

Abstract
The involvement of adenosine on the development of time-dependent reversal of long-term potentiation (LTP) by low-frequency stimulation (LFS) was investigated at Schaffer collateral–CA1 synapses of rat hippocampal slices. A train of LFS (2 Hz, 10 min, 1200 pulses) had no long-term effects on synaptic transmission but produced lasting depression of previously potentiated responses. This reversal of LTP (depotentiation) was observed when the stimulus was delivered ≤3 min after induction of LTP. However, application at 10 min after induction had no detectable effect on potentiation. This time-dependent reversal of LTP by LFS appeared to be mediated by extracellular adenosine, because it was mimicked by bath-applied adenosine and was specifically inhibited by the selective A1 adenosine receptor antagonist 8-cyclopentyl-1,3-dipropylxanthine (100 nm). The effect of adenosine could be mimicked by 5-HT1A receptor agonist buspirone, but the LFS-induced depotentiation could not be antagonized by 5-HT1A receptor antagonist NAN-190. The source of extracellular adenosine in response to LFS appeared to be attributable to the efflux of cAMP. In addition, this LFS-induced depotentiation was blocked by bath application of adenylyl cyclase activator forskolin or injection of a cAMP analog Sp-adenosine cAMP (10 mm) into postsynaptic neurons. Moreover, the selective protein phosphatase 1 and 2A inhibitors okadaic acid and calyculin A prevented the LFS-induced depotentiation. These results thus suggest that increasing extracellular adenosine appears to underlie the LFS-induced depotentiation via acting on the A1 receptor subtype to interrupt the cAMP-dependent biochemical processes leading to the LTP expression.
Keywords: adenosine, long-term potentiation (LTP), depotentiation, protein phosphatase, adenylyl cyclase, hippocampus
Long-term potentiation (LTP) is a form of activity-dependent increase in synaptic efficacy that has been considered as a possible elementary basis for learning and memory in the brain (Bliss and Collingridge, 1993). Although LTP is remarkable for its stability, recent work has provided evidence that it is vulnerable to disruption for several minutes after its induction. For example, it has been shown that a brief period of hypoxia reversed LTP in the CA1 region of hippocampal slices if applied within 1–2 min of induction but not at time thereafter (Arai et al., 1990). The time-dependent reversal of LTP was also effectively induced by low-frequency afferent stimulation (1–5 Hz) delivered within 10 min of LTP induction, both in vivo (Barrionuevo et al., 1980;Stäubli and Lynch, 1990) and in vitro (Fujii et al., 1991; Bashir and Collingridge, 1992). The phenomenon is termed “depotentiation.” In addition, antagonists that block cell–cell and cell–matrix interactions have also been demonstrated to reverse the LTP in a time-dependent manner (Bahr et al., 1997;Stäubli et al., 1998). These observations have led to a general belief that the biochemical processes that contribute to convert the initial potentiation into a persistent and not readily disrupted state require many minutes to reach completion (Bahr et al., 1997;Stäubli et al., 1998).
Adenosine, an endogenous purine, is well known to play an important role in the modulation of central synaptic transmission and neuronal excitability (Ribeiro, 1995; De Mendonca and Ribeiro, 1997). With respect to electrophysiological actions, adenosine can act presynaptically to decrease neurotransmitter release by inhibiting calcium influx into the presynaptic terminal (Gerber et al., 1989) or act postsynaptically to decrease neuronal excitability by activating potassium conductance and thus hyperpolarizing neurons (Gerber et al., 1989). In addition, there has been increasing evidence showing that adenosine and its derivatives modulated several forms of short-term and long-term activity-dependent synaptic plasticity (De Mendonca and Ribeiro, 1997). For example, both adenosine analog 2-chloroadenosine and adenosine were seen to decrease LTP at Schaffer collateral–CA1 synapses (Arai et al., 1990). However, the role of adenosine in the depotentiation of LTP is controversial at present time. De Mendonca et al. (1997) have reported that endogenous adenosine, acting through A1 adenosine receptors, is able to limit depotentiation in the hippocampus, because the A1adenosine receptor antagonist 8-cyclopentyl-1,3-dipropylxanthine (DPCPX) applied during low-frequency stimulation (LFS) application resulted in a facilitation of depotentiation of LTP. In contrast, recently, another A1 adenosine receptor antagonist, 8-cyclopentyltheophylline (8-CPT), has been shown to inhibit the depotentiation in hippocampal CA1 neurons, indicating that activation of A1 adenosine receptor by endogenous adenosine enhances the depotentiation of LTP (Fujii et al., 1997). The reason for this discrepancy in adenosine contribution to induction of depotentiation remains unclear. In an attempt to make some sense out of this confusing literature, we have therefore reinvestigated the role of adenosine in the development of LFS-induced depotentiation of LTP at Schaffer collateral–CA1 synapses in rat hippocampal slices using extracellular and intracellular recording methods. We have also investigated the cellular and molecular basis by which adenosine contributes to depotentiation.
MATERIALS AND METHODS
Slice preparation. Hippocampal slices (400-μm-thick) were obtained from 4- to 5-week old male Sprague Dawley rats for extracellular and intracellular synaptic recordings by the procedures described previously (Huang et al., 1996; Hsu and Huang, 1997). In brief, the rats were killed by stunning by cervical dislocation and decapitation, and transverse slices were cut from a tissue block of the brain using Vibroslice (Campden Instruments, Silbey, UK). The slices were placed in a storage chamber of artificial CSF (ACSF) oxygenated with 95% O2–5% CO2 and kept at room temperature for at least 1 hr before recording. The composition of the ACSF solution was (in mm): NaCl 117, KCl 4.7, CaCl2 2.5, MgCl2 1.2, NaHCO3 25, NaH2PO4 1.2, and glucose 11 at pH 7.3–7.4 and equilibrated with 95% O2–5% CO2.
Electrophysiological recordings. A single slice was then transferred to the recording chamber in which it was held submerged between two nylon nets and maintained at 32.0 ± 0.5°C. The chamber consisted of a circular well of a low volume (1–2 ml) and was perfused constantly at a rate of 2–3 ml/min. Standard extracellular field recording techniques were used. Extracellular recordings of field EPSPs (fEPSPs) were obtained from the stratum radiatum using microelectrodes filled with 1 m NaCl (resistance of 2–3 MΩ). A bipolar stainless steel stimulating electrode was placed in stratum radiatum to activate Schaffer collateral/commissural afferents at 0.033 Hz. The stimulation strength was set to elicit responses equivalent to 30–40% of the maximal fEPSP. In all experiments, baseline synaptic transmission was monitored for 30 min before drug administration or delivering either high- or low-frequency stimulation. The strength of synaptic transmission was quantified by measuring the slope of fEPSP. The fEPSP slopes were measured from ∼20–70% of the rising phase using a least-squares regression. LTP was induced by high-frequency stimulation, at the test pulse intensity, consisting of two 1 sec trains of stimuli at 100 Hz, delivered with an interval of 20 sec. Depotentiation was induced by application of 10 min low-frequency trains of stimuli at 2 Hz, and the stimulation intensity was the same as the test pulse intensity. The responses during the trains were not recorded, and for convenience, these periods are not shown on the graph. All values of residual potentiation reported here were calculated as the changes in fEPSP slope measured 40 min after the end of LFS. Intracellular recordings were made from CA1 pyramidal neurons using glass microelectrodes filled with 4m potassium acetate (80–100 MΩ). Microelectrodes were pulled from microfiber 1.0 mm capillary tubing on a Brown-Flaming electrode puller (Sutter Instruments, San Rafael, CA). Electrical signals were collected with an Axoclamp-2B (Axon Instruments, Foster City, CA) filtered at 1 kHz, sampled at 10 kHz, and an IBM 586-based computer with pCLAMP software (version 7.0; Axon Instruments) was used to on-line acquire and analyze the data. Sp-adenosine cAMP (Sp-cAMPS) dissolved in 4 mpotassium acetate solution was administered intracellularly by hyperpolarization current injection (0.1–0.3 nA) applied through the recording microelectrode for 30–40 min before application of tetanic stimulation (TS).
Drug application. All drugs were applied by dissolving them to the desired final concentrations in the ACSF and by switching the perfusion from control ACSF to drug-containing ACSF. Appropriate stock solutions of drugs were made and diluted with ACSF just before application. DPCPX, forskolin, 1,9-dideoxy-forskolin, probenecid, 4-[(3-butoxy-4-methoxyphenyl)methyl]-2-imidazolidinone (Ro 20–1724), nimodipine, and NAN-190 were dissolved in dimethylsulfoxide (DMSO) stock solutions and stored at −20°C until the day of experiment. The concentration of DMSO in the perfusion medium was 0.05%, which alone had no effect on the induction of either LTP or depotentiation of LTP in the CA1 region of rat hippocampus. Adenosine, DPCPX, 3,7-dimethyl-1-propargylxanthine (DMPX), Ro 20–1724, FPL-67156, Sp-cAMPS, okadaic acid, calyculin A, buspirone, and NAN-190 were purchased from Research Biochemicals (Natick, MA); forskolin, 1,9-dideoxy-forskolin, α,β-methyleneadenosine 5′-diphosphate (AOPCP), GMP, and probenecid were obtained from Sigma (St. Louis, MO).
Statistical analysis. The data for each experiment were normalized relative to baseline. All figures show mean ± SEM. The significance of the differences between the means was calculated by a paired Student's t test. Numbers of experiments are indicated by n. Probability values of p < 0.05 were considered to represent significant differences.
RESULTS
Induction of LTP and depotentiation
In hippocampal slices from 4- to 5-week old rats, application of a brief tetanic stimulation delivered to the Schaffer collateral/commissural afferents to CA1 neurons produced an immediate potentiation of fEPSP that persisted for as long as recording was continued; a typical example is shown in Figure1A. The slope of fEPSP measured 40 min after high-frequency TS was 136.6 ± 6.8% of baseline (n = 12) (Fig. 1B). These experiments showed that our 100 Hz tetanization protocol could effectively induce LTP at Schaffer collateral–CA1 synapses. Accumulative studies have shown that several forms of LFS protocols can be used to induce depotentiation of LTP (Fujii et al., 1991; De Mendonca et al., 1997; Otmakhova and Lisman, 1998). To establish a reliable depotentiation, a stronger LFS protocol, 2 Hz/10 min stimulation, was used (Otmakhova and Lisman, 1998). In control slices, 10 min of 2 Hz stimulation delivered to Schaffer collateral–CA1 synapses had no long-term effect on synaptic transmission (Fig.1C). On average, the slope of fEPSP measured 40 min after the end of 2 Hz stimulation was 102.2 ± 7.8% (n= 4) of baseline (Fig. 1D). However, LFS applied 1–3 min after LTP induction caused an immediate depression of the potentiated synaptic responses; this was followed by recovery toward the control level within a few minutes with no further changes thereafter. To examine the time dependence of the LTP reversal effect by LFS, we varied the time interval between the induction of LTP and the delivery of LFS. Figure 2 summarizes experiments in which LFS was applied 1 (A, B), 3 (C, D) or 10 (E, F) min after the induction of LTP . As shown, when LFS was applied 1 or 3 min after LTP induction, LTP was reversed almost completely. The residual potentiation measured 40 min after the end of LFS was 101.2 ± 7.2 (n = 8) and 111.3 ± 6.7% (n = 8 of 10) of baseline, respectively. In contrast, when LFS was delivered 10 min after LTP induction, it caused no reliable change in potentiated synaptic responses (Fig.2E,F). The mean residual potentiation measured 40 min after the end of LFS was 150.4 ± 7.6% (n = 8) of baseline, which was not significantly different from the LTP measured in control slices without LFS administration (136.3 ± 7.1% of baseline; n = 12) (Fig. 1). Thus, these experiments generally confirmed previous studies showing that LTP is vulnerable to disruption by depotentiating stimuli within a brief period after its induction (Arai et al., 1990;Larson et al., 1993; Stäubli and Chun, 1996). Because the 2 Hz/10 min LFS starting 3 min after LTP induction could strongly reverse LTP, we chose this protocol to examine the mechanisms underlying the LFS-induced depotentiation of LTP at Schaffer collateral–CA1 synapses.
Fig. 1.

High-frequency TS but not LFS induces long-lasting changes in synaptic transmission. A, An example of the time course of homosynaptic long-term potentiation at Schaffer collateral–CA1 synapses. The slope of fEPSP exhibited ∼95% increase after TS that slowly decayed during the first 10 min and remained stable at 45% increase afterwards. B, Summary of data from 12 experiments performed as in A.C, Example of an experiment showing that the protocol of low-frequency stimulation (2 Hz, 10 min) had no lasting effect on synaptic transmission. D, Plots the pooled data from four experiments performed as in C. The superimposed fEPSP in the inset of each graph illustrates respective recordings from example experiments taken at the time indicated bynumber. Bar denotes the period of the delivery of LFS. Calibration: 0.5 mV, 10 msec.
Fig. 2.

Time-dependent reversal of LTP by LFS.A–F, Examples in which LTP was induced at Schaffer collateral–CA1 synapses. LFS (2 Hz, 10 min) was applied at various times after LTP induction: A, 1 min after;C, 3 min after; or E, 10 min after.B, D, F, Summary of experiments similar to that shown in A,C, and E. Note that LFS erased potentiation when delivered 1 or 3 min after TS but was without effect when applied 10 min after. Calibration: 0.5 mV, 10 msec.
The extracellular adenosine contributes to LFS-induced depotentiation
To assess the role of extracellular adenosine in the LFS-induced time-dependent reversal of LTP, the adenosine receptor antagonists were applied during the delivery of LFS. First, we tried to examine the effect of selective adenosine A1 receptor antagonist DPCPX (0.1 μm) on LFS-induced depotentiation. A typical example is shown in Figure3A. When DPCPX (0.1 μm) was applied during LFS, the depotentiation of LTP was significantly inhibited. The slope of fEPSP after LFS recovered close to the initial LTP level. The residual potentiation measured 40 min after the end of LFS was 155.5 ± 12.9% (n = 7) of baseline (Fig. 3B). We next asked whether adenosine A2 receptor contributes to the induction of depotentiation. As a typical example shown in Figure3C, the selective adenosine A2receptor antagonist DMPX (5 μm) possessed no significant inhibitory effect on the LFS-induced depotentiation; the level of depotentiation did not differ from the control values. The residual potentiation measured 40 min after the end of LFS was 96.8 ± 6.7% (n = 6) of baseline (Fig.3D). Only DPCPX could effectively block the LFS-induced depotentiation of LTP. These results suggest that the activation of adenosine A1 but not A2receptor subtype is involved in the induction of depotentiation by LFS at Schaffer collateral–CA1 synapses.
Fig. 3.

A1 adenosine receptor antagonist DPCPX prevents the LFS-induced depotentiation. A, Example of an experiment showing that LFS-induced depotentiation was inhibited when A1 adenosine receptor antagonist DPCPX (0.1 μm) was applied during TS and left until the end of LFS.B, Summary of data from seven experiments performed as in A. C, A2 adenosine receptor antagonist DMPX (5 μm) does not affect the LFS-induced depotentiation. D, Pooled data from six experiments performed as in C. Note that only DPCPX could effectively block the LFS-induced depotentiation. Calibration: 0.5 mV, 10 msec.
To further establish that the LFS-induced depotentiation is mediated through the extracellular adenosine, it is essential to demonstrate that LFS-induced depotentiation should be mimicked by the direct application of adenosine. For this purpose, the effect of adenosine on the development of LTP was investigated. In the first set of experiments, adenosine (0.2 mm) was applied immediately after the induction of LTP. As a typical example shown in Figure4A, application of adenosine for 3 min caused a very rapid suppression of evoked synaptic response. The slope of fEPSP was reduced to 8.7 ± 2.4% (n = 5) after 3 min. After washout of adenosine, the fEPSP recovered to near the pretetanus baseline. On average, the fEPSP slope measured 40 min after washout of adenosine was 104.3 ± 5.3% (n = 5) of baseline (Fig. 4B). As shown in Figure 4, C and D, the reversal of LTP similar to the above results was also obtained with administration of adenosine beginning 3 min after the LTP induction. In contrast, when adenosine was applied 10 min after LTP induction, the synaptic responses consistently recovered to the potentiated level (Fig.4E), i.e., the slope of fEPSP measured 40 min after adenosine washout was 145.3 ± 4.3% (n = 6) of baseline (Fig. 4F). These results confirmed the previous finding of Arai et al. (1990) showing that adenosine reveals a time-dependent reversal of LTP in the CA1 region of rat hippocampal slices.
Fig. 4.

Time-dependent reversal of LTP by adenosine application. A–F, Examples in which LTP was induced at Schaffer collateral–CA1 synapses. Adenosine (0.2 mm) was applied at various times after LTP induction: A, immediately; C, 3 min after; or E, 10 min after. B, D, F, Summary of experiments similar to that shown in A,C, and E. Note that adenosine erased potentiation when delivered immediately or 3 min after the TS but was without effect when applied 10 min after. Calibration: 0.5 mV, 10 msec.
Mechanisms underlying the increase of extracellular adenosine by LFS
The preceding results point to the involvement of extracellular adenosine in the LFS-induced depotentiation. The next question we want to ask is what is the mechanism underlying the increase in extracellular adenosine. Previous studies have reported that multiple mechanisms may lead to increase extracellular adenosine, including the efflux of cAMP from cells followed by extracellular conversion to adenosine (Rosenberg et al., 1994), the release of ATP followed by rapid extracellular conversion to adenosine (Craig and White, 1993), and direct efflux of adenosine itself (Lloyd et al., 1993). If the LFS-induced depotentiation was mediated by adenosine formed from cAMP efflux, then blocking cAMP efflux or inhibiting the conversion of cAMP to adenosine might be expected to eliminate the LFS-induced depotentiation. To test this possibility, three pharmacological tools were used. The first tool was probenecid (200 μm), a specific inhibitor of cAMP transporter, which was used to inhibit the release of cAMP into the extracellular space (Henderson and Strauss, 1991). The second tool was Ro 20–1724 (200 μm), a specific type IV phosphodiesterase inhibitor, to prevent the extracellular conversion of cAMP into 5′-AMP (Gereau and Conn, 1994). The third tool was a combination of AOPCP (200 μm) and GMP (2 mm) to inhibit the ecto-5′-nucleotidase that converts AMP into adenosine (MacDonald and White, 1985). As shown in Table 1, all treatment alone failed to affect the LTP induction but exerted a significant inhibition on the LFS-induced depotentiation. The fEPSP slope measured 40 min after LTP induction was 134.8 ± 5.6 (n = 6), 157.6 ± 7.4 (n = 7), and 135.8 ± 4.7% (n= 6) of baseline, respectively. The residual potentiation measured 40 min after the end of LFS was 132.0 ± 4.8 (n = 9), 169.4 ± 5.3 (n = 6), and 127.1 ± 6.8% (n = 6) of baseline, respectively. These results suggest that extracellular conversion of cAMP is a potential source of extracellular adenosine underlying the LFS-induced depotentiation.
Table 1.
Effects of drugs treatments on LTP and LFS-induced depotentiation at hippocampal CA1 synapses
| Drug treatment | LTP 40 min after TS | The residual LTP 40 min after LFS following TS |
|---|---|---|
| Control | 136.6 ± 6.8% (n = 12)* | 111.3 ± 6.7% (n = 10) |
| Probenecid (200 μm) | 134.8 ± 5.6% (n = 6)* | 132.0 ± 4.8% (n = 10)* |
| Ro 20-1724 (200 μm) | 157.6 ± 7.4% (n = 7)* | 169.4 ± 5.3% (n = 6)* |
| AOPCP (200 μm) | 135.8 ± 4.7% (n = 6)* | 127.1 ± 6.8% (n = 6)* |
| +GMP (2 mm) | ||
| FPL 67156 (100 μm) | 130.7 ± 4.9% (n = 6)* | 103.4 ± 6.8% (n = 6) |
The data represent the percentage of baseline for fEPSP slope. Number of experiments is in parentheses.
*Significantly different from baseline (Student t test;p < 0.05).
Because the source of extracellular adenosine can be the extracellular catabolism of released ATP (White and MacDonald, 1990), we next evaluated the role of extracellular ATP as a source of extracellular adenosine. To test this idea, hippocampal slices were superfused with the ecto-ATPase inhibitor FPL 67156 (100 μm) to prevent the extracellular conversion of ATP to ADP, which could undergo subsequent dephosphorylation into adenosine (Crack et al., 1995). Application of FPL 67156 alone in the bath caused a significant reduction (34.6 ± 6.8% of baseline; n = 12) of the fEPSP slope but did not affect the degree of subsequent reversal of LTP induced by LFS. The residual potentiation measured 40 min after the end of LFS was 103.4 ± 6.8% (n = 6) of baseline (Table 1). Because the hippocampal CA1 LTP induction requires the depolarization of the postsynaptic membrane to relieve magnesium blockade of NMDA receptors and allows the entry of calcium (Nicoll and Malenka, 1995), a dramatic depression of glutamate release could impair LTP by failing to depolarize the postsynaptic neuron to a level that relieves the magnesium blockade. It is possible that FPL 67156 cannot block the LFS-induced depotentiation simply because TS fails to elicit LTP in the presence of this agent. To test this possibility, we directly examined the effect of FPL 67156 on the LTP induction. As shown in Table 1, the induction of LTP was not significantly affected by FPL 67156. In slices with FPL 67156 (100 μm) application for 10 min before and the TS, the slope of fEPSP measured 40 min after TS was 130.7 ± 4.9% (n = 6) of baseline. These results indicate that the machinery involved in the LTP induction is not inhibited by FPL 67156 and the extracellular conversion of ATP is not the major source of adenosine underlying the LFS-induced depotentiation.
Activation of adenylyl cyclase blocks LFS-induced depotentiation
The above results point to an increased activation of adenosine A1 receptors by the extracellular adenosine as mediating the LFS-induced depotentiation of LTP. Because it is well established that activation of A1 adenosine receptors couples to Gi-protein, which inhibits adenylyl cyclase and thereby reduces cAMP formation (Dunwiddie and Fredholm, 1989), experiments were designed to determine whether the LFS has led to inhibit the activity of adenylyl cyclase to reverse LTP. If the LFS-induced depotentiation is mediated via a decrease in cAMP-dependent process, the depotentiation should be blocked by the adenylyl cyclase activator. In agreement with this prediction, direct activation of adenylyl cyclase with forskolin (10 μm) completely blocked depotentiation, as seen in Figure5, A and B. Forskolin was applied starting 5 min before LTP induction and was washed out just after the end of LFS. The residual potentiation measured 40 min after the end of LFS was 192.8 ± 13.8% (n = 6) of baseline (Fig. 5B). Because forskolin has been reported to possess many cAMP-independent actions, including the blockade of several types of K+ currents, it is possible that the effect of forskolin on depotentiation is caused by its nonspecificity (Laurenza et al., 1989). To exclude this possibility, an analog of forskolin, 1,9-dideoxy-forskolin, which has no effect on adenylyl cyclase but does mimic many of the cAMP-independent actions of forskolin, was used. As shown in Figure 5, C andD, 1,9-dideoxy-forskolin did not affect the LFS-induced depotentiation in all seven slices tested. The residual potentiation measured 40 min after the end of LFS was 107.6 ± 9.6% (n = 7) of baseline (Fig. 5D). These results indicate that a reduction of cAMP-dependent process is involved in the mechanism underlying the LFS-induced depotentiation.
Fig. 5.

Selective activation of adenylyl cyclase prevents the LFS-induced depotentiation. A, Previous activation of adenylyl cyclase by forskolin (10 μm) produced a minor increase in the basal synaptic transmission and exerted a significant inhibition on the subsequent LFS-induced depotentiation. Forskolin was applied starting 5 min before the TS and left until the end of LFS.B, Summary of seven experiments performed as inA. C, Previous application of an inactive isomer of forskolin, 1,9-dideoxy-forskolin (10 μm), did not affect the LFS-induced depotentiation. D, Summary of five experiments performed as in C. Calibration: 0.5 mV, 10 msec.
Activation of 5-HT1A receptors mimics LFS-induced depotentiation
If the inhibition of adenylyl cyclase activity is a common route underlying the LFS-induced depotentiation, the activation of receptors coupled to inhibit adenylyl cyclase activity should reverse LTP in the same way as LFS. The 5-HT1A receptor is also coupled via a Gi-protein to inhibit adenylyl cyclase and has well documented physiological effect on the hippocampal CA1 neurons (Andrade and Nicoll, 1987). Attempts were made to see whether the activation of 5-HT1A receptors by direct application of the selective 5-HT1Areceptor agonist buspirone (0.1 mm) could effectively reverse LTP as LFS application. As shown in Figure6, A and B, the reversal of LTP similar to LFS or adenosine application was observed with administration of buspirone beginning 3 min after LTP induction. Application of buspirone for 10 min exerted a significant suppression of the slope of fEPSP. The fEPSP was reduced to 39.7 ± 4.7% (n = 5) of baseline after 10 min of buspirone. After washout of buspirone, the fEPSP recovered to near pretetanus level. In contrast, when buspirone was applied 10 min after LTP induction, the synaptic responses consistently recovered to the potentiated level (Fig. 6C,D), i.e., the slope of fEPSP recorded 40 min after buspirone washout was 145.6 ± 7.5% (n= 7) of baseline (Fig. 6D). These results suggest that the inhibition of adenylyl cyclase activity is a possible common signaling process underlying the induction of depotentiation in the hippocampal CA1 neurons.
Fig. 6.

Time-dependent reversal of LTP by 5-HT1A receptor agonist buspirone application.A–D, Examples in which LTP was induced at Schaffer collateral–CA1 synapses. Buspirone (0.2 mm) was applied at various times after LTP induction: A, 1 min; orC, 10 min after. B, D, Summary of experiments similar to that shown in A andC. Note that buspirone erased potentiation when delivered 1 min after the TS but was without effect when applied 10 min after. Calibration: 0.5 mV, 10 msec.
5-HT1A receptor antagonist cannot block depotentiation
Because application of 5-HT1A receptor agonist could effectively reverse LTP in the same way as adenosine, we then asked the question of whether adenosine is a unique signaling molecule released during LFS application to disrupt the stabilization of LTP. If the LFS-induced depotentiation is also mediated by the release of 5-HT, it is should be blocked by coapplication of 5-HT1A receptor antagonist during the LFS administration. To test this possibility, the effect of selective 5-HT1A receptor antagonist NAN-190 (1 μm) on the induction of LFS-induced depotentiation was examined. We found that NAN-190 possessed no significant inhibitory effect on the LFS-induced depotentiation; the degree of depotentiation did not differ from the control. The residual potentiation measured 40 min after the end of LFS was 106.3 ± 12.4% (n = 8) of baseline. These results suggest that the protocol used in the present study to elicit LFS-induced depotentiation of LTP is not attributable to the release of 5-HT, although 5-HT1A receptor agonist buspirone can mimic the inhibition of LFS of LTP development.
Postsynaptic loading of Sp-cAMPS blocks LFS-induced depotentiation
Our above experiments have shown that an inhibition of cAMP-gated pathways is critically involved in the mechanism for LFS-induced depotentiation. However, we do not know whether the presynaptic or postsynaptic blockade of cAMP-gated pathways is essential for the induction of depotentiation. If the blockade of postsynaptic cAMP-dependent process is responsible for LFS-induced depotentiation, direct activation of postsynaptic cAMP-dependent signaling cascades should inhibit the subsequent induction of LFS-induced depotentiation. To examine this possibility, CA1 neurons were recorded with microelectrodes filled with a nonhydrolizable cAMP analog, Sp-cAMPS (10 mm) (Yusta et al., 1988). The effectiveness of the diffusion of Sp-cAMPS into the cell was confirmed by the findings that the spike frequency accommodation and the afterhyperpolarization to a depolarization current pulse injection (1.5 nA for 300 msec) were abolished. We allowed 30–40 min for Sp-cAMPS diffuse into the cell before attempting to induce LTP. Figure7A shows a typical example of intracellular EPSP recorded in Sp-cAMPS-loaded cell. Extracellular fEPSP was also monitored simultaneously with intracellular EPSP to ensure that the LFS-induced depotentiation was induced in the population of the cells that were not subjected to the Sp-cAMPS. As a result, the LFS-induced depotentiation was completely blocked in the Sp-cAMPS-loaded cell; however, LFS-induced depotentiation was still observed in the field potential from neurons not loaded with Sp-cAMPS. The mean initial slope of EPSP recorded from the Sp-cAMPS-loaded cells measured 40 min after the end of LFS was 147.8 ± 6.4% (n = 6) of baseline. In contrast, the slope of fEPSP was 103.7 ± 6.9% (n = 6) of baseline measured 40 min after the end of LFS (Fig. 7B). These results suggest that the activation of postsynaptic cAMP-gated pathways blocks the LFS-induced depotentiation.
Fig. 7.
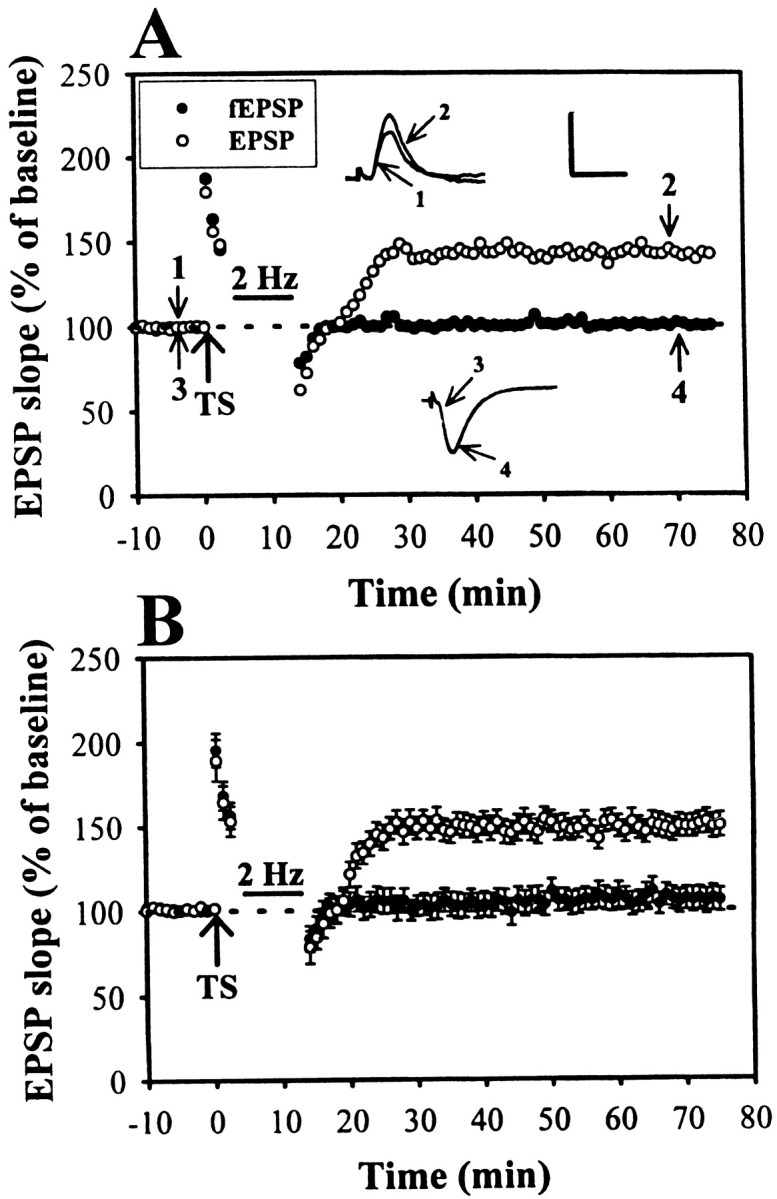
Activation of postsynaptic PKA prevents the LFS-induced depotentiation. A, Example of an experiment in which intracellular application of PKA activator, Sp-cAMPS (10 mm), prevented the LFS-induced depotentiation. In this experiment, extracellular fEPSP was also monitored simultaneously with intracellular EPSP. Note that LFS effectively erased potentiation of fEPSP when delivered 3 min after the TS, indicating that LFS-induced depotentiation could be induced in the population of cells that were not subjected to the Sp-cAMPS. B, Summary of six experiments performed as in A. Calibration: EPSP, 5 mV, 10 msec; fEPSP, 0.5 mV, 10 msec.
The role of protein phosphatases 1 and 2A on the LFS-induced depotentiation
Considerable evidence suggests that the activation of cAMP-dependent protein kinase A (PKA) could inhibit the function of protein phosphatase 1 (PP1) via its ability to phosphorylate the inhibitor 1 (I-1) (Mulkey et al., 1993; Lisman, 1994). Therefore, it is possible that LFS-induced depotentiation occurs via the indirect activation of PP1 by reducing the activation of PKA to phosphorylate I-1. The resulting activation of PP1 may lead to prevent LTP by returning Ca2+–calmodulin-dependent protein kinase II and its molecular targets to their unphosphorylated states and LTP fails (McGlade-McCulloh et al., 1993;Barria et al., 1997). To test this prediction, we examined the effect of protein phosphatases activity on the induction of depotentiation. Okadaic acid is a marine sponge toxin that is a potent and cell-permeable PP1/2A blocker (Cohen et al., 1990). After a 2–3 hr preincubation in okadaic acid (1 μm), the LFS-induced depotentiation was markedly inhibited. The residual potentiation measured 40 min after the end of LFS was 129.3 ± 8.2% (n = 6) of baseline (Fig.8A,B). Similar results were also obtained by preincubation of the slices with another potent inhibitor of PP1/PP2A, calyculin A (1 μm) (Ishihara et al., 1989). The residual potentiation measured 40 min after the end of LFS was 146.2 ± 4.8% (n = 5) of baseline (Fig.8C,D). These results indicate that the activation of PP1/PP2A plays an essential role in the development of LFS-induced depotentiation.
Fig. 8.

The protein phosphatase 1 and 2A inhibitors prevent the LFS-induced depotentiation. A,C, Example of an experiment showing that preincubation of slice for 2–3 hr in either 1 μm okadaic acid (PP1/2A inhibitor) or 1 μm calyculin A (PP1/2A inhibitor) effectively prevented the LFS-induced depotentiation. B,D, Summary of experiments similar to that shown inA and C. Calibration: 0.5 mV, 10 msec.
DISCUSSION
In the present study, we have made some progress in understanding the extracellular adenosine involved in the LFS-induced depotentiation at Schaffer collateral–CA1 synapses. There are four principal observations emerged from this work. First, the time-dependent reversal of LTP by LFS was mimicked by extracellular application of adenosine and was blocked by A1 adenosine receptor antagonist DPCPX but not by A2 receptor antagonist DMPX. Although transient extracellular application of 5-HT1A receptor agonist buspirone after LTP induction could also effectively reverse previously established LTP, 5-HT1A receptor antagonist NAN-190 did not affect the LFS-induced depotentiation. Second, the source of extracellular adenosine during LFS to exert depotentiation appeared to be attributable to the efflux of cAMP that is subsequently converted into adenosine by ecto-5′-nucleotidase. However, the extracellular conversion of ATP is not the major source of adenosine underlying the LFS-induced depotentiation. Third, previous activation of adenylyl cyclase by forskolin or injection of a cAMP analog Sp-cAMPS into postsynaptic neurons prevented the production of LFS-induced depotentiation. Fourth, protein phosphatase 1 and 2A inhibitors okadaic acid and calyculin A greatly reduced the depotentiation produced by LFS.
As mentioned in the introductory remarks, the present study was focused on the question of whether the extracellular adenosine contributes to the development of LFS-induced depotentiation. The experiments with DPCPX strongly suggest that LFS reverses LTP via a build-up of extracellular adenosine and activation of A1adenosine receptors (Fig. 3). These results contrast with those of De Mendonca et al. (1997), who showed that endogenous adenosine, acting through A1 adenosine receptors, exert inhibitory effect on LFS-induced depotentiation. The reason for this discrepancy is not clear but could be attributable partly to the use of stimulation paradigms for depotentiation (1 Hz, 900 stimuli vs 2 Hz, 1200 stimuli), resulting in activation of different cellular processes that may vary in their mode of action and produce different types of depressive effects. However, our results are generally in agreement with previous findings of Larson et al. (1993), who found that another A1 adenosine receptor antagonist, 8-CPT, could effectively inhibit the reversal of LTP by theta frequency stimulation, indicating that the endogenously released adenosine during LFS can act on A1 adenosine receptors to interrupt the biochemical processes leading to the expression of LTP. Similarly, Arai et al. (1990) also demonstrated that extracellular application of adenosine prevents LTP if applied within 1 min but not 5 min after LTP induction.
It appears that efflux of cAMP is the source for the increased extracellular adenosine underlying LFS-induced depotentiation. Evidence supporting this is that the perfusion of the hippocampal slices with either cAMP efflux transporter inhibitor probenecid or type IV phosphodiesterase inhibitor Ro 20–1724 was effective to inhibit the LFS-induced depotentiation. Furthermore, in agreement with this conclusion, we have also found that with a combination of AOPCP and GMP to inhibit the ecto-5′-nucleotidase that converts the AMP into adenosine markedly prevented the LFS-induced depotentiation. An open question that deviated from this observation is which cells in the hippocampal slices release cAMP into the extracellular space. The potential candidate is glial cells, because previous work has demonstrated that glial cells rather than neurons are capable of releasing significant amounts of cAMP induced by activation group II-like metabotropic glutamate receptors in the hippocampus (Winder et al., 1996). Another important issue that we did not address in the present study is whether the direct efflux of adenosine itself is a possible source of extracellular adenosine to underlie LFS-induced depotentiation. Several previous studies have suggested that both the interneurons (Manzoni et al., 1994) and CA1 pyramidal neurons (Brundege and Dunwiddie, 1996) can release enough adenosine induced by direct application of NMDA or electrical stimulation. Because the adenosine nucleoside transporters are bidirectional, the blockade of these transporters by currently available inhibitors (e.g., coapplication of dipyridamol and nitrobenzylthioinosine) may slow adenosine uptake and lead to more adenosine accumulation under equilibrium conditions. Under this condition, the recorded neurons did not undergo LTP after tetanic stimulation (our unpublished observations); it is therefore difficult to examine whether the direct adenosine efflux contributes to LFS-induced depotentiation. Although the release of ATP and its extracellular catabolism is one potential mechanism that can result in an increase in extracellular adenosine in hippocampal slices (White and MacDonald, 1990), the lack of effect of ecto-ATPase inhibitor FPL 67156 on LFS-induced depotentiation suggests that this mechanism was not involved. These findings are in excellent agreement with the current biochemical findings of Cunha et al. (1996), who showed that on electrical stimulation of rat hippocampal slices, the release of ATP, and the contribution of extracellular catabolism of adenine nucleotides to adenosine is greater at a high-frequency stimulation, whereas an LFS preferentially releases adenosine.
It has been repeatedly demonstrated that cAMP-dependent signaling pathways play a crucial role in several forms of hippocampal LTP. For example, the activation of cAMP–PKA pathways could potentially modulate the induction of LTP at Schaffer collateral, mossy fiber, and the medial perforant pathways (Frey et al., 1993; Blitzer et al., 1995;Abel et al., 1997). Unlike LTP, both theoretical (Lisman, 1994) and experimental (Mulkey et al., 1993) studies have revealed that the induction of long-term depression (LTD) can be inhibited by activation of cAMP-dependent signaling pathways. The use of adenylyl cyclase activator forskolin and PKA activator Sp-cAMPS in the present study provides strong evidence that the inhibition of postsynaptic cAMP–PKA-dependent signaling cascades is a plausible mechanism for LFS-induced depotentiation. These results are in complete agreement with a recent study that activation of D1/D5 dopaminergic receptors of hippocampal CA1 cells leading to activate adenylyl cyclase in turn triggering a cAMP–PKA signaling cascade greatly reduced the LFS (3 Hz/3 min)-induced depotentiation (Otmakhova and Lisman, 1998). We have, in addition, extended the findings of Otmakhova and Lisman (1998) by showing that postsynaptic cAMP pathways contribute to the conversion of the initial potentiation into a persistent and not readily disrupted state. This prediction is also supported by recent findings that early phase LTP can be inhibited by interfering with the cAMP pathway in the postsynaptic cell (Blitzer et al., 1995). The link between cAMP-dependent signaling cascade inhibition and the phenomenon of LTP reversal is also supported by buspirone experiment showing that activation of 5-HT1A receptors within 3 min after LTP induction consistently erases potentiation (Fig. 6). Although activation of 5HT1A receptor by buspirone can mimic the effect of adenosine and LFS to reverse the LTP, we could not inhibit depotentiation by applying 5-HT1Areceptor antagonist NAN-190 during the induction of depotentiation by LFS under the same condition in which A1adenosine receptor antagonist DPCPX could inhibit depotentiation. These data suggested that the LFS-induced depotentiation observed here might be merely a result of an increase of extracellular adenosine to activate A1 adenosine receptors rather than a change of 5-HT concentration, although both A1 adenosine receptors and 5-HT1A receptors are well known to couple to inhibit adenylyl cyclase via Gi-protein.
Given that LFS-induced depotentiation was blocked by selective PP1/PP2A inhibitors, okadaic acid, and calyculin A, this suggests a crucial role for PP1 or/and PP2A subtype(s) in the induction of depotentiation. There has been increasing evidence showing that LTD induction requires activation of a protein phosphatase cascade (Mulkey et al., 1993). However, the role of protein phosphatases in the depotentiation has not yet been understood. Although our data do not conclusively demonstrate the precise mechanisms whereby PP1 or PP2A are selectively activated by LFS, it appears that a diminution of cAMP–PKA system-mediated suppression of protein phosphatase activity may be involved. Indeed, previous studies have demonstrated that activation of PKA would inhibit PP1 function through phosphorylation of PP1 regulatory protein I-1 (Shenolikar and Nairn, 1991), because I-1, when phosphorylated, could inhibit the function of PP1. In contrast, PKA is inhibited, indirectly activates PP1, and leads to the dephosphorylation of a large set of target molecules, which are necessary for the expression of LTP, and LTP fails (Blitzer et al., 1995, 1998; Thomas et al., 1996). Our findings provided further evidence to support the hypothesis that the activation of PP signaling pathways is a potential component of biochemical cascades that subserve depotentiation induction at the central synapses (O'Dell and Kandel, 1994; Stäubli and Chun, 1996).
We conclude that the time-dependent reversal of LTP at Schaffer collateral–CA1 synapses by LFS is likely to be accounted for by the increase of extracellular adenosine acting on the A1 adenosine receptors to interrupt the cAMP–PKA-dependent signaling cascades leading to the development of LTP. Because LTP, in the mammalian brain, is generally assumed as a synaptic mechanism underlying learning and memory formation (Bliss and Collingridge, 1993) and the process that may selectively disrupt the formation of stable LTP may be the possible cause for the loss of memory, our findings imply a possibility that the extracellular adenosine is a potential common molecule to exert the mechanism of forgetting. If this is the case, the selective A1adenosine receptor antagonists might be useful in enhancing cognition in treatment of some dementing disorders (De Mendonca et al., 1997).
Footnotes
This work was supported by research grants from the Department of Health (88-HR-837) and the National Health Research Institute (GT-EX89S837C) of Taiwan.
Correspondence should be addressed to Dr. Kuei-Sen Hsu at the above address. E-mail: richard@mail.ncku.edu.tw.
REFERENCES
- 1.Abel T, Nguyen PV, Barad M, Deuel TAS, Kandel ER, Bourtchouladze R. Genetic demonstration of a role for PKA in the late phase of LTP and in hippocampus-based long-term memory. Cell. 1997;88:1–11. doi: 10.1016/s0092-8674(00)81904-2. [DOI] [PubMed] [Google Scholar]
- 2.Andrade R, Nicoll RA. Pharmacologically distinct actions of serotonin on single pyramidal neurons of the rat hippocampus recorded in vitro. J Physiol (Lond) 1987;394:99–124. doi: 10.1113/jphysiol.1987.sp016862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Arai A, Kessler M, Lynch G. The effects of adenosine on the development of long-term potentiation. Neurosci Lett. 1990;19:41–44. doi: 10.1016/0304-3940(90)90750-4. [DOI] [PubMed] [Google Scholar]
- 4.Bahr BA, Stäubli U, Xiao P, Chun D, Ji ZX, Esteban ET, Lynch G. Arg-Gly-Asp-Ser-selective adhesion and the stabilization of long-term potentiation: pharmacological studies and the characterization of a candidate matrix receptor. J Neurosci. 1997;17:1320–1329. doi: 10.1523/JNEUROSCI.17-04-01320.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Barria A, Muller D, Derkach V, Griffith LC, Soderling TR. Regulatory phosphorylation of AMPA-type glutamate receptors by CaM-KII during long-term potentiation. Science. 1997;276:2042–2045. doi: 10.1126/science.276.5321.2042. [DOI] [PubMed] [Google Scholar]
- 6.Barrionuevo G, Schottler F, Lynch G. The effects of repetitive low frequency stimulation on control and “potentiated” synaptic responses in the hippocampus. Life Sci. 1980;27:2385–2391. doi: 10.1016/0024-3205(80)90509-3. [DOI] [PubMed] [Google Scholar]
- 7.Bashir ZI, Collingridge GL. NMDA receptor-dependent transient homo- and heterosynaptic depression in picrotoxin-treated hippocampal slices. Eur J Neurosci. 1992;4:485–490. doi: 10.1111/j.1460-9568.1992.tb00898.x. [DOI] [PubMed] [Google Scholar]
- 8.Bliss TVP, Collingridge GL. A synaptic model of memory: long-term potentiation in the hippocampus. Nature. 1993;361:31–39. doi: 10.1038/361031a0. [DOI] [PubMed] [Google Scholar]
- 9.Blitzer RD, Wong T, Nouranifar R, Iyengar R, Landau EM. Postsynaptic cAMP pathway gates early LTP in hippocampal CA1 region. Neuron. 1995;15:1403–1414. doi: 10.1016/0896-6273(95)90018-7. [DOI] [PubMed] [Google Scholar]
- 10.Blitzer RD, Connor JH, Brown GP, Wong T, Shenolikar S, Iyengar R, Landau EM. Gating of CaMKII by cAMP-regulated protein phosphatase activity during LTP. Science. 1998;280:1940–1942. doi: 10.1126/science.280.5371.1940. [DOI] [PubMed] [Google Scholar]
- 11.Brundege JM, Dunwiddie TV. Modulation of excitatory synaptic transmission by adenosine released from single hippocampal pyramidal neurons. J Neurosci. 1996;16:5603–5612. doi: 10.1523/JNEUROSCI.16-18-05603.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cohen P, Holmes CFB, Tsukitani Y. Okadaic acid: a new probe for the study of cellular regulation. Trends Biochem Sci. 1990;15:98–102. doi: 10.1016/0968-0004(90)90192-e. [DOI] [PubMed] [Google Scholar]
- 13.Crack BE, Pollard CE, Beukers MW, Roberts SM, Hunt SF, Ingall AH, McKechnie KCW, Ijzerman AP, Leff P. Pharmacological and biochemical analysis of FPL 67156, a novel, selective inhibitor of ecto-ATPase. Br J Pharmacol. 1995;114:475–481. doi: 10.1111/j.1476-5381.1995.tb13251.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Craig CG, White TD. N-methyl-d-aspartate- and non-N-methyl-d-aspartate-evoked adenosine release from rat cortical slices: distinct purinergic sources and mechanisms of release. J Neurochem. 1993;60:1073–1080. doi: 10.1111/j.1471-4159.1993.tb03256.x. [DOI] [PubMed] [Google Scholar]
- 15.Cunha RA, Vizi ES, Ribeiro JA, Sebastião AM. Preferential release of ATP and its extracellular catabolism as a source of adenosine upon high- but not low-frequency stimulation of rat hippocampal slices. J Neurochem. 1996;67:2180–2187. doi: 10.1046/j.1471-4159.1996.67052180.x. [DOI] [PubMed] [Google Scholar]
- 16.De Mendonca A, Ribeiro JA. Adenosine and neuronal plasticity. Life Sci. 1997;60:245–251. doi: 10.1016/s0024-3205(96)00544-9. [DOI] [PubMed] [Google Scholar]
- 17.De Mendonca A, Almeida T, Bashir ZI, Ribeiro JA. Endogenous adenosine attenuates long-term depression and depotentiation in the CA1 region of the rat hippocampus. Neuropharmacology. 1997;36:161–167. doi: 10.1016/s0028-3908(96)00173-6. [DOI] [PubMed] [Google Scholar]
- 18.Dunwiddie TV, Fredholm BB. Adenosine A1 receptors inhibit adenylate cyclase activity and neurotransmitter release and hyperpolarize pyramidal neurons in rat hippocampus. J Pharmacol Exp Ther. 1989;249:31–37. [PubMed] [Google Scholar]
- 19.Frey U, Huang YY, Kandel ER. Effects of cAMP stimulate a late stage of LTP in hippocampal CA1 neurons. Science. 1993;260:1661–1664. doi: 10.1126/science.8389057. [DOI] [PubMed] [Google Scholar]
- 20.Fujii S, Saito K, Miyakawa H, Ito K, Kato H. Reversal of long-term potentiation (depotentiation) induced by tetanus stimulation of the input to CA1 neurons of guinea pig hippocampal slices. Brain Res. 1991;555:112–122. doi: 10.1016/0006-8993(91)90867-u. [DOI] [PubMed] [Google Scholar]
- 21.Fujii S, Sekino Y, Kuroda Y, Sasaki H, Ito KI, Kato H. 8-Cyclopentyltheophylline, an adenosine A1 receptor antagonist, inhibits the reversal of long-term potentiation in hippocampal CA1 neurons. Eur J Pharmacol. 1997;331:9–14. doi: 10.1016/s0014-2999(97)01024-8. [DOI] [PubMed] [Google Scholar]
- 22.Gerber U, Greene RW, Hass HL, Stevens DR. Characterization of inhibition mediated by adenosine in the hippocampus of the rat in vitro. J Physiol (Lond) 1989;417:567–578. doi: 10.1113/jphysiol.1989.sp017819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gereau RW, Conn PJ. Potentiation of cAMP responses by metabotropic glutamate receptors decreases excitatory synaptic transmission by a kinase-independent mechanism. Neuron. 1994;12:1121–1129. doi: 10.1016/0896-6273(94)90319-0. [DOI] [PubMed] [Google Scholar]
- 24.Henderson GB, Strauss BP. Evidence for cAMP and cholate extrusion in C6 rat glioma cells by a common anion efflux pump. J Biol Chem. 1991;266:1641–1645. [PubMed] [Google Scholar]
- 25.Hsu KS, Huang CC. Characterization of the anoxia-induced long-term synaptic potentiation in area CA1 of the rat hippocampus. Br J Pharmacol. 1997;122:671–681. doi: 10.1038/sj.bjp.0701409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Huang CC, Hsu KS, Gean PW. Isoproterenol potentiates synaptic transmission primarily by enhancing presynaptic calcium influx via P- and/or Q-type calcium channels in the rat amygdala. J Neurosci. 1996;16:1026–1033. doi: 10.1523/JNEUROSCI.16-03-01026.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ishihara H, Martin B, Brautingan D, Karaki H, Ozaki H, Kato Y, Fusetani N, Watabe S, Hashimoto K, Uemura D. Calyculin A and okadaic acid: inhibitors of protein phosphatase activity. Biochem Biophys Res Commun. 1989;159:871–877. doi: 10.1016/0006-291x(89)92189-x. [DOI] [PubMed] [Google Scholar]
- 28.Larson J, Xiao P, Lynch G. Reversal of LTP by theta frequency stimulation. Brain Res. 1993;600:97–102. doi: 10.1016/0006-8993(93)90406-d. [DOI] [PubMed] [Google Scholar]
- 29.Laurenza A, Sutkowski EM-H, Seamon KB. Forskolin: a specific stimulator of adenylyl cyclase or a diterpene with multiple sites of action. Trends Pharmacol Sci. 1989;10:442–447. doi: 10.1016/S0165-6147(89)80008-2. [DOI] [PubMed] [Google Scholar]
- 30.Lisman JE. The CaM kinase II hypothesis for the storage of synaptic memory. Trends Neurosci. 1994;17:406–412. doi: 10.1016/0166-2236(94)90014-0. [DOI] [PubMed] [Google Scholar]
- 31.Lloyd HGE, Lindström K, Fredholm BB. Intracellular formation and release of adenosine from rat hippocampal slices evoked by electrical stimulation and energy depletion. Neurochem Int. 1993;23:173–185. doi: 10.1016/0197-0186(93)90095-m. [DOI] [PubMed] [Google Scholar]
- 32.MacDonald WF, White TD. Nature of extrasynaptosomal accumulation of endogenous adenosine evoked by K+ and veratridine. J Neurochem. 1985;45:791–797. doi: 10.1111/j.1471-4159.1985.tb04062.x. [DOI] [PubMed] [Google Scholar]
- 33.Manzoni OJ, Manabe T, Nicoll RA. Release of adenosine by activation of NMDA receptors in the hippocampus. Science. 1994;265:2089–2101. doi: 10.1126/science.7916485. [DOI] [PubMed] [Google Scholar]
- 34.McGlade-McCulloh E, Yamamoto H, Tan S-E, Brickey DA, Soderling TR. Phosphorylation and regulation of glutamate receptors by calcium/calmodulin-dependent protein kinase II. Nature. 1993;362:640–642. doi: 10.1038/362640a0. [DOI] [PubMed] [Google Scholar]
- 35.Mulkey RM, Herron CE, Malenka RC. An essential role for protein phosphatases in hippocampal long-term depression. Science. 1993;261:1051–1055. doi: 10.1126/science.8394601. [DOI] [PubMed] [Google Scholar]
- 36.Nicoll RA, Malenka RC. Contrasting properties of two forms of long-term potentiation in the hippocampus. Nature. 1995;377:115–118. doi: 10.1038/377115a0. [DOI] [PubMed] [Google Scholar]
- 37.O'Dell T, Kandel E. Low-frequency stimulation erases LTP through an NMDA receptor-mediated activation of protein phosphatases. Learn Mem. 1994;1:129–139. [PubMed] [Google Scholar]
- 38.Otmakhova NA, Lisman JE. D1/D5 dopamine receptors inhibit depotentiation at CA1 synapses via cAMP-dependent mechanism. J Neurosci. 1998;18:1270–1279. doi: 10.1523/JNEUROSCI.18-04-01270.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ribeiro JA. Purinergic inhibition of neurotransmitter release in the central nervous system. Pharmacol Toxicol. 1995;77:299–305. doi: 10.1111/j.1600-0773.1995.tb01031.x. [DOI] [PubMed] [Google Scholar]
- 40.Rosenberg PA, Knowles R, Knowles KP, Li Y. Beta-adrenergic receptor-mediated regulation of extracellular adenosine in cerebral cortex in culture. J. Neurosci. 1994;14:2953–2965. doi: 10.1523/JNEUROSCI.14-05-02953.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Shenolikar S, Nairn AC. Protein phosphatases: recent progress. Adv Second Messenger Phosphoprotein Res. 1991;23:1–121. [PubMed] [Google Scholar]
- 42.Stäubli U, Chun D. Proactive and retrograde effects on LTP produced by theta pulse stimulation: mechanisms and characteristics of LTP reversal. Learn Mem. 1996;3:96–105. doi: 10.1101/lm.3.2-3.96. [DOI] [PubMed] [Google Scholar]
- 43.Stäubli U, Lynch G. Stable depression of potentiated synaptic responses in the hippocampus with 1–5 Hz stimulation. Brain Res. 1990;513:113–118. doi: 10.1016/0006-8993(90)91096-y. [DOI] [PubMed] [Google Scholar]
- 44.Stäubli U, Chun D, Lynch G. Time-dependent reversal of long-term potentiation by an integrin antagonist. J Neurosci. 1998;18:3460–3469. doi: 10.1523/JNEUROSCI.18-09-03460.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Thomas MJ, Moody TD, Makhinson M, O'Dell TJ. Activity-dependent β-adrenergic modulation of low-frequency stimulation induced LTP in the hippocampal CA1 region. Neuron. 1996;17:475–482. doi: 10.1016/s0896-6273(00)80179-8. [DOI] [PubMed] [Google Scholar]
- 46.White TD, MacDonald WF. Neural release of ATP and adenosine. Ann NY Acad Sci. 1990;603:287–299. doi: 10.1111/j.1749-6632.1990.tb37680.x. [DOI] [PubMed] [Google Scholar]
- 47.Winder DG, Ritch PS, Gereau RW, Conn PJ. Novel glial-neuronal signaling by coactivation of metabotropic glutamate and β-adrenergic receptors in rat hippocampus. J Physiol (Lond) 1996;494:743–755. doi: 10.1113/jphysiol.1996.sp021529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Yusta B, Ortiz-Caro J, Pascual A, Aranda A. Comparison of the effects of forskolin and dibutyryl cyclic AMP in neuroblastoma cells: evidence that some of the actions of dibutyryl cyclic AMP are mediated by butyrate. J Neurochem. 1988;51:1808–1818. doi: 10.1111/j.1471-4159.1988.tb01162.x. [DOI] [PubMed] [Google Scholar]