

Abstract
The inner ear can be permanently damaged by overexposure to high-level noise; however, damage can be decreased by previous exposure to moderate level, nontraumatic noise (Canlon et al., 1988). The mechanism of this “protective” effect is unclear, but a role for heat shock proteins has been suggested. The aim of the present study was to directly test protective effects of heat stress in the ear. For physiological experiments, CBA/CaJ mice were exposed to an intense octave band of noise (8–16 kHz) at 100 dB SPL for 2 hr, either with or without previous whole-body heat stress (rectal temperature to 41.5 °C for 15 min). The interval between heat stress and sound exposure varied in different groups from 6 to 96 hr. One week later, inner ear function was assessed in each animal via comparison of compound action potential thresholds to mean values from unexposed controls. Permanent threshold shifts (PTSs) were ∼40 dB in the group sound-exposed without previous heat stress. Heat-stressed animals were protected from acoustic injury: mean PTS in the group with 6 hr heat-stress–trauma interval was reduced to ∼10 dB. This heat stress protection disappeared when the treatment-trauma interval surpassed 24 hr. A parallel set of quantitative PCR experiments measured heat-shock protein mRNA in the cochlea and showed 100- to 200-fold increase over control 30 min after heat treatment, with levels returning to baseline at 6 hr after treatment. Results are consistent with the idea that upregulation of heat shock proteins protects the ear from acoustic injury.
Keywords: sound conditioning, acoustic trauma, cochlea, hearing, heat shock, quantitative PCR
Overexposure to intense sound damages inner ear sensory cells and can lead to permanent threshold shift (PTS) if exposure is sufficiently intense or prolonged. “Conditioning” the ear by pre-exposure to moderate level, nontraumatic sound can dramatically reduce PTS from a subsequent traumatic exposure (Canlon et al., 1988). Such conditioning-related protective effects have been demonstrated in several mammalian species, including humans (Miyakita et al., 1992), guinea pigs (Canlon et al., 1988), rabbits (Franklin et al., 1991), chinchillas (Subramaniam et al., 1996), gerbils (Ryan et al., 1994), and rats (Pukkila et al., 1997). Most studies have used conditioning exposures of many days duration; however, protective effects have been reported for conditioners as short as a few hours (Pukkila et al., 1997). Most studies have interposed an interval of several days between conditioner and traumatic exposure; however, the build-up and decay of these protective effects has not been systematically investigated.
The mechanism(s) whereby previous sound exposure can reduce subsequent acoustic injury are poorly understood. The effects of the sound conditioning per se on the structure and/or function of the cochlea have been evaluated in only a limited manner. One physiological study found significant enhancement of distortion product otoacoustic emissions (DPOAEs) in the sound-conditioned ear (Kujawa and Liberman 1996), suggesting that conditioning modifies the function of the electromotile outer hair cells (OHCs). An ultrastructural study of the conditioned ear reported elaboration of membranous tubules/vesicles in the basal pole of OHCs (Canlon et al., 1991, 1993); however, no other structures within the cochlear duct have been carefully examined.
In the search for mechanisms, it is useful to consider whether the protective effect of sound conditioning is a special case of the more generalized phenomenon whereby exposure to sublethal stressors protect against subsequent exposure to more severe conditions. The protective effects of stress-induced gene expression have been extensively studied in a number of systems from yeast to mammals, with a variety of stressors including heat shock, ischemia, and physical restraint (Lindquist and Craig, 1988; Welch 1992). Such protective effects involve multiple changes in gene expression. One pathway that has received considerable attention is the upregulation of inducible heat shock proteins (HSPs). HSPs are known to be expressed in the cochlea, and immunohistochemical studies suggest they are upregulated in response to acoustic overexposure and ischemia, as well as heat stress (Myers et al., 1992; Thompson and Neely, 1992; Lim et al., 1993;Akizuki et al., 1995). In the eye, whole-animal heat stress protects from light damage with elevation of inducible HSPs (Barbe et al., 1988;Tytell et al., 1993). Although the involvement of HSPs in the sound-conditioning of the cochlea has been proposed (e.g., Lim et al., 1993), the protective effects of heat stress on the ear have never been directly evaluated. Thus, the aims of this study were to assess the effects of whole-animal heat stress on vulnerability to acoustic injury and cochlear HSP production and thereby begin to address the idea that manipulation of stress-induced gene expression may underlie a variety of cochlear protective effects.
MATERIALS AND METHODS
Experimental groups and design
Experimental animals were male mice of the CBA/CaJ strain (The Jackson Laboratory, Bar Harbor, ME), aged 10–12 weeks (23–29 gm). All procedures were approved by the animal care committee of the Massachusetts Eye and Ear Infirmary.
Electrophysiological test groups. All animals in these groups were tested physiologically at 10–12 weeks of age in a terminal procedure to evaluate cochlear function. Before the final test, animals in different groups were subjected to different experimental manipulations, as schematized in Figure1. Each experimental manipulation is described in more detail below. The five groups were as follows: (1) aControl group to establish normal cochlear thresholds for the 10- to 12-week-old CBA/CaJ mouse; (2) a Trauma Onlygroup, exposed to the traumatic noise-band (without heat stress or anesthesia) 1 week before final test, to demonstrate the normal vulnerability of the ear; (3) five Anesthesia+Heat pre Trauma groups, anesthetized and heat-stressed (for 15 min) at varying time intervals (6, 12, 24, 48, or 96 hr) before the traumatic exposure to assess whether previous heat stress protects the ear; (4) four Anesthesia Only pre Trauma groups, anesthetized (without heat stress) at varying times (6, 12, 24, or 48 hr) before the acoustic trauma to differentiate effects of heat stress from effects of anesthetization; and (5) an Anesthesia+ Heat Only group, anesthetized and heat-stressed (without acoustic trauma) 6 hr before final test to evaluate whether the heat stress has compromised basic cochlear function at the time animals would have been traumatized. Numbers of animals in each experimental group are shown in Table1.
Fig. 1.

Schematic illustrating the timing of experimental manipulations for different groups.
Table 1.
Experimental groups

Molecular biology test groups. An additional six animals were tested at 10–12 weeks of age in a terminal procedure to evaluate cochlear mRNA levels. Before bilateral cochlear extraction, two groups (of two animals each) were anesthetized and heat-stressed before cochlear extraction, and a third group (also of two animals) served as a control (anesthesia without heat shock). The time interval between heat stress and extraction was 30 min for one group and 6 hr for the other. Two independent molecular analyses (described below) were run, each using one animal from each of the three groups.
Anesthesia and heat stress
Animals were heat-stressed while anesthetized with ketamine (60 mg/kg, i.p.) and xylazine (rompun) (6 mg/kg, i.p.). Immediately after injection, animals were placed in a pan on a heating pad in a warm room (ambient temperature ∼34 °C) to prevent hypothermia during the onset of anesthesia. Once surgical levels of anesthesia were reached (∼10 min later), a rectal thermometer was inserted; initial temperature was 37°C (± 0.5°C), as shown in Figure2. Each animal was then placed in an aluminum “boat” floating in a hot-water bath maintained at 46.5°C. To avoid direct thermal effects to the ear, the animal's head was placed on a gauze pad, left ear down, and only right ear function was ultimately measured. The rectal temperature, continuously monitored with a digital thermometer, rose at an average rate of 0.6°C/min, and within ∼8 min, reached 41.5°C at which it was maintained for 15 min (Fig. 2A). After the appropriate elapsed time, the animal was removed from the heat and placed in a small box within the warm room to fully recover from the anesthesia (∼30 min) before being returned to the animal facility when fully awake. As shown in Figure 2C, heat-stressed animals, on average, recovered from the anesthesia ∼60 min after initial injection.
Fig. 2.
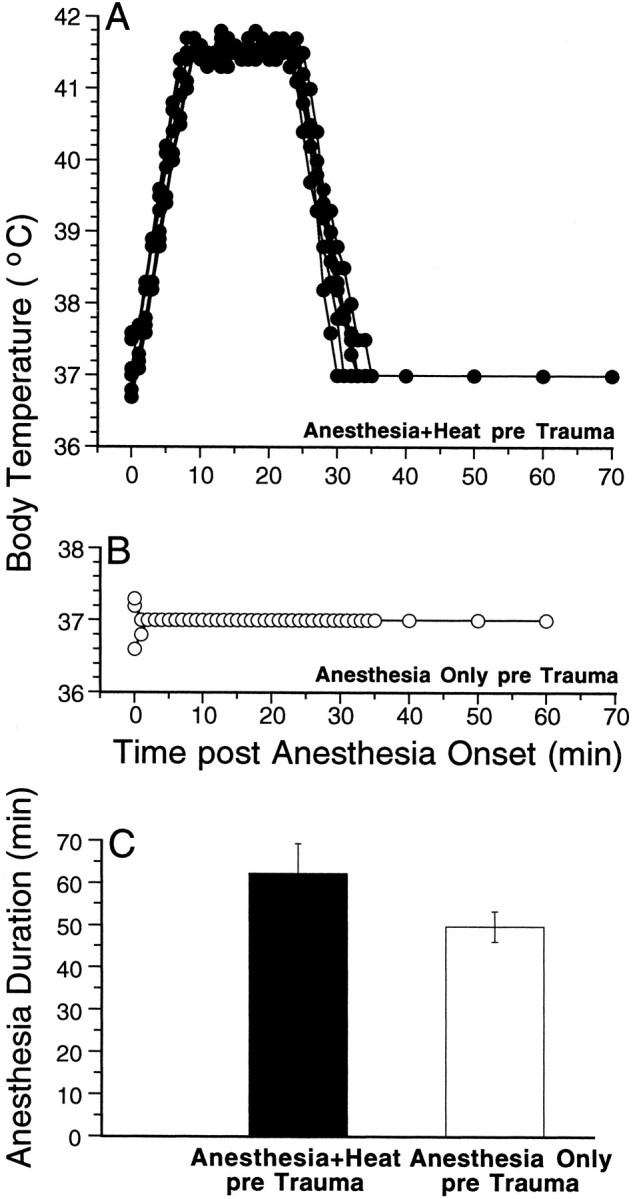
Temperature records during anesthesia are shown for one group of Anesthesia+Heat pre Trauma animals (A) and for the Anesthesia Only pre Trauma group (B); the mean ± SEM durations of anesthetization for the same two groups of animals are shown in C. For each animal, a rectal thermometer was inserted as soon as possible after injection of anesthetics and was removed only when the anesthesia began to wear off and the animal began to move. During heat stress, the temperature was read every minute under computer control (accuracy better than 0.1°C); after anesthesia plus heat or during the anesthesia only protocol, temperature was read by eye off a meter (to the nearest 0.25°C). TheAnesthesia+Heat pre Trauma group shown inA was the one with the shortest treatment–trauma interval (6 hr).
As a control for the effects of anesthetization, a separate group of animals was anesthetized without being heat-stressed Anesthesia Only pre Trauma. For this group, care was also taken that the rectal temperature was maintained near 37°C during the entire anesthetization period. Without such special precautions, the rectal temperature of an anesthetized mouse can quickly fall to 32°C at typical ambient temperatures. Temperature records for all these animals are shown in Figure 2B. As shown in Figure2C, without heat stress, animals recovered more quickly from the anesthesia (50 vs 60 min after injection).
Acoustic overexposure
Animals were exposed, unanesthetized and unrestrained, within cages (one per cage division) suspended inside a small reverberant sound exposure box (Liberman and Gao, 1995). The stimulus for the acoustic overexposure was an octave band of noise (8.0–16.0 kHz) presented at 100 dB SPL for 2 hr. The stimulus was generated by a custom-made white noise generator, filtered [Brick Wall (San Diego, CA) filter with a 60 dB/octave slope], amplified [Crown International (Elkhart, IN) power amplifier], and delivered [JBL Scientific (San Luis Obispo) compression driver] through an exponential horn fitted securely to a hole in the top of reverberant box. Sound exposure levels were measured at four positions within each cage using a ¼ inch Brüel and Kjaer (Atlanta, GA) condenser microphone; sound pressure was found to vary by <0.5 dB across these measurement positions.
Testing of compound action potential and DPOAEs
For the final test, animals were anesthetized with xylazine (rompun) (8 mg/kg, i.p.), followed by urethane (1.2 gm/kg, i.p.). Surgical levels of anesthesia were maintained with booster injection of rompun and urethane ( original dose), when necessary. Surgical preparation for the terminal experiment involved insertion of a tracheotomy tube and severing the right ear canal near the tympanic ring. DPOAEs were measured at this point. Then, the bulla was exposed on right side by removing the overlying muscle layers and was opened with a #11 scalpel blade for recording of compound action potentials (CAPs).
CAP was recorded from right cochlea of each case via fine silver wire on the round window referred to the tongue. The response was amplified (10,000×), filtered (100 Hz to 3 kHz), and averaged with and analog-to-digital board in a LabVIEW-driven data acquisition system. CAP thresholds were measured under computer control in response to 5 msec tone pips (0.5 msec rise–fall with a cos2 onset envelope, delivered at 10/sec). At each SPL, 32 responses were averaged (with stimulus polarity flipped on half of the presentations to remove microphonic potentials). Thresholds were defined as the sound pressure required to produce a peak-to-peak neural response of 3 μV.
The DPOAEs were measured using an ER-10C (Etymotics Research) acoustic system consisting of two sound sources and one microphone. The sensitivity of the microphone was measured by coupling a calibrated Brüel and Kjaer condenser microphone to the output port of the ER-10C system. Stimuli consisted of two primary tones (f1/f2 = 1:2), presented with f2 level always 10 dB < f1 level. Stimuli were generated digitally, but attenuation was provided with external analog attenuators. The ear canal sound pressure was filtered (high-pass at 1000 Hz), amplified, and digitized by a digital-to-analog board. A fast Fourier transform (FFT) was computed, and the sound pressures at f1, f2, and 2f1-f2 were extracted after spectral averaging from serial waveform traces. The noise floor also was measured (defined as the average of six points in the FFT on either side of the 2f1-f2frequency) and ranged between −20 and 5 dB SPL, depending on frequency.
Assessment of histopathology
For selected animals in each group, inner ears were harvested for histopathological analysis. After the final test, cochleas were dissected out, and tissue was fixed by intralabyrinthine perfusion of a buffered solution of glutaraldehyde and paraformaldehyde. Ears were post-fixed overnight (at 4°C) in the same solution, and then rinsed and osmicated for 1 hr (1% OsO4), dehydrated in graded ethanols, and infiltrated with epoxy resins. After polymerization, cochlear bone was removed with dental burrs, and the plasticized cochlear duct was dissected with razor blades into ∼12 pieces. Each piece was re-embedded in plastic (for easier handling), thinned with sanding disks, and mounted on microscope slides. In the final material, the entire organ of Corti could be viewed in the light microscope as a plastic-embedded surface preparation. Cochlear lengths were measured with a drawing tube, and the hair cells were counted with high-power Nomarski optics.
Quantitative PCR
For mRNA preparation, mice were anesthetized with ketamine–xylazine and decapitated. Both cochleas were dissected, cleaned of surrounding tissue, and placed on dry ice. Total RNA was isolated using Trizol Reagent (Life Technologies, Gaithersburg, MD). After the precipitated RNA was washed with 75% EtOH, it was resuspended in 25 μl of sterile RNase free water and quantified on a spectrophotometer. To remove residual genomic DNA, 1 μg of RNA was treated with 1 μl (1 U) of RNase free DNase I (Life Technologies) plus 2 μl of 10× reaction buffer, diluted to 20 μl of total volume. After 20 min at 21°C, DNase was inactivated by 1 μl (25 mm) of EDTA at 65°C for 10 min. To test for genomic DNA removal, 1 μl (∼ 50 ng) of DNased solution was added to 17 μl of PCR SuperMix (Life Technologies) and 1 μl each of mouse β-tubulin primers at 100 ng/μl. The nonreverse-transcribed RNA was subject to PCR as follows. Samples were kept at 94°C for 2 min and then subjected to thermocycling (40 cycles of 30 sec at 94°C, 1 min at 58°C, 1 min at 72°C, with a final extension at 72°C for 5 min). β-Tubulin primers were designed to amplify the 476–650 bp region (GenBank accession number X04663: 5′TTCAGCTGACCCACTCACTG3′ and 5′TGA- TGGACAGACAGGGTGGCAT3′).
Once the absence of genomic DNA was confirmed, cDNA was generated from 10 μl (500 ng) of total RNA using the TaqMan RT Reagents kit (Perkin-Elmer, Emeryville, CA). The RNA template was mixed with 10 μl of 10× buffer, 22 μl of MgCl, 20 μl of dNTPs, 5 μl of random hexamer primer, 2 μl of RNase inhibitor, 2.5 μl of MultiScribe reverse transcriptase, and 28.5 μl RNase free water for a total volume of 100 μl. The reactions were heated to 25°C for 10 min, then 48°C for 30 min, and finally [for reverse transcription (RT) enzyme inactivation] to 95°C for 5 min.
Primers for quantitative PCR were designed for mouse inducible HSP70 mRNA and 18S ribosomal RNA (rRNA) (as the endogenous control). The HSP70 primer was designed to amplify the 1225- 395 bp region of mouse HSP70 mRNA (GenBank accession number M12572: 5′AGGCCAGGGCTGGTATTACT3′ and 5′ AATGACCCGAGTTCAGGATG3′). The 18S primer was designed to amplify the 899–999 bp region of mouse 18S rRNA (GenBank accession numberX00686: 5′TTCGGAACTGAGGCCATGATT3′ and 5′ TTTCGCTCTGGTCCGTCTTG3′). To test the primers and the RT reaction, a preliminary (nonquantitative) PCR was performed on the cDNA; samples were kept at 94°C for 2 min and then thermocycled (40 cycles of 30 sec at 94°C, 1 min at 58°C, 1 min at 72°C, with a final extension at 72°C for 5 min). PCR bands, resolved on a 1.5% agarose gel, were confirmed to be present at the correct molecular weight compared with a Low Mass DNA Marker (Life Technologies).
Using the ABI Prism 7700 sequence detection system (Applied Biosystems, Foster City, CA), a quantitative SYBR Green assay was performed to measure relative amounts of HSP70 cDNA in the samples. One microliter of sample was combined with 2.5 μl of 10× buffer, 2 μl of 25 mm MgCl, 3 μl of dNTPs, 0.25 μl of AmpErase UNG, 0.125 μl of AmpliTaq Gold polymerase, 1 μl each of specific primers (for HSP70 or 18S), and 14.125 μl of water. All samples were run in triplicate in a 96-well microtiter plate. Thermocycle conditions were as follows: 1 cycle at 50°C 2 min, 1 cycle at 95°C 10 min, and 40 cycles 15 sec at 95°C and 1 min 60°C.
In the SYBR Green assay, fluorescence signal is measured after each thermocycle as it rises in proportion to the double-stranded DNA generated by PCR (Livak et al., 1995; Heid et al., 1996). To normalize for possible differences in sample volume, the signal arising from wells containing the HSP70 primers was compared with those with the primer for 18S rRNA. To compute the relative quantities of mRNA, we used the comparative ΔΔCt method [ABI Prism 7700 Sequence Detection System, User Bulletin #2 (1997), pp 23–27].
RESULTS
Effects of heat stress on vulnerability to acoustic injury
Threshold responses and histopathology
Overexposure to loud sound can damage sensory cells and cause permanent elevation of CAP thresholds (for review, see Saunders et al., 1985). The 2 hr exposure to octave-band noise at 100 dB SPL used in the present study caused frequency-dependent threshold elevations. As shown in Figure 3A, this 8–16 kHz noise band caused chronic threshold elevations in all nonheat-stressed (Trauma Only) mice examined, at all test frequencies above 8 kHz. The largest threshold elevations were seen at 17.5 kHz where they averaged ∼40 dB. The pattern and degree of PTSs were remarkably similar across all mice in this experimental group; for all test frequencies below 30 kHz, thresholds for all 10 Trauma Onlyanimals fell within a 20 dB range. The increased variance for test frequencies above 30 kHz is seen in both control and noise-exposed animals; it may arise, in part, from subtle cochlear damage from the surgery required to expose it.
Fig. 3.

The noise band used in the present study produces a reproducible pattern of permanent threshold shift in mice exposed without previous heat stress (A); hair cell loss is restricted to the extreme base (B). CAP threshold curves are shown in A for 10 Trauma Only animals compared with the mean CAP threshold curve for 10 control animals not exposed to the traumatic noise band.B shows the cytocochleogram for one of theTrauma Only cases, i.e., a plot of the percentage of hair cells remaining as a function of cochlear location. Cochlear location is translated into frequency using published data on the map for mouse (Ehret, 1983).
Hair cell loss was minimal in the Trauma Only animals. Figure 3B shows the loss pattern along the cochlear spiral in one typical case, aligning cochlear place to test frequency according to a cochlear map for mouse (Ehret, 1983). Significant hair cell loss in this case was restricted to the extreme basal regions of the cochlea where best frequencies are >50 kHz, i.e., above the highest frequency routinely tested. Throughout the cochlear region in which PTS was maximal, hair cell loss was insignificant. The absence of sensory cell loss is consistent with other studies in which noise-induced PTS on the order of 40 dB is associated with damage to the hair cell stereocilia rather than with the destruction of the sensory cells (Liberman and Dodds, 1984). Indeed, careful examination of the sensory hairs in these trauma only mice revealed clear-cut disarray and loss of stereocilia on inner hair cells (IHCs) and OHCs in a restricted cochlear region consistent with the frequency region showing maximal PTS (data not shown).
Pretreatment with whole-body heat stress significantly reduced PTS from the subsequent acoustic overexposure when the interval between heat stress and trauma was less than 24 hr. Mean PTSs for animals heat-stressed before the acoustic overexposure (Anesthesia+Heat pre Trauma) are compared in Figure4A with data from animals exposed without previous heat stress (Trauma Only). As described above, the mean PTS in the Trauma Only animals was close to 40 dB; in contrast, the mean PTS in animals heat stressed 6 hr before acoustic overexposure was only 13 dB, a protective effect of ∼25 dB. SEs have been left off Figure 4Afor clarity; however, they are included in Figure 4Bin which the maximum PTS (i.e., PTS at 17.5 kHz) is replotted for each of the groups shown in A. A statistical analysis (Fig.4B) showed highly significant differences (p ≤ 0.005 by ANOVA) between the Trauma Only group and the Anesthesia+Heat pre Trauma groups with the two shortest treatment–trauma interval groups, i.e., 6 hr (F = 31.55; p ≪ 0.0001 for the group effect; F = 2.932; p = 0.005 for the group–frequency interaction) and 12 hr (F = 11.32;p = 0.005 for the group effect; F = 2.351; p = 0.023 for the group–frequency interaction). Although the small protective effect suggested by the mean data in the 24 and 48 hr interval groups was not statistically significant (Fig.4B), the trend of the means suggests that protective effects of the heat stress may not have completely disappeared until 96 hr after the treatment. In a separate group of animals, the effects of a shorter heat stress, i.e., 5 rather than 15 min, were assessed for treatment–trauma intervals of 6, 12, 24, and 48 hr; although mean PTS for the 6 hr treatment–trauma interval group was 12 dB lower than for the Trauma Only group, for none of the 5 min heat stress groups was the protective effect statistically significant.
Fig. 4.

Heat stress significantly reduces PTS from a subsequent acoustic overexposure. A compares mean PTS for five Anesthesia+Heat pre Trauma groups with theTrauma Only group. All thresholds are normalized to the mean control thresholds seen in nontraumatized mice (Fig. 3).B examines the effect of treatment–trauma interval by replotting the maximum PTS (i.e., the value at 17.5 kHz) from each graph in A, arranged with increasing time interval between heat stress and trauma. SE bars are shown; **p < 0.005 indicates significant group difference between a given experimental group and the Trauma Onlygroup (ANOVA). Tests were performed as a series of two-way ANOVAs (factors are frequency and group), each of which compared all PTS values obtained at test frequencies between 7 and 40 kHz from a particular experimental group with comparable data from the control group.
Because the heat stress treatment in our experimental design included anesthetization, it was necessary to control for the possible effects of the anesthetization per se on susceptibility to subsequent acoustic injury. As illustrated in Figure5A, any protective effects of previous anesthetization alone were not statistically significant. That is to say, none of the four Anesthesia Only pre Traumagroups (with differing treatment–trauma intervals matching those tested in the heat stress groups) showed less PTS than the Trauma Only group.
Fig. 5.

Protective effects of heat stress are not caused by the associated anesthesia. A, Mean PTS values are compared for the four Anesthesia Only pre Trauma groups versus the Trauma Only group. Data show that anesthesia without heat stress has no protective effect at any of the four treatment–trauma intervals investigated (6, 12, 24, or 48 hr as shown in the key). B replots maximum PTS (value at 17.5 kHz) from each group, arranged according to treatment–trauma interval. Error bars show SEM.
Suprathreshold responses
Data in Figures 3-5 deal only with cochlear responses to stimuli at near-threshold sound pressure levels. In the present study, suprathreshold responses showed protective effects similar to those seen in the threshold responses described above. Mean amplitude versus level curves for CAP are shown in Figure6 for two Anesthesia+Heat pre Trauma groups compared with control and Trauma Onlyanimals. Data at six different test tone frequencies are illustrated inA–F, and the relation of those six test frequencies to the overall pattern of threshold shift in the appropriate experimental groups is shown in G. Consider first the data for the two highest test tone frequencies: 20.0 and 28.3 kHz (E andF, respectively). For both of these frequencies, positioned on the high-frequency edge of the damaged region (G), the suprathreshold responses are predicted by the near-threshold results: i.e., (1) the Anesthesia+Heat pre Trauma group with 6 hr treatment–trauma interval (filled circles) shows similar response magnitudes to the control group, consistent with almost complete protection from acoustic overexposure; and (2) theAnesthesia+Heat pre Trauma group with the 96 hr interval shows similar response magnitudes to the Trauma Only group, consistent with complete disappearance of protective effects. Indeed, damage appears, if anything, more severe in the 96 hr interval heat-stressed group; the significance of this trend is unclear. Similar results were obtained with the growth functions for DPOAEs (data not shown).
Fig. 6.
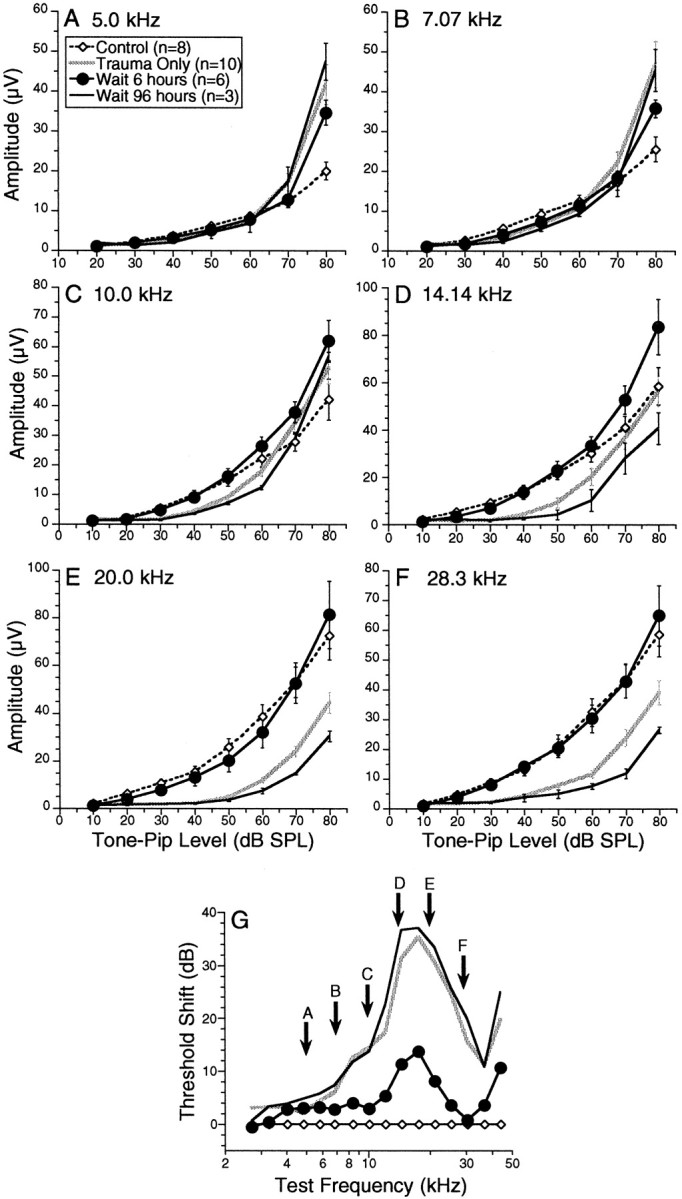
Average CAP amplitude (±SEM) versus level curves at six test frequencies for two of the Anesthesia+Heat pre Trauma groups (6 and 96 hr treatment–trauma intervals) compared with control and Trauma Only groups.A–F, Each compares, for a different tone pip frequency, the average value for the peak–peak CAP for the four groups indicated, as a function of tone pip intensity. The test frequency is noted at thetop of each panel (A–F) and also in G in which the mean PTS for each of these four groups are replotted from Figure 4A.
The data for the lower test tones frequencies (5.0, 7.07, and 10.0 kHz in A, B, and C, respectively) show supranormal response amplitudes at the highest stimulus levels. For these frequencies, positioned on the low-frequency side of the damaged region (G), the amplitude versus level curves of all three traumatized groups (i.e., Trauma Only and bothAnesthesia+Heat pre Trauma groups) cross the control group curves: i.e., the traumatic exposure has reduced CAP amplitudes at low stimulus levels but enhanced amplitudes at high stimulus levels. Such supranormal responses may arise from the phenomenon of hypersensitization (Liberman and Kiang, 1978); after cochlear insult, auditory nerve fibers often show decreased sensitivity at the characteristic frequency (CF) coupled with increased sensitivity for frequencies below CF (on the low-frequency “tail” of the tuning curve). Thus, high CF fibers in these traumatized ears may be more responsive to low-frequency tones, contributing to an enhanced CAP as the intensity of low-frequency test tones is raised. Consistent with this explanation, supranormal response amplitudes were not seen in the DPOAE (data not shown). Because DPOAEs are generated mainly near the CF region for the primary tones (Siegel et al., 1982), hypersensitization of basilar membrane response at frequencies well below CF should not add to the overall DPOAE amplitude.
Effects of heat stress on cochlear function before overexposure
In interpreting the effects of heat stress on vulnerability of the ear to acoustic injury, it is important to consider the effects of the heat stress per se on cochlear condition. Manipulations, which themselves elevate cochlear threshold while they protect the ear from acoustic injury, must be distinguished from other manipulations that protect the ear without also compromising basic auditory function. For example, if heat stress decreased middle ear transmission for 6–12 hr, it would produce a dramatic reduction in acoustic injury from an overexposure delivered within that time window.
To assess the direct effects of heat stress on cochlear function, cochlear thresholds were measured in a group of animals 6 hr after heat stress (Fig.7A,B). Because animals were anesthetized during heat stress, this group is referred to as Anesthesia+Heat pre Trauma. Cochlear responses in these heat-stressed animals (Fig. 7) were indistinguishable from those seen in control mice. Cochlear function (Fig. 7) was assessed in two ways for every animal; A shows CAP data, which provide a sensitive functional measure of the entire auditory periphery from sound conduction through synaptic transmission. An alternative measure of cochlear functioning is provided by DPOAEs, as shown in B, which assay the functional state of the middle ear and the contribution of the OHCs to cochlear mechanical amplification. By defining two different iso-response contours (5 and 25 dB SPL), we assess sound transmission and OHC function at both low and high sound levels. Heat stress per se did not affect any of these measures of basic cochlear function.
Fig. 7.
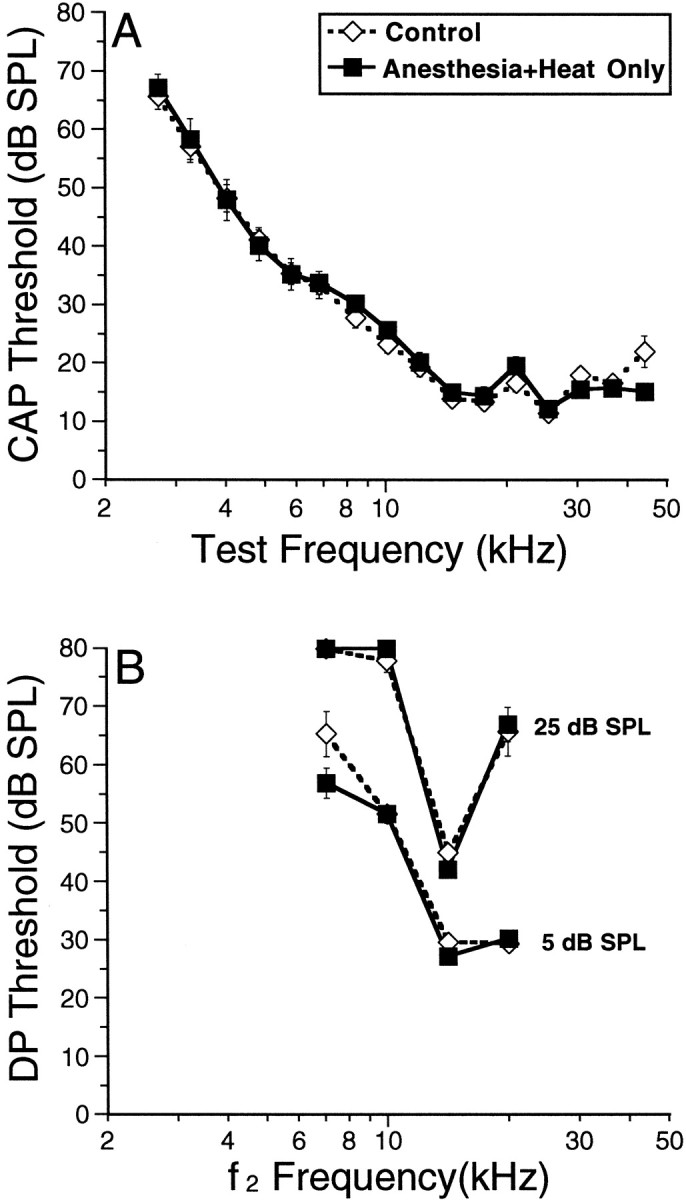
Heat stress does not affect basic cochlear function when measured at the time animals would have been exposed to traumatic sound. Average iso-response contours are shown for CAP (A) and DPOAE (B) for control versus Anesthesia+Heat Only. The iso-response amplitude defined as “threshold” for the CAP was 3 μV (peak-to-peak). Two iso-response values were used for the DPOAE: 5 and 15 dB SPL (as indicated).
Effects of heat stress on cochlear mRNA
A quantitative RT-PCR approach (Livak et al., 1995; Heid et al., 1996) was used to measure the changes in cochlear mRNA expression of the major inducible heat shock protein HSP70. HSP70 mRNA levels were assayed in the control group and in two groups of heat-stressed animals, one in which cochleas were extracted 30 min after heat stress and a second at 6 hr. As shown in Figure8A, the quantitative PCR approach assesses the growth of PCR reaction product after each thermocycle by the use of fluorescent markers. The greater the amount of a particular cDNA in the sample, the earlier the cycle number at which the fluorescent signal begins to rise rapidly out of the noise. To guarantee that overall RNA levels are comparable in each of the starting samples, results with probes for HSP70 were compared in every run with results with probes for 18S rRNA, which should be unaffected in expression level by the heat stress manipulation. Indeed, the quantitative PCR results for 18S rRNA were almost identical across experimental groups; in each case, the PCR signal rises steeply at approximately thermocycle 8.5 (Fig. 8A). This reproducibility of the 18S data allows a direct comparison of the HSP70 results in the three experimental groups.
Fig. 8.
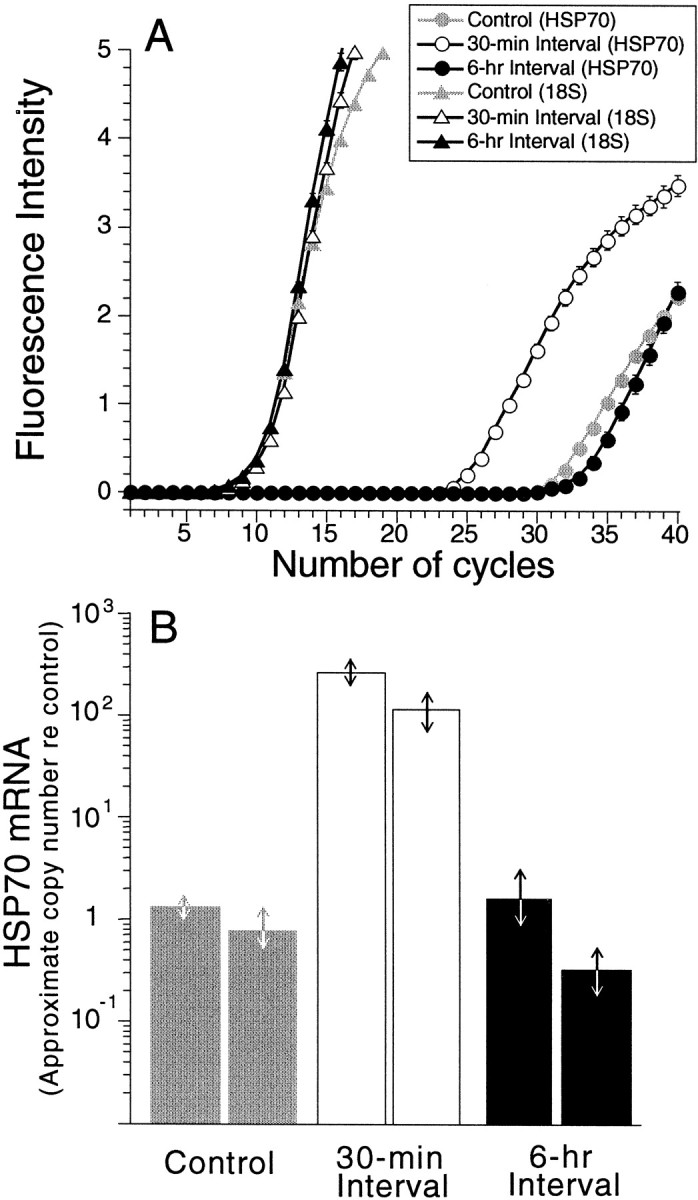
Quantitative PCR results on expression levels of HSP70 mRNA and 18S ribosomal RNA in three experimental groups demonstrate dramatic upregulation of HSP70 after heat stress.A replots raw output from the ABI Prism 7700 sequence detection system, i.e., the fluorescent signal intensity (proportional to amount of PCR reaction product) as a function of thermocycle number. Fluorescence intensity (ΔRn) is calculated by using the equation ΔRn = (Rn+) − (Rn−), whereRn+ equals the ratio of emission intensity of SYBR Green and emission intensity of passive reference during a reaction, and Rn− equals the ratio during cycles 1–7. This run was based on one animal (both cochleas) from each of the three experimental groups (control, heat shock wait 30 min, and heat shock wait 6 hr). Each point is the average ± SEM of three samples independently assayed.B extracts the approximate increase in HSP70 mRNA production re the control value for each of the six animals tested. Arrows indicate the range of results obtained across all samples assayed (3 or 4) from each animal. See Materials and Methods for further details.
This comparison suggests that HSP70 mRNA levels are increased ∼100-fold by 30 min after heat stress; the control and 30 min curves are shifted by 6.7 cycles, and, assuming a doubling of PCR product on each cycle, the approximate increase in mRNA levels is a factor of 26.7 (equal to 104). The data in Figure8A further suggest that HSP70 mRNA levels return toward baseline by 6 hr after heat stress. This spike of HSP70 mRNA production was confirmed in an independent set of PCR runs using a second set of cochleas for each of the three groups. Data from both sets of runs are summarized in Figure 8B in which the changes in HSP70 mRNA levels have been extracted from the raw data.
DISCUSSION
Functionally important structural changes in acoustic injury
Before considering mechanisms by which heat stress might reduce noise-induced PTS, it is important to consider the functionally important structural changes underlying PTS in the absence of heat stress. Threshold elevations seen immediately after an acoustic overexposure include both reversible and irreversible components (Miller et al., 1963). The reversible or temporary threshold shifts (TTSs) recover over a few days with an exponential time course (Miller et al., 1963), leaving a PTS. In the present study, we allowed a 1 week postexposure recovery so that thresholds would reflect PTSs.
Acoustic overexposure can lead to permanent damage in virtually all cochlear cell types if exposure level is high enough and/or duration is long enough (Saunders et al., 1985). However, the most vulnerable elements are hair cells. Although both IHCs and OHCs are often destroyed by acoustic overexposure, significant PTS (>40 dB) can be seen when all hair cells remain (Liberman and Dodds, 1984). In such cases, damage is found to hair cell stereocilia (Robertson, 1982;Engstrom et al., 1983; Liberman and Dodds, 1984), and the frequency pattern and magnitude of PTS is well explained by the cochlear pattern and degree of stereocilia damage (Liberman and Dodds, 1984). Given that stereocilia contain the mechanically sensitive transduction channels, it is not surprising that their loss or damage can lead to significant (even profound) PTS.
Although comprehensive histological analysis of the inner ear was beyond the scope of the present study, we analyzed hair cell loss patterns in a number of Trauma Only ears (Fig.3B). The only significant cell loss was seen at the cochlear base where best frequencies are >50 kHz. Although hair cell loss cannot account for PTS spanning cochlear frequencies from 7–40 kHz in the Trauma Only group, stereocilia disarray was seen in appropriate cochlear regions. Thus, the heat stress treatment, which decreased noise-induced PTS, is decreasing stereocilia damage rather than preventing hair cell degeneration.
Mechanism of heat stress protection
In this study, 15 min heat stress transiently increased mRNA levels of HSP70 in the cochlea (Fig. 8) and, in a separate group of animals, significantly decreased PTS from a subsequent acoustic overexposure (Fig. 4). Appropriate control groups were studied to demonstrate that this protective effect was caused by the heat per se and not the associated anesthetization (Fig. 5). Of course, care had to be taken to control body temperature during anesthetization (Fig. 2), because decreasing temperature during acoustic overexposure can decrease acoustic injury (Drescher, 1976). The demonstration of normal threshold and suprathreshold CAP and DPOAE responses, even as soon as 6 hr after heat stress (Fig. 7), shows that this treatment does not alter sound transmission through the middle ear, nor basic cochlear mechanics, transduction, or synaptic transmission. Rather, the protective effect must involve alterations in the initial resistance of the inner ear or in the effectiveness of its recovery–repair processes.
Our experimental design included a variety of treatment–trauma intervals to study the growth and decay of HSP70 gene expression in the cochlea as well as the accompanying heat stress-mediated protection. A dramatic increase in mRNA synthesis was seen by 30 min after heat stress and had decayed significantly 6 hr later. Previous nonquantitative studies have also reported upregulation of cochlear HSP70 after heat stress or noise exposure in rat (Lim et al., 1993) and guinea pig (Thompson and Neely, 1992; Akizuki et al., 1995). The cochlear protective effects observed here were maximal at the shortest interval tested (6 hr), were no longer statistically significant at 24 hr, and had disappeared completely by 96 hr (Fig. 4). These time courses are slightly more rapid than those reported in analogous studies of heat stress and retinal damage. In the eye, heat stress reduced light damage in vivo (Barbe et al., 1988; Tytell et al., 1993); mRNA for HSP70 peaked 6 hr after heat, and an 18 hr interval between heat stress and light exposure afforded maximum protection to retinal cells. Nevertheless, taken together, the physiological and molecular data are consistent with the hypothesis that HSPs mediate the protective effects seen in the present study. However, as discussed below, although the rapid rise in cochlear HSP mRNA is a necessary condition for the hypothesis, it is insufficient to prove it.
The role of HSPs in the cochlea is poorly understood; thus, it is difficult to provide a compelling hypothesis for mechanisms whereby HSPs might protect the ear from acoustic injury. Nevertheless, recent work suggests a possible line of investigation. A constitutive form of HSP, HSP27, has recently been reported in the cuticular plate and lateral wall of the rat cochlea (Leonova et al., 1998). This HSP is interesting in the context of acoustic injury. Although its role as a molecular chaperone is similar to other HSPs (Jakob et al., 1993), it associates specifically with F-actin filaments and controls their polymerization and depolymerization (Lavoie et al., 1993, 1995). F-Actin is particularly relevant to hearing and acoustic injury, because it is a major protein of stereocilia and the cuticular plate into which they insert (Raphael et al., 1994). As described above, stereocilia damage is the functionally important structural change underlying the type of PTS studied here. Ultrastructural studies have shown that noise-induced stereocilia damage is associated with degeneration–depolymerization of the rootlet, which anchors stereocilia into the cuticular plate (Liberman, 1987). Thus, heat stress-induced stabilization of stereocilia rootlets could underlie the protective effects seen. Of course, changes in gene expression induced by heat stress in particular, and stress in general, are not restricted to changes in HSPs. It is also possible, for example, that the heat stress leads to upregulation of enzymes, such as superoxide dismutase, or others involved in the control of cellular redox status, given recent studies of protection from acoustic injury via free-radical scavengers (Jacono et al., 1998; Sha et al., 1998).
Relationship between sound conditioning and heat stress protection
The phenomenon of sound “conditioning” (Canlon, 1996) in which pre-exposure to moderate level nontraumatic sound reduces PTS from subsequent traumatic exposures may be an ear-specific example of the more general observation that pre-exposure to sublethal insults affords significant protection from subsequent, more potent challenges. Challenges for which such protective effects are demonstrable also include hypoxia, ischemia, heat stress, and chemical toxins. In addition to such direct cellular insults, psychological stressors, such as physical restraint, can also induce changes in gene expression in the same “protective” pathways induced by the direct cellular challenges (Ghoshal et al., 1998). These psychological effects on gene expression are mediated via changes in circulating glucocorticoid levels and their actions on glucocorticoid receptors. Direct cellular stresses, such as changes in intracellular Ca2+, redox status, or protein structure, can lead to upregulation of the same protective pathways through modulation of a variety of other transcription factors.
Acoustic exposure of the type used in sound conditioning experiments (1) can affect cochlear blood flow (Axelsson and Vertes, 1982), (2) probably changes intracellular ion concentrations and redox status, (3) can elicit an array of psychological stress responses (Kryter, 1976), and (4) must involve some degree of physical restraint. Thus, the conditioning-mediated protective effects in the ear may involve changes in stress-induced gene expression mediated by a combination of systemic and local cellular signals. The recent demonstration of unilateral protection from acoustic injury after unilateral sound conditioning (Yamasoba et al., 1999) suggests that conditioning-related protection cannot be completely caused by modulation of systemic stress levels. Indeed, the existence of multiple mechanisms, with differing time constants for onset and decay, help explain contradictions in the conditioning literature. For example, Ryan et al. (1994) showed that protective effects of sound conditioning were absent when the trauma was presented immediately after conditioning, yet were clear-cut with a treatment–trauma interval of 1 week. On the other hand, Canlon and Fransson (1998) showed that a 2 hr conditioner–trauma interval afforded larger protection than at a 1 week interval. The decay of protective effects observed in the latter study is consistent with that seen in the present heat stress paradigm, whereas the former is not.
Although protection from sound conditioning has been reported in a number of mammalian species, it was not demonstra-ble in the one published study of the CBA/Ca mouse (Fowler et al., 1995). Of course, so many parameters are involved in choosing the conditioning and traumatic stimuli (duration, spectrum, and level for each), and the conditioner–trauma interval, a negative result from one particular parameter set, is not very meaningful. We have seen recently protective effects of sound conditioning in CBA/CaJ mice using a paradigm designed to parallel the heat stress experiments reported here, i.e., a short-term (15 min) sound conditioner followed 6–24 hr later by acoustic overexposure, and also designed to maximize stress by using the loudest conditioner for which there is no residual TTS 6 hr later, when the traumatic exposure is presented. The development of this mouse model of acoustic-injury protection will facilitate future studies of the molecular mechanisms involved, including direct measures of HSP production after sound conditioning procedures, which protect the ear from acoustic injury.
Footnotes
This work was supported by National Institute on Deafness and Other Communication Disorders Grant RO1 DC-0188. The skillful assistance of S. Hequembourg in the animal surgery is gratefully acknowledged.
Correspondence should be addressed to M. Charles Liberman, Eaton-Peabody Laboratory, Massachusetts Eye and Ear Infirmary, 243 Charles Street, Boston, MA 02114. E-mail: mcl@epl.meei.harvard.edu.
REFERENCES
- 1.Akizuki H, Yoshie H, Morita Y, Takahashi K, Hara A, Watanabe T, Uchiyama Y, Kusakari J. Nuclear transition of heat shock protein in guinea pig cochlea after hyperthermia. Hear Res. 1995;92:126–130. doi: 10.1016/0378-5955(95)00210-3. [DOI] [PubMed] [Google Scholar]
- 2.Axelsson A, Vertes D. Histological findings in cochlear vessels after noise. In: Hamernik RP, Henderson D, Salvi R, editors. New perspectives on noise-induced hearing loss. Raven; New York: 1982. pp. 49–68. [Google Scholar]
- 3.Barbe MF, Tytell M, Gower DJ, Welch WJ. Hyperthermia protects against light damage in the rat retina. Science. 1988;241:1817–1820. doi: 10.1126/science.3175623. [DOI] [PubMed] [Google Scholar]
- 4.Canlon B. The effects of sound conditioning on the cochlea. In: Salvi R, Hendserson D, editors. Auditory system plasticity and regeneration. Thieme; New York: 1996. pp. 118–127. [Google Scholar]
- 5.Canlon B, Fransson A. Reducing noise damage by using a mid-frequency sound conditioning stimulus. NeuroReport. 1998;9:269–274. doi: 10.1097/00001756-199801260-00017. [DOI] [PubMed] [Google Scholar]
- 6.Canlon B, Borg E, Flock A. Protection against noise trauma by pre-exposure to a low level acoustic stimulus. Hear Res. 1988;34:197–200. doi: 10.1016/0378-5955(88)90107-4. [DOI] [PubMed] [Google Scholar]
- 7.Canlon B, Borg E, Lofstrand P. Noise-induced hearing loss (Dancer DHA, Salvi RJ, Hamernik RP) Mosby Year Book; St. Louis: 1991. Physiologic and morphologic aspects of low-level acoustic stimulation. pp. 489–499. [Google Scholar]
- 8.Canlon B, Lofstrand P, Borg E. Ultrastructual changes in the presynaptic regions of outer hair cells after acoustic stimulation. Neurosci Lett. 1993;150:103–106. doi: 10.1016/0304-3940(93)90118-5. [DOI] [PubMed] [Google Scholar]
- 9.Drescher DG. Effect of temperature on cochlear responses during and after exposure to noise. J Acoust Soc Am. 1976;59:401–407. doi: 10.1121/1.380877. [DOI] [PubMed] [Google Scholar]
- 10.Ehret G. Peripheral anatomy and physiology II. In: Willott JF, editor. The auditory psychobiology of the mouse. Thomas; Springfield, IL: 1983. pp. 169–200. [Google Scholar]
- 11.Engstrom B, Flock A, Borg E. Ultrastructural studies of stereocilia in noise-exposed rabbits. Hear Res. 1983;12:251–264. doi: 10.1016/0378-5955(83)90110-7. [DOI] [PubMed] [Google Scholar]
- 12.Fowler T, Canlon B, Dolan D, Miller JM. The effect of noise trauma following training exposures in the mouse. Hear Res. 1995;88:1–13. doi: 10.1016/0378-5955(95)00062-9. [DOI] [PubMed] [Google Scholar]
- 13.Franklin DJ, Lonsbury Martin BL, Stagner BB, Martin GK. Altered susceptibility of 2f1-f2 acoustic-distortion products to the effects of repeated noise exposure in rabbits. Hear Res. 1991;53:185–208. doi: 10.1016/0378-5955(91)90053-c. [DOI] [PubMed] [Google Scholar]
- 14.Ghoshal K, Wang Y, Sheridan JF, Jacob ST. Metallothionein induction in response to restraint stress. J Biol Chem. 1998;273:27904–27910. doi: 10.1074/jbc.273.43.27904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Heid CA, Stevens J, Livak KJ, Williams PM. Real time quantitative PCR. Genome Res. 1996;6:986–994. doi: 10.1101/gr.6.10.986. [DOI] [PubMed] [Google Scholar]
- 16.Jacono AA, Hu B, Kopke RD, Henderson D, Van de Water TR, Steinman HM. Changes in cochlear antioxidant enzyme activity after sound conditioning and noise exposure in the chinchilla. Hear Res. 1998;117:31–38. doi: 10.1016/s0378-5955(97)00214-1. [DOI] [PubMed] [Google Scholar]
- 17.Jakob U, Gaestel M, Engel K, Buchner J. Small heat shock proteins are molecular chaperones. J Biol Chem. 1993;268:1517–1520. [PubMed] [Google Scholar]
- 18.Kryter KD. Extraauditory effects of noise. In: Henderson D, Dosanjh DS, Mills JH, editors. Effects of noise on hearing. Raven; New York: 1976. pp. 531–546. [Google Scholar]
- 19. Kujawa SG, Liberman MC. Sound conditioning enhances cochlear responses in guinea pig. Abstracts of the Association for Research in Otolaryngology Midwinter Meeting 1996. 19:34.
- 20.Lavoie JN, Hickey E, Weber LA, Landry J. Modulation of actin microfilament dynamics and fluid phase pinocytosis by phosphorylation of heat shock protein 27. J Biol Chem. 1993;268:24210–24214. [PubMed] [Google Scholar]
- 21.Lavoie JN, Lambert H, Hickey E, Weber LA, Landry J. Modulation of cellular thermoresistance and actin filament stability accompanies phosphorylation-induced changes in the oligomeric structure of heat shock protein 27. Mol Cell Biol. 1995;15:505–516. doi: 10.1128/mcb.15.1.505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Leonova EV, Fairfield D, Lomax MI, Altschuler RA. Expression of the HSP-27 in the organ of Corti of rat. Abstr Mol Biol Hear Deaf. 1998;3:177. [Google Scholar]
- 23.Liberman MC. Chronic ultrastructural changes in acoustic trauma: serial-section reconstruction of stereocilia and cuticular plates. Hear Res. 1987;26:65–88. doi: 10.1016/0378-5955(87)90036-0. [DOI] [PubMed] [Google Scholar]
- 24.Liberman MC, Dodds LW. Single-neuron labeling and chronic cochlear pathology. III. Stereocilia damage and alterations of threshold tuning curves. Hear Res. 1984;16:55–74. doi: 10.1016/0378-5955(84)90025-x. [DOI] [PubMed] [Google Scholar]
- 25.Liberman MC, Gao WY. Chronic cochlear de-efferentation and susceptibility to permanent acoustic injury. Hear Res. 1995;90:158–168. doi: 10.1016/0378-5955(95)00160-2. [DOI] [PubMed] [Google Scholar]
- 26.Liberman MC, Kiang NYS. Acoustic trauma in cats, cochlear pathology and auditory-nerve activity. Acta Otolaryngol (Stockh) 1978;358:5–63. [PubMed] [Google Scholar]
- 27.Lim HH, Jenkins OH, Myers MW, Miller JM, Altschuler RA. Detection of HSP72 synthesis after acoustic overstimulation in rat cochlea. Hear Res. 1993;69:146–150. doi: 10.1016/0378-5955(93)90102-7. [DOI] [PubMed] [Google Scholar]
- 28.Lindquist S, Craig EA. The heat-shock proteins. Annu Rev Genet. 1988;22:631–677. doi: 10.1146/annurev.ge.22.120188.003215. [DOI] [PubMed] [Google Scholar]
- 29.Livak KJ, Flood SJA, Marmaro J, Giusti W, Deetz K. Oligonucleotides with fluorescent dyes at opposite ends provide a quenched probe system for detecting PCR product and nucleic acid hybridization. PCR Methods Appl. 1995;4:357–362. doi: 10.1101/gr.4.6.357. [DOI] [PubMed] [Google Scholar]
- 30.Miller JD, Watson CS, Covell WP. Deafening effects of noise on the cat. Acta Otolaryngol Suppl (Stockh) 1963;176:1–89. [Google Scholar]
- 31.Miyakita T, Hellstrom P-A, Frimanson E, Axelsson A. Effect of low level acoustic stimulation on temporary threshold shift in young humans. Hear Res. 1992;60:149–155. doi: 10.1016/0378-5955(92)90017-h. [DOI] [PubMed] [Google Scholar]
- 32.Myers MW, Quirk WS, Rizk SS, Miller JM, Altschuler RA. Expression of the major mammalian stress protein in the rat cochlea following transient ischemia. Laryngoscope. 1992;102:981–987. doi: 10.1288/00005537-199209000-00005. [DOI] [PubMed] [Google Scholar]
- 33.Pukkila M, Zhai S, Virkkala J, Pirvola U, Ylikoski J. The “toughening” phenomenon in rat's auditory organ. Acta Otolaryngol Suppl (Stockh) 1997;529:59–62. doi: 10.3109/00016489709124081. [DOI] [PubMed] [Google Scholar]
- 34.Raphael Y, Athey BD, Wang Y, Lee MK, Altschuler RA. F-actin, tubulin and spectrin in the organ of Corti: comparative distribution in different cell types and mammalian species. Hear Res. 1994;76:173–187. doi: 10.1016/0378-5955(94)90098-1. [DOI] [PubMed] [Google Scholar]
- 35.Robertson D. Effects of acoustic trauma on stereocilia structure and spiral ganglion cell tuning properties in the guinea pig cochlea. Hear Res. 1982;7:55–74. doi: 10.1016/0378-5955(82)90081-8. [DOI] [PubMed] [Google Scholar]
- 36.Ryan AF, Bennett TM, Woolf NK, Axelsson A. Protection from noise-induced hearing loss by prior exposure to a nontraumatic stimulus: role of the middle ear muscles. Hear Res. 1994;72:23–28. doi: 10.1016/0378-5955(94)90201-1. [DOI] [PubMed] [Google Scholar]
- 37.Saunders JC, Dear SP, Schneider ME. The anatomical consequences of acoustic injury: a review and tutorial. J Acoust Soc Am. 1985;78:833–860. doi: 10.1121/1.392915. [DOI] [PubMed] [Google Scholar]
- 38.Sha SH, Dolan DF, Schacht J. Overexpression of superoxide dismutase protects against ototraumatic insults. Abstr Mol Biol Hear Deaf. 1998;3:130. [Google Scholar]
- 39.Siegel JH, Kim DO, Molnar CE. Effects of altering organ of Corti on cochlear distortion products f2–f1 and 2f1–f2. J Neurophysiol. 1982;47:303–328. doi: 10.1152/jn.1982.47.2.303. [DOI] [PubMed] [Google Scholar]
- 40.Subramaniam M, Henderson D, Henselman L. “Toughening” of the mammalian auditory system: spectral, temporal and intensity effects. In: Salvi R, Henderson D, editors. Auditory system plasticity and regeneration. Thieme; New York: 1996. pp. 128–142. [Google Scholar]
- 41.Thompson AM, Neely JG. Induction of heat shock protein in interdental cells by hyperthermia. Otolaryngol Head Neck Surg. 1992;107:769–774. doi: 10.1177/019459988910700611.1. [DOI] [PubMed] [Google Scholar]
- 42.Tytell M, Barbe MF, Brown IR. Stress (Heat shock) protein accumulation in the central nervous system. Adv Neurol. 1993;59:293–303. [PubMed] [Google Scholar]
- 43.Welch WJ. Mammalian stress response: cell physiology, structure/function of stress proteins, and implications for medicine and disease. Physiol Rev. 1992;72:1063–1081. doi: 10.1152/physrev.1992.72.4.1063. [DOI] [PubMed] [Google Scholar]
- 44.Yamasoba T, Dolan DF, Miller JM. Acquired resistance to acoustic trauma by sound conditioning is primarily mediated by changes restricted to the cochlea not by systemic responses. Hear Res. 1999;127:31–40. doi: 10.1016/s0378-5955(98)00178-6. [DOI] [PubMed] [Google Scholar]