

Abstract
The roles of Ca2+/calmodulin-dependent protein kinase II (CaM kinase II) and mitogen-activated protein kinase (MAPK) in long-term potentiation (LTP) were investigated in the CA1 area of hippocampal slices, using electrophysiological and biochemical approaches. A brief high-frequency stimulation, but not low-frequency stimulation, delivered to Schaffer collateral/commissural afferents produced a stable LTP and activated both CaM kinase II and 42 kDa MAPK. Different from the activity of CaM kinase II, the increase in MAPK activity was transient. At a concentration of 50 μm, but not of 30 μm,PD098059, a potent inhibitor of MAPK kinase, markedly inhibited the induction of LTP. Although the two concentrations had similar inhibitory effects on MAPK activity, only 50 μm PD098059 suppressed the activation of CaM kinase II. Application of calmidazolium, an antagonist of calmodulin, blocked both CaM kinase II activation and the LTP induction without affecting the increase in 42 kDa MAPK activity. Application of neurotrophin brain-derived neurotrophic factor (BDNF) promoted the induction of LTP, with concomitant activation of CaM kinase II. Under the same conditions, BDNF failed to activate MAPK in hippocampal slices. These results indicate that, although the LTP induction is accompanied by increases in two kinase activities, only CaM kinase II activation is required for this event.
Keywords: LTP, CaM kinase II, MAPK, hippocampal slice, PD098059, synaptic plasticity
Long-term potentiation (LTP) is considered to be a model of synaptic plasticity that may underlie memory and learning (Bliss and Collingridge, 1993; Malenka and Nicoll, 1993). Although the exact biochemical processes and molecular mechanisms remain unclear, several lines of evidence have shown that certain protein kinases are involved in LTP. A multifunctional Ca2+/calmodulin-dependent protein kinase II (CaM kinase II) in the brain has been intensively researched and was found to play a critical role in the induction of hippocampal LTP (Lisman and Goldring, 1988; Malinow et al., 1989; Malenka et al., 1989;Silva et al., 1992; Lledo et al., 1995; Miyamoto and Fukunaga, 1996;Barria et al., 1997). In the previous studies, we demonstrated that tetanic stimuli-induced LTP leads to a persistent increase in CaM kinase II activity in hippocampal slices (Fukunaga et al., 1993) accompanied with autophosphorylation of the kinase and phosphorylation of presynaptic and postsynaptic proteins (Fukunaga et al., 1995). This view has been confirmed by Ouyang et al. (1997), who visualized an increase in autophosphorylation of CaM kinase II in hippocampal slices after tetanic stimulation.
Recent studies have focused on another attractive protein kinase family, mitogen-activated protein kinase (MAPK). These enzymes may regulate a wide array of cellular processes (Roberts, 1992; Xia et al., 1996). In response to extracellular stimuli, such as growth factors, neurotrophins, and neurotransmitters, MAPK is activated by phosphorylation on both threonine and tyrosine residues (Bading and Greenberg, 1991; Seger and Krebs, 1995). In addition, the neuronal activity-dependent activation of 42 kDa MAPK has been reported by three groups of researchers, who demonstrated that high KCl pulse (Baron et al., 1996), electroconvulsive shock (Kang et al., 1994), or tetanic stimuli (English and Sweatt, 1996) induce increases in MAPK activity in the hippocampus. More recently, Atkins et al. (1998) reported that MAPK activity increased in the hippocampus during fear conditioning. Furthermore, studies using a pharmacological inhibitor showed that the MAPK translocation and signal cascade are required for LTP (English and Sweatt, 1997; Martin et al., 1997). However, the inhibition of synaptic potentiation in Aplysia neurons (Martin et al., 1997) and hippocampal slices (English and Sweatt, 1997) was mainly obtained, using a compound PD098059, an inhibitor for MAPK kinase (MEK) (Alessi et al., 1995), in which CaM kinase II activity was not detected.
Because our previous work showed that not only CaM kinase II but also 42 kDa MAPK was activated by stimulation of glutamate receptors with NMDA or glutamate (Fukunaga et al., 1992; Kurino et al., 1995; Fukunaga and Miyamoto, 1998) in cultured hippocampal neurons, we asked if MAPK activity also contributes as CaM kinase II does to regulation of synaptic plasticity. Here, we used in-gel kinase assay to simultaneously explore changes in activities of CaM kinase II and MAPK in hippocampal slices. We found that, although a brief high-frequency stimulation (HFS) produced increases in both 42 kDa MAPK and CaM kinase II activities, only CaM kinase II activation directly correlates with hippocampal LTP induction.
MATERIALS AND METHODS
Materials. The chemicals and reagents we used were obtained from the following sources: γ-[32P]ATP, DuPont NEN, Boston, MA; PD098059, Research Biochemicals, Natick, MA; calmidazolium (R24571), Sigma, St. Louis, MO; autocamtide-2-related inhibitory peptide, Bachem; and human recombinant brain-derived neurotrophic factor (BDNF), Calbiochem, La Jolla, CA. BDNF was dissolved in phosphate buffer (100 μg/ml) and stored at −30°C. Before every experiment, the stock solution was diluted with artificial CSF (ACSF) to the final concentration. PD098059 and calmidazolium were first dissolved in dimethylsulfoxide (DMSO) and then added to the ACSF perfusion. The final concentration of the solvent in the medium was kept at <0.01%. To ensure the complete solubility of the drugs, ACSF solutions were maintained at 32°C. The vehicle alone had no apparent effect on synaptic responses. Myelin basic protein (MBP) was purified from bovine brain (Deibler et al., 1972).
Preparation of hippocampal slices and electrophysiology.After decapitation, brains of male Sprague Dawley rats (∼5–8 weeks old) were rapidly removed, and the hippocampus was dissected out. Transverse hippocampal slices (450 μm thickness), prepared using a McIlwain tissue chopper, were incubated in continuously oxygenated ACSF, at room temperature and for at least 2 hr. The composition of ACSF contained (in mm): NaCl, 124; KCl, 3; CaCl2, 2.5; MgCl2, 1.5; NaH2PO4, 1.25; NaHCO3, 26; d-glucose, 10 (gassed with 95% O2 ± 5% CO2, pH 7.4). After a 2 hr recovery period, a slice was then transferred to an interface recording chamber and perfused at a flow rate of 2 ml/min with ACSF warmed at 32°C. The field EPSP (fEPSP) was evoked by 0.033 Hz test pulses through a bipolar stimulating electrode (twisted, 50 μm insulated tungsten wire) placed on the Schaffer collateral/commissural pathway, and recorded from the stratum radiatum of CA1 using a glass micropipette electrode filled with 3mNaCl. The stimulus intensity was adjusted to evoke a fEPSP of ∼1.5 mV amplitude, and the responses were monitored for at least 20 min to ensure a stable baseline. Then, this stimulus strength was maintained at the same level before and during the period of drug preincubation until each experiment was finished. LTP was induced by strong HFS, which consists of two 100 Hz, 1 sec trains delivered 20 sec apart. Low-frequency stimulation (LFS) which did not induce synaptic potentiation consisted of 20 pulses at 20 Hz. Shorter HFS (SHFS), a subthreshold stimulation for LTP induction, was applied at 100 Hz, 13 pulses to produce short-term potentiation (STP). After electrophysiological recording and stimulation, the slices were transferred to a glass slide at ice temperature, and the CA1 areas were dissected out and stored at −80°C until assay.
Autophosphorylation of CaM kinase II. Autophosphorylation of purified CaM kinase II was carried out at 0°C for 20 min. The reaction mixture contained 50 mm HEPES buffer, pH 7.5, 10 mm Mg2+, 0.2 mmCa2+, 1 μm calmodulin, and 50 μm ATP, and 3 μg of CaM kinase II in a total volume of 100 μl. After incubation for 20 min, the reaction was terminated by adding Laemmli’s sample solution.
Sample preparation. Hippocampal slice samples for kinase assay were prepared as previously described (Kurino et al., 1995;Fukunaga and Miyamoto, 1998). Briefly, the CA1 subregion from individual slices was homogenized at 4°C by sonication in solubilizing buffer containing 0.1% Triton X-100, 50 mmHEPES, 4 mm EGTA, 10 mm EDTA, 15 mmNa4P2O7, 100 mm β-glycerophosphate, 25 mm NaF, 0.1 mm leupeptin, 50 μg/ml trypsin inhibitor, 75 μm pepstatin A, 100 nm calyclin A, 1 mm sodium orthovanadate, and 1 mmdithiothreitol, pH 7.4. The insoluble material was removed by centrifugation at 15,000 × g for 5 min at 4°C, and the aliquots (5 μl) were used to determine protein content. Then, Laemmli’s sample buffer was added to supernatant fraction, and the samples were boiled at 100°C for 5 min.
Protein kinase assay. MAPK and CaM kinase II activities were determined by using in-gel kinase assay method of Geahlen et al. (1986), Kameshita and Fujisawa (1989), and Gotoh et al. (1990) with some modifications. Briefly, as a substrate for phosphorylation with kinases, MBP (0.5 mg/ml) was added to the separating SDS-PAGE before polymerization of acrylamide. Cell lysates (15–20 μg of protein/lane) were separated by 10% SDS-PAGE. After electrophoresis, SDS was removed by washing the gel with 20% 2-propanol in 50 mm Tris-HCl buffer, pH 7.5. Then, the gel was treated with 6 m guanidine HCl to denature the enzyme for 1 hr, followed by renaturation in 50 mm Tris-HCl buffer, pH 7.5, containing 0.04% Triton X-100, 5 mm mercaptoethanol, and 0.1 mm sodium orthovanadate for 16 hr at 4°C. After renaturation, the gel was preincubated at 22°C for 1 hr with 10 ml 40 mm HEPES buffer, pH 8.0, containing 2 mmdithiothreitol, 10 mm MgCl2, and 0.1 mm sodium orthovanadate. Phosphorylation of MBP was carried out by incubating the gel at 22°C for 1 hr with 10 ml 40 mm HEPES buffer, pH 8.0, containing 0.5 mmEGTA, 10 mm MgCl2, 0.1 mmsodium orthovanadate, and 40 μmγ-[32P] ATP (25 μCi). The gel was then rinsed with 5% (w/v) trichloroacetic acid solution containing 1% sodium pyrophosphate to remove noncovalently bound32P. Thereafter, the gel was dried, and the amount of 32P incorporation into MBP in the gel was quantified by a Bio-Imaging analyzer (BA100; Fujifilm, Tokyo, Japan) to determine the kinase activity.
Immunoprecipitation of CaM kinase II. Hippocampal slice cell lysates were prepared as described under “Sample preparation”. After centrifugation, the supernatant fraction was incubated with active-specific CaM kinase II antibody (10 μg of IgG protein) and 40 μl of protein A-Sepharose CL-4B suspension (50% v/v) at 4°C for 4 hr. After incubation, the immunocomplex immobilized on protein A was precipitated by centrifugation at 12,000 × g for 5 min. The immunocomplex was washed four times with a buffer containing 0.1% Triton X-100, and in mm: 50 HEPES, 4 EGTA, 10 EDTA, 15 Na4P2O7, 100 β-glycerophosphate, and 25 NaF. Laemmli’s sample buffer was added to the supernatant fraction and the immunocomplex, and then the samples were boiled at 100°C for 5 min before application to SDS-PAGE.
RESULTS
Identification of CaM kinase II and MAP kinase activities in MBP-contained polyacrylamide gels
To determine the activities of CaM kinase II and MAPK in hippocampal slices, we performed an in-gel kinase assay by using MBP as a good exogenous substrate for MAPK (Gotoh et al., 1990) as well as for CaM kinase II (Gupta et al., 1992). This method has been shown to be sensitive and useful for detection of a variety of protein kinases in complex mixtures (Geahlen et al., 1986; Kameshita and Fujisawa, 1989). Five major bands were detected in the hippocampal slices (Fig.1A). Among them, the band that matched CaM kinase II was identified by applying the purified CaM kinase II onto the gel. As shown in Figure 1A, the topmost band of hippocampal slice samples were seen to coincide with the purified CaM kinase II (lane 9).
Fig. 1.

Identification of CaM kinase II and MAPK activities in MBP-contained polyacrylamide gels. A,Autoradiographs of in-gel kinase assay. Lanes 1, 2, 7, and 8, Immunocomplexes from hippocampal slices incubated with (lanes 1 and 8) and without (lanes 2 and 7) anti-active CaM kinase II antibody; lanes 3–6, supernatants of the hippocampal slices after immunoprecipitation of CaM kinase II. Each sample was immunoprecipitated in the presence (lanes 3 and 5) and absence (lanes 4 and 6) of anti-active CaM kinase II antibody before application to SDS-PAGE. Lane 9, Autophosphorylation assay for purified CaM kinase II (1 μg) in the presence of Ca2+/calmodulin. B, Two lower bands corresponded to MAPK isoforms of 42 and 44 kDa and were markedly suppressed by 50 μm PD098059.
To gain further insight into the nature of the band of CaM kinase II in in-gel kinase assay, we next performed an experiment on immunoprecipitation of CaM kinase II in LTP-established and control slices. As seen in Figure 1A, samples of control and LTP slices were immunoprecipitated with anti-active CaM kinase II antibody or with the buffer lacking the antibody before separation by SDS-PAGE. The band of CaM kinase II was strongly detected in the antibody-untreated LTP (Fig. 1A, lane 6) but not in the control supernatant sample (Fig.1A, lane 4), whereas the solubilized CaM kinase II was much less visualized in anti-active CaM kinase II-treated LTP sample (Fig. 1A, lane 5), indicating that the band was able to be absorbed by anti-active CaM kinase II antibody. When the immunocomplexes were applied to SDS-PAGE, followed by autoradiography, the antibody-treated LTP (Fig. 1A, lane 8) and control (Fig.1A, lane 1) samples appeared to be detected in a band corresponding to CaM kinase II. Thus, changes in intensity of the topmost band may reflect a change in activity of CaM kinase II in slices, although the possibility is not excluded that a little activity of other unidentified kinase was present on background.
Two lower bands that corresponded to MAPK isoforms of 42 and 44 kDa had certainly been identified by Gotoh et al. (1990) and Kurino et al. (1995). Both bands were markedly suppressed by 50 μmPD098059, a specific inhibitor of MEK (Fig. 1B). In the following experiments, we used this approach to assay activities of autonomous CaM kinase II and MAPK in CA1 areas.
Transient activation of 42 kDa MAPK during LTP induction
In a previous study, we showed that stimulation of glutamate receptors by treatment with NMDA or glutamate produced a transient increase in 42 kDa MAPK activity as well as the Ca2+-independent form of CaM kinase II (Fukunaga et al., 1992; Kurino et al., 1995; Fukunaga and Miyamoto, 1998) in cultured hippocampal neurons, whereas in the hippocampal slices, HFS resulted in a long-lasting increase in activity of CaM kinase II (Fukunaga et al., 1993). These observations suggest that the activation of protein kinases can be regulated in an activity-dependent manner. A Western blotting study showed that 2 min after delivering HFS to Schaffer collateral pathway in hippocampal slices, the 42 kDa MAPK activity increased, and this activation was not persistent during the entire period of LTP induction (English and Sweatt, 1996, 1997). To confirm this result, we therefore investigated changes in MAPK activity in dissected CA1 areas of hippocampal slices, before and after HFS delivery. Figure 2 shows the time course of MAPK activities, under different conditions. Immediately after HFS, substantial increases in the activity of 42 kDa MAPK, but not 44 kDa MAPK were evident in LTP-established slices. A peak increase of 38.6 ± 3.7% (n = 8) in kinase activity was observed at 3 min after HFS, followed by a rapid recovery at 10 min and then reverting to nearly basal levels after 30 min. This result is similar to that reported by other investigators (English and Sweatt, 1996). However, in slices subjected to LFS, no increase in MAPK activity was observed (n = 6).
Fig. 2.
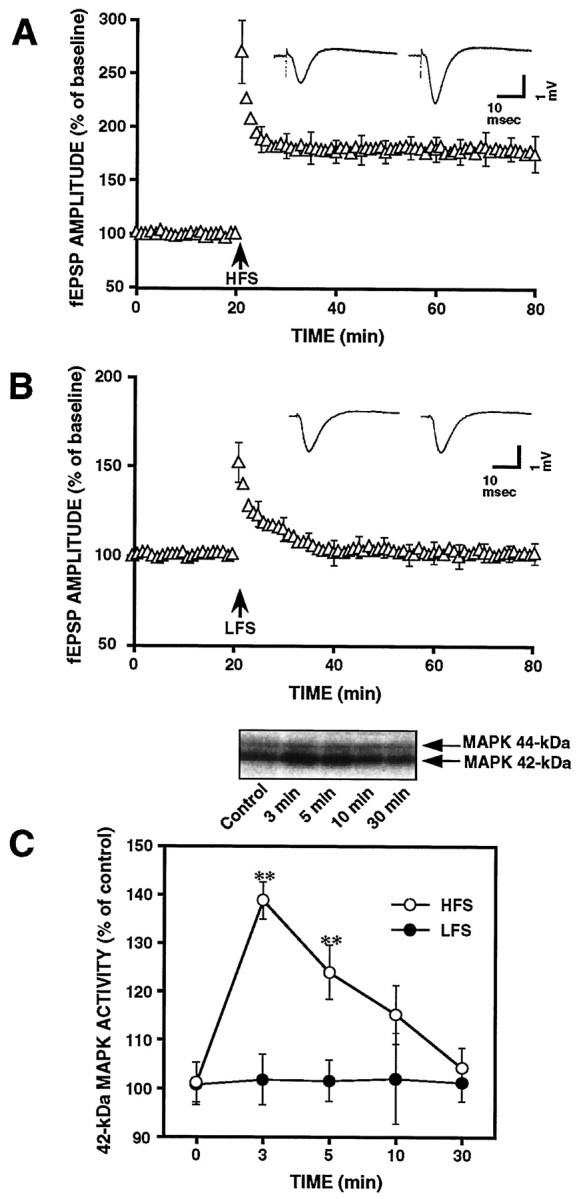
LTP induction is accompanied by a transient increase in 42 kDa MAPK but not 44 kDa MAPK activity. A,Electrophysiological recordings illustrate the average changes in synaptic efficacy in CA1 area of hippocampal slices (n = 12) produced by HFS. Average fEPSP amplitude for each time point was normalized to the average baseline response before HFS. Inset, Representative traces before and 1 hr after HFS. B, Normalized fEPSP amplitude from LFS-received slices. Inset, Representative traces before and 1 hr after LFS. C, Time course of 42 kDa MAPK activation in the slices treated with HFS or LFS. Top panel shows an autoradiograph of in-gel kinase assay for the time course of HFS-induced MAPK activity. Results are expressed as percentages of the activity in control slices at zero time. Data are mean ± SE values. The changes in the MAPK activity were statistically significant versus the control: **p< 0.01.
Concentration-dependent inhibitory effects of PD098059 on LTP
It has been well documented that the phosphorylation of MAPK is regulated by MEK, the MAPK kinase, which occurs upstream of MAPK. Thus, to test the physiological role of 42 kDa MAPK in LTP, we used the specific inhibitor of MEK, PD098059, to indirectly abolish the activation of MAPK. In the LTP experiments, we first used a relatively higher concentration of 50 μm PD098059 because ∼90% blocking effect on MAPK has been observed with use of this concentration (English and Sweatt, 1997). Before HFS delivery, PD098059 was applied to slices for 90 min in the preincubation period. During the drug application, we found no change in basal synaptic transmission of the slices (data not shown). In contrast, the potentiation of fEPSP after HFS delivery significantly decayed to nearly basal levels after 60 min in slices pretreated with 50 μm PD098059 (Fig.3A; n = 9), whereas a stable LTP was maintained for >1 hr in control slices (Fig. 3A; n = 12). However, at a concentration of 30 μm, the drug preincubation for the same period of 90 min did not attenuate the induction of LTP (Fig. 3B; n = 6). These results suggest that the inhibitory effect of PD098059 on LTP was concentration-dependent.
Fig. 3.

PD098059 caused concentration-dependent inhibitions of both LTP induction and CaM kinase II activation in CA1 areas. A, Compared with the control slices (open triangles; n = 12), preincubation of slices with 50 μm PD098059 markedly prevented HFS-sensitive induction of hippocampal LTP (filled triangles;n = 9). Average amplitude of fEPSP for each point was normalized to the average responses of baseline before HFS.B, At a lower concentration of 30 μm, PD098059 did not affect the induction of LTP (n = 6). C, Analysis of CaM kinase II and MAPK activities in the slices treated with different concentrations of PD098059.Left panel shows a representative autoradiograph of in-gel kinase assay for kinase activities. Right diagramillustrates statistical data. In HFS-receiving slices, kinase activity was assayed at 3 min after HFS. At 50 μm(n = 9), PD098059 suppressed both 42 and 44 kDa MAPK activity and prevented HFS-induced CaM kinase II activation. At 30 μm (n = 6) of the drug, only MAPK, not CaM kinase II was inhibited. Data are expressed as mean ± SE values. **p < 0.01.
Inhibitory effects of PD098059 on CaM kinase II and MAPK activation in response to LTP-inducing HFS
As described above, at concentrations of 30 and 50 μm, PD098059 appeared to have different effects on the LTP induction. To determine if the lack of inhibitory effect seen with 30 μm is caused by the weak blocking action of the drug on the activation of MAPK, we next examined effects of PD098059 on protein kinase activity in the hippocampal slices. Because a maximal increase in MAPK activity was observed at 3 min after HFS (Fig.2B), we designed an experimental protocol of collecting slices at 3 min after HFS to direct attention to this time point. In PD098059-treated slices, the activities of both 44 and 42 kDa MAPK were markedly inhibited by the drug (Fig. 3C). The extent of 42 kDa MAPK inhibition, expressed as a percentage of the control, was 40 ± 3.1% at 30 μm(n = 6) and 48 ± 4.3% at 50 μm (n = 9). Thus, at both concentrations, PD098059 similarly inhibited the HFS-induced transient activation of 42 kDa MAPK. These observations indicate that the effect of 50 μm PD098059 on LTP induction could not be simply explained by inhibitory effects of the drug on MAPK.
We then analyzed the changes in CaM kinase II activity detectable in the same gel. With the same protocol for MAPK assay, CaM kinase II activity was also examined at a time point of 3 min after HFS. In Figure 3C, the topmost band, which corresponds to CaM kinase II is so different in the presence of 30 or 50 μm PD098059. When the drug was applied at 50 μm, HFS did not produce any significant increase in CaM kinase II activity in the slices in which HFS failed to elicit the LTP. In contrast, in slices pretreated with 30 μm PD098059, a brief HFS markedly activated CaM kinase II as well as LTP. Statistical analysis showed that HFS delivery produced an increase of 78.6 ± 15.3 (n = 8) in CaM kinase II activity under condition of 30 μmdrug treatment, whereas no significant increase was detectable in 50 μm drug-treated slices (n = 9). Thus, in addition to MEK, its downstream target, PD098059, also has inhibitory effects on CaM kinase II activation of hippocampal slices.
A question arose here as to whether this effect occurs by directly inhibiting activity of CaM kinase II. To answer this question, we next tested the effect of PD098059 on the activity of purified CaM kinase IIin vitro. At concentrations of 10, 50, and 100 μm, PD098059 did not affect the activity of this kinase (data not shown), suggesting that PD098059 has no direct inhibitory effect on CaM kinase II.
Effects of calmidazolium on the activation of CaM kinase II and 42 kDa MAPK in LTP
CaM kinase II is a Ca2+/calmodulin-stimulated target enzyme. Because both Ca2+ influx and calmodulin activation are prerequisite for switching the kinase autophosphorylation, their inhibitors are capable of blocking activation of the kinase. Calmidazolium, a potent calmodulin antagonist that does not influence Ca2+ flux and PKC activity (Reymann et al., 1988), has been shown to block calmodulin-mediated LTP (Reymann et al., 1988; Fukunaga et al., 1995). Here, we repeated the experiments of calmidazolium on LTP and analyzed changes in activities of CaM kinase II and MAPK in the CA1 area of hippocampal slices. In accordance with our previous report, incubation of hippocampal slices with 50 μm calmidazolium for 1 hr strongly blocked post-tetanic potentiation and LTP induction (Fig.4C), whereas basal synaptic transmission and HFS-mediated depolarization were not affected (Fig.4A,B). The assay for protein kinase activity was then performed in HFS-received slices under calmidazolium-treated and untreated conditions, respectively. Slice samples for protein kinase assay were collected at 3 min after HFS. Compared with the LTP-established slices, in which both CaM kinase II and MAPK activities increased, bath-application of 50 μmcalmidazolium suppressed the activation of CaM kinase II that responded to HFS. However, delivery of the same HFS still produced a transient increase in MAPK activity from the control level to a higher activity of 132 ± 5.9 (n = 5), even in calmidazolium-treated slices (Fig. 4B). These results suggest that HFS-induced activation of MAPK per se may not lead to LTP induction.
Fig. 4.

Calmidazolium blocked the induction of LTP without affecting basal synaptic transmission, HFS-mediated depolarization, and HFS-induced 42 kDa MAPK activation. A, Normalized fEPSP amplitudes recorded before and during continuous calmidazolium application (n = 6). No change in basal synaptic transmission of the slices was found. B, Partial display of representative HFS-mediated depolarization responses in control and calmidazolium-treated slices. Calmidazolium had no effect on HFS-mediated depolarization. C, Normalized fEPSP amplitudes recorded before and after HFS. Pretreatment of slices with 50 μm calmidazolium for 1 hr strongly blocked the formation of LTP (filled triangles;n = 5). D, Kinase assay for activities of CaM kinase II and 42 kDa MAPK at 3 min after HFS. Both activities of CaM kinase II and 42 kDa MAPK increased in LTP-inducing slices (n = 8), whereas application of calmidazolium (50 μm) blocked HFS-induced increase in the activity of CaM kinase II but not of 42 kDa MAPK (n= 5). **p < 0.01. Calibration: 1 mV, 70 msec.
Strengthening the LTP induction and CaM kinase II activation by exogenous BDNF
It has been proposed that neurotrophic factors can regulate neuronal activity and synaptic efficacy via the activation of TrK family of tyrosine kinase receptors (Kang and Schuman, 1995; Levine et al., 1995; Figurov et al., 1996; Akaneya et al., 1997). One of these factors, BDNF, is synthesized predominantly in neurons, and is highly expressed in the hippocampus of the adult species brain. The binding of BDNF to the TrKB receptor triggers the kinase cascade and thereby activates MAPK in culture neurons (Finkbeiner et al., 1997; Fukunaga and Miyamoto, 1998). We then used BDNF as a modulator for MAPK to further explore the role of MAPK in LTP in hippocampal slices. As shown in Figure 5A, application of 50 ng/ml BDNF alone did not affect basal synaptic strength under our experimental conditions. This result is similar to that reported byFigurov et al. (1996) and by Gottschalk et al. (1998), which is contradictory to observations that BDNF induced a long-lasting enhancement of synaptic transmission (Kang and Schuman, 1995). Possible explanation for the discrepancy has been proposed by Kang et al. (1996), who reported that the perfusion rate is critical for the penetration of BDNF into the hippocampal slices. In Figure5C, subthreshold SHFS induced only an STP of fEPSP in control slices. However, when slices were preincubated with 50 ng/ml BDNF for >2 hr, the same SHFS triggered a typical LTP that lasted for >1 hr. This result is consistent with previous findings (Figurov et al., 1996).
Fig. 5.

BDNF promoted the induction of LTP and the activation of CaM kinase II without affecting basal synaptic transmission and MAPK activity. A, Normalized fEPSP amplitudes recorded before and during continuous application of 50 ng/ml BDNF (n = 6). BDNF alone did not affect basal synaptic strength of slices. B, Representative autoradiograph of MAPK activity assay in control and BDNF application slices. BDNF treatment did not result in MAPK activation.C, The fEPSP potentiation induced by a subthreshold SHFS in control (open triangles; n = 8) and BDNF-pretreated slices (filled triangles;n = 6). After 2 hr preincubation with BDNF (50 ng/ml), SHFS delivery converted STP to LTP. D, Kinase assay for CaM kinase II and MAPK activity in the presence or absence of BDNF. In contrast to control slices, the activity of CaM kinase II was increased in the slices treated with BDNF for 2 hr (n = 6), whereas no change in activity of MAPK was detectable. This effect was facilitated by delivering SHFS3(n = 6). **p < 0.01. An autoradiograph of kinase activities is shown in the left panel.
It has been shown that BDNF rapidly stimulates both 41 and 44 kDa MAPK with a time course reaching a maximum at 2 min and then returning toward baseline slowly over the next 60 min in cultured hippocampal pyramidal neurons (Marsh et al., 1993). To clarify whether the MAPK cascade is involved in BDNF-enhanced LTP, we measured the kinase activity in slices in the presence and absence of BDNF. We found no changes in both 44 and 42 kDa MAPK activity in BDNF-treated slices during the period of incubation with 50 ng/ml BDNF for 3, 10, 30, 60, and 120 min (Fig. 5B), except for a significant increase in activity of CaM kinase II at 2 hr (25 ± 7.7%; n= 6; Fig. 5D). Furthermore, delivery of SHFS to 2 hr BDNF-treated slices resulted in a further increase in CaM kinase II activity (43.4 ± 6.7%; n = 6; Fig.5D), which was detected at 3 min after SHFS (SHFS3). Based on these results, it seems unlikely that the effects of BDNF on SHFS-induced fEPSP potentiation are related to the activation of MAPK. Because the increase in activity of CaM kinase II is produced by BDNF, one can predict that the most possible candidate for LTP induction is CaM kinase II.
DISCUSSION
In the present study, using both electrophysiological and biochemical approaches we demonstrated that the induction of LTP is dependent on CaM kinase II activity rather than on MAPK activity in CA1 hippocampal slices. The transient activation of 42 kDa MAPK in HFS-receiving slices supports the view of activity-dependent regulation of this enzyme. A most interesting finding in our experiments is that we observed a heretofore unknown pharmacological effect of PD098059 that is commonly believed to be a specific inhibitor of MAPK kinase. Although earlier studies examined effects of this agent on a variety of protein kinases and found that only MAPK kinase is sensitive to it (Alessi et al., 1995; Dudley et al., 1995), information about its effect on CaM kinase II was not allied. Most recently, one research group suggested that MAPK activation is necessary for LTP because they found an inhibitory effect of PD098059 on LTP (English and Sweatt, 1997). In that paper, the authors said that they tested the effect of PD098059 on the activity of CaM kinase II in vitro and found no definite actions. This is consistent with our results. However, when we used it in HFS-induced slices, the data clearly showed that 50 μm PD098059 actually blocked HFS-induced activation of CaM kinase II in hippocampal slices. A similar result was also found in cultured hippocampal neurons (data not shown). This evidence suggests that there is an indirect effect of this compound on CaM kinase II, differing from its effect of binding to inactive form of MEK (Alessi et al., 1995), although the precise blocking mechanisms are unknown. Recently, PD098059 has been found to have a direct inhibitory effect on cyclooxygenase-1 and -2 (Borsch-Haubold et al., 1998). Therefore, one must be careful when using this drug as an MEK inhibitor at concentrations ≥50 μm.Additionally, these findings also suggest that the inhibition of hippocampal LTP induction with PD098059 was caused by its effects on CaM kinase II and not on the MEK/MAPK pathway. The parallel changes in hippocampal LTP and CaM kinase II activity indicate that CaM kinase II activation plays a critical role in LTP induction.
Recent studies have shown that the expression of BDNF is regulated by neuronal activity (Castren et al., 1992; Patterson et al., 1992;Dragunow et al., 1993) and that the synaptic efficacy and LTP induction were promoted by exogenous BDNF (Kang and Schuman, 1995; Figurov et al., 1996). Thus, BDNF may be a mediator of neuronal adaptive responses. Consistent with these findings, our data showed that BDNF enhanced the sensitivity of synapses to respond to SHFS. This effect may be caused by activation of CaM kinase II rather than to that of MAPK, because the active form of CaM kinase II alone has been found to be sufficient to augment synaptic strength with the same mechanism as LTP (Lledo et al., 1995), and BDNF can activate CaM kinase II without affecting MAPK activity in hippocampal slices. Other reports have recently shown that BDNF-induced CaM kinase II activation is dependent on a pathway of PLCγ and intracellular Ca2+ release (Blanquet and Lamour, 1997;Finkbeiner et al., 1997). Therefore, BDNF may be involved in the Ca2+/CaM-dependent kinase-mediated cascade of events that results in LTP construction. On the other hand, it has been proposed that a transcription factor, cAMP response element-binding protein (CREB), plays a key role in mediating neurotrophin responses because BDNF can activate CREB through a CaM kinase-dependent mechanism, and also BDNF itself can be regulated by a Ca2+/CaM kinase/CREB-dependent mechanism (Tsuda, 1996; Finkbeiner et al., 1997; Shieh et al., 1998; Tao et al., 1998). Therefore, the involvement of BDNF in the CaM kinase signaling cascade is important for the self control of the neuronal functions, including synaptic plasticity.
In addition to cytosolic localization, CaM kinase II was particularly enriched in postsynaptic densities (PSD), specializations of submembranous cytoskeleton that are believed to participate in the regulation of receptor function, structural modification, and synaptic plasticity (Kennedy et al., 1983; Wu et al., 1992; Kennedy, 1993). CaM kinase II in the PSD possesses regulatory properties and mechanisms of activation similar to the cytosolic CaM kinase II (Rich et al., 1989). Under our experimental conditions, only the soluble kinases were analyzed. However, our previous paper has demonstrated that >95% of CaM kinase II activity remained in the supernatant fraction (Fukunaga et al., 1993, 1995), i.e., only 5% of CaM kinase II activity was present in Triton X-100-insoluble cytoskeletal fraction, including PSD in hippocampal slices, which is consistent with the fact that most of CaM kinase II in the PSD is largely inactive (Rostas et al., 1986). Studies on association of soluble CaM kinase II with PSD showed that cytosolic CaM kinase II is reversibly translocated to the PSD and forms PSD–CaM kinase II complex in a phosphorylation-dependent manner (Yoshimura and Yamauchi, 1997).The translocated CaM kinase II may be collected in the Triton X-100-soluble fraction. When calcium influx increases in postsynapse, the cytosolic CaM kinase II is autophosphorylated and activated and then translocated to the PSD. Furthermore, other groups reported that the active CaM kinase II phosphorylates GluR1 and other functional proteins in PSD, resulting in an enhancement of postsynaptic response as occurs during LTP (McGlade-McCulloh et al., 1993). Thus, translocation of the soluble CaM kinase II to PSD in response to HFS-induced calcium influx was considered to be involved in LTP induction (Strack et al., 1997). In contrast to CaM kinase II in PSD, although 42 kDa MAPK and MAPK kinase were also found to be present in PSD (Suzuki et al., 1995), the roles of PSD-bound MAPK in regulation of synaptic response have not yet been surveyed.
Our results show that LTP-inducing electric stimuli rapidly increase the 42 kDa MAPK activity, and 50 μm PD098059 clearly inhibits the induction of LTP. However, we cannot say that MAPK activation is required for the LTP induction because inhibiting MAPK activity by 30 μm PD098059 did not affect the LTP expression. As MAPK participates in regulating diverse cellular processes, especially in gene transcription such as Elk-1 (Xia et al., 1996) and CREB transcriptional pathways (Xing et al., 1996; Finkbeiner et al., 1997), the activity-dependent activation of MAPK may underlie some important neurophysiological functions. Although we stated that the LTP induction cannot be attributed to MAPK activation, the maintenance of LTP cannot be excluded as the potential target of this kinase, because protein synthesis and gene expression are required for the late phase of LTP (Winder et al., 1998). Extensive studies are needed for understanding the signaling meaning of HFS-induced MAPK activation on the maintenance of LTP.
Footnotes
This work was supported in part by Grants-in-Aid for Scientific Research and for Scientific Research on Priority Areas from the Ministry of Education, Science, Sports, and Culture of Japan, and by a Research Grant from the Human Frontier Science Program. We thank Dr. H. Nakanishi and Prof. K. Yamamoto, Department of Pharmacology, Kyushu University Faculty of Dentistry, for technical advice.
Correspondence should be addressed to Prof. Eishichi Miyamoto, Department of Pharmacology, Kumamoto University School of Medicine, 2-2-1 Honjo, Kumamoto 860–0811, Japan.
REFERENCES
- 1.Akaneya Y, Tsumoto T, Kinoshita S, Hatanaka H. Brain-derived neurotrophic factor enhances long-term potentiation in rat visual cortex. J Neurosci. 1997;17:6707–6716. doi: 10.1523/JNEUROSCI.17-17-06707.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Alessi DR, Cuenda A, Cohen P, Dudley DT, Saltiel AR. PD 098059 is a specific inhibitor of the activation of mitogen-activated protein kinase kinase in vitro and in vivo. J Biol Chem. 1995;270:27489–27494. doi: 10.1074/jbc.270.46.27489. [DOI] [PubMed] [Google Scholar]
- 3.Atkins CM, Selcher JC, Petraitis JJ, Trzaskos JM, Sweatt JD. The MAPK cascade is required for mammalian associative learning. Nat Neurosci. 1998;1:602–608. doi: 10.1038/2836. [DOI] [PubMed] [Google Scholar]
- 4.Bading H, Greenberg ME. Stimulation of protein tyrosine phosphorylation by NMDA receptor activation. Science. 1991;253:912–4. doi: 10.1126/science.1715095. [DOI] [PubMed] [Google Scholar]
- 5.Baron C, Benes C, Van TH, Fagard R, Roisin MP. Potassium chloride pulse enhances mitogen-activated protein kinase activity in rat hippocampal slices. J Neurochem. 1996;66:1005–1010. doi: 10.1046/j.1471-4159.1996.66031005.x. [DOI] [PubMed] [Google Scholar]
- 6.Barria A, Muller D, Derkach V, Griffith LC, Soderling TR. Regulatory phosphorylation of AMPA-type glutamate receptors by CaM-KII during long-term potentiation. Science. 1997;276:2042–2045. doi: 10.1126/science.276.5321.2042. [DOI] [PubMed] [Google Scholar]
- 7.Blanquet PR, Lamour Y. Brain-derived neurotrophic factor increases Ca2+/calmodulin-dependent protein kinase II activity in hippocampus. J Biol Chem. 1997;272:24133–24136. doi: 10.1074/jbc.272.39.24133. [DOI] [PubMed] [Google Scholar]
- 8.Bliss TV, Collingridge GL. A synaptic model of memory: long-term potentiation in the hippocampus. Nature. 1993;361:31–39. doi: 10.1038/361031a0. [DOI] [PubMed] [Google Scholar]
- 9.Borsch-Haubold AG, Pasquet S, Watson SP. Direct inhibition of cyclooxygenase-1 and -2 by the kinase inhibitors SB 203580 and PD 98059. J Biol Chem. 1998;273:28766–28772. doi: 10.1074/jbc.273.44.28766. [DOI] [PubMed] [Google Scholar]
- 10.Castren E, Zafra F, Thoenen H, Lindholm D. Light regulates expression of brain-derived neurotrophic factor mRNA in rat visual cortex. Proc Natl Acad Sci USA. 1992;89:9444–9448. doi: 10.1073/pnas.89.20.9444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Deibler GE, Martenson RE, Kies MW. Large scale preparation of myelin basic protein from central nervous tissue of several mammalian species. Prep Biochem. 1972;2:139–165. doi: 10.1080/00327487208061467. [DOI] [PubMed] [Google Scholar]
- 12.Dragunow M, Beilharz E, Mason B, Lawlor P, Abraham W, Gluckman P. Brain-derived neurotrophic factor expression after long-term potentiation. Neurosci Lett. 1993;160:232–236. doi: 10.1016/0304-3940(93)90420-p. [DOI] [PubMed] [Google Scholar]
- 13.Dudley DT, Pang L, Decker SJ, Bridges AJ, Saltiel AR. A synthetic inhibitor of the mitogen-activated protein kinase cascade. Proc Natl Acad Sci USA. 1995;92:7686–7689. doi: 10.1073/pnas.92.17.7686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.English JD, Sweatt JD. Activation of p42 mitogen-activated protein kinase in hippocampal long term potentiation. J Biol Chem. 1996;271:24329–24332. doi: 10.1074/jbc.271.40.24329. [DOI] [PubMed] [Google Scholar]
- 15.English JD, Sweatt JD. A requirement for the mitogen-activated protein kinase cascade in hippocampal long term potentiation. J Biol Chem. 1997;272:19103–19106. doi: 10.1074/jbc.272.31.19103. [DOI] [PubMed] [Google Scholar]
- 16.Figurov A, Pozzo-Miller LD, Olafsson P, Wang T, Lu B. Regulation of synaptic responses to high-frequency stimulation and LTP by neurotrophins in the hippocampus. Nature. 1996;381:706–709. doi: 10.1038/381706a0. [DOI] [PubMed] [Google Scholar]
- 17.Finkbeiner S, Tavazoie SF, Maloratsky A, Jacobs KM, Harris KM, Greenberg ME. CREB: a major mediator of neuronal neurotrophin responses. Neuron. 1997;19:1031–1047. doi: 10.1016/s0896-6273(00)80395-5. [DOI] [PubMed] [Google Scholar]
- 18.Fukunaga K, Miyamoto E. Role of MAP kinase in neurons. Mol Neurobiol. 1998;16:79–95. doi: 10.1007/BF02740604. [DOI] [PubMed] [Google Scholar]
- 19.Fukunaga K, Soderling TR, Miyamoto E. Activation of Ca2+/calmodulin-dependent protein kinase II and protein kinase C by glutamate in cultured rat hippocampal neurons. J Biol Chem. 1992;267:22527–22533. [PubMed] [Google Scholar]
- 20.Fukunaga K, Stoppini L, Miyamoto E, Muller D. Long-term potentiation is associated with an increased activity of Ca2+/calmodulin-dependent protein kinase II. J Biol Chem. 1993;268:7863–7867. [PubMed] [Google Scholar]
- 21.Fukunaga K, Muller D, Miyamoto E. Increased phosphorylation of Ca2+/calmodulin-dependent protein kinase II and its endogenous substrates in the induction of long-term potentiation. J Biol Chem. 1995;270:6119–6124. doi: 10.1074/jbc.270.11.6119. [DOI] [PubMed] [Google Scholar]
- 22.Geahlen RL, Anostario M, Jr, Low PS, Harrison ML. Detection of protein kinase activity in sodium dodecyl sulfate-polyacrylamide gels. Anal Biochem. 1986;153:151–158. doi: 10.1016/0003-2697(86)90074-6. [DOI] [PubMed] [Google Scholar]
- 23.Gotoh Y, Nishida E, Yamashita T, Hoshi M, Kawakami M, Sakai H. Microtubule-associated-protein (MAP) kinase activated by nerve growth factor and epidermal growth factor in PC12 cells. Identity with the mitogen-activated MAP kinase of fibroblastic cells. Eur J Biochem. 1990;193:661–669. doi: 10.1111/j.1432-1033.1990.tb19384.x. [DOI] [PubMed] [Google Scholar]
- 24.Gottschalk W, Pozzo-Miller LD, Figurov A, Lu B. Presynaptic modulation of synaptic transmission and plasticity by brain-derived neurotrophic factor in the developing hippocampus. J Neurosci. 1998;18:6830–6839. doi: 10.1523/JNEUROSCI.18-17-06830.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gupta RP, Lapadula DM, Abou-Donia MB. Ca2+/calmodulin-dependent protein kinase II from hen brain. Purification and characterization. Biochem Pharmacol. 1992;43:1975–1988. doi: 10.1016/0006-2952(92)90641-u. [DOI] [PubMed] [Google Scholar]
- 26.Kameshita I, Fujisawa H. A sensitive method for detection of calmodulin-dependent protein kinase II activity in sodium dodecyl sulfate-polyacrylamide gel. Anal Biochem. 1989;183:139–143. doi: 10.1016/0003-2697(89)90181-4. [DOI] [PubMed] [Google Scholar]
- 27.Kang H, Schuman EM. Long-lasting neurotrophin-induced enhancement of synaptic transmission in the adult hippocampus. Science. 1995;267:1658–1662. doi: 10.1126/science.7886457. [DOI] [PubMed] [Google Scholar]
- 28.Kang H, Jia LZ, Suh KY, Tang L, Schuman EM. Determination of BDNF-induced hippocampal synaptic plasticity: role of the TrkB receptor and the kinetics of neurotrophin delivery. Learn Mem. 1996;3:188–196. doi: 10.1101/lm.3.2-3.188. [DOI] [PubMed] [Google Scholar]
- 29.Kang UG, Hong KS, Jung HY, Kim YS, Seong YS, Yang YC, Park JB. Activation and tyrosine phosphorylation of 44-kDa mitogen-activated protein kinase (MAPK) induced by electroconvulsive shock in rat hippocampus. J Neurochem. 1994;63:1979–1982. doi: 10.1046/j.1471-4159.1994.63051979.x. [DOI] [PubMed] [Google Scholar]
- 30.Kennedy MB. The postsynaptic density. Curr Opin Neurobiol. 1993;3:732–737. doi: 10.1016/0959-4388(93)90145-o. [DOI] [PubMed] [Google Scholar]
- 31.Kennedy MB, Bennett MK, Erondu NE. Biochemical and immunochemical evidence that the “major postsynaptic density protein” is a subunit of a calmodulin-dependent protein kinase. Proc Natl Acad Sci USA. 1983;80:7357–7361. doi: 10.1073/pnas.80.23.7357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kurino M, Fukunaga K, Ushio Y, Miyamoto E. Activation of mitogen-activation of mitogen-activated protein kinase in cultured rat hippocampal neurons by glutamate receptors. J Neurochem. 1995;65:1282–1289. doi: 10.1046/j.1471-4159.1995.65031282.x. [DOI] [PubMed] [Google Scholar]
- 33.Levine ES, Dreyfus CF, Black IB, Plummer MR. Brain-derived neurotrophic factor rapidly enhances synaptic transmission in hippocampal neurons via postsynaptic tyrosine kinase receptors. Proc Natl Acad Sci USA. 1995;92:8074–8077. doi: 10.1073/pnas.92.17.8074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lisman JE, Goldring MA. Feasibility of long-term storage of graded information by the Ca2+/calmodulin-dependent protein kinase molecules of the postsynaptic density. Proc Natl Acad Sci USA. 1988;85:5320–5324. doi: 10.1073/pnas.85.14.5320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lledo PM, Hjelmstad GO, Mukherji S, Soderling TR, Malenka RC, Nicoll RA. Calcium/calmodulin-dependent kinase II and long-term potentiation enhance synaptic transmission by the same mechanism. Proc Natl Acad Sci USA. 1995;92:11175–11179. doi: 10.1073/pnas.92.24.11175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Malenka RC, Kauer JA, Perkel DJ, Mauk MD, Kelly PT, Nicoll RA, Waxham MN. An essential role for postsynaptic calmodulin and protein kinase activity in long-term potentiation. Nature. 1989;340:554–557. doi: 10.1038/340554a0. [DOI] [PubMed] [Google Scholar]
- 37.Malenka RC, Nicoll RA. NMDA-receptor-dependent synaptic plasticity: multiple forms and mechanisms. Trends Neurosci. 1993;16:521–527. doi: 10.1016/0166-2236(93)90197-t. [DOI] [PubMed] [Google Scholar]
- 38.Malinow R, Schulman H, Tsien RW. Inhibition of postsynaptic PKC or CaMKII blocks induction but not expression of LTP. Science. 1989;245:862–866. doi: 10.1126/science.2549638. [DOI] [PubMed] [Google Scholar]
- 39.Marsh HN, Scholz WK, Lamballe F, Klein R, Nanduri V, Barbacid M, Palfrey HC. Signal transduction events mediated by the BDNF receptor gp 145trkB in primary hippocampal pyramidal cell culture. J Neurosci. 1993;13:4281–4292. doi: 10.1523/JNEUROSCI.13-10-04281.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Martin KC, Michael D, Rose JC, Barad M, Casadio A, Zhu H, Kandel ER. MAP kinase translocates into the nucleus of the presynaptic cell and is required for long-term facilitation in Aplysia. Neuron. 1997;18:899–912. doi: 10.1016/s0896-6273(00)80330-x. [DOI] [PubMed] [Google Scholar]
- 41.McGlade-McCulloh E, Yamamoto H, Tan SE, Brickey DA, Soderling TR. Phosphorylation and regulation of glutamate receptors by calcium/calmodulin-dependent protein kinase II. Nature. 1993;362:640–642. doi: 10.1038/362640a0. [DOI] [PubMed] [Google Scholar]
- 42.Miyamoto E, Fukunaga K. A role of Ca2+/calmodulin-dependent protein kinase II in the induction of long-term potentiation in hippocampal CA1 area. Neurosci Res. 1996;24:117–122. doi: 10.1016/0168-0102(95)00991-4. [DOI] [PubMed] [Google Scholar]
- 43.Ouyang Y, Kantor D, Harris KM, Schuman EM, Kennedy MB. Visualization of the distribution of autophosphorylated calcium/calmodulin-dependent protein kinase II after tetanic stimulation in the CA1 area of the hippocampus. J Neurosci. 1997;17:5416–5427. doi: 10.1523/JNEUROSCI.17-14-05416.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Patterson SL, Grover LM, Schwartzkroin PA, Bothwell M. Neurotrophin expression in rat hippocampal slices: a stimulus paradigm inducing LTP in CA1 evokes increases in BDNF and NT-3 mRNAs. Neuron. 1992;9:1081–1088. doi: 10.1016/0896-6273(92)90067-n. [DOI] [PubMed] [Google Scholar]
- 45.Reymann KG, Brodemann R, Kase H, Matthies H. Inhibitors of calmodulin and protein kinase C block different phases of hippocampal long-term potentiation. Brain Res. 1988;461:388–392. doi: 10.1016/0006-8993(88)90274-0. [DOI] [PubMed] [Google Scholar]
- 46.Rich DP, Colbran RJ, Schworer CM, Soderling TR. Regulatory properties of calcium/calmodulin-dependent protein kinase II in rat brain postsynaptic densities. J Neurochem. 1989;53:807–816. doi: 10.1111/j.1471-4159.1989.tb11777.x. [DOI] [PubMed] [Google Scholar]
- 47.Roberts TM. A signal chain of events. Nature. 1992;360:534–535. doi: 10.1038/360534a0. [DOI] [PubMed] [Google Scholar]
- 48.Rostas JA, Weinberger RP, Dunkley PR. Multiple pools and multiple forms of calmodulin-stimulated protein kinase during development: relationship to postsynaptic densities. Prog Brain Res. 1986;69:355–371. doi: 10.1016/s0079-6123(08)61070-5. [DOI] [PubMed] [Google Scholar]
- 49.Seger R, Krebs EG. The MAPK signaling cascade. FASEB J. 1995;9:726–735. [PubMed] [Google Scholar]
- 50.Shieh PB, Hu SC, Bobb K, Timmusk T, Ghosh A. Identification of a signaling pathway involved in calcium regulation of BDNF expression. Neuron. 1998;20:727–740. doi: 10.1016/s0896-6273(00)81011-9. [DOI] [PubMed] [Google Scholar]
- 51.Silva AJ, Stevens CF, Tonegawa S, Wang Y. Deficient hippocampal long-term potentiation in α-calcium-calmodulin kinase II mutant mice. Science. 1992;257:201–206. doi: 10.1126/science.1378648. [DOI] [PubMed] [Google Scholar]
- 52.Strack S, Choi S, Lovinger DM, Colbran RJ. Translocation of autophosphorylated calcium/calmodulin-dependent protein kinase II to the postsynaptic density. J Biol Chem. 1997;272:13467–13470. doi: 10.1074/jbc.272.21.13467. [DOI] [PubMed] [Google Scholar]
- 53.Suzuki T, Okumura-Noji K, Nishida E. ERK2-type mitogen-activated protein kinase (MAPK) and its substrates in postsynaptic density fractions from the rat brain. Neurosci Res. 1995;22:277–285. doi: 10.1016/0168-0102(95)00902-6. [DOI] [PubMed] [Google Scholar]
- 54.Tao X, Finkbeiner S, Arnold DB, Shaywitz AJ, Greenberg ME. Ca2+ influx regulates BDNF transcription by a CREB family transcription factor-dependent mechanism. Neuron. 1998;20:709–726. doi: 10.1016/s0896-6273(00)81010-7. [DOI] [PubMed] [Google Scholar]
- 55.Tsuda M. Cascade of gene expression induced by Ca2+ signals in neurons. Neurochem Int. 1996;29:443–451. doi: 10.1016/0197-0186(96)00014-9. [DOI] [PubMed] [Google Scholar]
- 56.Winder DG, Mansuy IM, Osman M, Moallem TM, Kandel ER. Genetic and pharmacological evidence for a novel, intermediate phase of long-term potentiation suppressed by calcineurin. Cell. 1998;92:25–37. doi: 10.1016/s0092-8674(00)80896-x. [DOI] [PubMed] [Google Scholar]
- 57.Wu K, Huang Y, Adler J, Black IB. On the identity of the major postsynaptic density protein. Proc Natl Acad Sci USA. 1992;89:3015–3019. doi: 10.1073/pnas.89.7.3015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Xia Z, Dudek H, Miranti CK, Greenberg ME. Calcium influx via the NMDA receptor induces immediate early gene transcription by a MAP kinase/ERK-dependent mechanism. J Neurosci. 1996;16:5425–5436. doi: 10.1523/JNEUROSCI.16-17-05425.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Xing J, Ginty DD, Greenberg ME. Coupling of the RAS-MAPK pathway to gene activation by RSK2, a growth factor-regulated CREB kinase. Science. 1996;273:959–963. doi: 10.1126/science.273.5277.959. [DOI] [PubMed] [Google Scholar]
- 60.Yoshimura Y, Yamauchi T. Phosphorylation-dependent reversible association of Ca2+/calmodulin-dependent protein kinase II with the postsynaptic densities. J Biol Chem. 1997;272:26354–26359. doi: 10.1074/jbc.272.42.26354. [DOI] [PubMed] [Google Scholar]