

Abstract
The spontaneous recessive mutant mouse stargazer(stg) begins to show ataxia around postnatal day 14 and display a severe impairment in the acquisition of classical eyeblink conditioning in adulthood. These abnormalities have been attributed to the specific reduction in brain-derived neurotrophic factor (BDNF) and the subsequent defect in TrkB receptor signaling in cerebellar granule cells (GCs). In the stg mutant cerebellum, we found that EPSCs at mossy fiber (MF) to GC synapses are devoid of the fast component mediated by AMPA-type glutamate receptors despite the normal slow component mediated by NMDA receptors. The sensitivity of stg mutant GCs to exogenously applied AMPA was greatly reduced, whereas that to NMDA was unchanged. Glutamate release from MF terminals during synaptic transmission to GCs appeared normal. By contrast, AMPA receptor-mediated EPSCs were normal in CA1 pyramidal cells of the stg mutant hippocampus. Thus, postsynaptic AMPA receptor function was selectively impaired instg mutant GCs, although the transcription of four AMPA receptor subunit genes in the stg GC was comparable to the wild-type GC. We also examined the cerebellum of BDNF knockout mice and found that their MF–GC synapses had a normal AMPA receptor-mediated EPSC component. Thus, the impaired AMPA receptor function in the stg mutant GC is not likely to result from the reduced BDNF–TrkB signaling. These results suggest that the defect in MF to GC synaptic transmission is a major factor that causes the cerebellar dysfunction in the stg mutant mouse.
Keywords: mutant mouse, ataxia, stargazer, cerebellum, AMPA receptor, granule cell, mossy fiber, synaptic transmission, BDNF
The stargazer(stg) mutant mouse is characterized by ataxia and head tossing as well as spike-wave seizures (Noebels et al., 1990; Qiao et al., 1996). It has recently been revealed that the gene disrupted instg and waggler (allelic to stg) encodes a 36 kDa brain-specific protein stargazin (Letts et al., 1998). This is a neuronal counterpart of the γ subunit of skeletal muscle voltage-gated Ca2+ channels and is expressed widely in the brain, including the cerebellum and hippocampus (Letts et al., 1998). Inappropriate Ca2+ entry resulting from this gene mutation may cause functional changes in cortical neurons, which is thought to contribute to the pronounced seizure phenotype in thestg mutant mouse (Di Pasquale et al., 1997; Letts et al., 1998). On the other hand, ataxia has been considered to result from cerebellar dysfunction. The stg mutant mouse displays a severe impairment in the acquisition of classical eyeblink conditioning (Qiao et al., 1998), a motor learning paradigm that is critically involved in the cerebellar function and plasticity (Thompson, 1986; Kim and Thompson, 1997; Yeo and Hesslow, 1998). Immature granule cell (GC)-like neurons persist in the adult stg cerebellum, which suggests retarded cytodifferentiation of stg mutant GCs (Qiao et al., 1998).
In addition to these cerebellar phenotypes, the stg mutant mouse displays near-total reduction of brain-derived neurotrophic factor (BDNF) mRNA expression in the GC layer of the cerebellum, despite normal BDNF expression in other brain regions, including the hippocampus (Qiao et al., 1996). The absence of BDNF mRNA in the GCs was observed at postnatal day 14 (P14), which is coincident with the onset of ataxia. In the cerebellum, expressions of both full-length and truncated TrkB, a neurotrophin receptor for BDNF, were also normal, whereas TrkB receptor-mediated tyrosine phosphorylation was reduced significantly (Qiao et al., 1998). Because BDNF is reported to enhance excitatory synaptic transmission (Lessmann et al., 1994; Kang and Schuman, 1995) and long-term potentiation (Figurov et al., 1996), it has been assumed that the impaired BDNF–TrkB signal transduction is the major cause of abnormalities in cerebellar physiology and development in the stg mutant mouse (Qiao et al., 1998).
In the present study, we found that EPSCs at mossy fiber (MF) to GC synapses of the stg mutant cerebellum are devoid of the AMPA receptor-mediated fast component without significant change in the NMDA receptor-mediated slow component. Our results indicate that this abnormality is attributable to defects in postsynaptic AMPA receptor function but not in presynaptic glutamate release. We also examined the cerebellum of the BDNF-deficient mouse and found that MF–GC synapses had a normal AMPA receptor-mediated EPSC component. Thus, the defect in AMPA receptor function in the stg mutant GC seems unlikely to result from the lack of BDNF production. We propose that the impaired AMPA receptor function at MF–GC synapses leads to functional deafferentation of the cerebellar circuit, which would cause cerebellar dysfunction in the stg mutant mouse.
MATERIALS AND METHODS
Animals. Wild-type (C57BL/6J; +/+) andstargazer mutant (C3B6Fe+; stg/stg) mice were obtained from the breeding colonies of the Jackson Laboratory. Mice were maintained in the vivarium of Jichi Medical School and Kanazawa University on a 12 hr light/dark cycle with free access to food and water. Heterozygous males (+/stg) and homozygous females (stg/stg) were mated to produce stg mutant mice. Because we have not found any electrophysiological difference so far between +/+ and +/stg, nonmutant littermates including both genotypes were used in electrophysiological analyses as wild-type controls. Null-mutant mice of BDNF were obtained from Jackson Laboratory. For the study of BDNF knockout mice, littermates including +/+ and +/− were used as wild-type controls.
Electrophysiology. Sagittal cerebellar slices of 200–250 μm thickness were prepared from stg mutant mice and wild-type mice (P30–P89) or from BDNF knockout mice and wild-type mice (P18–P20), as described previously (Edwards et al., 1989; Llano et al., 1991; Kano and Konnerth, 1992; Aiba et al., 1994). A whole-cell recording was made from visually identified GCs using a 40× water immersion objective attached to an Olympus (BH-2 or BX-50) upright microscope (Edwards et al., 1989; Farrant et al., 1994; Ebradlidze et al., 1996; Takahashi et al., 1996). The resistance of patch pipettes was 5–10 MΩ when filled with an intracellular solution composed of (in mm): 60 CsCl, 30 Cs d-gluconate, 20 TEA-Cl, 20 BAPTA, 4 MgCl2, 4 ATP, and 30 HEPES (pH 7.3, adjusted with CsOH). The composition of standard bathing solution was (in mm): 125 NaCl, 2.5 KCl, 2 CaCl2, 1 MgSO4, 1.25 NaH2PO4, 26 NaHCO3, and 20 glucose, bubbled continuously with a mixture of 95% O2 and 5% CO2. Bicuculline (10 μm) was always present in the saline to block spontaneous IPSCs. For the analysis of CA1 pyramidal cells, transverse hippocampal slices of 200–250 μm thickness were cut, and a whole-cell recording was made from visually identified CA1 pyramidal cells (Edwards et al., 1989). The composition of the pipette solution for recording from CA1 pyramidal cells was (in mm): 120 CsCl, 20 CsOH, 8 NaCl, 10 EGTA, 4 ATP, and 10 HEPES (pH 7.3, adjusted with CsOH). Ionic currents were recorded with an Axopatch-1D patch-clamp amplifier (Axon Instruments). The pipette access resistance was compensated as described previously (Llano et al., 1991). Stimulation and on-line data acquisition were performed using the PULSE program (version 7.5, HEKA). Signals were filtered at 3 kHz and digitized at 20 kHz. The decay phase of EPSCs was fitted with the PULSE-FIT program (version 7.5, HEKA). For stimulation of MFs in the cerebellum or Schaffer collateral/commissural afferents in the hippocampus, a glass pipette with a 5–10 μm diameter tip filled with standard saline was used. Square pulses (duration, 0.1 msec; amplitude, 1–10 V) were applied for focal stimulation. Most of the experiments were performed at a bath temperature of 32°C, unless indicated otherwise.
In situ hybridization. Two homozygous stgand two wild-type mice at 7 postnatal weeks were used for in situ hybridization analysis. Under deep pentobarbital anesthesia, adult mouse brains were excised from the skull and immediately frozen in powdered dry ice. Fresh frozen sections (20 μm in thickness) were prepared by cryostat and mounted on glass slides precoated with 3-aminopropyltriethoxisilane or poly-l-lysine (Sigma, St. Louis, MO). For isotopic detection of the mouse AMPA receptor subunit mRNAs, 45-mer antisense oligonucleotides were synthesized against C-terminal regions downstream to the transmembrane domain M4. The sequence is antisense to 5′-GGTTTCTGTTTGATTCCACAGCAATCCATCAATGAAGCCATACGGO-3′ of the mouse GluRα1 subunit cDNA (nucleotide residues 2467–2511; GenBank Accession No. X57497), 5′-GTGGCAAAGAATGCACAGAATATTAACCCATCTTCCTCGCAGAAT-3′ of the mouse GluRα2 subunit cDNA (nucleotide residues 2479–2523; GenBank Accession No. X57498), 5′-CTCACAAAGAACACCCAAAACTTTAAGCCTGCTCCTGCCACCAAC-3′ of the mouse GluRα3 subunit cDNA (nucleotide residues 2491–2535; GenBank Accession No. AB022342), and 5′-CTGACTTTTTCCGAAGCCATAAGAAACAAAGCCAGGTTATCCATC-3′ of the mouse GluRα4 subunit cDNA (nucleotide residues 2482–2526; GenBank Accession No. AB022913). These oligonucleotides were labeled with35S-dATP to a specific activity of 0.5 × 109 dpm/mg DNA, using terminal deoxyribonucleotidyl transferase (BRL, Bethesda, MD). Sections were processed for fixation, acetylation, prehybridization, and hybridization, as reported previously (Watanabe et al., 1993). The slides were washed twice at 55°C for 40 min in 0.1 × SSC containing 0.1% sarcosyl and were exposed to Hyperfilm-β max (Amersham, Buckinghamshire, England) for 1 month.
Antibody and immunoblot. Rabbit polyclonal antibody against the mouse GluRα4 was produced. A complementary DNA fragment encoding the C-terminal amino acid residues 828–881 of the mouse GluRα4 (TFSEAIRNKARLSITGSVGENGRVLTPDCPKAVHTGTAIRQSSGLAVIASDLP, GenBank Accession No. AB022913) was introduced into pGEX-4T-2 plasmid vector (Pharmacia Biotech AB, Uppsala, Sweden) to obtain glutathioneS-transferase (GST) fusion protein. The fusion proteins expressed in Escherichia coli BL21 were purified with glutathione Sepharose-4B (Pharmacia), and the polypeptide of GluRα4 was separated from GST by thrombin digestion followed by RP-HPLC. The purified polypeptide was injected into female New Zealand White rabbits at intervals of 2–4 weeks. From the antiserum sampled 2 weeks after the sixth injection, immunoglobulins were separated using Protein G-Sepharose (Pharmacia), and antibodies specific to the GluRα4 polypeptide were affinity-purified using fusion protein-coupled CNBr-activated Sepharose 4B (Pharmacia).
The stg mutant and wild-type mice (P21) were decapitated under anesthesia, and cerebella were removed rapidly. Each cerebellum was homogenized in 10 vol of buffer H (10 mm Tris-Cl, pH 7.2, 5 m MEDTA, 0.32 m sucrose, 1 mm phenylmethylsulfonyl fluoride, and 10 mg/l leupeptin) within 3 min of decapitation and centrifuged at 700 ×g for 10 min to obtain a postnuclear fraction. Protein determination was made by the method of Lowry et al. (1951). Equal amounts of the protein were fractionated by SDS-PAGE and electroblotted onto a nitrocellulose membrane (Schleicher and Schuell, Dassel, Germany). The blot was immunoreacted with anti-GluRα4 antibody at 1 μg/ml and visualized by chemiluminescence (ECL detection system, Amersham, Tokyo, Japan). For semi-quantitative analysis, the immunoreactive bands were scanned using a densitometer (Shimazu CS-9300PC).
RESULTS
Reduction of the non-NMDA receptor-mediated component of EPSCs at mossy fiber-granule cell synapses in stg mutant mice
In parasagittal cerebellar slices prepared from wild-type and stg mutant mice, GCs were recorded in the whole-cell configuration (Edwards et al., 1989; Farrant et al., 1994; Ebradlidze et al., 1996; Takahashi et al., 1996). In GCs from wild-type mice, EPSCs induced by MF stimulation (MF–EPSCs) were composed of a fast component followed by a slow component. The amplitude of the slow component at a holding potential of −70 mV was significantly smaller than that at +40 mV (Fig.1A). The current–voltage (I–V) relationship for the fast component was linear, whereas that for the slow component measured at 50 msec after the stimulus exhibited a characteristic outward rectification (Fig. 1C). The fast and slow components were selectively blocked by a non-NMDA receptor antagonist, 6-nitro-7-sulfamoylbenzo[f]quinoxaline-2,3-dione (NBQX, 5 μm), and an NMDA receptor antagonist, 3-(R-2-carboxypiperazin-4-yl)-propyl-1-phosphoninc acid (R-CPP, 10 μm), respectively, in 14 GCs examined (Fig.1E). These results indicate that the fast and slow components of MF–EPSCs in wild-type mice are mediated by non-NMDA and NMDA receptors, respectively.
Fig. 1.

Lack of non-NMDA receptor-mediated fast component in MF–GC excitatory synaptic transmission in the stgmutant mice. A, B, EPSCs elicited by MF stimulation in GCs from the wild-type (A) and stgmutant (B) mice at holding potentials of +40 mV (top panels) and −70 mV (bottom panels). Each trace is a single-sweep record, and several traces are superimposed for each record. C, D,I–V relationships of MF–EPSCs from wild-type (C) and stg mutant (D) mice measured at the peak (○) and 50 msec after (●) the stimulus. The EPSC amplitudes were normalized to the mean value at +40 mV in each experimental condition. Each data point and attached error bar represent mean and SEM. E, F, Effects of NBQX (5 μm) and CPP (10 μm) on MF–EPSCs in GCs from wild-type (E) andstg mutant (F) mice at a holding potential of +40 mV. Each trace is an average of 10 consecutive sweeps. Records in the control external solution, in the solution containing NBQX (5 μm), and in that containing CPP (10 μm) are superimposed. MF stimulation was repeated at 0.2 Hz.
In stg mutant GCs, MF–EPSCs were devoid of distinguishable fast components (Fig. 1B). The MF–EPSC amplitude was much smaller at a holding potential of −70 mV than at +40 mV (Fig.1B). The I–V relationship exhibited a characteristic outward rectification measured at both peak and 50 msec after the stimulus (Fig. 1D). MF–EPSCs were not affected by NBQX (5 μm) but were almost abolished by R-CPP (10 μm) in 15 stg mutant GCs examined (Fig. 1F). These results indicate that MF–EPSCs in stg mutant GCs are mediated by NMDA receptors and are devoid of the non-NMDA receptor-mediated fast component. The lack of non-NMDA receptor-mediated fast component was obvious at P14, which coincides with the onset of ataxia in the stg mutant mouse (Qiao et al., 1996). The defect was obvious also in younger stg mutant mice at P11. On the other hand, the lack of non-NMDA receptor-mediated fast component was found in MF–EPSCs of the stg mutant mouse around P80, indicating that the defect persists into adulthood. When measured at a holding potential of +40 mV, the amplitudes of MF–EPSCs in stg mutant GCs were similar to that of the slow component of MF–EPSCs in the wild-type GCs (Table 1). The 10–90% rise time and the decay time constants (τ1 and τ2, fitted with double exponentials) were also similar (Table 1). These results indicate that stgmutant GCs have a specific defect in non-NMDA receptor function at MF–GC synapses without apparent abnormality in NMDA-receptor function.
Table 1.
Properties of the NMDA receptor-mediated component of MF-EPSCs
| Genotype | Amplitude (pA) | Rise time (msec) | Decay (τ1) (msec) | Decay (τ2) (msec) |
|---|---|---|---|---|
| Wild type (n = 11) | 17.8 ± 9.3 | 2.5 ± 0.8 | 15.3 ± 8.5 | 61.0 ± 20.6 |
| stg mutant (n = 13) | 18.8 ± 9.5 | 2.3 ± 0.7 | 14.2 ± 5.6 | 56.9 ± 29.0 |
GCs near the surface of the slices were recorded in the presence of NBQX (5 μm) or CNQX (10 μm) at a holding potential of +40 mV. The bath temperature was 32°C. Data are expressed as mean ± SD.
Selective reduction of AMPA sensitivity in stg mutant granule cells
To examine whether the reduced non-NMDA receptor-mediated component of MF–EPSCs in the stg mutant mouse is caused by the defect in postsynaptic receptor function, we directly measured the whole-cell currents through AMPA receptors and NMDA receptors in GCs (Tempia et al., 1996). Leak-subtracted voltage ramps (from a holding potential of +40 to −110 mV, 2000 msec duration) were adopted to construct instantaneous I–V relationships of the current induced by bath-applied AMPA or NMDA. Wild-type GCs responded well to both AMPA (10 μm) and NMDA (100 μm). The I–V relationship during AMPA application was linear (Fig.2A), whereas that during NMDA application exhibited a clear outward rectification (Fig.2C). In contrast, stg mutant GCs had almost no response to the challenge of AMPA (Fig. 2B), whereas they exhibited an outwardly rectifying I–Vrelationship during NMDA application (Fig. 2D). These results indicate that postsynaptic AMPA receptor function is impaired in the stg mutant GCs without a significant change in NMDA receptor function.
Fig. 2.

Significantly reduced sensitivity to exogenous AMPA in GCs of the stg mutant mice. A, B, Instantaneous I–Vrelationships of the AMPA (10 μm)-induced current evoked in GCs from wild-type (A) and stgmutant (B) mice. Currents were measured during voltage ramp from +40 to −110 mV (duration, 2000 msec). C, D, Instantaneous I–Vrelationships of the NMDA (100 μm)-induced current evoked in GCs from wild-type (C) and stgmutant (D) mice. Currents were measured and illustrated as in A and B. Records were taken in the presence of tetrodotoxin (0.5 μm) and strychnine (1 μm). For leak subtraction, evoked currents measured in the control solution were subtracted from those in the presence of AMPA or NMDA. Each I–V curve is an average of data from 9–12 different GCs. Error bars at −100, −80, −60, −40, −20, 0, +20, and +40 mV represent SEM.
Because NMDA receptors have higher affinity to glutamate than AMPA receptors, it is possible that reduced presynaptic glutamate release could preferentially reduce the AMPA receptor-mediated component of EPSCs. To pursue this possibility, we estimated the time course of free glutamate concentration in the MF–GC synaptic cleft by application of α-amino pimelic acid (α-APA) (Fig.3A), a rapidly dissociating NMDA receptor antagonist that displaces synaptically released glutamate (Scanziani et al., 1997). To isolate NMDA receptor-mediated components, MF–EPSCs were recorded at a holding potential of +40 mV in the presence of a non-NMDA antagonist, 6-cyano-7-nitroquinoxaline-2,3-dion (CNQX, 10 μm). We found no significant difference in the inhibitory potency of α-APA on MF–EPSCs between the wild-type andstg mutant mice at all antagonist concentrations tested (10–1000 μm) (Fig. 3A,B), suggesting normal glutamate release from MF terminals in stg mutant mice.
Fig. 3.

Effects of a low-affinity NMDA receptor blocker, α-amino pimelic acid (α-APA), on the NMDA receptor-mediated components of MF–EPSCs. Specimen records (A) and summary graph show concentration–inhibition curves (B) for the wild-type (top record in A; open circles in B) and stg mutant (bottom record in A; closed circles in B) GCs. Each trace inA is an average of 20 consecutive sweeps. Theordinate in B indicates percentage of the control EPSC amplitude before application of α-amino pimelic acid (mean ± SEM). Records were taken at a holding potential of +40 mV and in the presence of CNQX (10 μm) and bicuculline (10 μm).
Non-NMDA receptor-mediated EPSC component is normal in CA1 pyramidal cells of the stg mutant mouse
To clarify whether the defect in AMPA receptor function is specific to GCs, we studied excitatory synaptic transmission in CA1 pyramidal cells (Fig. 4). In both wild-type and stg mutant pyramidal cells, stimulation of Schaffer collateral/commissural afferents evoked EPSCs at a holding potential of −65 mV (Fig. 4A) that were significantly blocked by a non-NMDA antagonist, CNQX (10 μm) (data not shown). When the holding potential was changed to +45 mV, the slow EPSC component became larger (Fig.4A), and this was blocked by an NMDA-blocker,dl-2-amino-5-phosphonopentanoate (AP-5, 50 μm) (data not shown). These results indicate that peak EPSC at −65 mV [EPSC(−65)] and EPSC 100 msec after the stimulus at +45 mV [EPSC(+45)]) represent non-NMDA and NMDA receptor-mediated components at respective holding potentials. The mean amplitudes of the peak currents at −65 mV were 39.9 + 25.9 pA and 49.1 ± 24.3 pA (mean ± SD) for the wild-type (n = 20) and the stg mutant (n = 22) mice, respectively. Those of the currents at 100 msec at +45 mV were 6.7 + 5.0 pA and 11.0 ± 11.8 pA (mean ± SD) for the wild-type (n = 20) and thestg mutant (n = 22) mice, respectively. These values were not significantly different between the wild-type andstg mutant mice. Moreover, the amplitude ratio of EPSC(+45) to EPSC(−65) in the stg mutant mouse was not different from that in the wild-type mouse (Fig. 4B). These results suggest that AMPA receptor function at Schaffer collateral/commissural afferents to CA1 pyramidal cell synapses is normal in thestg mutant mouse.
Fig. 4.

Normal excitatory synaptic responses in hippocampal CA1 pyramidal cells of the stg mutant mice.A, EPSCs elicited by stimulation of Schaffer collateral/commissural afferents in CA1 pyramidal cells from the wild-type (top traces) and stg mutant mice (bottom traces) measured at holding potentials of +45 mV (upward traces) and −65 mV (downward traces). B, Ratios of EPSC at +45 mV measured at 100 msec after the stimulus [EPSC(+45)] to peak EPSC at −65 mV [EPSC(−65)]. Error bars represent SEM.
Expressions of four AMPA receptor subunit mRNAs are normal
To clarify whether the selective AMPA receptor dysfunction in stg mutant GCs results from altered expressions of AMPA receptor subunits, we comparatively examined the expression of four AMPA receptor subunit mRNAs by in situ hybridization with35S-labeled subunit-specific antisense oligonucleotide probes (Fig. 5). In the brain of adult wild-type mice (Fig. 5A–D), characteristic distributions of the GluRα1–4 mRNAs were visualized in patterns consistent with previous results reported in the adult rat brain (Boulter et al., 1990;Keinänen et al., 1990). Briefly, the GluRα1–3 mRNAs were highly expressed in both the telencephalon and cerebellum, whereas the GluRα4 mRNA predominated in the cerebellum. Within the cerebellum, the granular layer displayed abundant signals for the GluRα2 and GluRα4 mRNAs but not for the GluRα1 or GluRα3 mRNA. In the hippocampus, the CA1 pyramidal cell layer expressed high levels of the GluRα1–3 mRNAs. The brain of the stg mutant mouse, including the granular layer of the cerebellum and CA1 pyramidal cell layer of the hippocampus, showed distribution patterns and comparable transcription levels similar to those of the wild-type mouse (Fig.5E–H). The hybridization and washing for the wild-type and stg mutant mice were performed in the same experiment under the same conditions, and glass slides were exposed to a single x-ray film. These characteristic signals disappeared when hybridization was performed in the presence of excess unlabeled antisense oligonucleotides or with use of sense probes (data not shown). Therefore, the expression of AMPA receptor subunits in thestg mutant brain is normal, at least at the transcription level.
Fig. 5.
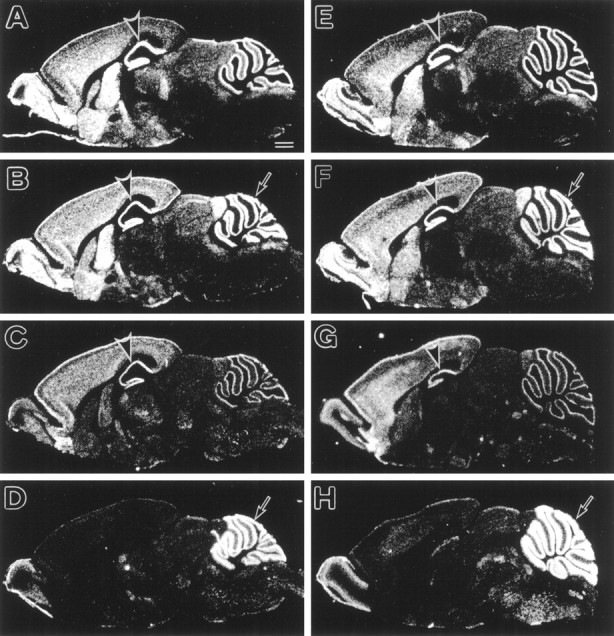
In situ hybridization showing the expressions of four AMPA receptor subunit mRNAs in the adult stg mutant (A–D) and wild-type (E–H) mice. A, E, GluRα1; B, F, GluRα2; C, G, GluRα3;D, H, GluRα4. No differences in the distribution and levels of each AMPA subunit mRNA are found between thestg mutant and wild-type mice. Rostral is to the left, and dorsal is to the top. Scale bar, 1 mm.
The GluRα4 protein level is normal in the stgmutant cerebellum
To further examine at the translational level, we compared the size and amount of GluRα4 in the cerebellum. This subunit is transcribed abundantly in the cerebellar GC but is almost absent in the hippocampal pyramidal cell layer (Fig. 5D). We raised polyclonal rabbit IgG against the C terminus of the mouse GluRα4, which is low in homology with other mouse AMPA receptor subunits (Sakimura et al., 1990) (GenBank Accession No.: GluRα3, AB022342; GluRα4, AB022913). Immunoblot detected a single protein band at 102 kDa, whose apparent size was slightly higher than that calculated from the amino acid sequence deduced from the cloned mouse GluRα4 cDNA, probably caused by glycosylation. In the wild-type andstg mutant mice, the 102 kDa protein bands were detected in similar size (Fig. 6), and their amounts were measured semiquantitatively by using a densitometer. The mean density of the band from the stg mutant cerebella (n = 5) was 93% of that from the wild-type cerebella (n = 5), showing no significant difference. Therefore, the GluRα4 protein level is normal in the stg mutant cerebella.
Fig. 6.

Immunoblot analysis of the GluRα4 subunit proteins from the cerebella of three wild-type (lanes 1–3) and three stg mutant (lanes 4–6) mice. Fifty micrograms each of the postnuclear proteins of the cerebellum were loaded on each lane of SDS-PAGE (7% gel). The anti-GluRα4 antibody was used at 1 μg/ml. Molecular mass markers are indicated on the left.
Defect in stg mutant cerebellum is not caused by impaired BDNF expression in granule cells
It has been reported previously that stg mutant GCs exhibit a selective reduction of BDNF mRNA expression in cerebellar GCs (Qiao et al., 1996), which has been assumed to be a major cause of cerebellar dysfunction leading to ataxia and impaired eyeblink conditioning (Qiao et al., 1998). To examine whether the aforementioned electrophysiological abnormalities of the AMPA receptor function can be ascribed to a defect in BDNF expression, we studied the cerebellum of the BDNF-deficient mouse (Ernfors et al., 1994; Jones et al., 1994).
In GCs from wild-type (Fig.7A) and BDNF-deficient mice (Fig. 7B), MF–EPSCs were composed of a fast component followed by a slow component. The I–Vrelationship for the fast component was linear, whereas that for the slow component measured at 50 msec from the stimulus exhibited a characteristic outward rectification (Fig. 7C,D). The fast and slow components were selectively blocked by CNQX (10 μm) or AP-5 (50 μm), respectively (data not shown). These results indicate that MF to GC excitatory synaptic transmission develops normally even in the absence of BDNF expression.
Fig. 7.

MF–GC excitatory synaptic transmission in BDNF knockout mice is normal. A, B, EPSCs elicited by MF stimulation in GCs from the wild-type (A) and BDNF knockout (B) mice at holding potentials of +40 mV (top panels) and −70 mV (bottom panels). Each trace is single-sweep record, and several traces are superimposed for each record. C, D,I–V relationships of MF–EPSCs from wild-type (C) and BDNF knockout (D) mice measured at the peak (○) and at 50 msec after the stimulus (●). The EPSC amplitudes were normalized to the mean value at +40 mV in each experimental condition. Each data point and attached error bar represent mean and SEM.
DISCUSSION
Selective defect of AMPA-receptor function instg mutant GCs
The present study has demonstrated that EPSCs at MF to GC synapses of the stg mutant cerebellum are devoid of the AMPA receptor-mediated fast component, whereas the NMDA receptor-mediated slow component is normal. The sensitivity to exogenously applied AMPA was also greatly reduced, whereas that to NMDA was normal. In contrast, the time course of free glutamate concentration in the MF–GC synaptic cleft was similar between the wild-type and stg mutant GCs, as assessed by using a rapidly dissociating NMDA receptor antagonist α-APA. These results suggest that the loss of AMPA receptor-mediated EPSCs in the stg mutant mouse is likely to result from impaired postsynaptic AMPA receptor function rather than reduced glutamate release at the MF–GC synapse. By contrast, we detected normal AMPA receptor-mediated responses at hippocampal synapses in thestg mutant mouse. Thus, it is clear that the loss of AMPA receptor function is not a common feature to excitatory synapses in thestg mutant brain but occurs preferentially at the MF–GC synapse.
Our in situ hybridization data indicate that the expression of four AMPA receptor subunits (GluRα1–4) in the stgmutant brain is normal at the transcription level. Our preliminary immunohistochemical data indicate that the wild-type and stgmutant cerebella exhibit similar patterns of immunoreactivity to antibodies against GluRα4. In both wild-type and stgmutant cerebella, the GluRα4 immunoreactivity was detected in the GC layer and the molecular layer (our unpublished data). The immunoblot analysis indicates that the size and amount of GluRα4 protein in thestg mutant cerebellum were normal. These results suggest that GluRα4 subunits are translated normally in the stgmutant GCs, but their function is lost almost completely presumably because of defects in post-translational modification.
Selective AMPA receptor defect is unlikely to result from BDNF deficiency in stg mutant GCs
It has been reported previously that the stgmutant mouse displays a selective and near-total reduction of BDNF mRNA expression in the cerebellar GC layer (Qiao et al., 1996) and that TrkB receptor-mediated tyrosine phosphorylation is downregulated (Qiao et al., 1998). Application of BDNF potentiates the efficacy of neuromuscular synapses in culture (Lohof et al., 1993), enhances synaptic strength in the hippocampus (Lessmann et al., 1994; Kang and Schuman, 1995), and facilitates the induction of long-term potentiation (Figurov et al., 1996), whereas it inhibits GABAA-receptor mediated inhibitory synaptic transmission (Tanaka et al., 1997). On the basis of these actions, it could be possible to postulate that the lack of BDNF in the stg mutant GCs causes impairment of excitatory synaptic transmission and plasticity. However, we found normal AMPA receptor-mediated responses in the MF–GC synapse in the BDNF knockout mouse. Moreover, we failed to observe any increment of the AMPA receptor-mediated EPSC component by incubating the cerebellar slices for up to 12 hr with external solution containing 100 ng/ml BDNF (n = 15; data not shown). The same concentration of BDNF is reported to rescue deficits in hippocampal LTP in BDNF knockout mice (Patterson et al., 1996). Therefore, BDNF deficiency by itself may not cause the abnormality of MF–GC synaptic transmission in thestg mutant mouse.
A recent study with cultured cerebellar cells has shown that AMPA receptor stimulation activates Lyn, an Src family nonreceptor protein tyrosine kinase, which then induces BDNF expression through the mitogen-activated protein kinase cascade (Hayashi et al., 1999). From the evidence, we rather speculate that the lack of AMPA receptor function may lead to the reduced BDNF mRNA expression in GCs. In this context, normal levels of BDNF mRNA in the stg mutant hippocampus (Qiao et al., 1996) can be interpreted as resulting from normal AMPA receptor-mediated excitatory transmission. It is possible, however, that AMPA receptor function and BDNF expression may be unrelated in vivo. Further studies are required to elucidate the presumed causal relationship between these two phenomena.
Motor dysfunction in stg mutant mice is caused by functional deafferentation of the cerebellum
Because the Mg2+ block of NMDA receptor channels is significant near the resting membrane potential (approximately −60 mV) (Mayer et al., 1984; Nowak et al., 1984;D’Angelo et al., 1994), usual excitatory synaptic transmission at low frequency is considered to be mediated mainly by AMPA receptors (D’Angelo et al., 1995). The lack of AMPA receptor-mediated synaptic currents at MF–GC synapses implies a virtual absence of massive afferent information to GCs in stg mutant mice in vivo. MFs convey various sensory signals to the cerebellum via the spinal cord and brain stem and also transmit information related to motor command to the cerebellum from the cerebral cortex (Ito, 1984). These signals are conveyed through GCs and their axons, parallel fibers (PFs), to various neurons in the cerebellar cortex, including Purkinje cells (PCs), basket cells, stellate cells, and Golgi cells. We also found that excitatory synaptic transmission from PFs to PCs and also those from climbing fibers (CFs) to PCs were significantly impaired in the stg mutant cerebellum; the amplitudes of PF–EPSCs and CF–EPSCs of stg mutant PCs were ∼60% of the wild-type PCs (K. Hashimoto and M. Kano, unpublished observations). Therefore, synapses relaying two excitatory inputs to the cerebellar cortex have either no (MF–GC synapses) or significantly reduced (PF–PC synapses, CF–PC synapses) AMPA receptor-mediated synaptic currents. Thus, the cerebellar cortex of the stg mutant mouse is functionally deafferented.
The stg mutant mice have been reported to display severe impairment of the acquisition of classical eyeblink conditioning (Qiao et al., 1998), which involves both cerebellar cortex and interpositus nucleus (Kim and Thompson, 1997). When a neutral conditioned stimulus (CS), such as tone, is repeatedly applied with an unconditioned stimulation (US), such as periorbital shock, the mouse gradually develops eyeblink response to the CS. In this learning paradigm, it is considered that signals related to CS and those related to US are conveyed to the cerebellum via MFs and CFs, respectively (Kim and Thompson, 1997; Yeo and Hesslow, 1998). Thus, the impaired acquisition of eyeblink conditioning in the stg mutant mouse can be interpreted as resulting from the failure to transmit both CS and US signals to the cerebellar cortex. Therefore, the functional deafferentation of the stg mutant cerebellum seems likely to cause motor learning deficits, leading to motor discoordination.
Defects in post-translational modification is a possible cause of impaired AMPA receptor function in stg mutant GCs
To seek the molecular basis for the selective loss of the AMPA receptor function in stg mutant GCs, we performedin situ hybridization and immunoblot analyses. At both the transcription and translation levels, however, we found no appreciable changes that could explain the selective loss. Thus, it is assumed that the defect of AMPA receptor function occurs in the post-translational process. After biosynthesis, AMPA receptor subunits undergo phosphorylation and glycosylation (Kawamoto et al., 1995; Roche et al., 1996). Subsequently, they are targeted to synaptic sites, where they interact with the PDZ domain-containing protein family, such as GRIP and ABP, and become clustered and anchored to the postsynaptic membrane (Dong et al., 1997; Srivastava et al., 1998).
The γ subunit is an auxiliary transmembrane subunit of voltage-gated Ca2+ channels (Walker and Waard, 1998). The mutated subunit stargazin increases steady-state inactivation of the α1 Ca2+ channels in vitro, implying inappropriate Ca2+ entry through modified Ca2+ channels (Letts et al., 1998). In cortical pyramidal cells, this may cause enhancement of cesium-sensitive inward rectifier current (Di Pasquale et al., 1997). Although it remains uncertain how Ca2+ channel function is involved in the regulation of AMPA receptor function in the cerebellum, the present results suggest the importance of the post-translational process for AMPA receptor subunits to become functional channels. In this regard, the stg mutant mouse is an intriguing model system to elucidate the mechanisms for the regulation of AMPA receptor function in future studies.
Footnotes
This work has been partly supported by grants from the Japanese Ministry of Education, Science, Sports and Culture (M.K., M.W.) and the Human Frontier Science Program (M.K.) and by Special Coordination Funds for Promoting Science and Technology from Science and Technology Agency (M.K., M.W.). We thank Dr. N. Kawai for continuous encouragement throughout the course of this study, E. Kushiya, R. Natume, K. Matsumoto, and Y. Okada for excellent technical assistance, and Drs. T. Ohno-Shosaku, T. Tabata, and N. Suzuki for critically reading this manuscript.
Correspondence should be addressed to Masanobu Kano, Department of Physiology, Kanazawa University School of Medicine, 13-1 Takara-machi, Kanazawa 920-8640, Japan.
REFERENCES
- 1.Aiba A, Kano M, Chen C, Stanto ME, Fox GD, Herrup K, Zwingman TA, Tonegawa S. Deficient cerebellar long-term depression and impaired motor learning in mGluR1 mutant mice. Cell. 1994;79:377–388. [PubMed] [Google Scholar]
- 2.Boulter J, Hollmann M, O’Shea-Greenfield A, Hartley M, Deneris E, Maron C, Heinemann S. Molecular cloning and functional expression of glutamate receptor subunit genes. Science. 1990;249:1033–1037. doi: 10.1126/science.2168579. [DOI] [PubMed] [Google Scholar]
- 3.D’Angelo E, Rossi P, Taglietti V. Voltage-dependent kinetics of N-methyl-d-aspartate synaptic currents in rat cerebellar granule cells. Eur J Neurosci. 1994;6:640–645. doi: 10.1111/j.1460-9568.1994.tb00309.x. [DOI] [PubMed] [Google Scholar]
- 4.D’Angelo E, De Filippi G, Rossi P, Taglietti V. Synaptic excitation of individual rat cerebellar granule cells in situ: evidence for the role of NMDA receptors. J Physiol (Lond) 1995;484:397–413. doi: 10.1113/jphysiol.1995.sp020673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Di Pasquale E, Keegan KD, Noebels JL. Increased excitability and inward rectification in layer V cortical pyramidal neurons in epileptic mutant mouse stargazer. J Neurophysiol. 1997;77:621–631. doi: 10.1152/jn.1997.77.2.621. [DOI] [PubMed] [Google Scholar]
- 6.Dong H, O’Brien RJ, Fung ET, Lanahan AA, Worley PF, Huganir RL. GRIP: a synaptic PDZ domain-containing protein that interacts with AMPA receptors. Nature. 1997;386:279–284. doi: 10.1038/386279a0. [DOI] [PubMed] [Google Scholar]
- 7.Ebradlidze AK, Rossi DJ, Tonegawa S, Slater NT. Modification of NMDA receptor channels and synaptic transmission by targeted disruption of the NR2C gene. J Neurosci. 1996;16:5014–5025. doi: 10.1523/JNEUROSCI.16-16-05014.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Edwards FA, Konnerth A, Sakmann B, Takahashi T. A thin slice preparation for patch-clamp recordings from neurons of the mammalian central nervous system. Pflügers Arch. 1989;414:600–612. doi: 10.1007/BF00580998. [DOI] [PubMed] [Google Scholar]
- 9.Ernfors P, Lee KF, Jaenisch R. Mice lacking brain-derived neurotrophic factor develop with sensory deficits. Nature. 1994;368:147–150. doi: 10.1038/368147a0. [DOI] [PubMed] [Google Scholar]
- 10.Farrant M, Feldmeyer D, Takahashi T, Cull-Candy SG. NMDA-receptor channel diversity in the developing cerebellum. Nature. 1994;368:335–339. doi: 10.1038/368335a0. [DOI] [PubMed] [Google Scholar]
- 11.Figurov A, Pozzo Miller LD, Olafsson P, Wang T, Lu B. Regulation of synaptic responses to high-frequency stimulation and LTP by neurotrophins in the hippocampus. Nature. 1996;381:706–709. doi: 10.1038/381706a0. [DOI] [PubMed] [Google Scholar]
- 12.Hayashi T, Umemori H, Mishina M, Yamamoto T. The AMPA receptor interacts with and signals through the protein tyrosine kinase Lyn. Nature. 1999;397:72–76. doi: 10.1038/16269. [DOI] [PubMed] [Google Scholar]
- 13.Ito M. The cerebellum and neural control. Raven; New York: 1984. [Google Scholar]
- 14.Jones KR, Farinas I, Backus C, Reichardt LF. Targeted disruption of the BDNF gene perturbs brain and sensory neuron development but not motor neuron development. Cell. 1994;76:989–999. doi: 10.1016/0092-8674(94)90377-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kang H, Schuman EM. Long-lasting neurotrophin-induced enhancement of synaptic transmission in the adult hippocampus. Science. 1995;267:1658–1662. doi: 10.1126/science.7886457. [DOI] [PubMed] [Google Scholar]
- 16.Kano M, Konnerth A. Cerebellar slices for patch clamp recording. In: Kettenmann H, Grantyn R, editors. Practical electrophysiological methods. Wiley-Liss; New York: 1992. pp. 54–57. [Google Scholar]
- 17.Kawamoto S, Hattori S, Sakimura K, Mishina M, Okuda K. N-linked glycosylation of the alpha-amino 3-hydroxy-5-methylisoxazole-4-propionate (AMPA)-selective glutamate receptor channel alpha 2 subunit is essential for the acquisition of ligand-binding activity. J Neurochem. 1995;64:1258–1266. doi: 10.1046/j.1471-4159.1995.64031258.x. [DOI] [PubMed] [Google Scholar]
- 18.Keinänen K, Wisden W, Sommer B, Werner P, Herb S, Verdoorn TA, Sakmann B, Seeburg PH. A family of AMPA-selective glutamate receptors. Science. 1990;249:556–560. doi: 10.1126/science.2166337. [DOI] [PubMed] [Google Scholar]
- 19.Kim JJ, Thompson RF. Cerebellar circuits and synaptic mechanisms involved in classical eyeblink conditioning. Trends Neurosci. 1997;20:177–181. doi: 10.1016/s0166-2236(96)10081-3. [DOI] [PubMed] [Google Scholar]
- 20.Lessmann V, Gottmann K, Heumann R. BDNF and NT-4/5 enhance glutamatergic synaptic transmission in cultured hippocampal neurones. NeuroReport. 1994;6:21–25. doi: 10.1097/00001756-199412300-00007. [DOI] [PubMed] [Google Scholar]
- 21.Letts VA, Felix R, Biddlecome GH, Arikkath J, Mahaffey CL, Valenzuela A, Bartlett FS, II, Mori Y, Campbell KP, Frankel WN. The mouse stargazer gene encodes a neuronal Ca2+-channel γ subunit. Nat Genet. 1998;19:340–347. doi: 10.1038/1228. [DOI] [PubMed] [Google Scholar]
- 22.Llano I, Marty A, Armstrong CM, Konnerth A. Synaptic- and agonist-induced excitatory currents of Purkinje cells in rat cerebellar slice. J Physiol (Lond) 1991;434:183–213. doi: 10.1113/jphysiol.1991.sp018465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lohof AM, Ip NY, Poo MM. Potentiation of developing neuromuscular synapses by the neurotrophins NT-3 and BDNF. Nature. 1993;363:350–353. doi: 10.1038/363350a0. [DOI] [PubMed] [Google Scholar]
- 24.Lowry OH, Rosebrough NJ, Farr AL, Randall RJ. Protein measurement with the folin phenol reagent. J Biol Chem. 1951;193:265–275. [PubMed] [Google Scholar]
- 25.Mayer ML, Westbrook GL, Guthrie PB. Voltage-dependent block by Mg2+ of NMDA responses in spinal cord neurones. Nature. 1984;309:261–263. doi: 10.1038/309261a0. [DOI] [PubMed] [Google Scholar]
- 26.Noebels JL, Qiao X, Bronson RT, Spencer C, Davisson MT. Stargazer: a new neurological mutant on chromosome 15 in the mouse with prolonged cortical seizures. Epilepsy Res. 1990;7:129–135. doi: 10.1016/0920-1211(90)90098-g. [DOI] [PubMed] [Google Scholar]
- 27.Nowak L, Bregestovski P, Ascher P, Herbet A, Prochiantz A. Magnesium gates glutamate-activated channels in mouse central neurones. Nature. 1984;307:462–465. doi: 10.1038/307462a0. [DOI] [PubMed] [Google Scholar]
- 28.Patterson SL, Abel T, Deuel TA, Martin KC, Rose JC, Kandel ER. Recombinant BDNF rescues deficits in basal synaptic transmission and hippocampal LTP in BDNF knockout mice. Neuron. 1996;16:1137–1145. doi: 10.1016/s0896-6273(00)80140-3. [DOI] [PubMed] [Google Scholar]
- 29.Qiao X, Hefti F, Knusel B, Noebels JL. Selective failure of brain-derived neurotrophic factor mRNA expression in the cerebellum of stargazer, a mutant mouse with ataxia. J Neurosci. 1996;16:640–648. doi: 10.1523/JNEUROSCI.16-02-00640.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Qiao X, Chen L, Gao H, Bao S, Hefti F, Thompson RF, Knusel B. Cerebellar brain-derived neurotrophic factor-TrkB defect associated with impairment of eyeblink conditioning in stargazer mutant mice. J Neurosci. 1998;18:6990–6999. doi: 10.1523/JNEUROSCI.18-17-06990.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Roche KW, O’Brien RJ, Mammen AL, Bernhardt J, Huganir RL. Characterization of multiple phosphorylation sites on the AMPA receptor GluR1 subunit. Neuron. 1996;16:1179–1188. doi: 10.1016/s0896-6273(00)80144-0. [DOI] [PubMed] [Google Scholar]
- 32.Sakimura K, Bujo H, Kushiya E, Araki K, Yamazaki M, Yamazaki M, Meguro H, Warashina A, Numa S, Mishina M. Functional expression from cloned cDNAs of glutamate receptor species responsive to kainate and quisqualate. FEBS Lett. 1990;272:73–80. doi: 10.1016/0014-5793(90)80452-o. [DOI] [PubMed] [Google Scholar]
- 33.Scanziani M, Salin PA, Vogt KE, Malenka RC, Nicoll RA. Use-dependent increases in glutamate concentration activate presynaptic metabotropic glutamate receptors. Nature. 1997;385:630–634. doi: 10.1038/385630a0. [DOI] [PubMed] [Google Scholar]
- 34.Srivastava S, Osten P, Vilim FS, Khatri L, Inman G, States B, Daly C, DeSouza S, Abagyan R, Valtschanoff JG, Weinberg RJ, Ziff EB. Novel anchorage of GluR2/3 to the postsynaptic density by the AMPA receptor-binding protein ABP. Neuron. 1998;21:581–591. doi: 10.1016/s0896-6273(00)80568-1. [DOI] [PubMed] [Google Scholar]
- 35.Takahashi T, Feldmeyer D, Suzuki N, Onodera K, Cull-Candy SG, Sakimura K, Mishina M. Functional correlation of NMDA receptor epsilon subunits expression with the properties of single-channel and synaptic currents in the developing cerebellum. J Neurosci. 1996;16:4376–4382. doi: 10.1523/JNEUROSCI.16-14-04376.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Tanaka T, Saito H, Matsuki N. Inhibition of GABAA synaptic responses by brain-derived neurotrophic factor (BDNF) in rat hippocampus. J Neurosci. 1997;17:2959–2966. doi: 10.1523/JNEUROSCI.17-09-02959.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tempia F, Kano M, Schneggenburger R, Schirra C, Garaschuk O, Plant T, Konnerth A. Fractional calcium current through neuronal AMPA-receptor channels with a low calcium permeability. J Neurosci. 1996;15:456–466. doi: 10.1523/JNEUROSCI.16-02-00456.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Thompson RF. The neurobiology of learning and memory. Science. 1986;233:941–947. doi: 10.1126/science.3738519. [DOI] [PubMed] [Google Scholar]
- 39.Walker D, Waard MD. Subunit interaction sites in voltage-dependent Ca2+ channels: role in channel function. Trends Neurosci. 1998;21:148–154. doi: 10.1016/s0166-2236(97)01200-9. [DOI] [PubMed] [Google Scholar]
- 40.Watanabe M, Inoue Y, Sakimura K, Mishina M. Distinct distributions of five NMDA receptor channel subunit mRNAs in the forebrain. J Comp Neurol. 1993;338:377–390. doi: 10.1002/cne.903380305. [DOI] [PubMed] [Google Scholar]
- 41.Yeo CH, Hesslow G. Cerebellum and conditioned reflexes. Trends Cognit Sci. 1998;2:322–330. doi: 10.1016/s1364-6613(98)01219-4. [DOI] [PubMed] [Google Scholar]