

Abstract
In this report, we examine how the Ras protein regulates neuronal survival, focusing on sympathetic neurons. Adenovirus-expressed constitutively activated Ras (RasV12) enhanced survival and the phosphorylation of Akt (protein kinase B) and MAP kinase (MAPK), two targets of Ras activity. Functional inhibition of endogenous Ras by adenovirus-expressed dominant-inhibitory Ras (N17Ras) decreased nerve growth factor (NGF)-dependent survival and both Akt and MAPK phosphorylation as well. To determine the signaling pathways through which Ras mediates survival, we used Ras effector mutants and pharmacological inhibitors that selectively suppress phosphatidylinositol 3-kinase (PI3-K)/Akt or MAP kinase kinase (MEK)/MAPK pathways. The Ras effector mutant RasV12Y40C, which selectively stimulates PI3-K and Akt, rescued survival in the absence of NGF, and the PI3-K inhibitor LY 294002 inhibited both Ras- and NGF-dependent survival. RasV12T35S, which activates MEK/MAPK but not PI3-K/Akt, was less effective at rescuing survival, whereas the MEK inhibitor PD 098059 also partially suppressed Ras-dependent survival. To investigate the mechanisms by which Ras suppresses neuronal death, we examined whether Ras functions by inhibiting the proapoptotic p53 pathway (Jun-N-terminal kinase/p53/BAX) that is necessary for neuronal death after NGF withdrawal and p75NTR activation. We found that RasV12 suppressed c-jun, BAX, and p53 levels, whereas inhibition of NGF-induced Ras-survival activity via N17Ras increased the levels of these proteins. Furthermore, the E1B55K protein, which suppresses p53 activity, blocked N17Ras-induced neuronal death. Together, these results indicate that Ras is, in part, both necessary and sufficient for survival of sympathetic neurons and that this effect is mediated by activation of both the PI3-K– and MEK-signaling cascades, which in turn suppress a proapoptotic p53 pathway.
Keywords: Ras, Ras effectors, sympathetic neurons, nerve growth factor, Trk, survival, apoptosis, adenovirus, Raf, PI3-kinase, p53, Bax
Nerve growth factor (NGF) is essential for maintaining the survival of different subsets of neurons, including sympathetic neurons from the rat superior cervical ganglia (Levi-Montalcini, 1987). NGF mediates survival by binding to TrkA (Kaplan et al., 1991a,b; Klein et al., 1991), an event that stimulates the activity of multiple signaling proteins, including the small GTP-binding protein p21Ras (Segal and Greenberg, 1996; Kaplan and Miller, 1997). Ras activates several downstream effectors, including Raf and phosphatidylinositol 3-kinase (PI3-K) (Rodriguez-Viciana et al., 1994; Vojtek and Der, 1998). Raf binds to and activates MAP kinase kinase 1 (MEK1) and MEK2, activators of the MAP kinases [MAPK or extracellular signal-regulated kinase (ERK)], which play important roles in the regulation of neuritogenesis in pheochromocytoma 12 (PC12) cells (Cowley et al., 1994; Fukuda et al., 1995; Pang et al., 1995). PI3-K is a key regulator of cell survival, morphology, and growth cone extension in neural cells (Yao and Cooper, 1995; Dudek et al., 1997; Crowder and Freeman, 1998; Klesse and Parada, 1998). A target of PI3-K activity, the serine/threonine kinase Akt (or protein kinase B), has been shown to be necessary and sufficient for stimulating the survival of sympathetic, cerebellar, and immortalized hippocampal neurons (Dudek et al., 1997, Philpott et al., 1997; Crowder and Freeman, 1998; Eves et al., 1998). The role of the Ras effectors has been best defined in non-neuronal cells via the use of Ras effector mutants. Distinct mutations in the protein recognition, or effector, domain result in the loss of the ability of constitutively activated Ras (RasV12) to stimulate Raf-, PI3-K–, or Ral-GDP dissociation stimulator (GDS)-signaling pathways and of selective biological responses (Rodriguez-Viciana et al., 1997). For example, the Ras effector mutant RasV12Y40C induces membrane ruffling in fibroblasts via its ability to activate selectively PI3-K and not Raf or RalGDS (Joneson et al., 1996;Rodriguez-Viciana et al., 1997). In several cases, such as the induction of DNA synthesis (Joneson et al., 1996), the concerted action of multiple Ras effectors is needed to mimic the biological actions of wild-type RasV12.
In primary neurons, the role of Ras in neurotrophin-regulated cell survival and differentiation varies depending on the cell type assayed. Ras activity is necessary and sufficient for survival and neuritogenesis of rat and chick sensory but not chick sympathetic neurons (Borasio et al., 1993; Vogel et al., 1995; Klesse and Parada, 1998). Paradoxically, Ras activity is required for the survival, although not neuritogenesis, of rat sympathetic neurons (Nobes and Tolkovsky, 1995; Nobes et al., 1996; Markus et al., 1997). In these cases, the mechanisms by which Ras stimulates survival and suppresses apoptosis remain to be determined.
In this report, we examine the signaling pathways used by Ras to stimulate sympathetic neuron survival and to suppress apoptosis. We show that Ras activity is required for a portion of neuronal survival and that Ras mediates its actions via both PI3-K and MEK. Ras exerts its effects by suppressing the levels or activities of c-jun, the p53 tumor suppressor, and Bax, which are key elements in the major apoptotic pathway in sympathetic neurons, and apoptosis resulting from suppression of NGF-induced Ras activity requires p53 activity.
MATERIALS AND METHODS
Preparation of recombinant adenoviruses. C-myc-tagged RasV12 and dominant-inhibitory Ras (N17Ras) constructs (kind gift of Dr. C. Bazenet) as well as the double mutants RasV12T35S, RasV12D38E, and RasV12E37G (kind gift of Dr. P. Warne) were cloned into the pAd-CMV-F1-IRES-EGFP bicistronic expression vector (Exogen Neurosciences, Montreal, Quebec, Canada). RasV12Y40C was cloned into the expression vector pAd-CMV5-F1 (Exogen Neurosciences). Expression of Ras and, for the bicistronic vectors, green fluorescent proteins (GFPs) was confirmed in transiently transfected 293 cells by Western blot analysis using anti-Ras [Pan Ras (Ab-4); 1:1000; Oncogene Sciences, Uniondale, NY] and anti-GFP (1:2000; Clontech, Palo Alto, CA) antibodies, respectively. Replication-defective recombinant adenoviruses were prepared and purified as described previously (Slack et al., 1996). Briefly, expression vectors containing the Ras mutants were cotransfected with replication-defective adenoviral DNA (Quantum Biotechnologies, Laval, Quebec, Canada) in 293 cells. Plaques were selected and tested for Ras expression by Western blot analysis. Positive plaques were repurified twice by limiting dilution. Finally, recombinant adenoviruses were amplified, purified on CsCl gradients, and titered by plaque assay in 293 cells. As controls, GFP- orEscherichia coli β-galactosidase-expressing recombinant adenoviruses (Exogen Neurosciences and Dr. F. Graham, McMaster University, Hamilton, Ontario, Canada, respectively) generated using the same viral backbone as above were used. The recombinant adenovirus expressing E1B55K was a generous gift of Phil Branton (McGill University, Montreal, Quebec, Canada).
Primary neuronal cultures. Mass cultures of pure sympathetic neurons derived from the superior cervical ganglia (SCG) were prepared from postnatal day 1 rats essentially as described previously (Ma et al., 1992). Cells were plated in rat tail collagen-coated 96 (for survival assays)- or 6 (for Western blot analysis)-well plates (Falcon Labware; Becton Dickinson, Lincoln Park, NJ) at densities of 3000–5000 or 0.5–1 × 105 cells, respectively. For nuclear staining, 5000–10,000 neurons were plated on poly-d-lysine (Sigma, St. Louis, MO) and laminin (Becton Dickinson)–treated glass coverslips. Neurons were maintained in Ultraculture medium containing 2 mm glutamine, 100 U/ml penicillin, 100 μg/ml streptomycin (all from BioWhittaker, Walkersville, MD), and 3% rat serum (Harlan Bioproducts, Madison, WI) and initially cultured for 5 d in the presence of 50 ng/ml NGF (Cedarlane, Hornby, Ontario, Canada) after which they were maintained in the same medium without serum. This concentration of 50 ng/ml NGF was used to maximally induce p75NTR, tyrosine hydroxylase, Talpha1 α-tubulin, and other differentiation-related genes (Ma et al., 1992). Thereafter, the NGF concentration was reduced to 10 ng/ml. We have shown previously that 10 ng/ml is the amount of NGF required for 100% survival of our neuronal cultures (Belliveau et al., 1997).
Viral infections. After 5 d in culture, a sample well was trypsinized, and cells were counted. Subsequently, medium was removed, and recombinant adenovirus diluted in Ultraculture and DMEM (50:50; DMEM from Life Technologies, Gaithersburg, MD) containing 2 mm glutamine, 100 U/ml penicillin, 100 μg/ml streptomycin, 10% fetal bovine serum, and 30 ng/ml NGF was added to the sympathetic neurons. The multiplicity of infection (MOI) indicates the number of plaque-forming units added per cell. After 20–24 hr, the virus-containing medium was removed and replaced with Ultraculture medium containing 2 mm glutamine, 100 U/ml penicillin, 100 μg/ml streptomycin, and 20 ng/ml NGF (serum-free). After an additional 24 hr, the medium was removed, and the cells were washed 4 times with NGF-free medium and then incubated in serum-free medium with or without the addition of 10 ng/ml NGF.
Survival assays and pharmacological treatments. Survival assays were performed 72–96 hr after viral infection (2–5 d after NGF withdrawal) as described previously (Slack et al., 1996), using the nonradioactive cell proliferation assay (MTT; Sigma). Briefly, MTT reagent (final concentration, 0.5 mg/ml) was added to each well for 2.5 hr. After removing the media and lysing the cells with 100 μl of solubilization solution [HCl acid and isopropanol (0.002:1)], the colorimetric reaction was measured in a spectrophotometer.
For survival assays after pharmacological treatments, neurons were infected where appropriate, and after 2 d, cultures were exposed to different concentrations of PD 98059 and LY 294002 in serum-free media (Calbiochem, San Diego, CA). We established that maximal effects on survival were attained at concentrations of 75 μm PD 98059 and 100 μm LY 294002 in NGF-treated cultures. Cells treated with the appropriate volume of diluted DMSO, used to solubilize the drug stocks, served as controls. The volume of DMSO never surpassed 2 μl/ml of media.
Immunocytochemistry, nuclear staining, and terminal deoxynucleotidyl transferase-mediated biotin-dUTP nick end labeling. For Ras and c-myc immunocytochemistry, cells maintained in the presence of 10 ng/ml NGF were fixed 48 hr after infection with 2% formalin in PBS for 10 min. Cells were then washed and blocked for 20 min with 5% goat serum in PBS and 0.02% Triton X-100, followed by a 90 min incubation with either rat anti-Ras (20 μl/ml; Ab-2; Oncogene Sciences) or mouse anti-c-myc (1:150; PharMingen, Mississauga, Ontario, Canada) antibodies. After washing, cells were incubated, where appropriate, with a cy3-conjugated anti-rat or anti-mouse secondary antibody (1:1000; Jackson ImmunoResearch, West Grove, PA) for 45 min, washed, mounted in mounting media (Sigma), and examined by fluorescence microscopy.
For terminal deoxynucleotidyl transferase-mediated dUTP nick-end labeling (TUNEL), cells fixed in ice-cold methanol and acetone (1:1) for 10 min were incubated with 1.5% terminal deoxynucleotidyl transferase (TdT; Promega, Madison, WI) and 1% biotin-16-dUTP (Boehringer Mannheim, Montreal, Quebec, Canada) in TdT buffer for 1 hr at 37°C. After washing, cells were incubated with cy3-streptavidin (1:3000; Jackson ImmunoResearch) for 45 min at room temperature. Cultures were counterstained with Hoechst 33258 stain (2 μg/ml; ICN Biomedicals, Costa Mesa, CA) for 1 min. Quantitation was done on randomly selected fields by counting the total number of Hoechst- and TUNEL-positive neurons.
Western blot analysis. Seventy-two to ninety-six hours after viral infections (2–3 d after NGF withdrawal, where appropriate), cells were washed twice in PBS and lysed in Tris-buffered saline (TBS) lysis buffer (Knusel et al., 1994) (137 mm NaCl and 20 mm Tris, pH 8.0) containing 1% NP-40, 10% (v/v) glycerol, 1 mm phenylmethylsulfonyl fluoride, 10 μg/ml aprotinin, 0.2 μg/ml leupeptin, 1.5 mm sodium vanadate, and 0.1% SDS for 10 min at 4°C. After a 5 min centrifugation, the protein concentration in the supernatant was determined using the BCA Protein Assay Reagent kit (Pierce, Rockford, IL). Equal amounts of proteins (50–100 μg) were separated on 7.5% (for MAPK, Akt, and p53 proteins) or 15% (for Ras proteins) polyacrylamide gels under denaturing conditions. After gel electrophoresis, proteins were transferred to nitrocellulose membranes. Membranes were then blocked in 5% nonfat milk in TBS containing 0.08% Tween 20 (TBST) for 90 min followed by an overnight incubation with the primary antibodies diluted in 5% nonfat milk in TBST as follows: rabbit anti-phosphorylated-MAPK (1:10,000; Promega), rabbit anti-phosphorylated-Akt (1:1000; New England Biolabs, Beverly, MA), rabbit anti-MAPK1 (which recognizes denatured MAPK2 as well; 1:10,000; Santa Cruz Biotechnologies, Santa Cruz, CA), mouse anti-Ras [Pan Ras (Ab-4); 1:1000; Oncogene Sciences], mouse monoclonal anti-c-jun (1:1000; Santa Cruz Biotechnologies), rabbit anti-Bax (1:500; Santa Cruz Biotechnologies), and rabbit anti-p53 (1:1000; Novocastra, Newcastle upon Tyne, United Kingdom). Membranes were then extensively washed and incubated for 90 min with HRP-conjugated goat anti-mouse (for Ras and anti c-jun) or goat anti-rabbit (for all other antibodies) IgG (Boehringer Mannheim) diluted 1:10,000 in TBST and 5% nonfat milk. Immunopositive bands were visualized using enhanced chemiluminescence (Amersham, Arlington Heights, IL) and XAR x-ray film (Eastman Kodak, Rochester, NY).
RESULTS
Constitutively activated Ras supports sympathetic neuron survival in the absence of NGF and stimulates phosphorylation of both MAPK and Akt
Previous studies have shown that Ras can mediate sympathetic neuron survival (Nobes and Tolkovsky, 1995; Nobes et al., 1996; Markus et al., 1997). However, the downstream effectors activated by Ras in these neurons remain unknown. To determine this, we generated a bicistronic recombinant adenovirus expressing RasV12 (Seeburg et al., 1984) and GFP from the same promoter and confirmed that it was appropriately expressed in sympathetic neurons by Western blot analysis (Fig. 1A) and fluorescence microscopy (Fig.2A). We then determined whether, as predicted, RasV12 could support sympathetic neuron survival in the absence of NGF. Specifically, sympathetic neurons were grown for 5 d in 50 ng/ml NGF, infected for 24 hr with the RasV12-expressing adenovirus, and 2 d later washed free of NGF. MTT assays revealed that control sympathetic neurons infected with GFP died within 2 d of NGF withdrawal (Fig. 1B), as observed previously (Chun and Patterson, 1977; Martin et al., 1988; Edwards and Tolkovsky, 1994; Bamji et al., 1998). In contrast, infection with RasV12 adenovirus rescued sympathetic neurons from death after NGF withdrawal in a concentration-dependent manner (Fig. 1C). This survival response was maximal at 100–200 MOI, where it was similar to survival induced by 5 ng/ml NGF (Fig. 1C). At 5 d after infection, survival induced by RasV12 was also similar to survival induced by 5 ng/ml NGF (data not shown), indicating that RasV12 appears to prevent and not just delay cell death in a portion of our neurons. RasV12 was never able to rescue survival to the levels seen for those neurons maintained in 10 ng/ml NGF, even though the level of RasV12 expression was high (Fig. 1A) and most of the neurons were expressing the mutated protein as determined by anti-c-myc immunocytochemistry (Fig. 2A,C). Typically, 77–85% of neurons expressed visible GFP after infection with Ras-expressing viruses.
Fig. 1.
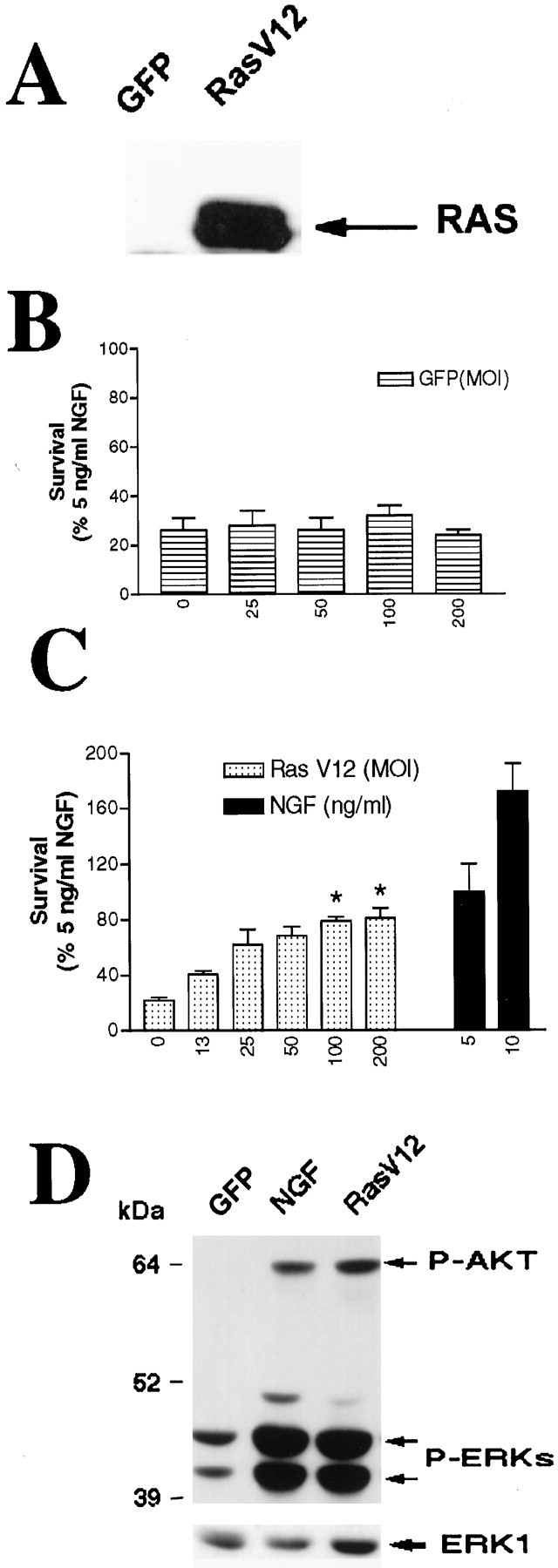
Ras is sufficient to sustain sympathetic neuronal survival and stimulates the phosphorylation of Akt and MAPK.A, Expression of RasV12 in sympathetic neurons is shown. NGF-selected sympathetic neurons were infected with 100 MOI of recombinant adenovirus-expressing GFP or RasV12; 3 d later, neurons were lysed, and equal amounts of protein were analyzed by Western blotting with an antibody specific for Ras. Note the high level of expression of RasV12. B, C, Ras is sufficient to prevent apoptotic neuronal death after NGF withdrawal. Sympathetic neurons were cultured for 5 d in the presence of 50 ng/ml NGF and infected with different MOIs of recombinant adenoviruses expressing either GFP (B) or RasV12 (C). Forty-eight hours after the onset of the infection, NGF was withdrawn for 2 d, and survival was assessed using MTT assays. As controls, uninfected sister cultures were maintained in 5 or 10 ng/ml NGF for the final 2 d (blackbars in C). Each point represents the mean ± SEM of quadruplicate wells from three representative experiments. Results are normalized to the amount of survival obtained with 5 ng/ml NGF. Values for RasV12 are significantly different from that of control (NGF-deprived) cells atp < 0.05 (*) (one-way ANOVA andposthoc Dunnett's multiple comparison test). D, RasV12 sustains high levels of phosphorylation of Akt and MAPK in the absence of NGF. After 5 d in 50 ng/ml NGF, cultures were infected with 100 MOI of RasV12 or GFP control virus. Twenty-four and forty-eight hours after the onset of the infection, cells were deprived of NGF. As a positive control, cultures were maintained in the continuous presence of 10 ng/ml NGF. After 2 d, cells were lysed, and Western blot analyses were performed using phosphorylation and activation state-specific antibodies against Akt or MAPK. Note that the levels of phosphorylation of these two Ras downstream effectors are similar in cultures infected with RasV12 and in those treated with NGF. To confirm that the amounts of protein were similar in eachlane, we reprobed the blots with anti-ERK1 antibody (bottom bands). P-AKT, Phospho-Akt;P-ERKs, phospho-MAPKs.
Fig. 2.

Immunohistochemistry of Ras mutants expressed in sympathetic neurons via recombinant adenovirus. Sympathetic neurons were infected with 100 MOI of c-myc-tagged RasV12 (A,C) or 200 MOI of c-myc-tagged N17Ras (B,D) in the presence of NGF, and 48 hr later cells were processed for immunocytochemistry using anti-myc (A,B). Note that the majority of neurons (phase contrast inC,D) express the Ras mutant proteins.
To confirm that the survival effect observed with RasV12 was caused by a rescue from NGF withdrawal-induced apoptosis, we also examined neurons by TUNEL staining (Fig.3A). Quantitative TUNEL analysis confirmed that these neurons were rescued from apoptosis; 82 ± 6% of neurons withdrawn from NGF for 2 d had TUNEL-positive nuclei, whereas only 28 ± 2% of neurons were TUNEL positive when they were infected with RasV12 and then withdrawn from NGF (Fig. 3A). Thus, RasV12 expression was able to rescue the majority of sympathetic neurons from NGF withdrawal-induced apoptosis, indicating that Ras signaling is sufficient for neurotrophin-mediated sympathetic neuron survival.
Fig. 3.

Ras regulates sympathetic neuron apoptosis.A, Quantitation of TUNEL-positive cells is shown. Cultures (n = 3) were infected with RasV12 (100 MOI) or N17Ras (200 MOI) in the presence of 10 ng/ml NGF. Twenty-four hours after infection, cultures infected with RasV12 were deprived of NGF, while those infected with N17Ras were maintained in 10 ng/ml NGF. Three days after infection, cells were processed for TUNEL and Hoechst staining. As controls, sympathetic neurons were infected with 200 MOI (control for N17 Ras) or 100 MOI (control for RasV12) of a recombinant adenovirus expressing GFP. Results are expressed as a percentage of TUNEL-positive neurons relative to the total number of Hoechst-labeled cells. B, C, Sympathetic neurons infected with N17Ras (200 MOI) and maintained in 10 ng/ml NGF for 3 d after infection were processed for TUNEL (B) and Hoechst staining (C). As determined by fluorescence microscopy, neurons that were TUNEL positive (B) had nuclei containing highly condensed or fragmented chromatin (C), confirming that they were undergoing apoptosis, whereas those that were TUNEL negative always had spherical and uniformly distributed chromatin.
We then determined whether expression of RasV12 led to the activation of multiple signaling pathways, focusing on the Raf/MEK/MAPK as well as on PI3-K/Akt pathways. Sympathetic neurons were infected with adenoviruses expressing either GFP or RasV12, maintained for an additional day in NGF, withdrawn from NGF for 2 d, and subsequently analyzed by Western blots for activation of MAPK and Akt using phosphorylation and activation state-specific antibodies (Fig.1D). As expected, the levels of phosphorylation of both Akt and the MAPKs were significantly higher in neurons maintained in NGF than in those infected with GFP adenovirus and withdrawn from NGF for 48 hr (Fig. 1D). In neurons maintained in the absence of NGF, expression of RasV12 led to strong phosphorylation of both Akt and the MAPKs to levels comparable with those seen in neurons maintained in 10 ng/ml NGF (Fig. 1D). Thus, these results show that RasV12 expression is sufficient to sustain sympathetic neuron survival and can activate both MAPK- and Akt-signaling pathways in these neurons.
Dominant-inhibitory Ras blocks NGF-induced survival of sympathetic neurons and Akt and MAPK phosphorylation
Neonatal sympathetic neurons require neurotrophin-mediated TrkA receptor activation for their survival, both in vivo(Levi-Montalcini and Brooker, 1960) and in culture (Levi-Montalcini and Angeletti, 1963; Coughlin and Collins, 1985; Belliveau et al., 1997). To determine whether this TrkA-dependent survival is mediated via Ras, we generated a bicistronic recombinant adenovirus expressing N17Ras (Sigal et al., 1986) and GFP from the same promoter. We then confirmed that this adenovirus generated the appropriate protein products. Sympathetic neurons were infected with N17Ras adenovirus in the presence of 10 ng/ml NGF, and 3 d later Western blots for Ras and immunocytochemistry for c-myc were performed. The biochemical analysis (Fig. 4A) demonstrated overexpression of a Ras-immunoreactive protein of ∼21 kDa in neurons infected with the N17Ras adenovirus. In control neurons, a similar-sized band was observed, corresponding to endogenous Ras, which was present at much lower levels (Fig. 4A). Fluorescence microscopy also indicated that the majority of sympathetic neurons expressed N17Ras (Fig. 2B,D).
Fig. 4.
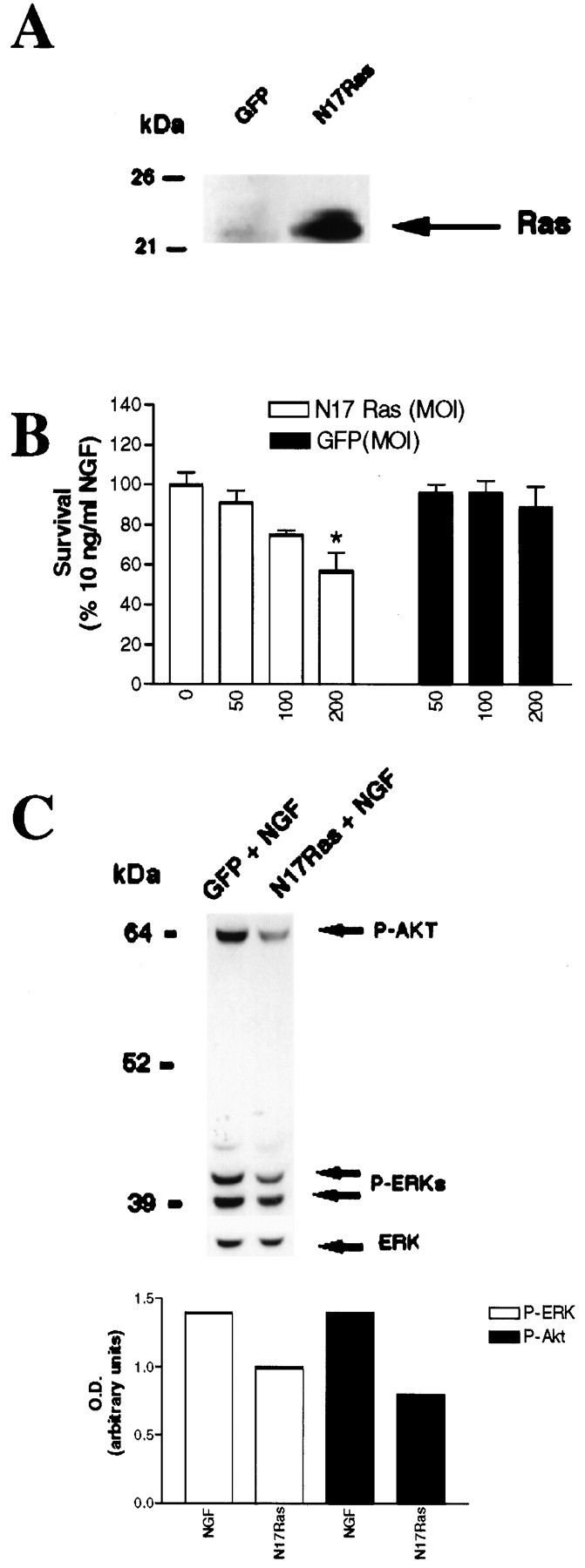
Dominant-inhibitory Ras suppresses neurotrophin-mediated sympathetic neuron survival. A,Expression of dominant-inhibitory Ras in sympathetic neurons is shown. NGF-selected sympathetic neurons were infected with adenovirus expressing GFP or N17Ras; 3 d later, neurons were lysed, and equal amounts of protein were separated by gel electrophoresis. After transfer, the blot was probed with anti-Ras. Note that endogenous Ras can be seen in the GFP-infected neurons (arrow), but that N17Ras is expressed at much higher levels. As reported previously, Ras migrates at a slightly higher apparent molecular weight than 21 kDa (Cox et al., 1995). B, MTT assays for sympathetic neurons infected with various MOIs of recombinant adenovirus expressing either N17Ras or GFP in the presence of NGF are shown. Neurons were selected in 50 ng/ml NGF for 5 d, infected with adenovirus, and, 48 hr after the onset of the infection, switched into media containing 10 ng/ml NGF for an additional 2 d, before the MTT assay was performed. Values are normalized to that of 10 ng/ml NGF, which supports 100% neuronal survival (Belliveau et al., 1997), and error bars represent the SEM. A value that is statistically different from that of 10 ng/ml NGF is denoted by an * at p< 0.01(one-way ANOVA and posthocDunnett's multiple comparison test). Results are the means from three independent experiments. The GFP adenovirus has no effect on neuronal survival, whereas N17Ras significantly reduces NGF-mediated survival. C,Top, N17Ras decreases the levels of phosphorylation of Akt and MAPK in cultures maintained in NGF. After 5 d in 50 ng/ml NGF, cultures were infected with N17Ras or GFP control virus (200 MOI) for 24 hr in the presence of 10 ng/ml NGF. Three days later, cells were lysed, and equal amounts of proteins were analyzed by Western blotting using phosphorylation and activation state-specific antibodies against Akt or MAPK. Blots were reprobed with an antibody directed against total ERK1 (bottom bands). Shown is a representative blot of the experiment performed at least three times.Bottom, The intensity of each band (top), normalized against those of total ERK1, is shown in the bar graph. Note the decrease in the levels of NGF-mediated phosphorylation of both phospho-Akt (P-Akt) and, to a lesser extent, phospho-MAPK (P-ERK) in cultures infected with N17Ras.
Having confirmed that N17Ras was expressed in neurons, we then asked whether Ras activation was necessary for NGF-mediated sympathetic neuron survival. Specifically, postnatal day 1 sympathetic neurons were cultured in 50 ng/ml NGF for 5 d, at which time they were infected for 24 hr with the N17Ras adenovirus at various MOIs. After infection, neurons were maintained in 10 ng/ml NGF for 3 additional days, and survival was then quantified using MTT assays (Manthorpe et al., 1986) (Fig. 4B). N17Ras expression decreased NGF-mediated sympathetic neuron survival by up to 43%, whereas infection with a control GFP-expressing adenovirus at the same MOIs had no effect (Fig.4B), indicating that Ras activation is required for at least a portion of the NGF-mediated survival.
To confirm that this decrease in neuronal survival was caused by apoptosis, we examined neurons by Hoechst staining and TUNEL analysis 3 d after infection with N17Ras (Fig. 3). Quantitative analysis revealed that 4 ± 1% of neurons infected with an adenovirus expressing only GFP and maintained in 10 ng/ml NGF were TUNEL positive (Fig. 3A). In contrast, infection with the N17Ras adenovirus increased the percentage of TUNEL-positive nuclei to 32 ± 2% (Fig. 3A). In addition, the TUNEL-negative neurons exhibited intact, rounded nuclei as revealed by Hoechst staining, whereas those that were TUNEL positive showed condensed and in some cases fragmented nuclei (Fig. 3B,C), the latter an indication of apoptotic death induced by growth factor deprivation (Darzynkiewicz et al., 1992; Deckwerth and Johnson, 1993; Edwards and Tolkovsky, 1994).
We then investigated whether infection with the N17Ras adenovirus led to inhibition of two of the signal transduction pathways activated by RasV12 in these neurons, Raf/MEK/MAPK and PI3-K/Akt (Fig.1D). In one set of experiments, sympathetic neurons were cultured in NGF for 5 d, infected with adenoviruses expressing either GFP alone or N17Ras for 24 hr, and then maintained in 10 ng/ml NGF for 3 additional days. Cells were subsequently lysed and analyzed for phosphorylation of MAPK and Akt using phosphorylation and activation state-specific antibodies. Western blot analysis revealed that in cultures maintained in 10 ng/ml NGF, N17Ras induced a 42% decrease in phosphorylation of Akt and a 30% decrease in phosphorylation of MAPKs relative to that in neurons maintained in NGF but infected with the GFP adenovirus (Fig. 4C). These results show that Ras is necessary for a portion of NGF-induced sympathetic neuron survival and that cell death induced by N17Ras is accompanied by a concomitant reduction in Akt and MAPK phosphorylation.
Ras sustains sympathetic neuron survival via multiple downstream effectors
Together, these results indicate (1) that Ras is a key regulator of sympathetic neuron survival, (2) that it signals via multiple pathways in sympathetic neurons, and (3) that at least one of these downstream pathways may mediate sympathetic neuron survival. To determine which of these pathways are involved in this response, we generated recombinant adenoviruses expressing Ras effector mutants that selectively activate downstream targets. Initially, we generated one Ras mutant, RasV12Y40C, that binds and activates PI3-K but not Raf, a second, RasV12T35S, that binds and activates Raf and RalGDS but not PI3-K, a third, RasV12E37E, that activates RalGDS but neither Raf nor PI3-K, and a fourth, RasV12D38E, that activates Raf (Rodriguez-Viciana et al., 1997). We then confirmed that these effector mutants were expressed in sympathetic neurons at similar levels. Specifically, sympathetic neurons were infected with one of these four viruses, and 3 d later, Western blot analysis was performed with anti-Ras. RasV12T35S, RasV12Y40C, and RasV12E37G adenoviruses expressed immunoreactive proteins of the appropriate size in sympathetic neurons (Fig. 5A). However, we could not obtain consistent high Ras levels with the RasV12D38E adenovirus. In view of this, we used RasV12T35S as a selective Raf/MEK/MAPK inducer.
Fig. 5.
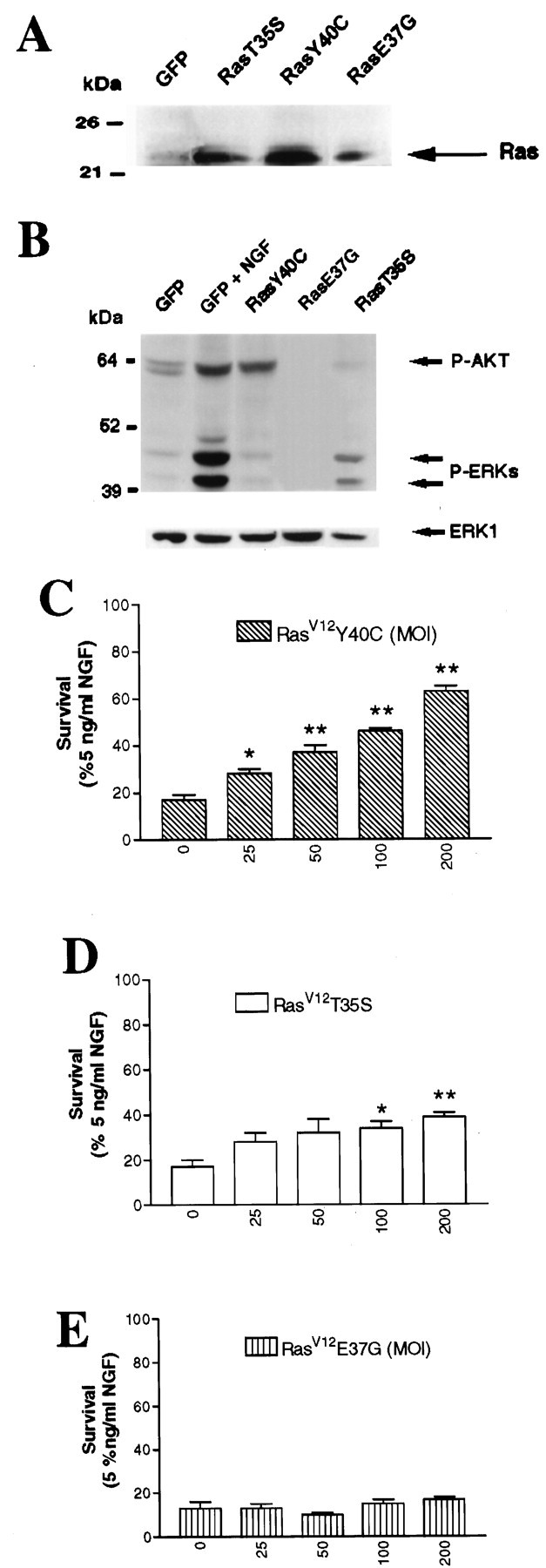
Ras sustains sympathetic neuronal survival via multiple signaling pathways. A, Expression of Ras effector mutants in sympathetic neurons is shown. Cells were infected with 100 MOI of adenoviruses expressing GFP, RasV12T35S (RasT35S), or RasV12E37G (RasE37G) or with 200 MOI of RasV12Y40C (RasY40C) adenovirus for 3 d. Equivalent amounts of protein were separated by gel electrophoresis, as shown by the bottom bands, and Western blots were probed with anti-Ras. Note that the levels of expression of the different Ras effector mutants are comparable. B, Ras effector mutants differentially activate Ras downstream targets. Sympathetic neurons were infected with 200 MOI of RasV12Y40C, 100 MOI of RasV12T35S, or 100 MOI of RasV12E37G in the presence of 30 ng/ml NGF, and 48 hr after the onset of the infection, NGF was withdrawn. As controls, neurons were infected with 100 MOI of GFP adenovirus and maintained in 10 ng/ml NGF (GFP + NGF). Two days after NGF withdrawal, cell lysates were prepared and normalized for equal amounts of proteins. Phosphorylation of MAPK and Akt was examined on Western blots using anti-phospho-MAPK (P-ERK) or anti-phospho-Akt (P-AKT). Note that RasV12Y40C only induces phosphorylation of Akt, whereas RasV12T35S is able to induce phosphorylation of MAPK (ERK) but not Akt. As expected, RasV12E37G did not stimulate the phosphorylation of these two Ras downstream effectors. C–E, Ras sustains sympathetic neuron survival via PI3-K and Raf but not RalGDS. Sympathetic neurons were prepared and after 5 d in vitro infected with increasing MOIs of adenovirus expressing RasV12Y40C (C), RasV12T35S (D), or RasV12E37G (E), which selectively activates PI3-K, Raf/MEK/MAPK, or RalGDS pathways, respectively. Results from the MTT assay performed 2 d after NGF withdrawal are expressed as the percentage of survival with 5 ng/ml NGF and are means ± SEM of quadruplicate wells from three independent experiments. Values are significantly different from that of NGF-deprived cultures (0 MOI) atp < 0.05 (*) or p < 0.01 (**) (one-way ANOVA and posthoc Dunnett's multiple comparison test).
We next confirmed that these effector mutants functioned as predicted in sympathetic neurons. Specifically, NGF-dependent sympathetic neurons were infected with adenoviruses expressing activated Ras or one of the three effector mutants and were maintained for an additional 24 hr in NGF. Two days after NGF withdrawal, Western blot analysis was performed for phosphorylated Akt, a downstream target of PI3-K, or for phosphorylated MAPKs, downstream targets of Raf-1 and MEK (Fig.5B). RasV12Y40C, which selectively activates PI3-K, induced phosphorylation of Akt but not phosphorylation of the MAPKs (Fig. 5B). Conversely, expression of RasV12T35S led to phosphorylation of the MAPKs but not Akt (Fig. 5B). Finally, as predicted, RasV12E37GE failed to induce phosphorylation of these downstream substrates. In the case of each mutant, the phosphorylation of MAPK and Akt was less than that observed with NGF treatment.
Because the Ras effector mutants selectively activated the predicted pathways in sympathetic neurons, we next determined whether these proteins were capable of mediating sympathetic neuron survival. Neurons were grown for 5 d in 50 ng/ml NGF and infected with one of the three Ras effector mutant-expressing adenoviruses, and 48 hr later they were withdrawn from NGF for 2 d. MTT assays revealed that RasV12Y40C maintained survival of sympathetic neurons in the absence of NGF in a concentration-dependent manner (Fig. 5C), with a statistically significant increase in survival first detected at 25 MOI. At the highest MOI used in this study (200 MOI), RasV12Y40C was able to mediate ∼60% of the survival mediated by RasV12 when these proteins were expressed at similar levels (data not shown). Similarly, RasV12T35S was able to promote sympathetic neuron survival (Fig. 5D), although to a much lesser degree than was RasV12Y40C (Fig. 5C). RasV12E37G was unable to mediate sympathetic neuron survival at any of the MOIs used either alone (Fig.5E) or in combination with other effector mutants (data not shown), in spite of the fact that it was expressed at levels similar to that of the other two effector mutants (Fig. 5A). These results suggest that the RalGDS pathway is not important in mediating sympathetic neuron survival and that Ras promotes sympathetic neuron survival via pathways originating primarily from PI3-K but also from Raf-1.
To confirm that Ras mediates neuronal survival via these two downstream pathways, we used two pharmacological agents: (1) PD 98059 that selectively inhibits MEK (which is immediately downstream of Raf) and (2) LY 294002 that selectively inhibits PI3-K. Initially, we confirmed biochemically the selectivity of these pharmacological agents in sympathetic neurons. Specifically, sympathetic neurons were selected in NGF, infected with adenoviruses expressing GFP or RasV12, and then switched to media without NGF containing 100 μm LY 294002 or 75 μm PD 98059 for 2 d. As a control, uninfected sister cultures were maintained in media containing 10 ng/ml NGF with or without one of these two drugs. We then analyzed these neurons for phosphorylation of Akt (downstream of PI3-K) and MAPKs (downstream of MEK) by Western blots. As shown previously in other cell types (Vlahos et al., 1994; Dudley et al., 1995), LY 294002 inhibited the NGF-induced phosphorylation of Akt but had no effect on the phosphorylation of MAPK, whereas PD 98059 inhibited NGF-induced phosphorylation of MAPKs but not of Akt (Fig.6A). These two drugs had similar effects on substrate phosphorylation in response to activated Ras. LY 294002 inhibited Ras-induced phosphorylation of Akt, with no effect on the phosphorylation of MAPK, whereas PD 98059 inhibited Ras-induced phosphorylation of MAPK, with no effect on Akt (Fig. 6A).
Fig. 6.
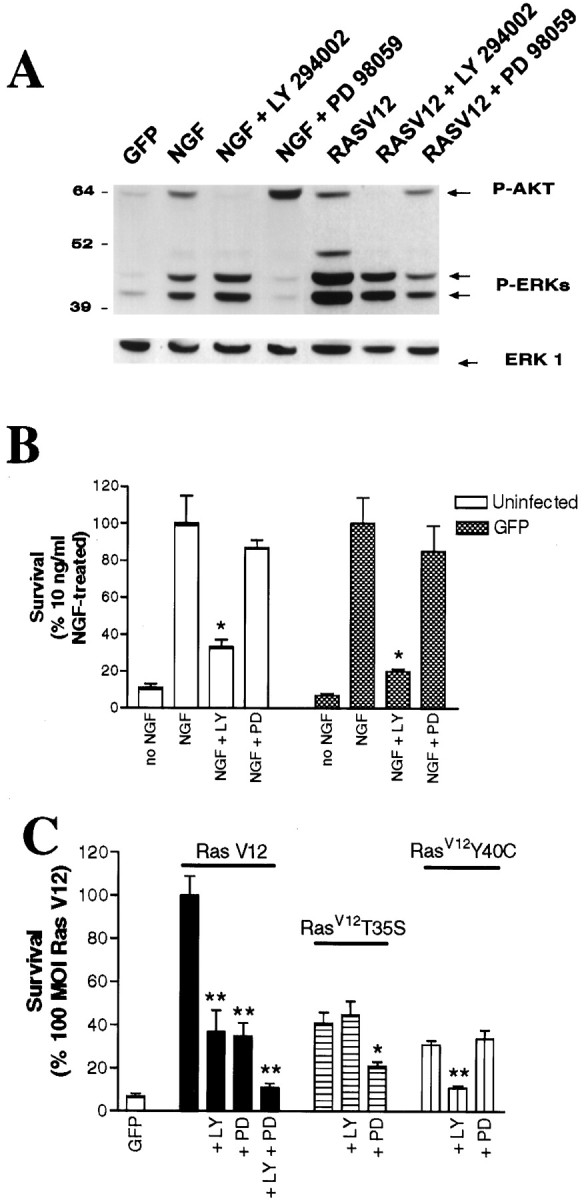
Inhibition of NGF- and Ras-mediated survival by the PI3-K inhibitor LY 294002 and the MEK inhibitor PD 98059.A, NGF-selected sympathetic neurons were infected with 100 MOI of adenoviruses expressing GFP or RasV12. Two days after the initiation of the infection, cultures were switched into media with no NGF but containing 100 μm LY 294002 or 75 μm PD 98059. As controls, mock-infected sister cultures were switched into media containing 10 ng/ml NGF plus one of the same two drugs. Two days later, neurons were lysed and analyzed by Western blotting with anti-phospho-Akt (P-AKT) and anti-phospho-MAPK (P-ERK). To ensure that similar amounts of proteins were present in all lanes, we reprobed the blot with an antibody against ERK1 (bottombands). Note that, as expected, LY 294002 inhibits phosphorylation of Akt, whereas PD 98059 only affects phosphorylation of MAPK. B, C, PI3-K and MEK are required to mediate Ras survival effects on sympathetic neurons. B, Sympathetic neurons were infected with 100 MOI of GFP adenovirus and were switched to media containing 10 ng/ml NGF plus 100 μm LY 294002 (LY) or 75 μm PD 98059 (PD) 48 hr after the onset of the infection. MTT assays were performed 2 d later. Mock-infected sister cultures were also analyzed after the same treatment. C, Neurons were infected with 100 MOI of adenoviruses expressing GFP, RasV12, RasV12T35S, or RasV12Y40C adenovirus (200 MOI) and 48 hr later switched to media containing no NGF and 100 μm LY 294002, 75 μm PD 98059, or both for 2 d before MTT assays. In B and C, results represent the means ± SEM of four to five replicates from three independent experiments and are expressed as the percentage of survival attained with 10 ng/ml NGF (B) or RasV12 (C). Results are different from their appropriate control (no drug treatment) at p < 0.05 (*) orp < 0.01 (**) (one-way ANOVA andposthoc Dunnett's multiple comparison test).
Having confirmed the selectivity of action of these two drugs, we next determined whether they inhibited the ability of RasV12 to mediate sympathetic neuron survival, as predicted by the Ras effector data. As a baseline, we determined the effects of these two drugs on NGF-induced survival of sympathetic neurons that were either mock infected or infected with an adenovirus expressing GFP (Fig. 6B). For this study, neurons were selected in 50 ng/ml NGF for 5 d, infected with GFP-expressing adenovirus (100 MOI), and after 48 hr switched for an additional 2 d to 10 ng/ml NGF with or without PD 98029 or LY 294002, after which neuronal survival was measured by MTT assays. The PI3-K inhibitor LY 294002 decreased by 70–80% the survival of uninfected or GFP-infected sympathetic neurons (Crowder and Freeman, 1998), whereas PD 98029 decreased sympathetic neuron survival by <20% (Fig. 6B). Our results showing suppression of survival by LY 294002 are similar to those of Crowder and Freeman (1998) but differ from those of Philpott et al. (1997).
We next determined the effects of these two drugs on the inhibition of sympathetic neuron survival as mediated by RasV12 or the two Ras effector mutants RasV12T35S and RasV12Y40C. NGF-dependent sympathetic neurons were infected with 100 MOI of each of these three viruses and were then switched into media without NGF, containing either PD 98059 or LY 294002. Two days later, we performed MTT assays to measure neuronal survival (Fig. 6C). Inhibition of MEK with PD 98059 completely abolished the ability of RasV12T35S, which selectively activates Raf-1 and MEK, to promote sympathetic neuron survival (Fig. 6C), whereas LY 294002 had no effect. This result further confirms that the effects of RasV12T35S are mediated via Raf/MEK/MAPK rather than, or in addition to, RalGDS. Similarly, inhibition of PI3-K activity with LY 290042 completely abolished the ability of RasV12Y40C, which selectively activates PI3-K, to mediate sympathetic neuron survival (Fig. 6C), whereas PD 98059 had no effect. Finally, as predicted by the Ras effector mutant data, LY 290042 and PD 98059 were both independently able to inhibit survival mediated by RasV12; inhibition of PI3-K by LY 290042 inhibited Ras-mediated survival by ∼60%, whereas both LY 290042 and PD 98059 together decreased survival to near background levels. Thus, as indicated by the effector mutant data, Ras mediates sympathetic neuron survival via activation of PI3-K/Akt and Raf/MEK/MAPK signal transduction pathways.
Activated Ras suppresses and dominant-inhibitory Ras induces the c-jun/p53/Bax pathway in sympathetic neurons
We have demonstrated recently that sympathetic neuron apoptosis after NGF withdrawal or p75 receptor activation is dependent on an apoptotic pathway that involves JNK, p53, and Bax (Aloyz et al., 1998). To determine whether activated Ras can suppress this pathway, we infected sympathetic neurons with RasV12 adenovirus and biochemically examined proteins in this pathway. Sympathetic neurons were infected with RasV12 and withdrawn from NGF, and 24–48 hr later, the levels of c-jun, p53, and Bax were examined. Withdrawal from NGF increased, as expected, the level and degree of phosphorylation of c-jun (Ham et al., 1995; Bamji et al., 1998; Eilers et al., 1998) and increased the levels of p53 and Bax protein (Aloyz et al., 1998) in GFP-expressing adenovirus-infected cultures relative to that in sister cultures treated with 10 ng/ml NGF (Fig.7A). RasV12 expression inhibited the increases in c-jun, p53, and Bax protein levels that are observed after NGF withdrawal (Fig. 7A), coincident with its ability to rescue sympathetic neuron apoptosis.
Fig. 7.

Dominant-inhibitory Ras induces and activated Ras suppresses the p53 apoptotic pathway. Sympathetic neurons were infected with 100 MOI of RasV12 (A) or GFP (A, labeled GFP − NGF) or 200 MOI of N17Ras (B). Forty-eight hours after infection, the media were changed, and cultures were maintained in the absence (GFP − NGF,RasV12 − NGF) or presence (NGF, N17Ras + NGF) of 10 ng/ml NGF. Two days later, cells were lysed, and equal amounts of proteins were separated electrophoretically and transferred onto nitrocellulose membranes. Western blots were probed with anti c-jun, anti-p53, anti-Bax, anti- P-Akt, anti-P-ERK, and anti-ERK 1. Note that in NGF-treated cultures, N17Ras induces an increase in the levels of expression of c-jun and a shift to a larger apparent molecular weight, indicative of increased phosphorylation (B). RasV12 suppressed (A) whereas N17Ras increased the levels of p53 and Bax protein (B). Shown are representative blots of experiments performed at least four times. Thelanes labeled ERK 1 indicate total protein amounts assayed. P-Akt, Phospho-Akt;P-ERK, phospho-MAPK.
To determine whether TrkA-induced activation of Ras mediates survival by suppressing the c-jun/p53/Bax apoptotic pathway, we inhibited Ras activity by infecting sympathetic neurons with N17Ras and examined the levels of c-jun, p53, and Bax. Specifically, neurons were maintained in NGF for 5 d, infected with either GFP or N17Ras-expressing adenovirus, and then maintained with or without 10 ng/ml NGF for an additional 2 d. Expression of N17Ras elevated the levels of these apoptotic proteins in NGF-treated neurons (Fig. 7B), coincident with its ability to induce apoptosis. Thus, our results suggest that in sympathetic neurons, TrkA-mediated activation of Ras normally suppresses the c-jun/p53/Bax apoptotic pathway.
To provide genetic evidence that p53 is required for apoptosis induced by suppression of the NGF-activated Ras-signaling pathway, we asked whether suppression of p53 could prevent N17Ras-induced sympathetic neuronal death in the presence of NGF. To suppress p53, we used an adenovirus vector that functionally ablates p53 via the actions of E1B55K and targets p53 for degradation via the adenovirus E4orf6 product (Yew and Berk, 1992; Querido et al., 1997; Teodoro and Branton, 1997; Aloyz et al., 1998). We have shown previously that this vector will suppress p53 levels and rescue sympathetic neurons from NGF withdrawal and p75NTR-induced cell death (Aloyz et al., 1998). As reported previously (Aloyz et al., 1998), after NGF withdrawal, E1B55K rescued up to ∼55% of the neurons relative to those maintained in 10 ng/ml NGF (Fig. 8). Maximal antiapoptotic effects of E1B55K were attained at 300 MOI of E1B55K, which was thus used for subsequent experiments. Expression of E1B55K significantly blocked the apoptotic effects of N17Ras in neurons maintained in the presence of NGF (Fig. 8). In data from three separate experiments, N17Ras expression inhibited neuronal survival by 42%, whereas coexpression of E1B55K with N17Ras reduced neuronal loss to 17%. Infection with control GFP adenovirus (300 MOI) did not affect the decrease in neuronal survival induced by N17Ras (Fig. 8). These results further suggest that the antiapoptotic effects of Ras are caused by its inhibition of the p53 proapoptotic protein in sympathetic neurons.
Fig. 8.

p53 is required for neuronal death induced by suppression of NGF-induced Ras activity. Sympathetic neurons maintained in the presence of 10 ng/ml NGF were infected with increasing MOI of E1B55K (which functionally ablates p53) and withdrawn from neurotrophin support 48 hr after infection. Neurons were also infected with 200 MOI of N17Ras either alone or with the simultaneous addition of either 300 MOI of E1B55K (E1B) or, as a control, 300 MOI of GFP and kept in the continuous presence of 10 ng/ml NGF. In all cases, neurons were processed for MTT assays 96 hr after infection. Values are normalized as the percentage of survival attained with 10 ng/ml NGF and represent means ± SEM of quadruplicate wells from three experiments. Values for N17Ras + GFP are significantly different from those for N17Ras + E1B55K at p < 0.05 (*) (one-way ANOVA and posthoc Dunnett's multiple comparison test). E1B55K significantly blocked the apoptotic effects induced by N17Ras in NGF-treated cultures.
DISCUSSION
In this study, we have examined the role of Ras and its multiple signal transduction pathways in neurotrophin-induced survival responses of postnatal rat sympathetic neurons, during the period corresponding to the time point when these neurons undergo naturally occurring cell death in vivo. Our results support several conclusions. First, activation of Ras is necessary and sufficient for a portion of NGF sympathetic neuron survival. Second, neurotrophins induce the phosphorylation of both Akt and MAPK in these neurons in a Ras-dependent manner. Third, the antiapoptotic effects of Ras are mediated primarily by PI3-K, and partially by MEK, but not by RalGDS. Fourth, Ras appears to regulate sympathetic neuron survival by suppressing the activity or levels of c-jun, p53, and Bax, which are components of the key pathway mediating naturally occurring cell death in these neurons (Deckwerth et al., 1996; Xiang et al., 1996, 1998;Aloyz et al., 1998; Bamji et al., 1998; Eilers et al., 1998). Fifth, p53 activity is required for apoptosis induced by suppression of the NGF-activated Ras pathway.
Ras as a major mediator of sympathetic neuron survival
In agreement with previous reports on the role of Ras in rat sympathetic SCG neurons (Nobes and Tolkovsky, 1995; Nobes et al., 1996), we show that Ras is a major mediator of NGF-induced sympathetic neuron survival. The authors of these previous reports used scrape loading of anti-Ras antibodies into newly isolated postnatal sympathetic neurons to show that Ras was responsible for approximately one-half of NGF-dependent neuronal survival. Similarly, we find that activated Ras supports, and N17Ras suppresses, approximately one-half of the survival responses of postnatal day 5 sympathetic neurons expressing these proteins via recombinant adenovirus. The observation that only one-half of the sympathetic neurons are affected by these manipulations of Ras activity could be attributable to low levels of expression of exogenously expressed Ras proteins or to the presence of both Ras- and non-Ras-dependent survival pathways in sympathetic neurons. It is unlikely that the expression of activated Ras via adenovirus was insufficient to promote complete survival responses, because (1) most cells in each experiment were infected by adenovirus, (2) increasing the virus particles per cell did not increase the ability of activated Ras to increase neuronal survival beyond 50% of the cells, and (3) activated Ras strongly stimulated MAPK and Akt phosphorylation, the latter a major mediator of sympathetic neuron survival (Crowder and Freeman, 1998). The same N17Ras adenovirus did fully block neurite outgrowth responses and induced apoptosis of NGF-dependent PC12 cells (I. E. Mazzoni, F. D. Miller, and D. Kaplan, unpublished observations), indicating that the N17Ras adenovirus that we use has the potential to inhibit NGF responses of cells completely. Therefore, the inability of activated Ras to fully stimulate and N17Ras to fully inhibit survival suggests that TrkA mediates at least one-half of neuronal survival in a Ras-independent manner. These results suggest to us that multiple signaling pathways regulate survival, with some pathways (PI3-K) being crucial for the survival of most cells and others (Ras) being necessary if completely and efficiently suppressed. It is also possible that the strength of PI3-K survival signaling depends on the relative importance of Ras-dependent and -independent signaling in a given neuron. The later may include Ras-independent mechanisms regulating PI3-K-derived survival, such as through Gab-1 (Holgado-Madruga et al., 1997) or TrkA-activated proteins such as phospholipase C-γ1, SRC, rAPS1, SH2-B, fibroblast receptor substrate-2, and SVC-associated neurotrophic factor target (Kaplan and Miller, 1997; Kouhara et al., 1997; Qian et al., 1998).
Studies in non-neuronal cells have shown that Ras has the potential to activate a large number of downstream effectors and may do so in a cell-specific manner (for review, see Quilliam et al., 1995; Marshall, 1996; Vojtek and Der, 1998). Furthermore, activation of different Ras-dependent pathways can lead to distinct biological responses (Joneson et al., 1996; Khosravi-Far et al., 1996; Rodriguez-Viciana et al., 1997). Our data provide evidence that, in sympathetic neurons, Ras can stimulate both MEK/MAPK and PI3-K/Akt pathways and, moreover, that Ras sustains neuronal survival via multiple signal transduction pathways as well. Both the MEK/MAPK- and PI3-K/Akt-signaling pathways, activated by RasV12T35S and RasV12Y40C, respectively, contributed ∼40 and 60% of the maximal survival effects induced by activated Ras. Similarly, treatment of neurons with the MEK inhibitor PD 98059, together with the PI3-K inhibitor LY 294002, completely abolished the survival-inducing effects of activated Ras. These results suggest that MEK and PI3-K activity account for most, if not all, of the survival effects induced by activated Ras in sympathetic neurons. PI3-K, however, appeared to be the dominant Ras effector protein for survival responses. In this regard, N17Ras blocked Akt phosphorylation more efficiently than MAPK phosphorylation. Furthermore, expression of RasV12T35S, which specifically induces MEK and MAPK activity, was less efficient at stimulating survival than was RasV12Y40C, which specifically stimulates PI3-K and Akt activity. Other investigators have also shown that MEK plays only a minor role in NGF-mediated survival of sympathetic neurons (Virdee and Tolkovsky, 1995a,b; Creedon et al., 1996). In our experiments, PI3-K was required for 60% of Ras-induced and 80% of NGF-induced neuronal survival. Although the activities of PI3-K and Akt have been shown to be sufficient to sustain sympathetic neuronal survival, there are contradictory reports on whether their activities are necessary to mediate NGF's antiapoptotic effects (Philpott et al., 1997; Crowder and Freeman, 1998). Our results agree with those of Crowder and Freeman (1998), who also used LY 294002, as well as dominant-inhibitory PI3-K and Akt, to inhibit sympathetic neuron survival. Our results also suggest that much, but not all, of PI3-K-induced neuronal survival is caused by Ras activity and agree with recent studies performed in sensory neurons showing that Ras-dependent activation of PI3-K is necessary to induce neuron survival (Klesse and Parada, 1998).
Another downstream effector stimulated by Ras, RalGDS, did not affect neuronal survival because expression of RasV12E37G, which activates this pathway (Rodriguez-Viciana et al., 1997), could not sustain, either in combination with RasV12Y40C or RasV12T35S (Mazzoni., Miller, and Kaplan, unpublished observations) or alone (Fig.5E), sympathetic neuron survival after NGF withdrawal. RasV12E37G expression did not augment or inhibit the survival effects observed with the other Ras effector mutant RasV12T35S (Mazzoni, Miller, and Kaplan, unpublished observations). Nevertheless, we cannot rule out a survival role for other Ras effector proteins, such as Rin1, p120GAP, and AF6 (for review, see Quilliam et al., 1995;Marshall, 1996; Vojtek and Der, 1998), whose expression and function in sympathetic neurons remain to be determined.
Mechanism of Ras-mediated suppression of apoptosis
What is the mechanism by which Ras mediates NGF-dependent survival? In postnatal sympathetic neurons, both in vivoduring the period of naturally occurring cell death and in vitro, the balance between survival and apoptosis depends on the relative strengths of TrkA prosurvival signals and p75NTR-mediated apoptotic signals (Aloyz et al., 1998; Bamji et al., 1998). A similar finding has been reported in oligodendrocytes (Yoon et al., 1998). Apoptosis of sympathetic neurons induced by both p75NTR and NGF deprivation is regulated via a common cell death sensor, p53. We have shown recently that p53 is necessary and sufficient for inducing apoptosis in postnatal sympathetic neurons both in vivo and in culture (Slack et al., 1996; Aloyz et al., 1998). In this regard, we demonstrated that p53 is upregulated in response to NGF withdrawal as well as after p75NTR activation, both of which involve signaling via the MEK kinase(MEKK)/JNK pathway (Estus et al., 1994; Ham et al., 1995; Casaccia-Bonnefil et al., 1996; Bamji et al., 1998). Furthermore, we found p53 to lie downstream of JNK and upstream of Bax (Aloyz et al., 1998). Here we show that expression of activated Ras suppresses, whereas N17Ras increases, c-jun, p53, Bax levels, anti-P-Akt, anti-P-ERK, and anti-ERK 1. In addition, functional ablation of p53 by E1B55K prevents apoptotic neuronal death induced by N17Ras in NGF-treated cultures. The observation that E1B55K could not completely rescue N17Ras-treated cultures to control (NGF-treated) values could mean that additional apoptotic pathways are operational in these cells. These might include Akt-induced phosphorylation of the Bad and forkhead apoptotic proteins (Datta et al., 1997; Brunet et al., 1999), although we have not found Bad to be phosphorylated in NGF-treated sympathetic neuron cultures. On the basis of our data, we propose that Ras and its effectors PI3-K/Akt, and to a lesser extent MEK/MAPK, are major signaling proteins used by TrkA to suppress apoptosis. Thus, sympathetic neuron survival may not depend on the relative strengths of independent pro- and antiapoptotic signaling pathways. Rather, neuronal survival may be dependent on the ability of TrkA-activated Ras, and on the Ras-activated effectors PI3-K/Akt and MEK, to suppress the MEKK/JNK/p53/Bax cell death pathway. Because p53 is a primary cell death sensor for PNS and CNS neurons undergoing cell death because of excitotoxicity, survival factor deprivation, and p75NTR activity (Morrison et al., 1996; Aloyz et al., 1998; Bamji et al., 1998; Xiang et al., 1998), Ras could function in many neuronal contexts as a general suppressor of p53 apoptotic actions.
Footnotes
This work was supported by grants from the Medical Research Council of Canada and from the Neuroscience Network to D.K. and F.D.M. D.K. is a recipient of the Harold Johns and Canadian Cancer Society Research Scientist Award, and F.D.M. is a Killam Scholar. I.E.M. is funded by a fellowship from the Natural Sciences and Engineering Research Council of Canada. We thank Dr. P. Warne (Imperial Cancer Research Fund, United Kingdom) and Dr. C. Bazenet (EISAI London Research Laboratories, United Kingdom) for providing us with the mutated Ras constructs, Dr. L. Paquet (Exogen Neurosciences, Montreal, Quebec, Canada) for his helpful advice, and B. Bourque and M. Ahkavan for technical assistance.
Correspondence should be addressed to Dr. D. Kaplan or Dr. F. Miller, Center for Neuronal Survival, Montreal Neurological Institute, 3801 rue University, Montreal, Quebec, Canada H3A 2B4. E-mail:mcdv@musica.mcgill.ca.
Dr. Mazzoni's present address: Caprion Pharmaceuticals, 6100 Royalmont Avenue, Montreal, Quebec, Canada H4P 2R2.
REFERENCES
- 1.Aloyz RS, Bamji SX, Pozniak CD, Toma JG, Atwal J, Kaplan DR, Miller FD. p53 is essential for developmental neuron death as regulated by the TrkA and p75 neurotrophin receptors. J Cell Biol. 1998;143:1691–1703. doi: 10.1083/jcb.143.6.1691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bamji SX, Majdan M, Pozniak CD, Belliveau DJ, Aloyz R, Kohn J, Causing CG, Miller FD. The p75 neurotrophin receptor mediates neuronal apoptosis and is essential for naturally occurring sympathetic neuronal death. J Cell Biol. 1998;140:911–923. doi: 10.1083/jcb.140.4.911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Belliveau DJ, Krivko I, Kohn J, Lachance C, Pozniak C, Rusakov D, Kaplan D, Miller FD. NGF and neurotrophin-3 both activate TrkA on sympathetic neurons but differentially regulate survival and neuritogenesis. J Cell Biol. 1997;136:375–388. doi: 10.1083/jcb.136.2.375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Borasio GD, Markus A, Wittinghofer A, Barde Y-A, Heumann R. Involvement of ras p21 in neurotrophin-induced response of sensory, but not sympathetic neurons. J Cell Biol. 1993;121:665–672. doi: 10.1083/jcb.121.3.665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Brunet A, Bonni A, Zigmond MJ, Lin MZ, Juo P, Hu LS, Anderson MJ, Arden KC, Blenis J, Greenberg ME. Akt promotes cell survival by phosphorylating and inhibiting a Forkhead transcription factor. Cell. 1999;96:857–868. doi: 10.1016/s0092-8674(00)80595-4. [DOI] [PubMed] [Google Scholar]
- 6.Casaccia-Bonnefil P, Carter BD, Dobrowsky RT, Chao MV. Death of oligodendrocytes mediated by the interaction of nerve growth factor with its receptor p75. Nature. 1996;383:716–719. doi: 10.1038/383716a0. [DOI] [PubMed] [Google Scholar]
- 7.Chun LY, Patterson PH. Role of NGF in the development of rat sympathetic neurons in vitro. I. Survival, growth and differentiation of catecholamine production. J Cell Biol. 1977;75:694–704. doi: 10.1083/jcb.75.3.694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Coughlin MD, Collins MB. Nerve growth factor-independent development of embryonic mouse sympathetic neurons in dissociated cell culture. Dev Biol. 1985;110:392–401. doi: 10.1016/0012-1606(85)90098-3. [DOI] [PubMed] [Google Scholar]
- 9.Cowley S, Paterson H, Kemp P, Marshall CJ. Activation of MAP kinase kinase is necessary and sufficient for PC12 differentiation and for transformation of NIH 3T3 cells. Cell. 1994;77:841–852. doi: 10.1016/0092-8674(94)90133-3. [DOI] [PubMed] [Google Scholar]
- 10.Cox AD, Solski PA, Jordan JD, Der CJ. Analysis of Ras protein expression in mammalian cells. Methods Enzymol. 1995;255:195–221. doi: 10.1016/s0076-6879(95)55023-2. [DOI] [PubMed] [Google Scholar]
- 11.Creedon DJ, Johnson EM, Jr, Lawrence JC., Jr Mitogen-activated protein kinase-independent pathways mediate the effects of nerve growth factor and cAMP on neuronal survival. J Biol Chem. 1996;271:20713–20718. doi: 10.1074/jbc.271.34.20713. [DOI] [PubMed] [Google Scholar]
- 12.Crowder RJ, Freeman RS. Phosphatidylinositol 3-kinase and Akt protein kinase are necessary and sufficient for the survival of nerve growth factor-dependent sympathetic neurons. J Neurosci. 1998;18:2933–2943. doi: 10.1523/JNEUROSCI.18-08-02933.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Darzynkiewicz Z, Bruno S, Del Bino G, Gorczyca W, Hotz MA, Lassota P, Traganos F. Features of apoptotic cells measured by flow cytometry. Cytometry. 1992;13:795–808. doi: 10.1002/cyto.990130802. [DOI] [PubMed] [Google Scholar]
- 14.Datta SR, Dudek H, Tao X, Masters S, Fu H, Gotoh Y, Greenberg ME. Akt phosphorylation of BAD couples survival signals to the cell-intrinsic death machinery. Cell. 1997;91:231–241. doi: 10.1016/s0092-8674(00)80405-5. [DOI] [PubMed] [Google Scholar]
- 15.Deckwerth TL, Johnson EM., Jr Temporal analysis of events associated with programmed cell death (apoptosis) of sympathetic neurons deprived of nerve growth factor (NGF). J Cell Biol. 1993;123:1207–1222. doi: 10.1083/jcb.123.5.1207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Deckwerth TL, Elliott JL, Knudson CM, Johnson EM, Jr, Snider WD, Korsmeyer SJ. Bax is required for neuronal death after trophic factor deprivation and during development. Neuron. 1996;17:401–411. doi: 10.1016/s0896-6273(00)80173-7. [DOI] [PubMed] [Google Scholar]
- 17.Dudek H, Datta SD, Franke TF, Birnbaum MJ, Yao R, Cooper GM, Segal RA, Kaplan DK, Greenberg ME. Regulation of neuronal survival by the serine-threonine kinase Akt. Science. 1997;275:661–665. doi: 10.1126/science.275.5300.661. [DOI] [PubMed] [Google Scholar]
- 18.Dudley DT, Pang L, Decker SJ, Bridges AJ, Saltiel AR. A synthetic inhibitor of the mitogen-activated protein kinase cascade. Proc Natl Acad Sci USA. 1995;92:7686–7689. doi: 10.1073/pnas.92.17.7686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Edwards SN, Tolkovsky AM. Characterization of apoptosis in cultured rat sympathetic neurons after nerve growth factor withdrawal. J Cell Biol. 1994;124:537–546. doi: 10.1083/jcb.124.4.537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Eilers A, Whitfield J, Babij C, Rubin LL, Ham J. Role of the jun kinase pathway in the regulation of c-jun expression and apoptosis in sympathetic neurons. J Neurosci. 1998;18:1713–1724. doi: 10.1523/JNEUROSCI.18-05-01713.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Estus S, Zaks WJ, Freeman RS, Gruda M, Bravo R, Johnson EM., Jr Altered gene expression in neurons during programmed cell death: identification of c-jun as necessary for neuronal apoptosis. J Cell Biol. 1994;127:1717–1727. doi: 10.1083/jcb.127.6.1717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Eves EM, Xiong W, Bellacosa A, Kennedy SG, Tsichlis PN, Rosner MR, Hay N. Akt, a target for phosphatidylinositol 3-kinase, inhibits apoptosis in a differentiating neuronal cell line. Mol Cell Biol. 1998;18:2143–2152. doi: 10.1128/mcb.18.4.2143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Fukuda M, Gotoh Y, Tachibana T, Dell K, Hattori S, Yoneda Y, Nishida E. Induction of neurite outgrowth by MAP kinase in PC12 cells. Oncogene. 1995;20:239–244. [PubMed] [Google Scholar]
- 24.Ham J, Babij C, Whitfield J, Pfarr CM, Lallemand D, Yaniv M, Rubin LL. A c-jun dominant negative mutant protects sympathetic neurons against programmed cell death. Neuron. 1995;14:927–939. doi: 10.1016/0896-6273(95)90331-3. [DOI] [PubMed] [Google Scholar]
- 25.Holgado-Madruga M, Moscatello DK, Emlet DR, Dieterich R, Wong AJ. Grb2-associated binder-1 mediates phosphatidylinositol 3-kinase activation and the promotion of cell survival by nerve growth factor. Proc Natl Acad Sci USA. 1997;94:12419–12424. doi: 10.1073/pnas.94.23.12419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Joneson T, White MA, Wigler MH, Bar-Sagi D. Stimulation of membrane ruffling and MAP kinase activation by distinct effectors of Ras. Science. 1996;271:810–812. doi: 10.1126/science.271.5250.810. [DOI] [PubMed] [Google Scholar]
- 27.Kaplan DR, Miller FD. Signal transduction by the neurotrophin receptors. Curr Opin Cell Biol. 1997;9:213–221. doi: 10.1016/s0955-0674(97)80065-8. [DOI] [PubMed] [Google Scholar]
- 28.Kaplan DR, Hempstead BL, Martin-Zanca D, Chao MV, Parada LF. The trk proto-oncogene product: a signal transducing receptor for the nerve growth factor. Science. 1991a;252:554–558. doi: 10.1126/science.1850549. [DOI] [PubMed] [Google Scholar]
- 29.Kaplan DR, Martin-Zanca D, Parada LF. Tyrosine phosphorylation and tyrosine kinase activity of the trk proto-oncogene product induced by NGF. Nature. 1991b;350:158–160. doi: 10.1038/350158a0. [DOI] [PubMed] [Google Scholar]
- 30.Khosravi-Far R, White MA, Westwick JK, Solski PA, Chrzanowska-Wodnicka M, Van Aelst L, Wigler MH, Der CJ. Oncogenic Ras activation of Raf/mitogen-activated protein kinase-independent pathways is sufficient to cause tumorigenic transformation. Mol Cell Biol. 1996;16:3923–3933. doi: 10.1128/mcb.16.7.3923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Klein R, Jing SQ, Nanduri V, O'Rourke E, Barbacid M. The trk proto-oncogene encodes a receptor for nerve growth factor. Cell. 1991;65:189–197. doi: 10.1016/0092-8674(91)90419-y. [DOI] [PubMed] [Google Scholar]
- 32.Klesse LJ, Parada LF. p21 Ras and phosphatidylinositol-3 kinase are required for survival of wild-type and NF-1 mutant sensory neurons. J Neurosci. 1998;18:10420–10428. doi: 10.1523/JNEUROSCI.18-24-10420.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Knusel B, Rabin SJ, Hefti F, Kaplan DR. Regulated neurotrophin receptor responsiveness during neuronal migration and early differentiation. J Neurosci. 1994;14:1542–1554. doi: 10.1523/JNEUROSCI.14-03-01542.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kouhara H, Hadari YR, Spivak-Kroizman T, Schilling J, Bar-Sagi D, Lax I, Schlessinger J. A lipid-anchored Grb2-binding protein that links FGF-receptor activation to the Ras/MAPK signaling pathway. Cell. 1997;89:693–702. doi: 10.1016/s0092-8674(00)80252-4. [DOI] [PubMed] [Google Scholar]
- 35.Levi-Montalcini R. The nerve growth factor 35 years later. Science. 1987;237:1154–1162. doi: 10.1126/science.3306916. [DOI] [PubMed] [Google Scholar]
- 36.Levi-Montalcini R, Angeletti PU. Essential role for nerve growth factor in the survival and maintenance of dissociated sensory and sympathetic embryonic nerve cells in vitro. Dev Biol. 1963;7:653–657. doi: 10.1016/0012-1606(63)90149-0. [DOI] [PubMed] [Google Scholar]
- 37.Levi-Montalcini R, Brooker B. Destruction of sympathetic ganglia in mammals by an antiserum to the nerve growth factor protein. Proc Natl Acad Sci USA. 1960;46:384–391. doi: 10.1073/pnas.46.3.384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ma Y, Campenot RB, Miller FD. Concentration-dependent regulation of neuronal gene expression by nerve growth factor. J Cell Biol. 1992;117:135–141. doi: 10.1083/jcb.117.1.135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Manthorpe M, Fagnani R, Skaper SD, Varon S. An automated colorimetric assay for neurotrophic factors. Dev Brain Res. 1986;25:191–198. doi: 10.1016/s0006-8993(86)80227-x. [DOI] [PubMed] [Google Scholar]
- 40.Markus A, von Holst A, Rohrer H, Heumann R. NGF-mediated survival depends on p21ras in chick sympathetic neurons from the superior cervical ganglia but not from lumbosacral ganglia. Dev Biol. 1997;191:306–310. doi: 10.1006/dbio.1997.8771. [DOI] [PubMed] [Google Scholar]
- 41.Marshall CJ. Ras effectors. Curr Opin Cell Biol. 1996;8:197–204. doi: 10.1016/s0955-0674(96)80066-4. [DOI] [PubMed] [Google Scholar]
- 42.Martin DP, Schmidt RE, Distefano PS, Lowry OH, Carter JG, Johnson EM. Inhibitors of protein synthesis and mRNA synthesis prevent neuronal cell death caused by NGF deprivation. J Cell Biol. 1988;106:829–844. doi: 10.1083/jcb.106.3.829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Morrison RS, Wenzel HJ, Kinoshita Y, Robbins CA, Donehower LA, Schwartzkroin PA. Loss of the p53 tumor suppressor gene protects neurons from kainate-induced cell death. J Neurosci. 1996;16:1337–1345. doi: 10.1523/JNEUROSCI.16-04-01337.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Nobes CD, Tolkovsky AM. Neutralising anti-p21ras Fabs suppress rat sympathetic neuron survival induced by NGF, LIF, CNTF and cAMP. Eur J Neurosci. 1995;7:344–350. doi: 10.1111/j.1460-9568.1995.tb01069.x. [DOI] [PubMed] [Google Scholar]
- 45.Nobes CD, Reppas JB, Markus A, Tolkovsky AM. Active p21Ras is sufficient for rescue of NGF-dependent rat sympathetic neurons. Neuroscience. 1996;70:1067–1079. doi: 10.1016/0306-4522(95)00420-3. [DOI] [PubMed] [Google Scholar]
- 46.Pang L, Sawada T, Decker SJ, Saltiel AR. Inhibition of MAP kinase kinase blocks the differentiation of PC-12 cells induced by nerve growth factor. J Biol Chem. 1995;270:13585–13588. doi: 10.1074/jbc.270.23.13585. [DOI] [PubMed] [Google Scholar]
- 47.Philpott KL, McCarthy MJ, Klippel A, Rubin LL. Activated phosphatidylinositol 3-kinase and Akt kinase promote survival of superior cervical neurons. J Cell Biol. 1997;139:809–815. doi: 10.1083/jcb.139.3.809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Qian X, Riccio A, Zhang Y, Ginty DD. Identification and characterization of novel substrates of Trk receptors in developing neurons. Neuron. 1998;21:1017–1029. doi: 10.1016/s0896-6273(00)80620-0. [DOI] [PubMed] [Google Scholar]
- 49.Querido E, Marcellus RC, Lai A, Charbonneau R, Teodoro JG, Ketner G, Branton PE. Regulation of p53 levels by the E1B 55-kilodalton protein and E4orf6 in adenovirus-infected cells. J Virol. 1997;71:3788–3798. doi: 10.1128/jvi.71.5.3788-3798.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Quilliam LA, Khosravi-Far R, Huff SY, Der CJ. Guanine nucleotide exchange factors: activators of the Ras superfamily of proteins. Bioessays. 1995;17:395–404. doi: 10.1002/bies.950170507. [DOI] [PubMed] [Google Scholar]
- 51.Rodriguez-Viciana P, Warne PH, Dhand R, Vanhaesebroeck B, Gout I, Fry MJ, Waterfield MD, Downward J. Phosphatidylinositol-3-OH kinase as a direct target of Ras. Nature. 1994;370:527–532. doi: 10.1038/370527a0. [DOI] [PubMed] [Google Scholar]
- 52.Rodriguez-Viciana P, Warne PH, Khwaja A, Marte BM, Pappin D, Das P, Waterfield MD, Ridley A, Downward J. Role of phosphoinositide 3-OH kinase in cell transformation and control of the actin cytoskeleton by Ras. Cell. 1997;89:457–467. doi: 10.1016/s0092-8674(00)80226-3. [DOI] [PubMed] [Google Scholar]
- 53.Seeburg PH, Colby WW, Capon DJ, Goeddel DV, Levinson AD. Biological properties of human c-Ha-ras1 genes mutated at codon 12. Nature. 1984;312:71–75. doi: 10.1038/312071a0. [DOI] [PubMed] [Google Scholar]
- 54.Segal RA, Greenberg ME. Intracellular signaling pathways activated by neurotrophic factors. Annu Rev Neurosci. 1996;19:463–489. doi: 10.1146/annurev.ne.19.030196.002335. [DOI] [PubMed] [Google Scholar]
- 55.Sigal IS, Gibbs JB, D'Alonzo JS, Temeles GL, Wolanski BS, Socher SH, Scolnick EM. Mutant ras-encoded proteins with altered nucleotide binding exert dominant biological effects. Proc Natl Acad Sci USA. 1986;83:952–956. doi: 10.1073/pnas.83.4.952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Slack RS, Belliveau DJ, Rosenberg M, Atwal J, Aloyz R, Lochmuller H, Hagighi A, Lach B, Seth P, Cooper E, Miller FD. Adenovirus-mediated gene transfer of the tumor suppressor p53 induces apoptosis in postmitotic neurons. J Cell Biol. 1996;135:1085–1096. doi: 10.1083/jcb.135.4.1085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Teodoro JG, Branton PE. Regulation of p53-dependent apoptosis, transcriptional repression, and cell transformation by phosphorylation of the 55-kilodalton E1B protein of human adenovirus type 5. J Virol. 1997;71:3620–3627. doi: 10.1128/jvi.71.5.3620-3627.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Virdee K, Tolkovsky AM. Activation of p44 and p42 MAP kinases is not essential for the survival of rat sympathetic neurons. Eur J Neurosci. 1995a;7:2159–2169. doi: 10.1111/j.1460-9568.1995.tb00637.x. [DOI] [PubMed] [Google Scholar]
- 59.Virdee K, Tolkovsky AM. Inhibition of p42 and p44 mitogen-activated protein kinase activity by PD98059 does not suppress nerve growth factor-induced survival of sympathetic neurones. J Neurochem. 1995b;67:1801–1805. doi: 10.1046/j.1471-4159.1996.67051801.x. [DOI] [PubMed] [Google Scholar]
- 60.Vlahos CJ, Matter WF, Hui KY, Brown RF. A specific inhibitor of phosphatidylinositol 3-kinase, 2-(4-morpholinyl)-8-phenyl-4H-1-benzopyran-4–1 (LY 294002). J Biol Chem. 1994;269:5241–5248. [PubMed] [Google Scholar]
- 61.Vogel KS, Brannan CI, Jenkins NA, Copeland NG, Parada LF. Loss of neurofibromin results in neurotrophin-independent survival of embryonic sensory and sympathetic neurons. Cell. 1995;82:733–742. doi: 10.1016/0092-8674(95)90470-0. [DOI] [PubMed] [Google Scholar]
- 62.Vojtek AB, Der CJ. Increasing complexity of the Ras signaling pathway. J Biol Chem. 1998;273:19925–19928. doi: 10.1074/jbc.273.32.19925. [DOI] [PubMed] [Google Scholar]
- 63.Xiang H, Hochman DW, Saya H, Fujiwara T, Schwartzkroin PA, Morrison RS. Evidence for p53-mediated modulation of neuronal viability. J Neurosci. 1996;16:6753–6765. doi: 10.1523/JNEUROSCI.16-21-06753.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Xiang H, Kinoshita Y, Knudson CM, Korsmeyer SJ, Schwartzkroin PA, Morrison RS. Bax involvement in p53-mediated neuronal cell death. J Neurosci. 1998;18:1363–1373. doi: 10.1523/JNEUROSCI.18-04-01363.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Yao RI, Cooper GM. Requirement for phosphatidylinositol-3 kinase in the prevention of apoptosis by nerve growth factor. Science. 1995;275:665–668. doi: 10.1126/science.7701324. [DOI] [PubMed] [Google Scholar]
- 66.Yew PR, Berk AJ. Inhibition of p53 transactivation required for transformation by adenovirus early 1B protein. Nature. 1992;337:82–85. doi: 10.1038/357082a0. [DOI] [PubMed] [Google Scholar]
- 67.Yoon SO, Casaccia-Bonnefil P, Carter B, Chao MV. Competitive signaling between TrkA and p75 nerve growth factor receptors determines cell survival. J Neurosci. 1998;18:3273–3281. doi: 10.1523/JNEUROSCI.18-09-03273.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]