

Abstract
Optic nerve formation in mouse involves interactions between netrin-1 at the optic disk and the netrin-1 receptor DCC (deleted in colorectal cancer) expressed on retinal ganglion cell (RGC) axons. Deficiency in either protein causes RGC pathfinding defects at the disk leading to optic nerve hypoplasia (Deiner et al., 1997). Here we show that further along the visual pathway, RGC axons in netrin-1- or DCC-deficient mice grow in unusually angular trajectories within the ventral hypothalamus. In heterozygousSeyneu mice that also have a small optic nerve, RGC axon trajectories appear normal, indicating that the altered RGC axon trajectories in netrin-1 and DCC mutants are not secondarily caused by optic nerve hypoplasia. Intrinsic hypothalamic patterning is also affected in netrin-1 and DCC mutants, including a severe reduction in the posterior axon projections of gonadotropin-releasing hormone neurons. In addition to axon pathway defects, antidiuretic hormone and oxytocin neurons are found ectopically in the ventromedial hypothalamus, apparently no longer confined to the supraoptic nucleus in mutants. In summary, netrin-1 and DCC, presumably via direct interactions, govern both axon pathway formation and neuronal position during hypothalamic development, and loss of netrin-1 or DCC function affects both visual and neuroendocrine systems. Netrin protein localization also indicates that unlike in more caudal CNS, guidance about the hypothalamic ventral midline does not require midline expression of netrin.
Keywords: optic chiasm, netrin, DCC, hypothalamus, gonadotropin-releasing hormone, luteinizing hormone-releasing hormone, antidiuretic hormone, vasopressin, supraoptic nucleus, oxytocin, visual system, retinal ganglion cell, axon guidance, cell migration, diencephalon, CD44, organum vasculosum of the lamina terminalis
Despite the importance of the hypothalamus in neuroendocrine and autonomic function, little is known of the developmental mechanisms that give rise to the complex patterns of neuronal connectivity in this anterior CNS region. Most neurons of the neuroendocrine hypothalamus are born starting at approximately embryonic day 10.5 (E10.5) and subsequently migrate away from the ventricular zone to form hypothalamic neuronal groups (nuclei) distinguished by expression of specific neuropeptides or hormone-releasing factors (Altman and Bayer, 1978a,b). Many of these neurons then send axons posteriorly to the median eminence (ME), infundibulum, or posterior pituitary to regulate hormonal release into the bloodstream (for review, see Swanson and Sawchenko, 1983; Ganten et al., 1986; Swanson, 1986). As these events occur, retinal ganglion cell (RGC) axons enter the hypothalamus from the optic nerves, cross the ventral midline to form a commissure called the optic chiasm, and then continue onward forming the optic tracts. Because development of hypothalamic neurons and RGC axon pathways are spatiotemporally intermixed, similar mechanisms and molecular cues may govern both visual and neuroendocrine development in this CNS region.
Caudal to the hypothalamus, cell differentiation and axon guidance about the ventral midline of the developing hindbrain and spinal cord appear to be organized by activities derived from midline structures such as the notochord and floor plate (for review, see Dodd et al., 1998). An important CNS ventral midline guidance cue is netrin-1, a diffusible protein secreted by ventral midline floor plate cells that can attract commissural axons (Kennedy et al., 1994; Serafini et al., 1994; Shirasaki et al., 1996), and animals deficient in netrin-1, or the netrin-1 receptor DCC (deleted in colorectal cancer), lack or have severely reduced ventral midline commissures (Serafini et al., 1996;Fazeli et al., 1997). Because the notochord and floor plate are not clearly morphologically defined at the ventral diencephalon midline in comparison with the hindbrain and spinal cord, it is unknown whether axon guidance and cell patterning about the ventral hypothalamic midline involve cues such as netrin-1 or use novel mechanisms.
Here we investigated whether netrin-1 or DCC are involved in mouse hypothalamic development. The results show netrin-1 and DCC are required for normal development of RGC axon trajectories during chiasm formation and for development of gonadotropin-releasing hormone (GnRH) axon pathways. Furthermore, in the absence of netrin-1 or DCC, antidiuretic hormone (ADH) and oxytocin neurons of the supraoptic nucleus (SON) appear to migrate abnormally into the chiasm region. Thus, specific features of neuronal connectivity about the ventral hypothalamic midline are governed by netrin-1 and DCC. However, unlike in spinal cord and hindbrain, netrin does not appear to be expressed at the ventral hypothalamic midline and therefore cannot be used by the CNS as a midline cue at this location. Hypothalamic developmental abnormalities can now be added to corpus callosum defects (Serafini et al., 1996; Fazeli et al., 1997) and optic nerve hypoplasia (Deiner et al., 1997) in netrin-1 and DCC mutant mice. This trio of seemingly disparate phenotypes is reminiscent of the human congenital multisystem syndrome of septo-optic dysplasia (Skarf and Hoyt, 1984;Brodsky et al., 1996).
MATERIALS AND METHODS
Netrin-1-, DCC-, Pax6-, and GAP-43-deficient mice.Production, breeding, and genotyping of the four mutant mouse strains used have been described previously. Netrin-1–deficient mice were produced via targeted deletion of the netrin-1 gene resulting in a fusion transcript comprised of all of domain VI and the first epidermal growth factor–like repeat of domain V of netrin-1 fused to the transmembrane domain of CD4 and the cytoplasmic domain of β-geo (Skarnes et al., 1995; Serafini et al., 1996). These mice were originally generated in a CD-1 background, and mice used in the present study were derived by mating with a number of outbred strains. DCC-deficient mutant mice were generated by a targeted deletion of exon 3 of the DCC gene reported to result in complete loss of DCC protein expression (Fazeli et al., 1997). These mice were originally generated in a 129/Sv background, and mice used in the present study were derived by mating with a number of different strains including C57/bl6 and CD-1. GAP-43–deficient mutant mice were produced by a targeted deletion of exon 2 of the GAP-43 gene that encodes 85% of the protein-coding sequence (Strittmatter et al., 1995), and the GAP-43 mice used in the present study were in a C57/bl6 background. Pax6Seyneu mice arose as a naturally occurring point mutation predicted to disrupt Pax6 gene function (Hill et al., 1991), and mice used in the present study were in a C57/bl6 background. The processing of tissue from these litters for immunostaining and for DiI labeling of RGC axons was performed as described below for wild-type C57/bl6 mice.
Orientation terminology. During mouse embryogenesis at ∼E8, a 90° turn occurs along the dorsal–ventral axis to create the cephalic flexure such that rostral CNS anterior to the flexure comes to lie ventrally. Because the present study describes results after the formation of the flexure and around birth, we have adopted the orientation terminology used conventionally in both the vision and the neuroendocrine literature. Anterior therefore refers to the direction toward the nose, and dorsal is toward the top of the head.
Antibodies. Anti-DCC [rabbit polyclonal; 1:3000; gift of Dr. E. Fearon, University of Michigan Medical Center (Reale et al., 1996)]; anti-DCC (mouse mAb; 5 μg/ml; PharMingen, San Diego, CA; 15041A); anti–pan-netrin [rabbit polyclonal 11760; 1:50; raised using a peptide antigen corresponding to sequence conserved in chick netrin-1, chick netrin-2, and mouse netrin-1 (a gift from T. Kennedy and M. Tessier-Lavigne; also see Deiner et al., 1997)]; anti-CD44 [rat mAb KM201 (IgG); 1:3 (Miyake et al., 1990)]; anti-GnRH (rabbit polyclonal; 1:2500; gift of W. Vale, Salk Institute; PBL, L45); anti–corticotropin-releasing hormone (CRH; rabbit polyclonal; 1:500; gift of W. Vale; PBL, rC70); anti-ADH (rabbit polyclonal; 1:250; Chemicon, Temecula, CA; AB937, AB1565); anti–adrenocorticotropic hormone [ACTH; rabbit polyclonal; 1:1200; gift of National Hormone and Pituitary Program (NHPP); lot AFP39013082]; anti–growth hormone (GH; guinea pig polyclonal; 1:1200; gift of Dr. A. F. Parlow, Harbor-University of California, Los Angeles, Medical Center; lot AFP11121390); anti–follicle-stimulating hormone (FSH; rabbit polyclonal; 1:1000; gift of Dr. A. F. Parlow; lot AFP131078); and anti–luteinizing hormone (LH; guinea pig polyclonal; 1:1000; gift of NHPP; lot AFP22238790GPOLHBB).
Immunohistochemistry. E12–E18 embryos (plug day = E0) were harvested from anesthetized C57/bl6 timed pregnant mice (Sretavan et al., 1994). For immunostaining using 10–20 μm coronal cryostat sections, E12–E18 heads were fixed in 4% paraformaldehyde in 0.1m phosphate buffer, pH 7.25 (PFA), for 1 hr at 4°C, infiltrated overnight in 30% sucrose in PBS, embedded in ornithine carbamyl transferase (OCT; Baxter, McGaw Park, IL), and stored at −80°C until use. Postnatal day 0 (P0) animals were first perfusion fixed with 4% PFA, immersion fixed in 4% PFA 1–2 d, and infiltrated 1–5 d in 30% sucrose in PBS before embedding in OCT. For immunostaining using paraffin sections, heads were fixed using Carnoy's fixative (Bancroft and Stevens, 1982) and then embedded in EM-400 wax (Surgipath, Richmond, IL) until use. For immunostaining using 100-μm-thick vibratome sections, heads were fixed in 4% PFA, RGC axons were labeled with DiI as described below, and heads were then embedded in 3% agarose in PBS.
Immunolabeling of DCC, CD44, ADH, GnRH, CRH, GH, ACTH, FSH, and LH was performed on cryostat or paraffin sections preblocked in heat-inactivated normal goat or donkey serum (HINGS or HINDS, respectively) for 20 min. Incubations with primary antibody were performed at room temperature (RT) for 1–2 hr followed by five washes in 0.1 m PBS (4 min/wash). Secondary antibody was applied for 1–2 hr at RT; the sections were then washed five times before coverslipping. All antibody incubations were performed in the presence of 0.1% Triton X-100 and either 1% HINDS or 1% HINGS for blocking. In some cases, a second 10% HINGS or 10% HINDS 20 min blocking step was performed before addition of the secondary antibody. Netrin immunolabeling in 5- to 10-μm-thick paraffin sections was performed as described previously (Deiner et al., 1997). The detection of primary antibodies was performed using the appropriate secondary antibodies conjugated to either Cy2 or Cy3 (Jackson ImmunoResearch, West Grove, PA). For visualization using alkaline phosphatase, sections were labeled with the appropriate alkaline phosphatase–tagged secondary antibody and incubated with the color substrate BM Purple (Boehringer Mannheim, Indianapolis, IN). CD44 immunostaining in whole mounts was as described in Sretavan et al. (1994) using the Vectostain-ABC kit (Vector Laboratories, Burlingame, CA). CD44 immunostaining using 100-μm-thick sagittal sections was also as described in Sretavan et al. (1994) except that all antibody incubations, washes, and reactions were performed in 24-well plates.
DiI labeling and angle measurements of RGC axons.Anterograde labeling of axons in the optic nerves with DiI (D-282; Molecular Probes, Eugene, OR) was performed as described in Sretavan et al. (1994). After overlying tissue was dissected to reveal labeled axons on the hypothalamic surface, preparations were aligned for imaging using the anterior edge of the pituitary and the midline as landmarks. Labeled axons were imaged as described below (Digital imaging). By the use of these images, the amount of axon turning at two sites (designated angles 1 and 2; see Fig. 3c for depiction) was measured, in degrees, using Adobe Photoshop. Measurements were made without knowledge of genotype, and similar values were obtained when an image was remeasured on separate days and by different observers. Angles 1 and 2 were measured in embryos from E12.5–E15.5. Individual results from each animal were pooled into wild-type, heterozygous, and homozygous categories to obtain a mean turning angle expressed in degrees for each genotype. The results from netrin-1– and DCC-deficient animals were combined, because the results were very similar. At all ages, the mean turning angle in mutants was always greater (sharper posterior turn in mutants for angle 1 and sharper anterior turn in mutants for angle 2) compared with mean turning angles measured in wild-type littermates [shown for E14–E15 (see Fig. 3d)].
Fig. 3.

Altered RGC axon trajectories at the ventral hypothalamus of DCC- or netrin-1–deficient mouse embryos. a, b, Each panel shows images of DiI-labeled RGC axons at the ventral hypothalamic region in wild-type (top, green), heterozygous (middle, green), and mutant (bottom, red) littermate E14 embryos. The retina of origin is toward the topleft,and the optic tract is toward the bottomright. The arrow indicates the orientation (anterior is at the top), and thewhitedot indicates the midline.c, The schematic diagram shows the location of the angles 1 and 2 measured in RGC axon projections, for example, shown here for the projections of wild-type (top) and mutant (bottom) embryos of a.d, The graph shows angles 1 and 2 in heterozygous (Het; columns 1 and 2) or mutant (Mut; columns 3 and 4) embryos compared with wild-type (WT) embryos at E14–E15. Data from netrin and DCC embryos have been pooled. The amount in degrees by which angles 1 and 2 in heterozygotes or in mutants exceed angles 1 and 2 in wild-type embryos is plotted on the y-axis.Asterisks indicate a significant difference was found in mutants (but not heterozygotes) compared with wild type (Student'st test; p < 0.002 for mutants;p > 0.2 for heterozygotes; n = 5 wild type, 9 heterozygous, and 10 mutants). Blackbars, angle 1; graybars, angle 2. e, In the chiasm of wild-type embryos, a subset of RGC axons (asterisk; shown as a negative image to highlight axons) crosses the midline posterior to the main RGC axon bundle. Theinset shows the outline of the main bundle in the chiasm (darkgray), and the redbox indicates the approximate region from which the image in e was taken. In many cases of netrin-1 or DCC mutant embryos, RGC axons crossed the midline at a point that correlates with this posterior region. f, DiI-labeled RGC axons at the ventral hypothalamus of E14 wild-type (green) and Pax6Seyneu/+ heterozygous (red) littermate embryos are shown. Although optic nerve size in Seyneu/+ heterozygous embryos is reduced, abnormally angular RGC axon trajectories such as those seen in netrin-1 or DCC mutants were not observed. g, The graph shows that angles 1 and 2 inSeyneu/+ heterozygous animals were not significantly different from those present in wild-type littermates (y-axis same as in d). Scale bars:a, b, f, 250 μm; e, 100 μm.
Digital imaging. DiI and Cy3 fluorescence was visualized using rhodamine fluorescence optics, and Cy2 was visualized using fluorescein optics. Images of the immunolabeling patterns in tissue sections and whole mounts were captured digitally using a Photometrics PXL2 cooled CCD camera or an Optronics color CCD camera and processed for presentation using Adobe Photoshop and/or Illustrator. For all wild-type and mutant image pairs, images were collected using the same camera exposure times and processed simultaneously in Adobe Photoshop, allowing comparison between images. For multiple serial section reconstructions, ADH neurons in every third 20 μm section were imaged digitally, and Adobe Illustrator was used to trace the position of labeled neurons. Tracings of serial sections were then assembled into one single image file.
GH and ACTH assays. P0 wild-type and mutant pups were decapitated, and 20–30 μl of blood was collected in the presence of 1 μl of anticoagulant. Older ages were not examined because mutants die soon after birth. Blood was immediately centrifuged for 5 min at 3000 × g; serum was collected, either diluted in water (ACTH assay) or undiluted (GH RIA), and snap frozen. ACTH levels were assayed at the University of California, San Francisco, Clinical Laboratories, Department of Laboratory Medicine using an antibody-based chemiluminescence assay system, the ACTH 100T kit from Nichols Institute Diagnostics (catalog #60-4175; San Juan Capistrano, CA). GH levels were measured by radioimmunoassay using125I-labeled mouse growth hormone, antiserum to rat growth hormone, and a mouse growth hormone standard generously supplied by Dr. A. F. Parlow (Harbor-University of California, Los Angeles, Medical Center).
RESULTS
Netrin-1 and DCC expression in the developing hypothalamus
Immunostaining using a polyclonal pan-netrin antibody [from T. E. Kennedy; see Deiner et al. (1997)] revealed netrin protein localization in discrete regions within the E12–E16 embryonic mouse hypothalamus. These included bilateral patches extending inward from the pial surface to deeper zones within the hypothalamus (Fig.1a,c,d) and in the ventricular zone (Fig. 1e). In contrast, DCC was much more widely expressed throughout the hypothalamus but was noticeably absent at a few locations including the ventricular zone (Fig. 1g–i). Below, we more fully describe the patterns of netrin and DCC expression in relation to the development of RGC, GnRH, ADH, and oxytocin neurons. This is followed by an analysis of how the development of these neurons and their axons is affected when netrin-1 or DCC function is disrupted.
Fig. 1.

DCC and netrin expression in the E12 mouse hypothalamus. The schematicdiagrams on the left show the eyes, optic nerves, and ventral hypothalamus. The redrectangle indicates the anterior–posterior hypothalamic level at which tissue sections were obtained for netrin (middle) and DCC (right) immunostaining. Tissue sections separated by 20–30 μm were used for netrin and DCC immunostaining at each level. In all panels, dorsal is at the top.a–f, Netrin (arrowheads) was found in bilateral patches at different sites along the anterior–posterior length of the hypothalamus. a, Netrin expression at the ventral medial septal and diagonal band region is shown. Thearea of the dottedrectangle is shown at higher magnification in theinset at the topright. Staining in blood cells and vessels is nonspecific. b,Note the absence of netrin at the ventral midline region. c, d, Netrin is expressed at the ventral–lateral hypothalamic regions corresponding approximately to where axons of chiasm neurons (see Fig. 2) grow dorsally to join the TPOC and where the initial portions of the optic tract will later develop. There is no apparent ventral midline netrin expression. e, Netrin expression in the ventricular zone is shown. The area of thedottedrectangle is shown at higher magnification in the inset at the topright. Such bilateral patches of netrin were found in the ventricular zone region scattered throughout the anterior–posterior length of the hypothalamus. f,Netrin expression in the presumptive lateral posterior ME region is shown. g–l, DCC expression at different levels along the anterior–posterior length of the hypothalamus is shown. DCC was found in specific domains distributed throughout the hypothalamus but was absent from the ventricular zones. D, Dorsal;DB, diagonal band; ME, median eminence;MS, medial septum; OC, future chiasm region; OT, future region of the optic tract;POA, preoptic area. Scale bar, 200 μm.
Relationship to RGC axons and chiasm neurons
At E12.5, the first RGC axons have arrived at the ventral hypothalamus and have begun to form the chiasm. As the RGC axons enter the midline region, they encounter optic chiasm neurons that are found along the posterior boundary of the future chiasm and are organized into an inverted V-shaped array (for review, see Mason and Sretavan, 1997). These embryonic optic chiasm neurons express the cell surface protein CD44 (Sretavan et al., 1994) and the SSEA-1 epitope (Marcus et al., 1995) and send processes dorsally into the tract of the postoptic commissure (TPOC) (Easter et al., 1993; Sretavan et al., 1994). During development, RGC axons grow through the chiasm midline region along the anterior edge of this CD44/SSEA-positive neuronal population (referred to in this paper as chiasm neurons), and RGC axons forming the optic tract grow immediately anterior to chiasm neuron axons suggesting chiasm neurons form a posterior border for the RGC axons (Sretavan et al., 1994; Marcus and Mason, 1995). Experimental evidence also shows that chiasm neurons are required for RGC axons to cross the chiasm midline (Sretavan et al., 1995).
Starting from E12 onward, netrin was expressed bilaterally along the optic nerves (see Fig. 2a) and just posterior to the attachment point of the optic nerves to the brain, lateral to the midline (see Fig. 1c). The location of this lateral zone of netrin protein expression approximately correlated with the location of the chiasm neuron axons and the future position of the initial portions of the optic tracts. By E14–E16, although similar in overall pattern, the levels of netrin expression were greatly reduced. During RGC axon ingrowth and chiasm formation, netrin was not detected at the ventral midline in the hypothalamus (Fig.1a–f). The early pattern of hypothalamic netrin protein expression reported here is similar to that of netrin mRNA (T. Serafini, personal communication), suggesting that the immunostaining pattern accurately reflects netrin protein distribution.
Fig. 2.
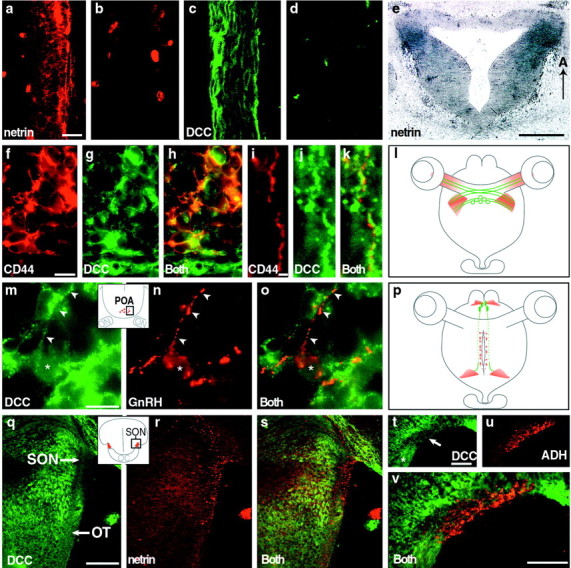
Relationship of netrin and DCC expression to RGC axons, chiasm neurons, and GnRH and ADH neurons. a,Netrin expression in the optic nerve at E14 (the retina is toward thetop). b, Immunostaining of an adjacent optic nerve section with anti-netrin antibody omitted. Note nonspecific staining of blood cells. c, DCC expression on RGC axons within the E14 optic nerve. d, Immunostaining of an adjacent optic nerve section with anti-DCC antibody omitted.e, E12.5 horizontal section dorsal to where the optic nerves attach to the hypothalamus showing netrin (darkblue) bilaterally in the approximate future region of the optic tract (see also Fig. 1c,d). f,E12.5 coronal section through the region of CD44-immunopositive chiasm neurons (red). g, The same section as inf showing that DCC (green) is expressed by many neurons in this area. h, Overlay of images in f and g showing colocalization of CD44 and DCC on neurons in this area. Most CD44-positive cells express DCC. i, CD44 expression (red) on a dorsolaterally projecting axon of a chiasm neuron. j,The same section as in i showing expression of DCC (green) in the same region. k,Overlay of images in i and j showing DCC expression on CD44-positive axons. l, Summary diagram of netrin expression (red) with respect to the developing RGC axon and chiasm neuron pathway. DCC expression on RGC axons and chiasm neurons and axons is depicted in green.m, E18 Sprague Dawley rat coronal sections through the POA showing DCC-immunopositive neurons and axons. Inset,The approximate region (boxedarea) shown in m–o. Similar results were seen in E16 mouse POA (data not shown). n, The same section as inm showing GnRH expression (red) in a neuron cell body (*) and axon (arrowheads).o, Overlay showing cellular colocalization of DCC and GnRH. p, Summary diagram showing netrin expression (red) in the position where many GnRH neuron cell bodies reside (the preoptic area) and along the pathway taken by many GnRH axons as they grow toward the ME (dorsal regions adjacent to the third ventricle). DCC expression on GnRH neurons and their axons is depicted in green. q, E14 coronal section at the level of the SON showing DCC expression (green) in the tissue surrounding but not within the SON. Inset,The approximate region (boxedarea) shown in higher magnification in q–s. r, A section adjacent to q showing netrin expression (red) within and just outside of the SON.s, Superimposition of q ands. t, E16 coronal section showing DCC expression in tissue surrounding the SON (arrow), including the region of the optic tract (asterisk).u, Adjacent section showing ADH-immunopositive neurons (red) within the SON. v, Superimposition of t and u. A, Anterior. Scale bars: a–d, 25 μm;e, 200 μm; f–h, 10 μm;i–k, 1 μm; m–o, 10 μm;q–s, 100 μm; t, u, 100 μm;v, 100 μm. OT, Future region of the optic tract; POA, preoptic area; SON, supraoptic nucleus.
RGC axons expressed DCC during their course within the optic nerve (Fig. 2c), the chiasm, and the optic tract (Fig.2q,t). Chiasm neurons and their axons also expressed DCC (Fig. 2f–k) and therefore, like RGC axons, may also be capable of responding to netrin. The expression data (summarized in Fig. 2l) indicate that RGC axons do not grow toward the midline and form the chiasm in response to netrin secreted from the hypothalamic midline. However, the bilateral patches of netrin expression in the optic nerve and the vicinity of the initial optic tract region suggest that netrin and DCC interactions could mediate some aspects of RGC or chiasm neuron axon pathway development in the ventral hypothalamus.
GnRH neurons and axon pathways
GnRH neurons originate in the olfactory placode and then migrate along olfactory axons to the mediobasal olfactory bulb where they enter the brain and take up final residence in several hypothalamic regions including the medial septum, diagonal band, and preoptic areas (Schwanzel-Fukuda and Pfaff, 1989; Wray et al., 1989; Livne et al., 1993). From ages E13.75 through birth (Wu et al., 1997), GnRH neurons residing in these regions send axons posteriorly along both ventro- and dorsomedial routes (Hoffman and Gibbs, 1982) to terminate in the ME lateral contact zone and subsequently regulate gonadotropin release. Netrin was found at the ventral medial septal and presumptive diagonal band regions (see Fig. 1a), where some migrating GnRH neurons take up final residence in the brain. In addition, bilateral patches of netrin protein were present in regions adjacent to the ventricles along the anterior–posterior length of the hypothalamus (see Fig. 1e) as well as in the lateral posterior regions of the presumptive ME (see Fig. 1f), a pattern reminiscent of the pathway taken by GnRH axons. Many GnRH neurons and their axons expressed DCC (Fig. 2m–o). The expression of DCC on GnRH neurons (see summary in Fig. 2p) together with the presence of netrin protein at the final destination of migrating GnRH neurons suggests that netrin and DCC interactions could play a role in directing cellular migration of GnRH neurons into the brain. In addition, netrin expression along GnRH axon pathways and in the axon target region (the presumptive ME) also suggests that netrin and DCC interactions could play a role in GnRH axon pathfinding.
Netrin, DCC, and SON development
ADH and oxytocin neurons destined to form the SON are thought to arise in the ventral diencephalon ventricular zone and migrate laterally to form a well-defined nucleus that is bounded along its ventromedial border by the optic tract (Karim and Sloper, 1980; Okamura et al., 1983). Axons of ADH and oxytocin neurons within the SON extend ventrocaudally to the midline region and then grow posteriorly through the ME internal zone into the posterior pituitary. There, ADH and oxytocin released into the circulation regulate blood pressure, osmolality, and volume (ADH), as well as parturition and lactation (oxytocin). At E14, when the SON is just beginning to form, netrin was expressed within the developing SON (see Fig. 2r), while DCC was expressed in the surrounding tissue (see Fig. 2q). This pattern was found in subsequent stages of development after many ADH neurons had appeared in the SON (see Fig. 2t–v). The absence of DCC expression in the SON was verified using both polyclonal and monoclonal anti-DCC antibodies. Thus developing neurons within the SON are notable for the absence of DCC even though netrin is apparently produced within the SON itself.
Abnormal RGC axon trajectories in netrin-1 and DCC mutants
We investigated and compared hypothalamic development between netrin-1– and DCC-deficient mice to identify developmental events dependent on netrin-1 and DCC interactions. On the basis of the pattern of netrin-1 and DCC expression and because RGC axons respond to netrin-1 in vitro (Wang et al., 1996; Deiner et al., 1997;de la Torre et al., 1997), we first examined chiasm formation. To do this, we labeled RGC axons with DiI and visualized their trajectories in ventral hypothalamus in whole-mount preparations. Images of wild-type and mutant RGC axon pathways were aligned for analysis using the anterior edge of the pituitary and the ventral midline as markers. In netrin-1–deficient and in DCC-deficient mice, a smaller number of RGC axons appeared to reach the chiasm, consistent with the fact that fewer RGC axons exit the eye in these embryos (Deiner et al., 1997). After entering the hypothalamus, RGC axons grew along abnormally angular trajectories in all netrin-1 and DCC mutant embryos compared with wild-type and heterozygous littermates (see Fig.3a,b) and sometimes appeared possibly to extend across the midline at a more posterior position.
To analyze the altered RGC axon trajectories, we measured two RGC axon-turning angles in wild-type, heterozygous, and mutant littermates (see Fig. 3c). Because this phenotype appeared quite similar in both the netrin-1 and DCC mutants (compare Fig. 3a withb), we pooled the results from both sets of mutants. At E14–E15, both turning angles were significantly greater in netrin-1 and DCC mutants compared with wild-type littermates [p< 0.002 using Student's t test; n = 5 wild type and 10 pooled netrin-1 and DCC mutants; similar results found at E12–E13 (data not shown)]. For example, at E14–E15, compared with those in wild type, the turning angles in mutants were on average 23° greater at angle 1 (Fig. 3d, black bars) and 15° greater at angle 2 (Fig. 3d, gray bars). In E14–E15 heterozygotes, both turning angles were not significantly different compared with those in wild-type littermates (p > 0.2 using Student's t test;n = 5 wild type and 9 heterozygotes).
During normal development, early arriving RGC axons run in close apposition to chiasm neurons. As many more RGC axons continue to enter the hypothalamus to form the chiasm, RGC axons appear to be added anteriorly such that the main bundle of RGC axons is increasingly separated from the chiasm neurons (Sretavan et al., 1994; Marcus and Mason, 1995). Of note, however, a number of RGC axons remain posteriorly (see Fig. 3e, asterisk) in between the main RGC axon bundle and the chiasm neurons. In some netrin-1 and DCC mutant embryos, the RGC axon bundle appeared to cross the midline at a posteriorly shifted position (Fig. 3a,b). This position most likely falls within the region where the small number of posterior RGC axons are normally found in the wild-type chiasm. This suggests that the apparent posterior position of the RGC axon bundle in mutants may have resulted from a reduced addition of axons anteriorly. Of note, RGC axons in netrin-1– or DCC-deficient embryos entered the hypothalamus at approximately the correct position, and despite the abnormally angular trajectories about the midline, RGC axons appeared to readjust and form the optic tract at a normal position in mutants (Fig. 3a,b). Thus, the loss of netrin-1 or DCC affects the RGC axon pathway specifically in the region of the chiasm. This defect could reflect either a direct or an indirect role for netrin-1 and DCC interactions. Because netrin-1 was not expressed at the midline region, netrin-1 interacting with DCC on RGC axons in the proximal portion of the optic nerve could directly affect RGC axon trajectories through the optic chiasm. Alternatively, netrin-1 could interact with DCC to regulate another aspect of CNS development that is altered in mutant mice, indirectly leading to the altered RGC axon trajectories. Such potential causes could include altered development of the cells making up this midline optic chiasm region or simply could be caused by the optic nerve hypoplasia present in the mutant mice. We further investigated these possibilities below.
Angular RGC axon trajectories are independent of a small optic nerve
Because a smaller number of RGC axons exit the eye in netrin-1 or DCC mutants, we examined whether abnormal RGC axon chiasm trajectories could simply be a consequence of optic nerve hypoplasia. To investigate, we studied chiasm development inSeyneu mice that have a mutated Pax6 gene (Hill et al., 1991), a paired box transcription factor expressed early in eye development (Walther and Gruss, 1991).Seyneu/+ heterozygotes have a small eye that we reasoned should lead to a reduced number of RGC axons in the optic nerve. This was confirmed in our analysis that showed that the diameter of the optic nerve in Seyneu/+heterozygotes was reduced by 20–30% compared with normal (see Fig.3f). In no case, however, did we observe abnormally angular RGC axon trajectories, and angles 1 and 2 were not significantly different in Seyneu/+heterozygotes compared with wild-type E14–E15 littermates (p > 0.2; Student's t test;n = 5 wild type and 10 heterozygotes; see Fig.3f,g). Therefore, a small optic nerve by itself does not appear to lead to the abnormal RGC axon trajectories in the chiasm region. Instead, abnormal RGC axon trajectories in netrin-1 and DCC mutants are more likely caused by the disruption of local hypothalamic developmental events dependent on netrin-1 and DCC function.
Development of chiasm neurons
One local neuronal population that can influence RGC axon trajectories is chiasm neurons. Because chiasm neurons express DCC, and netrin was expressed in the region traversed by their axons, it was possible that defects in chiasm neuron development in DCC or netrin–1 mutants may have secondarily caused the abnormal RGC axon trajectories. Analysis of chiasm neuron development after CD44 immunostaining and DiI labeling in E12 littermates showed, however, that these neurons were arrayed normally in an inverted V-shape (compare Fig.4a with b) and sent axons dorsally in mutants, similar to the pattern seen in wild-type embryos [compare Fig. 4c with d; DiI data not shown; also see Sretavan et al. (1994)]. The relationship of chiasm neurons to DiI-labeled RGC axons was analyzed at E14 in sagittal section. In midsagittal sections in both wild-type and mutant embryos, CD44-positive neurons at the midline appeared as curve-shaped cell groups (compare Fig. 4e with f,arrows). Furthermore, the distance between these cell groups and the most posterior extent of the RGC axon bundle was approximately the same in wild-type (Fig. 4g) and mutant (Fig.4h) littermates. These results suggested that altered RGC axon trajectories in mutants were not secondarily caused by grossly abnormal chiasm neuron development.
Fig. 4.

Chiasm neuron development in DCC- or netrin-1–deficient mouse embryos. a, b, E12 hypothalamic whole-mount preparations showing CD44 immunostaining of the inverted V-shaped array of chiasm neurons (arrowheads) in heterozygous (HET;a) and netrin-1 mutant (MUT;b) littermates. These inverted V-shaped patterns are similar to that present in wild-type embryos (Sretavan et al., 1994).c, d, CD44 immunostaining of E12.5 coronal sections in wild type (c) and netrin-1 mutant littermates (d) showing CD44-positive neuron cell bodies ventromedially (asterisks) and their axons (arrowheads) that project dorsolaterally. e, f, DiI-labeled RGC axons and CD44-labeled chiasm neurons in midsagittal sections of E14 wild type (e) and DCC mutant (f) littermates. In both, the RGC axons appear as red bundles, whereas the labeled CD44 neurons appear as a lightbrowncurve-shapedgroup(blackarrows). In e andf, sections through the posterior (singleasterisks) and anterior (doubleasterisks) pituitary are visible on theright. Verydarkblackcells are blood cells. In wild-type animals, the distance between the posterior edge of the CD44 neurons and the anterior edge of the pituitary was somewhat variable. A similar degree of variability was seen in the mutants. g, h,Tracings showing the relationship between the posterior portion of the RGC axon bundles and the CD44 neuron array in midline sagittal sections of several wild-type (g) and DCC-deficient (h) embryos. RGC axons in DCC mutants were found in approximately the same position and did not appear closer to CD44 neurons compared with wild type. Scale bars:a–f, 200 μm.
Reduced GnRH axon projections
At the ventral hypothalamus, in addition to angular RGC axon trajectories, the GnRH axon pathway was also severely affected in netrin-1 and DCC mutants. This was in agreement with a potential role for a direct involvement of netrin-1 and DCC interactions in the formation of the GnRH axon pathway because, as shown above, netrin was expressed along the route taken by developing GnRH axons. In wild-type animals, most anteriorly located GnRH neurons send axons posteriorly through the region of the organum vasculosum of the lamina terminalis (OVLT) just anterior to the chiasm and then along both ventral and dorsal routes adjacent to the third ventricle to reach the ME (Hoffman and Gibbs, 1982). In DCC mutants at P0, the location and numbers of GnRH axons in the OVLT appeared similar to that in wild-type animals (compare Fig. 5a withe), but the posterior axon projections to the ME were severely reduced (compare Fig. 5c with g). However, the few axons that did reach the ME appeared to terminate in the appropriate position of the lateral ME contact zone. Similar findings were seen in netrin-1 mutants, although the number of GnRH axons in the OVLT appeared reduced in some netrin-1 mutants (OVLT, compare Fig. 5b with f; ME, compare Fig.5d with h). In contrast to the GnRH axon pathway, axons of corticotropin-releasing hormone (CRH) neurons (compare Fig.5j with k) that also grow into the medial ME contact zone and axons of ADH neurons (compare Fig. 5l withm) that grow through the ME internal zone to the infundibulum were not noticeably affected. This appropriate CRH and ADH axon growth suggests that ME development was quite normal and eliminates the possibility of a general disruption of all axon growth through this region in mutants. A reduced GnRH projection to the ME was seen in netrin-1 and DCC mutants as early as E15 (data not shown) when the first GnRH axons are thought to reach the ME. This indicates that the reduced GnRH axon projection was not secondarily caused by axons dying back but more likely reflects an initial failure of the GnRH axons to reach their target.
Fig. 5.
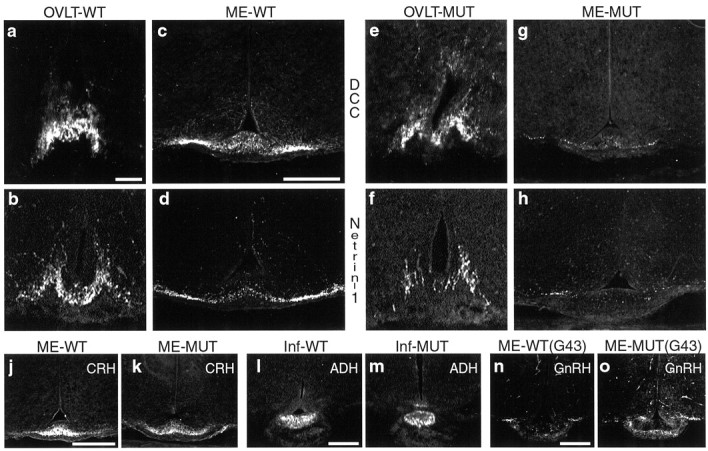
The axon pathways of GnRH neurons in wild-type and DCC- or netrin-1–deficient P0 littermates (coronal sections).a–h, Wild-type newborn animals (a–d) and their mutant littermates (e–h) are shown. a, b, Examples of GnRH axons in the OVLT of wild-type littermates from a DCC (a) and netrin-1 (b) litter are shown. c, d,Examples of GnRH axons in the ME regions of the same wild-type animals shown in a and b, respectively, are presented. e, f, Examples of GnRH axons in the OVLT of DCC (e)- and netrin-1 (f)–deficient animals are shown. The number of OVLT GnRH axons in DCC-deficient animals appears quite similar to that in wild type (compare with a, b). The number of OVLT GnRH axons in netrin-1–deficient animals appears reduced compared with that in wild type (compare with a,b).g, h, Examples of GnRH axon projections in the ME regions of DCC (g)- and netrin-1 (h)–deficient animals (same animals shown ine,f, respectively) are presented. The GnRH axon projections are severely reduced compared with that in wild type but terminate in the appropriate lateral ME contact zone regions (see c, d). j, k, Unlike GnRH axons, axons of CRH-positive neurons in wild type (j) and mutant (k) appear to grow equally well into the medial ME contact zone (k is same animal shown ing). l, m, Axons of ADH-positive neurons, which also grow through the ME internal zone to the infundibulum, appeared similar in wild-type (l) compared with DCC mutant (m) infundibulum. n, o,In comparison with wild type (n), GnRH-positive axon projections were not reduced in an E18 GAP-43–deficient littermate (o). RGC axon growth in the chiasm is severely disrupted in GAP-43–deficient embryos. In allpanels, dorsal is at the top.G43, GAP-43; Inf, infundibulum. Scale bars: a, b, e, f, 50 μm; c, d, g, h, 200 μm; j, k, 200 μm; l, m, 200 μm;n, o, 200 μm.
Because RGC axons forming the chiasm are positioned close to the path of GnRH axons, altered RGC axon trajectories in the ventral hypothalamus may have led to the reduced posterior GnRH axon projection. This possibility was examined in GAP-43–deficient mice in which RGC axon growth at the chiasm is disrupted to a greater extent than in netrin-1– or DCC-deficient embryos (Strittmatter et al., 1995;Sretavan and Kruger, 1998) due to an initial failure of RGC axon progression from the chiasm region into the optic tracts (Kruger et al., 1998). In GAP-43 mutants, GnRH axon projections to the ME were not reduced (compare Fig. 5n with o), suggesting that developmental events other than abnormal RGC axon growth at the chiasm were responsible for the reduced GnRH axon projections in netrin-1 and DCC mutants.
Ectopic ADH and oxytocin neurons
In addition to axon pathway defects, ectopic neurons were found in the ventral hypothalamus of both netrin-1– and DCC-deficient mice. In wild-type animals at P0, ADH- and oxytocin-immunopositive neurons are found within the SON (arrows in Fig.6a; oxytocin data not shown) that is bounded ventromedially by the optic tract. In both netrin-1– and DCC-deficient animals, we observed trails of ADH neurons just beneath the ventrolateral hypothalamus pial surface extending from the SON to the ventral midline (Fig. 6b, arrowheads). These ectopic ADH neurons had leading and trailing processes (Fig.6c) and resembled migrating neurons (for example, seeEdmondson and Hatten, 1987). Some ectopic ADH neurons were found immediately anterior to the chiasm, appearing as bilateral clusters centered about the ventral midline (see Fig. 6d,g). Of note, these ectopic cells lie approximately in the region of the optic chiasm and the altered RGC axon projections. Reconstructions in Figure 6,e and f, show the positions of all ADH-positive neurons at the ventral hypothalamic surface in a wild-type compared with a DCC mutant littermate. This pattern of ectopic ADH neurons was also seen at E15, during the period of active neuron migration into the developing SON (Altman and Bayer, 1978a,b; Karim and Sloper, 1980;Okamura et al., 1983), raising the likelihood that ectopic neurons represent a failure of ADH neurons to properly migrate to or properly terminate migration within the SON. As with ADH neurons, ectopic oxytocin neurons in P0 mutant embryos were also present as clusters centered at the ventral midline anterior to the mutant chiasm (see Fig.6i,j).
Fig. 6.

Ectopic ADH and oxytocin neurons in DCC- or netrin-1–deficient P0 littermates (coronal sections).a, In a typical P0 wild-type animal, ADH-positive cells are confined laterally in the SON (arrows).b, In a DCC mutant littermate, ADH-positive neurons are found within the SON region (arrows) and ectopically in a trail (arrowheads) extending from the SON to the ventral midline. c, Under higher magnification, ectopic ADH neurons (arrowheads) have leading and trailing processes reminiscent of migrating neurons. d, Ectopic ADH neurons are found as bilateral cell clusters centered about the midline (arrow) in the region anterior to the chiasm.e, f, The positions of ADH-positive neurons (red) in individual coronal sections from wild-type (e) and DCC mutant (f) littermates were traced, and tracings from serial sections were stacked. The stacked tracings were then oriented to show the pattern of ADH expression at the ventral surface of the brain. The pituitary is at the bottom of each of these images (posterior), and the chiasm is at the top(anterior). The asterisk marks the location of ectopic neurons with migrating profiles (shown in c). Thearrow points to the area containing ADH neuronal clusters at the midline (shown in d). g,Ectopic ADH neuron clusters are also found at the midline (arrow) in the region anterior to the chiasm in netrin-1 mutants. h, i, In contrast to a wild-type littermate (h), ectopic oxytocin neurons in the mutant (i) are found in the region anterior to the chiasm and form bilateral clusters at the midline (arrow). The arrowhead points to the optic nerve, just entering the brain. j, Higher magnification view of ectopic oxytocin neuron clusters in the region anterior to the chiasm in a DCC mutant (arrow indicates midline) is shown. k, m, The approximate number and distribution of ADH neurons in the PVN appeared similar in wild-type (k) and mutant (m) littermates. l, n, The approximate number and distribution of oxytocin neurons in the PVN appeared similar in wild-type (l) and mutant (n) littermates. o, p,ADH-positive neurons are normally restricted to the SON in E18 wild-type (o) or GAP-43–deficient (p) embryos even though RGC axon growth into the optic tracts in these mutant embryos is severely reduced. In allpanels (except e, f) dorsal is at the top. Ant., Anterior;Post., posterior. Scale bars: a, b, 500 μm; c, 50 μm; d, g, j, 25 μm;h, i, 250 μm; k–n, 250 μm; o, p, 250 μm.
Other than the presence of ectopic ADH and oxytocin neurons, hypothalamic neuronal migration and the positions of other hypothalamic nuclei did not appear to be generally disrupted in netrin-1– or DCC-deficient embryos. For example, as shown above, CD44 neurons were found in their appropriate location (see Fig. 4). Furthermore the paraventricular nucleus (PVN), which also normally contains ADH and oxytocin neurons (for review, see Swanson and Sawchenko, 1983; Swanson, 1986), appeared normal in size and location in wild type (Fig.6k,l) compared with mutants (Fig. 6m,n), and trails of ectopic ADH or oxytocin neurons originating from the PVN were not observed. This suggests that ADH and oxytocin neurons belonging to the PVN develop independently of netrin-1 and DCC interactions.
The optic tract and ectopic ADH neurons
The apposition of the SON and optic tract RGC axons (see Fig.2q–v) suggests that RGC axons might normally serve as a barrier against abnormal neuron migration out of the SON. Fewer optic tract RGC axons in netrin-1 or DCC mutants (because of optic nerve hypoplasia) might then allow ectopic migration ventromedially out of this nucleus. To investigate, we examined the distribution of ADH neurons in E18 GAP-43–deficient embryos that have only very few RGC axons in the optic tracts (Sretavan and Kruger, 1998). In these mutant animals, ADH neurons were confined to the SON as in wild-type littermates (compare Fig. 6o with p), indicating that a reduced number of optic tract RGC axons is probably not the cause of ectopic ADH neurons in netrin-1– or DCC-deficient embryos.
Pituitary development in netrin-1 and DCC mutants
Because normal pituitary development and function depend on proper innervation from specific hypothalamic neurons (for example, seeSchonemann et al., 1995; Michaud et al., 1998), we examined pituitary cell differentiation and hormone production as a means of revealing additional possible defects in hypothalamic neurons, especially neurons for which there were no convenient markers. Because newborn mutants usually died within 24 hr, we only examined pituitaries in animals up to age P0. In netrin-1–deficient and in DCC-deficient mutants, pituitary size and shape appeared grossly normal [compare Fig.7a,c (wild type) withb,d (mutant); also see Figure 4e vsf]. Innervation of the posterior pituitary by ADH-immunopositive axons revealed no obvious abnormalities (compare Fig. 5l with m). In pituitaries of wild-type compared with mutant littermates, the expression patterns of ACTH (Fig.7a,b) and GH (Fig. 7c,d) as well as luteinizing hormone and follicle-stimulating hormone (data not shown) appeared normal. In mutants at P0, circulating hormone levels of adrenocorticotropic hormone (ACTH) and growth hormone (GH) were not reduced compared with that of wild-type littermates (see Fig.7e,f). This finding suggested that although ACTH levels are thought to be affected in part by ADH (Molitch, 1995), the presence of ectopic ADH neurons did not noticeably affect ACTH levels at P0. Furthermore, although we did not directly examine growth-hormone releasing hormone (GHRH) neuron development in mutant animals, the observation of normal GH expression in the anterior pituitary and normal serum GH levels suggested that GHRH neuron differentiation, innervation, and regulation of GH levels were most likely not significantly affected in mutants (Godfrey et al., 1993; Lin et al., 1993; Li et al., 1996). In summary, although netrin-1– and DCC-deficient animals exhibit specific defects in hypothalamic development, this was not associated with obvious pituitary or neuroendocrine abnormalities within the scope of our analysis at E12–P0.
Fig. 7.
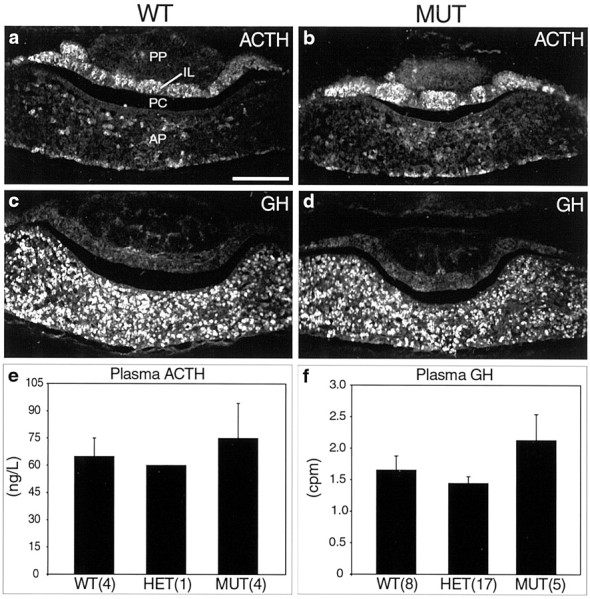
Pituitary development in DCC- or netrin-1–deficient mice. a–d, Coronal sections of the pituitary gland at P0 are shown. The overall size and shape of the pituitary is not different between wild-type (a, c) and mutant (b, d) animals. Mutants have normal organization of the posterior pituitary, intermediate lobe, pituitary cleft, and anterior pituitary. a, ACTH-positive neurons in the intermediate lobe of a wild-type animal are shown. b,ACTH neurons are found in a similar pattern in a netrin-1–deficient littermate. c, GH-positive neurons are located in the anterior pituitary in wild-type animals. d, GH-positive neurons are found in a similar pattern in a netrin-1–deficient littermate. e, Plasma ACTH levels in P0 animals are shown. Values (in ng/l) were obtained for animals from three different DCC litters, and the values obtained for animals of each genotype were pooled. The numbers in parenthesesindicate the number of animals of each genotype. Older ages were not examined because mutants die postnatally. Error bars indicate SEM.f, Plasma GH levels in P0 DCC litters are shown. Values (expressed as cpm) were obtained for animals from three different DCC litters, and the values for animals of each genotype were pooled.AP, Anterior pituitary; IL, intermediate lobe; PC, pituitary cleft; PP, posterior pituitary. Scale bar: a–d, 200 μm.
DISCUSSION
Results from this study demonstrate that specific aspects of mouse hypothalamus development require the guidance molecule netrin-1 and the netrin receptor DCC (see summary in Fig.8). The similarities in phenotype in both netrin-1 and DCC mutants suggest that this is via direct netrin-1 and DCC interactions. In the absence of netrin-1 and DCC, RGC axons are affected together with GnRH axons as well as ADH and oxytocin neurons. Although previous studies have identified important regulatory genes involved in hypothalamic cell fate specification (Schonemann et al., 1995; Kimura et al., 1996; Torres et al., 1996; Macdonald et al., 1997;Michaud et al., 1998; Dale et al., 1999), this present study describes some of the guidance molecules involved in the patterning of axon pathways and the determination of neuronal positions in the developing mouse hypothalamus.
Fig. 8.

Summary of defects in RGC axon trajectories and in development of GnRH, ADH, and oxytocin (OXY) neurons in DCC- or netrin-1-deficient animals.
Netrin-1 and DCC interactions and RGC growth in the developing hypothalamus
The mechanism by which the lack of netrin-1 and DCC interactions leads to the abnormally angular RGC axon trajectories remains unknown.Seyneu/+ heterozygous mice that have a small optic nerve do not exhibit abnormal RGC axon trajectories within the hypothalamus, indicating that optic nerve hypoplasia alone does not cause this phenotype. Previous studies show that chiasm neurons are involved in chiasm formation (for review, see Mason and Sretavan, 1997), and in netrin-1 and DCC mutants, abnormal trajectories could result if the development of these neurons was affected. However, this does not appear to be the case because chiasm neuron development and the relationship of RGC axons to these neurons appear unaffected in mutants. Although a simple model is that netrin-1 is required to guide DCC-expressing RGC axons in the chiasm region, this guidance model cannot involve a normal midline focus of netrin expression because netrin was not observed at the hypothalamic ventral midline during the period of RGC axon ingrowth and chiasm formation. It should be noted that netrin was present in regions flanking the midline area. It is therefore possible that disruption of direct netrin-1 and DCC interactions in the proximal portion of the optic nerve could affect growth of RGC axons within the chiasm region. Our findings support the notion that netrin-1 and DCC interactions govern axon guidance about the ventral midline in both anterior and posterior CNS commissures. However, although in the spinal cord netrin-1 is required for commissural axon guidance to the midline floor plate, in the hypothalamus deficient netrin-1 function alters RGC axon trajectories but is not required for RGC axons to reach or cross the midline.
Disrupted GnRH neuron axon projections
The results indicate that GnRH neuron migration and/or axon pathfinding require netrin-1 and DCC interactions. In DCC mutants, GnRH innervation of the OVLT appeared relatively unaffected, suggesting that a substantial number of GnRH neurons migrated appropriately into the hypothalamus and that GnRH axon pathfinding was not generally disrupted. Because individual GnRH axons reportedly form en passant synapses in the OVLT and then project into the ME (Hoffman and Gibbs, 1982), the relative lack of GnRH axons posteriorly in the ME suggests a disruption of GnRH axon pathfinding after growth through the OVLT. If so, this implies that GnRH axons require DCC function not for OVLT innervation but specifically for pathfinding in more posterior parts of the hypothalamus. Recent cell culture studies suggest that a diffusible guidance cue may attract GnRH axons to the ME (Rogers et al., 1997). Whether this cue is netrin-1 and this pathfinding process involves signaling through DCC remain to be tested. In comparison with DCC mutants, however, the GnRH innervation of the OVLT appeared reduced in some netrin-1 mutants. This suggests that netrin-1, independent of interactions with DCC, may also be required to ensure either proper GnRH axon targeting of the OVLT or proper GnRH neuron migration into the hypothalamus.
On the basis of the reduced GnRH axon projection, one might predict that netrin-1 or DCC deficiency would disrupt the regulation of gonadotropin release, leading to hypogonadism. If true, this would indicate that altered GnRH neuron development can lead to hypogonadism both via the failure of proper GnRH neuron migration into the CNS as in Kallman's syndrome (Schwanzel-Fukuda et al., 1989) as well as via the disruption of GnRH axon pathway formation.
Ectopic ADH and oxytocin neurons
During normal development, ADH and oxytocin neurons are thought to originate in the region of the ventral diencephalic sulcus and then to migrate laterally to the PVN and SON (Altman and Bayer, 1978a,b). In mutants, the SON was flanked ventromedially by a trail of ectopic ADH neurons, and both ADH and oxytocin neurons were abnormally located in the region of the chiasm. However, the PVN appeared unaffected with no apparent trails of ectopic neurons. Thus, ADH- and oxytocin-expressing neurons were not all affected equally in the netrin-1– and DCC-deficient state.
One model of ADH and oxytocin neuron migration is that netrin-1 in the SON attracts migrating ADH and oxytocin neurons, and in mutants some neurons cannot therefore find their way to the SON and migrate to ectopic positions instead. However, on the basis of the trail of ectopic neurons leading back to the SON, we believe it is more likely that netrin-1 and DCC interactions are required to confine ADH and oxytocin neurons to the SON, and the absence of these interactions in mutants leads to inappropriate migration of neurons out of the SON. However, a consideration is that DCC was not expressed by neurons in the SON. It is possible that DCC was expressed by the SON neurons and was downregulated early on or that an unconventional netrin-1 and DCC interaction occurs in which DCC in the surrounding tissue serves as a “barrier” guidance cue signaling via netrin-1 to prevent neuronal migration out of the SON. An alternative that cannot be eliminated is that an unknown cell type normally prevents migration out of the SON and that its development is somehow affected in netrin-1– and DCC-deficient mutants.
A shared feature of all developmental defects
Although netrin and DCC expression patterns fit with a simple model in which netrin guides GnRH axons posteriorly, expression patterns do not suggest simple models to explain the altered RGC axon projections and the ectopic ADH neurons in mutants. However, although the three phenotypes identified in mutants involve neurons originating from separate regions of the CNS, we note that they all exhibit developmental defects around the ventral hypothalamus/chiasm midline region. This includes the presence of ectopic ADH and oxytocin neuron clusters, angular RGC axon trajectories, and severely reduced GnRH axon projections. The fact that all these developmental defects overlap anatomically at the ventral hypothalamic midline chiasm region allows for the possibility that one particular developmental defect in this region may have secondarily caused one or more of the other defects. For example, the presence of ectopic ADH neurons at the hypothalamic midline might alter RGC axon trajectories at the chiasm or even the ability of GnRH neurons to send axons posteriorly through this region.
Relationship to septo-optic dysplasia
In the adult, acquired disorders affecting the hypothalamus often involve the visual system because of the location of the chiasm on the ventral hypothalamic surface (for review, see Siatkowski and Glaser, 1995). This study shows that development of the visual and hypothalamic neuroendocrine systems is also linked via shared guidance molecules and suggests that mutations affecting guidance molecules could lead to congenital disorders affecting both vision and neuroendocrine function. The occurrence of hypothalamic abnormalities (shown in this paper), optic nerve hypoplasia (Deiner et al., 1997), and an absent corpus callosum (Serafini et al., 1996; Fazeli et al., 1997) in the netrin-1– or DCC-deficient mice is phenotypically similar to the human congenital disorder of septo-optic dysplasia (SOD). This raises the possibility that SOD may be linked to mutations in developmental genes such as netrin-1 or DCC. Although most cases of SOD have been described in the literature as primarily sporadic in nature (Harris and Haas, 1972;Brodsky et al., 1996), a recent finding suggests that certain cases of SOD are caused by a mutation in the regulatory gene Hesx1 (Dattani et al., 1998). In the mouse, Hesx1 is expressed in presumptive forebrain and Rathke's pouch (which forms the anterior pituitary), and mice missing Hesx1 have severely affected eye, pituitary, and prosencephalon development with accompanying abnormalities of the corpus callosum, septum pellucidum, and anterior and hippocampal commissures. This phenotypic combination is remarkably similar to but more severe than that observed in DCC or netrin-1 mutant mice. This suggests it may be worth examining whether netrin-1 or DCC genes are regulated by Hesx1 and whether they may be mutated in certain cases of SOD.
Footnotes
This research was supported by National Institutes of Health (NIH) Grant EY 10688 and a grant from the That Man May See Foundation (D.S.), NIH Grant EY 02162 (Department of Ophthalmology), and NIH Grant EY 07120 (University of California, San Francisco). We thank Wylie Vale, Albert Parlow, Lawrence Frohman, Eric Fearon, Tim Kennedy, Mark Fishman, Tito Serafini, Amin Fazeli, and Marc Tessier-Lavigne for reagents and mice. Thanks to Selna Kaplan, Lita Ramos, Eric Birgbauer, Chris Severin, Judy Mak, and Amanda Kahn for technical assistance and contributions. The ACTH, LH, and FSH antibodies were a generous gift of the National Hormone and Pituitary Program, the National Institute of Diabetes and Digestive Kidney Research, the National Institute of Child Health and Human Development, and the United States Department of Agriculture. We also thank Michelle Bland, John Rubenstein, and Richard Weiner for their insightful comments.
Correspondence should be addressed to Dr. David Sretavan, Beckman Vision Center, Departments of Ophthalmology and Physiology, 10 Kirkham Street, Room K107, University of California, San Francisco, San Francisco, CA 94143-0730. E-mail: dws@itsa.ucsf.edu.
REFERENCES
- 1.Altman J, Bayer SA. Development of the diencephalon in the rat. I. Autoradiographic study of the time of origin and settling patterns of neurons of the hypothalamus. J Comp Neurol. 1978a;182:945–971. doi: 10.1002/cne.901820511. [DOI] [PubMed] [Google Scholar]
- 2.Altman J, Bayer SA. Development of the diencephalon in the rat. II. Correlation of the embryonic development of the hypothalamus with the time of origin of its neurons. J Comp Neurol. 1978b;182:973–993. doi: 10.1002/cne.901820512. [DOI] [PubMed] [Google Scholar]
- 3.Bancroft JD, Stevens A, editors. Theory and practice of histological techniques, 2nd Edition. Churchill Livingstone; New York: 1982. [Google Scholar]
- 4.Brodsky MC, Baker RS, Hamed LM. Pediatric neuro-ophthalmology. Springer; New York: 1996. [Google Scholar]
- 5.Dale K, Sattar N, Heemskerk J, Clarke JD, Placzek M, Dodd J. Differential patterning of ventral midline cells by axial mesoderm is regulated by BMP7 and chordin. Development. 1999;126:397–408. doi: 10.1242/dev.126.2.397. [DOI] [PubMed] [Google Scholar]
- 6.Dattani MT, Martinez-Barbera JP, Thomas PQ, Brickman JM, Gupta R, Mårtensson IL, Toresson H, Fox M, Wales JK, Hindmarsh PC, Krauss S, Beddington RS, Robinson IC. Mutations in the homeobox gene HESX1/Hesx1 associated with septo-optic dysplasia in human and mouse. Nat Genet. 1998;19:125–133. doi: 10.1038/477. [DOI] [PubMed] [Google Scholar]
- 7.Deiner MS, Kennedy T, Fazeli A, Serafini T, Tessier-Lavigne M, Sretavan DW. Netrin-1 and DCC mediate axon guidance locally at the optic disc: loss of function leads to optic nerve hypoplasia. Neuron. 1997;19:575–589. doi: 10.1016/s0896-6273(00)80373-6. [DOI] [PubMed] [Google Scholar]
- 8.de la Torre JR, Höpker VH, Ming GL, Poo MM, Tessier-Lavigne M, Hemmati-Brivanlou A, Holt CE. Turning of retinal growth cones in a netrin-1 gradient mediated by the netrin receptor DCC. Neuron. 1997;19:1211–1224. doi: 10.1016/s0896-6273(00)80413-4. [DOI] [PubMed] [Google Scholar]
- 9.Dodd J, Jessell TM, Placzek M. The when and where of floor plate induction. Science. 1998;282:1654–1657. doi: 10.1126/science.282.5394.1654. [DOI] [PubMed] [Google Scholar]
- 10.Easter SS, Jr, Ross LS, Frankfurter A. Initial tract formation in the mouse brain. J Neurosci. 1993;13:285–299. doi: 10.1523/JNEUROSCI.13-01-00285.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Edmondson J, Hatten ME. Glial-guided granule neuron migration in vitro: a high-resolution time-lapse video microscopic study. J Neurosci. 1987;7:1928–1934. doi: 10.1523/JNEUROSCI.07-06-01928.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fazeli A, Dickinson SL, Hermiston ML, Tighe RV, Steen RG, Small CJ, Stoeckli ET, Keino-Masu K, Masu M, Rayburn H, Simons J, Bronson RT, Gordon JI, Tessier-Lavigne M, Weinberg RA. Phenotype of mice lacking functional Deleted in colorectal cancer (Dcc) gene. Nature. 1997;386:796–804. doi: 10.1038/386796a0. [DOI] [PubMed] [Google Scholar]
- 13.Ganten D, Pfaff DW, Arai Y. Morphology of hypothalamus and its connections. Current topics in neuroendocrinology, Vol 7. Springer; New York: 1986. [Google Scholar]
- 14.Godfrey P, Rahal JO, Beamer WG, Copeland NG, Jenkins NA, Mayo KE. GHRH receptor of little mice contains a missense mutation in the extracellular domain that disrupts receptor function. Nat Genet. 1993;4:227–232. doi: 10.1038/ng0793-227. [DOI] [PubMed] [Google Scholar]
- 15.Harris RJ, Haas L. Septo-optic dysplasia with growth hormone deficiency (De Morsier syndrome). Arch Dis Child. 1972;47:973–976. doi: 10.1136/adc.47.256.973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Hill RE, Favor J, Hogan BL, Ton CC, Saunders GF, Hanson IM, Prosser J, Jordan T, Hastie ND, van Heyningen V. Mouse small eye results from mutations in a paired-like homeobox-containing gene. Nature 354 1991. 522 525[Erratum(1992)355:750]. [DOI] [PubMed] [Google Scholar]
- 17.Hoffman GE, Gibbs FP. LHRH pathways in rat brain: “deafferentation” spares a sub-chiasmatic LHRH projection to the median eminence. Neuroscience. 1982;7:1979–1993. doi: 10.1016/0306-4522(82)90012-4. [DOI] [PubMed] [Google Scholar]
- 18.Karim MA, Sloper JC. Histogenesis of the supraoptic and paraventricular neurosecretory cells of the mouse hypothalamus. J Anat. 1980;130:341–347. [PMC free article] [PubMed] [Google Scholar]
- 19.Kennedy TE, Serafini T, de la Torre JR, Tessier-Lavigne M. Netrins are diffusible chemotropic factors for commissural axons in the embryonic spinal cord. Cell. 1994;78:425–435. doi: 10.1016/0092-8674(94)90421-9. [DOI] [PubMed] [Google Scholar]
- 20.Kimura S, Hara Y, Pineau T, Fernandez-Salguero P, Fox CH, Ward JM, Gonzalez FJ. The T/ebp null mouse: thyroid-specific enhancer-binding protein is essential for the organogenesis of the thyroid, lung, ventral forebrain, and pituitary. Genes Dev. 1996;10:60–69. doi: 10.1101/gad.10.1.60. [DOI] [PubMed] [Google Scholar]
- 21.Kruger K, Tam AS, Lu C, Sretavan DW. Retinal ganglion cell axon progression from the optic chiasm to initiate optic tract development requires cell autonomous function of GAP-43. J Neurosci. 1998;18:5692–5705. doi: 10.1523/JNEUROSCI.18-15-05692.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Li H, Zeitler PS, Valerius MT, Small K, Potter SS. Gsh-1, an orphan Hox gene, is required for normal pituitary development. EMBO J. 1996;15:714–724. [PMC free article] [PubMed] [Google Scholar]
- 23.Lin SC, Lin CR, Gukovsky I, Lusis AJ, Sawchenko PE, Rosenfeld MG. Molecular basis of the little mouse phenotype and implications for cell type-specific growth. Nature. 1993;364:208–213. doi: 10.1038/364208a0. [DOI] [PubMed] [Google Scholar]
- 24.Livne I, Gibson MJ, Silverman AJ. Biochemical differentiation and intercellular interactions of migratory gonadotropin-releasing hormone (GnRH) cells in the mouse. Dev Biol. 1993;159:643–656. doi: 10.1006/dbio.1993.1271. [DOI] [PubMed] [Google Scholar]
- 25.Macdonald R, Scholes J, Strähle U, Brennan C, Holder N, Brand M, Wilson SW. The Pax protein Noi is required for commissural axon pathway formation in the rostral forebrain. Development. 1997;124:2397–2408. doi: 10.1242/dev.124.12.2397. [DOI] [PubMed] [Google Scholar]
- 26.Marcus R, Mason C. The first retinal axon growth in the mouse optic chiasm: axon patterning and the cellular environment. J Neurosci. 1995;15:6389–6402. doi: 10.1523/JNEUROSCI.15-10-06389.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Marcus RC, Blazeski R, Godement P, Mason CA. Retinal axon divergence in the optic chiasm: uncrossed axons diverge from crossed axons within a midline glial specialization. J Neurosci. 1995;15:3716–3729. doi: 10.1523/JNEUROSCI.15-05-03716.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mason CA, Sretavan DW. Glia, neurons, and axon pathfinding during optic chiasm development. Curr Opin Neurobiol. 1997;7:647–653. doi: 10.1016/s0959-4388(97)80084-0. [DOI] [PubMed] [Google Scholar]
- 29.Michaud JL, Rosenquist T, May NR, Fan CM. Development of neuroendocrine lineages requires the bHLH-PAS transcription factor SIM1. Genes Dev. 1998;12:3264–3275. doi: 10.1101/gad.12.20.3264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Miyake K, Underhill CB, Lesley J, Kincade PW. Hyaluronate can function as a cell adhesion molecule and CD44 participates in hyaluronate recognition. J Exp Med. 1990;172:69–75. doi: 10.1084/jem.172.1.69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Molitch ME. Neuroendocrinology. In: Becker KL, editor. Endocrinology and metabolism, 3rd Edition. McGraw-Hill; New York: 1995. pp. 221–283. [Google Scholar]
- 32.Okamura H, Fukui K, Koyama E, Tsutou HL, Tsutou T, Terubayashi H, Fujisawa H, Ibata Y. Time of vasopressin neuron origin in the mouse hypothalamus: examination by combined technique of immunocytochemistry and [3H]thymidine autoradiography. Brain Res. 1983;285:223–226. doi: 10.1016/0165-3806(83)90055-x. [DOI] [PubMed] [Google Scholar]
- 33.Reale MA, Reyes-Mugica M, Pierceall WE, Rubinstein MC, Hedrick L, Cohn SL, Nakagawara A, Brodeur GM, Fearon ER. Loss of DCC expression in neuroblastoma is associated with disease dissemination. Clin Cancer Res. 1996;2:1097–1102. [PubMed] [Google Scholar]
- 34.Rogers MC, Silverman AJ, Gibson MJ. Gonadotropin-releasing hormone axons target the median eminence: in vitro evidence for diffusible chemoattractive signals from the mediobasal hypothalamus. Endocrinology. 1997;138:3956–3966. doi: 10.1210/endo.138.9.5416. [DOI] [PubMed] [Google Scholar]
- 35.Schonemann MD, Ryan AK, McEvilly RJ, O'Connell SM, Arias CA, Kalla KA, Li P, Sawchenko PE, Rosenfeld MG. Development and survival of the endocrine hypothalamus and posterior pituitary gland requires the neuronal POU domain factor Brn-2. Genes Dev. 1995;9:3122–3135. doi: 10.1101/gad.9.24.3122. [DOI] [PubMed] [Google Scholar]
- 36.Schwanzel-Fukuda M, Pfaff DW. Origin of luteinizing hormone-releasing hormone neurons. Nature. 1989;338:161–164. doi: 10.1038/338161a0. [DOI] [PubMed] [Google Scholar]
- 37.Schwanzel-Fukuda M, Bick D, Pfaff DW. Luteinizing hormone-releasing hormone (LHRH)-expressing cells do not migrate normally in an inherited hypogonadal (Kallmann) syndrome. Brain Res Mol Brain Res. 1989;6:311–326. doi: 10.1016/0169-328x(89)90076-4. [DOI] [PubMed] [Google Scholar]
- 38.Serafini T, Kennedy TE, Galko MJ, Mirzayan C, Jessell TM, Tessier-Lavigne M. The netrins define a family of axon outgrowth-promoting proteins homologous to C. elegans UNC-6. Cell. 1994;78:409–424. doi: 10.1016/0092-8674(94)90420-0. [DOI] [PubMed] [Google Scholar]
- 39.Serafini T, Colamarino SA, Leonardo ED, Wang H, Beddington R, Skarnes WC, Tessier-Lavigne M. Netrin-1 is required for commissural axon guidance in the developing vertebrate nervous system. Cell. 1996;87:1001–1014. doi: 10.1016/s0092-8674(00)81795-x. [DOI] [PubMed] [Google Scholar]
- 40.Shirasaki R, Mirzayan C, Tessier LM, Murakami F. Guidance of circumferentially growing axons by netrin-dependent and -independent floor plate chemotropism in the vertebrate brain. Neuron. 1996;17:1079–1088. doi: 10.1016/s0896-6273(00)80241-x. [DOI] [PubMed] [Google Scholar]
- 41.Siatkowski RM, Glaser JS. The optic chiasm in endocrinologic disorders. In: Becker KL, editor. Principles and practice of endocrinology and metabolism, 2nd Edition. Lippincott; Philadelphia: 1995. pp. 193–207. [Google Scholar]
- 42.Skarf B, Hoyt CS. Optic nerve hypoplasia in children. Association with anomalies of the endocrine and CNS. Arch Ophthalmol. 1984;102:62–67. doi: 10.1001/archopht.1984.01040030046032. [DOI] [PubMed] [Google Scholar]
- 43.Skarnes WC, Moss JE, Hurtley SM, Beddington RS. Capturing genes encoding membrane and secreted proteins important for mouse development. Proc Natl Acad Sci USA. 1995;92:6592–6596. doi: 10.1073/pnas.92.14.6592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sretavan DW, Kruger K. Randomized retinal ganglion cell axon routing at the optic chiasm of GAP-43-deficient mice: association with midline recrossing and lack of normal ipsilateral axon turning. J Neurosci. 1998;18:10502–10513. doi: 10.1523/JNEUROSCI.18-24-10502.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sretavan DW, Feng L, Pure E, Reichardt LF. Embryonic neurons of the developing optic chiasm express L1 and CD44, cell surface molecules with opposing effects on retinal axon growth. Neuron. 1994;12:957–975. doi: 10.1016/0896-6273(94)90307-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sretavan DW, Pure E, Siegel MW, Reichardt LF. Disruption of retinal axon ingrowth by ablation of embryonic mouse optic chiasm neurons. Science. 1995;269:98–101. doi: 10.1126/science.7541558. [DOI] [PubMed] [Google Scholar]
- 47.Strittmatter SM, Fankhauser C, Huang PL, Mashimo H, Fishman MC. Neuronal pathfinding is abnormal in mice lacking the neuronal growth cone protein GAP-43. Cell. 1995;80:445–452. doi: 10.1016/0092-8674(95)90495-6. [DOI] [PubMed] [Google Scholar]
- 48.Swanson LW. Organization of mammalian neuroendocrine system. In: Mountcastle VB, Bloom FE, Geiger SM, editors. Handbook of physiology, Sec 1, The nervous system. American Physiological Society; Bethesda, MD: 1986. pp. 317–363. [Google Scholar]
- 49.Swanson LW, Sawchenko PE. Hypothalamic integration: organization of the paraventricular and supraoptic nuclei. Annu Rev Neurosci. 1983;6:269–324. doi: 10.1146/annurev.ne.06.030183.001413. [DOI] [PubMed] [Google Scholar]
- 50.Torres M, Gomez-Pardo E, Gruss P. Pax2 contributes to inner ear patterning and optic nerve trajectory. Development. 1996;122:3381–3391. doi: 10.1242/dev.122.11.3381. [DOI] [PubMed] [Google Scholar]
- 51.Walther C, Gruss P. Pax-6, a murine paired box gene, is expressed in the developing CNS. Development. 1991;113:1435–1449. doi: 10.1242/dev.113.4.1435. [DOI] [PubMed] [Google Scholar]
- 52.Wang L-C, Rachel RA, Marcus R, Mason CA. Chemosuppression of retinal axon growth by the mouse optic chiasm. Neuron. 1996;17:849–862. doi: 10.1016/s0896-6273(00)80217-2. [DOI] [PubMed] [Google Scholar]
- 53.Wray S, Grant P, Gainer H. Evidence that cells expressing luteinizing hormone-releasing hormone mRNA in the mouse are derived from progenitor cells in the olfactory placode. Proc Natl Acad Sci USA. 1989;86:8132–8136. doi: 10.1073/pnas.86.20.8132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Wu TJ, Gibson MJ, Rogers MC, Silverman AJ. New observations on the development of the gonadotropin-releasing hormone system in the mouse. J Neurobiol. 1997;33:983–998. doi: 10.1002/(sici)1097-4695(199712)33:7<983::aid-neu9>3.0.co;2-4. [DOI] [PubMed] [Google Scholar]