unlike mAb 5C3 (LeSauteur et al., 1996). Mab MNAC13 binds to the extracellular domain of TrkA and does not interact with the related receptors TrkB and TrkC. Mab MNAC13 binds to TrkA receptors exposed at the cell surface and also to soluble forms of its extracellular domain. Mab MNAC13 inhibits the binding of NGF to human or rat TrkA receptor on cells and is very effective in preventing the functional activation of TrkA by NGF in a variety of systems, including NGF-induced survival and differentiation of PC12 cells. On the other hand, the binding of MNAC13 to the soluble TrkA receptor is not inhibited by NGF, and vice versa. Therefore, the mode of inhibition of the antibody requires an entirely native receptor.
We have exploited this antibody to study the maintenance of the cholinergic phenotype in the rat basal forebrain in vivo, by the method of hybridoma implant. This method has been validated by a number of previous studies (Berardi et al., 1994; Domenici et al., 1994b; Molnar et al., 1997, 1998) to achieve a time-controlled, continuous infusion of neutralizing antibodies in the CNS. A detailed study of the effects on rat basal forebrain, induced by the implant of anti-TrkA secreting cells, revealed that BFCNs are greatly affected by the antibody treatment, both in terms of cholinergic cell number and of cell soma size. When antibody-secreting cells are implanted at P2, the effects (observed at P8) are at least as severe as those obtained with anti-NGF antibodies, and, interestingly, were observed also when anti-TrkA cells are implanted at P8, when anti-NGF antibodies, delivered by the same route, are no longer effective (Molnar et al., 1998). The effects induced by anti-TrkA, as those induced by anti-NGF, are transient and reversible by P22, but the time for recovery is longer after anti-TrkA treatment than it is for the anti-NGF one.
These results demonstrate that BFCNs are more sensitive to the block of TrkA activation than they are to the block of NGF, and that the critical period in the sensitivity of BFCNs to the functional inactivation of TrkA is longer than that observed when NGF is intercepted. We consider it unlikely that a different potency between the anti-NGF and the anti-TrkA antibodies would explain the results, because in vitro the two antibodies appear to have a comparable affinity for their targets, and in vivo the levels of the anti-TrkA (Table 1) were, if anything, lower than those of the anti-NGF (Molnar et al., 1998). Several other reasons could account for the observed difference in the effects of anti-TrkA and anti-NGF treatments. In the case of anti-TrkA, NGF is still free to interact with the p75 receptors on BFCNs, with no concomitant TrkA activation on the same cells. According to recent views (Van der Zee et al., 1996; Yoon et al., 1998), this may lead to cell death, given the proposed role of TrkA activation to negate cell death induced by the activation of the p75 receptor (Yoon et al., 1998). However, we found no evidence for cell death in control or treated brains. In the case of anti-NGF treatment, the TrkA receptor would still be available to interact with other putative factors that may substitute for NGF, such as, for instance, the neurotrophin NT-3, which is known to signal also through TrkA (Davies et al., 1995; Wyatt et al., 1997) and is found at reasonably high concentrations in the septum (Katoh-Semba et al., 1996).
A comparison with TrkA−/− knock-out mice shows that the effects on BFCNs, obtained by implanting anti-TrkA antibody-secreting cells, are quantitatively much stronger than the mild cholinergic deficit observed in TrkA−/− mice (Fagan et al., 1997), with a reduction of BFCNs at P9 of 75%, against no reduction at all, at the same age, in TrkA−/− mice. With anti-TrkA antibodies, a similar reduction is also seen up to P16, with the implant made at P2 or at P8. The cholinergic deficit (both cell number and cell body size) is completely reverted by P22. Conversely, at this age the TrkA−/− show a mild reduction of cholinergic cells (30%, only in the medial septum, none in the diagonal band), but unfortunately the TrkA −/− mice cannot be followed longer because of their short life span. Thus, there must be compensatory mechanisms called into play by the knock-out of the NGF or the TrkA genes.
The limited life spans of both NGF−/− and TrkA−/− mice limit their use in understanding the NGF–TrkA interaction in the developing and adult basal forebrain. The availability of the recombinant forms of neutralizing antibodies against NGF (Ruberti et al., 1993) and against TrkA (this work) will allow their use with the neuroantibody (Piccioli et al., 1991, 1995), or with other gene transfer approaches, to disrupt the NGF–TrkA interaction under time-controlled conditions.
In conclusion, the derivation of a new antagonistic antibody against TrkA has allowed to further define the complexity of the NGF–TrkA interactions in the basal forebrain and in other brain regions such as the visual cortex (Pesavento et al., 1999). More generally, the properties of mAb MNAC13 make this antibody a valuable tool for future studies in a broad range of different applications.
Footnotes
This work was supported by contributions from the Societa' Italiana per la Ricerca Scientifica srl (SIRS srl) and Howard Hughes Medical Institute (HHMI 75195–547401). P.P. was supported by SIRS srl, Roma, Italy. We are very grateful to Gabriella Rossi and Daniele Sblattero for their help in various parts of the work and to Sonia Covaceuszach for purifying the TrkA immunoadhesin.
Correspondence should be addressed to Antonino Cattaneo, Neuroscience Programme, International School for Advanced Studies, Via Beirut 2/4, 34014 Trieste, Italy. E-mail: cattaneo@sissa.it.
REFERENCES
- 1.Avignone E, Molnar M, Berretta N, Casamenti F, Prosperi C, Ruberti F, Cattaneo A, Cherubini E. Cholinergic function in the hippocampus of juvenile rats chronically deprived of NGF. Brain Res Dev Brain Res. 1998;109:137–147. doi: 10.1016/s0165-3806(98)00072-8. [DOI] [PubMed] [Google Scholar]
- 2.Batchelor PE, Armstrong DM, Blaker SN, Gage FH. Nerve growth factor receptor and choline acetyltransferase colocalization in neurons within the rat forebrain: response to fimbria-fornix transection. J Comp Neurol. 1989;284:187–204. doi: 10.1002/cne.902840204. [DOI] [PubMed] [Google Scholar]
- 3.Berardi N, Cellerino A, Domenici L, Fagiolini M, Pizzorusso T, Cattaneo A, Maffei L. Monoclonal antibodies to nerve growth factor affect the postnatal development of the visual system. Proc Natl Acad Sci USA. 1994;91:684–688. doi: 10.1073/pnas.91.2.684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bird RE, Hardman KD, Jacobson JW, Johnson S, Kaufman BM, Lee SM, Lee T, Pope SH, Riordan GS, Whitlow M. Single-chain antigen-binding proteins. Science. 1988;242:423–425. doi: 10.1126/science.3140379. [DOI] [PubMed] [Google Scholar]
- 5.Bradbury A, Cattaneo A, Hoogenboom H. Manual of the EMBO theoretical and practical course. Trieste, Italy, November; 1996. Selecting from phage display libraries. pp. 1–122. [Google Scholar]
- 6.Cattaneo A, Biocca S, Nasi S, Calissano P. Hidden receptors for nerve growth factors in PC12 cells. Eur J Biochem. 1983;135:285–290. doi: 10.1111/j.1432-1033.1983.tb07650.x. [DOI] [PubMed] [Google Scholar]
- 7.Cattaneo A, Rapposelli B, Calissano P. Three distinct types of monoclonal antibodies after long term immunization of rats with mouse nerve growth factor. J Neurochem. 1988;50:1003–1010. doi: 10.1111/j.1471-4159.1988.tb10565.x. [DOI] [PubMed] [Google Scholar]
- 8.Chamow SM, Ashkenazi A. Immunoadhesins: principles and applications. Trends Biotechnol. 1996;14:52–60. doi: 10.1016/0167-7799(96)80921-8. [DOI] [PubMed] [Google Scholar]
- 9.Chao MV, Hempstead BL. p75 and Trk: a two-receptor system. Trends Neurosci. 1995;18:321–326. [PubMed] [Google Scholar]
- 10.Chen KS, Nishimura MC, Armanini MP, Crowley C, Spencer SD, Phillips HS. Disruption of a single allele of the nerve growth factor gene results in atrophy of basal forebrain cholinergic neurons and memory deficits. J Neurosci. 1997;17:7288–7296. doi: 10.1523/JNEUROSCI.17-19-07288.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Clary DO, Weskamp G, Austin LR, Reichardt LF. TrkA cross-linking mimics neuronal responses to nerve growth factor. Mol Biol Cell. 1994;5:549–563. doi: 10.1091/mbc.5.5.549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Crowley C, Spencer SD, Nishimura MC, Chen KS, Pitts-Meek S, Armanini MP, Ling LH, MacMahon SB, Shelton DL, Levinson AD, Phillips HS. Mice lacking nerve growth factor display perinatal loss of sensory and sympathetic neurons yet develop basal forebrain cholinergic neurons. Cell. 1994;76:1001–1011. doi: 10.1016/0092-8674(94)90378-6. [DOI] [PubMed] [Google Scholar]
- 13.Davies AM, Minichiello L, Klein R. Developmental changes in NT3 signalling via TrkA and TrkB in embryonic neurons. EMBO J. 1995;14:4482–4489. doi: 10.1002/j.1460-2075.1995.tb00127.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Di Stefano PS, Friedman B, Radieijewski C, Alexander C, Boland P, Schik CM, Lindsay RM, Wiegand SJ. The neurotrophins BDNF, NT3, and NGF display distinct patterns of retrograde transport in peripheral and central neurons. Neuron. 1992;8:983–993. doi: 10.1016/0896-6273(92)90213-w. [DOI] [PubMed] [Google Scholar]
- 15.Domenici L, Fontanesi G, Cattaneo A, Bagnoli P, Maffei L. Nerve growth factor (NGF) uptake and transport following injection in the developing rat visual cortex. Vis Neurosci. 1994a;11:1093–1102. doi: 10.1017/s095252380000691x. [DOI] [PubMed] [Google Scholar]
- 16.Domenici L, Cellerino A, Berardi N, Cattaneo A, Maffei L. Antibodies to nerve growth factor (NGF) prolong the sensitive period for monocular deprivation in the rat. NeuroReport. 1994b;5:2041–2044. doi: 10.1097/00001756-199410270-00013. [DOI] [PubMed] [Google Scholar]
- 17.Dutar P, Bassant ME, Sen M, Lamour Y. The septohippocampal pathway: structure and function of a central cholinergic system. Physiol Rev. 1995;75:393–427. doi: 10.1152/physrev.1995.75.2.393. [DOI] [PubMed] [Google Scholar]
- 18.Fagan AM, Garber M, Barbacid M, Silos-Santiago I, Holtzman DM. A role for TrkA during maturation of striatal and basal forebrain cholinergic neurons in vivo. J Neurosci. 1997;17:7644–7654. doi: 10.1523/JNEUROSCI.17-20-07644.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Frade JM, Barde YA. Nerve growth factor: two receptors, multiple functions. BioEssays. 1998;20:137–145. doi: 10.1002/(SICI)1521-1878(199802)20:2<137::AID-BIES6>3.0.CO;2-Q. [DOI] [PubMed] [Google Scholar]
- 20.Galfre' G, Milstein C. Preparation of monoclonal antibodies: strategies and procedures. Methods Enzymol. 1981;73:3–45. doi: 10.1016/0076-6879(81)73054-4. [DOI] [PubMed] [Google Scholar]
- 21.Greene LA, Tischler AS. Establishment of a noradrenergic clonal line of rat adrenal pheochromocytoma cells which respond to nerve growth factor. Proc Natl Acad Sci USA. 1976;73:2424–2428. doi: 10.1073/pnas.73.7.2424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hanke T, Szawlowski P, Randall RE. Construction of solid matrix-antibody-antigen complexes containing simian immunodeficiency virus p27 using tag-specific monoclonal antibody and tag-linked antigen. J Gen Virol. 1992;73:653–660. doi: 10.1099/0022-1317-73-3-653. [DOI] [PubMed] [Google Scholar]
- 23.Hefti F. Nerve growth factor promotes survival of septal cholinergic neurons after fimbrial transections. J Neurosci. 1986;6:2155–2162. doi: 10.1523/JNEUROSCI.06-08-02155.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Holtzman DM, Li Y, Parada LF, Kinsman S, Chen C-K, Valletta JS, Zhou J, Long JB, Mobley WC. p140 trk RNA marks NGF-responsive forebrain neurons: evidence that Trk gene expression is induced by NGF. Neuron. 1992;9:465–478. doi: 10.1016/0896-6273(92)90184-f. [DOI] [PubMed] [Google Scholar]
- 25.Holtzman DM, Kilbridge J, Li Y, Cunningham E, Jr, Lenn NJ, Clary D, Reichardt LF, Mobley WC. TrkA expression in the CNS: evidence for the existence of several novel NGF-responsive CNS neurons. J Neurosci. 1995;15:1567–1576. doi: 10.1523/JNEUROSCI.15-02-01567.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kaplan DR, Hempstead BL, Martin-Zanca D, Chao MV, Parada LF. The trk proto-oncogene product: a signal transducing receptor for nerve growth factor. Science. 1991;252:554–558. doi: 10.1126/science.1850549. [DOI] [PubMed] [Google Scholar]
- 27.Katoh-Semba R, Kaisho Y, Shintani A, Nagahama M, Kato K. Tissue distribution and immunocytochemical localization of neurotrophin-3 in the brain and peripheral tissues of rats. J Neurochem. 1996;66:330–337. doi: 10.1046/j.1471-4159.1996.66010330.x. [DOI] [PubMed] [Google Scholar]
- 28.Kawaja MD, Rosenberg MB, Yoshida K, Gage FH. Somatic gene transfer of nerve growth factor promotes the survival of axotomized septal neurons and the regeneration of their axons in adult rats. J Neurosci. 1992;12:2849–2864. doi: 10.1523/JNEUROSCI.12-07-02849.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Klein R, Jing S, Nanduri V, O'Rourke E, Barbacid M. The trk proto-oncogene encodes a receptor for nerve growth factor. Cell. 1991;65:189–197. doi: 10.1016/0092-8674(91)90419-y. [DOI] [PubMed] [Google Scholar]
- 30.Koh S, Higgins GA. Differential regulation of the low affinity nerve growth factor receptor during postnatal development in the rat brain. J Comp Neurol. 1991;313:494–508. doi: 10.1002/cne.903130310. [DOI] [PubMed] [Google Scholar]
- 31.Korsching S. The role of nerve growth factor in the CNS. Trends Neurosci. 1986;9:570–573. [Google Scholar]
- 32.Krebber A, Bornhauser S, Burmester J, Honegger A, Willuda J, Bosshard HR, Pluckthun A. Reliable cloning of functional antibody variable domains from hybridomas and spleen cell repertoires employing a reengineered phage display system. J Immunol Methods. 1997;201:35–55. doi: 10.1016/s0022-1759(96)00208-6. [DOI] [PubMed] [Google Scholar]
- 33.LeSauteur L, Maliartchouk S, Le Jeune H, Quirion R, Saragovi HU. Potent human p140-TrkA agonists derived from an anti-receptor monoclonal antibody. J Neurosci. 1996;16:1308–1316. doi: 10.1523/JNEUROSCI.16-04-01308.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lucidi-Phillipi CA, Clary DO, Reichardt LF, Gage FH. TrkA activation is sufficient to rescue axotomized cholinergic neurons. Neuron. 1996;16:653–663. doi: 10.1016/s0896-6273(00)80084-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Maliartchouk S, Saragovi HU. Optimal nerve growth factor trophic signals mediated by synergy of TrkA and p75 receptor-specific ligands. J Neurosci. 1997;17:6031–6037. doi: 10.1523/JNEUROSCI.17-16-06031.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Molnar M, Ruberti F, Cozzari C, Domenici L, Cattaneo A. A critical period in the sensitivity of BF cholinergic neurons to NGF deprivation. NeuroReport. 1997;8:575–579. doi: 10.1097/00001756-199701200-00039. [DOI] [PubMed] [Google Scholar]
- 37.Molnar M, Tongiorgi E, Avignone E, Gonfloni S, Ruberti F, Domenici L, Cattaneo A. The effects of anti-nerve growth factor monoclonal antibodies on developing basal forebrain neurons are transient and reversible. Eur J Neurosci. 1998;10:3127–3140. doi: 10.1046/j.1460-9568.1998.00314.x. [DOI] [PubMed] [Google Scholar]
- 38.Novak M, Jakes R, Edwards PC, Milstein C, Wishik CM. Difference between the tau protein of Alzheimer's paired helical filament core and normal tau revealed by epitope analysis of monoclonal antibodies 423 and 7.51. Proc Natl Acad Sci USA. 1991;88:5837–5841. doi: 10.1073/pnas.88.13.5837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Paxinos G, Watson C. The rat brain in stereotaxic coordinates. Academic; London: 1986. [DOI] [PubMed] [Google Scholar]
- 40.Pesavento E, Margotti E, Righi M, Cattaneo A, Domenici L (1999) Blocking the NGF-TrkA interaction rescues the developmental loss of LTP in the rat visual cortex. Role of cholinergic system. Neuron, in press. [DOI] [PubMed]
- 41.Piccioli P, Ruberti F, Biocca S, Di Luzio A, Bradbury A, Cattaneo A. Neuroantibodies: molecular cloning of a monoclonal antibody against substance P for expression in the central nervous system. Proc Natl Acad Sci USA. 1991;88:5611–5615. doi: 10.1073/pnas.88.13.5611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Piccioli P, Di Luzio A, Amann R, Schuligoi R, Surani MA, Donnerer J, Cattaneo A. neuroantibodies: ectopic expression of a recombinant anti-substance P antibody in the central nervous system of transgenic mice. Neuron. 1995;15:373–384. doi: 10.1016/0896-6273(95)90041-1. [DOI] [PubMed] [Google Scholar]
- 43.Radeke MJ, Misko TP, Hsu C, Herzenberg LA, Shooter EM. Gene transfer and molecular cloning of the rat nerve growth factor receptor. Nature. 1987;325:593–597. doi: 10.1038/325593a0. [DOI] [PubMed] [Google Scholar]
- 44.Ruberti F, Bradbury A, Cattaneo A. Cloning and expression of an anti-nerve growth factor (NGF) antibody for studies using the neuroantibody approach. Cell Mol Neurobiol. 1993;13:559–568. doi: 10.1007/BF00711464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ruberti F, Cattaneo A, Bradbury A. The use of the RACE method to clone hybridoma cDNA when V region primers fail. J Immunol Methods. 1994;173:33–39. doi: 10.1016/0022-1759(94)90280-1. [DOI] [PubMed] [Google Scholar]
- 46.Sieler M, Schwab ME. Specific retrograde transport of nerve growth factor (NGF) from neocortex to nucleus basalis in the rat. Brain Res. 1984;300:33–39. doi: 10.1016/0006-8993(84)91338-6. [DOI] [PubMed] [Google Scholar]
- 47.Smeyne RJ, Klein R, Schnapp A, Long L K, Bryant S, Lewin A, Lira S A, Barbacid M. Severe sensory and sympathetic neuropathies in mice carrying a disrupted Trk/NGF receptor gene. Nature. 1994;368:246–249. doi: 10.1038/368246a0. [DOI] [PubMed] [Google Scholar]
- 48.Tuszynski MH, Armstrong DM, Gage FH. Basal forebrain cell loss following fimbria/fornix transection. Brain Res. 1990;508:241–248. doi: 10.1016/0006-8993(90)90402-w. [DOI] [PubMed] [Google Scholar]
- 49.Van der Zee CEEM, Ross GM, Riopelle RJ, Hagg T. Survival of cholinergic forebrain neurons in developing p75 NGFR-deficient mice. Science. 1996;274:1729–1732. doi: 10.1126/science.274.5293.1729. [DOI] [PubMed] [Google Scholar]
- 50.Williams LR, Varon S, Peterson GM, Wictorin K, Fisccher W, Bjorklund A, Gage FH. Continuous infusion of nerve growth factor prevents basal forebrain neuronal death after fimbria-fornix transection. Proc Natl Acad Sci USA. 1986;83:9231–9235. doi: 10.1073/pnas.83.23.9231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Winter G, Griffiths AD, Hawkins RE, Hoogenboom HR. Making antibodies by phage display technology. Annu Rev Immunol. 1994;12:433–455. doi: 10.1146/annurev.iy.12.040194.002245. [DOI] [PubMed] [Google Scholar]
- 52.Wyatt S, Pinon LG, Ernfors P, Davies AM. Sympathetic neuron survival and TrkA expression in NT3-deficient mouse embryos. EMBO J. 1997;16:3115–3123. doi: 10.1093/emboj/16.11.3115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Yoon SO, Casaccia-Bonnefil P, Carter B, Chao MV. Competitive signaling between TrkA and p75 nerve growth factor receptors determines cell survival. J Neurosci. 1998;18:3273–3281. doi: 10.1523/JNEUROSCI.18-09-03273.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]