

Abstract
The ventral tegmental area (VTA) is the origination zone for dopaminergic neurons involved in reward and addictive properties of a variety of abused substances. A major excitatory projection to VTA neurons originates in the medial prefrontal cortex, and several lines of evidence suggest that glutamatergic synapses on VTA neurons are activated and modified during exposure to psychostimulant drugs. Here, we report for the first time that amphetamine depresses excitatory glutamatergic synaptic transmission onto VTA neurons in the midbrain slice preparation. Unexpectedly, this depression is mediated not by activation of dopamine receptors, but instead by activation of serotonin receptors. Our findings suggest that an acute effect of amphetamine exposure is the release of serotonin in the VTA, which in turn modulates excitation of VTA neurons. This process may be an important early component of permanent changes occurring in the reward pathway that contribute to drug addiction.
Keywords: ventral tegmental area (VTA), amphetamine, psychostimulants, excitatory synaptic transmission, serotonin, glutamatergic
The ventral tegmental area (VTA) of the midbrain is the source of dopaminergic neurons that form the mesocorticolimbic projection, a major limb of the reward pathway and a target for drugs of abuse. Glutamatergic afferents originating in medial prefrontal cortex (PFC) drive VTA neurons and control their firing patterns (Gariano and Groves, 1988; Svensson and Tung, 1989), and thus glutamatergic synaptic transmission in VTA represents a possible site of action of addictive drugs. Here, we have directly tested the effects of amphetamine on excitatory synaptic transmission onto VTA neurons in vitro.
The increased drug craving that accompanies compulsive drug taking in human drug addiction has been modeled experimentally as behavioral sensitization, a progressive and permanent augmentation of behavioral responses to drugs of abuse, including psychostimulants such as amphetamine and cocaine (Kalivas and Stewart, 1991; Robinson and Berridge, 1993). Initiation of sensitization occurs in the VTA (Kalivas and Weber, 1988; Vezina and Stewart, 1990; Vezina, 1993). Moreover, glutamatergic input from the PFC to VTA is required for the initiation of sensitization to amphetamine (for review, see Kalivas, 1993; White, 1996; Wolf, 1998). Thus, excitatory afferents from the PFC and consequent excitatory synaptic transmission in the VTA are necessary and perhaps sufficient to trigger sensitization.
Substantial evidence supports the involvement of VTA dopamine cells and dopamine release in drug addiction and behavioral sensitization (DiChiara and Imperato, 1988; Kalivas, 1993; White, 1996). Amphetamine releases dopamine from somatodendritic sites in the VTA (Kalivas and Duffy, 1993) by inhibiting reuptake of dopamine via the transporter and by promoting nonvesicular release of dopamine (Seiden et al., 1993). Psychostimulants can also increase extracellular serotonin levels by binding to the serotonin transporter present on serotonergic terminals originating in the raphe nucleus (Ritz et al., 1987; Vertes, 1991), and there is growing evidence for an involvement of serotonin systems in drug addiction. Serotonin mediates some physiological effects of cocaine on VTA dopamine cells via presynaptic regulation of GABA release (Cameron and Williams, 1994, 1995) and methylenedioxy-methamphetamine (or “ecstasy”) on dopamine cell spontaneous activity (Obradovic et al., 1996). Moreover, 5-HT1B serotonin receptor knock-out mice have an increased likelihood to self-administer cocaine (Rocha et al., 1998b).
Despite considerable evidence that glutamatergic afferents control the firing of VTA neurons and that this pathway is essential in the development of sensitization, the effects of psychostimulants on excitatory synaptic transmission have never been tested. Here, we directly examined the acute effects of amphetamine on glutamatergic synaptic transmission, using whole-cell patch-clamp recordings from VTA neurons in midbrain slices. We also explored the hypothesis that dopamine and/or serotonin alter excitatory synaptic transmission in the VTA. We report that amphetamine directly depresses glutamatergic synaptic transmission onto VTA neurons. This is a consequence of serotonin release and does not involve dopamine receptors. The depression of excitatory synaptic transmission is a likely early event following psychostimulant administration in the naïve brain. Our results implicate serotonin in the modification of dopaminergic function in the reward pathway.
MATERIALS AND METHODS
Preparation of brain slices. Sprague Dawley rats, 16- to 23-d-old, were anesthetized with halothane and quickly decapitated. The brain was rapidly removed into ice-cold artificial CSF (ACSF) containing (in mm): NaCl 119, KCl 2.5, MgSO4 1.3, CaCl2 2.5, NaH2PO4 1.0, NaHCO3 26.0, and glucose 11, continuously gassed with 95% O2–5% CO2, pH 7.4, osmolarity of 290 mOsm. Kynurenic acid (1 mm) was included in the ACSF during slice preparation and storage. Horizontal midbrain slices (250 μm thick), dorsal to the interpeduncular fossa and containing the VTA, were cut using a vibratome, transferred to a submersion chamber at 30°C containing ACSF, and used for recordings from 1 to 6 hr after preparation.
Electrophysiology. Midbrain slices were continuously perfused with ACSF (without kynurenic acid) and warmed to 28–32°C at a flow rate of 2–4 ml/min. Picrotoxin (100 μm) was added to block GABAA receptors, to study excitatory synaptic transmission in isolation. Under low-power magnification, the location of the VTA was identified medial to the substantia nigra pars compacta and the medial terminal nucleus of the accessory optic tract. Using a 40× water immersion objective with differential interference contrast and a CCD camera, individual cells in the VTA were visualized on a monitor for whole-cell patch-clamp recordings.
Patch pipettes had resistances of 2–4 MΩ when filled with (in mm): potassium gluconate 100, MgCl25, HEPES 40, EGTA 0.6, Na2-ATP 2, and Na2-GTP 0.3, pH 7.25, osmolarity of 280 mOsm. Biocytin (0.4%) was added to allow post hoc identification of recorded cells for immunolabeling experiments (see below). Signals were amplified through an Axoclamp 2B amplifier (Axon Instruments, Foster City, CA) used in either bridge mode to record voltage signals or continuous single electrode voltage-clamp mode to record evoked EPSCs. In bridge mode, cells were allowed to fire spontaneous action potentials from their resting potential of between −50 and −30 mV for 1 sec sweeps to allow estimation of spontaneous firing rates. To investigate the presence of the hyperpolarization-activated nonselective cation conductance characteristic of dopamine cells (IH) (Grace and Onn, 1989;Johnson and North, 1992), cells were injected with sufficient negative current to set the membrane potential to −50 mV, and hyperpolarizing current steps (700 msec duration) were passed to activateIH. This conductance was considered to be present if, during hyperpolarizing current steps to between −90 and −110 mV, the membrane potential depolarized by the end of the step by >25%. This voltage “sag” response was taken to indicate the presence of IH, characteristic of VTA dopamine cells; many of these cells, when tested, were found to stain for tyrosine hydroxylase (TH) (see below). An end-of-step depolarization of <15% (no voltage sag) was considered to indicate a nondopamine cell, whereas an end-of-step depolarization of between 15 and 25% (intermediate voltage sag) was taken to indicate a third cell type.
The membrane potential was then voltage clamped at −70 mV, and EPSCs were evoked using a bipolar stainless steel stimulating electrode placed rostral to VTA at least 100 μm from the cell. Paired stimuli 50 msec apart (100–400 μA for 100 μsec) were delivered every 10 sec. Series resistance was measured at the beginning of the experiment and ranged from 15 to 25 MΩ. Test pulses acquired throughout the experiment enabled checks for series resistance stability. Experiments were discarded if changes in series resistance >10% occurred. Signals were filtered at 3 kHz and collected on-line to a computer using Axobasic software (Axon Instruments) and programs kindly donated by Drs. Daniel Madison (Molecular and Cellular Physiology, Stanford University, Stanford, CA) and Felix Schweizer (Neurobiology, University of California Los Angeles, Los Angeles, CA).
Pharmacology. All drugs were added directly to the ACSF perfusing the slice chamber. Controls for solution switches were routinely performed. All agonists were added for 10 min, allowing adequate time for equilibration. Dopamine, norepinephrine, and serotonin solutions were prepared immediately before use. All antagonists were added at least 10–15 min before agonist application. In dopamine depletion experiments, littermates were given either the tyrosine hydroxylase inhibitor α-methyl-para-tyrosine or saline vehicle in double-blind experiments [200–300 mg/kg, i.p., at least 2 hr before slice preparation (Spector et al., 1965)].
Drug effects were assessed by averaging EPSC amplitudes for 5–10 min during the peak response and comparing this value with 10 min of averaged data just before drug application and are reported as mean ± SEM throughout the text. Significance was measured using unpaired t tests.
Immunolabeling for tyrosine hydroxylase. Immediately after recording, slices were fixed in 4% paraformaldehyde for at least 24 hr. Slices were immersed in a 30% sucrose solution and resectioned at 50–90 μm on a freezing microtome. Slices were then preincubated for 2 hr in 0.5% BSA with 0.1% Triton X-100 and incubated overnight in mouse anti-tyrosine hydroxylase antibody (1:1000) at 4°C. This was followed by incubation for 1 hr in cy3-conjugated goat anti-mouse secondary antibody (1:500) and then a 1 hr incubation in FITC-conjugated extravidin (1:200).
Identification of dopamine neurons. To determine whether cells used for recordings were dopamine cells or VTA interneurons, a combination of physiology and immunohistochemistry was used (Table1; Fig. 1). Thirty-three percent (43 of 129) of biocytin-labeled cells were recovered, and of these, 63% (27 of 43 cells) were positive for TH (the rate-limiting enzyme in catecholamine synthesis). In general, these TH-positive cells had clear dopamine cell physiological characteristics (described below), although four cells with nondopamine cell characteristics were also TH-positive. All cells showing TH-positive staining were considered to be dopamine cells, regardless of physiological characteristics. Cells were classified into three groups based on the presence of a depolarizing sag in response to negative current step injections. Those showing a >25% end-of-step change in voltage were classed as dopamine cells (81%, 104 of 129 cells) (Fig. 1B). Those showing a <15% end-of-step change in voltage, or no sag (6%, 8 of 129 cells), were classed as nondopamine cells unless TH-positive (2 of 8 of these cells were TH-positive). Those showing end-of-step voltage changes of between 15 and 25% (intermediate sags; 9%, 12 of 129 cells) were classed as dopamine cells only if TH-positive (2 of 12 cells). In the dopamine cell class, mean spontaneous firing rate was 1.5 ± 0.2 Hz in 87 cells tested (22% of these cells showed no spontaneous activity). Cells showing no sag were either silent or fired at high frequency (>5 Hz). Intermediate cells spontaneously fired similarly to dopamine cells (1.6 ± 0.5 Hz; n = 11; 36% showed no spontaneous activity). All figure legends report the number of cells classed as dopamine cells for each experimental group.
Table 1.
Classification of VTA cells by tyrosine hydroxylase immunolabeling and physiology
| Tyrosine hydroxylase (TH) immunolabeling | Voltage sag (n = 104) | No voltage sag (n = 8) | Intermediate voltage sag (n = 12) |
|---|---|---|---|
| TH-positive (n = 27) | n = 23 | n = 2 | n = 2 |
| TH-negative (n = 16) | n = 13 | − | n = 3 |
| Soma not recovered (n = 13) | n = 12 | − | n = 1 |
| Not tested (n = 68) | n = 56 | n = 6 | n = 6 |
Characterization of VTA cells. Numbers of cells showing positive staining for tyrosine hydroxylase, where tested, and the presence of the conductance IH, indicated by a depolarizing voltage sag in response to injections of hyperpolarizing current (see Materials and Methods). Total number of cells, 124.
Fig. 1.
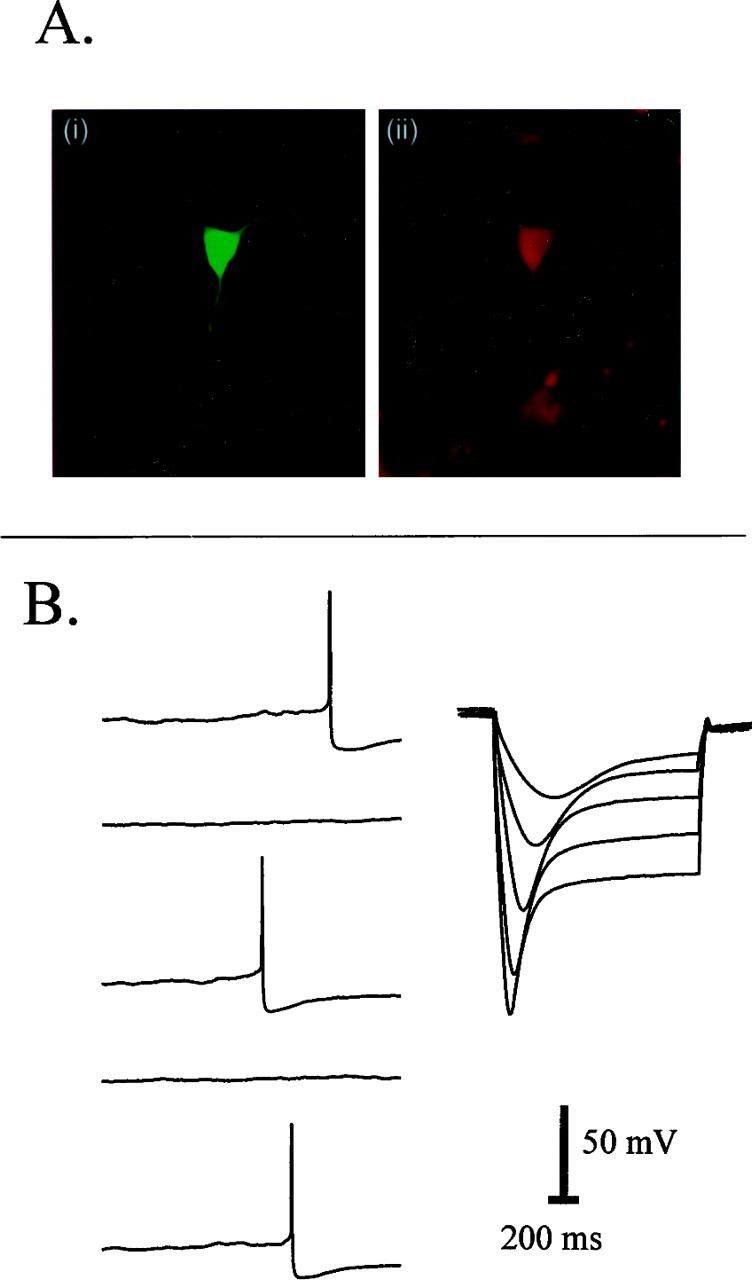
Characterization of VTA cells. A,Left (i) shows biocytin fill of the recorded cell, and right (ii) shows that the same cell is TH-positive. B, Physiological responses from the cell pictured in A showing typical dopamine cell characteristics: spontaneous firing and a distinctive voltage sag response to hyperpolarizing current.
Materials. Standard lab chemicals were obtained from Mallinckrodt (St. Louis, MO), Aldrich (Milwaukee, WI), or Fluka (Buchs, Switzerland). Biocytin, extravidin, and BSA were obtained from Sigma (St. Louis, MO), ATP and GTP were obtained from Boehringer Mannheim (Indianapolis, IN), TH monoclonal antibody was obtained from DiaSorin (Stillwater, MN), and goat anti-mouse IgG was obtained from Chemicon (Temecula, CA). All agonists, antagonists, and amphetamine were obtained from Research Biochemicals (Natick, MA).
RESULTS
Neurons recorded from within the VTA were classed as dopamine cells or nondopamine cells based on their physiological and morphological characteristics; a typical dopamine cell is shown in Figure 1. The data from all cells are summarized in Table 1. EPSCs recorded from dopamine neurons at −70 mV were primarily mediated by non-NMDA glutamate receptors (Fig. 2), because they were almost completely blocked by 6,7-dinitroquinoxaline-2,3-dione (DNQX; 10 μm). A small EPSC component remained at −70 mV; on depolarizing the cell to −20 mV, this component increased in amplitude and was blocked byd,l-2-amino-5-phosphonovalerate (APV; 100 μm), indicating that a portion of the EPSC is mediated by NMDA receptors. In the presence of both antagonists, the EPSC at −70 mV was blocked (data not shown).
Fig. 2.
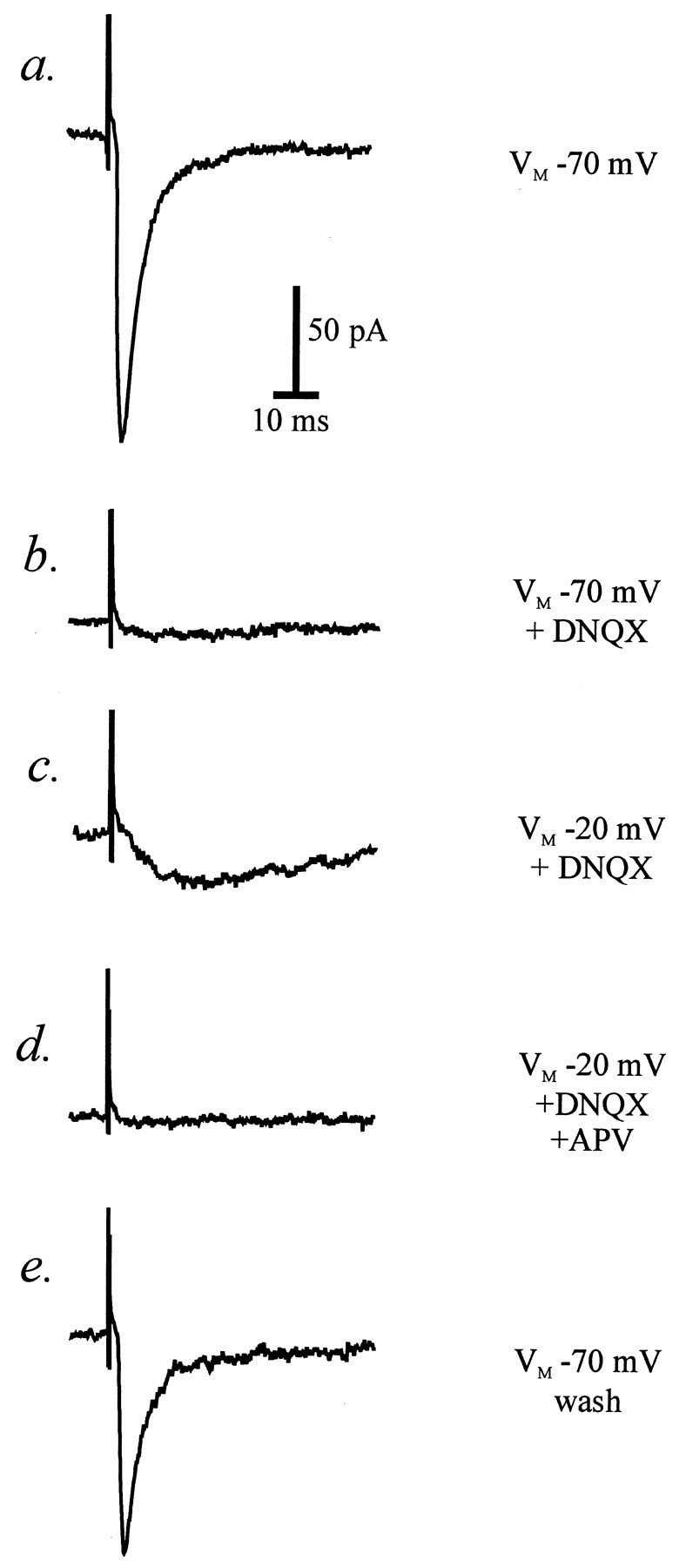
Excitatory postsynaptic currents in a VTA dopamine cell (TH-positive). Single records from one experiment showing (fromtop to bottom) a typical EPSC recorded at −70 mV in the presence of 100 μm picrotoxin (a), inhibition of the EPSC at −70 mV by 10 μm DNQX (b), a new, slow component of the EPSC in DNQX when the cell is held at −20 mV (c), blockade of the slow EPSC with 100 μm APV (d), and recovery of the fast EPSC recorded at −70 mV on washing out the glutamate receptor antagonists (e).
Psychostimulants increase extracellular glutamate levels in the VTA measured using in vivo microdialysis (Kalivas and Duffy, 1995; Xue et al., 1996). We therefore first tested whether amphetamine had any effect on glutamatergic synaptic transmission in the VTA. Surprisingly, we found that amphetamine caused a substantial depression of glutamate EPSCs (Fig. 3). Consecutive applications of 10 μm amphetamine elicited reproducible and reversible EPSC depression (Fig. 3A). The effect of amphetamine was concentration-dependent; the threshold for the response was 1 μm. In three cells tested, 0.1 μm amphetamine had no detectable effect on EPSC amplitude. A robust depression of EPSC amplitude was seen with 10 μm amphetamine (30 ± 4%) (Fig.3B), and because this response was on the linear part of the dose–response curve (Fig. 3C), 10 μm amphetamine was used in subsequent experiments to test the effects of antagonists.
Fig. 3.

Effect of amphetamine on EPSCs in VTA cells.Aa, Time course data from one dopamine cell showing reproducible depression of EPSC amplitude (normalized to predrug values) in response to repetitive applications of amphetamine (10 μm, 10 min; bars). Ab, Example records (average of 5–10 traces) showing control EPSCs and EPSCs in the presence of amphetamine at the indicated times during the experiment (1–3). B, Averaged time course data from seven experiments showing the reversible depression of the EPSC by amphetamine (10 μm, 10 min;bar). For this and all figures showing averaged data, only every third error bar is shown for clarity. Five of the seven cells were dopamine cells. C, Concentration–response graph for depression of EPSC amplitude (percentage) by amphetamine. Nineteen of 26 cells included in this graph were dopamine cells.
Having found that amphetamine decreases glutamate synaptic transmission, we next tested the idea that amphetamine causes this depression of the glutamatergic EPSC via the release of dopamine. When we examined the effect of exogenously applied dopamine, we found that, at high concentrations, dopamine mimicked amphetamine, depressing the amplitude of the EPSC (38 ± 5% depression) (Fig.4A). The threshold for the effect of dopamine was ∼10 μm, with 100 μm causing a robust response (Fig.4A, inset). To determine which dopamine receptors might mediate the effect of amphetamine, selective agonists and antagonists for D1 and D2 dopamine receptors were used. Although dopamine depressed the EPSC, the D1 antagonist SCH 23390 (2 μm), the D2antagonist sulpiride (10 μm), or a combination of both antagonists did not affect the ability of amphetamine to depress the EPSC (Fig. 4B,C); this result suggested that dopamine receptors are not involved in depressing excitatory synaptic transmission. In agreement with this interpretation, the D1 agonist SKF 82958 (10 μm) and the D2 agonist quinpirole (30 μm) evoked only very small depressions of the EPSC amplitude (SKF 82958, 8 ± 3%;n = 4; quinpirole, 14 ± 4%; n = 5) (data not shown).
Fig. 4.
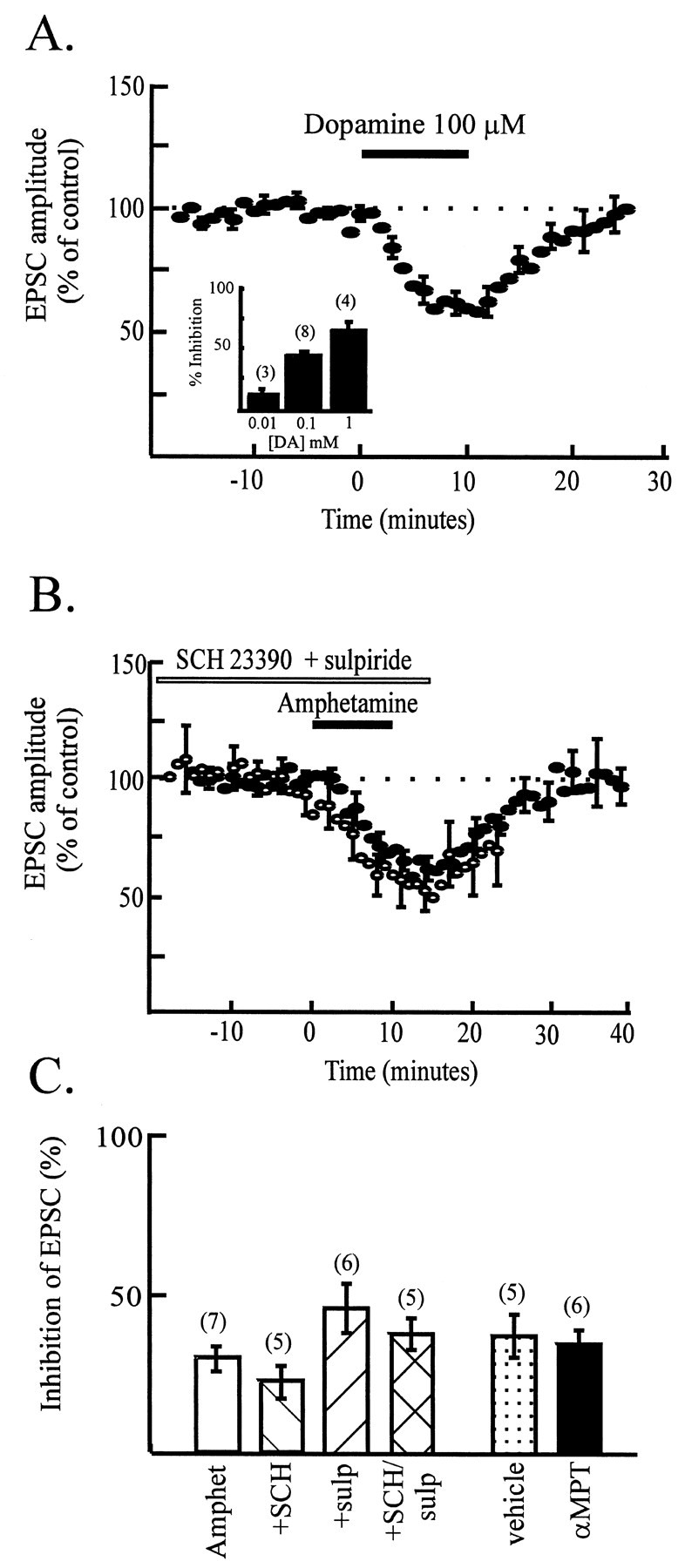
Dopamine does not mediate the effect of amphetamine. A, Time course data from eight experiments showing the reversible depression of EPSC amplitude (normalized to control values) by a high concentration of dopamine (100 μm, 10 min). Seven of the eight cells were dopamine cells. Inset shows concentration dependence of the depression of EPSC amplitude by dopamine. Thirteen of 15 cells used for this bar chart were dopamine cells. B, Time course data showing the combined data from four cells (open symbols) illustrating that the effect of amphetamine on EPSC amplitude in the presence of both 2 μm SCH 23390 and 10 μmsulpiride is superimposable with the control response (filled symbols; from Fig. 3A).C, Bar chart summarizing the lack of effect of the dopamine D1 and D2 antagonists SCH 23390 (2 μm) and sulpiride (10 μm) on the response to amphetamine (patterned bars). Thirteen of 16 cells used for the dopamine antagonist studies were dopamine cells. Thefilled bar illustrates that pretreatment of animals with the tyrosine hydroxylase inhibitor α-methyl-p-tyrosine has no effect on the response to amphetamine compared with saline pretreated animals (dotted bar). Eleven of 11 cells in this study were dopamine cells.
As a control to ensure that amphetamine was able to release dopamine in our slices, we monitored the effect of amphetamine on the spontaneous activity of dopamine cells, in either cell-attached patch mode or whole-cell current-clamp mode, allowing the cells to fire action potentials. In both configurations, amphetamine (10 μm) completely suppressed spontaneous firing in two cells (data not shown); in two other cells, this effect of amphetamine on spontaneous activity was prevented by sulpiride (10 μm) (data not shown), demonstrating that D2 receptors are responsible, as reported previously (Seutin et al., 1991). These data suggest that amphetamine can indeed release dopamine within our slices and also that sulpiride is effective at blocking D2 receptors under our recording conditions; however, the depression of excitatory synaptic transmission is not mediated by released dopamine. As a further test for the involvement of dopamine in mediating the effect of amphetamine, animals were given α-methyl-p-tyrosine to deplete dopamine. The effect of amphetamine was not significantly different in slices from animals given the dopamine depletor compared with littermate controls given saline injections (Fig. 4C). Together, these unexpected results indicate that endogenous dopamine released by amphetamine is unlikely to mediate the depression of excitatory synaptic transmission observed with amphetamine. We suspected, therefore, that exogenously added dopamine depresses the EPSC (Fig. 4A) via effects on nondopamine receptors. Consistent with this hypothesis, SCH 23390 (2 μm) and sulpiride (10 μm) also failed to block the depression of the EPSC produced by exogenously applied dopamine (data not shown).
Because the effect of amphetamine on glutamate synaptic transmission does not appear to be via dopamine release and activation of dopamine receptors, this suggested that it might be via the known effect of amphetamine on transporters for either norepinephrine or serotonin. Norepinephrine itself had little effect on EPSC amplitude, causing 11.3 ± 1.8% depression at 10 μm (n= 4). Furthermore, the effect of amphetamine was not significantly inhibited by α-adrenoceptor antagonists, causing 24 ± 5% depression in the presence of the α1 antagonist prazosin (1 μm; n = 5) and 30.3 ± 3.5% inhibition in the presence of the α2 antagonist yohimbine (1 μm; n = 3). Therefore, we tested the idea that amphetamine acts by releasing serotonin. Exogenously applied serotonin caused a reversible, concentration-dependent depression of EPSC amplitude (39 ± 6%) (Fig. 5).
Fig. 5.
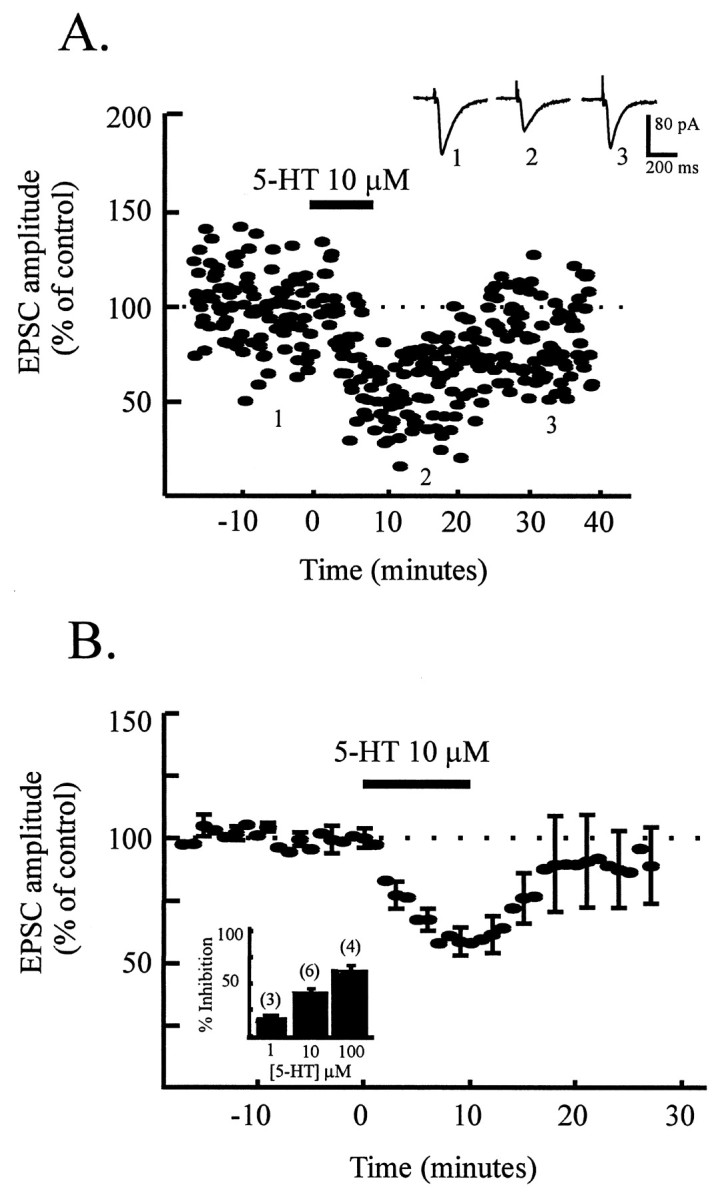
Serotonin mimics the effect of amphetamine.A, Time course data from one dopamine cell showing reversible depression of EPSC amplitude (normalized to predrug values) in response to serotonin (10 μm, 10 min;bar). Inset shows example records (average of 5–10 traces) showing control EPSCs and EPSCs in the presence of serotonin. B, Time course data from six cells showing the reversible depression of EPSC amplitude (normalized to control values) by serotonin (10 μm, 10 min;5-HT). Four of six cells were dopamine cells.Inset shows concentration dependence of the effect of serotonin in depressing EPSC amplitude (percentage). Nine of 13 cells used for this bar chart were dopamine cells.
To elucidate the involvement of serotonin receptors in mediating the effect of amphetamine, we examined the effects of amphetamine in the presence of serotonin receptor antagonists. Ketanserin (100 nm), a selective 5-HT2 receptor antagonist, did not block the effects of amphetamine (amphetamine-induced depression of the EPSC, 32 ± 4%;n = 3) (data not shown). However, two broad spectrum serotonin receptor antagonists prevented amphetamine from depressing excitatory synaptic transmission. Methysergide (10 μm) completely blocked the effect of amphetamine on the EPSC (amphetamine-induced depression of EPSC in the presence of methysergide, 3 ± 2%; n = 5) (Fig.6A). Methiothepin (1 μm), a structurally distinct serotonin antagonist, also significantly inhibited the effect of amphetamine on EPSCs (amphetamine-induced depression of EPSC in the presence of methiothepin, 11 ± 5%; n = 5) (Fig.6B); in two of five cells, methiothepin caused a complete block of the response to amphetamine. Methysergide also inhibited the depressant effect of 10 μmserotonin (to 18 ± 4%; n = 5). The effect of 100 μm dopamine was also blocked by methysergide (to 15 ± 5%; n = 3), supporting the idea that, at this high concentration, dopamine actually binds to serotonin receptors to reduce the EPSC. These results strongly suggest that the depression of excitatory synaptic transmission onto VTA dopamine neurons by amphetamine is mediated by serotonin receptors.
Fig. 6.
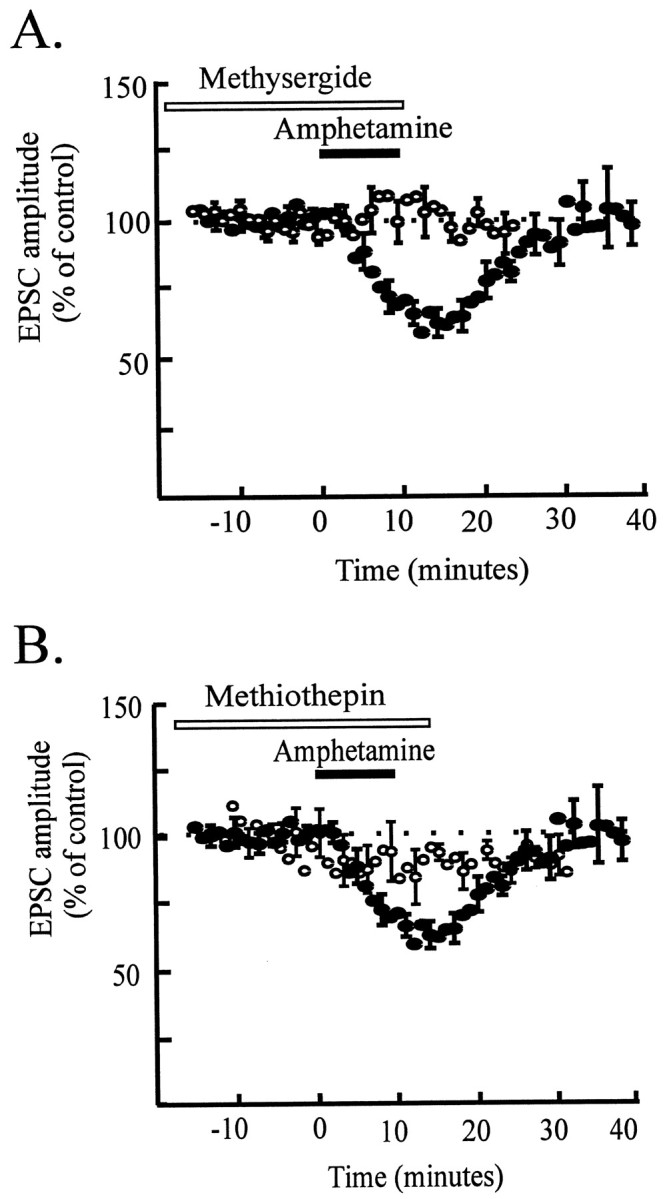
Serotonin receptors mediate the effect of amphetamine. A, Time course data from five cells (open symbols) illustrating that the effect of amphetamine is almost completely blocked in the presence of methysergide (10 μm) compared with control data (filled symbols; from Fig. 3A). Four of five cells were dopamine cells. B, Time course data from five cells (open symbols) showing that the effect of amphetamine is inhibited in the presence of methiothepin (1 μm) compared with control data (filled symbols; from Fig. 3A). Four of five cells were dopamine cells.
DISCUSSION
This work is the first demonstration of the acute effects of psychostimulants on excitatory synaptic transmission in the VTA. We report that amphetamine produces a rapid and reproducible decrease in excitatory synaptic transmission onto VTA neurons in slices from drug-naïve animals. In general, previous work on psychostimulant action has focused on dopaminergic mechanisms; our results demonstrate that psychostimulant-mediated release of serotonin importantly modulates excitatory synaptic transmission in the reward pathway.
Glutamatergic synaptic transmission is reduced by amphetamine
Based on previous findings using in vivo microdialysis, we expected amphetamine to increase glutamate release rather than to decrease it. Kalivas and Duffy (1995) reported an increase in extracellular glutamate levels in the VTA in response to systemic cocaine, and Xue et al. (1996) reported a delayed increase in glutamate efflux in response to systemic amphetamine. However, Wolf and Xue (1998) found that, when amphetamine was administered directly into the VTA, there was an initial decrease in glutamate efflux, followed by an increase on removing the stimulant. The decrease in glutamate release is in agreement with our observation that acute amphetamine decreases glutamatergic synaptic transmission. Repeated applications of amphetamine decrease the EPSC reproducibly, suggesting that glutamatergic synaptic transmission in the isolated VTA is not potentiated by amphetamine treatment, even after more than 1 hr. Together, these observations suggest the possibility that the increase in glutamate efflux observed in vivo results from alterations in glutamate reuptake rather than an increase in synaptically released glutamate.
Amphetamine does not depress EPSCs via dopamine receptors
Surprisingly, we found that dopamine receptors do not mediate the depression of excitatory synaptic transmission by amphetamine. Amphetamine does release dopamine in the midbrain slice preparation used because, in a separate experiment, dopamine evoked D2 receptor-mediated inhibition of spontaneous firing. Dopamine at high concentrations also mimics the effect of amphetamine on the EPSC amplitude. However, antagonists at D1-type dopamine receptors [including D1 and D5 subtypes; (Sibley and Monsma, 1992)] and D2-type receptors [including D2, D3, and D4 subtypes; (Sibley and Monsma, 1992)] did not attenuate the effect of amphetamine. This was surprising because it is well established that psychostimulants evoke changes in dopamine release and dopamine receptor sensitivity (Kalivas, 1993; White, 1996). We hypothesize that psychostimulants first alter glutamatergic activity that in turn produces downstream changes in the dopaminergic axis. In support of this, the NMDA glutamate receptor antagonist MK-801 prevents changes in dopamine receptor sensitivity that accompany behavioral sensitization (Wolf et al., 1994).
Amphetamine depresses glutamatergic transmission via serotonin
Amphetamine depresses excitatory synaptic transmission via activation of serotonin receptors, as two different serotonin receptor antagonists block the effect. To account for this, it is possible that amphetamine itself binds directly to serotonin receptors within the slice to reduce the EPSC; amphetamine is known to bind with low affinity to 5-HT2 receptors (Ritz and Kuhar, 1989). We think this explanation unlikely, because the 5-HT2 receptor antagonist ketanserin did not block the effect of amphetamine. Instead, a more likely explanation for our results is that amphetamine promotes serotonin release via its well documented direct effects on the serotonin transporter (Scheel-Kruger, 1971; Ritz et al., 1987) and that released serotonin depresses the EPSC via serotonin receptors. This is further substantiated by the finding that neither dopamine or norepinephrine antagonists attenuate the effect of amphetamine, ruling out an effect of amphetamine on transporters for these neurotransmitters as a mechanism for depressing EPSC amplitude. Historically, serotonin has received less attention than dopamine as a mediator of psychostimulant effects. However, lesions of serotonergic pathways increase rates of amphetamine self-administration, and increasing recent evidence suggests that serotonin may be an important player in the mechanisms underlying drug addiction (Lyness et al., 1980; Leccese and Lyness, 1984; Cameron and Williams, 1994; Obradovic et al., 1996; Parsons et al., 1998; Rocha et al., 1998a,b).
Our findings emphasize that one acute effect of amphetamine in the naïve VTA is activation of serotonin receptors, causing potent depression of glutamatergic synapses in the VTA. This result was unexpected, given the considerable evidence that dopamine mediates the rewarding effects of amphetamine in many animal models of drug addiction (Kalivas and Stewart, 1991; White, 1996). We speculate that, by modifying glutamatergic drive to VTA dopamine neurons, serotonin exerts important early modulatory effects on the mesocorticolimbic dopamine system after amphetamine administration.
Amphetamine modulates excitatory transmission onto both dopamine and nondopamine neurons
Previous work has demonstrated that prefrontal cortical afferents form excitatory synapses with both dopaminergic and nondopaminergic neurons within the VTA (Sesack and Pickel, 1992; Bonci and Malenka, 1999). In our study, evoked glutamatergic excitatory synaptic currents were recorded from both VTA cell types (identified physiologically and by TH immunoreactivity) after stimulation of presumed prefrontal cortical afferents to the VTA. Amphetamine depressed glutamatergic synaptic transmission onto both dopamine and nondopamine VTA neurons, indicating that overall excitatory drive to the VTA may be acutely depressed by psychostimulants.
Glutamatergic afferents drive VTA dopamine neurons
Glutamatergic afferents ordinarily have a profound effect on VTA dopamine cell activity. Stimulation of medial PFC elicits burst firing of midbrain dopamine cells in vivo (Gariano and Groves, 1988), and burst firing increases dopamine release at the terminal fields of VTA dopamine cells (Gonon, 1988; Bean and Roth, 1991). Conversely, blockade of glutamatergic synaptic transmission with kynurenic acid (Grenhoff et al., 1988) or cooling of the medial PFC (Svensson and Tung, 1989) changes the activity of dopamine cells from a burst firing pattern to a pacemaker pattern. Thus, stimulating excitatory transmission enhances dopaminergic activity in the mesocorticolimbic system, whereas depressing excitatory transmission reduces dopaminergic activity. Our results therefore suggest that a first exposure to amphetamine will reduce burst firing of dopamine neurons because of the blockade of excitatory transmission. This depression of excitatory transmission will act synergistically with the acute depression of dopamine cell spontaneous firing because of somatodendritic release of dopamine in the VTA (White, 1996) to decrease dopaminergic activity in this system.
Effects of amphetamine change with multiple exposures
It will be particularly interesting to test whether the inhibitory effects of an initial exposure to amphetamine will change during subsequent drug exposures, because evidence supports the idea that the effects of amphetamine are altered with repeated exposure. Thus, whereas a sensitizing regimen of amphetamine had no effect on midbrain dopamine cell firing or its modulation by PFC stimulation immediately after withdrawal, at 10 d after withdrawal, dopamine cell firing in response to PFC stimulation was potently enhanced (Tong et al., 1995). This correlates well with a report that amphetamine-induced dopamine release in the nucleus accumbens was not increased immediately after withdrawal from a sensitizing regimen but was increased 10–14 d later (Wolf et al., 1993). Intriguingly, a similar case has been presented for GABABreceptor-mediated inhibitory transmission in the VTA; D1 agonists enhance IPSPs in naïve animals but decrease IPSPs in sensitized animals (Cameron and Williams, 1993; Bonci and Williams, 1996). If depression of glutamatergic transmission was also reversed to potentiate glutamatergic transmission, this would provide a synergistic physiological mechanism to enhance dopamine cell excitability. Future studies will address the question of whether the effect of amphetamine changes as behavioral sensitization develops.
In conclusion, the present experiments demonstrate that amphetamine substantially depresses glutamatergic synaptic transmission to VTA cells via serotonin receptor activation. Our results suggest that serotonin is an important modulator of excitatory drive to VTA neurons and that a rapid early effect of psychostimulant exposure is release of this modulator. These experiments emphasize the importance of serotonergic systems in the complex modulation of elements in the reward pathway and provide new insight into the cellular effects of a drug abused by humans.
Footnotes
This work was supported by National Institutes of Health Grant DA11289.We thank Drs. Yong Li, Donald Lo, Lori McMahon, and Johanna Kornblum for helpful comments and discussion of this manuscript, and Andrew Pittman for excellent technical assistance.
Correspondence should be addressed to J. A. Kauer, Department of Neurobiology, Duke University Medical Center, Box 3209, Durham, NC 27710. E-mail: juliek@neuro.duke.edu.
REFERENCES
- 1.Bean AJ, Roth RH. Extracellular dopamine and neurotensin in rat prefrontal cortex in vivo: effects of median forebrain bundle stimulation frequency, stimulation pattern, and dopamine autoreceptors. J Neurosci. 1991;11:2694–2702. doi: 10.1523/JNEUROSCI.11-09-02694.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bonci A, Malenka RC. Properties and plasticity of excitatory synapses on dopaminergic and GABAergic cells in the ventral tegmental area. J Neurosci. 1999;19:3723–3730. doi: 10.1523/JNEUROSCI.19-10-03723.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bonci A, Williams JT. A common mechanism mediates long-term changes in synaptic transmission after chronic cocaine and morphine. Neuron. 1996;16:631–639. doi: 10.1016/s0896-6273(00)80082-3. [DOI] [PubMed] [Google Scholar]
- 4.Cameron DL, Williams JT. Dopamine D1 receptors facilitate transmitter release. Nature. 1993;366:344–347. doi: 10.1038/366344a0. [DOI] [PubMed] [Google Scholar]
- 5.Cameron DL, Williams JT. Cocaine inhibits GABA release in the VTA through endogenous 5-HT. J Neurosci. 1994;14:6763–6767. doi: 10.1523/JNEUROSCI.14-11-06763.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cameron DL, Williams JT. Opposing roles for dopamine and serotonin at presynaptic receptors in the ventral tegmental area. Clin Exp Pharmacol Physiol. 1995;22:841–845. doi: 10.1111/j.1440-1681.1995.tb01947.x. [DOI] [PubMed] [Google Scholar]
- 7.DiChiara G, Imperato A. Drugs abused by humans preferentially increase synaptic dopamine concentrations in the mesolimbic system of freely moving rats. Proc Natl Acad Sci USA. 1988;85:5274–5278. doi: 10.1073/pnas.85.14.5274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gariano RF, Groves PM. Burst firing induced in midbrain dopamine neurons by stimulation of the medial prefrontal and anterior cingulate cortices. Brain Res. 1988;462:194–198. doi: 10.1016/0006-8993(88)90606-3. [DOI] [PubMed] [Google Scholar]
- 9.Gonon FG. Nonlinear relationship between impulse flow and dopamine released by rat midbrain dopaminergic neurons as studied by in vivo electrochemistry. Neuroscience. 1988;24:19–28. doi: 10.1016/0306-4522(88)90307-7. [DOI] [PubMed] [Google Scholar]
- 10.Grace AA, Onn S-P. Morphology and electrophysiological properties of immunocytochemically identified rat dopamine neurons recorded in vitro. J Neurosci. 1989;9:3463–3481. doi: 10.1523/JNEUROSCI.09-10-03463.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Grenhoff J, Tung C-S, Svensson TH. The excitatory amino acid antagonist kynurenate induces pacemaker-like firing of dopamine neurons in rat ventral tegmental area in vivo. Acta Physiol Scand. 1988;134:567–568. doi: 10.1111/j.1748-1716.1998.tb08535.x. [DOI] [PubMed] [Google Scholar]
- 12.Johnson SW, North RA. Two types of neurone in the rat ventral tegmental area and their synaptic inputs. J Physiol (Lond) 1992;450:455–468. doi: 10.1113/jphysiol.1992.sp019136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kalivas PW. Neurotransmitter regulation of dopamine neurons in the ventral tegmental area. Brain Res Rev. 1993;18:75–113. doi: 10.1016/0165-0173(93)90008-n. [DOI] [PubMed] [Google Scholar]
- 14.Kalivas PW, Duffy P. Time course of extracellular dopamine and behavioral sensitization to cocaine. II. Dopamine perikarya. J Neurosci. 1993;13:276–284. doi: 10.1523/JNEUROSCI.13-01-00276.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kalivas PW, Duffy P. D1 receptors modulate glutamate transmission in the ventral tegmental area. J Neurosci. 1995;15:5379–5388. doi: 10.1523/JNEUROSCI.15-07-05379.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kalivas PW, Stewart J. Dopamine transmission in the initiation and expression of drug- and stress-induced sensitization of motor activity. Brain Res Rev. 1991;16:223–244. doi: 10.1016/0165-0173(91)90007-u. [DOI] [PubMed] [Google Scholar]
- 17.Kalivas PW, Weber B. Amphetamine injection into the ventral mesencephalon sensitizes rats to peripheral amphetamine and cocaine. J Pharmacol Exp Ther. 1988;245:1095–1101. [PubMed] [Google Scholar]
- 18.Leccese AP, Lyness WH. The effects of putative 5-hydroxy-tryptamine receptor active agents on d-amphetamine self-adminis-tration in controls and rats with 5,7-dihydroxytryptamine median forebrain bundle lesions. Brain Res. 1984;303:153–162. doi: 10.1016/0006-8993(84)90223-3. [DOI] [PubMed] [Google Scholar]
- 19.Lyness WH, Friedle NM, Moore KE. Increased self-administration of d-amphetamine after destruction of 5-hydroxytryptaminergic neurons. Pharmacol Biochem Behav. 1980;12:937–941. doi: 10.1016/0091-3057(80)90456-6. [DOI] [PubMed] [Google Scholar]
- 20.Obradovic T, Imel KM, White SR. Methylenedioxymeth-ampheatmine-induced inhibition of neuronal firing in the nucleus accumbens is mediated by both serotonin and dopamine. Neuroscience. 1996;74:469–481. doi: 10.1016/0306-4522(96)00135-2. [DOI] [PubMed] [Google Scholar]
- 21.Parsons LH, Weiss F, Koob GF. Serotonin1B receptor stimulation enhances cocaine reinforcement. J Neurosci. 1998;18:10078–10089. doi: 10.1523/JNEUROSCI.18-23-10078.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ritz MC, Kuhar MJ. Relationship between self-administration of amphetamine and monoamine receptors in brain: comparison with cocaine. J Pharmacol Exp Ther. 1989;248:1010–1017. [PubMed] [Google Scholar]
- 23.Ritz MC, Lamb RJ, Goldberg SR, Kuhar MJ. Cocaine receptors on dopamine transporters are related to self-administration of cocaine. Science. 1987;237:1219–1223. doi: 10.1126/science.2820058. [DOI] [PubMed] [Google Scholar]
- 24.Robinson TE, Berridge KC. The neural basis of drug craving: an incentive-sensitization theory of addiction. Brain Res Rev. 1993;18:247–291. doi: 10.1016/0165-0173(93)90013-p. [DOI] [PubMed] [Google Scholar]
- 25.Rocha BA, Fumagalli F, Gainetdinov RR, Jones SR, Ator R, Giros B, Miller GW, Caron MG. Cocaine self-administration in dopamine-transporter knockout mice. Nat Neurosci. 1998a;1:132–137. doi: 10.1038/381. [DOI] [PubMed] [Google Scholar]
- 26.Rocha BA, Scearce-Levie K, Lucas JJ, Hiroi N, Castanon N, Crabbe J C, Nestler ER, Hen R. Increased vulnerability to cocaine in mice lacking the serotonin-1B receptor. Nature. 1998b;393:175–178. doi: 10.1038/30259. [DOI] [PubMed] [Google Scholar]
- 27.Scheel-Kruger J. Comparative studies of various amphetamine analogues demonstrating different interactions with the metabolism of the catecholamines in the brain. Eur J Pharmacol. 1971;14:47–59. doi: 10.1016/0014-2999(71)90121-x. [DOI] [PubMed] [Google Scholar]
- 28.Seiden LS, Sabol KE, Ricuarte GA. Amphetamine: effects on catecholamine systems and behavior. Annu Rev Pharmacol Toxicol. 1993;32:639–677. doi: 10.1146/annurev.pa.33.040193.003231. [DOI] [PubMed] [Google Scholar]
- 29.Sesack SR, Pickel VM. Prefrontal cortical efferents in the rat synapse on unlabeled neuronal targets of catecholamine terminals in the nucleus accumbens septi and on dopamine neurons in the ventral tegmental area. J Comp Neurol. 1992;320:145–160. doi: 10.1002/cne.903200202. [DOI] [PubMed] [Google Scholar]
- 30.Seutin V, Verbanck P, Massotte L, Dresse A. Acute amphetamine-induced subsensitivity of A10 dopamine autoreceptors in vitro. Brain Res. 1991;558:141–144. doi: 10.1016/0006-8993(91)90731-a. [DOI] [PubMed] [Google Scholar]
- 31.Sibley DR, Monsma FJ. Molecular biology of dopamine receptors. Trends Pharmacol Sci. 1992;13:61–69. doi: 10.1016/0165-6147(92)90025-2. [DOI] [PubMed] [Google Scholar]
- 32.Spector S, Sjoerdsma A, Udenfriend S. Blockade of endogenous norepinephrine synthesis by α-methyl-tyrosine, an inhibitor of tyrosine hydroxylase. J Pharmacol Exp Ther. 1965;147:86–95. [PubMed] [Google Scholar]
- 33.Svensson TH, Tung C-S. Local cooling of pre-frontal cortex induces pacemaker-like firing of dopamine neurons in rat ventral tegmental area in vivo. Acta Physiol Scand. 1989;136:135–136. doi: 10.1111/j.1748-1716.1989.tb08640.x. [DOI] [PubMed] [Google Scholar]
- 34.Tong Z-Y, Overton PG, Clark D. Chronic administration of (+)-amphetamine alters the reactivity of midbrain dopaminergic neurons to prefrontal cortex stimulation in the rat. Brain Res. 1995;674:63–74. doi: 10.1016/0006-8993(94)01439-o. [DOI] [PubMed] [Google Scholar]
- 35.Vertes RP. A PHA-L analysis of ascending projections of the dorsal raphe nucleus in the rat. J Comp Neurol. 1991;313:643–668. doi: 10.1002/cne.903130409. [DOI] [PubMed] [Google Scholar]
- 36.Vezina P. Amphetamine injected into the ventral tegmental area sensitizes the nucleus accumbens dopaminergic response to systemic amphetamine: an in vivo microdialysis study in the rat. Brain Res. 1993;605:332–337. doi: 10.1016/0006-8993(93)91761-g. [DOI] [PubMed] [Google Scholar]
- 37.Vezina P, Stewart J. Amphetamine administered to the ventral tegmental area but not to the nucleus accumbens sensitizes rats to systemic morphine: lack of conditioned effects. Brain Res. 1990;516:99–106. doi: 10.1016/0006-8993(90)90902-n. [DOI] [PubMed] [Google Scholar]
- 38.White FJ. Synaptic regulation of mesocorticolimbic dopamine neurons. Annu Rev Neurosci. 1996;19:405–436. doi: 10.1146/annurev.ne.19.030196.002201. [DOI] [PubMed] [Google Scholar]
- 39.Wolf ME. The role of excitatory amino acids in behavioral sensitization to psychomotor stimulants. Prog Neurobiol. 1998;54:679–720. doi: 10.1016/s0301-0082(97)00090-7. [DOI] [PubMed] [Google Scholar]
- 40.Wolf ME, Xue C-J. Amphetamine and D1 dopamine receptor agonists produce biphasic effects on glutamate efflux in rat ventral tegmental area: modification by repeated amphetamine administration. J Neurochem. 1998;70:198–209. doi: 10.1046/j.1471-4159.1998.70010198.x. [DOI] [PubMed] [Google Scholar]
- 41.Wolf ME, White FJ, Nassar R, Brooderson RJ, Khansa MR. Differential development of autoreceptor subsensitivity and enhanced dopamine release during amphetamine sensitization. J Pharmacol Exp Ther. 1993;264:249–255. [PubMed] [Google Scholar]
- 42.Wolf ME, White FJ, Hu X-T. MK-801 prevents alterations in the mesoaccumbens dopamine system associated with behavioral sensitization to amphetamine. J Neurosci. 1994;14:1735–1745. doi: 10.1523/JNEUROSCI.14-03-01735.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Xue C-J, Ng JP, Li Y, Wolf ME. Acute and repeated systemic amphetamine administration: effects on extracellular glutamate, aspartate, and serine levels in rat ventral tegmental area and nucleus accumbens. J Neurochem. 1996;67:352–363. doi: 10.1046/j.1471-4159.1996.67010352.x. [DOI] [PubMed] [Google Scholar]