

Abstract
Tetanic stimuli to layer I–II afferents in rat prefrontal cortex induced long-term depression (LTD) of layer I–II to layer V pyramidal neuron glutamatergic synapses when tetani were coupled to bath application of dopamine. This LTD was blocked by the following metabotropic glutamate receptor (mGluR) antagonists coapplied with dopamine: (S)-α-methyl-4-carboxyphenylglycine (MCPG; group I and II antagonist), (RS)-1-aminoindan-1,5-dicarboxylic acid (AIDA; group I antagonist), or (RS)-α-methylserine-O-phosphate monophenyl ester (MSOPPE; group II antagonist). This suggests that the dopamine-facilitated LTD requires synaptic activation of groups I and II mGluRs during tetanus. LTD could also be induced by coupling tetani to bath application of groups I and II mGluR agonist (1S,3R)-1-aminocyclopentane-1,3-dicarboxylic acid (1S,3R-ACPD). In the next series of experiments, coapplication of dopamine and 1S,3R-ACPD, but not application of either drug alone, consistently induced LTD without tetani or even single test stimuli during drug application, suggesting that coactivation of dopamine receptors and the mGluRs is sufficient for LTD induction. Immunoblot analyses with anti-active mitogen-activated protein kinases (MAP-Ks) revealed that D1 receptors, D2 receptors, group I mGluRs, and group II mGluRs all contribute to MAP-K activation in prefrontal cortex, and that combined activation of dopamine receptors and mGluRs synergistically or additively activate MAP-Ks. Consistently, LTD by dopamine + 1S,3R-ACPD coapplication, as well as the two other forms of LTD (LTD by dopamine + tetani and LTD by 1S,3R-ACPD + tetani), was blocked by bath application of MAP-K kinase inhibitor PD98059. LTD by dopamine + 1S,3R-ACPD coapplication was also blocked by postsynaptic injection of synthetic MAP-K substrate peptide. Our results suggest that dopamine receptors and groups I and II mGluRs cooperate to induce LTD through converging postsynaptic activation of MAP-Ks.
Keywords: long-term depression, long-term potentiation, synaptic plasticity, prefrontal cortex, dopamine, metabotropic glutamate receptor, MAP kinase, learning and memory, schizophrenia
Critical modulation of prefrontal cognitive function by dopaminergic input arising from ventral tegmental area (Glowinski et al., 1984; Kolb, 1984; Kuroda et al., 1996) is well documented in rats (Simon et al., 1980; Zahrt et al., 1997), primates (Sawaguchi and Goldman-Rakic, 1994; Goldman-Rakic, 1995), and humans (Barchas et al., 1994; Iversen, 1995; Okubo et al., 1997). Dopaminergic modulation on synaptic models of neuronal plasticity, long-term potentiation (LTP), and long-term depression (LTD) (Hirsch and Crepel, 1990) (for review, see Bliss and Collingridge, 1993; Otani and Ben-Ari, 1993; Bear and Abraham, 1996) have been also reported in rat prefrontal cortex, where dopamine facilitates induction of LTD, but not LTP, through D1 and D2 receptor activation (Law-Tho et al., 1995;Otani et al., 1998b).
Many studies demonstrated roles of metabotropic glutamate receptors (mGluRs) (Nicoletti et al., 1996; Conn and Pin, 1997) in LTP and LTD (Aniksztejn et al., 1992; Bashir et al., 1993; Kato, 1993; Huang et al., 1997; Oliet et al., 1997; Manahan-Vaughan et al., 1998; Otani and Connor, 1998). In prefrontal cortex, LTP by theta burst stimulation is blockable by group I and II mGluR antagonist (S)-α-methyl-4-carboxyphenylglycine (MCPG) (Vickery et al., 1997). It is thought that MCPG blocks LTP through acting on postsynaptic group I mGluRs (Vickery et al., 1997; Morris et al., 1998). Indeed, group I mGluRs exist abundantly in postsynaptic elements throughout cortex and other structures (Romano et al., 1995). More specifically, the mGluR5 seems to exist on dendritic spines and shafts, as well as on the membrane between spines, of cortical pyramidal neurons (Romano et al., 1995), whereas the mGluR1 is present in nonpyramidal neurons in cortex (Fotuhi et al., 1993). A recent demonstration also suggests postsynaptic existence, as well as presynaptic existence as classically viewed, of group II mGluRs in cortex and other structures (Petralia et al., 1996). Localization of group III mGluRs is still largely regarded as presynaptic [Gereau and Conn (1995); Jin and Daw (1998); but see Bradley et al. (1996)].
Dopamine D1 receptors exist on dendritic spines and shafts of pyramidal neurons in monkey frontal cortex layers II and V (Smiley et al., 1994;Bergson et al., 1995). Importantly, these D1 receptors appear to be colocalized with functioning glutamate receptors within the same spines–shafts, although dopaminergic presynaptic terminals may not have a direct contact with these postsynaptic elements, suggesting that dopamine receptors extrasynaptically receive neurotransmitter dopamine (Smiley et al., 1994). Colocalization of dopamine (at least D1) and glutamate receptors in the same spines–shafts points to the possibility that these two types of receptors interact with each other postsynaptically. If it is the case, such an interaction may play a role in prefrontal LTD induction. We have shown that prefrontal LTD, whose induction is facilitated by dopamine, does not require activation of NMDA receptors (Otani et al., 1998b) but still requires postsynaptic Ca2+-dependent processes (Otani et al., 1998b). Therefore, in this paper, we investigated involvement of mGluRs in prefrontal LTD induction.
Preliminary data have been published previously in abstract form (Otani et al., 1998a,c).
MATERIALS AND METHODS
Slice preparation and electrophysiology. Male Sprague Dawley rats (23–30 d old) were decapitated, and their brains were removed. Coronal slices (300–400 μm; 2.2–3.7 mm from bregma) were sectioned by the use of a Campden Vibratome in chilled (∼0°C) oxygenated (95% O2/5% CO2) artificial CSF (ACSF) of the following composition (in mm): NaCl 124, KCl 2, NaHCO3 26, KH2PO4 1.15, MgCl2 1, CaCl2 2, andd-glucose 11. The slices were allowed to recover for at least 2 hr at room temperature (∼20°C) in a chamber filled with continuously oxygenated ACSF. A slice was then transferred to a submerged-type recording chamber where it was perfused with ACSF (28°C) at the rate of 1 ml/min.
Stable intracellular recordings were made with sharp, glass micropipettes filled with 3 m K-acetate (80–120 MΩ tip resistance) from the soma of >120 layer V pyramidal neurons in the prelimbic area of prefrontal cortex. Negative currents were initially injected by the use of an Axoclamp 2A amplifier, but after stabilization of the cells, most or all currents were removed. The cells had mean resting membrane potential of −71 ± 0.6 mV (SEM) with input resistance 60 ± 2.5 MΩ. Mean membrane potential held during experiments was −74 ± 0.5 mV. A spike height of at least 70 mV was required to continue experiments. Only cells that remained within 10% of changes from the initial values of membrane potential, spike height, and input resistance were included for later analysis. The mode of spike discharge was routinely examined before experiments by the application of a depolarizing current step (500 msec) from resting membrane potential. Amplitude of the depolarizing step was set so that a 30 msec application at that amplitude charges the cell to fire one action potential. Of the neurons tested, 59% were classified as regular spiking cells, and 18% were classified as bursting cells. Five percent of the neurons showed a burst firing followed by regular spiking with adaptation. The remaining 18% showed a few sporadic spikes before a strong adaptation ceased spiking. As in the study of Law-Tho (1995) and our previous study (Otani et al., 1998b), there was no correlation between a discharge mode and the degree of synaptic plasticity induction.
A bipolar, Teflon-coated tungsten stimulating electrode (external diameter 125 μm) was placed on layer I–II (immediately interior to pial surface) of the prelimbic area. The EPSP of 5–10 mV amplitude was evoked at 0.033 Hz by the application of monophasic square voltage pulses (100 μsec; Digitimer isolated stimulator). The responses were fed to an Axoclamp 2A amplifier at current-clamp mode, digitized at 5–10 kHz with a Labmaster interface, and stored in an on-line IBM computer for later analyses (ACQUIS1 program, developed by G. Sadoc, Institut Alfred Fessard, CNRS, Gif sur Yvette, France). Synaptic responses evoked by high-frequency stimulation were stored on a magnetic tape by the use of a SONY PCM-701ES and a Betamax SL-HF100F. LTD-inducing tetanic stimuli consisted of four trains of 50 Hz stimuli (100 pulses), delivered at 0.1 Hz. The 0.033 Hz test stimuli were resumed 30 sec after tetanic stimulation. All experiments were performed in the presence of the GABA-A antagonist bicuculline methiodide (1 μm) in bathing medium.
For the analysis of single EPSPs, we measured initial rising slope (the ≤1 msec period from its onset; millivolts per milliseconds), which contains only the monosynaptic component of the responses (Hirsch and Crepel, 1990). To express changes of the EPSP slope, we averaged responses from the 10 min period just before tetani–drug application (baseline) and also from the 35–40 min period after tetani–drug application. We calculated percentage decreases–increases of the initial slope from the baseline value. These percentage decreases–increases were compared among different groups. For the analysis of synaptic responses evoked by high-frequency stimuli, we measured the number of spikes, the number of the EPSPs whose amplitudes were >50% of the first EPSP in the given episode of high-frequency stimuli, and 90% decay time from peak membrane potential (Otani et al., 1998b). Statistical analyses (two-tailed Student's ttest) were performed with p < 0.05 considered as significant. All values were expressed as mean ± SEM.
In many experiments, biocytin (1.5%; Sigma, St. Louis, MO) was included in recording electrodes and injected into cells by passing positive current steps (∼0.5 nA, 500 msec at 1 Hz for at least 10 min) at the end of experiments. The slices were fixed in 4% paraformaldehyde dissolved in potassium PBS (0.01 m) for at least overnight. They were then washed in the PBS solution three times (10 min each) and placed in 1 ml of 0.1% PBS–Triton X-100 solution containing 25 μl of solutions A and B (peroxidase standard PK-4000; Vectastain ABC Kit, Vector Laboratories, Burlingame, CA) for up to 48 hr. The slices were washed again in PBS solution. They were then placed in diaminobenzidine tetrahydrochloride (DAB) solution (Peroxidase Substrate Kit, SK-4100, Vector) for 10 min. The slices were washed three times in PBS solution before being mounted on microscope slides.
Drugs used in the electrophysiological studies were (1S,3R)-1-aminocyclopentane-1,3-dicarboxylic acid (1S,3R-ACPD; Tocris Cookson, Bristol, UK), (RS)-1-aminoindan-1,5-dicarboxylic acid (AIDA; Tocris Cookson), ascorbic acid (Sigma), bicuculline methiodide (Tocris Cookson), BAPTA (Research Biochemicals International, Natick, MA), 2S,2′R,3′R-2-(2′,3′-dicarboxycyclopropyl)glycine (DCG IV; Tocris Cookson), S-3,5-dihydroxyphenylglycine (DHPG; Tocris Cookson), dopamine (Sigma), (S)-α-methyl-4-carboxyphenylglycine (MCPG; Tocris Cookson), (RS)-α-methylserine-O-phosphate monophenyl ester (MSOPPE; Tocris Cookson), mitogen-activated protein kinase substrate peptide Ala-Pro-Arg-Thr-Pro-Gly-Gly-Arg-Arg (Alexis Biochemicals), PD98059 (2′-amino-3′-methoxyflavone; Alexis Biochemicals). All drugs were applied in perfusing medium, except for BAPTA and MAP-K peptide, which were included in recording electrodes and injected to postsynaptic cells. Dopamine was dissolved in ascorbic acid solution (20 μm; Sigma) to reduce oxidization of the compound.
Biochemical analyses. For a separate series of experiments to bioassay MAP-Ks, coronal brain slices were prepared identically as described above. After a recovery period (>2 hr), a slice was transferred to a well (∼10 ml volume) filled with oxygenated ACSF (28°C) containing 1 μm bicuculline methiodide. After a 10 min incubation with bicuculline, the slice was transferred to a separate well that additionally contains dopamine alone, dopamine with SCH23390 (D1 antagonist; Research Biochemicals International) and/or (−)-sulpiride (D2 antagonist; Sigma), dopamine with one of the following mGluR agonists, 1S,3R-ACPD, DHPG, and DCG IV, or one of the mGluR agonists alone. There, the slice was incubated for either 2 or 5 min in the drug-containing experimental solution. Separate slices that were only incubated in bicuculline-containing ACSF (10 min) served as control. Bicuculline itself did not modify activity of MAP-Ks. Some other slices were incubated in the bicuculline–ACSF for 10 min and then in the experimental ACSF for 10 min, before they were retransferred to the bicuculline–ACSF for either 2, 15, or 60 min. At the end of a given incubation period, the prelimbic area of both hemispheres was removed and stored at −20°C for later analyses.
Immunoblot analysis of MAP-Ks was performed as described by Towbin et al. (1979). Frozen slices were rapidly thawed and homogenized at 4°C in 20 μl of lysis buffer containing 1% nonidet P-40, 1% deoxycholic acid, 0.1% SDS, 158 mm NaCl, 10 mm Tris, pH 7.8, 1 mm phenylmethylsulfonyl fluoride, and 1 mm Na3VO4 (RIPA buffer), stirred for 20 min at 4°C, and centrifuged for 15 min at 12,000 × g. The protein concentration was determined by the Bradford microassay method (Bradford, 1976) using γ-globulin as standard. Equal amounts of protein from lysates (50 μg) were separated by electrophoresis on SDS–10% polyacrylamide gel and transferred onto a nitrocellulose membrane. Membranes were incubated for 2 hr at room temperature with 0.25% gelatin in Tris-buffered saline (10 mm Tris-HCl, pH 7.5, 150 mm NaCl) containing 0.05% Tween 20 (TBST) to block non-specific binding sites, and then for 2 hr with the polyclonal anti-active MAP kinase antibody (anti-ERKs) at 1:20,000 (Promega, Madison, WI), which recognizes dually phosphorylated MAP-Ks p44 and p42. After extensive washing steps with TBST, the membranes were incubated for 2 hr with horseradish peroxidase-conjugated goat anti-rabbit IgG (Dako, Glostrup, Denmark) at a dilution of 1:5000, then washed with blocking buffer. Proteins were visualized after a chemiluminescence staining following the manufacturer's protocol (ECL, Amersham, Arlington Heights, IL). Immunoreactive bands were analyzed by means of densitometry with a scanner, and their intensity was quantified using NIH image 1.61 software.
Reprobing with specific antibodies was performed after incubation for 2 hr at 65°C in 200 mm glycine, pH 2.5, containing 1% SDS, followed by two washes with 1 m Tris-HCl, pH 8, and one wash with TBST. The membrane was then incubated with the polyclonal anti-ERK2 C-14 antibody (at 1:1000; Santa Cruz Biotechnology, Santa Cruz, CA), which recognizes ERK2/p42 and weakly ERK1/p44.
RESULTS
LTD induction in the presence of dopamine requires synaptic activation of mGluRs
Figure 1A shows a representative layer V pyramidal neuron stained with biocytin, and Figure 1B shows our experimental configuration. As we have shown previously (Otani et al., 1998b), tetanic stimuli (50 Hz, 100 pulses × 4 in 10 sec intervals) delivered to layer I–II fibers at the end of 10–15 min bath application of dopamine (100 μm in 20 μm ascorbic acid) induced LTD of the monosynaptic component of the EPSP recorded from the soma of layer V pyramidal neurons (Fig. 1E) (−22 ± 7.6% decrease over baseline, measured during the 35–40 min period after tetani; n = 14,p < 0.002 vs control group shown in Fig.1C). The tetani alone did not induce any lasting synaptic changes (−0.7 ± 1.1%, n = 11) (Fig.1C), whereas dopamine application alone only transiently depressed synaptic transmission, which recovered fully within 30 min (4.7 ± 7.0% measured 35–40 min after the beginning of washout,n = 5) (Fig. 1D).
Fig. 1.
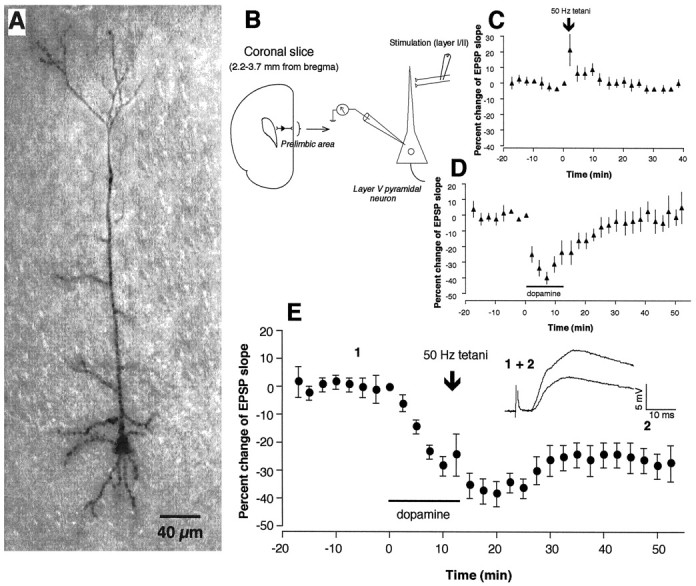
Dopaminergic facilitation of LTD induction in layer I-II to layer V pyramidal neuron glutamatergic synapses in rat prefrontal cortex. A, Photo image of a representative neuron stained with biocytin. B, Schematic representation of experimental protocol used for electrical measurement. C, Application of 50 Hz tetanic stimuli to layer I-II fibers did not induce lasting synaptic changes (−0.7 ± 1.0% over baseline measured at 35–40 min after tetani,n = 11). D, Bath application of dopamine (100 μm in 20 μm ascorbic acid, 10–15 min) acutely depressed the synaptic responses, but the depression fully recovered within 30 min after dopamine washout (4.7 ± 7.0% over baseline measured at 35–40 min after washout,n = 5). E, Application of tetanic stimuli in the presence of dopamine induced LTD [−22 ± 7.6% over baseline measured at 35–40 min after tetani–washout,n = 14, p < 0.002 vs control (C)]. Averaged synaptic responses taken from the indicated time points in this condition are superimposed and shown in the inset in E.
To test whether this LTD induction facilitated by dopamine requires synaptic activation of mGluRs, we first used the broad spectrum mGluR antagonist MCPG (300–500 μm), which acts on both group I and group II mGluRs. MCPG was coapplied to the bath with dopamine. At the end of the 10–15 min application period, during which dopamine exerted its acute depressant effect on the synaptic responses (Fig.2A), tetanic stimuli at 50 Hz were delivered (n = 5) (Fig.2A). Test stimulation was resumed 30 sec after the last train of tetanus. Synaptic responses were followed for at least 40 min. Tetani in the presence of dopamine + MCPG failed to induce LTD (Fig. 2A) [−5.2 ± 6.5% change over baseline, measured during the 35–40 min period after tetani–drug washout; n = 5, p > 0.3 vs control group in which tetani were delivered alone (−0.7 ± 1.1%)] (Fig. 1C). To test whether MCPG itself facilitates induction of LTP to mask LTD, in another group of neurons (n = 5) we applied MCPG alone for 10–15 min and delivered tetanic stimuli. In three of five neurons, MCPG facilitated short-term plasticity induction (potentiation in one case and depression in two cases), but never facilitated long-term plasticity induction. Mean percentage changes of the EPSP slope calculated from these five neurons are depicted in Figure 2B (−0.3 ± 3.8% over baseline at 35–40 min; n = 5, p > 0.9 vs control). Thus, MCPG genuinely blocked LTD induction in the presence of dopamine.
Fig. 2.

Dopaminergic facilitation of LTD is inhibited by the presence of MCPG, the antagonist of group I and group II mGluRs.A, MCPG (300–500 μm) was coapplied in the bath with dopamine. The rest of the protocols are identical to those in Figure 1E. MCPG blocked induction of LTD [−5.2 ± 6.5% 35–40 min after tetani–drug washout,n = 5, p > 0.3 vs control (Fig. 1C)]. Top traces are averaged responses taken from the indicated time points. B, The same tetanic stimuli in the presence of MCPG alone did not induce lasting synaptic changes (−0.3 ± 3.8% 35–40 min after tetani–MCPG, n = 5, p > 0.9 vs control), suggesting that the block of dopamine-facilitated LTD by MCPG (A) was not a masking of LTD. Top traces are averaged synaptic responses taken from the indicated time points.
Group I mGluRs are involved in LTD induction in the presence of dopamine
We next used the specific antagonist of group I mGluRs AIDA (200 μm) (Moroni et al., 1997). AIDA was bath-applied with dopamine for 10–15 min, and 50 Hz tetani were delivered at the end of the coapplication (n = 5). AIDA also blocked LTD induction in the presence of dopamine. We plotted average changes of the EPSP slope calculated from the five cells in Figure3A. Mean EPSP change occurring during the 35–40 min period after tetani–drug washout (1.8 ± 6.0%, n = 5) shows no significant difference from mean value calculated from the same period in the control group (−0.7 ± 1.1%; p > 0.1) (Fig. 1C), but it shows a significant difference from that of the group in which dopamine application alone was combined with tetani (−22 ± 7.6%;p < 0.025) (Fig. 1E). In another group of cells (n = 6), AIDA was applied alone to test whether AIDA itself facilitates LTP to mask LTD. We plotted mean percentage changes of the EPSP slope from six experiments under this condition in Figure 3B. Mean change at the 35–40 min period after tetani–drug washout is −1.3 ± 7.0% (p > 0.9 vs control). Thus, the failure of AIDA to induce lasting synaptic plasticity suggests that the block of LTD with AIDA (Fig. 3A) cannot be attributed to a masking of LTD.
Fig. 3.

Group I mGluR antagonist AIDA inhibits dopaminergic facilitation of LTD. A, AIDA (200 μm) was bath-applied with dopamine for 10–15 min, and tetanic stimuli were delivered (identical to Fig.1E except the presence of AIDA). No LTD was induced [−1.8 ± 6.0% 35–40 min after tetani–drug washout,n = 5, p > 0.1 vs control (Fig. 1C), but p < 0.025 versus dopamine + 50 Hz group (Fig. 1E)].B, In a separate group of neurons, AIDA alone was bath-applied, and tetani were delivered. Short-term changes occurred, but no lasting synaptic changes were induced (−1.3 ± 7.0% 35–40 min after tetani–AIDA, n = 6,p > 0.9 vs control), suggesting that the block of LTD by AIDA (A) was not a masking of LTD.Top traces are averaged synaptic responses taken from the indicated time points.
Group II mGluRs are also involved in LTD induction in the presence of dopamine
Next, we coapplied the selective group II mGluR antagonist MSOPPE (200 μm) (Thomas et al., 1996) in the bath with dopamine for 10–15 min before tetanic stimuli were delivered (n = 7). In Figure4A, we plotted mean EPSP changes from these experiments. The EPSP slope change occurring during the 35–40 min period after tetani–drug washout is −5.3 ± 3.1%, still showing no significant difference compared with the control group (p > 0.1), but showing a significant difference from the group in which dopamine application alone was combined with tetani (p < 0.04).
Fig. 4.

Group II mGluR antagonist MSOPPE inhibits dopaminergic facilitation of LTD. A, MSOPPE (200 μm) was bath-applied with dopamine for 10–15 min, and tetanic stimuli were delivered (identical to Fig.1E except the presence of MSOPPE). Tetani failed to induce LTD [−5.3 ± 3.1% 35–40 min after tetani–drug washout, n = 7, p > 0.1 vs control (Fig. 1C), but p < 0.04 versus dopamine + 50 Hz group (Fig. 1E)].B, Tetanic stimuli in the presence of MSOPPE alone induced some degree of post-tetanic changes in five of nine cases but did not induce LTP on average. Mean percentage increase 35–40 min after tetani–drug washout calculated from eight experiments was 0.6 ± 3.4% over baseline (recording of one cell that showed STP discontinued 25 min after tetani; p > 0.7 vs control depicted in Fig. 1C). Top tracesare averaged synaptic responses taken from the indicated time points.
In a separate group of cells (n = 9) (Fig.4B), the effect of MSOPPE alone on synaptic plasticity was tested. Tetani delivered at the end of 10–15 min application of MSOPPE were sometimes followed by potentiation (four of nine cells) or depression (one of nine cells). In both cases, the EPSP slope returned to control level within 30 min, except one case in which some degree of lasting changes was still observed at 35–40 min after tetani (16% increase). In Figure 4B, we plotted mean changes of the EPSP slope calculated from eight experiments in which intracellular recordings continued for 40 min after tetanic stimuli [the recording of one cell that showed short-term potentiation (STP) discontinued 25 min after tetani]. Because of the variability of the changes in the responses after tetani, the graph shows large SEMs after delivery of tetani. The variation and the post-tetanic effects, however, decreased toward the end of recording, generating a mean EPSP slope change of only 0.6 ± 3.4% during the 35–40 min period after tetani–drug washout (p > 0.7 compared with control, n = 8). These results suggest that the blockade of LTD with MSOPPE (Fig. 4A) cannot be explained by a masking of LTD.
Synaptic responses during 50 Hz stimuli in the presence of group II mGluR antagonist
Presynaptic group II mGluRs use-dependently inhibit glutamate release at least in certain synapses (Conn and Pin, 1997). Thus, the group II antagonist MSOPPE may facilitate synaptic transmission during tetanus. Postsynaptic depolarization then may exceed a degree optimal for LTD induction, leading to a blockade of LTD. To test this possibility, we analyzed synaptic responses during tetanus in the presence of MSOPPE (n = 8) (see Fig.4B for EPSP plots before and after tetani in this group. Responses during tetanus in one cell could not be recorded because of a technical failure). We measured the following three parameters in high-frequency synaptic responses: (1) the number of spikes per tetanus episode, (2) the number of the EPSPs per tetanus episode whose amplitudes remained larger than 50% of the initial EPSP evoked in that episode, and (3) 90% decay time from peak membrane potential (spike threshold was taken when a spike(s) was present). On average, MSOPPE increased the number of spikes (p < 0.05 in all four train episodes) and the number of EPSPs (p < 0.05 in the first train episode; p < 0.1 in the second and fourth episodes) evoked to a train of tetanus over control group (n = 12) (Fig. 5, insets). Ninety percent decay time value did not show a significant difference on average over control, but when the five cells in which a clear STP or short-term depression was induced are taken into account, MSOPPE increased 90% decay over control at least in the first tetanus episode (2217 ± 606 vs 726 ± 228 msec, p < 0.02). One typical example that shows increases in these three parameters is shown and compared with a control cell in Figure 5. MSOPPE, in contrast, did not change at least two postsynaptic parameters tested by direct somatic current injections (0.5–0.6 nA for 500 msec), i.e., spike-train adaptation and afterhyperpolarization (n = 3; data not shown).
Fig. 5.
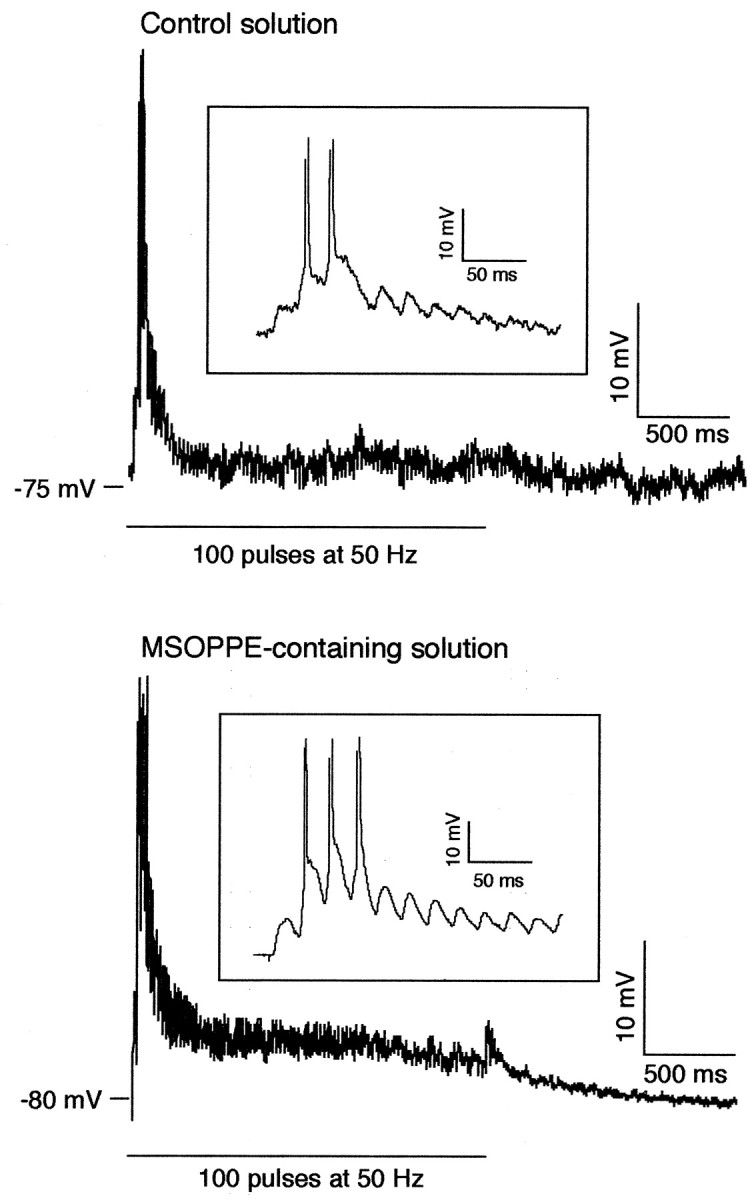
MSOPPE, the group II mGluR antagonist, augments synaptic responses during 50 Hz tetanic stimuli, compared with control condition (see Fig. 4B for plots of EPSP slope before and after tetani in this MSOPPE condition). Traces shown in this figure are the synaptic responses evoked by the first of four episodes of tetanic stimuli in two different cells. On average, MSOPPE (200 μm, n = 8) increased the number of spikes (p < 0.05 in all four train episodes) and the number of the EPSPs whose amplitudes were larger than 50% of the first EPSP evoked in the given train episode (p < 0.05 in the first train episode,p < 0.1 in the second and fourth episodes). Ninety percent decay time from peak membrane depolarization was, on average, not different from control, but if five cells that showed clear post-tetanic changes (see Results) are taken into account, MSOPPE increased 90% decay over control in the first tetanus episode (2217 ± 606 msec vs 726 ± 228 msec, p< 0.02). The MSOPPE-treated cell shown in this figure is a cell that showed clear increases in all three of these parameters. MSOPPE did not reduce spike-train adaptation and afterhyperpolarization tested with postsynaptic current injection (data not shown). It is unlikely, however, that this augmentation of synaptic responses during tetanus by MSOPPE per se is the mechanism by which MSOPPE blocked LTD in the presence of dopamine (Fig. 4A), because dopamine itself augmented synaptic responses during tetanus (Otani et al., 1998b) and occluded the effect of MSOPPE. Insets show the same responses by different scales of amplitude and time.
We next compared synaptic responses during tetanus in the presence of MSOPPE + dopamine (no LTD condition) with those in the presence of dopamine alone (LTD condition). Under this comparison, MSOPPE no longer exerted significant changes in the high-frequency synaptic responses in the above three parameters. In other words, the enhancing effects of dopamine on these parameters (Otani et al., 1998b) are sufficient to occlude the effects of MSOPPE.
In contrast to MSOPPE, the group I antagonist AIDA did not significantly increase postsynaptic responses during tetanus. Postsynaptic responses in the presence of AIDA + dopamine also did not differ from those in the presence of dopamine alone.
Effects of mGluR agonists on synaptic responses and LTD
Results so far indicate that synaptic activation of groups I and II mGluRs must occur for dopamine to facilitate LTD induction. In separate groups of cells, we tested whether pharmacological activation of mGluRs can also facilitate LTD by 50 Hz tetani, where 50 Hz tetani alone do not induce lasting synaptic changes (Fig. 1C).
First, 1S,3R-ACPD (100 μm), the groups I and II mGluR agonist, was applied for 10–15 min before delivery of tetani (n = 4). All four cells expressed LTD (Fig.6A). Mean change of the EPSP slope occurring during the 35–40 min period after tetani–drug washout was −37 ± 5.7% (n = 4,p < 0.001 vs control depicted in Fig. 1C). As shown in Figure 6B, and as is the case in hippocampus (Aniksztejn et al., 1992), sole application of 1S,3R-ACPD induced only a transient depression that fully recovers within 30 min (3.4 ± 4.2% mean change at 35–40 min after washout; n = 4). Thus, pharmacological stimulation of groups I and II mGluRs facilitates LTD induction.
Fig. 6.

Simultaneous activation of group I and group II mGluRs by 1S,3R-ACPD facilitates LTD induction. A, Tetanic stimuli in the presence of 1S,3R-ACPD (100 μm, 10–15 min) induced LTD (n = 4). Mean LTD occurring 35–40 min after tetani–drug washout was −37 ± 5.7% [(n = 4, p < 0.001 vs control (Fig. 1, top right inset)]. Top tracesare averaged responses taken from time points 1 and2. A superimposed representation of the responses (1+2) is also shown. B, Bath application of 1S,3R-ACPD (100 μm) alone only transiently depressed synaptic responses, which fully recovered within 30 min (n = 4; mean percentage change of the EPSP slope 35–40 min after drug washout, 3.4 ± 4.2%).
Second, involvement of group I mGluRs in LTD induction was tested by the use of specific group I mGluR agonist DHPG (100 μm) (Brabet et al., 1995; Sekiyama et al., 1996). First, as shown in Figure7A (n = 4), when applied alone (10–15 min), DHPG caused a transient depression of the EPSP (Gereau and Conn, 1995), which is accompanied by several postsynaptic parameter changes, including weak membrane depolarization and a reduction in spike-train adaptation and afterhyperpolarization (tested at membrane potential restored to resting level) (Fig.7A, top traces) (cf. Vickery et al., 1997). The depression of the EPSP fully recovered within 30 min (mean change at 35–40 min, 2.0 ± 4.5%, n = 4). In another group of cells (n = 5) (Fig. 7B), 50 Hz tetani were applied at the end of DHPG application. DHPG did not facilitate LTD induction (4.8 ± 7.1% at 35–40 min,n = 5, p > 0.1 vs control). Thus, sole activation of group I mGluRs is not sufficient to facilitate LTD.
Fig. 7.
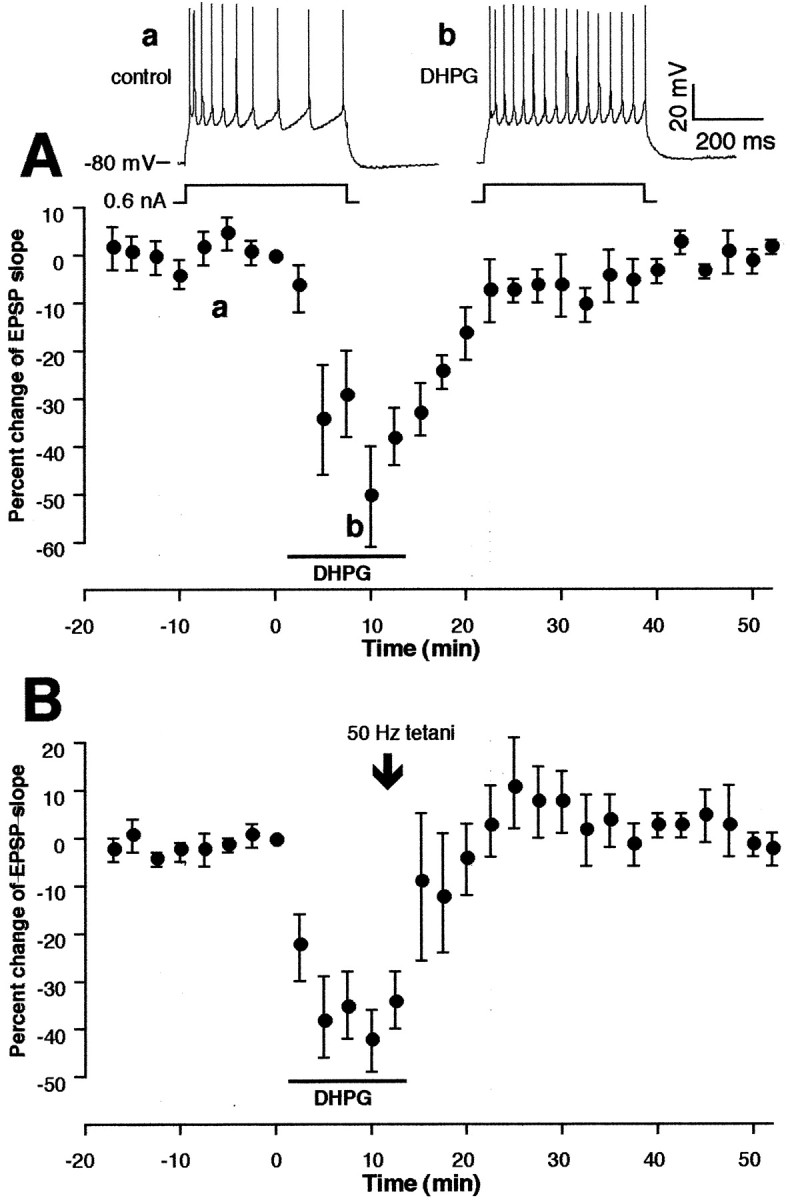
Sole activation of group I mGluRs with the agonist DHPG is insufficient to facilitate LTD. A, In the first group (n = 4), DHPG (100 μm, 10–15 min) was applied alone in the bath without tetani. DHPG induced a transient synaptic depression that was accompanied by a mild (a few millivolts) postsynaptic membrane depolarization and a reduction in spike-train adaptation and afterhyperpolarization (shown in top traces; membrane potential set at resting level). Mean change of the EPSP slope 35–40 min after drug washout was 2.0 ± 4.5% (n = 4). B, Tetanic stimuli in the presence of DHPG failed to facilitate LTD induction. Mean change of the EPSP slope 35–40 min after tetani–DHPG was 4.8 ± 7.1% [n = 5, p > 0.1 vs control (Fig. 1C)].
Third, effects of the potent group II mGluR agonist DCG IV (Ishida et al., 1993) on the synaptic responses and LTD were tested. Application of DCG IV [50–100 nm, concentrations two orders below that to activate NMDA receptors (>10 μm) (Ishida et al., 1993; Wilsch et al., 1994)] acutely depressed the synaptic responses (−25 ± 4.4% at the end of 10–15 min application,n = 6) (Fig.8A). The acute depression was not accompanied by postsynaptic membrane depolarization or changes in spike-train adaptation or afterhyperpolarization (Fig.8A, top traces). Interestingly, the synaptic depression remained after washout of DCG IV, showing a −21 ± 3.8% mean LTD during the 35–40 min period after washout (n = 6) (Fig. 8A). In another group of cells (n = 4), tetanic stimuli were applied at the end of 10–15 min DCG IV application. Tetani did not augment LTD expressed at 35–40 min after tetani–DCG IV washout (−19 ± 7.1%, p > 0.8 compared with DCG IV group; data not shown).
Fig. 8.

Sole activation of group II mGluRs with potent agonist DCG IV induces postsynaptic Ca2+-dependent LTD without tetanic stimuli. A, Bath application of DCG IV (50–100 nm) acutely depressed synaptic response that is followed by a lasting depression. Mean change of the EPSP slope 35–40 min after drug washout was −21 ± 3.8% (n = 6). Top traces show that DCG IV did not change spike-train adaptation and afterhyperpolarization. Superimposed averaged synaptic responses taken from time points 1 and2 are also shown. B, DCG IV-induced LTD was absent in cells injected with BAPTA. BAPTA did not block acute depressant action of DCG IV on the synaptic responses. Mean change of the EPSP slope 35–40 min after DCG IV washout was 0.1 ± 2.2% (n = 4, p < 0.005 vs above DCG IV group). In a separate group of neurons (n = 4), a late application of MSOPPE did not reverse the expression of DCG IV-induced LTD (see Results for details), further suggesting that DCG IV-induced LTD is not a result of insufficient washout of the drug.
Two lines of evidence suggest that LTD induced by DCG IV is not a result of insufficient washout from the acute depressant action of the drug. First, as shown in Figure 8B, a previous postsynaptic injection of Ca2+ chelator BAPTA (100 mm in recording electrodes) completely blocked LTD by DCG IV application (0.1 ± 2.2% mean EPSP change at 35–40 min after drug washout, n = 4,p < 0.005 vs DCG IV group), but not the acute depression by DCG IV (−22 ± 2.1% at the end of 10–15 min application). Diffusion of BAPTA was facilitated by steady negative current injections through the recording electrodes (0.2–0.4 nA for at least 30 min before drug application). Second, application of MSOPPE (200 μm) did not reverse the expression of DCG IV-induced LTD. MSOPPE (200 μm) was applied at 25 min after the beginning of DCG IV washout until the end of recording. Depression expressed at 35–40 min after DCG IV washout in the presence of MSOPPE was −17 ± 5.4% (n = 5,p > 0.5 vs DCG IV group; data not shown). These data suggest that strong pharmacological stimulation of group II mGluRs can by itself induce LTD. The BAPTA experiments further show that LTD induced by DCG IV involves postsynaptic Ca2+-dependent processes.
Coactivation of dopamine receptors and mGluRs is sufficient for LTD induction
Dopamine sole application or 1S,3R-ACPD sole application does not induce LTD (Figs. 1D,6B), whereas 50 Hz stimuli combined with either drug induce LTD (Figs. 1E, 6A). Does then coapplication of these two agonists activate biochemical cascades sufficient to induce LTD? To answer this question, dopamine (100 μm) and 1S,3R-ACPD (100 μm) were coapplied in the bath for 10–15 min without delivery of tetanic stimuli (n = 5). In five of five cases, clear LTD was induced. LTD occurring during the 35–40 min period after the end of coapplication was −27 ± 2.6% (Fig.9A) (n = 5, p < 0.005 vs the group in which dopamine was applied alone, and p < 0.001 vs the group in which ACPD was applied alone). In four other cells, we tested whether 0.033 Hz single test stimuli during the drug coapplication are necessary for this LTD. Thus, test synaptic stimulation was halted just before coapplication of dopamine + 1S,3R-ACPD until 30 min after the beginning of washout of the drugs. Under this condition, as shown in Figure9B, coapplication of dopamine + 1S,3R-ACPD still induced LTD (−29 ± 5.1% 35–40 min after washout, n = 4, p < 0.01 vs dopamine alone group, and p < 0.005 vs 1S,3R-ACPD alone group). In four other cells, we halted test synaptic stimuli for 45 min (equivalent to the 15 min drug application–stimulus omission period plus the 30 min post-drug stimulus omission period in the above condition) to test whether omission of test stimuli itself causes decreases of synaptic responses. This was not the case (data not shown; −0.3 ± 5.2% during the period equivalent to 35–40 min after washout, p < 0.01 vs the group depicted in Fig. 9B). These results suggest that LTD induced by coactivation of dopamine receptors and groups I and II mGluRs requires neither stimulation of presynaptic fibers nor the activation of other classes of postsynaptic receptors by evoked transmitter release.
Fig. 9.

Simultaneous activation of dopamine receptors and mGluRs is sufficient for LTD induction. A, Bath application (10–15 min) of dopamine (100 μm in 20 μm ascorbic acid) and 1S,3R-ACPD (100 μm) induced synaptic depression that remained after washout of the drugs [−27 ± 2.6% 35–40 min after drug washout,n = 5, p < 0.005 vs dopamine-alone group (Fig. 1D) andp < 0.001 vs 1S,3R-ACPD-alone group (Fig.6B)]. B, Induction of LTD by dopamine + ACPD coapplication did not require 0.033 Hz single test synaptic stimuli (−29 ± 4.2% 35–40 min after drug washout,n = 4, p < 0.01 vs dopamine-alone group and p < 0.005 vs 1S,3R-ACPD-alone group). Test stimuli were halted just before application of the drugs until 30 min after the beginning of drug washout. Halting of test stimuli itself did not change synaptic responses (see Results). C, LTD was also induced by coapplication of dopamine and group I agonist DHPG (100 μm) in four of five cells. The plots were made from all five experiments (−29 ± 8.9% 35–40 min after drug washout,n = 5, p < 0.025 vs both DHPG and dopamine groups). Thus, although LTD induced by 50 Hz tetani in the presence of dopamine requires synaptic activation of group Iand group II mGluRs (Figs. 2A, 3A, 4A), and although 50 Hz tetani in the presence of groups I and II agonist 1S,3R-ACPD, but not DHPG, induce LTD (Figs. 6A, 7B), pharmacological activation of group I mGluRs alone with DHPG, if combined with dopamine application, can induce LTD. However, this LTD induction is somewhat less consistent than that after groups Iand II mGluRs are coactivated with 1S,3R-ACPD (nine LTD of nine cases) (Fig.9A,B).
In an additional group (n = 5) (Fig. 9C), we tested whether sole stimulation of group I mGluRs with DHPG (100 μm) combined with dopamine application induces LTD. In four of five cases, dopamine + DHPG coapplication resulted in LTD (mean EPSP change −29 ± 8.9%, n = 5,p < 0.025 vs both DHPG and dopamine groups). Thus, although LTD induced by 50 Hz in the presence of dopamine (Fig.1E) requires synaptic activation of group Iand group II mGluRs (Figs. 2A,3A, and 4A), and although 50 Hz tetani in the presence of groups I and II agonist 1S,3R-ACPD, but not group I agonist DHPG, induce LTD (Figs. 6A, 7B), pharmacological activation of group I mGluRs alone with DHPG, if combined with dopamine bath application, can preclude the requirement for group II mGluR activation in LTD. However, it is important to note that one of the five cells in the dopamine + DHPG group showed a complete return to baseline level after drug washout (−0.4% at 35–40 min), suggesting that concurrent activation of group I andgroup II mGluRs with 1S,3R-ACPD facilitated LTD more consistently (clear LTD in nine of nine cases; groups depicted in Fig. 9A,B combined).
LTD in prefrontal cortex requires activation of MAP kinases
In the next series of experiments, we searched for a biochemical manner where dopamine receptors and mGluRs cooperate to induce LTD. We chose to examine the participation of mitogen-activated protein kinases MAP-Ks (also known as ERKs), because MAP-Ks occupy a position where activation of many other second messengers, including protein kinase C (PKC) and cAMP-dependent protein kinase A (PKA), converge to exert their effects (Nestler and Greengard, 1994; Cobb and Goldsmith, 1995; Robinson and Cobb, 1997; Roberson et al., 1999). We used PD98059, a cell-permeable specific inhibitor of MAP-K kinases 1 and 2 (MEK1 and MEK2) (Alessi et al., 1995; Dudley et al., 1995). The MEKs specifically activate ERK1 and ERK2, respectively (Cohen, 1997). The effect of PD98059 was tested on three forms of LTD reported in this paper: (1) LTD induced by 50 Hz stimuli in the presence of dopamine (Fig.1E), (2) LTD induced by 50 Hz stimuli in the presence of 1S,3R-ACPD (Fig. 6A), and (3) LTD induced by coapplication of dopamine and 1S,3R-ACPD (Fig. 9A). PD98059 (20 μm) was bath-applied 15 min before application of dopamine (100 μm) or 1S,3R-ACPD (100 μm), or both, until the end of the application–tetani. PD98059 neither changed baseline synaptic responses during the preincubation period nor affected the acute depression of synaptic responses by dopamine or 1S,3R-ACPD (Fig.10). PD98059, however, completely blocked induction of LTD by 50 Hz tetani in the presence of dopamine (Fig. 10A) (0.3 ± 4.1% EPSP change 35–40 min after drugs–tetani, n = 6,p < 0.02 vs dopamine + tetani group depicted in Fig.1E). Large post-tetanic potentiation was noted in some cells of this group. Second, PD98059 completely blocked LTD by 50 Hz tetani in the presence of 1S,3R-ACPD (Fig.10B) (−0.6 ± 3.9% 35–40 min after drugs–tetani, n = 4, p < 0.002 vs 1S,3R-ACPD + tetani group). Third, PD98059 completely blocked LTD by dopamine + 1S,3R-ACPD (Fig. 10C) (−4.9 ± 5.8% 35–40 min after drug washout, n = 6, p < 0.02 vs dopamine + 1S,3R-ACPD group shown in Fig. 9A). In addition, 50 Hz tetani in the presence of PD98059 alone did not induce lasting synaptic changes (data not shown; −2.6 ± 3.9%,n = 4).
Fig. 10.

LTD induction in prefrontal cortex requires activation of MAP-Ks. A, LTD by 50 Hz tetani in the presence of dopamine (Fig. 1E) was blocked by bath application of PD98059 (20 μm), the specific inhibitor of MAP-K kinases (MEK1 and MEK2, which phosphorylate ERK1 and ERK2). Mean change of the EPSP slope measured 35–40 min after drug washout–tetani was 0.3 ± 4.1% (n = 6,p < 0.02 vs dopamine + tetani group depicted in Fig. 1E). B, LTD by 50 Hz tetani in the presence of 1S,3R-ACPD (Fig.6A) was also blocked by bath application of PD98059. Mean change of the EPSP slope measured 35–40 min after drug washout–tetani was −0.6 ± 3.9% [n = 4,p < 0.002 vs 1S,3R-ACPD + tetani group (Fig.6A)]. C, LTD by coactivation of dopamine receptors and groups I and II mGluRs with 1S,3R-ACPD (Fig. 9A) is blocked by PD98059. Mean change of the EPSP slope measured 35–40 min after drug washout was −4.9 ± 5.8% (n = 6,p < 0.02 vs the group depicted in Fig.9A). D, LTD by dopamine + 1S,3R-ACPD coapplication is also blocked by postsynaptic injection of specific synthetic MAP-K substrate peptide Ala-Pro-Arg-Thr-Pro-Gly-Gly-Arg-Arg (1 mm in electrode). This peptide, when abundantly present, acts as a specific competitive inhibitor against endogenous MAP-K substrates. Mean change of the EPSP slope 35–40 min after drug washout was 8.4 ± 6.2% [n = 5, p < 0.001 vs dopamine + 1S,3R-ACPD group (Fig.9A)]. Thus, critical MAP-K activation for LTD induction after coactivation of dopamine receptors and mGluRs occurs in postsynaptic sites.
In a separate group of neurons (n = 5) (Fig.10D), we tested postsynaptic locus of critical MAP-K activation for induction of LTD by dopamine + 1S,3R-ACPD coapplication. This form of LTD was chosen, because it does not require any presynaptic stimulation for its induction (Fig. 9B) and because it is this form of LTD for which we bioassayed MAP-K activity during induction (see next section). We used synthetic MAP-K substrate peptide Ala-Pro-Arg-Thr-Pro-Gly-Gly-Arg-Arg (Baron et al., 1996), which acts under physiological conditions as a specific competitive inhibitor against endogenous MAP-K substrates. The peptide was postsynaptically injected through recording electrodes (1 mm in electrodes). Diffusion was allowed for at least 45 min before the application of dopamine and 1S,3R-ACPD. In five of five cases, dopamine + 1S,3R-ACPD failed to induce LTD under this condition. Mean changes of the EPSP slope calculated from all five experiments are depicted in Figure10D (8.4 ± 6.2% 35–40 min after drugs,n = 5, p < 0.001 vs dopamine + 1S,3R-ACPD group). Thus, critical MAP-K activation for LTD induction occurs in postsynaptic sites.
Synergistic–additive activation of MAP kinases by dopamine and mGluR agonists
Provided with the above results, we decided to biochemically isolate converging activation of MAP-Ks by dopamine receptors and mGluRs in prefrontal tissue. Activation of MAP-Ks occurs through a dual phosphorylation of threonine and tyrosine residues (Her et al., 1993). We measured MAP-K activation by the use of antibody anti-active ERKs recognizing only the dually phosphorylated active forms of ERK1 and ERK2, after exposure of prefrontal tissue to the agonists and antagonists of dopamine and mGlu receptors. As depicted in Figure11A,B(top traces), in the control condition, no phosphorylated ERK1 (44 kDa) band was detected despite the presence of a modest level of phosphorylated ERK2 (42 kDa). The top traces also show that both dopamine (100 μm) and 1S,3R-ACPD (100 μm) increase the phosphorylation of ERK2 (42 kDa) within 2 min after the beginning of bath application. Coapplication of dopamine + 1S,3R-ACPD (a principal LTD condition) (Figs.9A,B, 10C,D) resulted in an apparent additive increase of the level of ERK2 phosphorylation. Similar patterns of ERK2 activity increases were seen with the other mGluR agonists DHPG (100 μm) and DCG IV (100 nm) (Fig.11A,B). In the case of ERK1, dopamine does not increase its phosphorylation, whereas 1S,3R-ACPD weakly increases it. Interestingly, when dopamine and 1S,3R-ACPD are applied together (an LTD condition), an increase in the phosphorylation exceeds that achieved by 1S,3R-ACPD alone, showing a clear synergism (Fig. 11A,B). Again, similar patterns of activity increases were obtained with DHPG and DCG IV. As shown in the bottom traces of Figure11A,B, the two phosphorylated bands were demonstrated to correspond to p44/ERK1 and p42/ERK2, by the use of antibody anti-ERK2, which recognizes nonphosphorylated p42/ERK2 and, weakly, p44/ERK1.
Fig. 11.

Dopamine and mGluR agonists additively or synergistically activate MAP-Ks as detected with anti-active MAP-K antibody. Slices were incubated at 28°C in buffer containing 1 μm bicuculline without other drugs (CNT) or with dopamine (DA, 100 μm) or an mGluR agonist (1S,3R-ACPD, 100 μm; DHPG, 100 μm; DCG IV, 100 nm) or both for 2 or 5 min. Bicuculline itself did not change MAP-K activity (data not shown). Equal amounts of lysates (50 μg) were resolved on a 10% acrylamide gel. Activated MAP-Ks were detected with an antibody anti-active MAP-Ks (anti-active ERKs) that recognizes dually phosphorylated ERKs. After stripping, the immunoblot was reprobed with polyclonal anti-ERK2 (see Materials and Methods). A,B, Results with 2 and 5 min application, respectively. Note that phosphorylated protein bands are always denser after dopamine plus an mGluR agonist than either drug alone (see Results).C, Quantification by scanning densitometry of the autoradiogram of anti-active ERKs using NIH Image 1.6. The data are representative of at least three independent experiments. For the ERK2 activity (2 and 5 min), every drug condition showed statistical significance of at least p < 0.05 over control. For the ERK1 activity (5 min), the increase seen in the presence of dopamine and a given mGluR agonist was always significantly larger than the increase seen in the presence of that mGluR agonist alone (p < 0.05). The graph is plotted as percentage changes relative to CNT levels for p42/ERK2, and raw arbitrary numbers for p44/ERK1 because of the lack of p44/ERK1 phosphorylation in CNT.
In an additional experiment (n = 4), we determined whether the group II mGluR antagonist MSOPPE affects MAP-K activity. This experiment was performed for the following reason. Group II mGluRs are coupled negatively to cAMP (Tanabe et al., 1992, 1993). Therefore, their block might increase cAMP levels and lead to the activation of MAP-Ks. If this is the case, then it would suggest that the blockade by MSOPPE of LTD induction (Fig. 4A) may have resulted from a previous saturation of MAP-K activation rather than an inhibition of MAP-K activation through antagonism against synaptic stimulation of group II mGluRs. Prefrontal tissues were exposed to MSOPPE (200 μm in the presence of bicuculline) for 20 min, and ERK1 and ERK2 phosphorylations were determined. MSOPPE did not increase MAP-K phosphorylation at all (data not shown), rejecting the possibility that there is a tonic inhibition of MAP-K activation by group II mGluRs. Rather, stimulation of group II mGluRsactivates MAP-Ks (Fig. 11, DCG IV results).
Quantification by scanning densitometry (Fig. 11C) shows that in all drug conditions, maximal effect on the phosphorylation of p44/ERK1 and p42/ERK2 was obtained at 5 min after drug application. Activation of p42/ERK2 with mGluR agonists (1S,3R-ACPD, DHPG, or DCG IV) was approximately fourfold over that of control condition (p < 0.05). With dopamine, activation of p42/ERK2 was approximately 2.5-fold over control (p < 0.05). When dopamine and one of the mGluR agonists were coapplied, the increase of p42/ERK2 reached approximately sixfold activation over control (p< 0.05). Activation of p44/ERK1 was undetectable in control and dopamine conditions, as shown in Figure11A,B. The mGluR agonists stimulated p44/ERK1 phosphorylation. The combination of a mGluR agonist with dopamine caused further increases of p44/ERK1 phosphorylation, which are significantly different from the increases seen in the presence of the mGluR agonist alone (p < 0.05). We observed that the activation of MAP-Ks by these drugs declines to basal level within 60 min after drug washout (data not shown).
In another set of experiments, we tested which subtype of dopamine receptors is involved in the dopaminergic activation of MAP-Ks (Fig.12). As shown in Figure12A, dopamine (100 μm) increased p42/ERK2 phosphorylation, and also in this case the phosphorylation of p44/ERK1. These increases were reduced by the presence of D1 antagonist SCH23390 (1 μm) or D2 antagonist sulpiride (50 μm) and blocked by the co-presence of two antagonists. Group results obtained from quantification by scanning densitometry (Fig. 12B) revealed a synergistic effect of D1 and D2 receptor activation on MAP-K phosphorylation. First, dopamine increased p42/ERK2 phosphorylation more than twofold (p < 0.005). The presence of either SCH23390 or sulpiride was sufficient to significantly reduce the dopamine effect on p42/ERK2 phosphorylation (p < 0.001 for the dopamine + SCH23390 group and p < 0.05 for the dopamine + sulpiride group, compared with dopamine group). Co-presence of SCH23390 and sulpiride blocked the effect of dopamine on ERK2 phosphorylation (p < 001). Similar patterns of the effects of dopamine and dopamine antagonists were seen for p42/ERK1 phosphorylation. These results show that sole activation of either D1 or D2 receptors by dopamine is insufficient to activate MAP-Ks; two subtypes of dopamine receptors must be stimulated to bring about MAP-K activation.
Fig. 12.
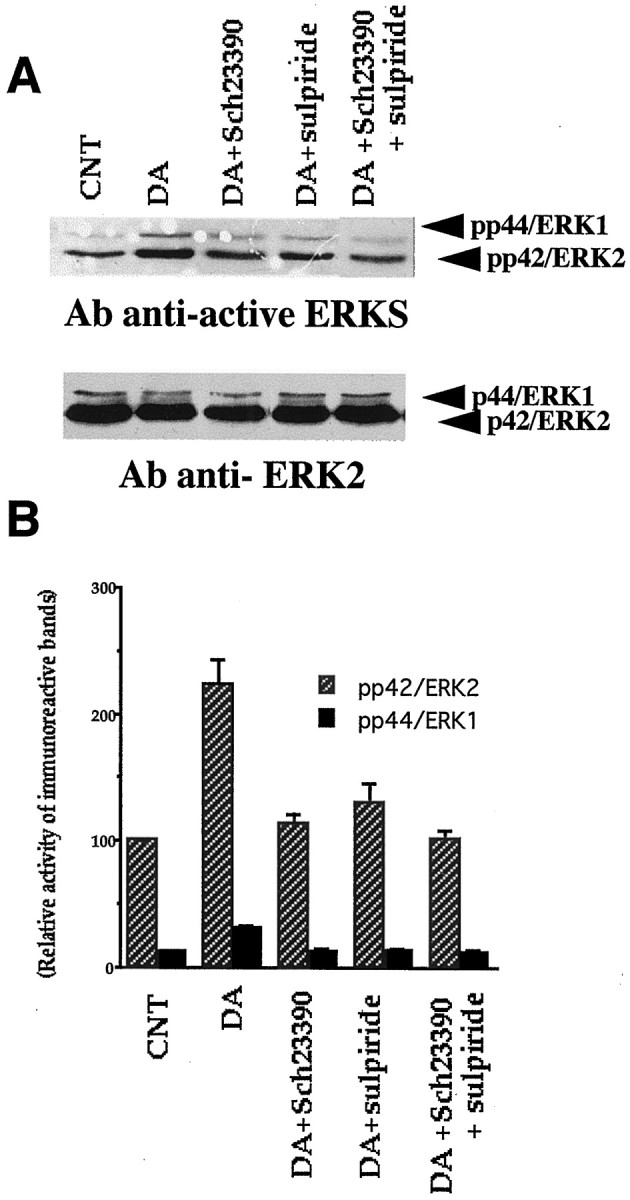
Dopamine D1 and D2 receptors synergistically activate MAP-Ks as detected with anti-active MAP-K antibody. Methods are similar to those depicted in Figure 11 except the use of D1 antagonist SCH23390 (1 μm) and D2 antagonist sulpiride (50 μm). These antagonists were preincubated for 10 min before dopamine (100 μm) was applied. In all results in the figure, the duration of dopamine application was 2 min, but similar results were obtained after 5 min application. A, Phosphorylation bands of ERK1 and ERK2 after various conditions. Control tissue showed a relatively dense band of ERK2 as in Figure 11and also a light ERK1 band in this case. Dopamine increased phosphorylation of both ERK1 and ERK2. Note that phosphorylation bands are lighter in the presence of SCH23390 or sulpiride, and the band in the presence of two antagonists is at control level. B, Synergism between D1 and D2 receptors revealed by quantification by scanning densitometry of the autoradiogram of anti-active ERKs using NIH Image 1.6. Each group contains four observations. Dopamine increased ERK1 and ERK2 phosphorylation more than twofold (ERK1,p < 0.001; ERK2, p < 0.005). This dopamine effect was highly significantly reduced by the presence of the D1 antagonist SCH23390 (ERK1, p < 0.002; ERK2, p < 0.001) or the D2 antagonist sulpiride (ERK1, p < 0.002; ERK2, p < 0.05). Co-presence of SCH23390 and sulpiride blocked dopamine phosphorylation of MAP-Ks (ERK1 and ERK2, p < 0.001). These results suggest that sole activation of either D1 or D2 receptors by dopamine is insufficient to bring about activation of MAP-Ks. Two subtypes of dopamine receptors must be stimulated for MAP-K activation. The graph is plotted as percentage changes relative to CNT levels for both p42/ERK2 and p44/ERK1. For this purpose, control was taken as 100 for ERK2 and 12.5 for ERK1, to reflect roughly the ERK2/ERK1 densitometry proportion obtained by scanning.
DISCUSSION
Figure 13 schematically shows possible mechanisms underlying LTD induction in layer I–II to layer V pyramidal neuron glutamatergic synapses of rat prefrontal cortex, based on the present results and those obtained in previous studies in this laboratory. First (Fig. 13(a)), LTD induced by 50 Hz in the presence of dopamine (depicted as a dopaminergic axon terminal) does not require NMDA receptor activation (Otani et al., 1998b), consistent with the fact that dopamine acutely depresses the NMDA component, as well as the non-NMDA component, of synaptic transmission through presynaptic or postsynaptic action, or both (Low-Tho et al., 1994;Low-Tho, 1995; our unpublished observations). Second (Fig.13(b)), dopamine, however, augments synaptic responses during the LTD-inducing high-frequency drive to enhance postsynaptic depolarization, which permits Ca2+ flow through voltage-gated channels and facilitates LTD (Otani et al., 1998b). At least L-type channels are not involved in the Ca2+ flow, because the L-channel blocker nifedipine did not block LTD (our unpublished observation). The present study showed that (Fig. 13(c)) LTD induction by 50 Hz stimuli in the presence of dopamine is blockable by the application of MCPG (groups I and II antagonist), AIDA (group I antagonist), or MSOPPE (group II antagonist). Thus, for this LTD induction, concurrent synaptic activation of group I and group II mGluRs during tetanus is required. Furthermore, it was shown that 1S,3R-ACPD (groups I and II agonist), but not DHPG (group I agonist), facilitates LTD induction by 50 Hz stimuli. Moreover, coapplication of dopamine and 1S,3R-ACPD induced LTD without any presynaptic electrical stimulation. Coapplication of dopamine and DHPG also induced LTD, but in a somewhat less consistent fashion, and interestingly, the potent group II agonist DCG IV by itself induced LTD in a postsynaptic Ca2+-dependent manner. The important involvement of group II mGluRs in LTD induction in prefrontal cortex might explain why the recovery from the depression after sole application of 1S,3R-ACPD is slower than that after sole application of DHPG (Fig. 6B vs 7A).
Fig. 13.

Schematic drawing of possible mechanisms for LTD induction in prefrontal layer I–II to layer V pyramidal neuron glutamatergic synapses. The coexistence of D1-like and D2-like dopamine receptors and that of D2-like and glutamate in the same synapse are still hypothetical. In classical protocol (Fig.1E), LTD is induced by 50 Hz electrical stimulation to the glutamatergic fibers in the presence of dopamine in the bath (shown in the figure as a dopaminergic synaptic terminal).(a) Dopamine alone acutely and reversibly depresses low-frequency glutamatergic transmission, including the NMDA receptor-mediated component (Law-Tho et al., 1994; Law-Tho, 1995; our unpublished observation), by presynaptic or postsynaptic action, or both, and accordingly, LTD induced by the classical protocol does not require NMDA receptor activation (Otani et al., 1998b).(b) During high-frequency (50 Hz) drive to glutamatergic synapses, dopamine augments the synaptic responses and increases postsynaptic depolarization (Otani et al., 1998). This effect of dopamine serves as a critical factor for the NMDA-independent, postsynaptic Ca2+-dependent induction of LTD (Otani et al., 1998b). (c) In the present study, it was shown that for this LTD induction, synaptic activation of both group I and group II mGluRs is necessary (Figs. 2A, 3A, 4A). Moreover, coapplication of dopamine and 1S,3R-ACPD consistently induced LTD without tetanus or single synaptic stimuli, suggesting that coactivation of dopamine receptors and the mGluRs is sufficient for LTD induction (Fig. 9A,B). We propose that a biochemical mechanism underlying this LTD induction is converging postsynaptic activation of MAP-Ks (ERK1 and ERK2) by groups I and II mGluRs and D1-like and D2-like dopamine receptors. Thus, dopamine D1 and D2 receptors synergistically activate MAP-Ks (Fig. 12), whereas group I mGluRs alone or group II mGluRs alone can activate MAP-Ks (Fig. 11). A combined activation of the dopamine receptors and mGluRs causes synergistic (ERK1) or additive (ERK2) increases of MAP-K activity (Fig. 11). Second messenger pathways involved in this converging MAP-K activation in prefrontal neurons (shown as pathwaysi, ii, iii, andiv) are yet to be demonstrated. However, the following lines of evidence support our hypothesis. (i andii) Activation of MAP-Ks by PKC and PKA has been demonstrated in various cell types including hippocampal cells (Nestler and Greengard 1994; Cobb and Goldsmith, 1995; Robinson and Cobb, 1997;Roberson et al., 1999). (iii) Lines of evidence suggest that D2 receptors are coupled positively to phospholipase A2 and production of arachidonic acid, which activates PKC (Piomelli et al., 1991; Vial and Piomelli, 1995; Nilsson et al., 1998). There may be a synergistic action between D1 and D2 receptors for arachidonic acid production (Piomelli et al., 1991). (iv) It has been reported that group II mGluR activation with DCG IV, as well as group I mGluR activation with DHPG, stimulates the phospholipase D pathway, which also leads to PKC activation (Klein et al., 1997).
Figure 13 also shows that a biochemical consequence of metabotropic receptor cooperation (i.e., D1 and D2 receptors and groups I and II mGluRs) is the postsynaptic converging activation of ERK1 and ERK2. Our biochemical analyses showed that prefrontal MAP-Ks are activated by dopamine, 1S,3R-ACPD, DHPG, and DCG IV. Importantly, when dopamine and one of the mGluR agonists were coapplied, there were synergistic (ERK1) or additive (ERK2) increases of MAP-Ks. Synergistic activation of MAP-Ks was also observed between dopamine D1 and D2 receptors. In this case, blockade of either D1 or D2 receptors was sufficient to block dopamine activation of MAP-Ks. This dopaminergic synergism is reminiscent of the synergistic action of D1 and D2 receptors in the production of arachidonic acid (Piomelli et al., 1991), the potent PKC activator that can in turn activate MAP-Ks (see below and Fig. 13 legend).
Consistent with the above results showing that MAP-Ks can be activated through dopamine receptors and mGluRs, three forms of LTD reported in this paper, i.e., LTD by 50 Hz stimuli in the presence of dopamine, LTD by 50 Hz stimuli in the presence of 1S,3R-ACPD, and LTD by coapplication of dopamine and 1S,3R-ACPD, were all blocked by bath application of the specific ERK1 and ERK2 phosphorylation inhibitor PD98059. Moreover, LTD by dopamine + 1S,3R-ACPD coapplication, whose induction may be purely postsynaptic (Fig.9B), was indeed blocked by a previous postsynaptic inhibition of MAP-K-mediated phosphorylation with the specific synthetic substrate peptide, showing that the critical converging activation of MAP-Ks takes place in postsynaptic sites. The involvement of MAP-Ks in synaptic plasticity is in line with other results obtained in hippocampus (Baron et al., 1996; English and Sweatt, 1996; Coogan et al., 1999).
In this paper, we did not investigate the question regarding which second messengers are involved in LTD and MAP-K activation. However, the following lines of evidence support our hypotheses depicted in Figure 13 (shown as pathways i, ii,iii, and iv). First, it is well known that dopamine D1 receptors are positively coupled to the adenylate cyclase–cAMP–PKA pathway (Jaber et al., 1996) and group I mGluRs are coupled to the phospholipase C–diacylglycerol–PKC pathway (Abe et al., 1992; Joly et al., 1995). These two major protein kinases activate MAP-Ks in various cell types including hippocampal neurons (pathwaysi and ii) (Nestler and Greengard, 1994; Cobb and Goldsmith, 1995; Robinson and Cobb, 1997; Roberson et al., 1999). Dopamine D2 receptors are classically known to downregulate the PKA pathway (Jaber et al., 1996), but it is also known that D2 receptors are positively coupled to phospholipase A2, whose activation produces arachidonic acid, a potent PKC activator (pathway iii) (Piomelli et al., 1991; Vial and Piomelli, 1995; Nilsson et al., 1998). In this respect, D1 receptors might act with D2 receptors and synergistically produce arachidonic acid (Piomelli et al., 1991), to bring about at least partially the synergistic activation of MAP-Ks (Fig. 12). Similar to D2 receptors, group II mGluRs are classically known to be negatively coupled to cAMP (Tanabe et al., 1992, 1993). Although this appears to be the case for presynaptic group II receptors (see Figs. 5 and 8A for acute effects of MSOPPE and DCG IV), postsynaptic group II mGluRs might be also positively coupled to phospholipase D (Klein et al., 1997), whose activation leads to PKC activation (pathway iv) (Tanaka and Nishizuka, 1994). Postsynaptic localization of Ca2+-dependent processes involving group II mGluRs was suggested by our result that DCG IV alone postsynaptically induces LTD (Fig.8A,B).
In some neurons treated with the group II antagonist MSOPPE, 50 Hz tetanic stimuli induced large STP (three of nine) or an STP followed by some degree of lasting potentiation (16%, one of nine). Analyses of synaptic responses in the presence of MSOPPE revealed that the drug enhances postsynaptic activity during tetanus. It is likely that this effect of MSOPPE on synaptic responses, probably attributable to its presynaptic action, underlies the plasticity induction in the MSOPPE condition. This notion further suggests that under normal conditions, presynaptic activation of group II mGluRs by released glutamate and a resulting downregulation of glutamate release inhibit LTP induction (cf. Huang et al., 1997). It is important, however, to note that the abnormal enhancement of synaptic responses during tetanus by MSOPPE is not a mechanism by which MSOPPE inhibited dopaminergic facilitation of LTD induction. This is because dopamine itself enhances synaptic activity during tetanus (Otani et al., 1998b), which is sufficient to occlude the effect of MSOPPE (see Results).
Modification of prefrontal glutamatergic transmission by dopamine appears critical for normal cognitive function. For example, the spatial specificity in working memory-related discharge of layer III neurons in monkey dorsolateral prefrontal cortex depends on a normal functioning of local D1 receptors (Williams and Goldman-Rakic, 1995). In fact, injection of D1 antagonists to dorsolateral prefrontal cortex in monkeys (Sawaguchi and Goldman-Rakic, 1994), or abnormal hyperactivation of D1 receptors in prefrontal area in rats (Zahrt et al., 1997), disrupts working memory performance. Furthermore, the psychotomimetic NMDA antagonists ketamine and phencyclidine (PCP) increase dopamine outflow in rat prefrontal cortex (Nishijima et al., 1994; Adams and Moghaddam, 1998) and impair working memory in rats (Moghaddam et al., 1997; Moghaddam and Adams, 1998). Importantly, PCP was also found to increase glutamate outflow in rat prefrontal cortex (Moghaddam and Adams, 1998), and a pharmacological block of this glutamate increase, even when dopamine level is still high, ameliorates the PCP-induced impairment of working memory (Adams and Moghaddam, 1998; Moghaddam and Adams, 1998), confirming that abnormality in glutamatergic transmission is probably the direct cause of cognitive impairment. It is yet to be demonstrated whether synaptic depression of prefrontal glutamatergic transmission is related to cognitive function. However, mechanistically, LTD in prefrontal cortex is readily induced when glutamatergic synapses are stimulated simultaneously with dopamine receptors. Thus, under physiological or pathological conditions, LTD might result from a coincident transmission at glutamatergic and dopaminergic synapses located on the same neuron. The present study revealed that for this LTD induction, cooperation between groups I and II mGluRs and dopamine receptors serves as a critical mechanism.
Footnotes
This work was financially supported by Biotech (CT96-0049). We thank O. Blond for his contribution to some of the experiments described in this paper.
Correspondence should be addressed to Dr. Satoru Otani, Laboratoire de Neurobiologie et Neuropharmacologie du Développement, Institut des Neurosciences case8, Université de Paris VI, Building B, 6th floor, 7 quai Saint Bernard, 75005 Paris, France. E-mail:satoru.otani@snv.jussieu.fr.
REFERENCES
- 1.Abe T, Sugihara H, Nawa H, Shigemoto R, Mizuno N, Nakanishi S. Molecular characterization of a novel metabotropic glutamate receptor mGluR5 coupled to inositol phophate/Ca2+ signal transduction. J Biol Chem. 1992;267:13361–13368. [PubMed] [Google Scholar]
- 2.Adams B, Moghaddam B. Corticolimbic dopamine neurotransmission is temporally dissociated from the cognitive and locomotor effects of phencyclidine. J Neurosci. 1998;18:5545–5554. doi: 10.1523/JNEUROSCI.18-14-05545.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Alessi DR, Cuenda A, Cohen P, Dudley DT, Saltiel AR. PD98059 is a specific inhibitor of the activation of mitogen-activated protein kinase kinase in vitro and in vivo. J Biol Chem. 1995;270:27489–27494. doi: 10.1074/jbc.270.46.27489. [DOI] [PubMed] [Google Scholar]
- 4.Aniksztejn L, Otani S, Ben-Ari Y. Quisqualate metabotropic receptors modulate NMDA currents and facilitate induction of long-term potentiation through protein kinase C. Eur J Neurosci. 1992;4:500–505. doi: 10.1111/j.1460-9568.1992.tb00900.x. [DOI] [PubMed] [Google Scholar]
- 5.Barchas JD, Faull KF, Quinn B, Elliott GR. Biochemical aspects of the psychotic disorders. In: Siegel GJ, Agranoff BW, Albers RW, Molinoff PB, editors. Basic neurochemistry, Ed 5. Raven; New York: 1994. pp. 959–1001. [Google Scholar]
- 6.Baron C, Benes C, Huynh VT, Fagard R, Roisin MP. Potassium chloride pulse enhances MAP kinase activity in rat hippocampal slices. J Neurochem. 1996;66:1005–1010. doi: 10.1046/j.1471-4159.1996.66031005.x. [DOI] [PubMed] [Google Scholar]
- 7.Bashir ZI, Bortolotto ZA, Davies CH, Berretta N, Irving AJ, Seal AJ, Henley JM, Jane DE, Watkins JC, Collingridge GL. Induction of LTP in the hippocampus needs synaptic activation of glutamate metabotropic receptors. Nature. 1993;363:347–350. doi: 10.1038/363347a0. [DOI] [PubMed] [Google Scholar]
- 8.Bear MF, Abraham WC. Long-term depression in hippocampus. Annu Rev Neurosci. 1996;19:437–462. doi: 10.1146/annurev.ne.19.030196.002253. [DOI] [PubMed] [Google Scholar]
- 9.Bergson C, Mrzljak L, Smiley JF, Pappy M, Levenson R, Goldman-Rakic PS. Regional, cellular, and subcellular variations in the distributions of D1 and D5 dopamine receptors in primate brain. J Neurosci. 1995;15:7821–7836. doi: 10.1523/JNEUROSCI.15-12-07821.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bliss TVP, Collingridge GL. A synaptic model of memory: long-term potentiation in the hippocampus. Nature. 1993;361:31–39. doi: 10.1038/361031a0. [DOI] [PubMed] [Google Scholar]
- 11.Brabet I, Mary S, Bockaert J, Pin J-P. Phenylglycine derivatives discriminate between mGluR1- and mGluR5-mediated responses. Neuropharmacology. 1995;34:895–903. doi: 10.1016/0028-3908(95)00079-l. [DOI] [PubMed] [Google Scholar]
- 12.Bradford M. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem. 1976;72:248–254. doi: 10.1006/abio.1976.9999. [DOI] [PubMed] [Google Scholar]
- 13.Bradley SR, Levey AI, Hersch SM, Conn PJ. Immunocytochemical localization of group III metabotropic glutamate receptors in the hippocampus with subtype-specific antibodies. J Neurosci. 1996;16:2044–2056. doi: 10.1523/JNEUROSCI.16-06-02044.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cobb MH, Goldsmith EJ. How MAP kinases are regulated. J Biol Chem. 1995;270:14843–14846. doi: 10.1074/jbc.270.25.14843. [DOI] [PubMed] [Google Scholar]
- 15.Coogan AN, O'Leary DM, O'Connor JJ. P42/44 MAP kinase inhibitor PD98059 attenuates multiple forms of synaptic plasticity in rat dentate gyrus in vitro. J Neurophysiol. 1999;81:103–110. doi: 10.1152/jn.1999.81.1.103. [DOI] [PubMed] [Google Scholar]
- 16.Cohen P. The search for physiological substrates of MAP and SAP kinases in mammalian cells. Trends Cell Biol. 1997;7:353–361. doi: 10.1016/S0962-8924(97)01105-7. [DOI] [PubMed] [Google Scholar]
- 17.Conn PJ, Pin J-P. Pharmacology and functions of metabotropic glutamate receptors. Annu Rev Pharmacol Toxicol. 1997;37:205–237. doi: 10.1146/annurev.pharmtox.37.1.205. [DOI] [PubMed] [Google Scholar]
- 18.Dudley DT, Pang L, Decker SJ, Bridges AJ, Saltiel AR. A synthetic inhibitor of the mitogen-activated PKC cascade. Proc Natl Acad Sci USA. 1995;92:7686–7689. doi: 10.1073/pnas.92.17.7686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.English JD, Sweatt JD. Activation of p42 mitogen-activated protein kinase in hippocampal long term potentiation. J Biol Chem. 1996;271:24329–24332. doi: 10.1074/jbc.271.40.24329. [DOI] [PubMed] [Google Scholar]
- 20.Fotuhi M, Sharp AH, Glatt CE, Hwang PM, von Krosigk M, Snyder H, Dawson TM. Differential localization of phosphoinositide-linked metabotropic glutamate receptor (mGluR1) and the inositol 1,4,5-trisphosphate receptor in rat brain. J Neurosci. 1993;13:2001–2012. doi: 10.1523/JNEUROSCI.13-05-02001.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gereau RW, IV, Conn PJ. Multiple presynaptic metabotropic glutamate receptors modulate excitatory and inhibitory synaptic transmission in hippocampal area CA1. J Neurosci. 1995;15:6879–6889. doi: 10.1523/JNEUROSCI.15-10-06879.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Glowinski J, Tassin JP, Thierry AM. The mesocortico-prefrontal dopaminergic neurons. Trends Neurosci. 1984;7:415–418. [Google Scholar]
- 23.Goldman-Rakic P. Cellular basis of working memory. Neuron. 1995;14:477–485. doi: 10.1016/0896-6273(95)90304-6. [DOI] [PubMed] [Google Scholar]
- 24.Her JH, Lakhani S, Zu K, Vila J, Dent P, Sturgill TW, Weber MJ. Dual phosphorylation and autophosphorylation in mitogen-activated protein (MAP) kinase activation. Biochem J. 1993;296:25–31. doi: 10.1042/bj2960025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hirsch JC, Crepel F. Use-dependent changes in synaptic efficacy in rat prefrontal neurons in vitro. J Physiol (Lond) 1990;427:31–49. doi: 10.1113/jphysiol.1990.sp018159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Huang L-Q, Rowan MJ, Anwyl R. mGluR II agonist inhibition of LTP induction, and mGluR II antagonist inhibition of LTD induction, in the dentate gyrus in vitro. NeuroReport. 1997;8:687–693. doi: 10.1097/00001756-199702100-00022. [DOI] [PubMed] [Google Scholar]
- 27.Ishida M, Saitoh T, Shimamoto K, Ohfune Y, Shinozaki H. A novel metabotropic glutamate receptor agonist: marked depression of monosynaptic excitation in the newborn rat isolated spinal cord. Br J Pharmacol. 1993;109:1169–1177. doi: 10.1111/j.1476-5381.1993.tb13745.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Iversen SD. Interactions between excitatory amino acids and dopamine systems in the forebrain: implications for schizophrenia and Parkinson's disease. Behav Pharmacol. 1995;6:478–491. [PubMed] [Google Scholar]
- 29.Jaber M, Robinson SW, Missale C, Caron MG. Dopamine receptors and brain function. Neuropharmacology. 1996;35:1503–1519. doi: 10.1016/s0028-3908(96)00100-1. [DOI] [PubMed] [Google Scholar]
- 30.Jin X, Daw NW. The group III metabotropic glutamate receptor agonist, l-AP4, reduces EPSPs in some layers of rat visual cortex. Brain Res. 1998;797:218–224. doi: 10.1016/s0006-8993(98)00378-3. [DOI] [PubMed] [Google Scholar]
- 31.Joly C, Gomeza J, Brabet I, Curry K, Bockaert J, Pin J-P. Molecular, functional, and pharmacological characterization of the metabotropic glutamate receptor type 5 splice variants: comparison with mGluR1. J Neurosci. 1995;15:3970–3981. doi: 10.1523/JNEUROSCI.15-05-03970.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kato N. Dependence of long-term depression on postsynaptic metabotropic glutamate receptors in visual cortex. Proc Natl Acad Sci USA. 1993;90:3650–3654. doi: 10.1073/pnas.90.8.3650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Klein J, Iovino M, Vakil M, Shinozaki H, Löffelholz K. Ontogenetic and pharmacological studies on metabotropic glutamate receptors coupled to phospholipase D activation. Neuropharmacology. 1997;36:305–311. doi: 10.1016/s0028-3908(97)00024-5. [DOI] [PubMed] [Google Scholar]
- 34.Kolb B. Functions of the frontal cortex of the rat: a comparative review. Brain Res Rev. 1984;8:65–98. doi: 10.1016/0165-0173(84)90018-3. [DOI] [PubMed] [Google Scholar]
- 35.Kuroda M, Murakami K, Igarashi H, Okada A. The convergence of axon terminals from the mediodorsal thalamic nucleus and ventral tegmental area on pyramidal cells in layer V of the rat prelimbic cortex. Eur J Neurosci. 1996;8:1340–1349. doi: 10.1111/j.1460-9568.1996.tb01596.x. [DOI] [PubMed] [Google Scholar]
- 36.Law-Tho D. PhD thesis. Université de Paris-Sud; 1995. Etude in vitro des effets des catecholamines (dopamine et noradrenaline) sur la transmission et la plasticite synaptiques dans le cortex prefrontal du rat. [Google Scholar]
- 37.Law-Tho D, Hirsch JC, Crepel F. Dopamine modulation of synaptic transmission in rat prefrontal cortex: an in vitro electrophysiological study. Neurosci Res. 1994;21:151–160. doi: 10.1016/0168-0102(94)90157-0. [DOI] [PubMed] [Google Scholar]
- 38.Law-Tho D, Desce JM, Crepel F. Dopamine favours the emergence of long-term depression versus long-term potentiation in slices of rat prefrontal cortex. Neurosci Lett. 1995;188:125–128. doi: 10.1016/0304-3940(95)11414-r. [DOI] [PubMed] [Google Scholar]
- 39.Manahan-Vaughan D, Braunewell K-H, Reymann KG. Subtype-specific involvement of metabotropic glutamate receptors in two forms of long-term potentiation in the dentate gyrus of freely moving rats. Neuroscience. 1998;86:709–721. doi: 10.1016/s0306-4522(98)00111-0. [DOI] [PubMed] [Google Scholar]
- 40.Moghaddam B, Adams BW. Reversal of phencyclidine effects by a group II metabotropic glutamate receptor agonist in rats. Science. 1998;281:1349–1352. doi: 10.1126/science.281.5381.1349. [DOI] [PubMed] [Google Scholar]
- 41.Moghaddam B, Adams BW, Verma A, Daly D. Activation of glutamatergic neurotransmission by ketamine: a novel step in the pathway from NMDA receptor blockade to dopaminergic and cognitive disruptions associated with the prefrontal cortex. J Neurosci. 1997;17:2921–2927. doi: 10.1523/JNEUROSCI.17-08-02921.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Moroni F, Lombardi G, Thomsen C, Leonardi P, Attucci S, Peruginelli F, Albani-Torregrossa S, Pellegrini-Giampietro DE, Luneia R, Pellicciari R. Pharmacological characterization of 1-aminoindan-1,5-ducarboxylic acid, AIDA, a potent mGluR1 antagonist. J Pharmacol Exp Ther. 1997;281:721–729. [PubMed] [Google Scholar]
- 43.Morris SH, Keelan J, Patterson L, Duchen MR, Bindman LJ. Group I metabotropic glutamate receptor (mGluR) agonists raise intracellular [Ca++] in medial frontal cortical neurons of the rat. Soc Neurosci Abstr. 1998;24:2024. [Google Scholar]
- 44.Nestler EJ, Greengard P. Protein phosphorylation and the regulation of neuronal function. In: Siegel GJ, Agranoff BW, Albers RW, Molinoff PB, editors. Basic neurochemistry. Raven; New York: 1994. pp. 449–474. [Google Scholar]
- 45.Nicoletti F, Bruno V, Copani A, Casabona G, Knöpfel T. Metabotropic glutamate receptors: a new target for the therapy of neurodegenerative disorders? Trends Neurosci. 1996;19:267–271. doi: 10.1016/S0166-2236(96)20019-0. [DOI] [PubMed] [Google Scholar]
- 46.Nilsson CL, Hellstrand M, Ekman A, Eriksson E. Direct dopamine D2-receptor-mediated modulation of arachidonic acid release in transfected CHO cells without the concomitant administration of a Ca2+-mobilizing agent. Br J Pharmacol. 1998;124:1651–1658. doi: 10.1038/sj.bjp.0702025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Nishijima K, Kashiwa A, Nishikawa T. Preferential stimulation of extracellular release of dopamine in rat frontal cortex to striatum following competitive inhibition of the N-methyl-d-aspartate receptor. J Neurochem. 1994;63:375–378. doi: 10.1046/j.1471-4159.1994.63010375.x. [DOI] [PubMed] [Google Scholar]
- 48.Okubo Y, Suhara T, Suzuki K, Kobayashi K, Inoue O, Terasaki O, Someya Y, Sassa T, Sudo Y, Matsushima E, Iyo M, Tateno Y, Toru M. Decreased prefrontal dopamine D1 receptors in schizophrenia revealed by PET. Nature. 1997;385:634–636. doi: 10.1038/385634a0. [DOI] [PubMed] [Google Scholar]
- 49.Oliet S, Malenka RC, Nicoll RA. Two distinct forms of long-term depression coexist in CA1 hippocampal pyramidal cells. Neuron. 1997;18:969–982. doi: 10.1016/s0896-6273(00)80336-0. [DOI] [PubMed] [Google Scholar]
- 50.Otani S, Ben-Ari Y. Biochemical correlates of long-term potentiation in hippocampal synapses. Int Rev Neurobiol. 1993;35:1–41. doi: 10.1016/s0074-7742(08)60567-x. [DOI] [PubMed] [Google Scholar]
- 51.Otani S, Connor JA. Requirement of rapid Ca2+ entry and synaptic activation of metabotropic glutamate receptors for the induction of long-term depression in adult rat hippocampus. J Physiol (Lond) 1998;511:761–770. doi: 10.1111/j.1469-7793.1998.761bg.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Otani S, Auclair N, Desce J-M, Crépel F. Dopaminergic receptors and metabotropic glutamate receptors cooperate to induce LTD in rat prefrontal glutamatergic synapses. Soc Neurosci Abstr. 1998a;24:2024. [Google Scholar]
- 53.Otani S, Blond O, Desce J-M, Crépel F. Dopamine facilitates long-term depression of glutamatergic transmission in rat prefrontal cortex. Neuroscience. 1998b;85:669–676. doi: 10.1016/s0306-4522(97)00677-5. [DOI] [PubMed] [Google Scholar]
- 54.Otani S, Desce J-M, Blond O, Crépel F. Interaction between dopamine receptors and group II mGluRs for LTD induction in rat prefrontal cortex. J Physiol (Lond) 1998c;507:26P. [Google Scholar]
- 55.Petralia RS, Wang Y-X, Niedzielski AS, Wenthold RJ. The metabotropic glutamate receptors, mGluR2 and mGluR3, show unique postsynaptic, presynaptic and glial localizations. Neuroscience. 1996;71:949–976. doi: 10.1016/0306-4522(95)00533-1. [DOI] [PubMed] [Google Scholar]
- 56.Piomelli D, Pilon C, Giros B, Sokoloff P, Martres MP, Schwartz JC. Dopamine activation of the arachidonic acid cascade as a basis for D1/D2 receptor synergism. Nature. 1991;353:164–167. doi: 10.1038/353164a0. [DOI] [PubMed] [Google Scholar]
- 57.Roberson ED, English JD, Adams JP, Selcher JC, Kondratick C, Sweatt JD. The mitogen-activated protein kinase cascade couples PKA and PKC to cAMP response element binding protein phosphorylation in area CA1 of hippocampus. J Neurosci. 1999;19:4337–4348. doi: 10.1523/JNEUROSCI.19-11-04337.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Robinson MJ, Cobb MH. Mitogen-activated protein kinase pathways. Curr Opin Cell Biol. 1997;9:180–186. doi: 10.1016/s0955-0674(97)80061-0. [DOI] [PubMed] [Google Scholar]
- 59.Romano C, Sesma MA, McDonald CT, O'Malley K, van del Pol AN, Olney JW. Distribution of metabotropic glutamate receptor mGluR5 immunoreactivity in rat brain. J Comp Neurol. 1995;355:455–469. doi: 10.1002/cne.903550310. [DOI] [PubMed] [Google Scholar]
- 60.Sawaguchi T, Goldman-Rakic PS. The role of D1-dopamine receptor in working memory: local injections of dopamine antagonists into the prefrontal cortex of Rhesus monkeys performing an oculomotor delayed-response task. J Neurophysiol. 1994;71:515–528. doi: 10.1152/jn.1994.71.2.515. [DOI] [PubMed] [Google Scholar]
- 61.Sekiyama N, Hayashi Y, Nakanishi S, Jane DE, Tse H-W, Birse EF, Watkins JC. Structure-acticity relationships of new agonists and antagonists of different metabotropic glutamate receptor subtypes. Br J Pharmacol. 1996;117:1493–1503. doi: 10.1111/j.1476-5381.1996.tb15312.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Simon H, Scatton B, Le Moal M. Dopaminergic A10 neurons are involved in cognitive functions. Nature. 1980;286:150–151. doi: 10.1038/286150a0. [DOI] [PubMed] [Google Scholar]
- 63.Smiley JF, Levey AL, Ciliax BJ, Goldman-Rakic PS. D1 dopamine receptor immunoreactivity in human and monkey cerebral cortex: predominant and extrasynaptic localization in dendritic spines. Proc Natl Acad Sci USA. 1994;91:5720–5724. doi: 10.1073/pnas.91.12.5720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Tanabe Y, Masu M, Ishii T, Shigemoto R, Nakanishi S. A family of metabotropic glutamate receptors. Neuron. 1992;8:169–179. doi: 10.1016/0896-6273(92)90118-w. [DOI] [PubMed] [Google Scholar]
- 65.Tanabe Y, Nomura A, Masu M, Shigemoto R, Mizuno N, Nakanishi S. Signal transduction, pharmacological properties, and expression patterns of two rat metabotropic glutamate receptors, mGluR3 and mGluR4. J Neurosci. 1993;13:1372–1378. doi: 10.1523/JNEUROSCI.13-04-01372.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Tanaka C, Nishizuka Y. The protein kinase C family for neuronal signaling. Annu Rev Neurosci. 1994;17:551–567. doi: 10.1146/annurev.ne.17.030194.003003. [DOI] [PubMed] [Google Scholar]
- 67.Thomas NK, Jane DE, Tse H-W, Watkins JC. α-Methyl derivatives of serine-O-phosphate as novel, selective competitive metabotropic glutamate receptor antagonists. Neuropharmacology. 1996;35:637–642. doi: 10.1016/0028-3908(96)84635-1. [DOI] [PubMed] [Google Scholar]
- 68.Towbin H, Staehelin T, Gordon J. Electrophoretic transfer of proteins from polyacrylamide gels to nitrocellulose sheets: procedure and some applications. Proc Natl Acad Sci USA. 1979;76:4350–4354. doi: 10.1073/pnas.76.9.4350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Vial D, Piomelli D. Dopamine D2 receptors potentiate arachidonate release via activation of cytosolic, arachidonate-specific phospholipase A2. J Neurochem. 1995;64:2765–2772. doi: 10.1046/j.1471-4159.1995.64062765.x. [DOI] [PubMed] [Google Scholar]
- 70.Vickery RM, Morris SH, Bindman LJ. Metabotropic glutamate receptors are involved in long-term potentiation in isolated slices of rat medial frontal cortex. J Neurophysiol. 1997;78:3039–3046. doi: 10.1152/jn.1997.78.6.3039. [DOI] [PubMed] [Google Scholar]
- 71.Williams GV, Goldman-Rakic PS. Modulation of memory fields by dopamine D1 receptors in prefrontal cortex. Nature. 1995;376:572–575. doi: 10.1038/376572a0. [DOI] [PubMed] [Google Scholar]
- 72.Wilsch VW, Pidoplichko VI, Opitz T, Shinozaki H, Reymann KG. Metabotropic glutamate receptor agonist DCG-IV as NMDA receptor agonist in immature rat hippocampal neurons. Eur J Pharmacol. 1994;262:287–291. doi: 10.1016/0014-2999(94)90743-9. [DOI] [PubMed] [Google Scholar]
- 73.Zahrt J, Taylor JR, Mathew RG, Arnsten AFT. Supranormal stimulation of D1 dopamine receptors in the rodent prefrontal cortex impairs spatial working memory performance. J Neurosci. 1997;17:8528–8535. doi: 10.1523/JNEUROSCI.17-21-08528.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]