

Abstract
Neuregulin is a neural factor implicated in upregulation of acetylcholine receptor (AChR) synthesis at the neuromuscular junction. Previous studies have demonstrated that the extracellular signal–regulated kinase (ERK) subgroup of MAP kinases is required for neuregulin-induced AChR gene expression. We report here that the neuregulin-mediated increase in AChR ε-subunit mRNA was a delayed response in C2C12 muscle cells. Neuregulin induced expression of immediate early genes c-jun and c-fos, which followed and depended on the ERK activation. Treatment of muscle cells with cycloheximide to inhibit c-JUN synthesis at the protein level and suppression of c-JUN function by a dominant-negative mutant blocked neuregulin-induced expression of the ε-subunit gene, indicating an essential role of c-JUN in neuregulin signaling. Furthermore, neuregulin activated c-JUN N-terminal kinase (JNK) in C2C12 muscle cells. Blockade of JNK activation by overexpressing dominant-negative MKK4 inhibited ε-promoter activation. Moreover, overexpression of the JNK dominant-negative mutant inhibited neuregulin-mediated expression of the εtransgene and endogenous ε-mRNA. Taken together, our results demonstrate important roles of c-JUN and JNK in neuregulin-mediated expression of the AChR ε-subunit gene and suggest that neuregulin activates multiple signaling cascades that converge to regulate AChR ε-subunit gene expression.
Keywords: neuregulin, AChR, c-JUN, JNK, neuromuscular junction, synapse, immediate early gene
Synaptic transmission at the neuromuscular junction is guaranteed by a high density of acetylcholine receptor (AChR) at the postsynaptic membrane. AChR expression is regulated both spatially and temporally during development. AChR synthesis is confined to the adult neuromuscular junction (Froehner, 1993; Hall and Sanes, 1993; Sanes and Lichtman, 1999). Studies using transgenic mice have shown that the regulatory elements of AChR genes can direct the expression of reporter genes at the neuromuscular junction (Klarsfeld et al., 1987; Sanes et al., 1991; Simon et al., 1992; Gundersen et al., 1993), indicating that transcription of AChR genes is most active in nuclei beneath the postsynaptic membrane, contributing to the high density of AChR at the neuromuscular junction.
Synaptic expression of AChR genes is believed to be mediated by neuregulin, a family of factors that have been originally identified as acetylcholine receptor–inducing activity (ARIA) that stimulates muscle AChR synthesis (Fischbach and Cohen, 1973; Jessell et al., 1979; Falls et al., 1993), as ligands of the receptor tyrosine kinase erbB2 (neu differentiation factor and heregulin) (Wen et al., 1992), and as neuronal factors essential for the proliferation and survival of Schwann cells (glia growth factor) (Marchionni et al., 1993). They are products of the nrg-1 gene generated by alternative splicing. Four lines of evidence indicate that neuregulin is a trophic factor released from motoneurons to regulate AChR synthesis at the neuromuscular junction. First, neuregulin heterozygous mutant mice that produce a low amount of neuregulin have a decreased postsynaptic AChR density and are myasthenic (Sandrock et al., 1997). Second, neuregulin mRNA is expressed in motoneurons before the onset of the neuromuscular junction and persists through adulthood (Fischbach and Rosen, 1997;Sandrock et al., 1997). Third, neuregulin accumulates in synaptic basal lamina in adult skeletal muscle (Chu et al., 1995; Goodearl et al., 1995; Jo et al., 1995; Sandrock et al., 1995), and ErbB proteins, neuregulin’s receptors, are localized at the neuromuscular junction (Altiok et al., 1995; Zhu et al., 1995). Last, in cultured muscle cells, neuregulin promotes AChR synthesis both at the protein level (Jessell et al., 1979; Usdin and Fischbach, 1986; Sandrock et al., 1997) and at the level of mRNA (Martinou et al., 1991; Tang et al., 1994; Chu et al., 1995).
Until recently, little was known about the signaling mechanism of neuregulin in the regulation of AChR gene expression. The ErbB2 and ErbB3 proteins become tyrosine phosphorylated in response to neuregulin (Corfas et al., 1993; Altiok et al., 1995; Jo et al., 1995; Si et al., 1996). Concomitantly, extracellular signal–regulated kinase (ERK) activity (Si et al., 1996; Tansey et al., 1996; Altiok et al., 1997) and phosphatidylinositol 3 (PI 3)-kinase activity (Tansey et al., 1996) are increased. Activation of ERK kinase is required for neuregulin regulation of AChR gene expression in cultured muscle cells (Si et al., 1996; Tansey et al., 1996; Altiok et al., 1997) and for synapse-specific expression of the ε-transgene in vivo (Si and Mei, 1999). Here we report that the neuregulin-mediated increase in AChR ε-subunit mRNA was a delayed response. Neuregulin induced expression of immediate early genes c-jun andc-fos and activated c-JUN N-terminal kinase (JNK), both of which were required for the ε-promoter transcription.
MATERIALS AND METHODS
Materials. Recombinant neuregulin (rHRGβ1177–244, a peptide of HRGβ1 residues 177–244) was generously provided by Dr. M. Sliwkowski (Holmes et al., 1992). Anti-ERK kinase antibody was a gift from Dr. G. S. Feng. The mouse AChR ε-subunit cDNA was provided by Dr. R. Huganir. The c-fosand c-jun cDNAs were provided by Drs. T. Curran and A. Langley (Curran et al., 1987). Dr. R. Davis provided JNK1, MKK4, and p38 constructs (Whitmarsh et al., 1997), and Dr. M. Birrer provided the c-JUN mammalian expression constructs (Brown et al., 1994). PD98059, AG1478, and SB202190 were purchased from Calbiochem (La Jolla, CA). Rapamycin was from Research Biochemicals (Natick, MA). Cell culture media were purchased from Life Technologies (Gaithersburg, MD). Anti-FLAG antibody and all other chemicals were from Sigma (St. Louis, MO).
Cell culture and transfection procedures. Mouse muscle C2C12 cells were maintained as undifferentiated myoblasts in a nutrient-rich growth medium containing DMEM supplemented with 20% fetal bovine serum, 0.5% chicken embryo extract, 2 mml-glutamine, 100 U/ml penicillin, and 100 μg/ml streptomycin at 37°C in an atmosphere of 5% CO2 and 95% humidity. To induce differentiation, we cultured myoblasts at 50–70% confluence in the differentiation medium (DM), DMEM supplemented with 4% horse serum, 2 mml-glutamine, 100 U/ml penicillin, and 100 μg/ml streptomycin. For transient transfection, C2C12 myoblasts at ∼50% confluence in six-well plates were cotransfected with the ε416-Luc transgene (1 μg of DNA) and p-cytomegalovirusβ (pCMVβ; 0.1 μg of DNA) with or without 4 μg of either wild-type or mutant c-JUN, MKK4, or JNK by the calcium phosphate technique (Sambrook et al., 1989). Twenty-four hours later cells were switched to DM. Myotubes then were treated with neuregulin for 24 hr. Stable C2C12 cell lines carrying wild-type or mutant JNK were generated as described previously (Si et al., 1996).
Treatment of cells. Forty-eight hours after switching to DM, the C2C12 myoblasts formed myotubes and were then treated with neuregulin. In experiments studying inhibitory effects, chemicals were added into the culture medium 30 min before neuregulin treatment and remained in the medium for the entire neuregulin stimulation period. The volume of solvent vehicles was kept ≤0.1% of the culture medium and did not significantly change the biological outcome. Culture medium was changed every 24 hr to keep myotubes healthy.
Northern blot analysis. Total RNA was extracted from C2C12 muscle cells by the use of a single-step RNA-isolating method modified from that of Chomczynski and Sacchi (1987). Briefly, cells of a 10 cm dish were lysed in 10 ml of the RNA-extracting buffer containing 2m guanidinium thiocyanate, 12.5 mm sodium citrate, pH 7.0, 50 mm β-mercaptoethanol, 0.25%N-lauroylsarcosine, 0.1 m sodium acetate, and 50% phenol. After the addition of 2 ml of chloroform, the lysate was mixed vigorously and subjected to a centrifugation at 8000 × g at 4°C for 20 min. The aqueous phase was mixed with an equal volume of isopropanol, and total RNA was isolated in the pellet of another centrifugation at 8000 × g at 4°C for 20 min. Unless otherwise indicated, 20 μg of total RNA was resolved on a 1% agarose gel by electrophoresis in 3-(N-morpholino)propanesulfonic acid–formaldehyde buffer as described by Lehrach et al. (1977). RNA was transferred to a nylon membrane in 20× SSC by capillary transfer and cross-linked to the membrane by the use of a UV cross-linker. The membrane filters were probed with a cDNA fragment of the ε-subunit or glyceraldehyde-3-phosphate dehydrogenase (GAPDH), both of which were labeled with [α-32P]dCTP by a random-prime method. RNA blots were hybridized in a buffer containing 6× SSC, 5× Denhardt’s solution, 0.5% SDS, 50% formamide, and 200 μg/ml salmon sperm DNA at 42°C for 48 hr. The membrane filters were washed with 0.1× SSC and 0.5% SDS at 42°C four times, each for 15 min, and exposed to Kodak X-Omat AR film (Eastman Kodak, Rochester, NY) at −80°C with an intensifying screen.
Quantitative image analysis. The quantification was performed by image analysis of radioautographic films by scanning the film with the Personal Densitometer (Molecular Dynamics, Sunnyvale, CA), and the captured image was analyzed with the ImageQuant software (Molecular Dynamics).
ERK kinase assay. The ERK kinase assay was performed essentially as described previously (Si et al., 1996).
JNK and p38 MAP kinase assays. C2C12 myotubes were cultured in serum-free DMEM medium for 24 hr before the stimulation with neuregulin. Myotubes were harvested in the lysis buffer containing 137 mm NaCl, 1% NP-40, 5 mm EDTA, 20 mm Tris-HCl, pH 7.5, 1 μm pepstatin, 1 μg/ml leupeptin, 0.2 mm PMSF, 2 μg/ml aprotinin, and 2 mm sodium vanadate. JNK was purified as described previously (Whitmarsh et al., 1997). Briefly, an aliquot of lysate (400 μg of protein) was incubated with 10 μg of glutathioneS-transferase (GST)–c-JUN (human c-JUN amino acids 1–79) fusion protein immobilized on agarose beads in a final volume of 0.4 ml at 4°C for 2 hr. After being washed three times with the lysis buffer and twice with a buffer containing 20 mm HEPES, pH 7.5, and 20 mm MgCl2, the beads were then incubated in 30 μl of the kinase assay buffer containing 20 mm HEPES, pH 7.5, 20 mm MgCl2, 20 mm β-glycerophosphate, 0.1 mm sodium vanadate, 20 μmATP, 2 mm DTT, and 5 μCi of [γ-32P]ATP at 30°C for 15 min. The p38 MAP kinase was purified from the cell lysate using 50 μl of protein G–agarose beads (1:1 slurry) preloaded with goat anti-p38 MAP kinase antibodies (Santa Cruz Biotechnology, Santa Cruz, CA) and assayed using as a substrate 2 μg of the GST–ATF2 fusion protein in the same phosphorylation buffer. The kinase reaction was stopped by the addition of 10 μl of 4× sample-loading buffer and subjected to electrophoresis on a 10% SDS-polyacrylamide gel that was then exposed to an x-ray film.
Luciferase and β-galactosidase assays. The luciferase assay was performed using a kit from Promega (Madison, WI) and following the manufacturer’s instructions. Briefly, 100 μl of cell lysate was mixed in an equal volume of luciferase substrate solution containing 20 mm Tricine, 1.07 mm(MgCO3)4Mg(OH)2·5H2O, 2.67 mm MgSO4, 0.1 mmEDTA, 33.3 mm DTT, 270 μm coenzyme A, 470 μm luciferin, and 530 μm ATP and was placed in a microβ luminometer (Wallac, Turku, Finland) to measure light production for 10 sec. β-Galactosidase activity was determined as described previously (Si et al., 1996). Luciferase activity of transgenes was normalized to β-galactosidase activity to correct for variation in transfection.
Protein concentration determination. Protein concentration was measured by the Bradford method using a Coomassie Protein Assay Reagent (Pierce, Rockford, IL) with BSA as a standard (Bradford, 1976).
Statistical analysis. Values were shown as mean ± SD. Statistical differences were analyzed using the one-tailed Student’st test. A value of p < 0.05 is considered statistically significant.
RESULTS
Delayed increase in AChR ε-subunit mRNA in neuregulin-stimulated C2C12 myotubes
The basal level of the ε-mRNA in C2C12 myoblasts was barely detectable and remained low until 3–4 d after the cells were switched to DM (data not shown). Neuregulin stimulation did not increase the ε-subunit expression in C2C12 myoblasts (Si et al., 1996). We studied the time course of neuregulin in the increase of AChR gene expression in C2C12 myotubes. Forty-eight hours after the medium switch when myotube formation was complete, cells were stimulated with 1 nm neuregulin for various times. In the presence of neuregulin, AChR ε-mRNA remained unchanged until 8–12 hr later when an increase was detectable. The neuregulin-induced increase in the ε-mRNA became robust 24 hr after stimulation (Fig.1). The increase in the ε-mRNA by neuregulin was specific because neuregulin had no effect on GAPDH mRNA (Fig. 1). The basal level of the ε-mRNA (in the absence of neuregulin) did not change significantly during the time of the experiment (Fig. 1A, top). Figure1B shows the means of two separate experiments. The expression of other AChR subunit genes in response to neuregulin followed a similar time course (data not shown).
Fig. 1.
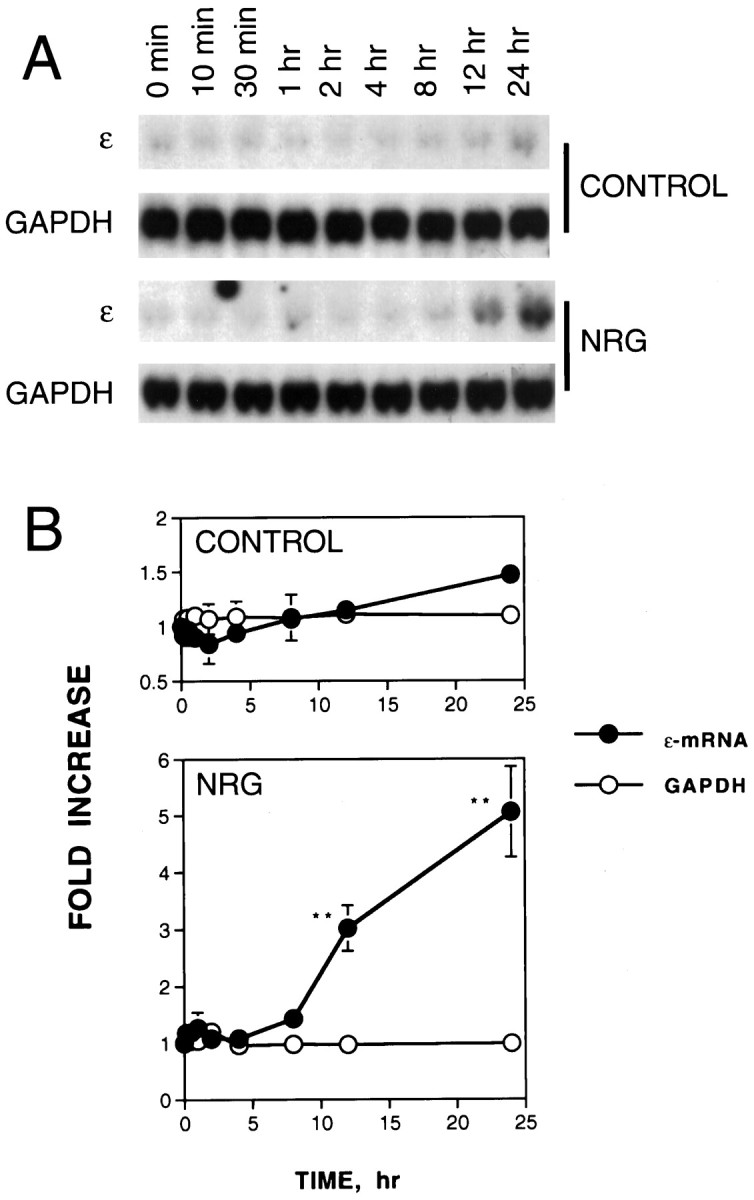
Delayed increase in the AChR ε-subunit mRNA by neuregulin in C2C12 myotubes. Stimulation of C2C12 myotubes was started by the addition of neuregulin (NRG) to a final concentration of 1 nm or an equal volume of vehicle (as a control). Cells were harvested for RNA isolation at the indicated times. Twenty micrograms of total RNA were resolved on a 1% agarose gel, transferred to nitrocellulose, and hybridized with [32P]-labeled DNA probes specific for the ε-subunit. The loading was uniform as evidenced by an equal amount of GAPDH mRNA. A, Representative Northern blot radiograms.B, Histograms showing mean ± SD of two different samples. The mRNA levels at 0 min were considered to be 100%. **p < 0.01.
Increase in the ε-mRNA by transient neuregulin stimulation
Neuregulin-induced expression of AChR genes requires activation of the ERK subgroup of MAP kinases in cultured muscle cells (Si et al., 1996; Tansey et al., 1996; Altiok et al., 1997). The increase in ERK kinase activity by neuregulin is transient with a peak at ∼5–8 min after neuregulin stimulation (Fig.2B) (Si et al., 1996). The ERK kinase activity returns to the basal level within 60 min of neuregulin stimulation. Yet the increase in the ε-mRNA was a delayed response (Fig. 1). These observations prompted us to determine the stimulation time sufficient for neuregulin to activate AChR gene expression. Previous experiments in the literature used a continuous stimulation protocol, in which neuregulin was present in the culture medium until cells were harvested.
Fig. 2.

Increase in the AChR ε-mRNA by transient stimulation with neuregulin. A, Stimulation protocol. Stimulation of myotubes with NRG began at time 0; after an incubation for the indicated time, the cells were washed and incubated in neuregulin-free medium. At 24 hr, cells were lysed to isolate total RNA, which was then blotted for AChR mRNA expression. B, Activation of ERK kinase by neuregulin. Top, C2C12 myotubes stimulated continuously without (control) or with 1 nm neuregulin for the indicated times. Bottom, C2C12 myotubes stimulated with 1 nm neuregulin for 1 min and then incubated in neuregulin-free medium for the indicated times. Control C2C12 myotubes were treated with an equal volume of DMEM (vehicle) without neuregulin for 5 min. ERK kinase was immunoprecipitated and assayed using MBP as a substrate in vitro with [γ-32P]ATP. The phosphorylated MBP was resolved on a 15% SDS gel and exposed to x-ray film. C, Northern blot radiograms. C2C12 myotubes were stimulated as described in A. Control C2C12 myotubes were treated with the same volume of DMEM (without neuregulin) for 24 hr. Twenty micrograms of total RNA were probed with [32P]-labeled ε-subunit probe. D,Histograms showing mean ± SD of three different samples. The mRNA levels in the absence of neuregulin were considered to be 100%.CONST, Continuous stimulation;ST, stimulation.
To study this, we have designed the protocol depicted in Figure2A. Neuregulin stimulation of myotubes began at time 0; after an incubation of the indicated time, cells were washed and incubated in neuregulin-free DM. At 24 hr, total RNA was isolated and assayed for AChR expression. As shown in Figure 2B, exposure of C2C12 myotubes to neuregulin for as short as 1 min was sufficient to activate ERK MAP kinases and subsequently increase the AChR ε-mRNA (Fig. 2C). The ε-mRNA level increased by transient neuregulin stimulation was comparable with that induced by the continuous stimulation (Fig. 2D). In vivo neuregulin binds to the extracellular matrix via the Ig-like domain in the N terminus (Loeb and Fischbach, 1995) and thus becomes concentrated at the neuromuscular junction. The recombinant neuregulin used in this study contained only the epidermal growth factor domain and should not bind to the extracellular matrix. Thus, ERK activation was more transient by neuregulin stimulation in the transient stimulation protocol. These results suggest that the initiation of the signal pathways required for neuregulin-increased AChR gene expression may be completed within 1 min. After being activated, AChR gene transcription machinery remained active even in the absence of neuregulin.
To determine whether the ε-mRNA elevation reflects an increase in transcription of the ε-subunit gene, we studied the effect of actinomycin D, an inhibitor of RNA polymerases, on neuregulin’s action. Treatment of C2C12 myotubes with actinomycin D decreased the basal level of the ε-mRNA (data not shown). Pretreatment with actinomycin D prevented the neuregulin-mediated increase in the ε-mRNA in C2C12 myotubes, suggesting an important role of active transcription in increasing the AChR mRNA (Fig.3A). Furthermore, the addition of actinomycin D after neuregulin stimulation decreased the ε-mRNA, suggesting that the sustained elevation also requires active transcription (Fig. 3B). These results demonstrated that both the initial increase and the maintenance of the ε-mRNA depended on active transcription in C2C12 myotubes.
Fig. 3.

Inhibition of the neuregulin-mediated increase in AChR ε-mRNA by actinomycin D. C2C12 myotubes were pretreated with 5 μg/ml actinomycin D for 30 min before 1 nm neuregulin stimulation for 12 hr. In the late-addition experiments, C2C12 cells were stimulated with 1 nm neuregulin for 12 hr to increase the ε-mRNA. Actinomycin D (5 μg/ml) was then added in the medium. After incubation for another 4 hr, C2C12 myotubes were collected for Northern blotting using 20 μg of total RNA. A, Representative Northern blot radiograms. B, Histograms showing mean ± SD of two different samples. The mRNA levels in the absence of neuregulin were considered to be the control (100%). **p < 0.01 in comparison with the neuregulin-mediated increase in the absence of actinomycin D.
Induction of c-fos and c-jun immediate early genes by neuregulin
To understand neuregulin’s signaling mechanism further, we sought to identify genes whose expression was upregulated by neuregulin in C2C12 myotubes. We and others have demonstrated previously that neuregulin activates ERK MAP kinase that is required for AChR subunit gene expression (Si et al., 1996; Tansey et al., 1996; Altiok et al., 1997). Moreover, evidence is compelling that the Ras-ERK pathway regulates transcription of immediate early genes such asc-jun and c-fos in a variety of cells (Segal and Greenberg, 1996). We determine whether neuregulin regulatesc-fos and c-jun expression in C2C12 cells. Neuregulin had no apparent effect on the level of c-jun andc-fos mRNAs in C2C12 myoblasts (Fig.4A). However, bothc-jun and c-fos mRNAs were increased by neuregulin within 5 min in C2C12 myotubes (Fig. 4B). These results indicate that, as observed with AChR gene expression in C2C12 cells (Si et al., 1996), the neuregulin induction ofc-jun and c-fos genes depended on cell differentiation. The c-jun expression peaked at ∼10 min, whereas the c-fos expression maximized at ∼30 min after the stimulation. Both mRNAs returned to the basal level (i.e., before stimulation) within 90 min. The neuregulin-induced expression ofc-jun and c-fos had a concentration–response relationship (Fig. 4C) similar to that of neuregulin-induced AChR gene expression (Si et al., 1996).
Fig. 4.

ERK kinase–dependent increase inc-jun and c-fos mRNAs in C2C12 myotubes by neuregulin. A, Effect of neuregulin onc-jun and c-fos mRNA levels in myoblasts.B, Time course of NRG-induced expression of c-jun and c-fos in myotubes.C, Concentration–response relationship of neuregulin-induced expression of c-jun andc-fos in myotubes. The fold increase by neuregulin in C2C12 cells was 5.2 ± 0.28 for c-fos and 2.4 ± 0.11 for c-jun mRNAs. D, Dependence of neuregulin-induced expression of c-jun andc-fos on ERK kinase activation. C2C12 myoblasts were treated with 10 nm neuregulin, 10% serum, or vehicle (control) for 30 min (A). Myotubes were treated with 1 nm neuregulin for the indicated times (B) or with various concentrations of neuregulin for 30 min (C). C2C12 myotubes were pretreated with 40 or 80 μm PD98059 (lower concentration of each treatment in the left-handlane), 200 nm or 1 μmrapamycin, 200 nm or 1 μm wortmannin, or 10 nm or 1 μm tyrphostin AG1478 for 30 min before stimulation with 1 nm neuregulin for 30 min (D). Twenty micrograms of total RNA isolated from neuregulin-treated myotubes were resolved on a 1% agarose gel, blotted to nitrocellulose, and probed with [32P]-labeledc-jun or c-fos cDNA fragments.
To determine the signaling mechanism of neuregulin in the regulation of the expression of c-jun and c-fos genes, we studied the effect of several chemicals known to affect the activity of ErbB protein tyrosine kinases, ERK, and PI 3-kinase in C2C12 myotubes (J. Si and L. Mei, unpublished observation). Pretreatment with tyrphostin AG1478, a potent and selective inhibitor of ErbB protein kinases (Levitzki and Gazit, 1995), and PD98059, an inhibitor of MEK (Dudley et al., 1995), had no effect on the basal levels ofc-jun and c-fos mRNAs (data not shown) yet blocked the neuregulin-induced increase in c-jun andc-fos mRNAs in a concentration-dependent manner (Fig.4D). The inhibitory effect of tyrphostin AG1478 and PD98059 was specific because they had no effect on the GAPDH mRNA. In contrast, the PI 3-kinase inhibitor wortmannin (Carraway et al., 1995) and the S6 kinase inhibitor rapamycin (Chung et al., 1992) did not appear to affect the basal or neuregulin-induced c-jun andc-fos mRNA. These results demonstrated that neuregulin-induced expression of c-jun and c-fosgenes in C2C12 myotubes, like neuregulin-mediated AChR subunit gene expression (Si et al., 1996; Altiok et al., 1997), requires the activation of the ERK subgroup of MAP kinases but not PI 3-kinase.
Inhibition of neuregulin-induced ε-mRNA expression by cycloheximide
The similar concentration–response curves of neuregulin in the induction of immediate early genes and in the upregulation of AChR genes (Fig. 4C) (Si et al., 1996) and their dependence on ERK activation (Fig. 4D) (Si et al., 1996; Tansey et al., 1996; Altiok et al., 1997) indicated a strong correlation between these two events. Immediate early gene expression peaked at 10–30 min, which was after ERK activation (5–10 min) but before the increase in AChR mRNAs (8–12 hr). These results raised the possibility that AChR transcriptional activation may require the previous induction and expression of immediate early genes. To address this possibility, we determined whether neuregulin was capable of increasing AChR mRNA expression in the presence of cycloheximide, a protein synthesis inhibitor that is known to block expression of the proteins encoded by immediate early genes (Greenberg et al., 1986). Treatment of C2C12 myotubes with 10 μg/ml cycloheximide had no effect on basal expression of the ε-subunit (Fig.5A). However, pretreatment with cycloheximide completely inhibited the neuregulin-mediated increase in the ε-mRNA (Fig. 5A,B), suggesting that the ε-subunit gene transcription required de novo protein synthesis. The inhibitory effect of cycloheximide did not appear to result from its potential toxicity because GAPDH mRNA did not change in these cells. Because cycloheximide alone did not affect the basal level of the ε-mRNA, it is unlikely that the decrease in neuregulin-induced ε-mRNA by cycloheximide is attributable to inhibiting synthesis of an mRNA-stabilizing factor. In the latter case, treatment with cycloheximide should lead to an increase in ε-mRNA in the absence of neuregulin. In contrast, cycloheximide blocked the neuregulin-mediated increase in the ε-mRNA. These results suggest that products of immediate early genes may be involved in regulation of ε-mRNA expression. It is worth noting that neuregulin was still able to induce immediate early gene c-fos mRNA expression in the presence of cycloheximide (Fig. 5C) that does not requirede novo protein synthesis. Furthermore, the c-fosmRNA was superinduced by neuregulin in the presence of cycloheximide (Fig. 5C). Cycloheximide has been shown to increase the stability of immediate early gene mRNAs and thus to lead to a superinduction (Greenberg and Ziff, 1984; Greenberg et al., 1986).
Fig. 5.

Inhibition by cycloheximide of the neuregulin-mediated increase in the ε-mRNA. C2C12 myotubes were treated with or without 10 μg/ml cycloheximide (CH) for 30 min before neuregulin stimulation for the indicated times. Total RNA was isolated for Northern blot analysis using the respective probe for the ε-subunit or c-fos. The loading was uniform as evidenced by an equal amount of GAPDH mRNA.A, Representative Northern blot radiograms of the ε-mRNA. B, Histograms showing mean ± SD of three different samples. The mRNA levels in the absence of neuregulin were considered to be 100% for the indicated time treatment. **p < 0.01 in comparison with the neuregulin-mediated increase in the absence of cycloheximide.C, Representative Northern blot radiograms of thec-fos mRNA.
Requirement of c-JUN in neuregulin-induced ε-subunit gene expression
Because c-FOS has to form a heterodimer with c-JUN to be functional whereas c-JUN can form functional homodimers (Angel and Karin, 1991), inhibition of c-JUN function should affect the neuregulin induction of the ε-subunit gene if it depends on the function of immediate early gene products. Therefore, we next investigated the role of c-JUN in the regulation of AChR gene expression using a dominant-negative (D/N) approach. The c-JUN dominant-negative mutant TAM-67 lacks amino acids 3–122 and has been shown previously to function as a potent inhibitor of c-JUN–mediated transcriptional activation (Brown et al., 1994, 1996). Because of a low transient transfection efficiency in C2C12 cells, which made it difficult to study the effect of transfected c-JUN on expression of the endogenous AChR gene, we studied the effect of TAM-67 on the promoter-driven luciferase gene expression using the transgene ε416-Luc. ε416-Luc contains the 416 nucleotides of the 5′-flanking region of the ε-subunit gene fused with the luciferase gene downstream of the transcription initiation site (Si et al., 1997). The 416 nucleotides contain cis-elements required for neuregulin induction in C2C12 cells (Si et al., 1997) as well as synapse-specific expression (Duclert et al., 1993, 1996). C2C12 myoblasts were cotransfected with the transgene ε416-Luc with the parental vector pCMV, wild type (WT) c-JUN , or TAM-67. The parental vector pCMV or c-JUN wild type had no effect on the neuregulin-induced ε-transgene expression. In contrast, the D/N mutant TAM-67 blocked the neuregulin-induced ε-subunit gene expression in the C2C12 cells (Fig.6). These results argue that c-JUN is required for neuregulin-induced expression of the ε-subunit gene. Coexpression of the c-JUN wild-type protein decreased the basal transgene expression, probably because of repressing transcriptional activation by myogenic transcription factors. There are two E box elements within the 416 nucleotide 5′-flanking region (Si et al., 1997). Mutation of the proximal E box decreased the basal but not the neuregulin-induced expression in Sol 8 muscle cells (Chu et al., 1995). c-JUN can interact physically with MyoD (Bengal et al., 1992) and thus inhibit trans-activation of the muscle creatine kinase enhancer by myogenic factors (Bengal et al., 1992; Li et al., 1992).
Fig. 6.

Dependence of the neuregulin-induced expression of the AChR ε-transgene on c-JUN. C2C12 myoblasts were transiently cotransfected with ε416-Luc and pCMVβ without (CONTROL) or with pCMV (pCMV), pCMV–c-JUN [c-JUN(WT)], or pCMV-TAM-67 [c-JUN(D/N)]. Twenty-four hours after transfection, myoblasts were incubated with the DM to induce myotube formation. After another 48 hr, myotubes were stimulated with 1 nmNRG for 24 hr. Luciferase activity and β-galactosidase activity were assayed as described in Methods and Materials. The ratios of the relative luciferase or β-galactosidase activity in neuregulin-stimulated cells over that in control cells are shown. Histograms are mean ± SD of four different samples.
Activation of JNK by neuregulin is required for its induction of the ε-subunit gene expression
The function of c-JUN is regulated by phosphorylation of the N-terminal–transactivating domain by JNK, a subgroup of MAP kinases structurally related to ERK (Foletta, 1996). We determined whether neuregulin activates JNK in muscle cells. JNK was purified from control and neuregulin-stimulated C2C12 cells and assayed using a GST–c-JUN fusion protein as a substrate. Stimulation of C2C12 myotubes with neuregulin indeed increased the JNK activity in manners dependent on both concentration and time (Fig. 7). The JNK activation by neuregulin peaked at 10 min after stimulation and at 100 pm, which were ∼2.2- to 2.6-fold above the basal (Fig. 7).
Fig. 7.
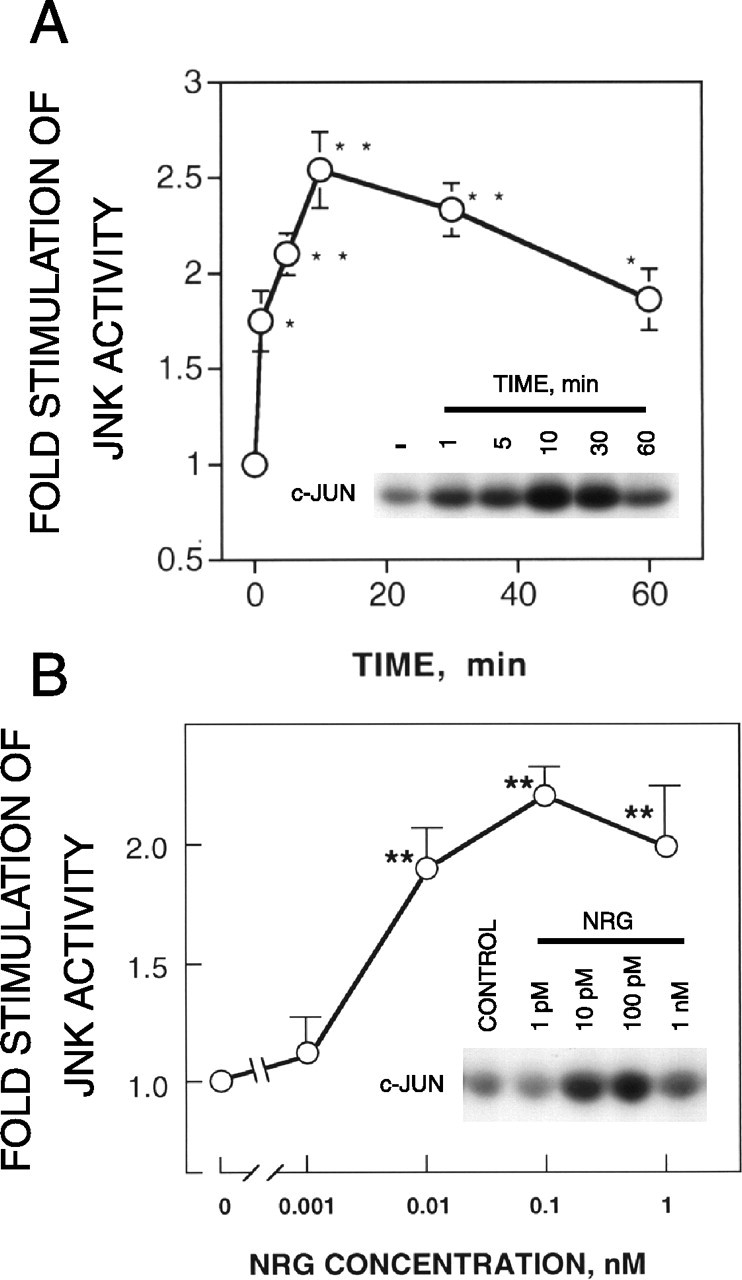
Activation of JNK in C2C12 myotubes by neuregulin. C2C12 myotubes were stimulated with 1 nm neuregulin for the indicated times (A) or with various concentrations of neuregulin for 10 min (B) and then harvested in the lysis buffer. To purify JNK, we incubated an aliquot of lysate with agarose beads containing the GST–c-JUN fusion protein at 4°C for 2 hr. After washing, the beads were incubated in the kinase assay buffer containing [γ-32P]ATP at 30°C for 15 min. The kinase reaction was stopped by the addition of the sample-loading buffer and subjected to electrophoresis on a 10% SDS-polyacrylamide gel. Shown are means ± SD of five different samples. The JNK activity levels in the absence of neuregulin were considered to be 100%. The inset of eachpanel shows a representative radiogram. *p < 0.05; **p < 0.01.
To explore the role of JNK in the regulation of AChR gene expression, we studied effects of a JNK mutant on ε416-Luc expression. The JNK1(ALF) mutant is kinase deficient because its tripeptide dual phosphorylation motif Thr-Pro-Tyr has been mutated to Ala-Leu-Phe and thus cannot be activated by its upstream kinase (Gupta et al., 1995). The mutant and wild-type JNK were cotransfected in C2C12 cells with ε416-Luc. As shown in Figure 8, the JNK kinase–deficient mutant blocked neuregulin-induced expression of the ε-transgene, whereas the wild type had no apparent effect, suggesting an essential role of JNK or a related kinase in neuregulin regulation of the ε-subunit gene. MKK4 is a kinase upstream of JNK that phosphorylates and thus activates JNK (Natali et al., 1992; Sanchez et al., 1994). To address further the importance of JNK or a related kinase in the regulation of AChR gene expression by neuregulin, a mutant MKK4 was introduced into C2C12 cells, and its effect on neuregulin-induced expression of ε416-Luc was assessed. The MKK4(Ala) mutant, in which Ser257 and Thr261 are converted to Ala residues, cannot be phosphorylated or activated and thus functions as a dominant-negative mutant of MKK4 (Whitmarsh et al., 1995). Overexpression of MKK4 inhibits activation of JNK in mammalian cells (Whitmarsh et al., 1995). When it was cotransfected with ε416-Luc, MKK4(Ala) effectively inhibited neuregulin-mediated induction of the ε-promoter activity (Fig. 8).
Fig. 8.
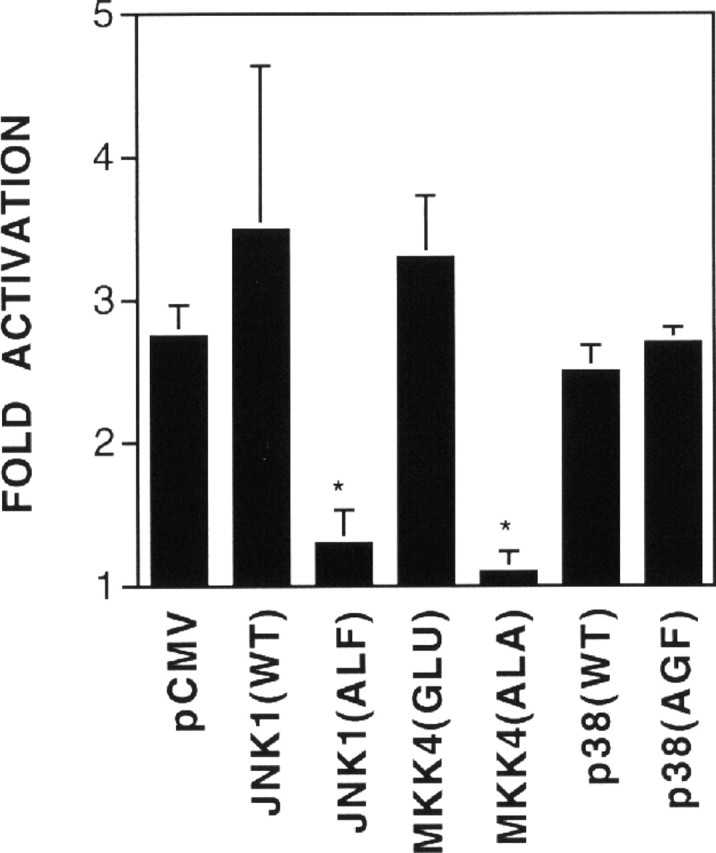
Requirement of JNK in neuregulin-induced AChR ε-transgene expression. C2C12 myoblasts were transiently transfected with ε416-Luc and pCMVβ with pCMV, pCMV–JNK1(WT), pCMV–JNK1(ALF), pCMV–MKK4(Glu), pCMV–MKK4(Ala), pCMV–p38(WT), or pCMV–p38(AGF). Twenty-four hours after transfection, myoblasts were incubated with the DM to induce myotube formation. After another 48 hr, myotubes were stimulated with 1 nm neuregulin for 24 hr and then harvested for assays of luciferase and β-galactosidase activity. The fold of the relative luciferase or β-galactosidase activity in neuregulin-stimulated cells over that in control cells is shown. Histograms are mean ± SD of four different samples. *p < 0.05.
In addition to JNK, p38 MAP kinase is another subgroup of MAP kinases whose activity is activated by stress, proinflammatory cytokines, and lipopolysaccharide (Han et al., 1994; Raingeaud et al., 1995). We tested whether p38 MAP kinase is a regulator of neuregulin-mediated expression of AChR genes. P38 kinase was measured in an immune complex kinase assay using ATF2 as a substrate. Neuregulin did not appear to activate the p38 MAP kinase in C2C12 myotubes (data not shown). Treatment of C2C12 myotubes with SB202190, an inhibitor of p38 (Lee et al., 1994), had no apparent effect on neuregulin-induced expression of the ε-mRNA (data not shown). Coexpression of the p38 dominant-negative mutant p38(AGF) had no apparent effect on the ε-transgene transcription activity (Fig. 8). These results suggest that p38 MAP kinase may not contribute to the regulation of AChR gene expression by neuregulin.
Having demonstrated the important role of JNK in the neuregulin-induced ε-transgene expression, we next asked whether JNK is also a critical regulator of the expression of the endogenous ε-subunit gene in response to neuregulin. To address this issue, the wild type or dominant-negative mutant JNK were introduced into C2C12 cells by transfection. Both constructs were tagged with a FLAG epitope. Neomycin-resistant clones were screened for expression of JNK proteins by Western blot analysis with anti-FLAG antibodies. We have obtained C2C12 cell lines that stably expressed the wild type or mutant of JNK1. Figure 9A shows a Western blot demonstrating expression of wild type and mutant JNK1 in two representative cell lines. Similar levels of expression were obtained in other stable cell lines (data not shown). No apparent morphological changes were observed when the JNK wild-type or mutant-expressing C2C12 cells were compared with the parental cells or the cells transfected with the empty vector. Stable expression of the JNK wild type or mutant did not appear to affect differentiation from myoblasts to myotubes. We examined neuregulin-induced ε-mRNA expression in two cell lines that stably expressed wild-type JNK1 and four cell lines that stably expressed mutant JNK1. As shown in Figure 9B, the wild-type JNK1 had no apparent effect on neuregulin’s effect; however, the dominant-negative mutant inhibited the neuregulin-induced expression of the endogenous ε-mRNA. A summary of three independent Northern blot analyses of parental, control (pCMV-transfected), JNK1(WT)#2, and JNK1(ALF)#1 cells is presented in Figure 9C. These results were in agreement with those with the ε-transgene transcription in transient expression experiments, supporting the notion that activation of JNK is required for neuregulin-induced expression of the ε-subunit gene.
Fig. 9.

Attenuation of neuregulin-induced expression of endogenous ε-mRNA by dominant-negative JNK1. C2C12 myoblasts were transfected with pCMV, pCMV–JNK1(WT), or pCMV–JNK1(ALF). All DNA constructs contained the G418-resistant gene neo. The FLAG epitope was tagged between the codons 1 and 2 of JNK. G418-resistant clones were isolated and characterized for JNK1 expression and AChR gene expression in response to NRG.A, Western blot analysis showing expression of wild-type and mutant JNK1 in the parental and stable cell lines JNK1(WT)#2 and JNK1(ALF)#1. Forty micrograms of proteins extracted from the indicated myotubes were resolved on a 10% SDS-PAGE gel, transferred, and blotted with anti-FLAG antibodies. Similar levels of JNK1 expression were observed in other stable cell lines described in B.B, Northern blot analysis. Twenty micrograms of total RNA were used. The loading was uniform as evidenced by an equal amount of GAPDH mRNA. C, Histograms showing mean ± SD of three different samples. The mRNA level of the parental cell line in the absence of neuregulin was considered to be 100%. **p < 0.01 in comparison with the neuregulin-mediated increase in parental, pCMV, or JNK1(WT)#2 cells.
DISCUSSION
Neuregulin-mediated expression of the AChR ε-subunit gene in muscle cells requires c-JUN and JNK in addition to the previously documented ERK. First, neuregulin induced expression of immediate early genes c-jun and c-fos. Expression ofc-jun and c-fos by neuregulin followed and depended on ERK activation, which has been demonstrated to be essential for neuregulin-upregulated AChR gene expression. Second, inhibition of protein synthesis or blockade of the c-JUN function by a c-JUN dominant-negative mutant attenuated or abolished neuregulin-mediated transcription from the ε-promoter. Third, neuregulin activated JNK in muscle cells. Blockade of JNK activation by overexpressing dominant-negative MKK4 inhibited ε-promoter activation. Moreover, overexpression of the JNK dominant-negative mutant inhibited neuregulin-mediated expression of the ε-transgene and endogenous ε-mRNA in muscle cells. These results suggest that neuregulin activates multiple signaling cascades that converge to regulate AChR subunit gene expression.
In contrast to the transient induction of immediate early genes likec-fos and c-jun, the induction of the ε-mRNA was delayed and persisted after neuregulin treatment in C2C12 myotubes. This time course is similar to that of other delayed-response gene products such as VGF (Levi et al., 1985; Salton et al., 1991), GAP43 (Federoff et al., 1988), transin (Machida et al., 1989), and COS-1 (Kaplan et al., 1997) whose expression is induced by NGF or basic FGF (bFGF) in pheochromocytoma 12 cells. Expression of these messages is also inhibited by cycloheximide. The mechanisms concerning how these delayed-response genes are regulated remain primarily unknown. NGF and bFGF both cause a prolonged activation of Ras resulting in a prolonged increase of ERK activity (Kaplan et al., 1997). The prolonged ERK activation leads to the phosphorylation of the cAMP regulatory element-binding protein for up to several hours that, along with immediate early gene products, leads to subsequent delayed-response gene expression (Bonni et al., 1995; Segal and Greenberg, 1996). The regulatory mechanisms of the AChR ε-subunit gene may be different from those induced by NGF or bFGF because neuregulin activated ERK transiently (Fig. 2).
The c-JUN and c-FOS proteins are the cellular homologs of viral oncogenes. The c-JUN proteins are able to dimerize among themselves or form a heterodimer with c-FOS, whereas c-FOS has to dimerize with c-JUN to form a dimer. These homo- or heterodimers are major components of the activator protein-1 (AP-1) transcription factor (Rauscher et al., 1989; Angel and Karin, 1991; Foletta, 1996). AP-1 plays a role in the regulation of both basal and inducible transcription of various genes in response to growth factors, cytokines, tumor promoters, and carcinogens. c-JUN may directly regulate neuregulin-induced AChR gene expression by binding to the promoter region of the ε-subunit gene. That c-JUN affects the neuregulin-induced ε416-Luc transgene expression in C2C12 myotubes may suggest that the element(s) required for the c-JUN effects, either direct or indirect, lies within the 416 nucleotides in the 5′-flanking region of the ε-subunit gene. A careful examination that failed to identify the palindromic sequence 5′TGAg/cTCA, the consensus binding site for AP-1, in this region does not support the notion that c-JUN regulates AChR gene expression by binding to a typical AP-1 element in the 5′-flanking region. An alternative hypothesis is that c-JUN interacts with other transcription factors to regulate AChR gene expression and thus does not require a direct interaction with a cis-element.
The ETS family of transcription factors may be candidate factors whose function is regulated by c-JUN. Recent studies indicate that ETS2 and GABP, both members of the ETS family, are required for neuregulin-induced and synapse-specific expression of the ε-subunit gene (Fromm and Burden, 1998; Sapru et al., 1998; Schaeffer et al., 1998). In a recent report, ETS2 has been found to associate strongly with the c-JUN–c-FOS dimer, and such an interaction is enhanced by promoter DNA containing the ets sites but does not require the AP-1 site (Basuyaux et al., 1997). Thus, c-JUN may regulate the ε-promoter transcription via interacting with the ETS family of proteins. It is worth noting that overexpression of the wild-type c-JUN or the MKK4 or JNK active mutant did not increase the basal level of ε-promoter transcription, suggesting that c-JUN, MKK4, or JNK is not sufficient to upregulate AChR gene expression. Previous studies have demonstrated that angiotensin II stimulates AP-1–driven transcription and c-JUN–c-FOS heterodimer DNA-binding activity in C2C12 cells (Puri et al., 1995). Treatment of C2C12 myotubes with angiotensin II had no effect on the basal or neuregulin-upregulated expression of the AChR ε-subunit (data not shown). These results suggest that c-JUN and/or c-FOS and JNK are required for but not sufficient to mediate the upregulation of AChR gene expression by neuregulin. It is also possible that c-JUN activates transcription of another factor that in turn activates ε-subunit gene expression.
Synaptic nuclei continue to express the AChR ε-subunit many weeks after the nerve has been removed (Brenner et al., 1990), suggesting an imprint signal capable of inducing AChR expression in the absence of the nerve. This imprint signal is believed to be neuregulin concentrated in the extracellular matrix at the neuromuscular junction. However, denervation decreases dramatically the neuregulin-like immunoreactivity at the neuromuscular junction (Sandrock et al., 1995). Yet the accumulation of AChR mRNA, especially the ε-mRNA, is not dependent on the continued presence of the nerve terminal (Goldman and Staple, 1989; Brenner et al., 1990). Our observation of the sustained increase in the AChR mRNA by a brief neuregulin stimulation supports the “imprinting” hypothesis. Clearly, the elevation was not caused by a continuous activation of the ErbB kinases because their activity, monitored by tyrosine phosphorylation, decreased within 15 min after and returned to the basal level within 60 min of neuregulin stimulation (Si et al., 1996). Concomitantly, the ERK kinase activation was transient by either continuous or transient stimulation of C2C12 cells with neuregulin. A simple explanation of this phenomenon is that the AChR mRNA may be stable and, after being synthesized, remains intact in the muscle cells for a longer period of time. Although this possibility cannot be ruled out in the present study; the fact that actinomycin D, an inhibitor of transcription, inhibited the neuregulin-elevated AChR mRNAs suggests a role of continuous active transcription in the maintenance of the AChR mRNA. Presumably, the intracellular neuregulin pathway, after being activated, may remain functional without further stimulation from extracellular neuregulin. The prolonged transcription should be mediated by a covalent modulation of transcription regulators in nuclei because the activation of ERK by neuregulin was transient and the phosphorylated MAP kinases were quickly translocated into nuclei in C2C12 myotubes (Si and Mei, unpublished observation). Potential candidate transcription factors are members of the ETS family, whose phosphorylation regulates expression of AChR genes (Sapru et al., 1998;Schaeffer et al., 1998). Alternatively and/or probably in addition, neuregulin may activate novel signaling events to sustain the transcription of AChR genes. The finding that c-JUN and its phosphorylation are required for neuregulin-upregulated expression of the AChR ε-subunit gene suggests that the immediate early gene products play an role in maintenance of the AChR ε-mRNA in the absence of neuregulin.
Footnotes
This work was supported by the National Institutes of Health and the National Institute of Neurological Disorders and Stroke Grant NS34062 and grants from the Muscular Dystrophy Association and the March of Dimes Birth Defects Foundation to L.M. We are grateful to Drs. M. Sliwkowski, G. S. Feng, R. Huganir, T. Curran, A. Langley, R. Davis, and M. Birrer for valuable reagents and to Dr. Wen C. Xiong and Sandra Won for discussion and suggestions.
Correspondence should be addressed to Dr. Lin Mei, Department of Neurobiology, University of Alabama at Birmingham, CIRC 5th Floor, 1530 3rd Avenue South, Birmingham, AL 35294-0021.
REFERENCES
- 1.Altiok N, Bessereau J-L, Changeux J-P. ErbB3 and ErbB2/neu mediate the effect of heregulin on acetylcholine receptor gene expression in muscle: differential expression at the endplate. EMBO J. 1995;14:4258–4266. doi: 10.1002/j.1460-2075.1995.tb00100.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Altiok N, Altiok S, Changeux JP. Heregulin-stimulated acetylcholine receptor gene expression in muscle: requirement for MAP kinase and evidence for a parallel inhibitory pathway independent of electrical activity. EMBO J. 1997;16:717–725. doi: 10.1093/emboj/16.4.717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Angel P, Karin M. The role of Jun, Fos and the AP-1 complex in cell-proliferation and transformation. Biochim Biophys Acta. 1991;1072:129–157. doi: 10.1016/0304-419x(91)90011-9. [DOI] [PubMed] [Google Scholar]
- 4.Basuyaux JP, Ferreira E, Stehelin D, Buttice G. The Ets transcription factors interact with each other and with the c-Fos/c-Jun complex via distinct protein domains in a DNA-dependent and -independent manner. J Biol Chem. 1997;272:26188–26195. doi: 10.1074/jbc.272.42.26188. [DOI] [PubMed] [Google Scholar]
- 5.Bengal E, Ransone L, Scharfmann R, Dwarki VJ, Tapscott SJ, Weintraub H, Verma I. Functional antagonism between c-Jun and MyoD proteins: a direct physical association. Cell. 1992;68:507–519. doi: 10.1016/0092-8674(92)90187-h. [DOI] [PubMed] [Google Scholar]
- 6.Bonni A, Ginty DD, Dudek H, Greenberg ME. Serine 133-phosphorylated CREB induces transcription via a cooperative mechanism that may confer specificity to neurotrophin signals. Mol Cell Neurosci. 1995;6:168–183. doi: 10.1006/mcne.1995.1015. [DOI] [PubMed] [Google Scholar]
- 7.Bradford MM. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem. 1976;72:248–254. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- 8.Brenner HR, Witzemann V, Sakmann B. Imprinting of acetylcholine receptor messenger RNA accumulation in mammalian neuromuscular synapses. Nature. 1990;344:544–547. doi: 10.1038/344544a0. [DOI] [PubMed] [Google Scholar]
- 9.Brown PH, Chen TK, Birrer MJ. Mechanism of action of a dominant-negative mutant of c-Jun. Oncogene. 1994;9:791–799. [PubMed] [Google Scholar]
- 10.Brown PH, Kim SH, Wise SC, Sabichi AL, Birrer MJ. Dominant-negative mutants of cJun inhibit AP-1 activity through multiple mechanisms and with different potencies. Cell Growth Differ. 1996;7:1013–1021. [PubMed] [Google Scholar]
- 11.Carraway KL, Soltoff SP, Diamonti AJ, Cantley LC. Heregulin stimulates mitogenesis and phosphatidylinositol 3-kinase in mouse fibroblasts transfected with erbB2/neu and erbB3. J Biol Chem. 1995;270:7111–7116. doi: 10.1074/jbc.270.13.7111. [DOI] [PubMed] [Google Scholar]
- 12.Chomczynski P, Sacchi N. Single-step method of RNA isolation by acid guanidinium thiocyanate-phenol-chloroform extraction. Anal Biochem. 1987;162:156–159. doi: 10.1006/abio.1987.9999. [DOI] [PubMed] [Google Scholar]
- 13.Chu GC, Moscoso LM, Sliwkowski MX, Merlie JP. Regulation of the acetylcholine receptor e subunit gene by recombinant ARIA: an in vitro model for transynaptic gene regulation. Neuron. 1995;14:329–339. doi: 10.1016/0896-6273(95)90289-9. [DOI] [PubMed] [Google Scholar]
- 14.Chung J, Kuo CJ, Crabtree GR, Blenis J. Rapamycin-FKBP specifically blocks growth-dependent activation of and signaling by the 70 kd S6 protein kinase. Cell. 1992;69:1227–1237. doi: 10.1016/0092-8674(92)90643-q. [DOI] [PubMed] [Google Scholar]
- 15.Corfas G, Falls DL, Fischbach GD. ARIA, a protein that stimulates acetylcholine receptor synthesis, also induces tyrosine phosphorylation of a 185-kDa muscle transmembrane protein. Proc Natl Acad Sci USA. 1993;90:1624–1628. doi: 10.1073/pnas.90.4.1624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Curran T, Gordon MB, Rubino KL, Sambucetti LC. Isolation and characterization of the c-fos(rat) cDNA and analysis of post-translational modification in vitro. Oncogene. 1987;2:79–84. [PubMed] [Google Scholar]
- 17.Duclert A, Savatier N, Changeux JP. An 83-nucleotide promoter of the acetylcholine receptor epsilon-subunit gene confers preferential synaptic expression in mouse muscle. Proc Natl Acad Sci USA. 1993;90:3043–3047. doi: 10.1073/pnas.90.7.3043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Duclert A, Savatier N, Schaeffer L, Changeux J-P. Identification of an element crucial for the sub-synaptic expression of the acetylcholine receptor epsilon-subunit gene. J Biol Chem. 1996;271:17433–17438. doi: 10.1074/jbc.271.29.17433. [DOI] [PubMed] [Google Scholar]
- 19.Dudley DT, Pang L, Decker SJ, Bridges AJ, Saltiel AR. A synthetic inhibitor of the mitogen-activated protein kinase cascade. Proc Natl Acad Sci USA. 1995;92:7686–7689. doi: 10.1073/pnas.92.17.7686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Falls DL, Rosen KM, Corfas G, Lane WS, Fischbach GD. ARIA, a protein that stimulates acetylcholine receptor synthesis, is a member of the Neu ligand family. Cell. 1993;72:801–815. doi: 10.1016/0092-8674(93)90407-h. [DOI] [PubMed] [Google Scholar]
- 21.Federoff HJ, Grabczyk E, Fishman MC. Dual regulation of GAP-43 gene expression by nerve growth factor and glucocorticoids. J Biol Chem. 1988;263:19290–19295. [PubMed] [Google Scholar]
- 22.Fischbach GD, Cohen SA. The distribution of acetylcholine sensitivity over uninnervated and innervated muscle fibers grown in cell culture. Dev Biol. 1973;31:147–162. doi: 10.1016/0012-1606(73)90326-6. [DOI] [PubMed] [Google Scholar]
- 23.Fischbach GD, Rosen KM. ARIA: a neuromuscular junction neuregulin. Annu Rev Neurosci. 1997;20:429–458. doi: 10.1146/annurev.neuro.20.1.429. [DOI] [PubMed] [Google Scholar]
- 24.Foletta VC. Transcription factor AP-1, and the role of Fra-2. Immunol Cell Biol. 1996;74:121–133. doi: 10.1038/icb.1996.17. [DOI] [PubMed] [Google Scholar]
- 25.Froehner SC. Regulation of ion channel distribution at synapses. Annu Rev Neurosci. 1993;16:347–368. doi: 10.1146/annurev.ne.16.030193.002023. [DOI] [PubMed] [Google Scholar]
- 26.Fromm L, Burden SJ. Synapse-specific and neuregulin-induced transcription require an Ets site that binds GABPa/GABPb. Genes Dev. 1998;12:3074–3083. doi: 10.1101/gad.12.19.3074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Goldman D, Staple J. Spatial and temporal expression of acetylcholine receptor RNAs in innervated and denervated rat soleus muscle. Neuron. 1989;3:219–228. doi: 10.1016/0896-6273(89)90035-4. [DOI] [PubMed] [Google Scholar]
- 28.Goodearl ADJ, Yee AG, Sandrock AW, Jr, Corfas G, Fischbach GD. ARIA is concentrated in the synaptic basal lamina of the developing chick neuromuscular junction. J Cell Biol. 1995;130:1423–1434. doi: 10.1083/jcb.130.6.1423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Greenberg ME, Ziff EB. Stimulation of 3T3 cells induces transcription of the c-fos proto-oncogene. Nature. 1984;311:433–438. doi: 10.1038/311433a0. [DOI] [PubMed] [Google Scholar]
- 30.Greenberg ME, Hermanowski AL, Ziff EB. Effect of protein synthesis inhibitors on growth factor activation of c-fos, c-myc, and actin gene transcription. Mol Cell Biol. 1986;6:1050–1057. doi: 10.1128/mcb.6.4.1050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gundersen K, Sanes JR, Merlie JP. Neural regulation of muscle acetylcholine receptor epsilon- and alpha-subunit gene promoters in transgenic mice. J Cell Biol. 1993;123:1535–1544. doi: 10.1083/jcb.123.6.1535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gupta S, Campbell D, Derijard B, Davis RJ. Transcription factor ATF2 regulation by the JNK signal transduction pathway. Science. 1995;267:389–393. doi: 10.1126/science.7824938. [DOI] [PubMed] [Google Scholar]
- 33.Hall ZW, Sanes JR. Synaptic structure and development: the neuromuscular junction. Cell [Suppl] 1993;72:99–121. doi: 10.1016/s0092-8674(05)80031-5. [DOI] [PubMed] [Google Scholar]
- 34.Han J, Lee JD, Bibbs L, Ulevitch RJ. A MAP kinase targeted by endotoxin and hyperosmolarity in mammalian cells. Science. 1994;265:808–811. doi: 10.1126/science.7914033. [DOI] [PubMed] [Google Scholar]
- 35.Holmes WE, Sliwkowski MX, Akita RW, Kenzel WJ, Lee J, Park JW, Yansura D, Abadi N, Raab H, Lewis GD, Shepard HM, Huang WJ, Wood WJ, Goeddel DV, Vandlen RL. Identification of heregulin, a specific activator of p185erbB2. Science. 1992;256:1205–1210. doi: 10.1126/science.256.5060.1205. [DOI] [PubMed] [Google Scholar]
- 36.Jessell TM, Siegel RE, Fischbach GD. Induction of acetylcholine receptors on cultured skeletal muscle by a factor extracted from brain and spinal cord. Proc Natl Acad Sci USA. 1979;76:5397–5401. doi: 10.1073/pnas.76.10.5397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Jo SA, Zhu X, Marchionni MA, Burden SJ. Neuregulins are concentrated at nerve-muscle synapses and activate ACh-receptor gene expression. Nature. 1995;373:158–161. doi: 10.1038/373158a0. [DOI] [PubMed] [Google Scholar]
- 38.Kaplan MD, Olschowka JA, O’Banion MK. Cyclooxygenase-1 behaves as a delayed response gene in PC12 cells differentiated by nerve growth factor. J Biol Chem. 1997;272:18534–18537. doi: 10.1074/jbc.272.30.18534. [DOI] [PubMed] [Google Scholar]
- 39.Klarsfeld A, Daubas P, Bourachot B, Changeux JP. A 5′ flanking region of the chicken acetylcholine receptor alpha-subunit gene confers tissue-specificity and developmental control of expression in transfected cells. Mol Cell Biol. 1987;7:951–955. doi: 10.1128/mcb.7.2.951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lee JC, Laydon JT, McDonnell PC, Gallagher TF, Kumar S, Green D, McNulty D, Blumenthal MJ, Heys JR, Landvatter SW, Strickler JE, McLaughlin MM, Siemens IR, Fisher SM, Livi GR, White JR, Adams JL, Young PR. A protein kinase involved in the regulation of inflammatory cytokine biosynthesis. Nature. 1994;372:739–746. doi: 10.1038/372739a0. [DOI] [PubMed] [Google Scholar]
- 41.Lehrach H, Diamond D, Worney JM, Boedtker H. RNA molecular weight determinations by gel electrophoresis under denaturing conditions. Biochemistry. 1977;16:4743–4751. doi: 10.1021/bi00640a033. [DOI] [PubMed] [Google Scholar]
- 42.Levi A, Eldridge JD, Paterson BM. Molecular cloning of a gene sequence regulated by nerve growth factor. Science. 1985;229:393–395. doi: 10.1126/science.3839317. [DOI] [PubMed] [Google Scholar]
- 43.Levitzki A, Gazit A. Tyrosine kinase inhibition: an approach to drug development. Science. 1995;267:1782–1788. doi: 10.1126/science.7892601. [DOI] [PubMed] [Google Scholar]
- 44.Li L, Chambard JC, Karin M, Olson EN. Fos and Jun repress transcriptional activation by myogenin and MyoD: the amino terminus of Jun can mediate repression. Genes Dev. 1992;6:676–689. doi: 10.1101/gad.6.4.676. [DOI] [PubMed] [Google Scholar]
- 45.Loeb JA, Fischbach GD. ARIA can be released from extracellular matrix through cleavage of a heparin-binding domain. J Cell Biol. 1995;130:127–135. doi: 10.1083/jcb.130.1.127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Machida CM, Rodland KD, Matrisian L, Magun BE, Ciment G. NGF induction of the gene encoding the protease transin accompanies neuronal differentiation in PC12 cells. Neuron. 1989;2:1587–1596. doi: 10.1016/0896-6273(89)90047-0. [DOI] [PubMed] [Google Scholar]
- 47.Marchionni MA, Goodearl ADJ, Chen MS, Bermingham-McDonogh O, Kirk C, Hendricks M, Danehy F, Misumi D, Sudhalter J, Kobayashi K, Wroblewski D, Lynch C, Baldassare M, Hiles I, Davis JB, Hsuan JJ, Totty NF, Otsu M, McBurney RN, Waterfield MD, Stroobant P, Gwynne D. Glial growth factors are alternatively spliced erbB2 ligands expressed in the nervous system. Nature. 1993;362:312–318. doi: 10.1038/362312a0. [DOI] [PubMed] [Google Scholar]
- 48.Martinou JC, Falls DL, Fischbach GD, Merlie JP. Acetylcholine receptor-inducing activity stimulates expression of the epsilon-subunit gene of the muscle acetylcholine receptor. Proc Natl Acad Sci USA. 1991;88:7669–7673. doi: 10.1073/pnas.88.17.7669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Natali PG, Nicotra MR, Sures I, Mottolese M, Botti C, Ullrich A. Breast cancer is associated with loss of the c-kit oncogene product. Int J Cancer. 1992;52:713–717. doi: 10.1002/ijc.2910520508. [DOI] [PubMed] [Google Scholar]
- 50.Puri PL, Avantaggiati ML, Burgio VL, Chirillo P, Collepardo D, Natoli G, Balsano C, Levrero M. Reactive oxygen intermediates mediate angiotensin II-induced c-Jun·c-Fos heterodimer DNA binding activity and proliferative hypertrophic responses in myogenic cells. J Biol Chem. 1995;270:22129–22134. doi: 10.1074/jbc.270.38.22129. [DOI] [PubMed] [Google Scholar]
- 51.Raingeaud J, Gupta S, Rogers JS, Dickens M, Han J, Ulevitch RJ, Davis RJ. Pro-inflammatory cytokines and environmental stress cause p38 mitogen-activated protein kinase activation by dual phosphorylation on tyrosine and threonine. J Biol Chem. 1995;270:7420–7426. doi: 10.1074/jbc.270.13.7420. [DOI] [PubMed] [Google Scholar]
- 52.Rauscher FJ, III, Voulalas PJ, Franza BR, Jr, Curran T. Fos and Jun bind cooperatively to the AP-1 site: reconstitution in vitro. Genes Dev. 1988;2:1687–1699. doi: 10.1101/gad.2.12b.1687. [DOI] [PubMed] [Google Scholar]
- 53.Salton SR, Fischberg DJ, Dong KW. Structure of the gene encoding VGF, a nervous system-specific mRNA that is rapidly and selectively induced by nerve growth factor in PC12 cells. Mol Cell Biol. 1991;11:2335–2349. doi: 10.1128/mcb.11.5.2335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Sambrook J, Fritsch EF, Maniatis T. Molecular cloning: a laboratory manual. Cold Spring Harbor Laboratory; Cold Spring Harbor, NY: 1989. [Google Scholar]
- 55.Sanchez I, Hughes RT, Mayer BJ, Yee K, Woodgett JR, Avruch J, Kyriakis JM, Zon LI. Role of SAPK/ERK kinase-1 in the stress-activated pathway regulating transcription factor c-Jun. Nature. 1994;372:794–798. doi: 10.1038/372794a0. [DOI] [PubMed] [Google Scholar]
- 56.Sandrock AW, Dryer SE, Rosen KM, Gozani SN, Kramer R, Theill LE, Fischbach GD. Maintenance of acetylcholine receptor number by neuregulins at the neuromuscular junction in vivo. Science. 1997;276:599–603. doi: 10.1126/science.276.5312.599. [DOI] [PubMed] [Google Scholar]
- 57.Sandrock AW, Jr, Goodearl AD, Yin QW, Chang D, Fischbach GD. ARIA is concentrated in nerve terminals at neuromuscular junctions and at other synapses. J Neurosci. 1995;15:6124–6136. doi: 10.1523/JNEUROSCI.15-09-06124.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Sanes JR, Lichtman JW. Development of the vertebrate neuromuscular junction. Annu Rev Neurosci. 1999;22:389–442. doi: 10.1146/annurev.neuro.22.1.389. [DOI] [PubMed] [Google Scholar]
- 59.Sanes JR, Johnson YR, Kotzbauer PT, Mudd J, Hanley T, Martinou JC, Merlie JP. Selective expression of an acetylcholine receptor-lacZ transgene in synaptic nuclei of adult muscle fibers. Development. 1991;113:1181–1191. doi: 10.1242/dev.113.4.1181. [DOI] [PubMed] [Google Scholar]
- 60.Sapru MK, Florance SK, Kirk C, Goldman D. Identification of a neuregulin and protein-tyrosine phosphatase response element in the nicotinic acetylcholine receptor e subunit gene: regulatory role of an ets transcription factor. Proc Natl Acad Sci USA. 1998;95:1289–1294. doi: 10.1073/pnas.95.3.1289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Schaeffer L, Duclert N, Huchet-Dymanus M, Changeux J-P. Implication of a multisubunit Ets-related transcription factor in synaptic expression of the nicotinic acetylcholine receptor. EMBO J. 1998;17:3078–3090. doi: 10.1093/emboj/17.11.3078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Segal RA, Greenberg ME. Intracellular signaling pathways activated by neurotrophic factors. Annu Rev Neurosci. 1996;19:463–489. doi: 10.1146/annurev.ne.19.030196.002335. [DOI] [PubMed] [Google Scholar]
- 63.Si J, Mei L. ERK MAP kinase activation is required for ARIA-induced increase in all five AChR subunit mRNAs as well as synapse-specific expression of the AChR epsilon-transgene. Mol Brain Res. 1999;67:18–27. doi: 10.1016/s0169-328x(99)00028-5. [DOI] [PubMed] [Google Scholar]
- 64.Si J, Luo Z, Mei L. Induction of acetylcholine receptor gene expression by ARIA requires activation of mitogen-activated protein kinase. J Biol Chem. 1996;271:19752–19759. doi: 10.1074/jbc.271.33.19752. [DOI] [PubMed] [Google Scholar]
- 65.Si J, Miller DS, Mei L. Identification of an element required for acetylcholine receptor-inducing activity (ARIA)-induced expression of the acetylcholine receptor epsilon subunit gene. J Biol Chem. 1997;272:10367–10371. doi: 10.1074/jbc.272.16.10367. [DOI] [PubMed] [Google Scholar]
- 66.Simon AM, Hoppe P, Burden SJ. Spatial restriction of AChR gene expression to subsynaptic nuclei. Development. 1992;114:545–553. doi: 10.1242/dev.114.3.545. [DOI] [PubMed] [Google Scholar]
- 67.Tang J, Jo SA, Burden SJ. Separate pathways for synapse-specific and electrical activity-dependent gene expression in skeletal muscle. Development. 1994;120:1799–1804. doi: 10.1242/dev.120.7.1799. [DOI] [PubMed] [Google Scholar]
- 68.Tansey MG, Chu GC, Merlie JP. ARIA/HRG regulates AChR epsilon subunit gene expression at the neuromuscular synapse via activation of phosphatidylinositol 3-kinase and Ras/MAPK pathway. J Cell Biol. 1996;134:465–476. doi: 10.1083/jcb.134.2.465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Usdin TB, Fischbach GD. Purification and characterization of a polypeptide from chick brain that promotes the accumulation of acetylcholine receptors in chick myotubes. J Cell Biol. 1986;103:493–507. doi: 10.1083/jcb.103.2.493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Wen D, Peles E, Cupples R, Suggs SV, Bacus SS, Luo Y, Trail G, Hu S, Silbiger SM, Levy RB, Koski RA, Lu HS, Yarden Y. Neu differentiation factor: a transmembrane glycoprotein containing an EGF domain and an immunoglobulin homology unit. Cell. 1992;69:559–572. doi: 10.1016/0092-8674(92)90456-m. [DOI] [PubMed] [Google Scholar]
- 71.Whitmarsh AJ, Shore P, Sharrocks AD, Davis RJ. Integration of MAP kinase signal transduction pathways at the serum response element. Science. 1995;269:403–407. doi: 10.1126/science.7618106. [DOI] [PubMed] [Google Scholar]
- 72.Whitmarsh AJ, Yang SH, Su MSS, Sharrocks AD, Davis RJ. Role of p38 and JNK mitogen-activated protein kinases in the activation of ternary complex factors. Mol Cell Biol. 1997;17:2360–2371. doi: 10.1128/mcb.17.5.2360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Zhu X, Lai C, Thomas S, Burden SJ. Neuregulin receptors, erbB3 and erbB4, are localized at neuromuscular synapses. EMBO J. 1995;23:5842–5848. doi: 10.1002/j.1460-2075.1995.tb00272.x. [DOI] [PMC free article] [PubMed] [Google Scholar]