

Abstract
Iron chelators are pluripotent neuronal antiapoptotic agents that have been shown to enhance metabolic recovery in cerebral ischemia models. The precise mechanism(s) by which these agents exert their effects remains unclear. Recent studies have demonstrated that iron chelators activate a hypoxia signal transduction pathway in non-neuronal cells that culminates in the stabilization of the transcriptional activator hypoxia-inducible factor-1 (HIF-1) and increased expression of gene products that mediate hypoxic adaptation. We examined the hypothesis that iron chelators prevent oxidative stress–induced death in cortical neuronal cultures by inducing expression of HIF-1 and its target genes. We report that the structurally distinct iron chelators deferoxamine mesylate and mimosine prevent apoptosis induced by glutathione depletion and oxidative stress in embryonic cortical neuronal cultures. The protective effects of iron chelators are correlated with their ability to enhance DNA binding of HIF-1 and activating transcription factor 1(ATF-1)/cAMP response element–binding protein (CREB) to the hypoxia response element in cortical cultures and the H19-7 hippocampal neuronal cell line. We show that mRNA, protein, and/or activity levels for genes whose expression is known to be regulated by HIF-1, including glycolytic enzymes, p21waf1/cip1, and erythropoietin, are increased in cortical neuronal cultures in response to iron chelator treatment. Finally, we demonstrate that cobalt chloride, which also activates HIF-1 and ATF-1/CREB in cortical cultures, also prevents oxidative stress–induced death in these cells. Altogether, these results suggest that iron chelators exert their neuroprotective effects, in part, by activating a signal transduction pathway leading to increased expression of genes known to compensate for hypoxic or oxidative stress.
Keywords: iron chelators, oxidative stress, glutathione, apoptosis, HIF-1, ATF-1/CREB
Disrupted iron homeostasis has been linked to a number of neurodegenerative states (Connor and Menzies, 1995) including Alzheimer's disease (Smith et al., 1997; Lovell et al., 1998), Parkinson's disease (Michel et al., 1992; Hartley et al., 1993; Gassen and Youdim; 1997; Olanow, 1997; Hirsch and Faucheux, 1998), stroke (Lipscomb et al., 1998), multiple sclerosis (Levine, 1997), and Friedreich's ataxia (Babcock et al., 1997; Rotig et al., 1997). The link between iron and the pathogenesis of several neurodegenerative conditions provides a firm rationale for the therapeutic use of small molecules, such as deferoxamine (DFO), which chelate iron and prevent it from participating in deleterious redox reactions. Indeed, DFO has been reported to slow progression of Alzheimer's disease (Crapper McLachlan et al., 1991), to enhance metabolic recovery in animal models of stroke (Hurn et al., 1995), and to attenuate the severity and duration of experimental allergic encephalomyelitis in rats (Bowern et al., 1984). Despite these promising clinical and preclinical observations, the precise mechanisms by which iron chelators prevent neuronal injury remain unclear.
Iron or other transition metals such as copper are believed to induce neuronal injury by converting superoxide (O2.−) and hydrogen peroxide (H2O2) into highly reactive, toxic hydroxyl radicals (OH.) in a sequence of reactions that are referred to cumulatively as the Haber–Weiss reaction (Winterbourn, 1995). According to this scheme, iron chelators diminish hydroxyl radical formation and oxidative stress by sequestering redox active iron. The abilities of structurally distinct chelators to inhibit apoptosis induced by depletion of superoxide dismutase in pheochromocytoma 12 (PC12) cells (Troy et al., 1996) or by depletion of glutathione in oligodendrocytes (Yonezawa et al., 1996) are consistent with the notion that iron chelators prevent apoptotic cell death by reducing hydroxyl radical formation and oxidative stress.
Recent studies, however, have identified novel pathways by which iron chelators may enhance survival (Beckman et al., 1990). Apoptosis induced by serum or growth factor deprivation of PC12 cells or sympathetic neurons can be prevented by pretreatment with higher concentrations of the iron chelators DFO and mimosine (MIM) than are required to prevent oxidative stress–induced death (Farinelli and Greene, 1996). In this paradigm, the protective effects of iron chelators cannot be mimicked by antioxidants, including vitamin E. Rather, the protective effects of chelating iron correlate with inhibition of proliferation. In this context, the antiproliferative effects of iron chelators have been attributed to a decrease in cyclin A/p34cdc2 kinase activity and mRNA levels (Terada et al., 1993; Feldman and Schonthal, 1994).
Iron chelators have also been shown to activate a hypoxia stress response pathway. DFO induces expression of hypoxia-inducible factor-1 (HIF-1) and transcription of downstream target genes including erythropoietin (Wang and Semenza, 1993), glycolytic enzymes (Semenza et al., 1994), and vascular endothelial growth factor (Levy et al., 1995; Forsythe et al., 1996; Jiang et al., 1997). HIF-1 is a heterodimer composed of HIF-1α (∼120 kDa) and HIF-1β (91–94 kDa) subunits that bind to the consensus sequence 5′-RCGTG-3′ (Semenza, 1998). These studies highlight the diversity of cellular responses to iron chelators and suggest that these pluripotent antiapoptotic agents may act to enhance survival by mechanisms other than simply suppressing hydroxyl radical formation. In this manuscript, we examine the hypothesis that iron chelators exert their neuroprotective effects, in part, by the enhancement of DNA binding of HIF-1 to the hypoxia response element and the consequent upregulation of HIF-1–regulated genes that provide resistance to oxidative or hypoxic stress.
MATERIALS AND METHODS
Primary neurons. Cell cultures were obtained from the cerebral cortex of fetal Sprague Dawley rats [embryonic day 17 (E17)] as described previously (Murphy et al., 1990). All experiments were initiated 24 72 hr after plating. Under these conditions the cells are not susceptible to glutamate-mediated excitotoxicity. For cytotoxicity studies, cells were rinsed with warm PBS and then placed in Minimum Essential Medium (MEM; Life Technologies, Gaithersburg, MD) with 5.5 gm/l glucose, 10% FCS, 2 mml-glutamine, and 100 μmcystine, containing the glutamate analog homocysteate (HCA; 1 mm). HCA was diluted from 100-fold concentrated solutions that were adjusted to pH 7.5. Viability was assessed by phase-contrast microscopy, lactate dehydrogenase (LDH) release (Koh and Choi, 1987;Ratan et al., 1994a,b), calcein AM/ethidium homodimer-1 staining (Molecular Probes, Eugene, OR) under fluorescence microscopy, or trypan blue exclusion. To evaluate the effects of iron chelators on cytotoxicity, we added deferoxamine mesylate (1–500 μm; Sigma, St. Louis, MO) or mimosine (1–500 μm; Sigma) at the time cortical neurons were exposed to HCA or up to 10 hr after HCA treatment. In parallel, cortical neurons were exposed to HCA and cobalt chloride (10–500 μm). Similar quantitative results were obtained independent of the viability assay used.
Glutathione levels. Total glutathione levels were measured by the method of Tietze (1969) as described in Ratan et al. (1994b)with the following modifications. At 0, 6, 9, and 12 hr after exposure to potential toxins ± inhibitors, the cells were washed with PBS, lysed with cold 3% perchloric acid, and centrifuged at 4°C at 7400 × g. The supernatants were diluted with 9 vol of 0.1 mNa2HPO4, and the remaining steps of the glutathione assay were performed as described (Ratan et al., 1994b). Glutathione levels were normalized to total protein levels that were determined using the bicinchoninic acid reagent (Pierce, Rockford, IL) method (Smith et al., 1985).
Immunoblot analysis. Cell lysates were obtained by rinsing cortical neurons with cold PBS and adding 0.1 m potassium phosphate containing 0.5% Triton X-100. Protein (2.5 μg) from cell lysates was boiled in Laemmli buffer and electrophoresed under reducing conditions on 12% polyacrylamide gels. Proteins were then transferred to a polyvinylidene difluoride membrane (Bio-Rad, Hercules, CA). Nonspecific binding was inhibited by incubation in Tris-buffered saline with Tween 20 (TBST; 50 mm Tris-HCl, pH 8.0, 0.9% NaCl, and 0.1% Tween 20) containing 5% nonfat dried milk for 1.5 hr. Primary antibodies against LDH (rabbit muscle; Rockland, Gilbertsville, PA), aldolase (rabbit muscle; Rockland), α-tubulin (Sigma), HIF-1α (Novus Biologicals, Littleton, CO), or HIF-1β (Affinity Bioreagents, Golden, CO) were diluted 1:1000, 1:1000, and 1:2000 in TBST containing 1% milk and exposed to membranes overnight at 4°C. For LDH and aldolase, membranes were incubated with HRP-conjugated anti-goat secondary antibody (Sigma), diluted 1:5000 in TBST containing 1% milk for 1.5 hr at room temperature. For the α-tubulin immunoblot, the membrane was incubated with HRP-conjugated anti-rabbit secondary antibody (Amersham, Arlington Heights, IL), diluted 1:5000 in TBST for 1.5 hr at room temperature. Immunoreactive proteins were detected according to the enhanced chemiluminescent protocol (Amersham).
Immunofluorescence staining and confocal microscopy.Indirect labeling methods were used to determine the levels of HIF-1α, HIF-1β, and neurofilament (NF; 200 kDa) in cortical neuronal cultures. Dissociated cells from the cerebral cortex (3–5 × 105) were seeded onto poly-d-lysine–coated eight-well culture slides (Becton Dickinson, Bedford, MA) and treated with 100 μm DFO ± HCA as described above for 4–5 hr. The cells were washed with warm PBS and fixed at room temperature for 15 min with 4% paraformaldehyde. After washing with PBS, fixed cells were incubated with blocking solution containing 0.3% Triton X-100, 5% bovine serum albumin, and 3% goat serum for 1 hr, followed by incubation with mouse anti-HIF-1α monoclonal antibody (1:200 dilution), rabbit anti-HIF-1β polyclonal antibody (1:200–1000), or a mouse anti-neurofilament antibody (1:100) overnight at 4° C. After three washes with PBS, the cells were incubated for 1 hr with FITC-conjugated goat anti-rabbit IgG antibody (1:200 dilution) and Texas Red–conjugated goat anti-mouse IgG antibody (1:200 dilution; Vector Laboratories, Burlingame, CA) or vice versa. All antibodies were diluted in PBS. The slides were washed three times with PBS and mounted with fluorochrome mounting solution (Vector Laboratories). Images were analyzed using a confocal microscope (MRC-1024; Bio-Rad). Control experiments were performed in the absence of primary antibody.
Aldolase activity. After 8 and 24 hr of exposure to the HCA ± antioxidants ± protein synthesis inhibitors, cells were washed with cold PBS and lysed with 0.5% Triton X-100 in 0.1m potassium phosphate buffer, pH 7.0. The lysates were divided for measurements of LDH activity (Ratan et al., 1994b), aldolase activity (catalog #752-A; Sigma), and protein. The aldolase assay is based on the spectrophotometric determination (λ = 550 nm) of the levels of hydrazone formed when 2,4-dinitrophenylhydrazine (a chromagen) is reacted with a hydrolysis product of dihydroxyacetone phosphate. Dihydroxyacetone phosphate is formed from fructose 1,6-diphosphate in a reaction catalyzed by aldolase, and thus the quantity of hydrazone is proportional to aldolase activity. The kit we used was designed for use in serum; its sensitivity and specificity for use in lysates of primary cultures were validated by use of serial dilutions of a standard concentration of aldolase as well as by addition of known quantities of aldolase to control lysates. Aldolase activity is expressed as absolute absorbance values normalized to total protein.
Electrophoretic mobility shift assays and supershift analysis. We performed electrophoretic mobility shift assays (EMSAs) on nuclear extracts from cortical neurons using a32P-labeled oligonucleotide containing a wild-type (W18) or mutant (M18) HIF-1–binding site (Wang and Semenza, 1995). The sense strand sequences of the double-stranded W18 and M18 oligonucleotides are 5′-GCCCTACGTGCTGTCTCA-3′ and 5′-GCCCTAAAAGCTGTCTCA-3′, respectively. Parallel EMSAs were performed using a radiolabeled Oct-1 (5′-TGTCGAATGCAAATGACTAGAA-3′; Santa Cruz Biotechnology, Santa Cruz, CA)–binding site. Embryonic cortical neurons were lightly trypsinized, pelleted, and resuspended in cold PBS. All subsequent steps were performed as described previously at 4°C (Lin et al., 1995). The cells were suspended in 10 mm Tris, pH 7.5, 1.5 mmMgCl2, 10 mm KCl, 10 μg/ml aprotinin, 0.5 μg/ml leupeptin, 3 mm PMSF, 3 mm DTT, and 1 mmNa3VO4 and lysed by 15 strokes in a Dounce homogenizer using a type B pestle, and the nuclei were pelleted at 4500 × g for 5 min, resuspended in 3–4 packed cell vol of buffer C (420 mm KCl, 20 mm Tris-HCl, pH 7.8, 1.5 mmMgCl2, 10 μg/ml aprotinin, 0.5 μg/ml leupeptin, 3 mm PMSF, 3 mmDTT, and 1 mmNa3VO4), and incubated for 30 min with gentle agitation. The absence of cytoplasmic contamination of the purified nuclei was verified by the absence of detectable LDH activity in the nuclear extract. The nuclear extract was centrifuged at 10,000 × g for 30 min, and the supernatant was dialyzed twice against 25–50 ml of buffer D (20 mm Tris-HCl, pH 7.8, 100 mmKCl, 0.2 mm EDTA, and 20% glycerol). The dialysate was centrifuged at 10,000 × g for 10 min at 4°C, and the supernatants were aliquoted, snap frozen in liquid N2, and stored at −80°C. Perchloric acid–precipitated protein was measured from a representative aliquot of each sample, and equal amounts were used for binding. Binding reactions were performed at 4°C for 15 min using 3–15 μg of nuclear protein (HIF-1 or Oct-1) and 0.25 ng (10,000–40,000 cpm) of labeled oligonucleotide in 30 μl of binding buffer containing 10 mm Tris-HCl, pH 7.5, 50 mmNaCl, 50 mm KCl, 1 mmMgCl2, 1 mm EDTA, 5 mm DTT, 5% glycerol, and 0.1 μg of sonicated denatured calf thymus DNA. For HIF-1 DNA-binding studies, a 10-fold excess of unlabeled M18 HIF-1 oligonucleotide was included in the binding reaction. DNA–protein complexes were separated from unbound probe on native 6% polyacrylamide gels at 195 V for 2 hr. The gel was vacuum dried and exposed to Kodak film (Eastman Kodak, Rochester, NY) for 8–15 hr at −80°C. Visual inspection of the free probe band at the bottom of the gel confirmed that equivalent amounts of radiolabeled probe were used for each sample (data not shown). Supershifts were performed with antibodies to HIF-1α (Wang et al., 1995a), HIF-1β (MA1–515; Affinity Bioreagents), ATF-1/cAMP response element–binding protein (CREB) (25C10G; Santa Cruz Biotechnology), or CREB-1 (24H4B; Santa Cruz Biotechnology). The antibody was added to the binding mixture immediately after the addition of the radiolabeled HIF-1 probe. The reaction mixture was incubated for 20 min, and the complexes were resolved as described above.
Northern blot analysis. Cells (2.5 × 106) derived from E17 cortices were plated in six-well dishes. After 1–2 d in culture, the cells were treated with or without iron chelators (± homocysteate). After a 4–6 hr exposure, total RNA was extracted from each treatment group using a kit (RNAeasy; Qiagen, Valencia, CA). For each sample, 5 μg of total RNA was electrophoresed in a 1% agarose and formaldehyde gel, transferred to a positively charged nylon membrane, and hybridized with enolase-1 (Semenza et al., 1996), LDH A (Semenza et al., 1996), erythropoietin (Wang and Semenza, 1996; Juul et al., 1998), p21/waf1/cip1 (Carmeliet et al., 1998), and brain-derived neurotrophic factor (Masonpierre et al., 1990) cDNA probes. β-actin was used as a negative control (Marquis and Demple, 1998). After washing, the blots were autoradiographed, and the bands were quantitatively analyzed using a phosphorimager (GS-525 Molecular Image Analyzer; Bio-Rad).
RESULTS
Iron chelators inhibit glutathione depletion–induced apoptosis in embryonic cortical neurons
In previous studies, we demonstrated that exposure of immature cortical neurons to glutamate or the glutamate analog HCA results in depletion of the antioxidant glutathione and in oxidative stress–induced cell death with the morphological and biochemical characteristics of apoptosis (Ratan et al., 1994a,b). To determine whether iron participates in glutathione depletion–induced apoptosis, we examined the protective effects of the iron chelator DFO in this paradigm. At concentrations ≥10 μm, DFO completely inhibited glutathione depletion–induced death. Concentrations of DFO ≥100 μm significantly inhibited not only the HCA-induced death but also the small and reproducible level of cell death seen in control cultures (Fig.1A). The ability of DFO to suppress the chromatin condensation and nuclear fragmentation characteristic of apoptosis in both HCA-treated and control cultures was verified by Hoechst 33258 staining (data not shown) and phase-contrast microscopy (Fig. 2). MIM, an iron chelator structurally distinct from DFO, also prevented glutathione depletion–induced death in cortical neurons (Fig.1B).
Fig. 1.
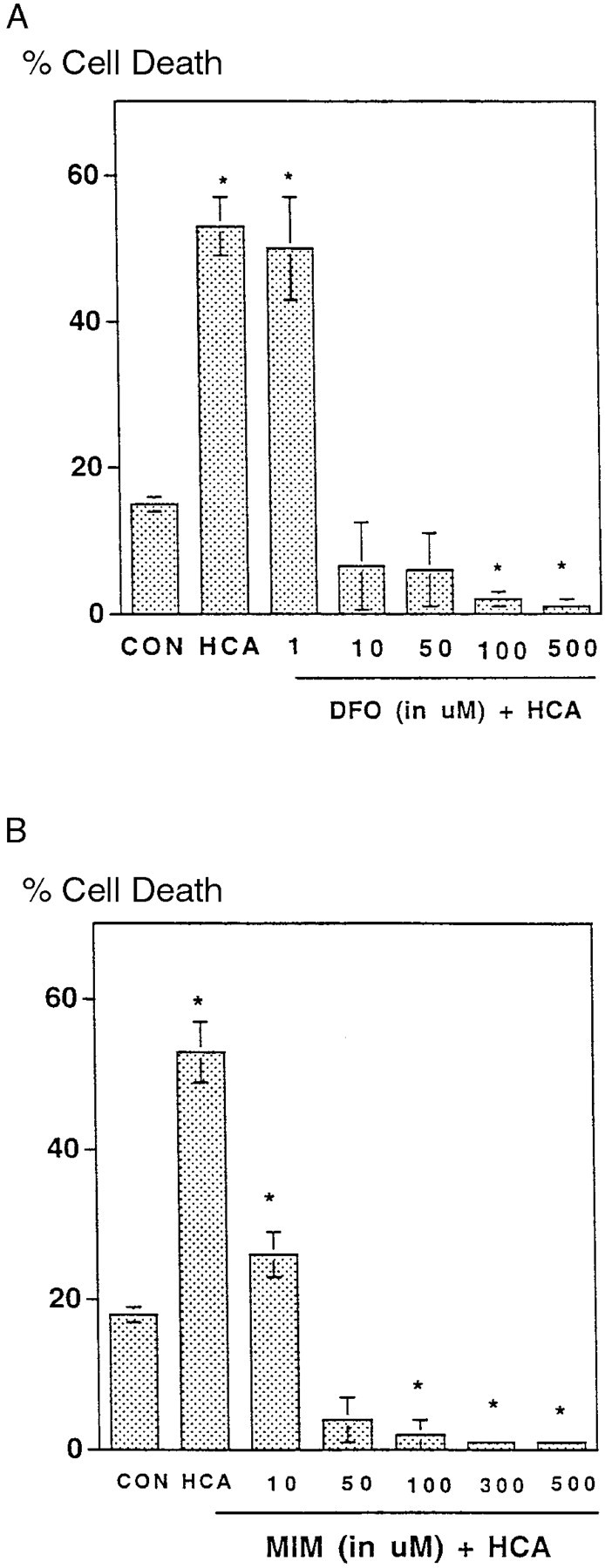
Effect of deferoxamine and mimosine on survival of cultured cortical neurons treated with homocysteate. A,Cultures were exposed to 1 mm HCA as described in Materials and Methods with varying concentrations of DFO. The cells were harvested at 24 hr and processed for LDH activity. Data are means ± SEM (expressed as a percentage of the total LDH activity in the culture released into the medium) from three to five experiments performed in triplicate. B, Effect of MIM on LDH release in cultures exposed to 1 mm HCA for 24 hr is shown. *p < 0.05 by ANOVA. CON, Control.
Fig. 2.
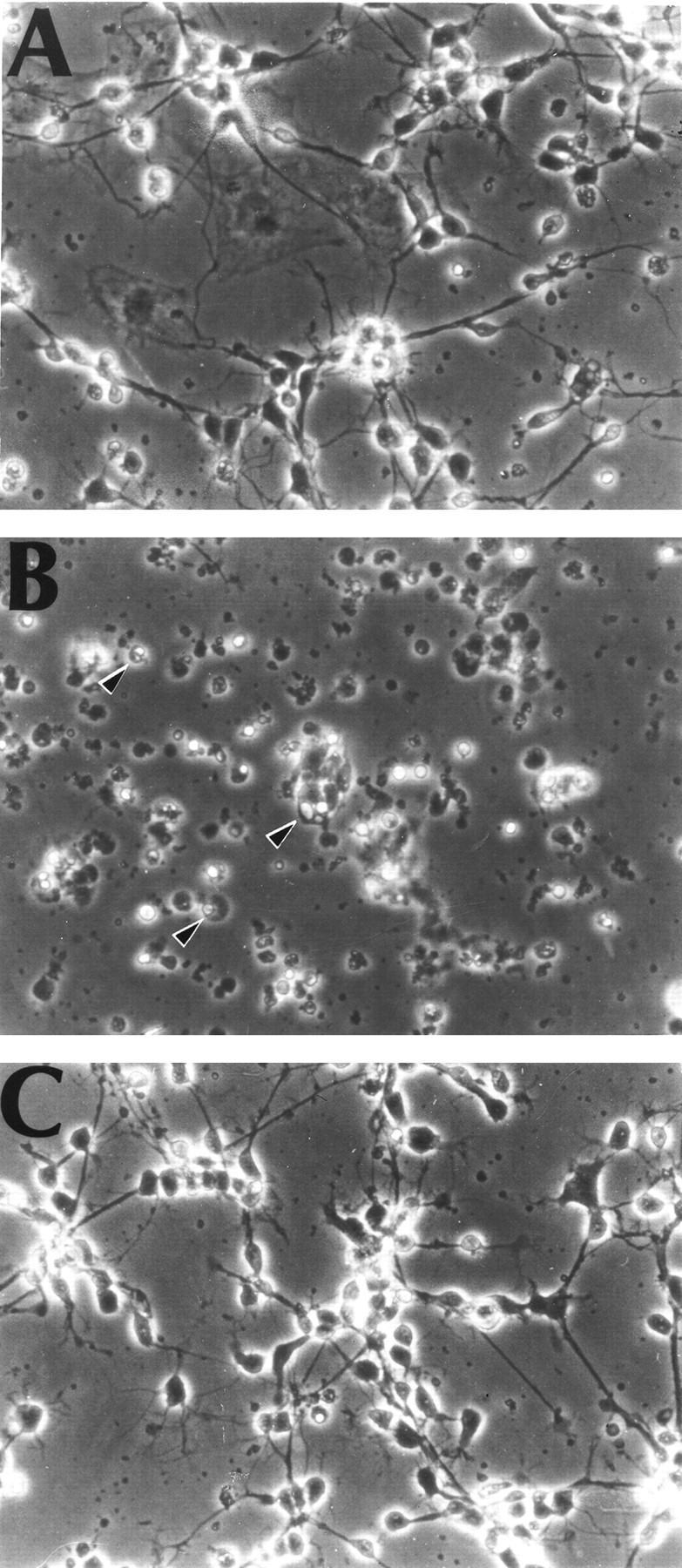
Phase-contrast microscopy of primary cortical neurons cultured for 2 d. A, Control (2 d in vitro). B, Cells 24 hr after 1 mmHCA to induce glutathione depletion. Arrowheads indicate cells with morphological features characteristic of apoptosis: nuclear and cytoplasmic condensation and phase-bright clumping of nuclear chromatin. C, Cells exposed to 1 mm HCA and 100 μm DFO. Magnification, 200×.
DFO acts distal to glutathione depletion in preventing cell death
HCA induces glutathione depletion by competitively inhibiting the uptake of cystine by its plasma membrane transporter (Murphy et al., 1989, 1990). Inhibition of cystine uptake or removal of cystine from the bathing medium leads to depletion of the antioxidant glutathione and death because of oxidative stress. To determine whether iron chelators act to suppress HCA-induced death by preventing cystine deprivation and glutathione depletion, we measured total glutathione levels [reduced glutathione (GSH) + oxidized glutathione (GSSG)] at several time points after HCA addition (Fig.3). Although the rate at which total glutathione levels are depleted by HCA exposure is slowed in the presence of 100 μm DFO, total glutathione levels are depleted by >65% by 10 hr after treatment with HCA alone or HCA and DFO in cortical neurons (Fig. 3). In contrast, there is little difference between the total glutathione levels in control and DFO-treated cultures, and levels remain unchanged throughout the 10 hr period of observation (Fig. 3). These results suggest that iron chelators act to inhibit glutathione depletion–induced apoptosis in cortical neurons distal to glutathione depletion. Consistent with this notion, both DFO and MIM could be added to cultures up to 10 hr after HCA addition (4–7 hr after glutathione depletion) and completely prevent cell death (data not shown).
Fig. 3.

Protection by iron chelators occurs distal to glutathione depletion in the HCA-induced cell death pathway. Total glutathione (nanograms of GSSG + GSH) per microgram of protein was measured in cells exposed for 0, 6, or 10 hr to HCA, DFO, or both. Values represent means ± SEM based on three to five experiments performed in triplicate.
Iron chelators induce several hypoxia response element–binding activities in cortical neuronal cultures
To determine whether iron chelators can induce enhanced HIF-1 DNA binding in cortical neuronal cultures, we performed EMSAs on nuclear extracts from control and HCA-treated cells in the presence or absence of DFO using a radiolabeled W18 oligonucleotide containing an HIF-1 DNA-binding site. Four hours of DFO treatment (100 μm) significantly increased three DNA-binding activities in cortical neurons in the presence or absence of HCA (Fig.4A, the three DNA binding activities are designated a, b, c). The induction of these DNA-binding activities by DFO seemed to be related to iron chelation because it also occurred in the presence of the structurally distinct iron chelator MIM (100 μm) (Fig. 4B). All three DNA-binding activities were inhibited in a concentration-dependent manner by cold W18 oligonucleotide, but only the fastest mobility complex was inhibited by M18 oligonucleotide containing mutations in the HIF-1–binding site (data not shown). These results demonstrate that the two slower mobility complexes (Fig. 4, complexes a, b) bind the HIF-1 site specifically, whereas the fastest-migrating complex (Fig. 4, complex c) represents nonspecific DNA binding. Enhanced binding to the HIF-1 DNA recognition sequence can occur within 2 hr after DFO treatment, permitting sufficient time for putative protective genes to be expressed before the commitment point of cells to die (∼12 hr after HCA treatment).
Fig. 4.
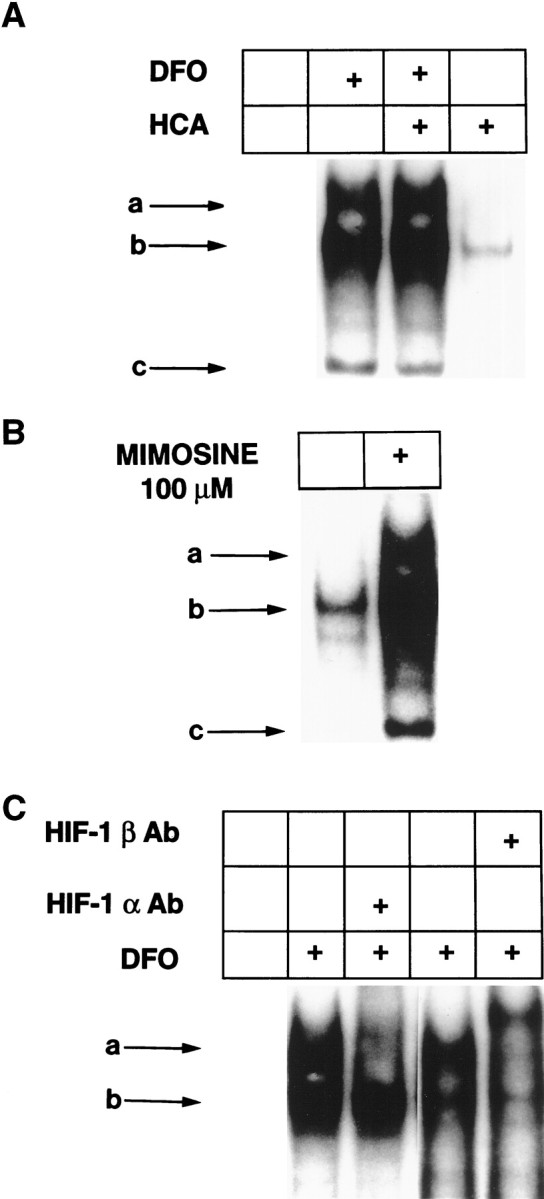
DFO and MIM induce HIF-1 DNA binding in embryonic cortical neurons. A, EMSA performed with 15 μg of nuclear extracts from control (leftlane) and DFO (100 μm)-, DFO (100 μm) + HCA (1 mm)-, and HCA (1 mm)-treated cortical neurons, using a 32P-labeled HIF-1–binding site oligonucleotide.B, EMSA performed as described in A with 15 μg of nuclear extracts from control and MIM (100 μm)-treated cortical neurons. C,Identification of HIF-1 in complexa. Antibodies to HIF-1α or HIF-1β were added after the addition of radiolabeled oligonucleotide to the nuclear-binding reaction as described in Materials and Methods. Ab, Antibody.
Identification of HIF-1 and ATF-1/CREB in nuclear extracts after iron chelator treatment
To determine whether HIF-1 was present in either of the DFO-induced DNA–protein complexes detected by EMSA, we used antibodies specific for the HIF-1α and HIF-1β subunits. Addition of individual subunit-specific antibodies during in vitro DNA binding revealed that the HIF-1α and HIF-1β were present in the slower-migrating complex (Fig. 4C). Addition of HIF-1α antibody disrupted complex formation, whereas HIF-1β antibody resulted in a DNA–protein–antibody complex of retarded mobility (supershift).
In addition to HIF-1, a faster-migrating, specific complex b(Fig. 4) was also induced by DFO. The electrophoretic mobility of complex b corresponds to a constitutive binding activity in non-neuronal cells whose levels of binding are independent of oxygen tension or iron chelation (Wang and Semenza, 1993). Analysis of HeLa cells identified the constitutive binding activity to the HIF-1 recognition sequence as ATF-1 and CREB, which also bind to the cAMP response element (CRE) (Kvietikova et al., 1995). The unexpected induction of complex b in cortical neurons by iron chelators suggested the possibility that it might have an identity distinct from ATF-1 and CREB. To explore this possibility further, we performed a competition experiment using increasing molar excesses of unlabeled CRE oligonucleotide containing the consensus binding site for ATF-1 and CREB. This oligonucleotide has been shown previously not to inhibit DNA binding by HIF-1 (Kvietikova et al., 1995). Unlabeled CRE oligonucleotide competitively inhibited the formation of complex b at lower concentrations of oligonucleotide than were required to inhibit HIF-1 DNA binding (data not shown), suggesting thatcomplex b might contain ATF-1 and CREB. An antibody that recognized both ATF-1 and CREB completely supershifted complex b, whereas a CREB-specific antibody partially supershifted the same complex (Fig. 5A). Antibodies to HIF-1α and HIF-1β had no significant effect on this complex (Fig. 4). These results suggest that, in E17 rat embryonic cortical neurons, ATF-1 represents the primary ATF/CREB family member that binds to the HIF-1 recognition sequence and establish the identity of the two induced HIF-1 recognition sequence–binding activities as HIF-1 and ATF-1/CREB. The differences between nuclear HIF-1 and ATF-1/CREB activity in control and DFO-treated cortical neurons could not be attributed to global differences in nuclear proteins because levels of the transcription factor Oct-1 were similar in these two extracts (Fig. 5B) .
Fig. 5.

DFO induces ATF-1 and CREB DNA binding to the HIF-1 recognition sequence in embryonic cortical neurons.A, EMSAs were performed (see Fig. 4 for description) using 100 μm DFO. Complex bwas identified as ATF-1 and CREB. Antibodies recognizing ATF-1 and CREB or CREB alone were added to the binding reaction as described in Materials and Methods; d corresponds to a DNA–protein–antibody complex. HIF-1 is less apparent in this EMSA because the gel was exposed for shorter periods of time than were other gels (see Fig. 4A–C). fpcorresponds to free unbound radiolabeled probe; ccorresponds to nonspecific DNA-binding activity. B, The effect of DFO on Oct-1 DNA-binding activity is shown. The EMSA of nuclear extracts from control (leftlane) and DFO-treated cells using a radiolabeled oligonucleotide with a Oct-1 sequence is shown.
EMSAs were performed to determine whether 10 μm DFO, the lowest concentration of chelator required to prevent glutathione depletion–induced death (Fig. 1), could induce HIF-1 and/or ATF-1/CREB DNA-binding activity. Induction of both complexes was observed after 4 hr of treatment with 10 μm DFO, although higher levels of induction of both complexes were seen at higher concentrations of drug (data not shown). At the three concentrations of DFO tested, supershift analysis confirmed that the induced complexes were composed of HIF-1 and ATF-1/CREB (Fig. 4C; data not shown). Thus, protection from glutathione depletion–induced death by iron chelators in cortical neuronal cultures can be correlated with induction of HIF-1 and ATF-1/CREB.
Iron chelators induce HIF-1 DNA-binding activity in neurons
The studies described above used primary cultures that are 85–90% neuronal but contain glial cells as well (Murphy et al., 1990). In these cultures, the induction of HIF-1 and ATF-1/CREB may occur in neurons and/or glia. To determine whether HIF-1 and ATF-1/CREB can be induced in cortical neurons by iron chelators, we used a conditionally immortalized cell line (H19-7) derived from hippocampal neuroblasts (Eves et al., 1996). After differentiation, these cells express neuronal markers (neurofilament positive and glial fibrillary acidic protein negative) (Eves et al., 1996). Treatment of undifferentiated or differentiated H19-7 cells for 4 hr with 100 μm DFO resulted in induction of two specific complexes similar to those induced in embryonic cortical neurons (data not shown). Supershift analysis confirmed that the slower-migrating complex was HIF-1 and the faster-migrating complex was ATF-1/CREB (data not shown). To verify that HIF-1 expression can be induced in neurons in response to iron chelator treatment, we performed immunohistochemical staining with antibodies to HIF-1α (data not shown) and HIF-1β (Fig. 6) followed by confocal microscopy. These experiments confirmed that HIF-1 is activated in both neurons (Fig. 6) and glia (data not shown).
Fig. 6.

DFO induces HIF-1 levels in neurons. Immunocytochemical analysis of HIF-1β (a, d) and neurofilament (b, e) in mock- (a–c) or DFO-treated (d–f) mixed cortical neuronal cultures. The images in c andf are derived from superimposing the HIF-1β Ab fluorescence on the NF Ab fluorescence.
Iron chelators induce expression of HIF-regulated genes in cortical neuronal cultures
To determine whether known HIF-1–regulated genes are induced by iron chelators in cortical neuronal cultures, we exposed cultures to increasing concentrations of DFO, and total RNA was isolated for blot hybridization using cDNA probes for LDH A, enolase-1, p21waf1/cip1, or erythropoietin. LDH, enolase, p21, and erythropoietin are known to be upregulated by hypoxia via HIF-1 in non-neuronal cells. A β-actin cDNA probe was used as a control. Quantitation of LDH A mRNA revealed a concentration-dependent induction by DFO in the presence or absence of HCA (Fig.7A,B). The induction of LDH A mRNA by HCA is unlikely to be mediated by HIF-1 because we did not observe increases in HIF-1 DNA binding under these conditions (Fig.4A). In addition to mRNA for LDH A, mRNAs for enolase-1, p21waf1/cip1, and erythropoietin were also induced in a concentration-dependent manner by DFO (Fig. 7C), whereas expression of β-actin (Fig. 7) and BDNF mRNA (data not shown) was unchanged. Because brain erythropoietin is expressed primarily in glial cells (Masuda et al., 1994) and glial cells represent <20% of the cells in our culture (Murphy et al., 1990), it is not surprising that erythropoietin mRNA expression was induced to a lesser degree than was that of LDH A, enolase-1, or p21waf1/cip1.
Fig. 7.
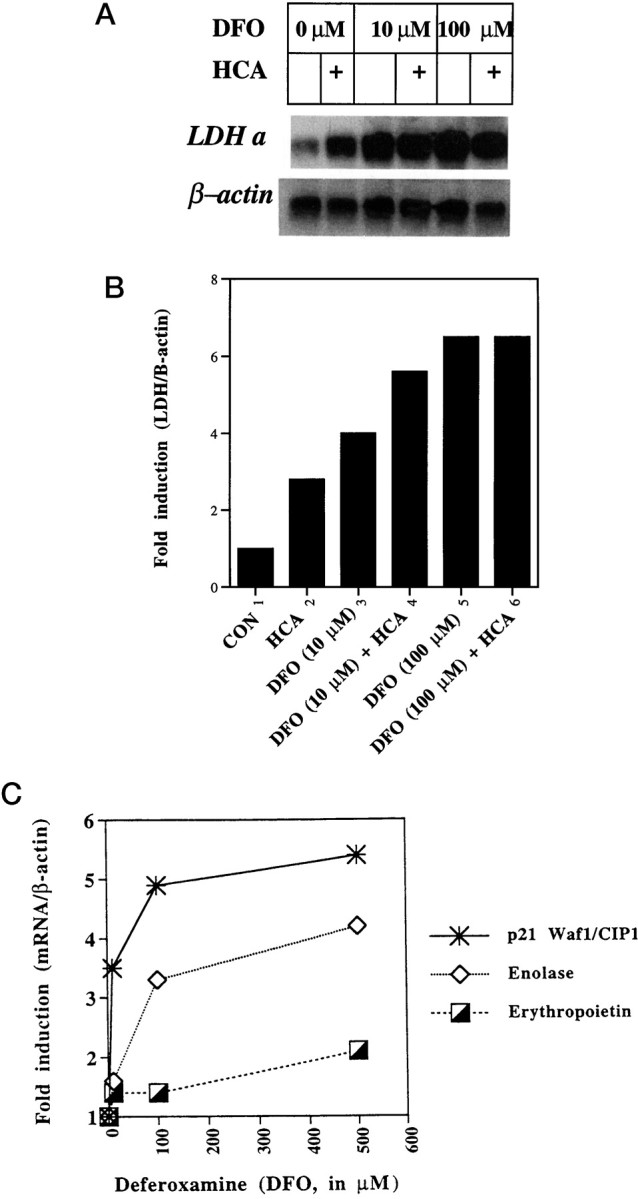
Effect of DFO exposure on gene expression.A, Cortical neuronal cultures were exposed to 0, 10, 100, or 500 μm DFO for 6 hr, and total RNA was isolated for blot hybridization with an LDH A cDNA probe. The blot was stripped and reprobed with a β-actin probe as a control. B,Hybridization signals were quantified by phosphorimager analysis. The graph shows the level of LDH A mRNA normalized to that of β-actin.C, Induction of p21waf1/cip1, enolase-1, and erythropoietin mRNA expression by exposure of cells to DFO (10, 100, and 500 μm) is shown. Each plot is a representative example of three separate experiments.
Iron chelators induce glycolytic enzymes via a transcriptional mechanism
Immunoblot assays revealed that 100 μm DFO increased LDH and aldolase protein levels, whereas levels of α-tubulin protein remained unchanged (data not shown). These results indicate that the increased mRNA levels observed after DFO treatment lead to corresponding increases in protein levels. Spectrophotometric measurements confirmed that induction of LDH and aldolase protein levels by DFO is associated with increases in enzyme activity (data not shown). To verify that the increases in activity we observed are the result of transcriptional mechanisms, we examined the effects of the protein synthesis inhibitor cycloheximide and the RNA synthesis inhibitor actinomycin-D on the induction of glycolytic enzyme activity by iron chelation. We used concentrations of cycloheximide (10 μg/ml) and actinomycin-D (2 μg/ml) that are known to inhibit incorporation of radiolabeled amino acids into protein by >90 and 60%, respectively, in cortical neurons (Ratan et al., 1994b). Cycloheximide and actinomycin-D almost completely inhibited the induction of LDH and aldolase activity by iron chelators (data not shown). These results suggest that iron chelators induce glycolytic enzyme activities via a transcriptional mechanism in cortical neurons, although we cannot exclude the possibility that actinomycin-D and cycloheximide attenuate HIF-1–regulated gene expression by abrogating DFO-induced HIF-1 induction. The antioxidant butylated hydroxyanisole did not induce LDH activity in cortical neurons at concentrations that prevent HCA-induced apoptosis. This result indicates that induction of LDH activity is not required for protection and that some (DFO and MIM) but not all antioxidants induce glycolytic enzyme activity.
Cobalt chloride, an activator of HIF-1, prevents glutathione depletion–induced death in cortical neurons
The results described above support the hypothesis that iron chelators protect neurons, in part, by activating a hypoxia signal transduction pathway leading to activation of HIF-1 and/or ATF-1/CREB and their protective target genes and predict that other known activators of this pathway will prevent glutathione depletion–induced death in cortical neurons. Divalent cations such as cobalt chloride have been shown to induce HIF-1 DNA binding and expression of hypoxia-inducible genes (Wang and Semenza, 1993; Semenza et al., 1994). We therefore examined the effects of cobalt chloride (10–500 μm) on HIF-1 activation and glutathione depletion–induced death in cortical neurons. Concentrations of cobalt chloride >200 μm significantly reduced glutathione depletion–induced death (Fig.8A). Zinc chloride (>200 μm) was not protective, and addition of 2 mm calcium chloride did not reverse protection by 250 μm cobalt chloride (data not shown). These results suggest that the protective effects are independent of cobalt's ability to block calcium influx. Moreover, similar to iron chelators, protective concentrations of cobalt chloride also induced HIF-1α (Fig. 8B) and HIF-1β (data not shown) protein expression and ATF-1/CREB (data not shown) as well as lactate dehydrogenase activity levels (Fig. 8C) in cortical cultures.
Fig. 8.

Effects of cobalt chloride on HIF-1α levels, LDH activity, and glutathione depletion–induced death. A,Cell viability was measured in cortical neuronal cultures (as described in Materials and Methods) 24 hr after HCA treatment and coadministration of vehicle (CON), CoCl2 (250 μm), or DFO (100 μm). *p < 0.05, different from control by ANOVA. B, Whole-cell lysates were prepared from cortical neurons treated for 5 hr with vehicle (control), CaCl2 (250 μm), or DFO (100 μm) treatment and subjected to immunoblot analysis using antibodies against HIF-1α and α-tubulin protein.C, LDH activity from cell lysates was measured (as described in Materials and Methods) 24 hr after control (CON), CoCl2 (250 μm), or DFO (100 μm) treatment. *p < 0.05 by ANOVA.
DISCUSSION
Protection from cell death by iron chelators has been widely attributed to their ability to prevent hydroxyl radical formation from the iron-catalyzed reaction of superoxide and hydrogen peroxide (the Haber–Weiss reaction) (Winterbourn, 1995). However, recent studies suggest additional mechanisms of protection by iron chelators (Beckman et al., 1990; Farinelli and Greene, 1996; Park et al., 1997a,b, 1998). In this manuscript, we present data supporting a novel mechanism by which iron chelators abrogate cell death in neurons. We correlate the protective effects of iron chelation with the induction of the transcription factors HIF-1 and ATF-1/CREB and with the enhanced expression of genes encoding glycolytic enzymes, p21waf1/cip1, and erythropoietin. Each of these genes has HIF-1– and ATF-1/CREB–binding sites in their promoters (Wang and Semenza, 1993; Semenza et al., 1994, 1996). That activation of HIF-1 and ATF-1/CREB may be causally linked to protection by iron chelators is supported by the ability of cobalt chloride (another activator of HIF-1 and ATF-1/CREB and their target genes in neurons) to prevent glutathione depletion–induced death (Fig. 8). This link is further strengthened because cobalt chloride, unlike DFO and MIM, is not known to have direct antioxidant-scavenging capacity.
Several plausible schemes can be envisaged by which the induction of HIF-1– and/or ATF-1/CREB–regulated genes, by exposure to iron chelators (or cobalt chloride), might prevent oxidative stress–induced death. Iron chelators may prevent oxidative stress–induced death by inducing glycolytic enzyme gene expression and consequent aerobic glycolysis. Indeed, recent studies have demonstrated that a shift in energy generation from glucose oxidation in mitochondria to aerobic glycolysis in proliferating thymocytes is associated with resistance to oxidative stress (Brand, 1997). The decrease in ROS resulting from aerobic glycolysis can be attributed to two cooperative effects: (1) the generation of the antioxidant pyruvate and (2) the prevention of excessive reactive oxygen species production by reducing the rate of mitochondrial glucose oxidation (Boveris and Chance, 1973;Dykens, 1997). The ability of chelators to stimulate aerobic glycolysis is supported by studies in muscle cells, in which iron chelators have been shown to increase glucose consumption, lactate production, and [14C]glucose incorporation into glycogen by approximately twofold, and these changes were associated with increases in levels of glucose transporter (GLUT-1) protein and mRNA concentration (Potashnik et al., 1995). Of note, the transcriptional induction of GLUT-1 by iron chelators or hypoxia has been shown to require the HIF-1 recognition site (Ebert et al., 1995), and HIF-1α–deficient embryonic stem cells express reduced levels of mRNAs encoding 13 different glucose transporters and glycolytic enzymes, including aldolase A, enolase-1, glut-1, and LDH A (Iyer et al., 1998). These data indicate that the effects of chelators on glucose metabolism were likely mediated, in part, via HIF-1. Thus, by inducing glycolytic enzyme expression, iron chelators may reduce the ambient free radical burden of neurons by enabling the cell to generate more energy (and antioxidant capacity) glycolytically and minimize the deleterious consequences of mitochondrial glucose oxidation (Tan et al., 1998). Future studies will clarify whether iron chelators enhance glucose use, pyruvate production, and lactate production in embryonic cortical neurons and whether these changes can be associated with reduced free radical production.
Other gene products regulated by HIF-1 that may mediate some of the salutary effects of iron chelators in neurons include erythropoietin, which has been shown to be synthesized in glial cells (Masuda et al., 1994) and to prevent cell death of serum-deprived cholinergic (Konishi et al., 1993) or ischemic hippocampal neurons (Sakanaka et al., 1998); heme oxygenase-1 (Lee et al., 1996, 1997), which has been shown to prevent ischemia-induced neuronal loss in the cortex (Takizawa et al., 1998) and to enhance resistance of neuronal cells to β-amyloid and hydrogen peroxide (Le et al., 1999); and the cyclin-dependent kinase inhibitor p21/Waf1/Cip1 (Carmeliet et al., 1998), whose overexpression in sympathetic neurons protects these cells from death induced by nerve growth factor deprivation (Park et al., 1997a,b, 1998;Freeman, 1998). The observation that DFO induces p21/Waf1/Cip1 in cortical neurons (Fig. 7C) suggests that iron chelators may suppress cell death induced by growth factor deprivation, in part, by upregulating HIF-1 and/or ATF-1/CREB, leading to an induction of p21/Waf1/Cip1.
The hypothesis that iron chelators protect neurons from oxidative stress by inducing HIF-1 and/or ATF-1/CREB predicts that maximal suppression of free radical generation as well as maximal protection by iron chelators requires new mRNA and protein synthesis. Because inhibitors of RNA or protein synthesis can abrogate glutathione depletion–induced death (Ratan et al., 1994b; Esch et al., 1998), we could not use these inhibitors to determine whether transcription is required for the protective effects of iron chelators. Future studies using dominant-negative constructs of HIF-1 (Forsythe et al., 1996;Jiang et al., 1996) and ATF-1 or CREB (Ahn et al., 1998) will address this question directly. Of note, recent studies have documented a proapoptotic effect of HIF-1 in embryonic stem cells or cortical neurons under conditions of hypoxia (Carmeliet et al., 1998; Halterman et al., 1999). In this context, HIF-1 appears to mediate upregulation of p53 and downregulation of Bcl-2 leading to cell death. Although our data, along with previous studies (Iyer et al., 1998), suggest that HIF-1 mediates prosurvival responses, these observations suggest that the effect of HIF-1 induction may depend on the cell type, the developmental stage of the cell, or the death stimulus.
Our results indicate that as in non-neuronal cells, treatment with iron chelators induces HIF-1 activity and expression of HIF-1–regulated genes (Wang and Semenza, 1993: Semenza et al., 1994; Melilo et al., 1997). The precise mechanism by which iron chelators engage the hypoxia signal transduction pathway and activate HIF-1 remains unclear (Gleadle et al., 1995; Huang et al., 1995; Wang et al., 1995a,b). However, our studies point out some important differences in the nature of the DNA-binding complexes induced by chelators in neurons versus non-neuronal cells. Specifically, in neurons, iron chelators induce the binding of both HIF-1– and ATF-1/CREB–binding activities. These observations raise the possibility that HIF-1 and ATF-1/CREB act together at the HIF-1 recognition sequence or, alternatively, at different DNA sites to regulate glycolytic gene expression in neurons. Of note, recent studies suggest that the transcriptional activation of the LDH A gene in response to hypoxia requires a multiprotein complex that includes HIF-1, ATF-1/CREB, and the coactivator p300/CREB-binding protein (Firth et al., 1995; Ebert and Bunn, 1998). It is unlikely that the induction of ATF-1/CREB in neurons but not in non-neuronal cells by iron chelators can be attributed to the postmitotic state of these cells, because the induction of HIF-1 and ATF-1/CREB was also demonstrated in mitotic H19-7 hippocampal neuroblasts in response to iron chelation.
In summary, we demonstrate that structurally distinct iron chelators abrogate glutathione depletion–induced apoptosis in neurons by acting distal to glutathione depletion. We correlate the protective effects of chelators with their ability to induce the transcription factors HIF-1 and ATF-1/CREB and the upregulation of glycolytic enzymes, p21waf1/cip1, and erythropoietin. These observations suggest that the induction of genes known to compensate for hypoxic stress may mediate the neuroprotective effects of iron chelators (and cobalt chloride) and identify novel potential applications for iron chelators (and cobalt chloride) in the treatment of neurodegenerative diseases associated with oxidative stress or disrupted energy metabolism (Beal, 1996; Sharp et al., 1998).
Footnotes
This work was supported by the National Institutes of Health Grants K08 NS01951, R29 NS34943, and R01 NS39170 to R.R. and a William Randolph Hearst Fund award to R.R. We would like to thank Bruce Demple and Ed Monuki for helpful suggestions on this manuscript, Shu-Hua Gu for expert technical assistance, Bert Vogelstein for the p21/waf1/cip1 cDNA, Rosalind Segal for the BDNF cDNA, and Eva Eves for the H19-7 cell line.
Correspondence should be addressed to Dr. Rajiv R. Ratan, Neurology Labs at The Beth Israel Deaconess Medical Center, Harvard Institutes of Medicine, Room 857, 77 Avenue Louis Pasteur, Boston, MA 02115. E-mail:rratan@caregroup.harvard.edu.
REFERENCES
- 1.Ahn S, Olive M, Aggarwal S, Krylov D, Ginty DD, Vinson C. A dominant-negative inhibitor of CREB reveals it is a general mediator of stimulus-dependent transcription of c-fos. Mol Cell Biol. 1998;18:967–977. doi: 10.1128/mcb.18.2.967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Babcock M, de Silva D, Oaks R, Davis-Kaplan S, Jiralerspong S, Montermini L, Pandolfo M, Kaplan J. Regulation of mitochondrial iron accumulation by Yfh1p, a putative homolog of frataxin. Science. 1997;276:1709–1712. doi: 10.1126/science.276.5319.1709. [DOI] [PubMed] [Google Scholar]
- 3.Beal MF. Mitochondria, free radicals and neurodegeneration. Curr Opin Neurobiol. 1996;6:661–666. doi: 10.1016/s0959-4388(96)80100-0. [DOI] [PubMed] [Google Scholar]
- 4.Beckman JS, Beckman TW, Chen J, Marshall PA, Freeman BA. Apparent hydroxyl radical production by peroxynitrite: implications for endothelial injury from nitric oxide and superoxide. Proc Natl Acad Sci USA. 1990;87:1620–1624. doi: 10.1073/pnas.87.4.1620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Boveris A, Chance B. The mitochondrial generation of hydrogen peroxide. General properties and effect of hyperbaric oxygen. Biochem J. 1973;134:707–716. doi: 10.1042/bj1340707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bowern N, Ramshaw IA, Clark IA, Doherty PC. Inhibition of autoimmune neuropathological process by treatment with an iron chelating agent. J Exp Med. 1984;160:1532–1543. doi: 10.1084/jem.160.5.1532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Brand K. Aerobic glycolysis by proliferating cells: protection against oxidative stress at the expense of energy yield. J Bioenerg Biomembr. 1997;29:355–364. doi: 10.1023/a:1022498714522. [DOI] [PubMed] [Google Scholar]
- 8.Carmeliet P, Dor Y, Herbert J-M, Fukumura D, Brusselmans K, Dewerchin M, Neeman M, Bono F, Abramovitch R, Maxwell P, Koch CJ, Ratcliffe P, Moons L, Jain RK, Collen D, Keshet E. Role of HIF-1 α in hypoxia-mediated apoptosis, cell proliferation and tumour angiogenesis. Nature. 1998;394:485–490. doi: 10.1038/28867. [DOI] [PubMed] [Google Scholar]
- 9.Connor JR, Menzies SL. Cellular management of iron in the brain. J Neurol Sci [Suppl] 1995;134:33–44. doi: 10.1016/0022-510x(95)00206-h. [DOI] [PubMed] [Google Scholar]
- 10.Crapper McLachlan DR, Dalton AJ, Kruck TP, Bell MY, Smith WL, Kalow W, Andrews DF. Intramuscular desferrioxamine in patients with Alzheimer's disease. Lancet. 1991;338:324–326. doi: 10.1016/0140-6736(91)92978-b. [DOI] [PubMed] [Google Scholar]
- 11.Dykens JA. Mitochondrial free radical production and the etiology of neurodegenerative disease. In: Beal MF, Bodis-Wollner I, Howell N, editors. Neurodegenerative diseases: mitochondria and free radicals in pathogenesis. Wiley; New York: 1997. pp. 319–334. [Google Scholar]
- 12.Ebert BL, Bunn F. Regulation of transcription by hypoxia requires a multiprotein complex that includes hypoxia-inducible factor 1, an adjacent transcription factor, and p300/CREB binding protein. Mol Cell Biol. 1998;18:4089–4096. doi: 10.1128/mcb.18.7.4089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ebert BL, Firth JD, Ratcliffe PJ. Hypoxia and mitochondrial inhibitors regulate expression of glucose transporter-1 via distinct Cis-acting sequences. J Biol Chem. 1995;270:29083–29089. doi: 10.1074/jbc.270.49.29083. [DOI] [PubMed] [Google Scholar]
- 14.Esch F, Lin K-I, Hills A, Zaman K, Baraban JM, Chatterjee S, Rubin L, Ash DE, Ratan RR. Purification of a multipotent antideath activity from bovine liver and its identification as arginase: nitric oxide-independent inhibition of neuronal apoptosis. J Neurosci. 1998;18:4083–4095. doi: 10.1523/JNEUROSCI.18-11-04083.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Eves EM, Boise LH, Thompson CB, Wagner AJ, Hay N, Rosner MR. Apoptosis induced by differentiation or serum deprivation in an immortalized central nervous system neuronal cell line. J Neurochem. 1996;67:1908–1920. doi: 10.1046/j.1471-4159.1996.67051908.x. [DOI] [PubMed] [Google Scholar]
- 16.Farinelli SE, Greene LA. Cell cycle blockers mimosine, ciclopirox, and deferoxamine prevent the death of PC12 cells and post-mitotic sympathetic neurons after removal of trophic support. J Neurosci. 1996;16:1150–1162. doi: 10.1523/JNEUROSCI.16-03-01150.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Feldman ST, Schonthal A. Negative regulation of histone H1 kinase expression by mimosine, a plant amino acid. Cancer Res. 1994;54:494–498. [PubMed] [Google Scholar]
- 18.Firth JD, Ebert BL, Ratcliffe PJ. Hypoxic regulation of lactate dehydrogenase A: interaction between hypoxia inducible factor 1 and cAMP response elements. J Biol Chem. 1995;270:21021–21027. doi: 10.1074/jbc.270.36.21021. [DOI] [PubMed] [Google Scholar]
- 19.Forsythe JA, Jiang B-H, Iyer NV, Agani F, Leung SW, Koos RD, Semenza GL. Activation of vascular endothelial growth factor gene transcription by hypoxia-inducible factor 1. Mol Cell Biol. 1996;16:4604–4613. doi: 10.1128/mcb.16.9.4604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Freeman RS. The cell cycle and neuronal death. In: Koliatsos VE, Ratan RR, editors. Cell death and diseases of the nervous system. Humana; Totawa, NJ: 1998. pp. 103–119. [Google Scholar]
- 21.Gassen M, Youdim MB. The potential role of iron chelators in the treatment of Parkinson's disease and related neurological disorders. Pharmacol Toxicol. 1997;80:159–166. doi: 10.1111/j.1600-0773.1997.tb00390.x. [DOI] [PubMed] [Google Scholar]
- 22.Gleadle JM, Ebert BL, Ratcliffe PJ. Diphenylene iodonium inhibits the induction of erythropoietin and other mammalian genes by hypoxia. Implications for mechanisms of oxygen sensing. Eur J Biochem. 1995;234:92–99. doi: 10.1111/j.1432-1033.1995.092_c.x. [DOI] [PubMed] [Google Scholar]
- 23.Halterman MW, Miller CC, Federoff HJ. Hypoxia-inducible factor-1 a mediates hypoxia-induced delayed neuronal death that involves p53. J Neurosci. 1999;19:6818–6824. doi: 10.1523/JNEUROSCI.19-16-06818.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hartley A, Cooper JM, Schapira AHV. Iron induced oxidative stress and mitochondrial dysfunction: relevance to Parkinson's disease. Brain Res. 1993;627:349–353. doi: 10.1016/0006-8993(93)90341-j. [DOI] [PubMed] [Google Scholar]
- 25.Hirsch EC, Faucheux BA. Iron metabolism and Parkinson's disease. Mov Disord. 1998;13[Suppl 1]:39–45. [PubMed] [Google Scholar]
- 26.Huang LE, Arany Z, Livingston DM, Bunn HF. Activation of hypoxia-inducible transcription factor depends primarily on redox-sensitive stabilization of its alpha subunit. J Biol Chem. 1995;271:32253–32259. doi: 10.1074/jbc.271.50.32253. [DOI] [PubMed] [Google Scholar]
- 27.Hurn PD, Koehler RC, Blizzard KK, Traystman RJ. Deferoxamine reduces early metabolic failure associated with severe cerebral ischemic acidosis in dogs. Stroke. 1995;26:688–694. doi: 10.1161/01.str.26.4.688. [DOI] [PubMed] [Google Scholar]
- 28.Iyer NV, Kotch LE, Agani F, Leung SW, Laughner E, Wenger RH, Gassman M, Gearhart JD, Lawler AM, Yu AY, Semenza GL. Cellular and developmental control of O2 homeostasis by hypoxia-inducible factor 1 alpha. Genes Dev. 1998;12:149–162. doi: 10.1101/gad.12.2.149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Jiang BH, Rue E, Wang GL, Roe R, Semenza GL. Dimerization, DNA binding, and tansactivation properties of hypoxia-inducible factor 1. J Biol Chem. 1996;271:17771–17778. doi: 10.1074/jbc.271.30.17771. [DOI] [PubMed] [Google Scholar]
- 30.Jiang BH, Agani F, Passantini A, Semenza GL. V-SRC induces expression of hypoxia-inducible factor 1 (HIF-1) and transcription of genes encoding vascular endothelial growth factor and enolase-1: involvement of HIF-1 in tumor progression. Cancer Res. 1997;57:5328–5335. [PubMed] [Google Scholar]
- 31.Juul SE, Anderson DK, Li Y, Christensen RD. Erythropoeitin and erythropoietin receptor in the developing human central nervous system. Pediatr Res. 1998;43:40–49. doi: 10.1203/00006450-199801000-00007. [DOI] [PubMed] [Google Scholar]
- 32.Koh J, Choi DW. Quantitative determinants of glutamate mediated cortical neuronal injury in cell culture by lactate dehydrogenase efflux assay. J Neurosci Methods. 1987;20:83–90. doi: 10.1016/0165-0270(87)90041-0. [DOI] [PubMed] [Google Scholar]
- 33.Konishi Y, Chui DH, Hirose H, Kunishita T, Tabira T. Trophic effect of erythropoietin and other hematopoietic factors on the cholinergic neurons in vitro and in vivo. Brain Res. 1993;609:29–35. doi: 10.1016/0006-8993(93)90850-m. [DOI] [PubMed] [Google Scholar]
- 34.Kvietikova I, Wenger RH, Marti HH, Gassman M. The transcription factors ATF-1 and CREB bind constitutively to the hypoxia-inducible factor-1 (HIF-1) DNA recognition site. Nucleic Acids Res. 1995;23:4542–4550. doi: 10.1093/nar/23.22.4542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Le WD, Xie WJ, Appel SH. Protective role of heme oxygenase-1 in oxidative stress-induced neuronal injury. J Neurosci Res. 1999;56:652–658. doi: 10.1002/(SICI)1097-4547(19990615)56:6<652::AID-JNR11>3.0.CO;2-5. [DOI] [PubMed] [Google Scholar]
- 36.Lee PJ, Alam J, Wiegand GW, Choi AM. Overexpression of heme oxygenase-1 in human pulmonary epithelial cells results in cell growth arrest and increased resistance to hyperoxia. Proc Natl Acad Sci USA. 1996;93:10393–10398. doi: 10.1073/pnas.93.19.10393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lee PJ, Jiang BH, Chin BY, Iyer NV, Alam J, Semenza GL, Choi AM. Hypoxia-inducible factor-1 mediates transcriptional activation of the heme oxygenase gene in response to hypoxia. J Biol Chem. 1997;272:5375–5381. [PubMed] [Google Scholar]
- 38.Levine SM. Iron deposits in multiple sclerosis and Alzheimer's disease brains. Brain Res. 1997;760:298–303. doi: 10.1016/s0006-8993(97)00470-8. [DOI] [PubMed] [Google Scholar]
- 39.Levy AP, Levy NS, Wegner S, Goldberg MA. Transcriptional regulation of the rat vascular endothelial factor gene by hypoxia. J Biol Chem. 1995;270:13333–13340. doi: 10.1074/jbc.270.22.13333. [DOI] [PubMed] [Google Scholar]
- 40.Lin K-I, Lee S-H, Narayanan R, Barban JM, Hardwick JM, Ratan RR. Thiol agents and Bcl-2 identify an alphavirus-induced pathway that requires activation of the transcription factor, NF-kappa B. J Cell Biol. 1995;131:1149–1161. doi: 10.1083/jcb.131.5.1149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lipscomb DC, Gorman LG, Traystman RJ. Low molecular weight iron in cerebral ischemic acidosis in vivo. Stroke. 1998;29:487–492. doi: 10.1161/01.str.29.2.487. [DOI] [PubMed] [Google Scholar]
- 42.Lovell MA, Robertson JD, Teesdale WJ, Campbell JL, Markesbery WR. Copper, iron, and zinc in Alzheimer's disease senile plaques. J Neurol Sci. 1998;158:47–52. doi: 10.1016/s0022-510x(98)00092-6. [DOI] [PubMed] [Google Scholar]
- 43.Marquis JC, Demple B. Complex genetic response of human cells to sublethal levels of pure nitric oxide. Cancer Res. 1998;58:3435–3440. [PubMed] [Google Scholar]
- 44.Masonpierre PC, Belluscio L, Friedman B, Alderson RF, Furth ME, Lindsay RM, Yancopoulos GD. NT-3, BDNF, and NGF in the developing rat nervous system: parallel as well as reciprocal patterns of expression. Neuron. 1990;5:501–509. doi: 10.1016/0896-6273(90)90089-x. [DOI] [PubMed] [Google Scholar]
- 45.Masuda S, Okano M, Yamagishi K, Nagao M, Ueda M, Sasaki R. A novel site of erythropoietin production. Oxygen-dependent production in cultured rat astrocytes. J Biol Chem. 1994;269:19488–19493. [PubMed] [Google Scholar]
- 46.Melilo G, Taylor LS, Brooks A, Musso T, Cox GW, Varesio L. Functional requirement of the hypoxia-responsive element in the activation of inducible nitric oxide synthase promoter by the iron chelator, desferrioxamine. J Biol Chem. 1997;272:12236–12243. doi: 10.1074/jbc.272.18.12236. [DOI] [PubMed] [Google Scholar]
- 47.Michel PP, Vyas S, Agid Y. Toxic effects of iron for cultured mesencephalic dopaminergic neurons derived from rat embryos. J Neurochem. 1992;59:118–127. doi: 10.1111/j.1471-4159.1992.tb08882.x. [DOI] [PubMed] [Google Scholar]
- 48.Murphy TH, Miyamoto M, Sastre A, Schnaar RL, Coyle JT. Glutamate toxicity in a neuronal cell line involves inhibition of cystine transport leading to oxidative stress. Neuron. 1989;2:1547–1558. doi: 10.1016/0896-6273(89)90043-3. [DOI] [PubMed] [Google Scholar]
- 49.Murphy TH, Schnaar RL, Coyle JT. Immature cortical neurons are uniquely sensitive to glutamate toxicity by inhibition of cystine uptake. FASEB J. 1990;4:1624–1633. [PubMed] [Google Scholar]
- 50.Olanow CW. Attempts to obtain neuroprotection in Parkinson's disease. Neurology. 1997;49[Suppl 1]:S26–S33. doi: 10.1212/wnl.49.1_suppl_1.s26. [DOI] [PubMed] [Google Scholar]
- 51.Park DS, Morris EJ, Greene LA, Geller HM. G1/S cell cycle blockers and inhibitors of cyclin-dependent kinases suppress camptothecin-induced neuronal apoptosis. J Neurosci. 1997a;17:1256–1270. doi: 10.1523/JNEUROSCI.17-04-01256.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Park DS, Levine B, Ferrari G, Greene LA. Cyclin-dependent kinase inhibitors and dominant negative cyclin-dependent kinase 4 and 6 promote survival of NGF-deprived sympathetic neurons. J Neurosci. 1997b;17:8975–8983. doi: 10.1523/JNEUROSCI.17-23-08975.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Park DS, Morris EJ, Padmanabhan J, Shelanski ML, Geller HM, Greene LA. Cyclin-dependent kinases participate in death of neurons evoked by DNA-damaging agents. J Cell Biol. 1998;143:457–467. doi: 10.1083/jcb.143.2.457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Potashnik R, Kozlovsky N, Ben-Ezra S, Rudich A, Bashan N. Regulation of glucose transport and GLUT-1 expression by iron chelators in muscle cells in culture. Am J Physiol. 1995;269:E1052–E1058. doi: 10.1152/ajpendo.1995.269.6.E1052. [DOI] [PubMed] [Google Scholar]
- 55.Ratan RR, Murphy TH, Baraban JM. Oxidative stress induces apoptosis in embryonic cortical neurons. J Neurochem. 1994a;62:376–379. doi: 10.1046/j.1471-4159.1994.62010376.x. [DOI] [PubMed] [Google Scholar]
- 56.Ratan RR, Murphy TH, Baraban JM. Macromolecular synthesis inhibitors prevent oxidative stress-induced apoptosis in embryonic cortical neurons by shunting cysteine from protein synthesis to glutathione. J Neurosci. 1994b;14:4385–4392. doi: 10.1523/JNEUROSCI.14-07-04385.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Rotig A, de Lonlay P, Chretien D, Foury F, Koenig M, Sidi D, Munnich A, Rustin P. Aconitase and mitochondrial iron-sulfur protein deficiency in Friedreich ataxia. Nat Genet. 1997;17:215–217. doi: 10.1038/ng1097-215. [DOI] [PubMed] [Google Scholar]
- 58.Sakanaka M, Wen TC, Matsuda S, Masuda S, Morishita E, Nagao M, Sasaki R. In vivo evidence that erythropoietin protects neurons from ischemic damage. Proc Natl Acad Sci USA. 1998;95:4635–4640. doi: 10.1073/pnas.95.8.4635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Semenza GL. Hypoxia-inducible factor 1 and the molecular physiology of oxygen homeostasis. J Lab Clin Med. 1998;131:207–214. doi: 10.1016/s0022-2143(98)90091-9. [DOI] [PubMed] [Google Scholar]
- 60.Semenza GL, Roth PH, Fang H-M, Wang GL. Transcriptional regulation of genes encoding glycolytic enzymes by hypoxia-inducible factor 1. J Biol Chem. 1994;269:23757–23763. [PubMed] [Google Scholar]
- 61.Semenza GL, Jiang BH, Leung SW, Passantino R, Concordet JP, Maire P, Giallongo A. Hypoxia response elements in the aldolase A, enolase 1, and lactate dehydrogenase A gene promoters contain essential sites for hypoxia-inducible factor-1. J Biol Chem. 1996;271:32529–32537. doi: 10.1074/jbc.271.51.32529. [DOI] [PubMed] [Google Scholar]
- 62.Sharp FR, Honkamniemi J, Massa SM. Molecular approaches to the therapy of stroke. In: Martin JB, editor. Molecular neurology. Scientific American; New York: 1998. pp. 115–133. [Google Scholar]
- 63.Smith MA, Harris PL, Sayre LM, Perrt G. Iron accumulation in Alzheimer's disease is a source of redox-generated free radicals. Proc Natl Acad Sci USA. 1997;94:9866–9868. doi: 10.1073/pnas.94.18.9866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Smith PK, Krohn RI, Hemronson GT, Mallin AK, Gortner FH, Prosenano MD, Fujimoto EK, Goche NM, Olson BJ, Klenk DC. Measurement of protein using bicinchoninic acid. Anal Biochem. 1985;150:76–85. doi: 10.1016/0003-2697(85)90442-7. [DOI] [PubMed] [Google Scholar]
- 65.Takizawa S, Hirabayashi H, Matsushima K, Tokuoka K, Shinohara Y. Induction of heme oxygenase protein protects neurons in cortex and striatum, but not in hippocampus, against transient forebrain ischemia. J Cereb Blood Flow Metab. 1998;18:559–569. doi: 10.1097/00004647-199805000-00011. [DOI] [PubMed] [Google Scholar]
- 66.Tan S, Sagara Y, Liu Y, Maher P, Schubert D. The regulation of reactive oxygen species production during programmed cell death. J Cell Biol. 1998;141:1423–1432. doi: 10.1083/jcb.141.6.1423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Terada N, Or R, Szepesi A, Lucas JJ, Gelfand EW. Definition of the roles for iron and essential fatty acids in cell cycle progression of normal human T lymphocytes. Exp Cell Res. 1993;204:260–267. doi: 10.1006/excr.1993.1032. [DOI] [PubMed] [Google Scholar]
- 68.Tietze F. Enzymatic method for quantitative determination of nanogram amounts of total and oxidized glutathione: applications to mammalian blood and other tissues. Anal Biochem. 1969;27:502–522. doi: 10.1016/0003-2697(69)90064-5. [DOI] [PubMed] [Google Scholar]
- 69.Troy CM, Derrossi D, Prochiantz A, Greene LA, Shelanski ML. Downregulation of Cu/Zn superoxide dismutase leads to cell death via the nitric oxide-peroxynitrite pathway. J Neurosci. 1996;16:253–261. doi: 10.1523/JNEUROSCI.16-01-00253.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Wang GL, Semenza GL. Desferrioxamine induces erythropoietin gene expression and hypoxia-inducible factor-1 DNA-binding activity: implications for models of hypoxia signal transduction. Blood. 1993;82:3610–3615. [PubMed] [Google Scholar]
- 71.Wang GL, Semenza GL. Purification and characterization of hypoxia-inducible factor 1. J Biol Chem. 1995;270:1230–1237. doi: 10.1074/jbc.270.3.1230. [DOI] [PubMed] [Google Scholar]
- 72.Wang GL, Semenza GL. Oxygen sensing and response to hypoxia by mammalian cells. Redox Report. 1996;2:89–96. doi: 10.1080/13510002.1996.11747034. [DOI] [PubMed] [Google Scholar]
- 73.Wang GL, Jiang B-H, Semenza GL. Effect of altered redox states on expression and DNA binding of hypoxia-inducible factor 1. Biochem Biophys Res Commun. 1995a;212:550–556. doi: 10.1006/bbrc.1995.2005. [DOI] [PubMed] [Google Scholar]
- 74.Wang GL, Jiang B-H, Rue EA, Semenza GL. Hypoxia-inducible factor 1 is a basic-helix-loop-helix-PAS heterodimer regulated by cellular oxygen tension. Proc Natl Acad Sci USA. 1995b;92:5510–5514. doi: 10.1073/pnas.92.12.5510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Winterbourn CC. Toxicity of iron and hydrogen peroxide: the Fenton reaction. Toxicol Lett. 1995;82–83:969–974. doi: 10.1016/0378-4274(95)03532-x. [DOI] [PubMed] [Google Scholar]
- 76.Yonezawa M, Back SA, Gan X, Rosenberg PA, Volpe JJ. Cystine deprivation induces oligodendroglial death: rescue by free radical scavengers and by a diffusible glial factor. J Neurochem. 1996;67:566–573. doi: 10.1046/j.1471-4159.1996.67020566.x. [DOI] [PubMed] [Google Scholar]