

Abstract
Apoptosis is a very general phenomenon, but only a few reports concern astrocytes. Indeed, astrocytes express receptors for tumor necrosis factor (TNF) α, a cytokine demonstrated on many cells and tissues to mediate apoptosis after recruitment of adaptor proteins containing a death effector domain (DED). PEA-15 is a DED-containing protein prominently expressed in the CNS and particularly abundant in astrocytes. This led us to investigate if PEA-15 expression could be involved in astrocytic protection against deleterious effects of TNF. In vitro assays evidence that PEA-15 may bind to DED-containing protein FADD and caspase-8 known to be apical adaptors of the TNF apoptotic signaling. After generation of PEA-15 null mutant mice, our results demonstrate that PEA-15 expression increases astrocyte survival after exposure to TNF.
Keywords: astrocytes, TNFα, apoptosis, death effector domain, PEA-15, FADD, caspase
Programmed cell death is extensively reported in the CNS for neurons and oligodendrocytes in several pathological conditions, but not for astrocytes. Inflammation and trauma of the adult CNS result in the production of several cytokines, among which tumor necrosis factor (TNF) α seems to play a major role (St. Pierre et al., 1996). Astrocytes produce TNF and express apoptosis-inducing receptors belonging to the TNF receptor superfamily, such as TNFR1 or Fas (Dopp et al., 1997; Becher et al., 1998), but are not susceptible for example to Fas-mediated cytotoxicity (Becher et al., 1998), suggesting a self-protective mechanism.
Apoptosis results from an orderly cascade of cellular events. It involves the sequential activation of aspartate-specific cysteine proteases named caspases (Alnemri et al., 1996). Extracellular signals such as FasL or TNF are well known to trigger such cascades. After binding to their respective ligands, Fas and TNFR1 receptors initiate apoptosis by recruiting the cytosolic adaptor molecule FADD/MORT (FADD) to the plasma membrane (Fraser and Evans, 1996). The N-terminal part of FADD contains a death effector domain (DED) that binds to homologous domains located in the N-terminal part of caspase-8 (FLICE, MACH, Mch5), allowing the autocleavage of the latter and its activation (Boldin et al., 1996; Muzio et al., 1996). DEDs also exist in another procaspase, caspase-10 (Mch4, FLICE2), where they might play the same protein-binding function (Vincenz and Dixit, 1997). Mutants of FADD lacking DED or mutants of caspase-8 with only its DEDs can act as dominant-negative inhibitors, suggesting that endogenous inhibitors of early steps of apoptosis could exist. Indeed, a family of DED-containing proteins expressed by γ-type herpes virus was characterized and shown to block the cascade after binding to either FADD or caspase-8 (Bertin et al., 1997; Thome et al., 1997). A mammalian homolog of these proteins (CASH/CASPER/CLARP/FLAME1/FLIP/I-FLICE/MRIT/Usurpin) (Golstev et al., 1997; Han et al., 1997; Hu et al., 1997; Inohara et al., 1997;Irmler et al., 1997; Shu et al., 1997; Srinivasula et al., 1997;Wallach, 1997b) is also recruited as FADD and caspase-8 in the death-inducing signaling complex and could modulate its efficacy (Scaffidi et al., 1999).
The prominently brain-expressed protein PEA-15 also contains a DED domain. PEA-15 was first characterized because of its enrichment in astrocytes, but it can be detected at lower levels in other cell types and out of the CNS (Danziger et al., 1995; Condorelli et al., 1998). This protein is phosphorylated on two different seryl residues, Ser104 being identified as a site of regulation for protein kinase C (PKC) (Araujo et al., 1993; Estelles et al., 1996), whereas Ser116 is regulated by calcium–calmodulin kinase II (CaMKII) (Kubes et al., 1998). PEA-15 is particularly abundant in astrocytes (Danziger et al., 1995), raising the hypothesis that it could play a role against cytokine-triggered apoptosis. Indeed, we demonstrate here that PEA-15 interacts in vitro with two other DED domain-containing proteins, FADD and caspase-8. Its function was further investigatedin vivo in astrocytes from wild-type versus PEA-15 null mutant mice. Expression of PEA-15 protects astrocytes from TNF-induced apoptosis.
MATERIALS AND METHODS
Glial primary cultures. Glial cultures were prepared as previously described (Araujo et al., 1993). Briefly, 16 mm wells or 100 mm dishes were coated with poly(l-ornithine) (1.5 μg/ml; Mr 40,000). Striatal cells from 16-d-old wild-type (WT) or PEA-15-null (KO) mouse embryos were dissociated and plated (80,000 cells per well) in a culture medium (Life Technologies, Gaithersburg, MD) consisting of a 1:1 mixture of MEM and F-12 nutrient supplemented with glucose (33 mm), glutamine (2 mm), sodium bicarbonate (3 mm), and 10% Nu-Serum (Collaborative Research, Bedford, MA). The culture medium was changed every 3 d for 3 weeks until glial elements had formed a confluent monolayer devoid of neuronal cells. As previously described, using morphological and immunohistochemical criteria, cells were shown to be virtually mature astrocytes (Birman et al., 1989).
cDNA constructs. Coding region of PEA-15 was cloned into pGEX-4T1 (Pharmacia Biotech) at EcoRI–SalI sites or into pcDNA3 (Invitrogen, San Diego, CA) tagged with a FLAG epitope at its C terminus at KpnI–BamHI sites. PEA-15 ORF was cloned into pEGFP-C1 (Clontech, Cambridge, UK). A PCR-generated fragment corresponding to the DED of PEA-15 (amino acids 1–79) was obtained using the following primers containingEcoRI and SalI sites, respectively: DEDPE, 5′-CGAGGAATTCGGCGGAATGG-CAGAGTACGGAACT-3′ and DDPEs, 5′-CGGCGTCGACTCAAACCACCATAGTG-AGGAG-3′. The PCR product was cloned into pGEX-4T1 or pEGFP-C1 vectors.
Glutathione S-transferase pull-down assay. Using a TNT-coupled reticulocyte lysate system (Promega, Madison, WI), PEA-15, FADD, and caspase-8 (Casp8) were translated in vitroin the presence of 35S-methionine. Five microliters of the reaction product were incubated with 5 μg of GST fusion proteins coupled to glutathione Sepharose beads in the following buffer: Tris 10 mm, pH 7.5, MgCl2 1 mm, Triton X-100 1%, NaCl 50 mm, and bovine serum albumin (BSA) 0.1% for 1.5 hr at room temperature. Beads were then washed twice in the same buffer and once in this buffer without BSA. Associated proteins were analyzed by a 4–20% SDS-PAGE followed by transfer on a nitrocellulose membrane and analysis in an Instantimager (Packard).
Deletion of PEA-15 gene. A full-length PEA-15 cDNA clone was used to screen a mouse SV129 genomic library (Stratagene, La Jolla, CA). Two overlapping phage clones were isolated, which were recloned into a bluescript vector, and restriction mapping was performed. A targeting vector was constructed by deleting a 4.5 kbBamHI–EcoRI genomic fragment that contains the entire PEA-15 gene. This was performed using a pPNT vector (Tybulewicz et al., 1991). A 10 kb BamHI genomic fragment was cloned 5′ of the PGKneo cassette, and a 4 kbEcoRI–BglII genomic fragment was inserted 3′ to the PGKneo cassette and 5′ of the PGK-TK cassette.
Homologous recombination in embryonic stem cells. A newly established embryonic stem (ES) cell line (derived from a 129/SvEvTacfBR mouse), TC1, was transfected with the targeting vector linearized with NotI and selected with G418 and FIAU. The cell culture, electroporation, and selection procedures were performed essentially as previously described (Deng et al., 1994). ES colonies that were resistant to both FIAU and G418 were isolated, expanded, and genomic DNA was prepared for Southern blot analysis to identify clones that had undergone homologous recombination. Genomic DNA was digested with DraIII, run on a 0.7% agarose gel, blotted onto Genescreen plus nylon membrane (NEN) and probed with a 1.3 kbBglII–DraIII fragment isolated from the genomic clone. The probe hybridized with a 15 kb wild-type band. However, DNA of cells in which homologous recombination had occurred demonstrated both a 15 kb wild-type band and an 8 kb mutated band. ES cells heterozygous for the targeted mutation were microinjected into C57BL/6J blastocysts to obtain germline transmission. The injected blastocysts were implanted into the uteri of pseudopregant Swiss Webster (Taconic, Germantown, NY) foster mothers and were allowed to develop to term. Male chimeras were identified on the basis of their agouti coat color and were mated with National Institutes of Health Black Swiss females (Taconic). Germ line transmission was confirmed by agouti coat color in the F1 animals, and all agouti mice were tested for the presence of the mutated PEA-15 allele by Southern blot analysis using the same conditions as those used for the detection of the homologous recombination event in ES cells. Mice carrying the heterozygote mutation were crossed to obtain homozygote mice, and genotypes were determined by Southern blotting.
Northern blot analysis. Total RNA was extracted from cultured astrocytes from WT or KO mice. RNA samples were electrophoresed though 1% agarose (Appligene, Heidelberg, Germany) gels following standard procedures (Ausubel et al., 1992) and transferred to Hybond-N membranes (Amersham, Arlington Heights, IL). A specific probe for the PEA-15 coding region (ATG to TGA) was synthesized using PCR (Estelles et al., 1996) and was labeled with α-32P-dCTP using the RadPrime DNA labeling kit (Life Technologies). Specific binding was analyzed and quantified in a Packard InstantImager.
Western blots. Electrotransfer of gels on Hybond C-Super nitrocellulose membrane was performed with a Bio-Rad (Hercules, CA) semidry blotting apparatus (15 V, 1 hr). Nitrocellulose blots were fixed and stained with Ponceau Red (0.2% in 1% acetic acid) and blocked with 2.5% casein for 1 hr (20°C). After overnight incubation at 4°C with antiserum diluted in 2.5% casein, bound antibodies (Danziger et al., 1995) were detected by anti-rabbit IgG coupled to horseradish peroxidase and revealed by chemiluminescence (Renaissance kit; DuPont, Billerica, MA).
32P labeling of astrocytes. Labeling was performed by preincubating cells for 4 hr with32PO43(0.15 mCi) in 250 μl of phosphate-free medium. Labeling was stopped as previously described (Araujo et al., 1993), and samples were prepared for two-dimensional PAGE electrophoresis according to Garrels (1979) with a few modifications (Araujo et al., 1993). Isoelectric focusing gels contained 2% total ampholines, pH 4–6.5 (Pharmacia, Piscataway, NJ). The second dimension was performed on 13% polyacrylamide gels according to Laemmli (1970). To detect proteins, gels were silver-stained, dried, exposed for autoradiography (1–3 d) with Kodak (Eastman Kodak, Rochester, NY) XAR-5 films, and counted and digitalized in an InstantImager (Packard).
In vitro analysis of genomic DNA fragmentation. After TNFα treatment, cells were pelleted and incubated in 350 μl of a lysis buffer (Tris 100 mm, NaCl 100 mm, EDTA 50 mm, SDS 1%, proteinase K 1 mg/ml, pH 7.8) for 2 hr at 55°C. DNA was then extracted with phenol–chloroform as previously described (Moyse and Michel, 1996). DNAs (1 μg for each sample) were32P-end-labeled for 10 min at room temperature using Kleenow polymerase (5 U) as previously described (Rösl, 1992). One-tenth of the sample was then electrophoresed on a 1.8% agarose gel for 2 hr at 100 V. After drying, the gel was analyzed in InstantImager.
Cell death assay. Astrocytes were replated (2000/cm2) on cover glass slides coated with polyornithine and allowed to recover for 4 d. After 2 hr in the presence of actinomycin D (10 μg/ml), TNFα [recombinant human TNF (Calbiochem, La Jolla, CA), 300 U/16 mm dish, 5 × 107 U/mg] was added for the indicated time. Cells were then stained with annexin V using a kit (Bender Medsystems), and/or fixed and stained with DAPI, and/or labeled with a terminal deoxynucleotidyl transferase-mediated biotinylated UTP nick end labeling (TUNEL) assay kit (Biovation), according to each manufacturer’s protocol with a few modifications.
Using a polyethylenimine (PEI)-based protocol, astrocytes cultured from KO mice were transiently transfected with pCMV-GFP in the presence of a fourfold excess of pcDNA3 expression constructs encoding PEA-15 or with the pEGFP-PEA-15 construct. Forty-eight hours after transfection, cells were treated with TNF, and apoptosis was evaluated using the same tests as above. The data (mean ± SEM) shown are the percentage of green fluorescent apoptotic cells among the total number of green cells.
RESULTS
Previous studies have shown that DED domains mediate the binding of the adaptor molecule FADD to the effector protease caspase-8 (Boldin et al., 1996; Muzio et al., 1996). Because PEA-15 is also a protein with a DED domain (Fig. 1), we investigated the ability of PEA-15 to interact with FADD and/ or caspase-8 using in vitro binding assays.35S-radiolabeled FADD, caspase-8, or PEA-15 was precipitated with PEA-15 (Fig.2A) or FADD (Fig.2B) GST fusion proteins immobilized on glutathione–Sepharose beads. FADD and caspase-8 associated specifically with GST-PEA-15, although the interactions of FADD and caspase-8 with GST-PEA-15 were weaker than those observed with GST-FADD. As PEA-15 bound, to some extent, to GST alone, no specific binding was observed with GST-PEA-15, suggesting that PEA-15 did not associate with itself. Simultaneous addition of FADD and caspase-8 neither prevented nor enhanced their interaction with PEA-15 (Fig.2A). In addition, GST-DED-PEA15 was also able to coprecipitate with FADD and caspase-8 but to a lower extent than the whole PEA-15 protein (data not shown). These results demonstrated that PEA-15 may interact with several DED domain-containing molecules and led us to further investigate the role of PEA-15 in cytokine-induced apoptosis in astrocytes.
Fig. 1.

Schematic representation of PEA-15 and comparison of death effector domains. A, The two identified sites of phosphorylation, Ser104 for PKC and Ser116 for CaMKII, are located outside of DED. B, Alignment of DEDs (sequence-based). Hydrophobic residues known in FADD to contribute to its structure (arrangement of six helices α1–α6, see Eberstadt et al., 1998) are shown in bold and are shaded. Theboxed and asterisked area is the F25 reported as critical for the proapoptotic function of FADD. Mouse PEA-15 (GenBank accession number X86804); mouse FADD (GenBank accession number U43184); human caspase-8 (GenBank accession number U60520); human caspase-10 (GenBank accession number 1498324); cFLIP (GenBank accession number U97076); and equine herpes virus-2 E8 protein (GenBank accession number U20824).
Fig. 2.

In vitro interactions of PEA-15. The indicated in vitro translated35S-labeled proteins were precipitated with GST (A, B, lanes1), GST-PEA-15 (A, lanes 2), or GST-FADD (B, lanes 2) fusion proteins immobilized on glutathione–Sepharose beads. The bound proteins were then analyzed by SDS-PAGE, visualized, and counted in a Packard InstantImager. Data presented are taken from one individual experiment of three or more giving similar results.
For this purpose, using homologous recombination, PEA-15 null mutant mice were created to investigate apoptosis in astrocytes that no longer express PEA-15. We designed a targeting vector that replaced a 4.5 kbBamHI–EcoRI genomic fragment containing the entire coding region of the PEA-15 gene with the neomycin resistance gene (neo) (Fig. 3A). The correct homologous event in ES cells was confirmed by Southern blotting (Fig. 3B), and mice were genotyped by the same procedure (Fig. 3B). The absence of PEA-15 expression in homozygous animals was analyzed by Northern and Western blotting (Fig.3C,D) and further confirmed by32P labeling of astrocytes grown in primary culture followed by two-dimensional PAGE and autoradiography (Fig. 3E). Heterozygous mutant mice (PEA-15+/−) were healthy and normal in size as were the homozygous PEA-15−/−. Body weight was decreased in PEA-15−/− animals in comparison with their wild-type littermates, but brain sizes were similar in wild-type, heterozygous, and homozygous animals. Gross examination of brain sections from 2-month-old animals revealed no difference between WT PEA-15+/+ and KO PEA-15−/− mice. Because PEA-15 is highly expressed in astrocytes, GFAP labeling that indicated no obvious difference between WT and KO in the number, morphology, and organization of glial cells (Fig. 4) was performed. All subsequent experiments were performed on astrocytes grown in primary culture.
Fig. 3.
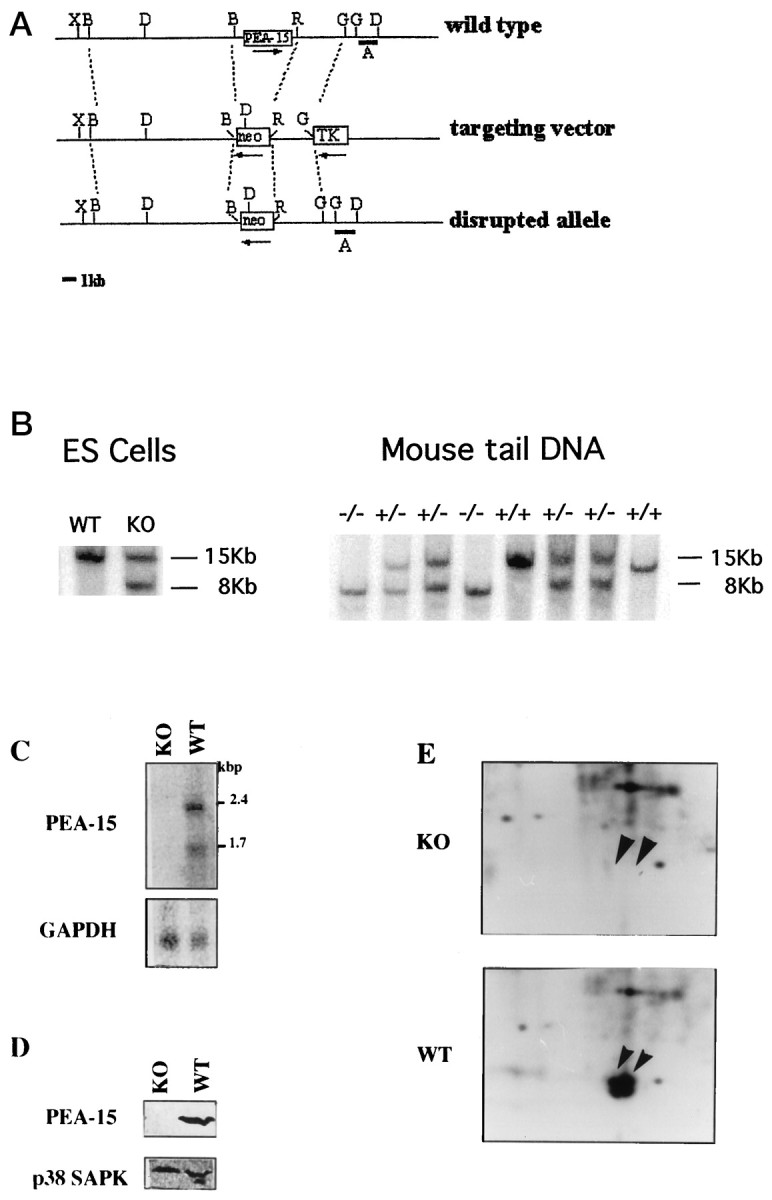
Generation of PEA-15 mutant mice by gene targeting. A, A schematic diagram of the PEA-15 locus, the targeting vector, and the targeted allele. Restriction enzyme sites are indicated as R,EcoRI;B, BamHI; D,DraIII;G, BglII; and X, XhoI. The entire PEA15 gene lies within a 4.5 kb BamHI–EcoRI fragment. In the targeting construct, this fragment is deleted and is replaced by the neomycin gene driven by the PGK promoter (neo). A 10 kbBamHI fragment flanks one side of the neo gene, and a 3 kb EcoRI–BglII fragment flanks the other side. On the other side of this fragment lies the thymidine kinase gene also driven by the PGK promoter (TK). Homologous recombination results in the disrupted allele as shown. Fragment A represents the 1.3 kb DraIII–BglII fragment that was used as a probe to differentiate between the wild-type and the disrupted alleles. The arrows mark the direction of transcription of the various genes. B,Southern blot analysis of DraIII digested DNA isolated from the parental ES cells (WT) as compared with DNA from an ES cell clone that had undergone homologous recombination (KO). Using a 1.3 kbDraIII–BglII probe, a shift down from a 15 kb wild-type band to an 8 kb mutated band is observed.C, Southern blot analysis of DNA isolated from the tails of a litter of pups born from a heterozygote × heterozygote mating. As predicted by Mendelian genetics, two wild-type mice, two homozygotes, and four heterozygotes are observed with the expected band pattern following DraIII digestion and hybridization with the 1 kb DraIII–BglII probe.D, Northern (RNA) blot demonstrating the absence of PEA-15 messages in −/− mutant astrocytes derived from these mice.E, Western blot demonstrating the absence of PEA-15 protein in the brains of −/− mice and in cultured astrocytes from these mice. F, Details of two-dimensional PAGE autoradiograms performed after 32P-labeling of cultured astrocytes and demonstrating the absence of PEA-15 in −/− mice.
Fig. 4.

Astrocytes from mice lacking PEA-15 seem normal in their number and morphology. Staining with an anti-GFAP antibody demonstrate similar population of astrocytes in hippocampal sections from PEA-15 null mutants (KO) and their wild-type littermates (WT). Original magnification 200× (top figures, scale bar, 300 μm) and 400× (bottom figures).
Under usual primary culture conditions, no difference was observed between WT and KO cells with respect to their growth rate and differentiation, i.e., GFAP expression. In both cell types, TNFα stimulated proliferation as observed after [3H]thymidine incorporation (128 ± 6 and 135 ± 8% stimulation over control for WT and KO astrocytes respectively; 24 hr; n = 3).
However, differences were observed between WT and KO cells after TNFα treatment in the presence of the transcription inhibitor actinomycin D. Astrocytes not expressing PEA-15 began to retract after a few hours, and many cells died within the first 24 hr. In contrast, no major morphological changes could be observed under the same conditions in WT cells (Fig. 5A ). The cell death observed in KO cells was further analyzed by several classical apoptotic tests. The TUNEL assay using fluorescent d-UTP, clearly labeled KO cell nuclei after 6 hr of treatment with TNF. After 24 hr of TNF treatment, >60% of the KO nuclei were labeled compared with <20% of nuclei from WT cells (Fig. 5B,D). In KO cells, a rapid labeling could be observed with Annexin V, already maximal at 6 hr. This was correlated with the typical condensation and fragmentation alterations of the nuclei of the corresponding cells as observed with DAPI staining (Fig. 5C,D). Finally, after DNA extraction, an in vitro 32P-labeling assay allowed the identification of ladders corresponding to DNA fragmentation in KO cells treated with TNF, whereas a minor effect could be observed in WT astrocytes (Fig. 5E). These data clearly demonstrated that PEA-15 KO cells were more sensitive to TNF-induced apoptosis than WT astrocytes.
Fig. 5.


Increased susceptibility to TNF-induced apoptosis of cultured astrocytes from −/− PEA-15 mice. A, Phase contrast analysis of astrocytes grown for 3 weeks in primary culture without or with a 24 hr treatment of TNF (300 U/well). KO = PEA−/− cells, WT = PEA+/+ cells. B, KO or WT astrocytes were incubated with TNF for 6 or 24 hr and analyzed byin situ polymerase assay (TUNEL) using d-UTP coupled to fluorescein. C, TNF-treated KO or WT cells were stained an annexin V labeling kit associated with nucleus staining with DAPI. AnnexinV-positive cells also present nucleus condensation and fragmentation usually observed during apoptosis. D,Quantification of results obtained with the different staining methods: DAPI, Annexin V, and TUNEL. Data are from four or more independent experiments. Statistically significant at **p < 0.01 versus control according to Student’s t test.E, After a 24 hr incubation with TNF, astrocytes were harvested, and their DNA was extracted and analyzed on a 1.8% agarose gels directly or after in vitro end labeling as described in Materials and Methods.
To confirm that increased susceptibility to TNF-induced apoptosis in KO astrocytes was a direct consequence of the lack of expression of PEA-15, cells were transiently transfected with vector pcDNA3-PEA-15 tagged with a FLAG epitope and with pcDNA3-GFP in a 4:1 ratio. After 48 hr, the expression of the transfected protein was checked by Western blotting with an anti-FLAG antibody. Cells were then treated with TNF and apoptosis assayed by Annexin V and DAPI staining. GFP expression alone did not promote apoptosis neither did it protect cells from the effects of TNF. In contrast, after cotransfection with PEA-15, expressing cells exhibited no labeling with annexin V, and their nuclei appeared normal after DAPI staining, indicating that PEA-15 expression protected the cell from TNF (Fig. 6). Similar results were obtained using a single vector pEGFP.C1 where PEA-15 was fused at the C terminus of GFP.
Fig. 6.

Expression of PEA-15 in KO cells protects them from TNF-induced apoptosis. PEA-15−/− astrocytes were transiently transfected with pcDNA3-GFP alone or pcDNA3-PEA-15-FLAG associated with pcDNA3-GFP in a 4:1 ratio. After 48 hr, expression of GFP +/− PEA-15 transfected cells was checked by immunocytochemistry and immunoblotting against the flag epitope (data not shown). Transfection protocol resulted in 4–15% of cultured astrocytes expressing GFP (n = 5). Cells were then treated for 24 hr with TNF. A, Expression of GFP alone does not protect astrocytes from TNF-induced apoptosis as evidenced by Annexin V labeling and DAPI staining (left panels). In contrast, after cotransfection of GFP and PEA-15 fluorescent cells are not labeled by Annexin V, and their nuclei appear normal after DAPI staining (right panels). B, Annexin V and DAPI staining were used to measure the percentage of apoptotic cells. Results are from five independent experiments, and significant at **p < 0.01 using Student’s ttest.
DISCUSSION
Proteins containing a DED, such as FADD and caspase-8, occupy an apical position in the apoptosis cascade triggered by TNF. We demonstrate that the astrocytic DED-containing protein PEA-15 interactsin vitro with FADD and caspase-8, and that its expression decreases astrocyte susceptibility to TNF-induced cell death.
In vitro interaction of PEA-15 with FADD appeared weaker than that observed between FADD and caspase 8. This could suggest that post-translational modifications not occurring during bacterial expression of the protein are important. Indeed, in astrocytes PEA-15 was always essentially observed under its phosphorylated forms (Araujo et al., 1993; Danziger et al., 1995). On the other hand, a low affinity for interaction could also explain the high level of PEA-15 expression in glial cells, whereas FADD or caspase-8 are expressed at very low levels (data not shown).
TNF triggers both cytotoxic and protective mechanisms, respectively independent and dependent on protein synthesis: whereas it hardly affects viability of normal cells, TNF destroys diseased cells, such as virus-infected or transformed cells, and cells treated with inhibitors of transcription or of protein synthesis (Wallach, 1997a). To balance these effects, several gamma-type herpes viruses express DED proteins demonstrated to bind FADD and/or caspase-8 and act as dominant negative effectors of cytokine-induced apoptosis (Bertin et al., 1997; Thome et al., 1997). I-FLICE, presenting essentially two DEDs and no caspase domain, was also shown to bind FADD and/or caspase-8 and/or caspase-10, and to inhibit apoptosis through a potential dominant negative mechanism. However I-FLICE did not seem sufficient to block apoptosis (Scaffidi et al., 1999), and even amplified cell death in other reports (Wallach, 1997b). It would be of interest to see if PEA-15 could also bind I-FLICE and enhance its efficacy to protect cells. In addition, a novel DED-containing molecule, DEDD, was proposed to transduce the death signaling from FADD to the nucleolus (Stegh et al., 1998). PEA-15 is a cytosolic protein, and does not translocate to the nucleus. It would be of interest to see if PEA-15 binds DEDD and prevents it to contribute its part in the apoptotic process initiated by CD95.
Among the specific features of the PEA-15 DED is a serine residue found at position 25, whereas the recently reported three-dimensional structure of the DED domain of FADD suggests a critical proapoptotic function for a phenylalanine at this position, an amino acid conserved in the DEDs of caspase-8 and caspase-10 as well as in viral and mammalian I-FLICE and in DEDD (Eberstadt et al., 1998). This may confer novel properties for the DED of PEA-15, in addition to an oligomerization with other DEDs. Indeed, this domain was recently demonstrated necessary for a newly discovered function of the protein, the reversal of the inhibitory effect of H-Ras on integrin signaling (Ramos et al., 1998). Work in progress in our group, based on a yeast two-hybrid screen of a brain and an astrocytic cDNA library will unravel specific partners of PEA-15 and allow the mapping of interacting regions.
Why should PEA-15 protect astrocytes from apoptosis? Programmed cell death is a very general phenomenon, but only a few reports concern astrocytes. Astrocytes grown in primary cultures were shown to undergo apoptosis under pharmacological treatments such as staurosporine or ceramide, a process involving the activation of caspase-3 (Keane et al., 1997). However, astrocytes seem rather protected in several pathological situations such as glutamate-triggered excitotoxicity (Choi, 1988) or calcium overload (Rzigalinski et al., 1997). Neuronal apoptosis is well documented in CNS damage resulting from trauma or ischemia, but in these situations astrocytes seem to be essentially involved in repair and regeneration at the site of injury. In response to a brain lesion or infection, glial cells have been demonstrated to secrete and respond to numerous cytokines (Merrill and Benveniste, 1996). Astrocytes produce TNF and express TNF receptors triggering several responses after TNF stimulation: cell proliferation, upregulation of TNF mRNAs, production of interleukin 8, macrophage-, granulocyte-, and granulocyte–macrophage colony stimulating factors (St. Pierre et al., 1996). In addition, TNF primes astrocytes to render them immunocompetent through the induction of the expression of several surface molecules, including MHC class II molecules, ICAM-1, VLA-1, and VLA-2, allowing recruitment of lymphocytes and monocytes (Merrill and Benveniste, 1996). Considering the time course of these events, astrocytes need to be protected against apoptosis that TNF could also trigger, and PEA-15 seems to play an important role in this process.
Footnotes
This work was supported by funds from the Institut National de la Santé et de la Recherche Médicale, European Science Foundation/European Neuroscience Program European research Grant 245, and VEGA Grant 2/5098/98. Requests concerning the PEA-15 knock-out mice should be submitted directly to Prof. Leder at Harvard Medical School. We thank Dr. E. Moyse for his help, Prof. Philip Leder for his encouragement at early steps of this project and for constant support, and Dr. Chris Henderson for critical reading of this manuscript and helpful suggestions.
Drs. Kitsberg and Formstecher contributed equally to this work.
Correspondence should be addressed to Dr. Hervé Chneiweiss, Institut National de la Santé et de la Recherche Médicale U114, Chaire de Neuropharmacologie, Collège de France, 11 Place M. Berthelot 75231 Paris cedex 05, France.
Dr. Pan’s present address: Genentech, South San Franscisco, CA 94080-4990.
Dr. Kitsberg’s present address: Alamone Laboratories, Jerusalem, Israel.
REFERENCES
- 1.Alnemri ES, Livingston DJ, Nicholson DW, Salvesen G, Thornberry NA, Wong WW, Yuan J. Human ICE/CED-3 protease nomenclature. Cell. 1996;87:171. doi: 10.1016/s0092-8674(00)81334-3. [DOI] [PubMed] [Google Scholar]
- 2.Araujo H, Danzinger N, Cordier J, Glowinski J, Chneiweiss H. Characterization of PEA-15, a major substrate for protein kinase C in astrocytes. J Biol Chem. 1993;268:5911–5920. [PubMed] [Google Scholar]
- 3.Ausubel FM, Brent R, Kingston RE, Moore DD, Seidman JG, Smith JA, Struhl K. Short Protocols in Molecular Biology. Greene and Wiley; New York: 1992. [Google Scholar]
- 4.Becher B, D’Souza SD, Troutt AB, Antel JP. Fas expression on human fetal astrocytes without susceptibility to fas-mediated cytotoxicity. Neuroscience. 1998;84:627–634. doi: 10.1016/s0306-4522(97)00455-7. [DOI] [PubMed] [Google Scholar]
- 5.Bertin J, Armstrong RC, Ottilie S, Martin DA, Wang Y, Banks S, Wang G-H, Senkevich TG, Alnemri ES, Moss B, Lenardo MJ, Tomaselli KJ, Cohen JI. Death effector domain-containing herpesvirus and poxvirus proteins inhibit both Fas- and TNFR1-induced apoptosis. Proc Natl Acad Sci USA. 1997;94:1172–1176. doi: 10.1073/pnas.94.4.1172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Birman S, Cordier J, Glowinski J, Chneiweiss H. CyclicAMP dependent protein kinase in mouse striatal neurones and astrocytes in primary culture: development, subcellular distribution and stimulation of endogenous phosphorylation. Neurochem Int. 1989;14:25–34. doi: 10.1016/0197-0186(89)90005-3. [DOI] [PubMed] [Google Scholar]
- 7.Boldin M, Goncharov TM, Goltsev YV, Wallach D. Involvement of MACH, a novel MORT1/FADD-interacting protease, in Fas/APO1- and TNF receptor-induced cell death. Cell. 1996;85:803–815. doi: 10.1016/s0092-8674(00)81265-9. [DOI] [PubMed] [Google Scholar]
- 8.Choi DW. Glutamate neurotoxicity and diseases of the nervous system. Neuron. 1988;1:623–634. doi: 10.1016/0896-6273(88)90162-6. [DOI] [PubMed] [Google Scholar]
- 9.Condorelli G, Vigliotta G, Iavarone C, Crauso M, Tocchetti CG, Andreozzi F, Cafieri A, Tecce MF, Formisano P, Beguinot L, Beguinot F. PED/PEA-15 gene controls glucose transport and is overexpressed in type 2 diabetes mellitus. EMBO J. 1998;17:3858–3866. doi: 10.1093/emboj/17.14.3858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Danziger N, Yokoyama M, Jay T, Cordier J, Glowinski J, Chneiweiss H. Cellular expression, developmental regulation, and phylogenic conservation of PEA15: the major substrate for protein kinase C in astrocytes. J Neurochem. 1995;64:1016–1025. doi: 10.1046/j.1471-4159.1995.64031016.x. [DOI] [PubMed] [Google Scholar]
- 11.Deng CX, Wynshaw-Boris A, Shen MM, Daugherty C, Ornitz DM, Leder P. Murine FGFR-1 is required for early postimplantation growth and axial organization. Genes Dev. 1994;8:3045–3057. doi: 10.1101/gad.8.24.3045. [DOI] [PubMed] [Google Scholar]
- 12.Dopp J, Mackenzie-Graham A, Otero G, Merrill J. Differential expression, cytokine modulation and specific functions of type-1 and type-2 tumor necrosis factor receptors in rat glia. J Neuroimmunol. 1997;75:104–112. doi: 10.1016/s0165-5728(97)00009-x. [DOI] [PubMed] [Google Scholar]
- 13.Eberstadt M, Huang B, Chen Z, Meadows RP, Ng S-C, Zheng L, Lenardo MJ, Fesik SW. NMR structure and mutagenesis of the FADD(Mort1) death-effector domain. Nature. 1998;392:941–945. doi: 10.1038/31972. [DOI] [PubMed] [Google Scholar]
- 14.Estelles A, Yokoyama M, Nothias F, Vincent J, Glowinski J, Vernier P, Chneiweiss H. The major astrocytic phosphoprotein PEA-15 is encoded by two mRNAs conserved on their full length in mouse and human. J Biol Chem. 1996;271:14800–14806. doi: 10.1074/jbc.271.25.14800. [DOI] [PubMed] [Google Scholar]
- 15.Fraser A, Evans G. A license to kill. Cell. 1996;85:781–784. doi: 10.1016/s0092-8674(00)81005-3. [DOI] [PubMed] [Google Scholar]
- 16.Garrels JI. Two-dimensional gel electrophoresis and computer analysis of proteins synthesized by clonal cell lines. J Biol Chem. 1979;254:7961–7977. [PubMed] [Google Scholar]
- 17.Golstev YV, Kovalenko AV, Arnold E, Varfoloveev EE, Brodianski VM, Wallach D. CASH, a novel caspase homologue with death effector domains. J Biol Chem. 1997;272:19641–19644. doi: 10.1074/jbc.272.32.19641. [DOI] [PubMed] [Google Scholar]
- 18.Han DKM, Chaudhary PM, Wright M, Friedman C, Trask B, Riedel RT, Baskin DG, Schwartz ST, Hood L. MRIT, a novel death-effector domain-containing protein, interacts with caspases and BclXL, and initiates cell death. Proc Natl Acad Sci USA. 1997;94:11333–11338. doi: 10.1073/pnas.94.21.11333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hu S, Vincenz C, Ni J, Gentz R, Dixit V. I-FLICE, a novel inhibitor of tumor necrosis factor-1 and CD-95-induced apoptosis. J Biol Chem. 1997;272:17255–17257. doi: 10.1074/jbc.272.28.17255. [DOI] [PubMed] [Google Scholar]
- 20.Inohara N, Koseki T, Hu Y, Chen S, Nunez G. CLARP, a death effector domain-containing protein interacts with caspase-8 and regulates apoptosis. Proc Natl Acad Sci USA. 1997;94:10717–10722. doi: 10.1073/pnas.94.20.10717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Irmler M, Thome M, Hahme M, Schneider P, Hofman K, Steiner V, Bodmer J-L, Schöter M, Burns K, Mattman C, Rimoldi D, French LE, Tschopp J. Inhibition of death receptor signals by cellular FLIP. Nature. 1997;388:190–195. doi: 10.1038/40657. [DOI] [PubMed] [Google Scholar]
- 22.Keane RW, Srinivasan A, Foster LM, Testa M-P, Ord T, Nonner D, Wang H-G, Reed JC, Bredesen DE, Kayalar C. Activation of CPP32 during apoptosis of neurons and astrocytes. J Neurosci Res. 1997;48:168–180. doi: 10.1002/(sici)1097-4547(19970415)48:2<168::aid-jnr9>3.0.co;2-a. [DOI] [PubMed] [Google Scholar]
- 23.Kubes M, Cordier J, Glowinski J, Girault J-A, Chneiweiss H. Endothelin induces a calcium-dependent phosphorylation of PEA-15 in intact astrocytes. J Neurochem. 1998;71:1307–1314. doi: 10.1046/j.1471-4159.1998.71031307.x. [DOI] [PubMed] [Google Scholar]
- 24.Laemmli UK. Cleavage of structural proteins during the assembly of the head of the bacteriophage T4. Nature. 1970;227:680–685. doi: 10.1038/227680a0. [DOI] [PubMed] [Google Scholar]
- 25.Merrill JE, Benveniste ET. Cytokines in inflammatory brain lesions: helpful and harmful. Trends Neurosci. 1996;19:331–338. doi: 10.1016/0166-2236(96)10047-3. [DOI] [PubMed] [Google Scholar]
- 26.Moyse E, Michel D. Analyses of apoptosis-associated DNA fragmentation in vivo during neurodegeneration in the peripheral olfactory system in adult mammals. In: Poirier J, editor. Neuromethods: apoptosis. Humana; Totowa, NJ: 1996. pp. 135–161. [Google Scholar]
- 27.Muzio M, Chinnaiyan AM, Kischkel FC, O’Rourke K, Shevchenko A, Ni J, Scaffidi C, Bretz JD, Zhang M, Gentz R, Mann M, Krammer PH, Peter M, Dixit VM. FLICE, a novel FADD-homologous ICE/Ced3-like protease, is recruited to the CD95 (Fas/APO1) death-inducing signaling complex. Cell. 1996;85:817–827. doi: 10.1016/s0092-8674(00)81266-0. [DOI] [PubMed] [Google Scholar]
- 28.Ramos J, Kojima TJ, Hughues PE, Fenczik CA, Ginsberg M. The death effector domain of PEA-15 is involved in its regulation of integrin activation. J Biol Chem. 1998;273:33897–33900. doi: 10.1074/jbc.273.51.33897. [DOI] [PubMed] [Google Scholar]
- 29.Rzigalinski BA, Liang S, McKinney JS, Willoughby KA, Ellis EF. Effect of Ca2+ on in vitro astrocyte injury. J Neurochem. 1997;68:289–296. doi: 10.1046/j.1471-4159.1997.68010289.x. [DOI] [PubMed] [Google Scholar]
- 30.Rösl F. A simple and rapid method for detection of apoptosis in human cells. Nucleic Acids Res. 1992;20:5243. doi: 10.1093/nar/20.19.5243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Scaffidi C, Schmitz I, Krammer PH, Peter ME. The role of c-FLIP in modulation of the CD95-induced apoptosis. J Biol Chem. 1999;274:1541–1548. doi: 10.1074/jbc.274.3.1541. [DOI] [PubMed] [Google Scholar]
- 32.Shu HB, Halpin DR, Goeddel D. Casper is a FADD- and caspase-related inducer of apoptosis. Immunity. 1997;6:751–763. doi: 10.1016/s1074-7613(00)80450-1. [DOI] [PubMed] [Google Scholar]
- 33.Srinivasula SM, Ahamad M, Ottilie S, Bullrich F, Banks S, Wang Y, Fernandez-Alnemri T, Croce CM, Litwak G, Tomaselli KJ, Alnemri A. FLAME-1, a novel FADD-like anti-apoptotic molecule that regulates Fas/TNFR1-induced apoptosis. J Biol Chem. 1997;272:18542–18545. doi: 10.1074/jbc.272.30.18542. [DOI] [PubMed] [Google Scholar]
- 34.St. Pierre BA, Merrill JE, Dopp JM. Effects of cytokines on CNS cells: glia. In: Merrill R, Benveniste E, editors. Cytokines and the CNS. CRC; Boca Raton: 1996. pp. 151–168. [Google Scholar]
- 35.Stegh AH, Schickling O, Ehret A, Scaffidi C, Peterhänsel C, Hofmann TG, Grummt I, Kramer PH, Peter ME. DEDD, a novel death effector domain-containing protein, targeted to the nucleolus. EMBO J. 1998;17:5974–5986. doi: 10.1093/emboj/17.20.5974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Thome M, Schneider P, Hofman K, Fickenscher H, Meinl E, Neipel F, Mattman C, Burns K, Bodmer J-L, Schöter M, Scaffidi C, Krammer PH, Peter ME, Tschopp J. Viral FLICE-inhibitory proteins (FLIPs) prevent apoptosis induced by death receptors. Nature. 1997;386:517–521. doi: 10.1038/386517a0. [DOI] [PubMed] [Google Scholar]
- 37.Tybulewicz VL, Crawford CE, Jackson PK, Bronson RT, Mulligan RC. Neonatal lethality and lymphopenia in mice with a homozygous disruption of the c-abl proto-oncogene. Cell. 1991;65:1153–1163. doi: 10.1016/0092-8674(91)90011-m. [DOI] [PubMed] [Google Scholar]
- 38.Vincenz C, Dixit V. Fas-associated death domain protein interleukin-1β-converting enzyme 2 (FLICE2), an ICE/Ced3 homologue, is proximally involved in CD95- and p55-mediated death signaling. J Biol Chem. 1997;272:6578–6583. doi: 10.1074/jbc.272.10.6578. [DOI] [PubMed] [Google Scholar]
- 39.Wallach D. Cell death induction by TNF: a matter of self control. Trends Biochem Sci. 1997a;22:107–109. doi: 10.1016/s0968-0004(97)01015-3. [DOI] [PubMed] [Google Scholar]
- 40.Wallach D. Placing death under control. Nature. 1997b;388:123–126. doi: 10.1038/40516. [DOI] [PubMed] [Google Scholar]