

Abstract
Methamphetamine neurotoxicity has been demonstrated in rodents and nonhuman primates. These neurotoxic effects may be associated with mechanisms involved in oxidative stress and the activation of immediate early genes (IEG). It is not clear, however, whether these IEG responses are involved in a methamphetamine-induced toxic cascade or in protective mechanisms against the deleterious effects of the drug. As a first step toward clarifying this issue further, the present study was thus undertaken to assess the toxic effects of methamphetamine in heterozygous and homozygous c-fos knock-out as well as wild-type mice. Administration of methamphetamine caused significant reduction in [125I]RTI-121–labeled dopamine uptake sites, dopamine transporter protein, and tyrosine hydroxylase–like immunohistochemistry in the striata of wild-type mice. These decreases were significantly exacerbated in heterozygous and homozygous c-fos knock-out mice, with the homozygous showing greater loss of striatal dopaminergic markers. Moreover, in comparison with wild-type animals, both genotypes of c-fos knock-out mice showed more DNA fragmentation, measured by the number of terminal deoxynucleotidyl transferase–mediated dUTP nick-end–labeled nondopaminergic cells in their cortices and striata. In contrast, wild-type mice treated with methamphetamine demonstrated a greater number of glial fibrillary acidic protein–positive cells than did c-fos knock-out mice. These data suggest that c-fos induction in response to toxic doses of methamphetamine might be involved in protective mechanisms against this drug-induced neurotoxicity.
Keywords: methamphetamine, neurotoxicity, c-fos mutant, glial fibrillary acidic protein, DNA fragmentation, cell death, apoptosis
Methamphetamine (METH) is an illicit drug that is abused throughout the world (Miller, 1991; Greberman and Wada, 1994; Shaw, 1999). The acute administration of this agent can cause neuropsychiatric complications including psychosis, coma, and death (Lan et al., 1998). Abrupt cessation of use can cause withdrawal symptoms akin to a suicidal depressive state (Murray, 1998). Although the acute effects of the drug might be caused by increases in the levels of synaptic dopamine (DA) (Stephans and Yamamoto, 1995), the long-term effects might be secondary to persistent perturbations in monoaminergic systems (Cadet and Brannock, 1998). For example, the administration of METH can cause marked depletion of dopaminergic or serotonergic markers in rodents (Ricaurte et al., 1982; Matsuda et al., 1988; O'Callaghan and Miller, 1994; Hirata and Cadet, 1997a,b;Fukumura et al., 1998), nonhuman primates (Villemagne et al., 1998), and human METH users (Wilson et al., 1996; McCann et al., 1998).
Several laboratories are actively seeking to decipher the cellular and molecular mechanisms of METH-induced neurotoxicity. A consensus has been built that indicates that superoxide radicals, hydrogen peroxide, and hydroxyl radicals (De Vito and Wagner, 1989; Cadet et al., 1994;Giovanni et al., 1995; Hirata et al., 1996, 1998b; Fumagalli et al., 1998, 1999; Itzhak et al., 1998a; Jayanthi et al., 1998; Yamamoto and Zhu, 1998) as well as nitric oxide (Itzhak and Ali, 1996; Sheng et al., 1996c; Itzhak et al., 1998b) might play major roles in METH toxicity. However, because the formation of reactive species is associated with complex pathophysiological changes, much remains to be done to understand fully the mechanisms involved in the pathobiological substrates induced by this drug. This laboratory has conducted studies aimed at elucidating possible roles of immediate early genes (IEGs) and their relationships to METH-mediated free radical production (Sheng et al., 1996a,b; Asanuma and Cadet, 1998; Hirata et al., 1998a). These investigations have documented an association between METH-induced superoxide radical production and IEG activation because transgenic mice that overexpress the antioxidant enzyme CuZn superoxide dismutase (SOD) showed marked attenuation of these IEG responses, including c-fos– (Hirata et al., 1998a) and AP-1–binding activity (Sheng et al., 1996a,b). On the basis of these results, however, it is not clear whether METH-associated c-fos induction results in either protoxic or protective events. A protective role for c-fos is supported by the demonstration that cells, which do not express c-fos, are more sensitive to DNA-damaging agents (Haas and Kaina, 1995; Kaina et al., 1997). c-fos has also been shown to be important for activating some neurotrophic and/or neuroprotective factors (Herdegen and Leah, 1998).
The existence of a null mutation of c-fos in mice (Johnson et al., 1992; Wang et al., 1992) offers a system in which the role of c-fos in drug-induced neurodegeneration can be investigated. We, thus, used these mutant mice to assess METH-induced deleterious effects in the brain. We now report that the neurotoxicity of METH on both dopaminergic and nondopaminergic systems is exacerbated in heterozygous (+/−) and homozygous (−/−) c-fos mutant mice.
MATERIALS AND METHODS
Animals, drug treatment, and temperature measurement.Homozygous (Homo; −/−) and heterozygous (Het; +/−) c-fos knock-out as well as wild-type (WT; +/+) mice obtained from The Jackson Laboratory (Bar Harbor, ME) were used in these experiments. The generation and derivation of these mice have been described previously in detail (Johnson et al., 1992). All animal use procedures were according to the National Institutes of Health Guide for the Care and Use of Laboratory Animals and were approved by the local animal care committee.
Homo, Het, and WT mice were given four injections of 10 mg/kg METH or saline at 2 hr intervals. Similar protocols of METH administration have been used extensively by us (Hirata et al., 1996) and others (Pu and Vorhees, 1995; Fumagalli et al., 1998). Core body temperature was able to be recorded in WT and +/− c-fos mice at 30 min intervals by the use of a mouse rectal probe (YSI, Yellow Springs, OH), whereas −/− c-fos mice were very susceptible to rectal injury and did not survive this procedure in conjunction with METH administration. The mice were killed at various time points after drug treatment. Brain tissues were processed for the various assays as described below.
Autoradiographic assays. Binding assays for DA transporters (DAT) were performed essentially as described previously by this laboratory with [125I]RTI-121 (specific activity, 2200 Ci/mmol) and 10 μmGBR-12909 to determine nonspecific binding (Hirata et al., 1996;Asanuma et al., 1998). [125I]RTI-121 binding in striatum was quantified using a Macintosh computer-based image analysis system (Image, NIH) with standard curves generated from125I microscales.
Tyrosine hydroxylase and glial fibrillary acidic protein immunohistochemistry. The animals were perfused transcardially, under deep pentobarbital anesthesia, first with saline followed by 40 ml of 4% paraformaldehyde in 0.1 m phosphate buffer at 4°C. The brains were removed, post-fixed overnight in 4% paraformaldehyde, and then allowed to equilibrate in 30% sucrose for 24 hr. Thirty micrometer coronal sections were then cut in a cryostat (Bright Instrument Company, Huntindon, United Kingdom). Free-floating sections containing striatal areas were used for tyrosine hydoxylase (TH) and glial fibrillary acidic protein (GFAP) immunostaining. Briefly, sections were exposed to 1% hydrogen peroxide for 20 min and then incubated for 30 min in 1% bovine serum albumin and 0.3% Triton X-100, followed by incubation with either the TH (Calbiochem, La Jolla, CA; polyclonal; 1:5000) or GFAP (Novo Laboratories; monoclonal; 1:100) primary antibody. Subsequent processing with biotinylated secondary antibody and ABC complex was performed according to the manufacturer's procedures described in the ABC kit (Vector Laboratories, Burlingame, CA). The free-floating sections were then reacted with 3,3′-diaminobenzidine (DAB) and hydrogen peroxide to visualize the peroxidase reaction. At the end of the reaction, the sections were mounted on microscope slides for further visualization and analysis.
To measure the relative intensity of TH-immunoreactivity (TH-IR), we collected striatal image from each photomicrograph using Adobe Photoshop. Image analysis of optical density (arbitrary unit) used the computer-based program NIH Image. A similar approach to the assessment of TH-IR fiber density has been validated previously using 6-hydroxydopamine–induced destruction of nigrostriatal DA pathways in the rat (Burke et al., 1990). In that report, it was shown that there was a significant correlation between circling behavior and the density of TH fibers measured by image analysis. Subsequently, that approach was used to demonstrate increased TH-IR fiber density after perinatal asphyxia (Burke et al., 1991). GFAP-positive astrocytes were counted in randomly chosen striatal subfields (600 × 900 μm). TH and GFAP immunohistochemistry data were obtained from six sections per animal (five to eight animals per group). Statistical analyses are described below.
Western blot. Analysis of DAT protein concentration in the striata of c-fos knock-out and WT mice was performed by Western blot (Fumagalli et al., 1999). Briefly, dissected striata were homogenized in a buffer containing 320 mm sucrose, 5 mmHEPES, 1 μg/ml leupeptin, 1 μg/ml aprotinin, and 1 μg/ml pepstatin. Homogenates were centrifuged at 2000 × gfor 5 min, and the supernatant fraction was subsequently centrifuged at 30,000 × g for 30 min. The resulting pellet was resuspended in the sample buffer (62.5 mmTris-HCl, 20% glycerol, 2% SDS, 0.01% bromphenol blue, and 1 mm dithiothreitol) and subjected to SDS-PAGE (10%). Proteins were electrophoretically transferred to a polyvinylidene difluoride (PVDF) membrane, and nonspecific sites were blocked in 5% nonfat dry milk in Tris-buffered saline (135 mm NaCl, 2.5 mm KCl, 50 mm Tris, and 0.1% Tween 20, pH 7.4). Membranes were then incubated in the presence of a polyclonal antibody to the N terminus of DAT (DAT-Nt) (Chemicon, Temecula, CA; 1:500) in Tris-buffered saline. DAT antibody binding and chemiluminescence enhancement were performed using the ECL Western blotting analysis system (Pharmacia, Piscataway, NJ). Densitometric analysis was performed and calibrated to coblotted dilutional standards of control striatum. Blots were then stripped for 20 min at 80°C (8m urea, 100 mm2-mercaptoethanol, and 62.5 mm Tris, pH 6.8) and reprobed with an antibody to α-tubulin (Sigma, St. Louis, MO; 1:2000).
Terminal deoxynucleotidyl transferase–mediated dUTP nick-end labeling histochemistry. A standard terminal deoxynucleotidyl transferase-mediated dUTP nick-end labeling (TUNEL) procedure for frozen tissue sections was performed according to the manufacturer's manual (Boehringer Mannheim, Indianapolis, IN). Briefly, slide-mounted sections were rinsed in 0.3% hydrogen peroxide–methanol to block endogenous peroxidase. They were then rinsed in 0.1% Triton X-100 in 0.1% sodium citrate for 2 min on ice to increase permeabilization of the cells. To label damaged nuclei, 50 μl of the TUNEL reaction mixture was added onto each sample in a humidified chamber followed by a 60 min incubation at 37°C. The peroxidase reaction was visualized with DAB-substrate solution. Procedures for negative controls were performed as described in the manufacture's manual and consisted of not adding the label solution (terminal deoxynucleotidyl transferase) to the TUNEL reaction mixture. No TUNEL-positive cells were observed in the negative controls. TUNEL-positive cells were counted in the frontal cortex and striatum using a Zeiss microscope (600 × 900 μm).
Statistical analyses. All data are presented as means ± SEM. The data were analyzed by ANOVA followed by Fisher's protected least significant difference test using the statistical program Statview 4.02. Criteria for significance were set at the 0.05 level.
RESULTS
[125I]RTI-121–binding autoradiography and Western blotting for DAT
To test the long-term toxic effects of METH on DA terminals in these mice, we performed DAT binding on coronal sections using receptor autoradiographic technique and Western blot analysis of DAT protein.
Representative photomicrographs of [125I]RTI-121–labeled DAT in the striata are shown in Figure 1. The intensity of binding is similar in saline-injected animals from the three genotypes (Fig. 1A–C). As expected from our previous results (Hirata and Cadet, 1997b; Tsao et al., 1998), METH injections caused marked decreases in the intensity of labeling in the striata of mice killed 1 week after drug treatment. The METH-induced decreases were more apparent in the c-fos knock-out mice (compare Fig.1F with D,E). Figure 1Gshows the quantitative data obtained from the image analysis. There was no significant difference in [125I]RTI-121–labeled DAT among animals of the three genotypes treated with saline. Injections of METH caused marked decreases in striatal [125I]RTI-121–labeled DAT in the three genotypes, with the Het and Homo c-fos knock-out mice showing somewhat greater loss of binding. For example, METH caused ∼72.7, 79.1, and 87.0% loss of DAT binding in WT, Het, and Homo c-fos mice, respectively.
Fig. 1.

A–F, Effects of METH on [125I]RTI-121–labeled striatal DA uptake sites in WT (A, D), heterozygous (B, E), and homozygous (C, F) c-fos mutant mice. Animals received saline (A–C) or METH (D–F) as described in Materials and Methods. They were killed 1 week after drug treatment. [125I]RTI-121–binding density is similar in the three saline-treated genotypes (A–C). METH administration caused marked reduction of DA uptake sites (D–F), with the greatest decreases occurring in c-fos −/− mice (F). G, The results of the statistical analyses of the quantitative data obtained from the image analyses. Values represent means ± SEM of five to eight animals per group. Key to statistics: *p < 0.0001 in comparison with saline-treated mice of similar genotypes; !p < 0.05, and !!p < 0.0001 in comparison with METH-treated WT mice.
DAT binding reflects interaction of the radioactive ligand [125I]RTI-121 with its binding site on the DAT protein. This might not necessarily reflect the concentration of protein or loss of DA terminals because a number of factors including redox status can affect the interaction of receptors with their ligands. Thus, a rapid decrease in DAT function has been reported after METH administration, at a time when there is no evidence of loss of DA terminals (Fleckenstein et al., 1997). This loss of function has been attributed to the production of superoxide radicals (Fleckenstein et al., 1997). Therefore, the concentration of DAT protein was measured by the use of Western blotting. These experiments showed no significant differences in DAT protein levels between the saline-injected WT, +/−, and −/− c-fos knock-out mice (Fig. 2). Administration of METH reduced the DAT protein expression by 44.6, 57.9, and 78.8% in WT, +/−, and −/− c-fos mutant mice, respectively. Fumagalli et al. (1999) have also reported significant METH-induced decreases in DAT protein.
Fig. 2.

Representative immunoblot of the effects of METH on DAT protein concentration. Lanes 1, 3, 5, From saline-injected +/+, +/−, and −/− c-fos mutant mice, respectively.Lanes 2, 4, 6, From METH-injected +/+, +/−, and −/− c-fos knock-out mice, respectively. There were significant changes in DAT concentration consisting of 44.6 ± 4.7, 57.9 ± 6.8, and 78.8 ± 5.3% decreases in +/+, −/−, and −/− c-fos knock-out mice, respectively. α-Tubulin is also shown and reveals similar loading for all groups (6 mice per group). Nt, Antibody against N terminus of the protein.
TH immunohistochemistry
The use of DAT binding and Western blotting, described above, reflects events at the level of striatal DA terminals that might not necessarily reflect the integrity (or lack thereof) of DA axons. Thus, to test further the toxic effects of METH on the striatal dopaminergic system, we also used TH immunohistochemistry to examine the architecture and density of DA fibers in mice killed 1 week after treatment.
Representative striatal sections for TH-IR are shown in Figure3. In the control groups, densely stained and closely packed TH-positive nerve processes are observed throughout the striata of all three genotypes (Fig. 3A,C,E). In mice killed 1 week after the last injection of the drug, there was a marked reduction in TH staining that led to a looser-distributed TH fiber network (Fig. 3B,D,F). These changes were much more evident in −/− c-fos knock-out mice, which showed a very substantial reduction in TH-IR in their striata (Fig. 3F). Figure3G shows the quantification of METH-induced TH staining by the Image program. There was no significant difference in the density of TH fibers between animals from the three genotypes injected with saline. However, there were marked differences in the toxic effects of METH between the three genotypes, with the −/− c-fos knock-out showing the greatest decreases. For example, in comparison with mice of the same genotype injected with saline, the optical density of fibers that remained TH-positive after METH treatment were ∼79.0, 60.3, and 23.6% in +/+, +/−, and −/− c-fos knock-out mice, respectively. Those results are somewhat parallel to those obtained for DAT protein, as reported above.
Fig. 3.
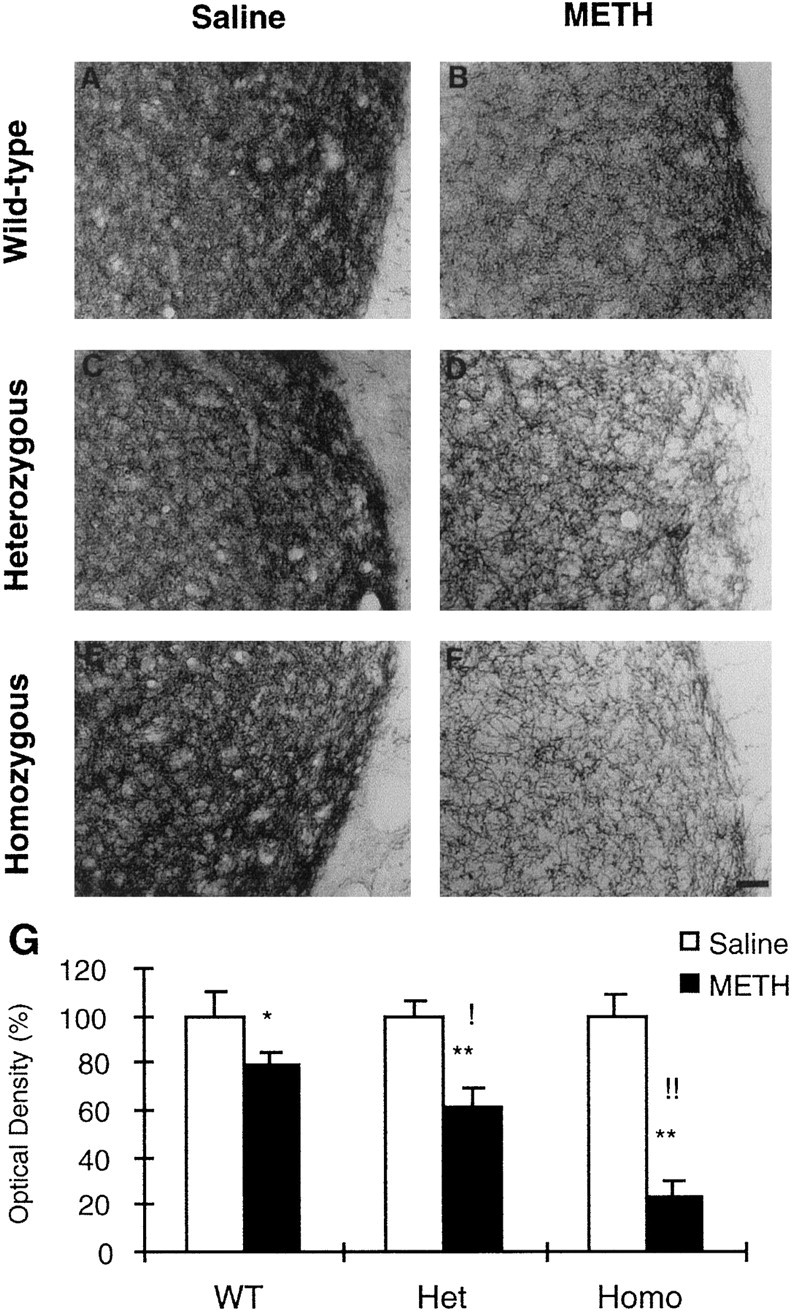
Effects of METH on TH-like immunoreactivity in mice. A–F, Animals were treated with either saline (A, C, E) or METH (B, D, F) 1 week after drug treatment as described in Materials and Methods. The intensity of staining is comparable in the saline-treated mice of the three genotypes (A, C, E). METH administration caused a visually obvious reduction of TH staining (B, D, F), which was more severe in homozygous c-fos knock-out mice (F). G, The results of the statistics for the semiquantitative data obtained using image analysis are shown. Values represent means ± SEM of five to eight animals (6 sections per animal) per group. Key to statistics: *p < 0.001, and **p < 0.0001 in comparison with saline-treated mice of similar genotypes; !p < 0.001, and !!p < 0.0001 in comparison with METH-treated WT mice. Scale bar, 100 μm.
TUNEL histochemistry in the frontal cortex and the striatum
The data about DAT and TH reflect the typical toxic effects of METH on striatal dopaminergic systems. Recently, however, it has been reported that METH can cause apoptosis both in vitro (Cadet et al., 1997; Stumm et al., 1999) and in vivo (Iwasa et al., 1996). Thus, to assess the time course of possible METH-induced DNA fragmentation, detected by the TUNEL reaction, we used animals killed at 3 d and at 1 week after drug treatment. These time points correspond to times used previously by us (Hirata and Cadet, 1997a,b) and others (Pu et al., 1996; Sonsalla et al., 1996) to assess the toxic effects of METH mostly on dopaminergic systems. In the case of apoptotic DNA fragmentation, it is important to assess early time points because cells undergoing apoptosis might be removed by endogenous phagocytes and microglias before the damaged cells can lyse and spill their contents into surrounding areas. Removal of these cells helps to preserve the functional integrity of the surrounding tissue (Ferrer et al., 1995; Sonnenfeld and Jacobs, 1995). Thus, attempts to detect and quantify cells with double-stranded DNA breaks at much later time points might be unsuccessful because the presence of these changes might have occurred much earlier after a course of drug treatment.
As is observed in Figures 4,5, and 6, very few TUNEL-positive cells are seen in the frontal cortices and the striata of animals from the three genotypes injected with saline. However, the administration of doses of METH that are known to cause significant perturbation in dopaminergic systems (Hirata and Cadet, 1997b) (see above) caused marked increases in TUNEL-positive staining in nondopaminergic cells in the cortex and striatum at both time points used in the present study. In addition, the increases were more prominent at the 3 d time point (Figs. 4-6). Counterstaining with toluidine blue indicated that the TUNEL-positive cells were of neuronal origin (data not shown).
Fig. 4.
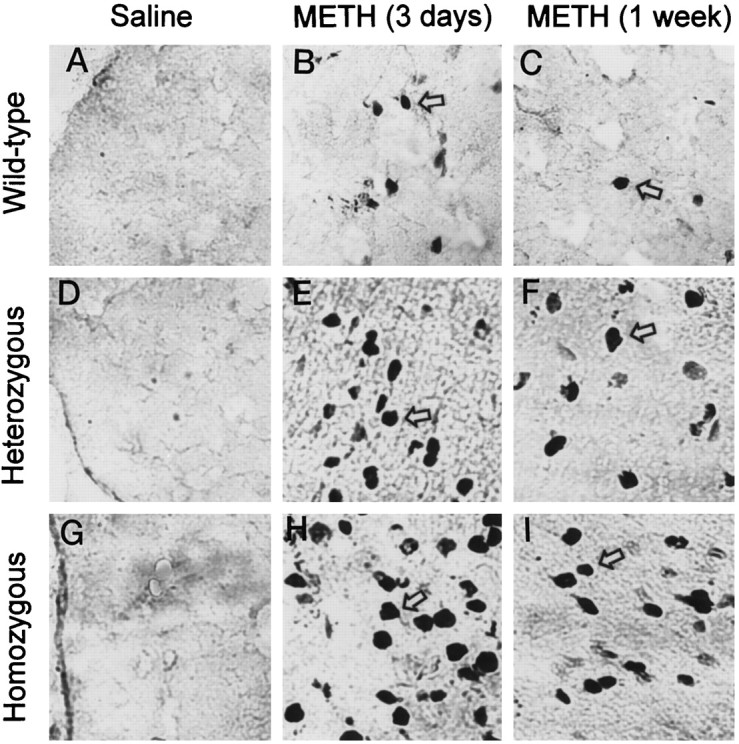
Representative photomicrographs of TUNEL-stained frontal cortices of mice. Very few positive cells appeared in the frontal cortices of saline-treated mice of the three genotypes (A, D, G). METH caused marked increases in TUNEL-positive cells at 3 d (B, E, H) and 1 week (C, F, I) after drug treatment. Thearrows point to typical positive cells. These photomicrographs were generated by using a Carl Zeiss Laser Scanning Confocal System with Axiovert 135–inverted microscopy. The objective lens was 40×. Quantitative data are provided below (see Fig.6).
Fig. 5.

Representative photomicrographs of TUNEL-stained striata of mice. Very few positive cells could be seen in the striata of saline-treated mice (A, D, G). As in the cortex, METH caused marked increases in TUNEL-positive cells at 3 d (B, E, H) and 1 week (C, F, I). Thearrows point to typical positive cells. The photomicrographs were generated as described in Figure 4. Quantitative data are provided below (see Fig. 6).
Fig. 6.

METH caused greater increases in TUNEL-positive cells in the frontal cortex and striatum of c-fos knock-out than in WT mice. The animals were treated and the brains were processed as described in Materials and Methods. Values represent means ± SEM of five to eight mice per group. Key to statistics: *p < 0.05, **p < 0.001, and ***p < 0.0001 in comparison with METH-treated WT mice killed at similar time points; !p < 0.05, !!p < 0.001, and !!!p < 0.0001 in comparison with saline-treated mice of similar genotypes.
Quantification of these changes revealed that, at the 3 d time point, METH caused ∼5.2-, 10.2-, and 17.1-fold increases in TUNEL-positive cells in the cortices of +/+, +/−, and +/− c-fos mice, respectively; these increases at 1 week were ∼2.7-, 5.0-, and 9.3-fold, respectively. A recent paper has also reported that degenerating Fluoro-Jade–positive nondopaminergic neurons in the rat cortex occurred in the greatest number at 3 d after METH treatment (Eisch et al., 1998). In the striatum, at the 3 d time point, the changes were ∼5.0-, 7.3-, and 10.8-fold in the +/+, +/−, and −/− c-fos knock-out mice, respectively; at 1 week, these increases were 2.8-, 4.3- and 6.0-fold, respectively (Fig. 6).
GFAP immunohistochemistry
Increased gliosis has been reported after a number of toxic injuries to brain (Norton et al., 1992). Similar results have been observed after METH administration to rodents (Pu and Vorhees, 1995;Fukumura et al., 1998). We, thus, sought to determine whether METH-induced toxicity would be associated with reactive gliosis in the present model. A few small GFAP-positive astrocytes were observed in the brains of mice from all three genotypes injected with saline (Fig.7A,C,E). These astrocytes are characterized by small cell bodies and very fine and short processes. As reported previously (Pu and Vorhees, 1995; Fukumura et al., 1998), METH treatment causes marked increases in the number of astrocytes in the striata of mice killed 1 week after drug treatment (Fig.7B,D,F). The METH-induced astrocytes in the wild-type mice were characterized by large densely stained cell bodies as well as longer and extensive processes (Fig. 7B). However, the METH-induced astrocytes in the +/− and −/− c-fos knock-out mice did not show much of an increase in their size, in the number of processes, or in the intensity of GFAP staining (compare Fig.7B with D,F). Moreover, the number of METH-induced astrocytes in the striata of the c-fos knock-out mice was less than the number in the WT mice. Figure 7G shows the quantitative data and revealed that the increases in METH-induced glial cells showed a gene-dosage phenomenon, with the heterozygous and homozygous c-fos knock-out mice showing respective changes that were 77.4 and 48.1% of those observed in wild-type mice.
Fig. 7.
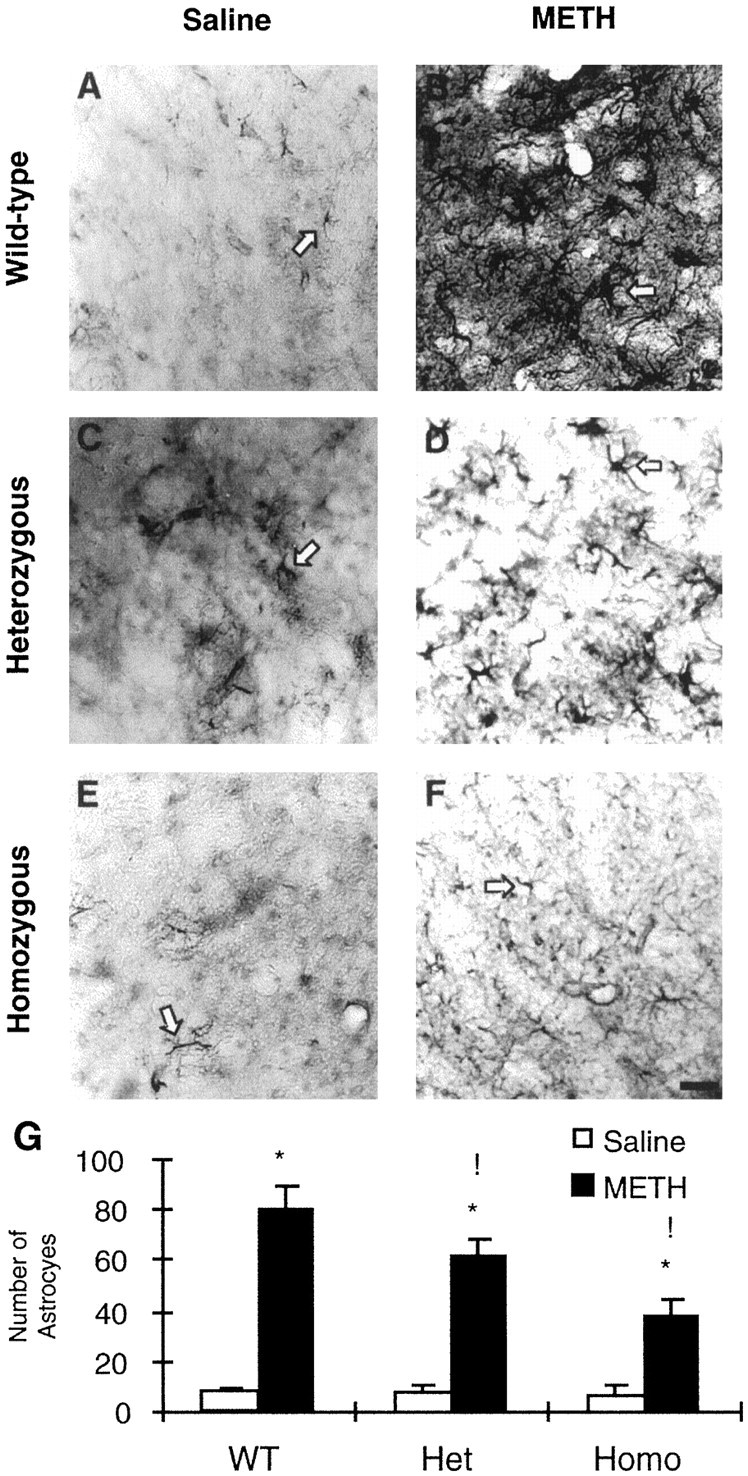
A–F, Effects of METH on GFAP-like immunohistochemistry in the striata of +/+ (A, B), +/− (C, D), and −/− (E, F) c-fos knock-out mice. A few small positive astrocytes are seen in the striata of saline-treated mice (A, C, E). METH caused marked increases in the number of astrocytes in the striatum in mice killed 1 week after drug treatment (B, D, F). METH-induced astrocytes were hypertrophic and densely stained in the WT mice (B). However, in the c-fos +/− (D) and −/− (F) mice, METH treatment caused a measurable increase in the number of astrocytes, but these were small in size and weakly stained. Thearrows point to typical positive cells.G, The statistical analyses of the data obtained from the counts of GFAP-positive cells in the striatum. Values represent means ± SEM from five to eight animals (6 sections per mouse) per group. Key to statistics: *p < 0.0001 in comparison with saline-treated mice of similar genotypes; !p < 0.01 in comparison with METH-treated WT mice. Scale bar, 100 μm.
Temperature fluctuation
Because it has been reported that METH neurotoxicity is generally related to increases in core body temperature (Bowyer et al., 1992;Miller and O'Callaghan, 1994), we measured the effects of METH on these mice. Figure 8 shows that METH caused increases of core body temperature from 37.2 to ∼39.5°C both in WT and +/− c-fos knock-out mice. Statistical analyses revealed no significant differences between the wild-type and heterozygous mice.
Fig. 8.

Effects of METH on core temperature in WT and heterozygous c-fos mutant mice. Core temperature was recorded in animals every 30 min during the administration of METH. Values represent means ± SEM from seven mice per group. There were no statistical differences in the METH-induced temperature elevation between the two genotypes of mice. Saline-treated animals did not exhibit changes in core body temperature over time (data not shown for the sake of clarity). Recorded ambient temperature was between 20.5 and 21.2°C.
DISCUSSION
The main finding of this study is that METH neurotoxicity is exacerbated in c-fos mutant mice. This is supported by the following observations. First, METH caused greater depletion of markers of dopaminergic systems in the striata of +/− and −/− c-fos knock-out mice, with markers at the levels of DA terminals (DAT binding and DAT protein) being more severely affected by METH. Second, METH-induced DNA fragmentation, measured by TUNEL-positive cells, was also more prominent in nondopaminergic cells of the cortices and striata of these c-fos mutant mice. It should be noted that this is the first demonstration, using the TUNEL approach, that METH can cause apoptotic DNA fragmentation in the brain. These results are consistent with bothin vitro (Cadet et al., 1997; Stumm et al., 1999) andin vivo (Iwasa et al., 1996) studies showing that METH can cause cell death via an apoptotic process.
Increased core body temperature has been reported to play a role in METH neurotoxicity (Bowyer et al., 1992, 1994; Miller and O'Callaghan, 1994; LaVoie and Hastings, 1999). Although most drugs that cause hypothermia tend to offer protection against METH-induced damage (Ali et al., 1994; Albers and Sonsalla, 1995), treatment with reserpine, a well known hypothermic agent, has actually been reported to exacerbate METH-induced toxicity (Wagner et al., 1983). When taken together, these data suggest that increased temperature is one participant in a complex series of events that cause the toxicity of METH. In the present experiments, we did examine the possible contribution of temperature in the exacerbation of METH-induced damage in the c-fos mutant mice; however, because there were no significant differences in temperature elevation between WT and +/− c-fos mice, it is very likely that the observed increased toxicity in c-fos mutants is not secondary to a temperature effect.
Another mechanism for the potentiation of METH-induced neurotoxicity involves the possibility that METH metabolism might have been altered in the c-fos mutant mice in such a way that METH might have accumulated to a higher concentration in the brains of these mice. This accumulation in METH might have occurred because the absence of c-fos could have led to downregulation of the cytochrome P450 isoenzymes that are involved in the metabolism of the drug (Baba et al., 1988; Lin et al., 1995, 1997) because of a lack of stimulation of putative AP-1–binding sites that have been found in some cytochrome P450 genes (Shaw et al., 1996; Quattrochi et al., 1998). This hypothesized increase in METH levels might have caused greater increases in DA release, higher temperature, as well as increased toxicity in the mutant mice. However, we failed to find any evidence of differences in METH-induced temperature regulation in the WT and c-fos mutants, thus suggesting that changes in METH metabolism might not be sufficient to explain the exacerbation in toxicity observed in these mice. Nevertheless, it could still be argued that, even in the absence of increased METH levels in the brains of c-fos mutants, it is still possible that METH might have caused greater displacement of DA from storage vesicles into the cytosol with secondary generation of reactive oxygen species (ROS) within that compartment. However, this argument would hold for only the exacerbations observed in the deleterious effects of METH on striatal DA terminals of the c-fos knock-out mice.
Possible role of oxidative stress in METH-induced apoptosis
Although this discussion of METH-mediated toxicity via ROS had been applied mainly to the pathological changes observed in monoaminergic terminals (for review, see Cadet and Brannock, 1998), it is becoming quite clear that METH can exert its nefarious effects beyond these systems. In the present study, we have shown that METH administration is associated with DNA fragmentation in intrinsic nondopaminergic neurons located in cortical and striatal regions of mouse brains. The observations in the cortex are compatible with other reports that METH can damage cortical cell bodies in rats (Pu et al., 1996; Eisch et al., 1998). Although similar findings are now being reported in mice for the first time, these results are consistent with observations that METH can alter the dynamics of antioxidant enzymes in cortical regions of mice (Jayanthi et al., 1998). It is also consistent with the recent demonstration that METH can cause apoptosis and activation of cell death–related genes in cortical cell cultures (Stumm et al., 1999). The observations of DNA fragmentation in intrinsic striatal cell bodies are also consistent with our recent suggestions that METH can damage cells located postsynaptic to DA terminals (Cadet et al., 1998). Although this remains to be fully clarified, it is very likely that there are links between the pathways that cause perturbations in DA terminals and DA axons and the mechanisms that damage intrinsic cell bodies. Therefore, when taken together, the exacerbation of METH-induced toxic effects on both dopaminergic (terminal and fiber damage) and nondopaminergic (apoptosis) systems suggests that c-fos might be involved in stimulating coordinated protective pathways against toxic injuries.
Possible role of c-fos in protective mechanisms
The IEG c-fos, a component of the AP-1 transcription factor (Rauscher et al., 1988), is widely distributed in the CNS (Senba and Ueyama, 1997) and is easily induced by multiple stimuli (Herdegen and Leah, 1998). In addition to its activation being dependent on cellular redox status (Chakraborti and Chakraborti, 1998), c-fos can also activate some antioxidant enzymes. For example, c-fos can interact with the promoter of the glutathione S-transferase gene and cause an increase in enzymatic activity (Moffat et al., 1994; Pinkus et al., 1995). Glutathione S-transferase is known to act in the glutathione pathway as a detoxifying enzyme, which gets rid of free radicals (Hayes and Strange, 1995). c-fos can also stimulate the antioxidant response element in the human NAD(P)H:quinone oxidoreductase gene (Li and Jaiswal, 1992). Moreover, c-fos can increase the levels of trophic or scaffolding factors via interaction with AP-1–binding sites located on the promoters of their genes (for review, see Herdegen and Leah, 1998). These include nerve growth factor, basic FGF, and BDNF (Shibata et al., 1991; Herdegen and Leah, 1998). Some of these factors can increase the activity of antioxidant enzymes in addition to acting as survival factors (Mattson et al., 1995; Skaper et al., 1998). Thus, c-fos induction by toxic doses of METH (Sheng et al., 1996a,b; Hirata et al., 1998a) might serve as an oxidative stress response that could activate downstream molecular and cellular events that participate in a coordinated protective response against METH. These arguments for c-fos as a protective agent are supported by reports that cells lacking c-fos are more sensitive to some DNA-damaging agents in vitro (Haas and Kaina, 1995;Kaina et al., 1997), in concordance with our present observations of increased METH toxicity in c-fos knock-out mice. Thus, the prolonged absence of c-fos could have rendered the brain more susceptible to METH-induced injury by altering cellular redox status in the mice.
Possible role of c-fos in reactive gliosis
An unexpected finding in this report is the observation that, although c-fos mutants are more susceptible to the toxicity of METH, these mice show much less of a reactive gliotic response to the drug. The attenuated gliotic response in the c-fos mutant might be caused by a lack of stimulation of the AP-1–binding site located on the promoter of the GFAP gene (Sarid, 1991) and suggests that transcription control of the GFAP gene might be dependent mostly on c-fos. This line of reasoning is compatible with previous data that had implicated c-fos expression in astrocytic replication (Masood et al., 1993; Pennypacker et al., 1996). Thus, it might not be unreasonable to suggest that, because of the blunted glial response in the c-fos mutant mice, METH-induced astrogliosis might actually be involved in mechanisms aimed at protecting the brain or meant to help in axonal regeneration, in addition to being a marker of neurotoxicity. This notion, although speculative, is supported by reports that astrocytes can synthesize and release neurotrophic factors that protect neurons from injury and death (Ridet et al., 1997; Kariko et al., 1998). For example, conditioned media from nigral astrocytes have been shown to improve the survival of dopaminergic neurons (O'Malley et al., 1992, 1994). Moreover, these ideas are also supported by observations that astrocytes contain high intracellular concentrations of antioxidants (Juurlink, 1997; Wilson, 1997). These ideas will need to be evaluated in these mutants using various neurotoxic models that feature reactive gliosis.
Summary
In conclusion, this is the first report showing that c-fos mutant mice are more sensitive to the toxic effects of METH on both dopaminergic and nondopaminergic systems in the brains. These data indicate that METH-induced c-fos activation is probably involved in a cascade that stimulates pleiotropic protective mechanisms against the deleterious effects of the drug. The present study also provides more evidence to support the view that METH not only causes degeneration of monoaminergic terminals but also causes damage to intrinsic striatal and cortical cells via an apoptotic process. Further studies are also needed to dissect the roles that METH-induced activated astrocytes might play, via epigenetic mechanisms, in the repair of brain regions affected by the drug.
Footnotes
We thank the staff of the Animal Care Facility at the Division of Intramural Research of the National Institutes of Health–National Institute on Drug Abuse for the impeccable care of the animals. We also thank Dr. Teruo Hayashi for help with the Western and confocal microscopy. We are very grateful to Ann Rea for help with the figures. We are also very thankful to the reviewers whose suggestions helped to improve this paper.
Correspondence should be addressed to Dr. Jean Lud Cadet, Molecular Neuropsychiatry Section, National Institutes of Health–National Institute on Drug Abuse Intramural Research program, 5500 Nathan Shock Drive, Baltimore, MD 21224. E-mail: JCADET@intra.nida.nih.gov.
REFERENCES
- 1.Albers DS, Sonsalla PK. Methamphetamine-induced hyperthermia and dopaminergic neurotoxicity in mice: pharmacological profile of protective and nonprotective agents. J Pharmacol Exp Ther. 1995;275:1104–1114. [PubMed] [Google Scholar]
- 2.Ali SF, Newport GD, Holson RR, Slikker W, Jr, Bowyer JF. Low environmental temperatures or pharmacologic agents that produce hypothermia decrease methamphetamine neurotoxicity in mice. Brain Res. 1994;658:33–38. doi: 10.1016/s0006-8993(09)90007-5. [DOI] [PubMed] [Google Scholar]
- 3.Asanuma M, Cadet JL. Methamphetamine-induced increase in striatal NF-kappaB DNA-binding activity is attenuated in superoxide dismutase transgenic mice. Brain Res Mol Brain Res. 1998;60:305–309. doi: 10.1016/s0169-328x(98)00188-0. [DOI] [PubMed] [Google Scholar]
- 4.Asanuma M, Hirata H, Cadet JL. Attenuation of 6-hydroxydopamine-induced dopaminergic nigrostriatal lesions in superoxide dismutase transgenic mice. Neuroscience. 1998;85:907–917. doi: 10.1016/s0306-4522(97)00665-9. [DOI] [PubMed] [Google Scholar]
- 5.Baba T, Yamamda H, Oguri K, Yoshimura H. Participation of cytochrome P-450 isozymes in N-demethylation, N-hydroxylation and aromatic hydroxylation of methamphetamine. Xenobiotica. 1988;18:475–484. doi: 10.3109/00498258809041684. [DOI] [PubMed] [Google Scholar]
- 6.Bowyer JF, Tank AW, Newport GD, Slikker W, Jr, Ali SF, Holson RR. The influence of environmental temperature on the transient effects of methamphetamine on dopamine levels and dopamine release in striatum. J Pharmacol Exp Ther. 1992;260:817–824. [PubMed] [Google Scholar]
- 7.Bowyer JF, Davied DL, Schmued L, Broening HW, Newport GD, Slikker W, Jr, Holson RR. Further studies of the role of hyperthermia in methamphetamine neurotoxicity. J Pharmacol Exp Ther. 1994;268:1571–1580. [PubMed] [Google Scholar]
- 8.Burke RE, Cadet JL, Kent JD, Karanas AL, Jackson-Lewis V. An assessment of the validity of densitometric measures of striatal tyrosine hydroxylase-positive fibers: relationship to apomorphine-induced rotations in 6-hydroxydopamine lesioned rats. J Neurosci Methods. 1990;35:63–73. doi: 10.1016/0165-0270(90)90095-w. [DOI] [PubMed] [Google Scholar]
- 9.Burke RE, Kent J, Kenyon N, Karanas A. Unilateral hypoxic-ischemic injury in neonatal rat results in a persistent increase in the density of striatal tyrosine hydroxylase immunoperoxidase staining. Brain Res Dev Brain Res. 1991;58:171–179. doi: 10.1016/0165-3806(91)90003-2. [DOI] [PubMed] [Google Scholar]
- 10.Cadet JL, Brannock C. Free radicals and the pathobiology of brain dopamine systems. Neurochem Int. 1998;32:117–131. doi: 10.1016/s0197-0186(97)00031-4. [DOI] [PubMed] [Google Scholar]
- 11.Cadet JL, Sheng P, Ali S, Rothman R, Carlson E, Epstein C. Attenuation of methamphetamine-induced neurotoxicity in copper/zinc superoxide dismutase transgenic mice. J Neurochem. 1994;62:380–383. doi: 10.1046/j.1471-4159.1994.62010380.x. [DOI] [PubMed] [Google Scholar]
- 12.Cadet JL, Ordonez SV, Ordonez JV. Methamphetamine induces apoptosis in immortalized neural cells: protection by the proto-oncogene, bcl-2. Synapse. 1997;25:176–184. doi: 10.1002/(SICI)1098-2396(199702)25:2<176::AID-SYN8>3.0.CO;2-9. [DOI] [PubMed] [Google Scholar]
- 13.Cadet JL, Ladeheim B, Hirata H. Effects of toxic doses of methamphetamine (METH) on dopamine D1 receptors in the mouse brain. Brain Res. 1998;786:240–242. doi: 10.1016/s0006-8993(97)01432-7. [DOI] [PubMed] [Google Scholar]
- 14.Chakraborti S, Chakraborti T. Oxidant-mediated activation of mitogen-activated protein kinases and nuclear transcription factors in the cardiovascular system: a brief overview. Cell Signal. 1998;10:675–683. doi: 10.1016/s0898-6568(98)00014-x. [DOI] [PubMed] [Google Scholar]
- 15.De Vito MJ, Wagner GC. Methamphetamine-induced neuronal damage: a possible role for free radicals. Neuropharmacology. 1989;28:1145–1150. doi: 10.1016/0028-3908(89)90130-5. [DOI] [PubMed] [Google Scholar]
- 16.Eisch AJ, Schmued LC, Marshall JF. Characterizing cortical neuron injury with Fluoro-Jade labeling after a neurotoxic regimen of methamphetamine. Synapse. 1998;30:329–333. doi: 10.1002/(SICI)1098-2396(199811)30:3<329::AID-SYN10>3.0.CO;2-V. [DOI] [PubMed] [Google Scholar]
- 17.Ferrer I, Olive M, Blanco R, Ballabriga J, Cinos C, Planas AM. Amoeboid microglial response following x-ray-induced apoptosis in the neonatal rat brain. Neurosci Lett. 1995;193:109–112. doi: 10.1016/0304-3940(95)11679-q. [DOI] [PubMed] [Google Scholar]
- 18.Fleckenstein AE, Metzger RR, Wilkins DG, Gibb JW, Hanson GR. Rapid and reversible effects of methamphetamine on dopamine transporters. J Pharmacol Exp Ther. 1997;282:834–838. [PubMed] [Google Scholar]
- 19.Fukumura M, Cappon GD, Pu C, Broening HW, Vorhees CV. A single dose model of methamphetamine-induced neurotoxicity in rats: effects on neostriatal monoamines and glial fibrillary acidic protein. Brain Res. 1998;806:1–7. doi: 10.1016/s0006-8993(98)00656-8. [DOI] [PubMed] [Google Scholar]
- 20.Fumagalli F, Gainetdinov RR, Valenzano KJ, Caron MG. Role of dopamine transporter in methamphetamine-induced neurotoxicity: evidence from mice lacking the transporter. J Neurosci. 1998;18:4861–4869. doi: 10.1523/JNEUROSCI.18-13-04861.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Fumagalli F, Gainetdinov RR, Wang YM, Valenzano KJ, Miller GW, Caron MG. Increased methamphetamine neurotoxicity in heterozygous vesicular monoamine transporter 2 knock-out mice. J Neurosci. 1999;19:2424–2431. doi: 10.1523/JNEUROSCI.19-07-02424.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Giovanni A, Liang LP, Hastings TG, Zigmond MJ. Estimating hydroxyl radical content in rat brain using systemic and intraventricular salicylate: impact of methamphetamine. J Neurochem. 1995;64:1819–1825. doi: 10.1046/j.1471-4159.1995.64041819.x. [DOI] [PubMed] [Google Scholar]
- 23.Greberman SB, Wada K. Social and legal factors related to drug abuse in the United States and Japan. Public Health Rep. 1994;109:731–737. [PMC free article] [PubMed] [Google Scholar]
- 24.Haas S, Kaina B. c-Fos is involved in the cellular defense against the genotoxic effect of UV radiation. Carcinogenesis. 1995;16:985–991. doi: 10.1093/carcin/16.5.985. [DOI] [PubMed] [Google Scholar]
- 25.Hayes JD, Strange RC. Potential contribution of the glutathione S-transferase supergene family to resistance to oxidative stress. Free Radic Res. 1995;22:193–207. doi: 10.3109/10715769509147539. [DOI] [PubMed] [Google Scholar]
- 26.Herdegen T, Leah JD. Inducible and constitutive transcription factors in the mammalian nervous system: control of gene expression by Jun, Fos and Krox, and CREB/ATF proteins. Brain Res Brain Res Rev. 1998;28:370–490. doi: 10.1016/s0165-0173(98)00018-6. [DOI] [PubMed] [Google Scholar]
- 27.Hirata H, Cadet JL. Methamphetamine-induced serotonin neurotoxicity is attenuated in p53-knockout mice. Brain Res. 1997a;768:345–348. doi: 10.1016/s0006-8993(97)00798-1. [DOI] [PubMed] [Google Scholar]
- 28.Hirata H, Cadet JL. p53-knockout mice are protected against the long-term effects of methamphetamine on dopaminergic terminals and cell bodies. J Neurochem. 1997b;69:780–790. doi: 10.1046/j.1471-4159.1997.69020780.x. [DOI] [PubMed] [Google Scholar]
- 29.Hirata H, Ladenheim B, Carlson E, Epstein C, Cadet JL. Autoradiographic evidence for methamphetamine-induced striatal dopaminergic loss in mouse brain: attenuation in CuZn-superoxide dismutase transgenic mice. Brain Res. 1996;714:95–103. doi: 10.1016/0006-8993(95)01502-7. [DOI] [PubMed] [Google Scholar]
- 30.Hirata H, Asanuma M, Cadet JL. Superoxide radicals are mediators of the effects of methamphetamine on Zif268 (Egr-1, NGFI-A) in the brain: evidence from using CuZn superoxide dismutase transgenic mice. Brain Res Mol Brain Res. 1998a;58:209–216. doi: 10.1016/s0169-328x(98)00055-2. [DOI] [PubMed] [Google Scholar]
- 31.Hirata H, Asanuma M, Cadet JL. Melatonin attenuates methamphetamine-induced toxic effects on dopamine and serotonin terminals in mouse brain. Synapse. 1998b;30:150–155. doi: 10.1002/(SICI)1098-2396(199810)30:2<150::AID-SYN4>3.0.CO;2-B. [DOI] [PubMed] [Google Scholar]
- 32.Itzhak Y, Ali SF. The neuronal nitric oxide synthase inhibitor, 7-nitroindazole, protects against methamphetamine-induced neurotoxicity in vivo. J Neurochem. 1996;67:1770–1773. doi: 10.1046/j.1471-4159.1996.67041770.x. [DOI] [PubMed] [Google Scholar]
- 33.Itzhak Y, Martin JL, Black MD, Ali SF. Effect of melatonin on methamphetamine- and 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine-induced dopaminergic neurotoxicity and methamphetamine-induced behavioral sensitization. Neuropharmacology. 1998a;37:781–791. doi: 10.1016/s0028-3908(98)00067-7. [DOI] [PubMed] [Google Scholar]
- 34.Itzhak Y, Gandia C, Huang PL, Ali SF. Resistance of neuronal nitric oxide synthase-deficient mice to methamphetamine-induced dopaminergic neurotoxicity. J Pharmacol Exp Ther. 1998b;284:1040–1047. [PubMed] [Google Scholar]
- 35.Iwasa M, Maeno Y, Inoue H, Koyama H, Matoba R. Induction of apoptotic cell death in rat thymus and spleen after a bolus injection of methamphetamine. Int J Legal Med. 1996;109:23–28. doi: 10.1007/BF01369597. [DOI] [PubMed] [Google Scholar]
- 36.Jayanthi S, Ladenheim B, Cadet JL. Methamphetamine-induced changes in antioxidant enzymes and lipid peroxidation in copper/zinc-superoxide dismutase transgenic mice. Ann NY Acad Sci. 1998;844:92–102. [PubMed] [Google Scholar]
- 37.Johnson RS, Spiegelman BM, Papaioannou V. Pleiotropic effects of a null mutation in the c-fos proto-oncogene. Cell. 1992;71:577–586. doi: 10.1016/0092-8674(92)90592-z. [DOI] [PubMed] [Google Scholar]
- 38.Juurlink BH. Response of glial cells to ischemia: roles of reactive oxygen species and glutathione. Neurosci Biobehav Rev. 1997;21:151–166. doi: 10.1016/s0149-7634(96)00005-x. [DOI] [PubMed] [Google Scholar]
- 39.Kaina B, Haas S, Kappes H. A general role for c-Fos in cellular protection against DNA-damaging carcinogens and cytostatic drugs. Cancer Res. 1997;57:2721–2731. [PubMed] [Google Scholar]
- 40.Kariko K, Harris VA, Rangel Y, Duvall ME, Welsh FA. Effect of cortical spreading depression on the levels of mRNA coding for putative neuroprotective proteins in rat brain. J Cereb Blood Flow Metab. 1998;18:1308–1315. doi: 10.1097/00004647-199812000-00005. [DOI] [PubMed] [Google Scholar]
- 41.Lan KC, Lin YF, Yu FC, Lin CS, Chu P. Clinical manifestations and prognostic features of acute methamphetamine intoxication. J Formos Med Assoc. 1998;97:528–533. [PubMed] [Google Scholar]
- 42.LaVoie MJ, Hastings TG. Dopamine quinone formation and protein modification associated with the striatal neurotoxicity of methamphetamine: evidence against a role for extracellular dopamine. J Neurosci. 1999;19:1484–1491. doi: 10.1523/JNEUROSCI.19-04-01484.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Li Y, Jaiswal AK. Regulation of human NAD(P)H:quinone oxidoreductase gene. Role of AP1 binding site contained within human antioxidant response element. J Biol Chem. 1992;267:15097–15104. [PubMed] [Google Scholar]
- 44.Lin LY, Kumagai Y, Hiratsuka A, Narimatsu S, Suzuki T, Fanae Y, Distefano EW, Cho AK. Cytochrome P4502D isozymes catalyze the 4-hydroxylation of methamphetamine enantiomers. Drug Metab Dispos. 1995;23:610–614. [PubMed] [Google Scholar]
- 45.Lin LY, Di Stefano EW, Schmitz DA, Hsu L, Ellis SW, Lennard MS, Tucker GT, Cho AK. Oxidation of methamphetamine and methylenedioxymethamphetamine by CYP2D6. Drug Metab Dispos. 1997;25:1059–1064. [PubMed] [Google Scholar]
- 46.Masood K, Besnard F, Su Y, Brenner M. Analysis of a segment of the human glial fibrillary acidic protein gene that directs astrocyte-specific transcription. J Neurochem. 1993;61:160–166. doi: 10.1111/j.1471-4159.1993.tb03551.x. [DOI] [PubMed] [Google Scholar]
- 47.Matsuda LA, Schmidt CJ, Gibb JW, Hanson GR. Effects of methamphetamine on central monoaminergic systems in normal and ascorbic acid-deficient guinea pigs. Biochem Pharmacol. 1988;37:3477–3484. doi: 10.1016/0006-2952(88)90700-9. [DOI] [PubMed] [Google Scholar]
- 48.Mattson MP, Lovell MA, Furukawa K, Markesbery WR. Neurotrophic factors attenuate glutamate-induced accumulation of peroxides, elevation of intracellular Ca2+ concentration, and neurotoxicity and increase of antioxidant enzyme activities in hippocampal neurons. J Neurochem. 1995;65:1740–1751. doi: 10.1046/j.1471-4159.1995.65041740.x. [DOI] [PubMed] [Google Scholar]
- 49.McCann UD, Wong DF, Yokoi F, Villemagne V, Dannals RF, Ricaurte GA. Reduced striatal dopamine transporter density in abstinent methamphetamine and methcathinone users: evidence from positron emission tomography studies with [11C]WIN-35,428. J Neurosci. 1998;18:8417–8422. doi: 10.1523/JNEUROSCI.18-20-08417.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Miller DB, O'Callaghan JP. Environment-, drug- and stress-induced alterations in body temperature affect the neurotoxicity of substituted amphetamines in the C57BL/6J mouse. J Pharmacol Exp Ther. 1994;270:752–760. [PubMed] [Google Scholar]
- 51.Miller MA. Trends and patterns of methamphetamine smoking in Hawaii. NIDA Res Monogr. 1991;115:72–83. [PubMed] [Google Scholar]
- 52.Moffat GJ, McLaren AW, Wolf CR. Involvement of Jun and Fos proteins in regulating transcriptional activation of the human pi class glutathione S-transferase gene in multidrug-resistant MCF7 breast cancer cells. J Biol Chem. 1994;269:16397–16402. [PubMed] [Google Scholar]
- 53.Murray JB. Psychophysiological aspects of amphetamine-methamphetamine abuse. J Psychol. 1998;132:227–237. doi: 10.1080/00223989809599162. [DOI] [PubMed] [Google Scholar]
- 54.Norton WT, Aquino DA, Hozumi I, Chiu FC, Brosnan CF. Quantitative aspects of reactive gliosis: a review. Neurochem Res. 1992;17:877–885. doi: 10.1007/BF00993263. [DOI] [PubMed] [Google Scholar]
- 55.O'Callaghan JP, Miller DB. Neurotoxicity profiles of substituted amphetamines in the C57BL/6J mouse. J Pharmacol Exp Ther. 1994;270:741–751. [PubMed] [Google Scholar]
- 56.O'Malley EK, Sieber BA, Black IB, Dreyfus CF. Mesencephalic type I astrocytes mediate the survival of substantia nigra dopaminergic neurons in culture. Brain Res. 1992;582:65–70. doi: 10.1016/0006-8993(92)90317-3. [DOI] [PubMed] [Google Scholar]
- 57.O'Malley EK, Sieber BA, Morrison RS, Black IB, Dreyfus CF. Nigral type I astrocytes release a soluble factor that increases dopaminergic neuron survival through mechanisms distinct from basic fibroblast growth factor. Brain Res. 1994;647:83–90. doi: 10.1016/0006-8993(94)91401-x. [DOI] [PubMed] [Google Scholar]
- 58.Pennypacker KR, Hong JS, Mullis SB, Hudson PM, McMillian MK. Transcription factors in primary glial cultures: changes with neuronal interactions. Brain Res Mol Brain Res. 1996;37:224–230. doi: 10.1016/0169-328x(95)00318-m. [DOI] [PubMed] [Google Scholar]
- 59.Pinkus R, Weiner LM, Daniel V. Role of quinone-mediated generation of hydroxyl radicals in the induction of glutathione S-transferase gene expression. Biochemistry. 1995;34:81–88. doi: 10.1021/bi00001a010. [DOI] [PubMed] [Google Scholar]
- 60.Pu C, Vorhees CV. Protective effects of MK-801 on methamphetamine-induced depletion of dopaminergic and serotonergic terminals and striatal astrocytic response: an immunohistochemical study. Synapse. 1995;19:97–104. doi: 10.1002/syn.890190205. [DOI] [PubMed] [Google Scholar]
- 61.Pu C, Broening HW, Vorhees CV. Effect of methamphetamine on glutamate-positive neurons in the adult and developing rat somatosensory cortex. Synapse. 1996;23:328–334. doi: 10.1002/(SICI)1098-2396(199608)23:4<328::AID-SYN11>3.0.CO;2-T. [DOI] [PubMed] [Google Scholar]
- 62.Quattrochi LC, Shih H, Pickwell GV. Induction of the human CYP1A2 enhancer by phorbol ester. Arch Biochem Biophys. 1998;350:41–48. doi: 10.1006/abbi.1997.0491. [DOI] [PubMed] [Google Scholar]
- 63.Rauscher FJ, III, Voulalas PJ, Franza BR, Jr, Curran T. Fos and Jun bind cooperatively to the AP-1 site: reconstitution in vitro. Genes Dev. 1988;2:1687–1699. doi: 10.1101/gad.2.12b.1687. [DOI] [PubMed] [Google Scholar]
- 64.Ricaurte GA, Guillery RW, Seiden LS, Schuster CR, Moore RY. Dopamine nerve terminal degeneration produced by high doses of methylamphetamine in the rat brain. Brain Res. 1982;235:93–103. doi: 10.1016/0006-8993(82)90198-6. [DOI] [PubMed] [Google Scholar]
- 65.Ridet JL, Malhotra SK, Privat A, Gage FH. Reactive astrocytes: cellular and molecular cues to biological function. Trends Neurosci. 1997;20:570–577. doi: 10.1016/s0166-2236(97)01139-9. [DOI] [PubMed] [Google Scholar]
- 66.Sarid J. Identification of a cis-acting positive regulatory element of the glial fibrillary acidic protein gene. J Neurosci Res. 1991;28:217–228. doi: 10.1002/jnr.490280209. [DOI] [PubMed] [Google Scholar]
- 67.Senba E, Ueyama T. Stress-induced expression of immediate early genes in the brain and peripheral organs of the rat. Neurosci Res. 1997;29:183–207. doi: 10.1016/s0168-0102(97)00095-3. [DOI] [PubMed] [Google Scholar]
- 68.Shaw KP. Human methamphetamine-related fatalities in Taiwan during 1991–1996. J Forensic Sci. 1999;44:27–31. [PubMed] [Google Scholar]
- 69.Shaw PM, Edigkaufer M, Dochmer J, Adesnik M. Sequence of the rat PB-inducible CYP2B1 promoter. Biochim Biophys Acta. 1996;1305:54–58. doi: 10.1016/0167-4781(95)00222-7. [DOI] [PubMed] [Google Scholar]
- 70.Sheng P, Wang XB, Ladenheim B, Epstein C, Cadet JL. AP-1 DNA-binding activation by methamphetamine involves oxidative stress. Synapse. 1996a;24:213–217. doi: 10.1002/(SICI)1098-2396(199611)24:3<213::AID-SYN2>3.0.CO;2-H. [DOI] [PubMed] [Google Scholar]
- 71.Sheng P, Ladenheim B, Moran TH, Wang XB, Cadet JL. Methamphetamine-induced neurotoxicity is associated with increased striatal AP-1 DNA-binding activity in mice. Brain Res Mol Brain Res. 1996b;42:171–174. doi: 10.1016/s0169-328x(96)00192-1. [DOI] [PubMed] [Google Scholar]
- 72.Sheng P, Cerruti C, Ali S, Cadet JL. Nitric oxide is a mediator of methamphetamine (METH)-induced neurotoxicity. In vitro evidence from primary cultures of mesencephalic cells. Ann NY Acad Sci. 1996c;801:174–186. doi: 10.1111/j.1749-6632.1996.tb17440.x. [DOI] [PubMed] [Google Scholar]
- 73.Shibata F, Baird A, Florkiewicz RZ. Functional characterization of the human basic fibroblast growth factor gene promoter. Growth Factors. 1991;4:277–287. doi: 10.3109/08977199109043913. [DOI] [PubMed] [Google Scholar]
- 74.Skaper SD, Floreani M, Negro A, Facci L, Giusti P. Neurotrophins rescue cerebellar granule neurons from oxidative stress-mediated apoptotic death: selective involvement of phosphatidylinositol 3-kinase and the mitogen-activated protein kinase pathway. J Neurochem. 1998;70:1859–1868. doi: 10.1046/j.1471-4159.1998.70051859.x. [DOI] [PubMed] [Google Scholar]
- 75.Sonnenfeld MJ, Jacobs JR. Macrophages and glia participate in the removal of apoptotic neurons from the Drosophila embryonic nervous system. J Comp Neurol. 1995;359:644–652. doi: 10.1002/cne.903590410. [DOI] [PubMed] [Google Scholar]
- 76.Sonsalla PK, Jochnowitz ND, Zeevalk GD, Oostveen JA, Hall ED. Treatment of mice with methamphetamine produces cell loss in the substantia nigra. Brain Res. 1996;738:172–175. doi: 10.1016/0006-8993(96)00995-x. [DOI] [PubMed] [Google Scholar]
- 77.Stephans SE, Yamamoto BY. Effect of repeated methamphetamine administrations on dopamine and glutamate efflux in rat prefrontal cortex. Brain Res. 1995;700:99–106. doi: 10.1016/0006-8993(95)00938-m. [DOI] [PubMed] [Google Scholar]
- 78.Stumm G, Schlegel J, Schafer T, Wurz C, Mennel HD, Krieg JC, Vedder H. Amphetamines induce apoptosis and regulation of bcl-x splice variants in neocortical neurons. FASEB J. 1999;13:1065–1072. doi: 10.1096/fasebj.13.9.1065. [DOI] [PubMed] [Google Scholar]
- 79.Tsao LI, Ladenheim B, Andrews AM, Chieueh CC, Cadet JL, Su TP. Delta opioid peptide [d-Ala2, d-Leu5]enkephalin blocks the long-term loss of dopamine transporters induced by multiple administration of methamphetamine: involvement of opioid receptors and reactive oxygen species. J Pharmacol Exp Ther. 1998;287:322–331. [PubMed] [Google Scholar]
- 80.Villemagne V, Yuan J, Wong DF, Dannals RF, Hatzidimitriou G, Mathews WB, Ravert HT, Musachio J, McCann UD, Ricaurte GA. Brain dopamine neurotoxicity in baboons treated with doses of methamphetamine comparable to those recreationally abused by humans: evidence from [11C]WIN-35,428 positron emission tomography studies and direct in vitro determinations. J Neurosci. 1998;18:419–427. doi: 10.1523/JNEUROSCI.18-01-00419.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Wagner GC, Lucot JB, Schuster CR, Seiden LS. Alpha-methyltyrosine attenuates and reserpine increases methamphetamine-induced neuronal changes. Brain Res. 1983;270:285–288. doi: 10.1016/0006-8993(83)90602-9. [DOI] [PubMed] [Google Scholar]
- 82.Wang ZQ, Ovitt C, Grigoriadis AE, Mohle-Steinlein U, Ruther U, Wagner EF. Bone and haematopoietic defects in mice lacking c-fos. Nature. 1992;360:741–745. doi: 10.1038/360741a0. [DOI] [PubMed] [Google Scholar]
- 83.Wilson JM, Kalasinsky KS, Levey AI, Bergeron C, Reiber G, Anthony RM, Schmunk GA, Shannak K, Haycock JW, Kish SJ. Striatal dopamine nerve terminal markers in human, chronic methamphetamine users. Nat Med. 1996;2:699–703. doi: 10.1038/nm0696-699. [DOI] [PubMed] [Google Scholar]
- 84.Wilson JX. Antioxidant defense of the brain: a role for astrocytes. Can J Physiol Pharmacol. 1997;75:1149–1163. [PubMed] [Google Scholar]
- 85.Yamamoto BK, Zhu W. The effects of methamphetamine on the production of free radicals and oxidative stress. J Pharmacol Exp Ther. 1998;287:107–114. [PubMed] [Google Scholar]