

Abstract
Nitric oxide (NO) is a new intercellular messenger that occurs naturally in the brain without causing overt toxicity. Yet, NO has been implicated as a mediator of cell death in cell death. One explanation is that ischemia causes overproduction of NO, allowing it to react with superoxide to form the potent oxidant peroxynitrite. To address this question, we used immunohistochemistry for citrulline, a marker for NO synthase activity, and 3-nitrotyrosine, a marker for peroxynitrite formation, in mice subjected to reversible middle cerebral artery occlusion. We show that ischemia triggers a marked augmentation in citrulline immunoreactivity but more so in the peri-infarct than the infarcted tissue. This increase is attributable to the activation of a large population (∼80%) of the neuronal isoform of NO synthase (nNOS) that is catalytically inactive during basal conditions, indicating a tight regulation of physiological NO production in the brain. In contrast, 3-nitrotyrosine immunoreactivity is restricted to the infarcted tissue and is not present in the peri-infarct tissue. In nNOSΔ/Δ mice, known to be protected against ischemia, no 3-nitrotyrosine immunoreactivity is detected. Our findings provide a cellular localization for nNOS activation in association with ischemic stroke and establish that NO is not likely a direct neurotoxin, whereas its conversion to peroxynitrite is associated with cell death.
Keywords: citrulline, ischemial, peroxynitrite, 3-nitrotyrosine, nitric oxide, nitric oxide synthase, peroxynitrite
Nitric oxide (NO) is a putative neurotransmitter in the brain and peripheral nervous system (Holscher, 1997). It is formed by the enzyme NO synthase (NOS), which generates NO from the guanidine nitrogen of arginine giving rise to NO and stoichiometric amounts of citrulline. NO functions in blood vessels in which it accounts for endothelial-derived relaxing factor activity (Moncada, 1994), in macrophages in which it accounts for the ability of activated macrophages to kill tumor cells and bacteria (MacMicking et al., 1997), and in neurons. For these three functions and localizations, NO is formed by three different types of NOS derived from three distinct genes referred to, respectively, as neuronal NOS (nNOS; NOS-1), macrophage or inducible NOS (iNOS; NOS-2), and endothelial NOS (eNOS; NOS-3) (Moncada et al., 1997). nNOS is localized to discrete populations of neurons in the brain and the peripheral nervous system, as well as to some forms of skeletal muscle and certain epithelial tissues (Yun et al., 1996). In the brain, iNOS can occur in astrocytes and microglia but usually not in neurons (Jaffrey and Snyder, 1995). There is some evidence that eNOS can occur in certain neuronal populations (Dinerman et al., 1994; O’Dell et al., 1994;Doyle and Slater, 1997), but this is controversial (Vincent and Kimura, 1992; Stanarius et al., 1997) so that it is likely that the predominant, if not sole, form of NOS in neurons in the brain is nNOS.
Initial evidence for a specific function of NO in the brain came from the demonstration that glutamate, acting through NMDA receptors, stimulates the conversion of arginine to citrulline with a concentration–response relationship similar to its stimulation of cGMP formation and that NOS inhibitors block both NOS activity and cGMP formation (Bredt and Snyder, 1989; Garthwaite et al., 1989). Thus, under presumed physiological conditions, NMDA transmission activates NO formation in the brain.
Excessive glutamate release can be toxic, because exposure of cortical cultures to glutamate or NMDA results in the delayed killing of neuronal cells (Choi, 1988). NO appears to be involved in mediating NMDA neurotoxicity in cortical cultures. Thus, NMDA neurotoxicity is markedly reduced after treatment with NOS inhibitors (Dawson et al., 1993) and in cultures from mice with targeted deletion of nNOS (nNOSΔ/Δ) (Dawson et al., 1996). Although NMDA activates NOS, the NOS-containing neurons resist toxic effects of NMDA and form NO that is released to kill adjacent non-NOS neurons (Dawson et al., 1993). The unique resistance of NOS neurons to NMDA toxicity appears to be associated with their very high content of manganese superoxide dismutase, which presumably prevents interactions of NO with superoxide to form the potent oxidant peroxynitrite (Gonzalez-Zulueta et al., 1998).
Glutamate neurotoxicity acting via NMDA receptors has been implicated in neural damage after ischemic stroke (Choi, 1988; Yun et al., 1996). Focal ischemia associated with middle cerebral artery occlusion (MCAo) leads to massive neuronal release of glutamate. Evidence that this glutamate accounts for neural damage comes from studies showing that NMDA antagonists block neural damage in a variety of stroke models (Choi, 1994). Because NO mediates NMDA neurotoxicity in brain cultures, NO may mediate glutamate neurotoxicity in ischemic stroke. Supporting evidence includes the demonstration that relatively specific nNOS inhibitors reduce stroke damage (Iadecola, 1997) and that such damage is also diminished in nNOSΔ/Δ mice (Huang et al., 1994). Although several reports of increased NO production after ischemia have been published (Malinski et al., 1993; Iadecola, 1997), little evidence exists regarding the source responsible for this increased production. This has precluded definitive conclusions regarding the role of NO in the pathogenesis of stroke damage. Indeed, in certain circumstances, NO confers protection against stroke because mice deficient in eNOS suffer greater damage than control mice after reversible ischemia (Huang et al., 1996). One explanation is that NO is not toxic in itself but becomes toxic only when it reacts with superoxide and is converted to peroxynitrite (Beckman and Koppenol, 1996) and that the likelihood that this will occur is dependent on where NO is being produced.
Because of difficulties in measuring and localizing NO itself, we focused on citrulline. Citrulline immunoreactivity (cit-IR) is localized exclusively to nNOS-containing neurons and is abolished after treatment with NOS inhibitors (Pasqualotto et al., 1991; Demas et al., 1997; Eliasson et al., 1997). Moreover, no other enzyme capable of synthesizing citrulline has been found in the brain (Pasqualotto et al., 1991; Eliasson et al., 1997). Thus, staining for citrulline provides a useful approach to evaluating NO turnover. In the present study, we have examined citrulline formation associated with MCAo. We demonstrate that citrulline levels are markedly augmented after ischemia but more so in the peri-infarct rather than the infarcted tissue. In contrast, 3-nitrotyrosine immunoreactivity (3NT-IR), which reflects the formation of peroxynitrite, is restricted to the infarcted area and is not present in the peri-infarct tissue. Our findings provide the first cellular localization of NOS activation in association with ischemic stroke and establish that NO is not likely a direct neurotoxin, whereas its conversion to peroxynitrite leads to cell death.
MATERIALS AND METHODS
Ischemic model. Wild-type mice (129/Sv and C57BL/6NcrlBR) and nNOSΔ/Δ mice (Huang et al., 1993) (20–25 gm) were housed under diurnal lighting conditions and allowed access to food and water ad libitum. For surgery, mice were anesthetized with 2% halothane for induction and maintained on 1% halothane in 70:30% nitrous oxide/oxygen by mask. Rectal temperature was maintained between 36.5 and 37.5°C with a homeothermic blanket system (YSI, Yellow Springs, OH). Focal cerebral ischemia was induced by unilateral MCAo (right side) using the intraluminal filament technique (Longa et al., 1989; Huang et al., 1994). Through a ventral midline incision, the right common and external carotid arteries were isolated and ligated. A microvascular clip (Zen temporary clip; Ohwa Tsusho, Tokyo, Japan) was temporarily placed on the internal carotid artery and the pterygopalatine artery. An 8–0 nylon monofilament (Ethicon, Somerville, NJ) coated with silicone was introduced into the internal carotid artery through a small incision in the external carotid artery and advanced 10 mm distal to the carotid bifurcation to occlude the MCA. The wound was sutured, and the animal was returned to its cage. At the indicated durations of MCAo, animals were anesthetized deeply with an overdose of pentobarbital, the filament was removed, and the common carotid artery was reopened. Reperfusion into the previously ischemic brain was allowed for 1 min, after which the animals were perfused for immunohistochemistry through the left ventricle.
NMDA administration. NMDA was administered by either intrastriatal perfusion or intraperitoneal injections. Either application caused similar increases in cit-IR, as determined by quantification of cit-IR perikarya. For intrastriatal NMDA perfusion, wild-type 129/Sv mice (20–27 gm) were anesthetized with 1.5% halothane in 2:1 air and oxygen. Mice were placed into a custom-designed stereotactic frame (David Kopf, Tujunga, CA), and microdialysis probes (CMA10; 1 mm membrane length, 0.5 mm diameter) were placed in the cerebral cortex. Stereotactic coordinates from bregma were as follows: 0.5 mm anterior, 2 mm lateral, and 1 mm deep from the brain surface (Sidman et al., 1971). Microdialysis probes were perfused with artificial CSF [containing (mm): NaCl 125, KCl 2.5, CaCl2 1.2, NaH2PO4·H2O 0.5, Na2HPO4 5, MgCl2·6H2O 1, and ascorbic acids 0.2, pH 7.4] at a flow rate of 2 μl/min using a microinfusion pump. After implantation, stabilization of the tissue was allowed by waiting for 1 hr, during which aCSF was superfused. A 30 min baseline control was obtained with aCSF superfusion, followed by 10 min of 1 mm NMDA superfusion. Six minutes later, the animals were perfused for immunohistochemistry. For intraperitoneal NMDA injections, mice were given 75 mg/kg NMDA dissolved in 0.9% NaCl. Mice were perfused for immunohistochemistry 30 min after administration.
3-Nitrotyrosine immunohistochemistry. Mice were deeply anesthetized and transcardially perfused with cold (4°C) saline, followed by 0.1 m sodium phosphate buffer, pH 7.4, containing a 4% paraformaldehyde solution. Brains were removed directly after perfusion, post-fixed for 2 hr, washed in 0.1m sodium phosphate buffer, and cryoprotected in increasing concentrations of 10 and 20% glycerol–2% DMSO solution. Frozen serial coronal sections of the entire brain were made at 50 μm intervals. Sections were subsequently stained for Nissl substance using cresyl violet to identify the lesioned locus and for immunohistochemical localization of 3-nitrotyrosine (monoclonal antibody; 1:500 dilution; Upstate Biotechnology, Lake Placid, NY) using a previously reported conjugated secondary antibody method (Ferrante et al., 1993). Tissue sections were preincubated in absolute methanol–0.3% hydrogen peroxide solution for 30 min, washed (three times) in PBS, pH 7.4, 10 min each, placed in 10% normal goat serum (Life Technologies, Gaithersburg, MD) for 1 hr, incubated free floating in primary antiserum at room temperature for 12–18 hr (all dilutions of primary antisera above included 0.3% Triton X-100 and 10% normal goat serum), washed (three times) in PBS for 10 min each, incubated with peroxidase-conjugated goat anti-rabbit IgG (1:300 in PBS; Boehringer Mannheim, Mannheim, Germany), washed (three times) in PBS 10 min each, and reacted with 3,3′-diaminobenzidine-HCl (1 mg/ml) in Tris-HCl buffer with 0.005% hydrogen peroxide. Specificity of antibody binding was established by preincubation of tissue sections for 3-nitrotyrosine immunohistochemistry for 6 hr at room temperature with either 20 mm nitrotyrosine or 1 mg/ml nitrated BSA containing ∼30 μm nitrotyrosine. For the data in Figure6, glutaraldehyde-fixed tissue was used to allow comparison between cit-IR and 3NT-IR in adjacent sections. Glutaraldehyde-fixed tissue was reduced with 0.5% NaBH4 to increase permeability and affinity of the citrulline immunohistochemistry (Campistron et al., 1986). This raises the possibility that nitrotyrosine would be reduced to aminotyrosine and not recognized by the monoclonal antibody. In our experiment on glutaraldehyde-fixed tissue, the staining was comparable in distribution with that seen in paraformaldehyde-fixed tissue using the same dilution of the antisera. Specificity of antibody binding in glutaraldehyde-fixed tissue was established by omission of the primary antibody and preincubation of tissue sections for 3-nitrotyrosine immunohistochemistry for 4 hr at room temperature with 1 mg/ml nitrated BSA containing ∼30 μm nitrotyrosine. This supports the preservation of the high-affinity epitope recognized by the monoclonal antibody after glutaraldehyde fixation and NaBH4treatment.
Fig. 6.
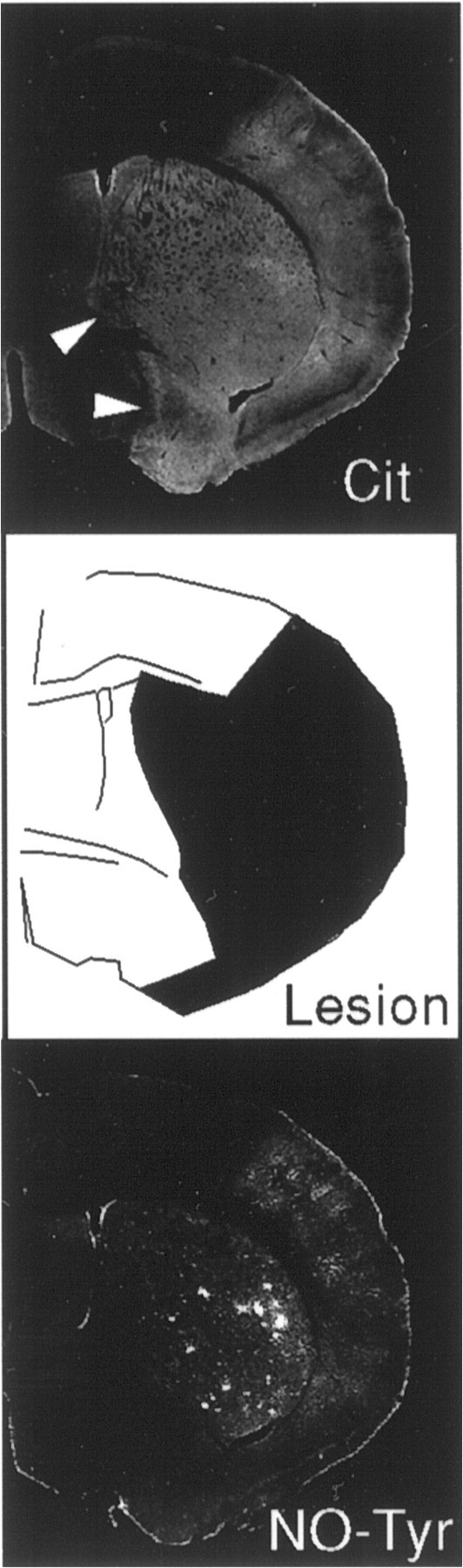
Comparison of 3NT-IR and cit-IR in relation to the ischemic lesion determined by delineating pallor in Nissl staining in contiguous tissue sections after 3 hr of MCAo. Whereas 3NT-IR is restricted to the ischemic lesion, cit-IR is high in other areas as well (arrowhead). CC, Cerebral cortex;St, striatum; −/−, nNOSΔ/Δmouse. Data are from a typical experiment that was replicated three times.
Citrulline and NOS immunohistochemistry. Citrulline as a free amino acid is not immunogenic. By cross-linking citrulline with glutaraldehyde, highly specific and sensitive antisera can be raised and used for immunohistochemistry (Eliasson et al., 1997). However, the use of glutaraldehyde has limitations because it gives rise to strong autofluorescence and precludes the observation of specific immunofluorescence staining necessary for double-labeling experiments using immunofluorescent antibodies. Previous studies by others (Pasqualotto et al., 1991) and us (Eliasson et al., 1997) have tried to overcome this limitation either by combining immunohistochemistry with NADPH diaphorase histochemistry or denaturing the antibody complex to allow multiple immunoenzyme staining. The former method is unreliable because NADPH staining is unsuitable as a marker for NOS in glutaraldehyde-fixed tissue (see Discussion). In the latter method, the use of two chromogens enabled reliable distinction between cells containing only citrulline (yellow), and nNOS/citrulline (dark blue), but made it difficult to distinguish between cells containing nNOS/citrulline (dark blue), and cells containing only nNOS (blue).
The citrulline antiserum has been characterized previously (Eliasson et al., 1997) and was used at a 1:12,000 dilution of purified crude antiserum. Animals were perfused through the left ventricle with 37°C oxygenated Krebs’–Henseleit buffer, followed by 250 ml of 37°C 5% glutaraldehyde–0.5% paraformaldehyde containing 0.2% Na2S2O5 in 0.1 m sodium phosphate, pH 7.4. Glutaraldehyde fixation is necessary because the citrulline antiserum is specific for glutaraldehyde-linked citrulline and does not detect free citrulline (Eliasson et al., 1997). Brains were post-fixed in the same buffer for 2 hr at room temperature and cryoprotected for 2 d at 4°C in 50 mm sodium phosphate, pH 7.4, 0.1 m NaCl, 20% (vol/vol) glycerol. Brain sections (40 μm) were cut on a sliding microtome. Free-floating brain sections were reduced for 20 min with 0.5% NaBH4 and 0.2% Na2S2O5 in PBS (10 mm, pH 7.4, and 0.19 m NaCl) to reduce background staining caused by the glutaraldehyde fixation. After washing for 45 min at room temperature in PBS containing 0.2% Na2S2O5, sections were blocked with 4% normal goat serum for 1 hr in the presence of 0.2% Triton X-100 and incubated overnight at 4°C with the primary antiserum diluted in PBS containing 2% goat serum and 0.1% Triton X-100. Immunoreactivity was visualized with the Vectastain ABC Elite kit (Vector Laboratories, Burlingame, CA) using 3,3′-diaminobenzidine-HCl as chromogen. The specificity of the antisera and the detection assay were tested by preabsorption with the antigen and omission of the primary antibody, respectively. The distribution of cit-IR neurons in immunostained sections was mapped using a computer–microscope mapping system equipped with video frame grabber, microscope stage digitizer, and software designed for quantitative analysis of labeled cells. This system (Wilson and Molliver, 1991) uses digitized video images and tracks microscope stage coordinates to create digital brain maps that maintain the location of labeled cells. Cit-IR neurons were mapped in coronal sections of the forebrain at the level of the decussation of the anterior commissure, and the density of labeled cells was determined in defined areas. A polyclonal antiserum to the C-terminal region of human nNOS (residues 1419–1433) was kindly provided by Jeffrey Spangenberg (IncStar, Stillwater, MN) and used at a 1:15,000 dilution of crude serum. Antiserum for iNOS (1 mg/ml to 10 μg/ml) (Transduction Laboratories, Lexington, KY) did not show any specific immunoreactivity of brain sections. Specificity of iNOS antibody binding was examined by omission of the primary antibody.
RESULTS
Cit-IR is stimulated by ischemia
Mice were subjected to MCAo and killed at a variety of time points, and their brains were stained for citrulline or nNOS. Both wild-type and nNOSΔ/Δ mice were used. Focal ischemia markedly enhances cit-IR (Fig.1). One hour after MCAo, examination at low magnification reveals markedly increased cit-IR in the areas supplied by the occluded MCA, including the corpus striatum, basal forebrain, and cerebral cortex. At 1 hr, cit-IR is also augmented in the contralateral cerebral cortex (Fig. 1b,arrow), whereas at 3 hr it has declined (Fig.1c,d). Presumably, the contralateral staining reflects a transient increase in global cortical firing.
Fig. 1.

Low-power inverted photomicrographs of cit-IR in representative coronal sections after no ischemia (a), 1 hr of unilateral MCAo (n = 3) (b), 3 hr (n = 4) (c), 3 hr with intense IR in the peri-infarct tissue (n = 5) (d), 3 hr nNOSΔ/Δ mouse (n = 3) (e), and 6 hr wild-type mouse (n = 3) (f). The circled Xindicates infarcted hemisphere. The increase in cit-IR ipsilateral to the occlusion is readily apparent in all wild-type brains subjected to MCAo for 30 min to 6 hr (n = 58). Increased cit-IR is not restricted to the infarct, because staining of either equal (b, c) or higher (d,f) intensity is always seen in peri-infarct areas (arrowheads). One hour of cerebral ischemia also elicits increased cit-IR in the contralateral cerebral cortex (n = 3) (b, arrow). This is transient, because a similar increase is not apparent in any brain subjected to >1 hr of MCAo (n = 28). For nNOSΔ/Δ mice, an ipsilateral increase in cit-IR is only seen after ≥3 hr ischemia (n = 3) but not for 2 hr (n = 9) or 30 min (n = 4). All sections in this figure were processed identically for immunohistochemistry and photographic reproduction to enable comparison of IR. Rectangles in a indicate area of photomicrographs shown in Figure 2.
At 3 hr, cit-IR augmentation in the infarcted hemisphere is nonuniform. In 60% of the brains, the medial striatum and basal forebrain stain much more intensely than lateral areas, with a clear demarcation between the two areas (Fig. 1d). In the remaining brains, the medial and lateral areas show similar staining (Fig.1c). This medial area receives blood supply from the anterior cerebral artery, whereas the MCA supplies the lateral area. Thus, the medial areas represent nonischemic, peri-infarct tissue, although the more lateral areas are infarcted (Lo et al., 1996). This suggests that sustained citrulline production is restricted to areas that are still receiving blood supply and are fully viable, whereas ischemic areas, supplied by the occluded MCA, have diminished citrulline production.
Higher magnification confirms the augmented cit-IR after focal ischemia and shows that it involves both neuronal perikarya and neuropil (Fig.2). At 30 min, increases in perikaryal and neuropil cit-IR are evident in the ischemic areas, in contrast to much less staining in contralateral areas (Fig.2a,b). At 1 hr, staining is further increased in the ischemic areas (Fig. 2c). In contrast, at 3 hr, peri-infarct tissue stains much more intensely than ischemic tissue (Fig. 2d). This differentiation in cit-IR between peri-infarct and infarcted tissue staining is also evident in the striatum (Fig. 2g).
Fig. 2.

High-power photomicrographs of cit-IR neurons from coronal sections shown in Figure 1. a–d, cit-IR in basal forebrain of wild-type mice after unilateral MCAo. In the peri-infarct zone (P), the longer the duration of ischemia, the more pronounced the IR (b–d). After 3 hr, a subset of wild-type mice (5 of 9) exhibits much more intense IR in the peri-infarct than the infarct zone (I) (d). In the infarct zone, IR increases in intensity initially but is diminished at 3 hr (d). In the striatum, the same pattern holds true (g). In nNOSΔ/Δ mice, after 3 hr of ischemia, IR is evident in both the basal forebrain (e) and striatum (h). The IR cells exhibit neuronal morphology (i), are most abundant in the peri-infarct tissue (P), and are blocked by incubating the citrulline antiserum with 200 μm soluble antigen (f). nNOS-IR cells are also seen in the nNOSΔ/Δ mice (i). Images are from typical immunohistochemistry that were replicated at least three times for each duration of occlusion. BF, Basal forebrain; St, striatum; ip, ipsilateral; co, contralateral. Scale bar (in i), 25 μm.a–h, Bright-field, 200×; i, phase contrast. Locations of areas magnified are indicated in Figure1a.
Cit-IR is stimulated by ischemia to a substantially lesser extent in nNOSΔ/Δ mice
Augmented cit-IR is evident in nNOSΔ/Δ mice after MCAo but to a substantially lesser extent than wild-type animals (Fig. 1e). Higher magnification examination in the basal forebrain and striatum at 3 hr reveals more cit-IR in the peri-infarct than infarcted tissue (Fig. 2e,h). This staining is blocked by preabsorption of the antiserum with the citrulline antigen (Fig. 2f). In nNOSΔ/Δmice, both nNOS and cit-IR involve neurons with well defined perikarya and processes (Fig. 2i). For occlusions <3 hr, no cit-IR is detected in the nNOSΔ/Δ mice (n= 6; data not shown). To ascertain a potential role of iNOS after focal ischemia, we conducted iNOS staining but detected no iNOS-IR in brains of wild-type or nNOSΔ/Δ mice after 3 hr of MCAo (n = 5; data not shown).
Ischemia causes a fourfold increase in the number of cit-IR perikarya
To quantify the neuronal responses in greater detail, we counted cit-IR perikarya (Fig. 3). We conducted cell counting in the corpus striatum and basal forebrain in brains subjected to 30 min of unilateral MCAo, a time point at which cit-IR is increased but cell loss as a result of necrosis is minimal. The striatum was differentiated into an area with a large amount of infarction (designated A), the striatal peri-infarct area (designated B), and the basal forebrain area (designated C) (Fig. 3b). Increased numbers of cit-IR perikarya are most apparent in the peri-infarct striatum with a substantially lesser augmentation in the infarcted area (Fig. 3a, histogram). The increase in the basal forebrain is similar to the augmentation in the peri-infarct striatum. Contralateral to MCAo, no increase in cit-IR compared with control is evident in any of the examined areas. We did not conduct cell counting in the cerebral cortex because the increased cit-IR in the cortex primarily reflects staining of neuropil, consistent with localization of cortical nNOS primarily in processes rather than in perikarya (Aoki et al., 1993).
Fig. 3.

Ischemia increases the number of cit-IR perikarya.a, A representative coronal section showing the distribution of cit-IR perikarya after 30 min of MCAo in a wild-type mouse. +, Cit-IR perikaryon; X, infarcted hemisphere.b, Three areas were selected for quantification: striatum, A; medial striatum, B; and basal forebrain, C. The histogram shows the number of cit-IR perikarya per square millimeter for each area. Unilateral MCAo increases the density of cit-IR cells in all areas examined ipsilateral (MCAo-Ipsi) but not contralateral (MCAo-Contra). This includes the medial striatum (area B), an area devoid of major necrosis after MCAo. Data are presented as means ± SEM. Untreated, n = 8; ipsilateral MCAo, n = 4; contralateral MCAo,n = 4. Significance was determined by comparing untreated or contralateral MCAo with ipsilateral MCAo by using one-way ANOVA with Fisher’s post hoc test. †p < 0.05; ‡p < 0.001. There was no statistically significant difference between untreated and contralateral MCAo in either area. c, A representative cit-IR neuron ipsilateral to MCAo displays extensive cit-IR of perikaryon and processes, whereas a representative neuron contralateral to MCAo displays much less extensive IR (d). This shows that quantification by counting cit-IR perikarya underestimates the overall increase in citrulline production. The adjacent endothelium is devoid of cit-IR, presumably because eNOS is not activated.c, d, Bright-field, 400×.
The fourfold increase in the number of cit-IR cells triggered by ischemia is less striking than the more dramatic overall enhancement of cit-IR in ischemic areas observed at low magnification (Fig. 1). This discrepancy reflects the greater increase of citrulline in neuronal processes than perikarya. Low-magnification staining of cit-IR reveals citrulline in neuronal processes, as well as perikarya. Higher magnification examination reveals substantial cit-IR in extensively branched processes in addition to perikaryal staining ipsilateral to MCAo, whereas contralateral staining is both of lesser intensity and extent (Fig. 3c,d). Some cit-IR occurs in neurons that overlay blood vessels, but staining is confined to the neurons with no endothelial staining (Fig. 3c). We see only occasional cit-IR in the endothelium with little difference between ischemic and nonischemic tissue.
Only a minority of nNOS cells stain for citrulline: evidence for “quiescent” nNOS
To clarify the relationship between cells that express citrulline and nNOS, we stained contiguous sections for nNOS and citrulline and quantified nNOS-IR and cit-IR in contiguous coronal sections (Fig.4). In untreated brains, the density of cit-IR neurons in the striatum is only 23% of the density of nNOS-positive neurons, similar to previous observations (Pasqualotto et al., 1991; Eliasson et al., 1997). This suggests that, under basal conditions, most nNOS neurons are not actively synthesizing citrulline at detectable levels. To evaluate whether they retain the capacity to form citrulline, we administered NMDA into the striatum by microdialysis (Fig. 4). NMDA administration, which activates nNOS by stimulating calcium entry (Bredt and Snyder, 1990), augments the number of cit-IR perikarya to levels comparable with the number of nNOS-IR perikarya. Ischemia also augments the number of cit-IR perikarya to levels comparable with those of nNOS-IR perikarya. In contrast, cit-IR contralateral to MCAo is similar to the levels observed under basal conditions.
Fig. 4.

Photomicrographs illustrating the presence of quiescent nNOS in mice. The density of cit-IR neurons in the striatum (a) is less than the density of nNOS-IR in a contiguous section (b). NMDA administration markedly increases cit-IR (c) to levels comparable with nNOS-IR. Similarly, in the striatum of mice subjected to 30 min of unilateral MCAo, contralateral cit-IR density (d) is less than nNOS-IR (e), although ipsilateral (f), they are comparable.Cit, Citrulline; ip, ipsilateral;co, contralateral; −/−, mouse deficient in nNOS expression; ctrl, control. a–e, Bright-field, 100×. Data are from typical immunohistochemistry that was repeated at least three times for each condition. g, Quantification of citrulline and nNOS-IR in the striatum demonstrates the presence of a large pool of quiescent nNOS neurons that can be activated by either NMDA application or 30 min of MCAo. Each data point is mean ± SEM. Control, n = 8; NMDA,n = 3; ipsilateral MCAo, n = 6; contralateral MCAo, n = 6. p < 0.001 for either NMDA or ipsilateral MCAo compared with either control or contralateral MCAo using one-way ANOVA with Bonferroni’spost hoc test; there is no statistically significant difference between either control and contralateral, or NMDA and ipsilateral.
The enhancement of nNOS activity after focal ischemia could be caused by increased enzymatic activity in all nNOS neurons, stimulation of previously inactive nNOS cells, or to stimulation of NOS activity in cells other than nNOS neurons. The intensity of cit-IR appears approximately the same in the minority of nNOS neurons displaying cit-IR under basal conditions as in neurons after NMDA treatment or focal ischemia. Thus, the mechanism of increased NO formation in ischemic stroke seems to primarily involve activation of previously “quiescent” nNOS neurons.
Different localizations of 3-nitrotyrosine and citrulline
A variety of evidence indicates that NO itself does not elicit cell death. Rather, the rapid reaction between NO and superoxide to form peroxynitrite, a powerful reactive oxygen species, is thought to mediate NO-triggered cell death (Beckman and Koppenol, 1996). Peroxynitrite mediates nitration of tyrosine to form 3-nitrotyrosine, which has been used as a biochemical marker for peroxynitrite formation. Although other chemical species can nitrate tyrosine, in vivo peroxynitrite is the major source, particularly in the absence of neutrophils (Beckman and Koppenol, 1996). Because of these considerations, one might expect 3NT to predominantly label infarcted tissue. To examine this possibility, we compared 3NT-IR and cit-IR after MCAo (Fig. 5). After 3 hr of MCAo, we observed a marked increase in 3NT-IR restricted to the ischemic infarcted area in wild-type mice (Fig. 5). IR occurs in neurons, the surrounding neuropil, and vascular endothelium in the striatum and the cerebral cortex. In nNOSΔ/Δmice, the ischemic lesion is reduced, as reported previously (Huang et al., 1994; Hara et al., 1996a), and devoid of enhanced 3NT-IR. In contiguous sections, we compared cit-IR and 3NT-IR (Fig. 6). Whereas 3NT-IR is restricted to the infarcted area, cit-IR extends more medially into the peri-infarcted zone.
Fig. 5.

3-Nitrotyrosine immunoreactivity in the ischemic lesion. Nissl staining (a, b) and 3NT-IR (c, d) of ischemic lesions of the right hemisphere in wild-type (wt) and nNOSΔ/Δ (−/−) mice at the level of the decussation of the anterior commissure. Nissl staining delineates the ischemic lesion of the wild-type mouse by staining pallor. Note the marked swelling of the right hemisphere with a gross shift to the left side. In comparison, except for pallor in the MCA territory on Nissl stain, little gross damage is evident in the nNOSΔ/Δ brains. There is a marked increase of 3NT-IR in a contiguous tissue section within the wild-type mouse in relation to the zone of cortical and subcortical damage observed in the Nissl section. No such increase is found in a serial cut tissue section from the nNOSΔ/Δ mouse. Although the contralateral hemisphere of the wild-type mouse appears darker than the nNOSΔ/Δ mouse, that staining was not specific. Photomicrographs of 3-nitrotyrosine immunohistochemistry in the cerebral cortex (e,f) and striatum (g,h) of wild-type and nNOSΔ/Δmouse, respectively, from the coronal sections shown above. There is a significant increase in 3NT-IR within cortical and striatal neurons and neuropil from tissue sections of wild-type mice compared with nNOSΔ/Δ mice. e–h, Bright-field, 200×.
DISCUSSION
One of the main findings of the present study is that nNOS is selectively activated in specific brain areas associated with focal ischemia. Abundant evidence has implicated excess release of NO in the etiology of ischemic brain damage (Malinski et al., 1993; Iadecola, 1997), which is substantially reduced in nNOSΔ/Δmice (Huang et al., 1994) and after treatment with NOS inhibitors (Dawson and Dawson, 1997). However, the poor spatial resolution in these studies has limited conclusions regarding sources of NO production after focal cerebral ischemia. Our study shows selective patterns of nNOS activation that clarify mechanisms whereby NO elicits ischemic damage.
Thus, one of our notable findings is that, after initial cit-IR increases in both the peri-infarcted and infarcted area, cit-IR, in most animals, is prominent only in the peri-infarct area, which continues to be supplied by branches of the anterior cerebral artery. Transient depolarizations associated with spreading depression could be responsible for the increased cit-IR in peri-infarct tissue. These depolarizations are known to occur in peri-infarct tissue, triggered by extracellular release of potassium and excitatory amino acids in the infarcted tissue (Hossmann, 1996). The triggering of the spreading depolarization by ischemia is variable, as a recent study in mice eliciting focal ischemia by the same procedure used here, revealed such events only in a subset of animals (Zaharchuk et al., 1997). Presumably, spreading depolarization is triggered in animals in which the medial peri-infarct zones stain intensely for citrulline.
The continued viability of tissue in an area associated with the most prominent augmentation of cit-IR indicates that NO itself is not sufficient to elicit neuronal death. Instead, it is likely that the combination of NO with superoxide, formed by hypoxic mitochondria, leads to the formation of peroxynitrite, which most likely kills cells by direct reaction with cellular targets (Beckman and Koppenol, 1996). The absence of 3NT-IR in nNOSΔ/Δ mice, which coincides with the protection against stroke damage in these animals, and the selective localization of 3NT-IR to areas of infarcted tissue in wild-type mice further substantiates the conclusion that peroxynitrite rather than NO itself is responsible for cell death. 3NT-IR is also selectively augmented in neurodegenerative diseases, such as amyotrophic lateral sclerosis and Parkinson’s disease (Schulz et al., 1995; Beal et al., 1997; Ferrante et al., 1997), suggesting a prominent role for peroxynitrite in neurotoxicity. The lack of any 3NT-IR in the nNOSΔ/Δ mice most likely is attributable to insufficient production of NO to compete with superoxide dismutase for superoxide. It is estimated that micromolar concentrations of NO are necessary to generate peroxynitrite. Whereas micromolar levels of NO have been recorded after ischemia (Malinski et al., 1993), physiological concentrations of NO in wild-type mice are in the nanomolar range. In nNOSΔ/Δ mice brains, Ca2+-dependent NOS activity and [3H]l-NG-nitro-arginine autoradiography are reduced by 95% compared with wild type (Huang et al., 1993; Hara et al., 1996b, 1997). The residual 5% NOS activity may not generate enough 3-nitrotyrosine for detection by immunohistochemistry.
Cit-IR was present in nNOSΔ/Δ mice brains, although at lower levels than wild-type brains, and was augmented after focal ischemia. This persistent cit-IR is unlikely to be attributable to eNOS. We (Dinerman et al., 1994; O’Dell et al., 1994) and others (Doyle and Slater, 1997) initially reported evidence for eNOS staining by immunohistochemistry and NADPH diaphorase in neurons, although studies by others only identified eNOS in blood vessels in the brain (Seidel et al., 1997; Stanarius et al., 1997; Topel et al., 1998). One limitation of immunohistochemistry involves the possibility of eNOS antibodies recognizing nonspecific antigens. NADPH diaphorase staining reflects oxidative enzyme activity using NADPH as an electron donor and so can involve enzymes other than NOS. This is supported by observations that changes in fixation protocols alter the distribution of NADPH diaphorase staining (Dinerman et al., 1994; Buwalda et al., 1995). In situ hybridization using appropriate probes and high-stringency conditions can be highly specific. In a recent study using a cRNA probe for eNOS, we failed to detect any eNOS in neuronal populations (G. E. Demas, L. J. Kriegsfeld, S. Blackshaw, P. Huang, S. C. Gammie, R. J. Nelson, and S. H. Snyder, unpublished observations). Furthermore, we failed to detect any increase in cit-IR in the endothelium after ischemia in both wild-type and nNOSΔ/Δ mice, suggesting that there is no accumulation of citrulline in the endothelium after ischemia. Thus, eNOS does not contribute substantially to the increase in cit-IR observed in post-ischemic wild-type and nNOSΔ/Δbrains.
iNOS has also been implicated in focal ischemia but only at >24 hr after onset of ischemia (Iadecola, 1997). No iNOS activity was detected by calcium-independent conversion ofl-[3H]arginine tol-[3H]citrulline in normal or ischemic mice brains 0–4 d after MCAo (Yoshida et al., 1995). In this study, using immunohistochemistry for iNOS, no labeling of neuronal populations in either wild-type or nNOSΔ/Δ mice was observed 22 hr after MCAo. Thus, iNOS is not responsible for the augmented cit-IR in the post-ischemic wild-type and nNOSΔ/Δ brains.
Because there are nNOS-positive neurons in the nNOSΔ/Δ mice (Fig. 2i), the persistent cit-IR presumably reflects alternatively spliced forms of nNOS, designated nNOSβ and nNOSγ, which do not use exon 2 for transcription (Brenman et al., 1996). Because the nNOSΔ/Δ mice were created by inserting a stop codon in exon 2 (Huang et al., 1993), nNOSβ and nNOSγ persist in these mice. Brenman et al. (1996) observed catalytic activity with nNOSβ but not with nNOSγ, whereas Marsden and colleagues (Wang et al., 1997) have detected some catalytic activity in nNOSγ cloned independently from the testes. Whether nNOSβ and nNOSγ contribute in intact organisms to NO formation has not been clear. Previously, we showed by in situ hybridization that nNOSβ mRNA is prominent in areas of the brain displaying residual NOS catalytic activity in nNOSΔ/Δ mice and that modest levels of cit-IR are retained in the mutant mice (Eliasson et al., 1997). Our present findings establish more definitively that the alternatively spliced residual forms of nNOS are catalytically active and capable of stimulation after focal ischemia. However, the level of NOS activity is substantially less than in wild-type animals, and no 3NT-IR is detectable, explaining the marked protection of nNOSΔ/Δ mice from stroke damage.
Our comparisons of proportions of cells staining for citrulline and nNOS reveal a pattern for activation of neurotransmitter formation that differs markedly from classical transmitters. Most neurotransmitters are stored in synaptic vesicles, and their effects are elicited after exocytosis. In contrast, NO, because of its lipid solubility (Yun et al., 1996), cannot be stored in vesicles, and transmitter release directly follows activation of nNOS (Jaffrey and Snyder, 1995). Under basal conditions, we find that only 20% of nNOS neurons stain for citrulline and hence are actively synthesizing NO. NMDA treatment or focal ischemia augments the proportion of cit-IR cells markedly. This activation of at baseline quiescent nNOS fits with abundant other evidence supporting a tight regulation of NOS activity necessary to enable NO to function as a neurotransmitter (Yun et al., 1996).
Footnotes
This work was supported by a Gustavus and Louise Pfeiffer Scholarship (M.J.L.E.), the Veterans Administration, National Institutes of Health Awards NS37102, NS35255, and AG12992 (R.J.F.), Interdepartmental Stroke Program Project P50 NS10828 (M.A.M.), United States Public Health Service Grant MH18501 (S.H.S), and Research Scientist Award DA-00074 (S.H.S.). We thank Eng H. Lo for help with intrastriatal NMDA perfusion, Paul L. Huang for assistance with the nNOSΔ/Δ mice, and Michael J. Schell for helpful advice.
Correspondence should be addressed to Dr. Solomon H. Snyder, Department of Neuroscience, Johns Hopkins University School of Medicine, 725 North Wolfe Street/813 Wood Basic Science Building, Baltimore, MD 21205.
REFERENCES
- 1.Aoki C, Fenstemaker S, Lubin M, Go CG. Nitric oxide synthase in the visual cortex of monocular monkeys as revealed by light and electron microscopic immunocytochemistry. Brain Res. 1993;620:97–113. doi: 10.1016/0006-8993(93)90275-r. [DOI] [PubMed] [Google Scholar]
- 2.Beal MF, Ferrante RJ, Browne SE, Matthews RT, Kowall NW, Brown RH., Jr Increased 3-nitrotyrosine in both sporadic and familial amyotrophic lateral sclerosis. Ann Neurol. 1997;42:644–654. doi: 10.1002/ana.410420416. [DOI] [PubMed] [Google Scholar]
- 3.Beckman JS, Koppenol WH. Nitric oxide, superoxide, and peroxynitrite: the good, the bad, and the ugly. Am J Physiol. 1996;271:C1424–C1437. doi: 10.1152/ajpcell.1996.271.5.C1424. [DOI] [PubMed] [Google Scholar]
- 4.Bredt DS, Snyder SH. Nitric oxide mediates glutamate-linked enhancement of cGMP levels in the cerebellum. Proc Natl Acad Sci USA. 1989;86:9030–9033. doi: 10.1073/pnas.86.22.9030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bredt DS, Snyder SH. Isolation of nitric oxide synthase, a calmodulin-requiring enzyme. Proc Natl Acad Sci USA. 1990;87:682–685. doi: 10.1073/pnas.87.2.682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Brenman JE, Chao DS, Gee SH, McGee AW, Craven SE, Santillano DR, Wu Z, Huang F, Xia H, Peters MF, Froehner SC, Bredt DS. Interaction of nitric oxide synthase with the postsynaptic density protein PSD-95 and α1-syntrophin mediated by PDZ domains. Cell. 1996;84:757–767. doi: 10.1016/s0092-8674(00)81053-3. [DOI] [PubMed] [Google Scholar]
- 7.Buwalda B, Nyakas C, Gast J, Luiten PG, Schmidt HH. Aldehyde fixation differentially affects distribution of diaphorase activity but not of nitric oxide synthase immunoreactivity in rat brain. Brain Res Bull. 1995;38:467–473. doi: 10.1016/0361-9230(95)02017-l. [DOI] [PubMed] [Google Scholar]
- 8.Campistron G, Buijs RM, Geffard M. Specific antibodies against aspartate and their immunocytochemical application in the rat brain. Brain Res. 1986;365:179–184. doi: 10.1016/0006-8993(86)90737-7. [DOI] [PubMed] [Google Scholar]
- 9.Choi DW. Glutamate neurotoxicity and diseases of the nervous system. Neuron. 1988;1:623–634. doi: 10.1016/0896-6273(88)90162-6. [DOI] [PubMed] [Google Scholar]
- 10.Choi DW. Glutamate receptors and the induction of excitotoxic neuronal death. Prog Brain Res. 1994;100:47–51. doi: 10.1016/s0079-6123(08)60767-0. [DOI] [PubMed] [Google Scholar]
- 11.Dawson TM, Dawson VL. Protection of the brain from ischemia. In: Batjer HH, editor. Cerebrovascular disease. Lippincott-Raven; Philadelphia: 1997. pp. 319–325. [Google Scholar]
- 12.Dawson VL, Dawson TM, Bartley DA, Uhl GR, Snyder SH. Mechanisms of nitric oxide-mediated neurotoxicity in primary brain cultures. J Neurosci. 1993;13:2651–2661. doi: 10.1523/JNEUROSCI.13-06-02651.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dawson VL, Kizushi VM, Huang PL, Snyder SH, Dawson TM. Resistance to neurotoxicity in cortical cultures from neuronal nitric oxide synthase-deficient mice. J Neurosci. 1996;16:2479–2487. doi: 10.1523/JNEUROSCI.16-08-02479.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Demas GE, Eliasson MJL, Dawson TM, Dawson VL, Kriegsfeld LJ, Nelson RJ, Snyder SH. Inhibition of neuronal nitric oxide synthase increases aggressive behavior in mice. Mol Med. 1997;3:610–619. [PMC free article] [PubMed] [Google Scholar]
- 15.Dinerman JL, Dawson TM, Schell MJ, Snowman A, Snyder SH. Endothelial nitric oxide synthase localized to hippocampal pyramidal cells: implications for synaptic plasticity. Proc Natl Acad Sci USA. 1994;91:4214–4218. doi: 10.1073/pnas.91.10.4214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Doyle CA, Slater P. Localization of neuronal and endothelial nitric oxide synthase isoforms in human hippocampus. Neuroscience. 1997;76:387–395. doi: 10.1016/s0306-4522(96)00297-7. [DOI] [PubMed] [Google Scholar]
- 17.Eliasson MJ, Blackshaw S, Schell MJ, Snyder SH. Neuronal nitric oxide synthase alternatively spliced forms: prominent functional localizations in the brain. Proc Natl Acad Sci USA. 1997;94:3396–3401. doi: 10.1073/pnas.94.7.3396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ferrante RJ, Kowall NW, Cipolloni PB, Storey E, Beal MF. Excitotoxin lesions in primates as a model for Huntington’s disease: histopathologic and neurochemical characterization. Exp Neurol. 1993;119:46–71. doi: 10.1006/exnr.1993.1006. [DOI] [PubMed] [Google Scholar]
- 19.Ferrante RJ, Shinobu LA, Schulz JB, Matthews RT, Thomas CE, Kowall NW, Gurney ME, Beal MF. Increased 3-nitrotyrosine and oxidative damage in mice with a human copper/zinc superoxide dismutase mutation. Ann Neurol. 1997;42:326–334. doi: 10.1002/ana.410420309. [DOI] [PubMed] [Google Scholar]
- 20.Garthwaite J, Garthwaite G, Palmer RM, Moncada S. NMDA receptor activation induces nitric oxide synthesis from arginine in rat brain slices. Eur J Pharmacol. 1989;172:413–416. doi: 10.1016/0922-4106(89)90023-0. [DOI] [PubMed] [Google Scholar]
- 21.Gonzalez-Zulueta M, Ensz LM, Mukhina G, Lebovitz RM, Zwacka RM, Engelhardt JF, Oberley LW, Dawson VL, Dawson TM. Manganese superoxide dismutase protects nNOS neurons from NMDA and nitric oxide-mediated neurotoxicity. J Neurosci. 1998;18:2040–2055. doi: 10.1523/JNEUROSCI.18-06-02040.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hara H, Huang PL, Panahian N, Fishman MC, Moskowitz MA. Reduced brain edema and infarction volume in mice lacking the neuronal isoform of nitric oxide synthase after transient MCA occlusion. J Cereb Blood Flow Metab. 1996a;16:605–611. doi: 10.1097/00004647-199607000-00010. [DOI] [PubMed] [Google Scholar]
- 23.Hara H, Waeber C, Huang PL, Fujii M, Fishman MC, Moskowitz MA. Brain distribution of nitric oxide synthase in neuronal or endothelial nitric oxide synthase mutant mice using [3H]l-NG-nitro-arginine autoradiography. Neuroscience. 1996b;75:881–890. doi: 10.1016/0306-4522(96)00313-2. [DOI] [PubMed] [Google Scholar]
- 24.Hara H, Ayata C, Huang PL, Waeber C, Ayata G, Fujii M, Moskowitz MA. [3H]l-NG-nitroarginine binding after transient focal ischemia and NMDA-induced excitotoxicity in type I and type III nitric oxide synthase null mice. J Cereb Blood Flow Metab. 1997;17:515–526. doi: 10.1097/00004647-199705000-00005. [DOI] [PubMed] [Google Scholar]
- 25.Holscher C. Nitric oxide, the enigmatic neuronal messenger: its role in synaptic plasticity. Trends Neurosci. 1997;20:298–303. doi: 10.1016/s0166-2236(97)01065-5. [DOI] [PubMed] [Google Scholar]
- 26.Hossmann KA. Periinfarct depolarizations. Cerebrovasc Brain Metab Rev. 1996;8:195–208. [PubMed] [Google Scholar]
- 27.Huang PL, Dawson TM, Bredt DS, Snyder SH, Fishman MC. Targeted disruption of the neuronal nitric oxide synthase gene. Cell. 1993;75:1273–1286. doi: 10.1016/0092-8674(93)90615-w. [DOI] [PubMed] [Google Scholar]
- 28.Huang Z, Huang PL, Panahian N, Dalkara T, Fishman MC, Moskowitz MA. Effects of cerebral ischemia in mice deficient in neuronal nitric oxide synthase. Science. 1994;265:1883–1885. doi: 10.1126/science.7522345. [DOI] [PubMed] [Google Scholar]
- 29.Huang Z, Huang PL, Ma J, Meng W, Ayata C, Fishman MC, Moskowitz MA. Enlarged infarcts in endothelial nitric oxide synthase knockout mice are attenuated by nitro-l-arginine. J Cereb Blood Flow Metab. 1996;16:981–987. doi: 10.1097/00004647-199609000-00023. [DOI] [PubMed] [Google Scholar]
- 30.Iadecola C. Bright and dark sides of nitric oxide in ischemic brain injury. Trends Neurosci. 1997;20:132–139. doi: 10.1016/s0166-2236(96)10074-6. [DOI] [PubMed] [Google Scholar]
- 31.Jaffrey SR, Snyder SH. Nitric oxide: a neural messenger. Annu Rev Cell Dev Biol. 1995;11:417–440. doi: 10.1146/annurev.cb.11.110195.002221. [DOI] [PubMed] [Google Scholar]
- 32.Lo EH, Hara H, Rogowska J, Trocha M, Pierce AR, Huang PL, Fishman MC, Wolf GL, Moskowitz MA. Temporal correlation mapping analysis of the hemodynamic penumbra in mutant mice deficient in endothelial nitric oxide synthase gene expression. Stroke. 1996;27:1381–1385. doi: 10.1161/01.str.27.8.1381. [DOI] [PubMed] [Google Scholar]
- 33.Longa EZ, Weinstein PR, Carlson S, Cummins R. Reversible middle cerebral artery occlusion without craniectomy in rats. Stroke. 1989;20:84–91. doi: 10.1161/01.str.20.1.84. [DOI] [PubMed] [Google Scholar]
- 34.MacMicking J, Xie QW, Nathan C. Nitric oxide and macrophage function. Annu Rev Immunol. 1997;15:323–350. doi: 10.1146/annurev.immunol.15.1.323. [DOI] [PubMed] [Google Scholar]
- 35.Malinski T, Bailey F, Zhang ZG, Chopp M. Nitric oxide measured by a porphyrinic microsensor in rat brain after transient middle cerebral artery occlusion. J Cereb Blood Flow Metab. 1993;13:355–358. doi: 10.1038/jcbfm.1993.48. [DOI] [PubMed] [Google Scholar]
- 36.Moncada S. Nitric oxide. J Hypertens Suppl. 1994;12:S35–S39. [PubMed] [Google Scholar]
- 37.Moncada S, Higgs A, Furchgott R. International union of pharmacology nomenclature in nitric oxide research. Pharmacol Rev. 1997;49:137–142. [PubMed] [Google Scholar]
- 38.O’Dell TJ, Huang PL, Dawson TM, Dinerman JL, Snyder SH, Kandel ER, Fishman MC. Endothelial NOS and the blockade of LTP by NOS inhibitors in mice lacking neuronal NOS. Science. 1994;265:542–546. doi: 10.1126/science.7518615. [DOI] [PubMed] [Google Scholar]
- 39.Pasqualotto BA, Hope BT, Vincent SR. Citrulline in the rat brain: immunohistochemistry and coexistence with NADPH-diaphorase. Neurosci Lett. 1991;128:155–160. doi: 10.1016/0304-3940(91)90250-w. [DOI] [PubMed] [Google Scholar]
- 40.Schulz JB, Matthews RT, Muqit MM, Browne SE, Beal MF. Inhibition of neuronal nitric oxide synthase by 7-nitroindazole protects against MPTP-induced neurotoxicity in mice. J Neurochem. 1995;64:936–939. doi: 10.1046/j.1471-4159.1995.64020936.x. [DOI] [PubMed] [Google Scholar]
- 41.Seidel B, Stanarius A, Wolf G. Differential expression of neuronal and endothelial nitric oxide synthase in blood vessels of the rat brain. Neurosci Lett. 1997;239:109–112. doi: 10.1016/s0304-3940(97)00912-9. [DOI] [PubMed] [Google Scholar]
- 42.Sidman RL, Angevine JB, Pierce ET. Atlas of the mouse brain and spinal cord. Harvard UP; Cambridge, MA: 1971. [Google Scholar]
- 43.Stanarius A, Topel I, Schulz S, Noack H, Wolf G. Immunocytochemistry of endothelial nitric oxide synthase in the rat brain: a light and electron microscopical study using the tyramide signal amplification technique. Acta Histochem. 1997;99:411–429. doi: 10.1016/S0065-1281(97)80034-7. [DOI] [PubMed] [Google Scholar]
- 44.Topel I, Stanarius A, Wolf G. Distribution of the endothelial constitutive nitric oxide synthase in the developing rat brain: an immunohistochemical study. Brain Res. 1998;788:43–48. doi: 10.1016/s0006-8993(97)01506-0. [DOI] [PubMed] [Google Scholar]
- 45.Vincent SR, Kimura H. Histochemical mapping of nitric oxide synthase in the rat brain. Neuroscience. 1992;46:755–784. doi: 10.1016/0306-4522(92)90184-4. [DOI] [PubMed] [Google Scholar]
- 46.Wang Y, Goligorsky MS, Lin M, Wilcox JN, Marsden PA. A novel, testis-specific mRNA transcript encoding an NH2-terminal truncated nitric-oxide synthase. J Biol Chem. 1997;272:11392–11401. [PubMed] [Google Scholar]
- 47.Wilson MA, Molliver ME. The organization of serotonergic projections to cerebral cortex in primates: retrograde transport studies. Neuroscience. 1991;44:555–570. doi: 10.1016/0306-4522(91)90077-2. [DOI] [PubMed] [Google Scholar]
- 48.Yoshida T, Waeber C, Huang Z, Moskowitz MA. Induction of nitric oxide synthase activity in rodent brain following middle cerebral artery occlusion. Neurosci Lett. 1995;194:214–218. doi: 10.1016/0304-3940(95)11752-i. [DOI] [PubMed] [Google Scholar]
- 49.Yun HY, Dawson VL, Dawson TM. Neurobiology of nitric oxide. Crit Rev Neurobiol. 1996;10:291–316. doi: 10.1615/critrevneurobiol.v10.i3-4.20. [DOI] [PubMed] [Google Scholar]
- 50.Zaharchuk G, Hara H, Huang PL, Fishman MC, Moskowitz MA, Jenkins BG, Rosen BR. Neuronal nitric oxide synthase mutant mice show smaller infarcts and attenuated apparent diffusion coefficient changes in the peri-infarct zone during focal cerebral ischemia. Magn Reson Med. 1997;37:170–175. doi: 10.1002/mrm.1910370204. [DOI] [PubMed] [Google Scholar]