

Abstract
Rab3A and rab3C are GTP-binding proteins of synaptic vesicles that regulate vesicle exocytosis. Rabphilin is a candidate rab3 effector at the synapse because it binds to rab3s in a GTP-dependent manner, it is co-localized with rab3s on synaptic vesicles, and it dissociates with rab3s from the vesicles during exocytosis. Rabphilin contains two C2 domains, which could function as Ca2+sensors in exocytosis and is phosphorylated as a function of stimulation. However, it is unknown what essential function, if any, rabphilin performs. One controversial question regards the respective roles of rab3s and rabphilin in localizing each other to synaptic vesicles: although rabphilin is mislocalized in rab3A knock-out mice, purified synaptic vesicles were shown to require rabphilin for binding of rab3A but not rab3A for binding of rabphilin. To test whether rabphilin is involved in localizing rab3s to synaptic vesicles and to explore the functions of rabphilin in regulating exocytosis, we have now analyzed knock-out mice for rabphilin. Mice that lack rabphilin are viable and fertile without obvious physiological impairments. In rabphilin-deficient mice, rab3A is targeted to synaptic vesicles normally, whereas in rab3A-deficient mice, rabphilin transport to synapses is impaired. These results show that rabphilin binds to vesicles via rab3s, consistent with an effector function of rabphilin for a synaptic rab3-signal. Surprisingly, however, no abnormalities in synaptic transmission or plasticity were observed in rabphilin-deficient mice; synaptic properties that are impaired in rab3A knock-out mice were unchanged in rabphilin knock-out mice. Our data thus demonstrate that rabphilin is endowed with the properties of a rab3 effector but is not essential for the regulatory functions of rab3 in synaptic transmission.
Keywords: long-term potentiation, synaptic vesicles, exocytosis, mossy fiber terminals, C2 domain, Ca2+-binding proteins, rab3, synaptotagmin, protein kinase A
Secretion of neurotransmitters by synaptic vesicle exocytosis initiates synaptic transmission. Exocytosis is triggered by Ca2+ in a rapid and restricted reaction: exocytosis is complete within 1 msec after Ca2+ influx, occurs only at the active zone, is triggered with low reliability, and when triggered, is limited to only one or two of multiple docked vesicles ready to fuse (Korn et al., 1994; Sabatini and Regehr, 1996; Goda and Südhof, 1997). In addition to its acute actions, Ca2+ also modulates synaptic exocytosis during synaptic plasticity, for example, when synapses are stimulated repetitively (Stevens and Wesseling, 1998; Wang and Kaczmarek, 1998). Thus Ca2+ has multiple synaptic actions that either promote or inhibit exocytosis. The balance between these actions is of great importance for shaping the properties of neural circuits.
The different, often opposing actions of Ca2+ at the synapse imply that there must be multiple targets for Ca2+. Several potential Ca2+-signaling proteins on synaptic vesicles have been identified (schematically diagrammed in Fig.1; for review, see Südhof, 1995). One of these proteins is rabphilin, which was discovered because of its interaction with the synaptic GTP-binding protein rab3A (Shirataki et al., 1993). Rabphilin is composed of an N-terminal rab3-interacting domain, a central phosphorylation domain, and two C-terminal C2 domains that may bind Ca2+ (Fykse et al., 1995). Six observations suggest that rabphilin performs a major regulatory function in exocytosis:
Fig. 1.
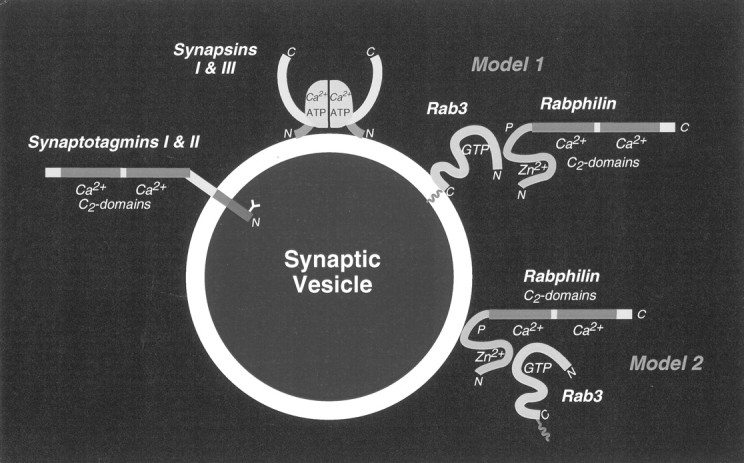
Model of Ca2+-binding proteins on synaptic vesicles: mode of rabphilin attachment to synaptic vesicles. A schematic view of the three known Ca2+-binding proteins of synaptic vesicles is shown: synaptotagmins I and II bind multiple Ca2+ ions via two C2 domains (Südhof and Rizo, 1996); synapsins I and III are directly regulated by Ca2+(Hosaka and Südhof, 1998a,b); and rabphilin, which also has two C2 domains that probably bind Ca2+. Two models for the role of the GTP-dependent binding of rabphilin to rab3s are presented: the hypothesis of Stahl et al. (1996) that rab3A is bound to vesicles and serves to recruit rabphilin (Model 1); or the hypothesis of Shirataki et al. (1994) that rabphilin is primarily bound to vesicles and is responsible for the vesicle-specific binding of rab3s (Model 2).
(1) Rabphilin binds in a GTP-dependent manner to rab3s, which in turn regulate exocytosis (for review, see Geppert and Südhof, 1998). At least two rab3s bind to rabphilin: the abundant rab3A and the less abundant rab3C (Li et al., 1994). In rab3A knock-out mice, synaptic exocytosis is enhanced, and long term potentation (LTP) at mossy fiber synapses in the hippocampal CA3 region is defective (Castillo et al., 1997; Geppert et al., 1997). Furthermore, rabphilin is mislocalized and partly degraded (Li et al., 1994). These findings suggest that rab3A may use rabphilin as an effector in regulating exocytosis.
(2) Rabphilin, rab3A, and rab3C are co-localized on synaptic vesicles and coordinately dissociate from the vesicles during or after exocytosis (Fischer von Mollard et al., 1994; Stahl et al., 1996). These results indicate that the association of rab3s and rabphilin with synaptic vesicles is regulated in conjunction with exocytosis.
(3) Rabphilin is phosphorylated by Ca2+-, calmodulin-dependent protein kinase II and by cAMP-dependent protein kinase (PKA) (Fykse et al., 1995; Lonart and Südhof, 1998). Phosphorylation is regulated in a region-specific manner; as a result, rabphilin phosphorylation is higher in mossy fiber terminals than in CA1 synaptic terminals. Because mossy fiber LTP is induced by PKA activation and requires rab3A (Castillo et al., 1997), the region-specific phosphorylation of rabphilin suggests that it may mediate the effects of PKA during induction of mossy fiber LTP.
(4) The two C2 domains of rabphilin are similar to those of synaptotagmin I and contain the residues that mediate Ca2+ binding in synaptotagmins (Südhof and Rizo, 1996). Recent nuclear magnetic resonance experiments confirmed that Ca2+ binds to rabphilin with a relatively high affinity (J. Ubach, T. C. Südhof, and J. Rizo, unpublished results). Thus rabphilin is a bona fide Ca2+-binding protein.
(5) In vitro binding studies have uncovered a number of potentially interesting interactions for rabphilin in addition to rab3 binding. Purported binding partners of rabphilin include phosphoinositides (Chung et al., 1998), rabaptin (Ohya et al., 1998) and α-actinin (Kato et al., 1996). The multiple interactions of rabphilin implicate rabphilin in regulating exocytosis, endocytosis, and the cytoskeleton.
(6) In agreement with the many binding properties of rabphilin, various overexpression and injection experiments of rabphilin resulted in multiple effects. The same N-terminal fragment of rabphilin inhibited exocytosis in cortical granule exocytosis in oocytes and in squid nerve terminals (Masumoto et al., 1996; Burns et al., 1998) but enhanced exocytosis in PC12 cells, chromaffin cells, and pancreatic β cells (Chung et al., 1995; Komuro et al., 1996; Arribas et al., 1997). Furthermore, rabphilin also inhibited endocytosis (Burns et al., 1998;Ohya et al., 1998).
Thus a large amount of information on rabphilin is available; however, there is no consensus on what rabphilin might actually do. The observations of opposite effects of rabphilin on secretion is puzzling, and it is difficult to imagine that a single protein mediates regulation of both exocytosis and endocytosis and of the cytoskeleton. A further controversial point is how rabphilin and rab3s are bound to synaptic vesicles for which two models have been presented (Fig. 1). Model 1 proposes that rab3s are attached directly to synaptic vesicles and secondarily recruit rabphilin as a putative effector. This suggests that hydrolysis of GTP bound to rab3s during exocytosis leads to the dissociation of rabphilin and then of rab3s (for review, seeSüdhof, 1997). Model 2 proposes that rabphilin is attached directly to the membrane, with rab3s being indirectly bound to rabphilin. According to this view, rabphilin functions in localizing rab3s to synaptic vesicles (Shirataki et al., 1994). Each of the two models is supported by independent evidence. The first model agrees well with the findings that rabphilin dissociates with rab3s during exocytosis and that rabphilin is degraded in rab3A knock-outs (Li et al., 1994; Stahl et al., 1996). The second model is based on the demonstration that rabphilin could be bound to synaptic vesicles after they had been biochemically stripped of rab3s (Shirataki et al., 1994).
In the present study, we have attempted to address these questions. Because biochemical approaches and overexpression methods led to conflicting results even in the same laboratory (e.g., Chung et al., 1995; Komuro et al., 1996; Masumoto et al., 1996; Burns et al., 1998), we thought that a different experimental strategy was required. For this purpose, we have generated and analyzed mice that lack rabphilin. We show that rabphilin is not essential for any investigated process, e.g., fertilization, endocrine regulation, or neurotransmission. Abnormalities in the regulation of synaptic transmission, which are typical for rab3A mutants, were absent from rabphilin mutants, and rab3A was normally targeted to synapses. Thus rabphilin is neither alone responsible for the regulatory effects of rab3s nor essential for rab3 targeting to synapses.
MATERIALS AND METHODS
Cloning of the murine rabphilin gene, construction of a targeting vector, and generation of mutant mice lacking rabphilin.Genomic clones containing four exons coding for residues 25–201 of rabphilin were isolated from a λFixII 129SV/J (Stratagene, La Jolla, CA) genomic library and characterized by restriction enzyme mapping and sequencing (Sambrook et al., 1989). Using the genomic DNA, we constructed a targeting vector, which deletes two exons encoding residues 74–146 of rabphilin (Fig. 2). First, an 11 kbXhoI–NotI fragment was cloned into theNotI and BamHI sites of pTK-Neo3A (Rosahl et al., 1995). Then the short arm was generated as a PCR fragment with the primers 185 (GCGATCGATTAGGGCAGGGTGGCACTTGGA) and 188 (GCGGTCGACTTTCCCACTAAGGAAGATGGC), cut with ClaI andSalI, and inserted over intermediate cloning steps into theClaI and a newly created unique XhoI site of pTK-Neo3A containing the long arm. The vector was linearized withNotI and electroporated into E14.1 mouse embryonic stem cells (Hooper et al., 1987) maintained on feeder layers (Genomesystems, St. Louis, MO). Cells were selected for homologous recombination with 1-[2-deoxy-2-fluoro-d-arabinofuranosyl]-5-iodouracil (FIAU) and G418 and passaged clonally (Thomas and Capecchi, 1987;Soriano et al., 1991). Clones were screened for homologous recombination by Southern blotting using HindIII-digested genomic DNA probed with a radiolabeled PCR probe (primers: 182, GCGGGATCCGGGAAAGGAGGGTAAAAG; 183, GCGGATCCTTAGTGGGAAATTGAAAAG) (outside probe is OP in Fig. 2). Two correctly targeted clones were expanded and microinjected into C57Bl6/J blastocysts (Biological Research Laboratories, Füllinsdorf, Switzerland). Chimeric males were crossed with C57Bl6/J female mice, and the heterozygous agouti offsprings were bred to homozygosity. Genotyping of mice was performed by PCR: Wild type allele = primer pairs 685 (GCCCAACTCCCGCTGGTTCCAGG) versus 671 (GGCTCTGCAAGATCTGCCTTGAGC) (1.3 kb product) or 1107 (GGGCTTCCATGACTTTGGTCCTGTGGC) versus 1108 (GACCTCCAACAACCGTCCGCATCCGG) (0.4 kb product). Mutant allele = 685 versus 428 (GAGCGCGCGCGGCGGAGTTGTTGAC) (1.3 kb product) or 1107 versus 428 (0.4 kb product). Mice were bred and maintained using standard mouse husbandry procedures.
Fig. 2.

Partial structure of the murine rabphilin gene and gene targeting strategy for the rabphilin knock-out. The restiction map on top indicates positions of enzyme sites (N, NheI; B,BamHI; H, HindIII;E, EcoRI; K,KpnI; X, XhoI;Xb, XbaI; C, ClaI;S, SalI). Exons are shown as black boxeswith the corresponding residue numbers. In the targeting vector on thebottom, the long arm (LA) and short arm (SA) for recombination are marked by hatched boxes, and the gene cassettes for positive selection (neo, neomycin resistance gene) and for negative selection (TK, thymidine kinase gene) are marked byopen boxes. Positions of oligonucleotide primers for PCR genotyping are indicated by arrows, and the location of the outside probe used for Southern blotting is indicated byOP. Note that homologous recombination replaces genomic sequences containing two exons encoding residues 74–146 with the neomycin gene.
Quantitative immunoblotting analysis. Mouse brains were removed after cervical dislocation and homogenized in 10 ml of ice-cold 0.32 m sucrose with a glass–Teflon homogenizer. Aliquots were boiled in SDS sample buffer (Sambrook et al., 1989) and stored at −80°C until further use. Proteins (10–80 μg; determined using the BCA protein assay; Pierce, Rockford, IL) were analyzed by SDS-PAGE and immunoblotting on Immobilon-P membranes (Millipore, Bedford, MA) using semidry transfer. Membranes were incubated with the primary antibodies as described (Towbin et al., 1979) with 5% dry milk instead of BSA as a blocking agent. Signals were detected with 125I-labeled secondary goat anti-rabbit antibodies and measured in a Fujix BAS5000 (Ray Test, Straubenhardt, Germany). Monoclonal antibodies were analyzed with a rabbit anti-mouse bridging antibody. Three groups of mice (wild type, rabphilin knock-outs, and rab3Aknock-outs), each with three adult animals, were analyzed on the same immunoblot. Signals were normalized to internal standards (Cl54.1 = NMDA receptor; or I370 = munc18-1) and plotted as percentage of wild type ± SD.
Subcellular fractionation. Cerebral cortex was dissected from five mice killed by cervical dislocation, homogenized in 30 ml of ice-cold 0.32 m sucrose, and subfractionated as described (Jones and Matus, 1974; Huttner et al., 1983). Determinations of protein concentrations and immunoblotting were performed as described above, except that HRP-labeled secondary antibodies were used with ECL detection (Amersham, Braunschweig, Germany).
Morphological analyses. Anesthetized adult mice (>6 weeks) were perfused with 4% paraformaldehyde in PBS. Brains and retina were cryoprotected and and cut into 5 μm sections on a cryostat as described (Mandell et al., 1990). The sections were incubated with the primary antibodies overnight at 4°C followed by Cy2- and Cy3-labeled secondary antibodies at room temperature for 2–4 hr (Mandell et al., 1990). Samples covered with Fluoromount G (Southern Biotechnology Association, Birmingham, AL) were viewed in a confocal microscope (MR1024; Bio-Rad, Hercules, CA).
Antibodies. The antibodies used were described previously: I372, I373, and I733 (McMahon and Südhof, 1995); L668 and I379 (McMahon et al., 1995); E765 (Robinson et al., 1993); D633 (Shisheva et al., 1994), I370 (Ullrich and Südhof, 1994), Cl54.1 (Brose et al., 1994), Q703 (Wang et al., 1997), Cl69.1 (Edelmann et al., 1995), V216 (Perin et al., 1990), Cl42.2 (Matteoli et al., 1991), 3H5 (Betz et al., 1998), P610, E028, and I735 (Rosahl et al., 1995); P932 (Betz et al., 1998); P180 (Castillo et al., 1997), I734, and I731 (Li et al., 1994); SA3613 (Telemenakis et al., 1997), SA3452, and SA3450 (Augustin et al., 1999); and α-p37 (Fykse et al., 1993) and T210 (Mignery et al., 1989). Commercially available antibodies: DOC2 and rabphilin (Transduction Laboratories, Lexington, KY); EA-53, NR4, DM1a, and Tub2.1 (Sigma, Deisenhofen, Germany); P942, P918, P936, P938, P939, P925, 580, and 583 (Synaptic Systems, Göttingen, Germany); GAD65/clone 11, Cy2-, and Cy3-conjugated goat anti-mouse (Biotrend, Köln, Germay); HRP-conjugated goat anti-mouse and anti-rabbit (BioRad, München, Germany); 125I-labeled goat-anti rabbit (Amersham, Braunschweig, Germay); rabbit anti-mouse (Jackson ImmunoResearch, West Grove, PA); and MAP-2 (Boehringer Mannheim, Mannheim, Germay). Unpublished: 4F9 (J. Song, K. Ichtchenko, T. C. Südhof, and N. Brose, personal communication); 4C12 (J. Song, K. Ichtchenko, T. C. Südhof, and N. Brose, personal communication); CSP (S. Tobaben, T. C. Südhof, and B. Stahl, personal commununication); Cl78.2 and Cl81.1 (R. Jahn, personal communication); Q698 (R. Janz and T. C. Südhof, unpublished data); and R321 (Y. Wang and T. C. Südhof, unpublished data).
Electrophysiological analysis. Standard procedures were used to prepare hippocampal slices (0.4–0.5 mm) from 4- to 9-week-old mice. For all experiments, the composition of the external solution was (in mm): 119 NaCl, 2.5 KCl, 1.2 MgSO4, 2.5 CaCl2, 1 NaHPO4, 26.2 NaHCO3, and 10 glucose (saturated with 95% O2 and 5% CO2 before superfusion of the slice at room temperature). For experiments on mossy fiber synapses, stimulation electrodes were placed in or adjacent to the dentate gyrus granule cell layer. Mossy fiber synaptic responses were recorded in the CA3 region using a pipette filled with the external solution and identified by the large facilitation that occurs when stimulation frequency is changed from 0.1 to 1 Hz (Salin et al., 1996) and by the depression of the synaptic response after application of L-CCG-1 (20 μm), a group 2 mGluR agonist that selectively blocks mossy fiber responses (Castillo et al., 1997). Experiments were included for data analysis only if L-CCG-1 caused a >80% reduction in the synaptic response. For all experiments, the baseline stimulation rate was 0.1 Hz, and the response that remained in the presence of L-CCG-1 was subtracted from the amplitudes of individual synaptic responses. Mossy fiber LTP was elicited using a 25 Hz, 5 sec tetanus in the presence of the NMDA receptor antagonist d-2 amino-5-phosphonovaleric acid (d-APV, 100 μm). For experiments that examined synaptic responses in the CA1 region, stimulation and recording electrodes were both placed in the stratum radiatum at approximately the same distance from the stratum pyramidale. All experiments were done in the presence of picrotoxin (100 μm) and d-APV (50 μm). LTP was elicited by three tetani of 100 Hz for 1 sec separated by 20 sec applied without d-APV after a 10 min stable baseline was obtained. Baseline stimulation rate was 0.1 Hz for all experiments. Data were filtered at 2 KHz, digitized at 10 KHz, and collected on-line using acquisition software developed in this laboratory by D. Selig. The amplitudes of mossy fiber synaptic responses were calculated by taking the mean of a 2–3 msec window around the peak and comparing this with the mean of a 5 msec window immediately before the stimulation artifact. For recordings in the CA1 region, initial slopes of the field EPSPs were measured. Mice were killed without knowledge of their identity (wild type vs knock-out), and data were acquired and analyzed blindly. Genotypes of individual mice were confirmed by PCR and immunoblot analysis using tail DNA and brain protein.
RESULTS
Generation of rabphilin knock-out mice by homologous recombination
After screening a murine genomic library with a probe from the 5′ end of the rabphilin cDNA, we isolated a genomic clone that contained four exons. The translated amino acid sequence of these four exons is 99% identical to residues 25–201 of rat rabphilin, indicating that the genomic clone includes part of the 5′ end of the murine rabphilin gene (Shirataki et al., 1993; Li et al., 1994). Using the genomic DNA, we constructed a targeting vector for homologous recombination in which two exons coding for residues 74–146 of rabphilin are replaced with a neomycin resistance cassette for positive selection with G418 (Fig.2). The neomycin resistance cassette is flanked by short and long arms of genomic rabphilin sequences for efficient recombination. In addition, adjacent to the short arm we placed two copies of the thymidine kinase gene for negative selection.
The linearized targeting vector was electroporated into E14.1 mouse embryonic stem cells (Hooper et al., 1987). Cells were selected with G418 and FIAU, and double-resistant clones were analyzed by Southern blotting and by PCR (Fig. 2). Clones with a homologously recombined rabphilin gene were injected into blastocysts. The resulting highly chimeric mice were used for breeding to generate heterozygous and homozygous mutant mice. Analysis of homozygous mutant mice showed that no rabphilin protein could be detected with antibodies to the N or C terminus of rabphilin (Fig. 3; data not shown). Thus the deletion we introduced into the rabphilin gene is a null mutant as expected. Nevertheless, mice lacking rabphilin were viable and fertile and exhibited no obvious phenotype. The mutant mice were capable of mating and caring for offspring and comparable in weight and longevity with their wild-type and heterozygous littermates.
Fig. 3.

PCR and immunoblotting analysis of wild-type and mutant mice. The top panels display a PCR analysis of DNA of the offspring from a heterozygous mating of rabphilin mutant mice. Brain proteins from the same animals were analyzed on thebottom panel with anti-rabphilin serum (Li et al., 1994). To show the absence of any additional bands in the mutant mice, a long exposure is shown. The genotype of the animals deduced from the analysis is indicated on top. KO, Knock-out; WT, wild-type.
Biochemical analysis of mutant mice lacking rabphilin
The viability and lack of an obvious phenotype of the rabphilin-deficient mice suggested that they are not subject to major developmental abnormalities or life-threatening impairments of brain function. However, potentially more subtle changes could occur in a knock-out [e.g., see the decreases in synaptic vesicle proteins in synapsin knock-out mice (Rosahl et al., 1995)]. Therefore, to test whether the composition of the brains is normal in the mutant mice, we performed a systematic quantitative analysis of the levels of 40 proteins in rabphilin- and rab3A-deficient mice (Table1). Rab3A knock-out mice were incorporated into this analysis to include several proteins that were not examined in the original description of the knock-out [e.g., RIM as the second rab3 effector or munc13 (Geppert et al., 1994)] and to directly compare rabphilin and rab3A knock-outs in the same experiment.
Table 1.
Quantification of protein levels in mutant and wild-type mice
| Protein | Rabphilin KO | RAB3A KO | Wild-type | Antibody |
|---|---|---|---|---|
| α-Actinin | 95 ± 13 | 86 ± 8 | 100 ± 8 | EA-53 |
| Complexin 1 | 93 ± 14 | 101 ± 11 | 100 ± 7 | P942 |
| 107 ± 11 | 104 ± 11 | 100 ± 18 | L668 | |
| Complexin 2 | 95 ± 12 | 100 ± 4 | 100 ± 7 | P942 |
| CSP | 95 ± 7 | 96 ± 5 | 100 ± 13 | αCSP |
| DOC2A | 97 ± 18 | 93 ± 16 | 100 ± 12 | αDOC2 |
| Dynamin | 117 ± 13 | 101 ± 5 | 100 ± 12 | E765 |
| GDI | 117 ± 16 | 114 ± 17 | 100 ± 5 | D633 |
| Mint | 97 ± 5 | 91 ± 6 | 100 ± 6 | P932 |
| Munc13-1 | 92 ± 3 | 98 ± 11 | 100 ± 4 | 3H5 |
| Munc13-1 (cortex) | 99 ± 6 | 105 ± 5 | 100 ± 7 | 3H5 |
| Munc13-2 | 98 ± 17 | 91 ± 11 | 100 ± 11 | SA3452 |
| Munc13-3 | 101 ± 9 | 83 ± 4 | 100 ± 4 | SA3450 |
| Munc18 | 96 ± 15 | 102 ± 11 | 100 ± 6 | I370 |
| Neurofilament-68 | 114 ± 17 | 96 ± 5 | 100 ± 2 | NR4 |
| Neuroligins I and III | 93 ± 9 | 91 ± 9 | 100 ± 7 | 4F9 |
| Neuroligin I | 100 ± 11 | 98 ± 10 | 100 ± 6 | 4C12 |
| NMDA-R1 | 99 ± 10 | 101 ± 12 | 100 ± 9 | C154.1 |
| NSF | 97 ± 9 | 94 ± 13 | 100 ± 6 | I372 |
| PSD95 | 93 ± 0 | 91 ± 3 | 100 ± 10 | P918 |
| Rab3A | 107 ± 13 | 4 ± 1 | 100 ± 11 | C142.2 |
| 102 ± 3 | 10 ± 3 | 100 ± 3 | C142.1 | |
| Rab3C | 108 ± 21 | 112 ± 12 | 100 ± 16 | P180 |
| Rabaptin | 107 ± 9 | 113 ± 6 | 100 ± 2 | R321 |
| Rabphilin | 4 ± 0 | 52 ± 6 | 100 ± 12 | I734 |
| 8 ± 1 | 51 ± 2 | 100 ± 7 | I731 | |
| RIM | 98 ± 7 | 91 ± 6 | 100 ± 8 | Q703 |
| SCAMP | 87 ± 10 | 97 ± 17 | 100 ± 16 | P936 |
| mSEC7 | 99 ± 7 | 98 ± 6 | 100 ± 9 | SA3613 |
| αSNAP | 104 ± 7 | 103 ± 4 | 100 ± 13 | I373 |
| SNAP-25 | 102 ± 15 | 106 ± 13 | 100 ± 10 | I733 |
| Synapsin Ia | 109 ± 14 | 99 ± 7 | 100 ± 12 | E028 |
| 100 ± 9 | 99 ± 8 | 100 ± 12 | P610 | |
| Synapsin Ib | 110 ± 16 | 98 ± 6 | 100 ± 10 | E028 |
| 110 ± 11 | 102 ± 8 | 100 ± 8 | P610 | |
| Synapsin IIa, b | 108 ± 17 | 97 ± 2 | 100 ± 6 | E028 |
| Synaptobrevin 1 | 118 ± 29 | 110 ± 20 | 100 ± 13 | P938 |
| Synaptobrevin 2 | 101 ± 11 | 104 ± 16 | 100 ± 8 | C169.1 |
| 97 ± 17 | 101 ± 16 | 100 ± 13 | P939 | |
| Synaptogyrin | 94 ± 15 | 108 ± 26 | 100 ± 13 | P925 |
| Synaptophysin I | 106 ± 12 | 105 ± 17 | 100 ± 9 | 580 |
| 99 ± 17 | 102 ± 21 | 100 ± 17 | P611 | |
| Synaptophysin II | 114 ± 4 | 100 ± 11 | 100 ± 7 | αp37 |
| Synaptotagmin I | 111 ± 9 | 100 ± 5 | 100 ± 9 | V216 |
| Synaptotagmin II | 102 ± 4 | 105 ± 10 | 100 ± 13 | I735 |
| α, β-Synuclein | 109 ± 10 | 106 ± 13 | 100 ± 6 | Q698 |
| Syntaxin | 109 ± 28 | 103 ± 16 | 100 ± 11 | I379 |
| 100 ± 2 | 96 ± 4 | 100 ± 8 | C178.2 | |
| α-Tubulin | 119 ± 18 | 106 ± 11 | 100 ± 14 | DM1a |
| β-Tubulin | 93 ± 7 | 98 ± 2 | 100 ± 8 | Tub2.1 |
Brain proteins from rabphilin knock-out (KO) mice, rab3A knock-out mice, and wild-type mice were analyzed by quantitative immunoblotting (see Materials and Methods) with125I-labeled secondary antibodies and phosphoimager detection. Three mice of each genotype were quantified on the same immunoblot. Signals were normalized to internal standards (NMDA receptor and munc18-1) and are expressed as percent of wild type ± SD. Statistically significant differences to wild-type levels are shown in bold (Student’s t test, p < 0.001). In addition to whole brain, Munc13-1 was separately quantified in cortex as indicated.
We quantified the relative levels of the various proteins by immunoblotting with 125I-labeled secondary antibodies and internal standards, analyzing the samples to be compared on the same gel. The NMDA receptor subunit 1 (antibody Cl54.1) and munc18-1 (antibody I370) were shown to be unchanged in numerous individual blots and served as internal standards, which were used to normalize the signals for the other proteins. Three mice of the same age were analyzed for each genotype.
As shown in Table 1, we detected no significant changes in the levels of any protein in the rabphilin knock-out mice. In particular, the levels of rab3A and rab3C were unchanged. GDP dissociation inhibitor (GDI, which is involved in the cycle of rab3s and rabphilin between vesicle-attached and dissociated forms) and RIM (the second brain rab3 effector) were similar in levels between wild-type and knock-out mice. Other proteins that have been proposed on the basis of in vitro interactions to be functionally related to rabphilin were also unaltered [e.g., rabaptin (Ohya et al., 1998) and α-actinin (Kato et al., 1996)]. Thus in the rabphilin knock-outs, there are no overall changes in the composition of the brain, nor are there selective alterations in the levels of selected proteins that functionally interact with rab3s. In rab3A knock-out mice, the overall composition of the brain also showed little change, although decreases in selected proteins were observed. As reported previously, the rabphilin level was significantly lower in therab3A mutant mice than in wild-type mice (Geppert et al., 1994). The level of the other putative rab3A effector, RIM, was unchanged, indicating that the presence of rab3A is essential for the maintenance of rabphilin but not for RIM. In addition to rabphilin, we observed changes in the level of Munc13-3, but the significance of this finding is unclear because the levels of the other Munc13 isoforms were unchanged (Table 1).
Normal synaptic architecture in brains lacking rabphilin
To investigate whether the rabphilin knock-out mice suffer from changes in the structure of the brain, we performed immunocytochemical analyses of wild type and rabphilin-deficient brains. As shown exemplarily in Figure 4 for the localizations of MAP-2, IP3 receptors, and synaptoporin/physin II (p37) in the cerebellum and hippocampus of rabphilin knock-out mice, the overall structure of the brain in mutant mice appeared normal. No significant differences between wild-type and knock-out brains were detected (Fig. 4; data not shown). The layered structures of the cerebellum and the hippocampus were preserved, as was the dendritic arborization of cerebellar Purkinje cells (visualized with an antibody to the IP3 receptor; Mignery et al., 1989). Similarly, the distributions of the cytoskeletal protein MAP2 (generally expressed in most dendrites) and the synaptic vesicle protein synaptoporin (p37; particularly enriched in mossy fiber terminals of the hippocampus) were unchanged.
Fig. 4.
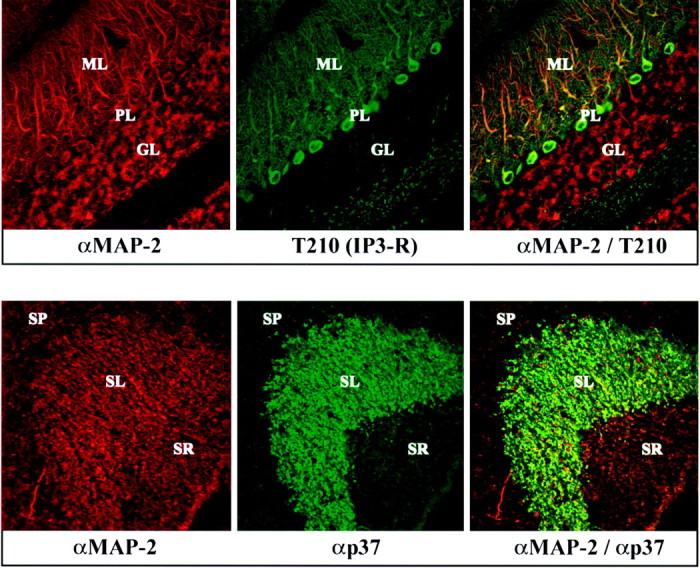
Confocal immunofluorescence analysis of brain sections from rabphilin knock-out mice. Frozen sections (5 μm) from the cerebellum (top panel) and hippocampus (bottom panel) of adult homozygous mutant mice were double-labeled with a monoclonal antibody to MAP-2 (MAP-2; red) and a polyclonal antibody to the IP3 receptor (T210; greenin top panel) or synaptophysin II (p37; green in bottom panel). Points of colocalization of the signals are shown in the right panels in yellow. The granule cell layer (GL), the Purkinje cell layer (PL), and the molecular layer (ML) of the cerebellum and the stratum radiatum (SR), the stratum lucidum (SL), and the stratum pyramidale (SP) of the hippocampal CA3 region are marked.
Rabphilin deficiency does not affect rab3A localization
The protein quantification (Table 1) showed that rab3A levels were normal in rabphilin knock-outs, whereas rabphilin was decreased in rab3A knock-outs. This suggests that the stability of rabphilin depends on rab3A but not vice versa. However, this result does not necessarily imply that the localization of rab3s is normal in the absence of rabphilin. To address this question, we analyzed the relative distributions of rabphilin and rab3s in wild-type and knock-out mice.
We first studied the cerebellum and hippocampus (Fig.5). In the cerebellum of wild-type mice, rabphilin appeared to be co-localized in all synapses of the molecular layer but only in a subset of synapses in the granule cell layer, suggesting that not all synapses contain rabphilin (Fig. 5, top panels). In the knock-out mice, no significant rabphilin signal was detected, but the distribution of rab3A was unchanged. A similar picture was observed in the hippocampus where mossy fiber terminals in the CA3 region were strongly positive for both rabphilin and rab3A but the distribution of rab3A was not changed in the rabphilin knock-out (Fig. 5, bottom panels). These findings indicate that rab3A does not depend in its synaptic localization on rabphilin, a conclusion that was confirmed in studies on the retina, which contains two well demarcated synaptic layers, the inner and outer plexiform layers (Fig.6). The thinner outer plexiform layer is composed of a single type of synapse, the ribbon synapses of the photoreceptor cells, whereas the thicker inner plexiform layer contains multiple synaptic zones. Analysis of wild-type synapses revealed that rabphilin was absent from outer plexiform layers, although rab3A was abundantly present (Fig. 6). This observation supports the conclusion reached above in the analysis of rabphilin in cerebellar sections that not all synapses that contain rab3s contain rabphilin. In itself, this finding implies that rabphilin cannot be essential for localizing rab3s to synapses, a conclusion that was confirmed in the analysis of retina sections from rabphilin and rab3A knock-out mice. In the rabphilin knock-outs, no change in the distribution of rab3A and its synaptic localization was observed. Conversely, rabphilin immunostaining was greatly decreased in the rab3A knock-out as expected (Li et al., 1994). Thus rab3A does not require rabphilin for its normal distribution in brain.
Fig. 5.
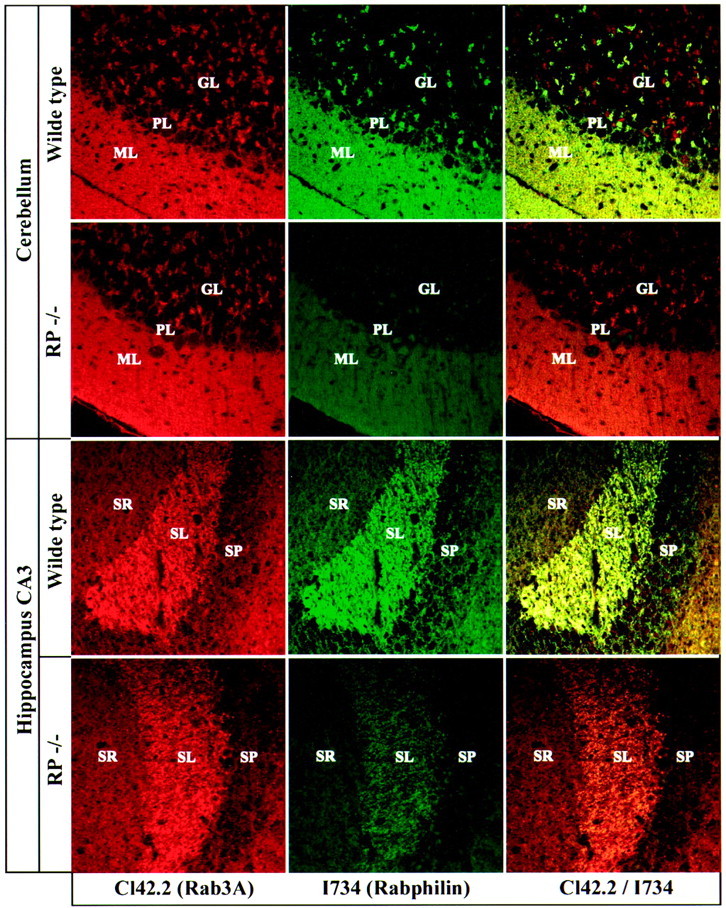
Comparative immunofluorescence analysis of rab3A and rabphilin in frozen sections from wild-type and rabphilin knock-out mice. Frozen sections (5 μm) were double-labeled with a monoclonal antibody against rab3A (Cl42.2; redsignal) and a polyclonal antibody against rabphilin (I734; green signal). Sections were viewed with a confocal microscope; signal colocalization is shown inyellow. GL, Granule cell layer;PL, Purkinje cell layer; ML, molecular layer; SR, stratum radiatum; SL, stratum lucidum; SP, stratum pyramidale.
Fig. 6.

Immunofluorescence analysis of retina sections from wild-type and rabphilin knock-out mice. Frozen sections (5 μm) from retina were double-labeled with a monoclonal antibody to rab3A (Cl42.2; red) and a polyclonal antibody to rabphilin (I734; green). Colocalization of the two proteins is shown in yellow. The inner plexiform layer (IPL) and outer plexiform layer (OPL) are identified.
Subcellular fractionation of rabphilin-deficient brain shows that rab3A is localized to synaptic vesicles
In presynaptic nerve terminals, rab3A and rabphilin are largely bound to free synaptic vesicles from which they dissociate during or after exocytosis (Stahl et al., 1996). This results in a cycle of rab3A and rabphilin between a GTP-bound synaptic vesicular state (∼70% at steady state) and a GDP-bound cytosolic state (∼30% at steady state) (for review, see Südhof 1995, 1997; Geppert and Südhof 1998). In addition to other functions, it has been proposed that rabphilin recruits rab3A to synaptic vesicles and then acts as a GTPase inhibitor (Shirataki et al., 1993, 1994). This proposal implies that even if rabphilin is not essential for rab3A localization to synaptic vesicles, it may still change the cycling of rab3A between soluble and membrane-bound forms. To test whether rab3A is still localized to synaptic vesicles in rabphilin-deficient neurons and whether the relative distribution of rab3A between the vesicles and the cytosol changes in the absence of rabphilin (as a measure of rab3A cycling), we analyzed rabphilin-deficient and wild-type brains by subcellular fractionation (Fig. 7).
Fig. 7.

Subcellular fractionation of wild-type and rabphilin knock-out brains. Brain cortex from rabphilin-deficient (−/−) or wild-type brains (+/+) were homogenized (H) and fractionated into nuclear pellets (P1), crude synaptosomes (P2), and a high-speed pellet (P3). The synaptosomes were then used to derive crude synaptic plasma membranes (LP1), crude synaptic vesicles (LP2), cytosol (S3), synaptosomal cytosol (LS2), and synaptic plasma membranes (SPM). The fractions were analyzed by SDS-PAGE and immunoblotting with the indicated antibodies:Cl54.1, NMDA-receptor 1; Cl81.1, GDI; αp37, synaptophysin II; Cl42.2, rab3A;Cl69.1, synaptobrevin 2.
Various subcellular fractions from rabphilin-deficient and wild-type mice were analyzed by immunoblotting. NMDA receptors (NR1) were found to be enriched in synaptic plasma membranes (SPMs) and in the heterogeneous P3 fraction, which contains vesicles derived from the endoplasmic reticulum, Golgi apparatus, lysosomes, and endosomes. This is an expected localization, because the NMDA receptor is thought to be present in intracellular vesicles in addition to postsynaptic densities attached to synaptic plasma membranes (Siegel et al., 1994). The synaptic vesicle proteins synaptoporin (αp37) and synaptobrevin (Syb2) were primarily found in the synaptic vesicle fraction (LP2) but also present in SPMs, presumably because docked vesicles are firmly attached to the active zone of the plasma membrane. As expected, rab GDI is a soluble protein of the synapse (LS2) and other cytosolic compartments (S3). Rab3A, however, was found in both the membrane-attached pattern of a synaptic vesicle protein (LP2 and SPM fractions) and as a soluble protein in the cytosolic fraction (LS2). This distribution resembles previous data showing that at steady state, ∼30% of rab3A is not membrane-associated but probably in a GDI-bound form in the cytosol (Fischer von Mollard et al., 1989; Südhof, 1997). Most importantly, however, there was no major change in the distribution of rab3A between the various fractions. In particular the relative abundance of rab3A in synaptic vesicles, cytosol, and synaptic plasma membranes was unchanged, confirming the morphological results that rab3A is not dependent on rabphilin for its localization.
Normal synaptic function in rabphilin knock-out mice
The absence of major changes in brain structure or composition in the rabphilin knock-out mice does not exclude the possibility that synaptic transmission is impaired in these mice. Rab3A knock-out mice, for example, display a similar lack of change in the structure and composition of the brain despite major changes in synaptic physiology (Li et al., 1994; Castillo et al., 1997; Geppert et al., 1997). To examine whether the deletion of rabphilin changes synaptic transmission, we used field potential recordings to study rabphilin knock-out mice physiologically.
We first examined mossy fiber synapses because they exhibited the most profound phenotype in rab3A knock-outs, namely a loss of LTP (Castillo et al., 1997). In addition to LTP, we studied two forms of short-term synaptic plasticity at mossy fiber synapses, paired pulse facilitation (PPF) (Zucker, 1989) and frequency facilitation (Regehr et al., 1994;Salin et al., 1996). All three forms of synaptic plasticity were found to be unchanged (Fig. 8). Paired pulse facilitation was tested at two interstimulus intervals (20 and 40 msec) with no differences in knock-out versus wild-type mice (Fig.8A1, A2) (knock-out PPF ratio: 20/40 msec interval, 2.3 + 0.3/2.2 + 0.2; n = 8 slices/4 animals; wild-type PPF ratio: 20/40 msec interval, 2.4 + 0.3/1.8 + 0.3; n = 9 slices/4 animals). Frequency facilitation was elicited by increasing the stimulation rate from 0.1 to 0.2 or 0.33 Hz; again, no alterations were found in the rabphilin knock-out mice (Fig.8B1,B2) (knock-out frequency facilitation: 0.33/0.2 Hz, 285 + 30/147 + 13% of baseline;n = 7 slices/ 4 animals; wild-type frequency facilitation: 257 + 11/163 + 14%; n = 8 slices/4 animals). Finally, in contrast to rab3A knock-out mice (Castillo et al., 1997), mossy fiber LTP was also normally induced in the rabphilin knock-out mice. As shown in Figure 8C, a 5 sec 25 Hz tetanus caused the same large initial potentiation and stable LTP in both wild-type and rabphilin knock-out mice (wild type: 189 + 20% of baseline measured 35–40 min after the tetanus; n = 6 slices/4 animals; knock-out: 184 + 16%, n = 8 slices/5 animals).
Fig. 8.

Short-term synaptic plasticity and LTP are normal at mossy fiber synapses in the hippocampal CA3 region in rabphilin knock-out mice. A1, A2, Magnitude of paired pulse facilitation at 20 msec (A1) and 40 msec (A2) interstimulus intervals in wild type (WT) and rabphilin knock-out mice (KO).B1, B2, Frequency facilitation induced by increasing the stimulation rate from 0.1 to 0.33 Hz (B1) or 0.2 Hz (B2) in wild-type and rabphilin knock-out mice.C, Induction of LTP by a 25 Hz, 5 sec tetanus (arrow) in wild-type and rabphilin knock-out mice.
Failing to find a significant abnormality at mossy fiber synapses, we examined synaptic transmission at excitatory synapses on CA1 pyramidal cells. At these synapses, an increase in synaptic depression during repetitive stimulation (14 Hz) was observed in rab3A knock-out mice (Geppert et al., 1994, 1997). Paired pulse facilitation was tested at intervals ranging from 20 to 250 msec and was not significantly altered in the rabphilin knock-out mice (Fig.9A) (wild-type,n = 16 slices/4 animals; knock-out, n = 14 slices/4 animals). Post-tetanic potentiation (PTP) was assessed by applying a 100 Hz, 1 sec tetanus (in the presence of 50 μmd-APV); again there was no difference between wild-type and rabphilin knock-out slices in the magnitude or time course of PTP (Fig. 9B) (peak enhancement in wild-type: 181.5 + 5.5% of baseline; n = 8 slices/4 animals; peak enhancement in knock-out: 177.1 + 5.0%, n = 12 slices/4 animals). We also examined NMDA receptor-dependent LTP; as shown in Figure 9C, LTP was essentially identical in the two populations of slices (wild-type: 148.9 + 8.4% of baseline measured 55–60 min after induction; n = 12 slice/4 animals; knock-out: 154.5 + 8.5%; n = 12 slice/4 animals). Finally, we measured synaptic depression during repetitive stimulation in the CA1 region. Again, the magnitude and time course of the depression caused by applying 100 stimuli at 14 Hz was not affected by the deletion of rabphilin (wild-type: 76.5 + 4.2% of baseline at end of train; n = 12 slices/4 animals; knock-out: 86.0 + 8.0%; n = 8 slices/4 animals) (Fig.10).
Fig. 9.
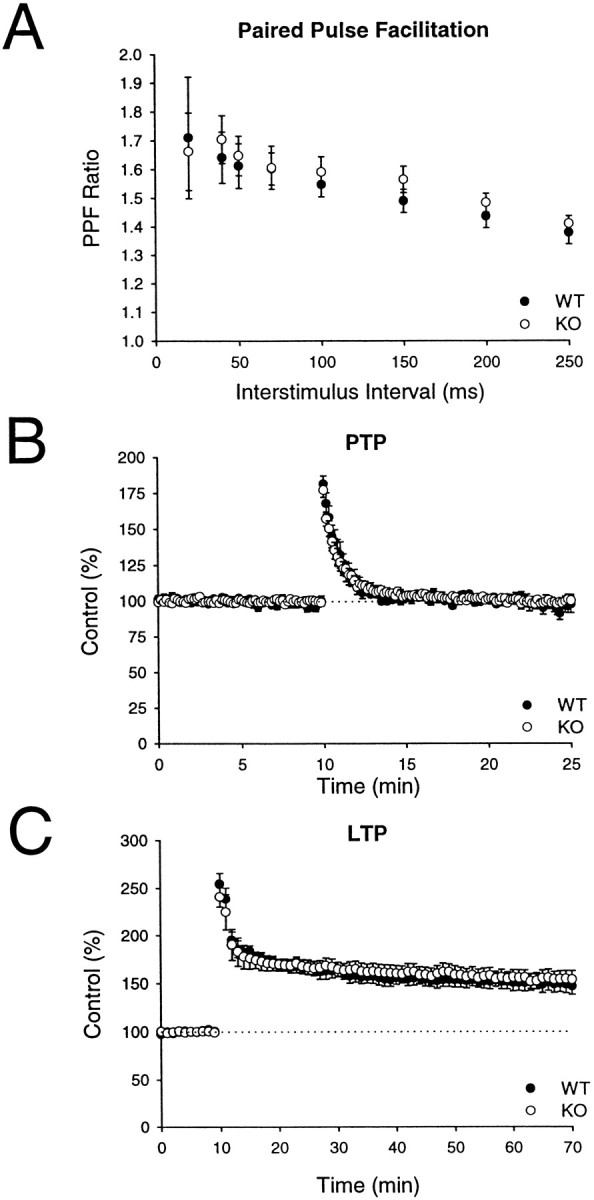
Short-term synaptic plasticity and LTP are normal at excitatory synapses in the hippocampal CA1 region in rabphilin knock-out mice. A, Paired pulse facilitation as a function of interstimulus interval in wild-type and rabphilin knock-out mice. B, Time course of posttetanic potentiation elicited by a 100 Hz, 1 sec tetanus in wild-type and rabphilin knock-out mice. C, Induction of LTP by three 100 Hz, 1 sec tetani in wild-type and rabphilin knock-out mice.
Fig. 10.
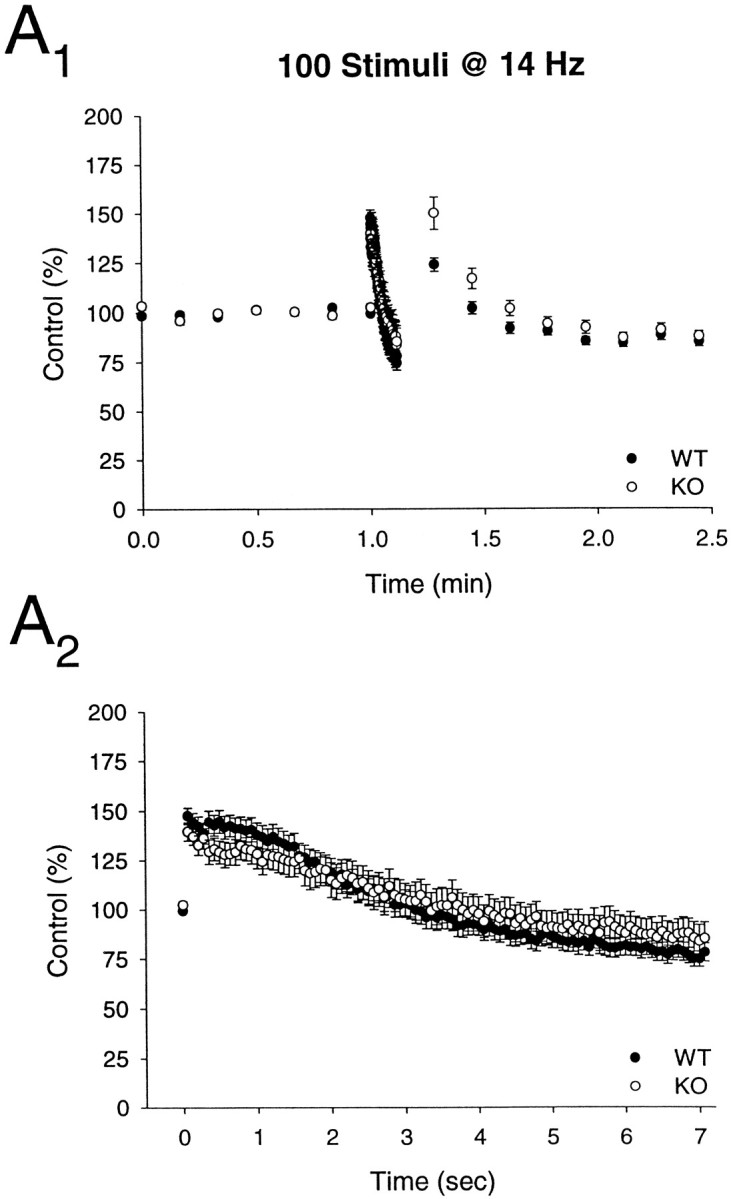
Synaptic depression is normal at excitatory synapses in the CA1 region of rabphilin knock-out mice. Time course of the effects of a stimulus train (100 stimuli at 14 Hz) is shown on a long (A1) and short (A2) time scale in wild type (WT) and rabphilin knock-out (KO) mice.
DISCUSSION
In the present study, we posed three questions about rabphilin that were raised by the previous biochemical characterization of rabphilin and the results of the rab3A knock-out analysis. These three questions are as follows. (1) Is rabphilin localized to synaptic vesicles via rab3s, or are rab3s localized to synaptic vesicles via rabphilin? In the first case, rabphilin would not be an effector protein but would mediate rab3 recruiting to synaptic vesicles. In the second case, one of the functions of rab3s would be to recruit rabphilin to synaptic vesicles. (2) Are the physiological functions of rab3s mediated by rabphilin as an effector? If rabphilin mediated some or all of the synaptic actions or rab3s, a molecular understanding of mossy fiber LTP would have to focus on rabphilin. (3) Is rabphilin a physiologically important Ca2+ sensor that mediates the actions of Ca2+ in regulating release? This is an important question independent of rab3 interactions in understanding the multiple regulatory actions of Ca2+ at a synapse.
Do rab3s recruit rabphilin, or does rabphilin recruit rab3s to synaptic vesicles?
Our data show that in the absence of rabphilin, rab3A is stable, is not decreased in levels, and is fully targeted to synapses. Previous studies had shown that in neurons that have no detectable levels of rab3A and other rab3 isoforms, rabphilin is not localized to synapses and becomes degraded (Li et al., 1994). Thus rabphilin needs rab3s for stability and synaptic localization, but rab3s do not require rabphilin. This result proves that in mice, rabphilin is recruited to synapses via rab3s and destabilized in the absence of rab3s (Fig. 1,Model 1).
Are the physiological functions of rab3s mediated by rabphilin as an effector?
The electrophysiological analysis shows that deleting rabphilin does not result in the same synaptic changes as deleting rab3A. In particular, the increased synaptic depression during repetitive stimulation that is characteristic of rab3A knock-out mice (Geppert et al., 1994) and is a consequence of the increased release in these mice (Geppert et al., 1997) was not observed in the rabphilin knock-out mice. Furthermore, mossy fiber LTP could be normally elicited in the rabphilin knock-out mice but not the rab3A knock-out mice. These results show surprisingly that none of the characterized impairments of rab3A-knock-out mice are due to the reduction of rabphilin in these mice. It is somewhat ironic that the knock-out of rabphilin, which has no currently known isoforms, should give no phenotype, whereas the knock-out of rab3A with multiple isoforms has severe consequences for synaptic function. However, it is still possible that rabphilin serves as an essential Ca2+ sensor in a subtle synaptic process, which was not tested in the current study, for example, the slow component of release.
Is rabphilin a major synaptic Ca2+ sensor?
We analyzed synaptic function in the rabphilin knock-out mice by a number of electrophysiological procedures. All parameters studied exhibited normal values. Specifically, we found no changes in various forms of synaptic plasticity, in synaptic depression elicited by high-frequency stimulation, and in any form of LTP. These results make it unlikely that rabphilin functions as a major synaptic Ca2+ sensor in exocytosis. In addition, our results suggest that the many binding reactions for rabphilin, most of which were characterized by only a single method, are unlikely to have a major physiological role.
The possibility of redundancy
A frequently evoked explanation for a lack of a phenotype in a knock-out experiment is that the deleted gene is functionally redundant. Three types of redundancy could be proposed. First, it is possible that multiple rabphilins are expressed in brain. Many synaptic proteins in mammals are expressed in multiple isoforms generated from distinct genes (e.g., synaptotagmins, rab3s, and synapsins). If there were multiple isoforms of rabphilin, and we deleted expression of only a single isoform, then other isoforms could compensate for the loss of rabphilin. However, no isoform of rabphilin has been discovered in brain by multiple approaches, e.g., expressed sequence tag data bank searches or low-stringency hybridization. This argues against the presence of rabphilin isoforms as a source of redundancy but does not exclude this possibility. Second, RIM as the other rab3 effector in brain could potentially substitute for rabphilin function. This hypothesis is unlikely, because RIM and rabphilin exhibit distinct localizations and properties. RIM is on the plasma membrane, whereas rabphilin is on vesicles. The C2 domains of RIM lack the residues involved in Ca2+ binding, whereas rabphilin C2 domains have these residues, suggesting major differences in regulation. Furthermore, only rabphilin but not RIM is unstable and degraded in the rab3A knock-out and is dependent in its synaptic targeting on rab3s. Howeever, other, as yet unidentified structurally unrelated rab3 effectors could function redundantly with rabphilin. Third, changes in other proteins might compensate for the loss of rabphilin. To address this, we analyzed the levels of 40 synaptic proteins in the knock-outs and observed no changes, making compensatory changes at least in a major synaptic protein unlikely.
The function of rabphilin
Taken together, our data argue against the notion that rabphilin has an essential function in basic release mechanisms. However, the lack of a striking phenotype in the rabphilin knock-out mice does not necessarily mean that rabphilin has no function. First, it is conceivable that rabphilin performs functions via its C2domains that are independent of rab3 binding. Because rabphilin shares its C2 domains with the Doc2 proteins that do not bind to rab3s (Sakaguchi et al., 1995; Verhage et al., 1997), it is possible that rabphilin performs a rab3-independent function via its C2 domains that is redundant with Doc2 function. Second, it is plausible that rabphilin may have no fundamental role in the execution or regulation of neurotransmitter release under normal conditions but could play a specialized role under as yet unidentified, possibly more extreme physiological conditions. This notion is supported by the finding that rabphilin is highly conserved in the nematode Caenorhabditis elegans, whereas other vesicle proteins (e.g., SV2 and synaptophysin) are not. Identification of these specialized conditions would aid considerably toward elucidating the biological significance of rabphilin.
Footnotes
This work was supported by the Sonderforschungsbereich and the Human Frontiers in Science Program. We thank Dr. Hermann Riedesel (Goettingen) for the support in breeding and analyzing knock-out mice, Drs. R. Jahn, B. Stahl, E. Link, and N. Brose for gifts of antibodies, and Drs. M. S. Brown, and J. L. Goldstein for invaluable discussions.
Correspondence should be addressed to Tom Südhof, Room Y5.322, University of Texas Southwestern Medical Center, 5323 Harry Hines Boulevard, Dallas, TX 75235.
Dr. Verhage’s present address: Department of Medical Pharmacology, Utrecht University, 3584 CG Utrecht, The Netherlands.
Dr. Geppert’s present address, 42 Abbotswood Gardens, Clayhall, Ilford, Essex IG5 0BG, UK.
REFERENCES
- 1.Arribas M, Regazzi R, Garcia E, Wollheim CB, De Camilli P. The stimulatory effect of rabphilin 3a on regulated exocytosis from insulin-secreting cells does not require an association-dissociation cycle with membranes mediated by rab3. Eur J Cell Biol. 1997;74:209–216. [PubMed] [Google Scholar]
- 2.Augustin I, Betz A, Herrmann C, Jo T, Brose N. Differential expression of two novel munc13 proteins in Rat Brain. Biochem J. 1999;337:363–371. [PMC free article] [PubMed] [Google Scholar]
- 3.Betz A, Ashery U, Rickmann M, Augustin I, Neher E, Südhof TC, Rettig J, Brose N. Munc13–1 is a presynaptic phorbol ester receptor that enhances neurotransmitter release. Neuron. 1998;21:123–136. doi: 10.1016/s0896-6273(00)80520-6. [DOI] [PubMed] [Google Scholar]
- 4.Brose N, Huntley GW, Stern-Bach Y, Sharma G, Morrison JH, Heinemann S. Differential assembly of coexpressed glutamate receptor subunits in neurons of rat cerebral cortex. J Biol Chem. 1994;269:16780–16784. [PubMed] [Google Scholar]
- 5.Burns ME, Sasaki T, Takai Y, Augustine GJ. Rabphilin-3A: a multifunctional regulator of synaptic vesicle traffic. J Gen Physiol. 1998;111:243–255. doi: 10.1085/jgp.111.2.243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Castillo PE, Janz R, Südhof TC, Tzonopoulos T, Malenka RC, Nicoll RA. Rab3A is essential for mossy fibre long-term potentiation in the hippocampus. Nature. 1997;388:590–593. doi: 10.1038/41574. [DOI] [PubMed] [Google Scholar]
- 7.Chung SH, Takai Y, Holz RW. Evidence that the rab3a-binding protein, rabphilin3a, enhances regulated secretion. J Biol Chem. 1995;270:16714–16718. doi: 10.1074/jbc.270.28.16714. [DOI] [PubMed] [Google Scholar]
- 8.Chung SH, Song WJ, Kim K, Bednarski JJ, Chen J, Prestwich GD, Holz RW. The C2 domains of rabphilin3A specifically bind phosphatidylinositol 4,5-bisphosphate containing vesicles in a Ca2+ dependent manner. J Biol Chem. 1998;273:10240–10248. doi: 10.1074/jbc.273.17.10240. [DOI] [PubMed] [Google Scholar]
- 9.Edelmann L, Hanson PI, Chapman ER, Jahn R. Synaptobrevin binding to synaptophysin: a potential mechanism for controlling the exocytotic fusion machine. EMBO J. 1995;14:224–231. doi: 10.1002/j.1460-2075.1995.tb06995.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fischer von Mollard G, Mignery GA, Baumert M, Perin MS, Hanson TJ, Burger PM, Jahn R, Südhof TC. Rab3 is a small GTP-binding protein exclusively localized to synaptic vesicles. Proc Natl Acad Sci USA. 1989;87:1988–1992. doi: 10.1073/pnas.87.5.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Fischer von Mollard G, Stahl B, Khokhlatchev A, Südhof TC, Jahn R. Rab3C is a synaptic vesicle protein that dissociates from synaptic vesicles after stimulation of exocytosis. J Biol Chem. 1994;269:10971–10974. [PubMed] [Google Scholar]
- 12.Fykse EM, Takei K, Walch-Solimena C, Geppert M, Jahn R, De Camilli P, Südhof TC. Relative properties and localizations of synaptic vesicle protein isoforms: the case of the synaptophysins. J Neurosci. 1993;13:4997–5007. doi: 10.1523/JNEUROSCI.13-11-04997.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fykse EM, Li C, Südhof TC. Phosphorylation of rabphilin-3A by Ca2+/calmodulin- and cAMP-dependent protein kinases in vitro. J Neurosci. 1995;15:2385–2395. doi: 10.1523/JNEUROSCI.15-03-02385.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Geppert M, Südhof TC. Rab3 and synaptotagmin: the yin and yang of synaptic membrane fusion. Annu Rev Neurosci. 1998;21:75–95. doi: 10.1146/annurev.neuro.21.1.75. [DOI] [PubMed] [Google Scholar]
- 15.Geppert M, Bolshakov VY, Siegelbaum SA, Takei K, De Camilli P, Hammer RE, Südhof TC. The role of Rab3A in neurotransmitter release. Nature. 1994;369:493–497. doi: 10.1038/369493a0. [DOI] [PubMed] [Google Scholar]
- 16.Geppert M, Goda Y, Stevens CF, Südhof TC. The small GTP-binding protein Rab3A regulates a late step in synaptic vesicle fusion. Nature. 1997;387:810–814. doi: 10.1038/42954. [DOI] [PubMed] [Google Scholar]
- 17.Goda Y, Südhof TC. Calcium regulation of neurotransmitter release: reliably unreliable? Curr Opin Cell Biol. 1997;9:513–518. doi: 10.1016/s0955-0674(97)80027-0. [DOI] [PubMed] [Google Scholar]
- 18.Hooper M, Hardy K, Handyside A, Hunter S, Monk M. HPRT-deficient (Lesch-Nyhan) mouse embryos derived from germline colonization by cultured cells. Nature. 1987;326:292–295. doi: 10.1038/326292a0. [DOI] [PubMed] [Google Scholar]
- 19.Hosaka M, Sudhof TC. Synapsins I and II are ATP-binding proteins with differential Ca2+ regulation. J Biol Chem. 1998a;273:1425–1429. doi: 10.1074/jbc.273.3.1425. [DOI] [PubMed] [Google Scholar]
- 20.Hosaka M, Sudhof TC. Synapsin III, a novel synapsin with an unusual regulation by Ca2+. J Biol Chem. 1998b;273:13371–13374. doi: 10.1074/jbc.273.22.13371. [DOI] [PubMed] [Google Scholar]
- 21.Huttner WB, Schiebler W, Greengard P, De Camilli P. Synapsin I (protein I), a nerve terminal-specific phosphoprotein. III. Its association with synaptic vesicles studied in a highly purified synaptic vesicle preparation. J Cell Biol. 1983;96:1374–1388. doi: 10.1083/jcb.96.5.1374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jones DH, Matus AI. Isolation of synaptic plasma membrane from brain by combined flotation-sedimentation density gradient centrifugation. Biochim Biophys Acta. 1974;356:276–287. doi: 10.1016/0005-2736(74)90268-5. [DOI] [PubMed] [Google Scholar]
- 23.Kato M, Sasaki T, Ohya T, Nakanishi H, Nishioka H, Imamura M, Takai Y. Physical and functional interaction of rabphilin-3A with α-actinin. J Biol Chem. 1996;271:31775–31778. doi: 10.1074/jbc.271.50.31775. [DOI] [PubMed] [Google Scholar]
- 24.Komuro R, Sasaki T, Orita S, Maeda M, Takai Y. Involvement of rabphilin-3A in Ca2+-dependent exocytosis from PC12 cells. Biochem Biophys Res Commun. 1996;219:435–440. doi: 10.1006/bbrc.1996.0251. [DOI] [PubMed] [Google Scholar]
- 25.Korn H, Sur C, Charpier S, Legendre PI, Faber DS. The one-vesicle hypothesis and multivesicular release. Adv Second Messenger Phosphoprotein Res. 1994;29:301–322. doi: 10.1016/s1040-7952(06)80022-4. [DOI] [PubMed] [Google Scholar]
- 26.Li C, Takei K, Geppert M, Daniell L, Stenius K, Chapman ER, Jahn R, De Camilli P, Südhof TC. Synaptic targeting of rabphilin-3A, a synaptic vesicle Ca2+/phospholipid-binding protein, depends on rab3A/3C. Neuron. 1994;13:885–898. doi: 10.1016/0896-6273(94)90254-2. [DOI] [PubMed] [Google Scholar]
- 27.Lonart G, Südhof TC. Region-specific phosphorylation of rabphilin in mossy fiber nerve terminals of the hippocampus. J Neurosci. 1998;18:634–640. doi: 10.1523/JNEUROSCI.18-02-00634.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mandell JW, Townes-Anderson E, Czernik AJ, Cameron R, Greengard P, De Camilli P. Synapsins in the vertebrate retina: absence from ribbon synapses and heterogeneous distribution among conventional synapses. Neuron. 1990;5:19–33. doi: 10.1016/0896-6273(90)90030-j. [DOI] [PubMed] [Google Scholar]
- 29.Masumoto N, Sasaki T, Tahara M, Mammoto A, Ikebuchi Y, Tasaka K, Tokunaga M, Takai Y, Miyake A. Involvement of rabphilin-3A in cortical granule exocytosis in mouse eggs. J Cell Biol. 1996;135:1741–1747. doi: 10.1083/jcb.135.6.1741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Matteoli M, Takei K, Cameron R, Hurlbut P, Johnston PA, Südhof TC, Jahn R, De Camilli P. Association of Rab3A with synaptic vesicles at late stages of the secretory pathway. J Cell Biol. 1991;115:625–633. doi: 10.1083/jcb.115.3.625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.McMahon HT, Südhof TC. Synaptic core complex of synaptobrevin, syntaxin, and SNAP25 forms high affinity α-SNAP binding site. J Biol Chem. 1995;270:2213–2217. doi: 10.1074/jbc.270.5.2213. [DOI] [PubMed] [Google Scholar]
- 32.McMahon HT, Missler M, Li C, Südhof TC. Complexins: cytosolic proteins that regulate SNAP receptor function. Cell. 1995;83:111–119. doi: 10.1016/0092-8674(95)90239-2. [DOI] [PubMed] [Google Scholar]
- 33.Mignery GA, Südhof TC, Takei K, De Camilli P. Putative receptor for inositol 1,4,5-trisphosphate similar to ryanodine receptor. Nature. 1989;342:192–195. doi: 10.1038/342192a0. [DOI] [PubMed] [Google Scholar]
- 34.Ohya T, Sasaki T, Kato M, Takai Y. Involvement of Rabphilin-3 in endocytosis through interaction with Rabaptin. J Biol Chem. 1998;273:613–617. doi: 10.1074/jbc.273.1.613. [DOI] [PubMed] [Google Scholar]
- 35.Perin MS, Fried VA, Mignery GA, Jahn R, Südhof TC. Phospholipid binding by a synaptic vesicle protein homologous to the regulatory region of protein kinase. C Nature. 1990;345:260–263. doi: 10.1038/345260a0. [DOI] [PubMed] [Google Scholar]
- 36.Regehr WG, Delaney KR, Tank DW. The role of presynaptic calcium in short-term enhancement at the hippocampal mossy fiber synapse. J Neurosci. 1994;14:523–537. doi: 10.1523/JNEUROSCI.14-02-00523.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Robinson PJ, Sontag JM, Liu JP, Fykse EM, Slaughter C, McMahon H, Südhof TC. Dynamin GTPase regulated by protein kinase C phosphorylation in nerve terminals [see comments]. Nature. 1993;365:163–166. doi: 10.1038/365163a0. [DOI] [PubMed] [Google Scholar]
- 38.Rosahl TW, Spillane D, Missler M, Herz J, Selig DK, Wolff JR, Hammer RE, Malenka RC, Südhof TC. Essential functions of synapsins I and II in synaptic vesicle regulation. Nature. 1995;375:488–493. doi: 10.1038/375488a0. [DOI] [PubMed] [Google Scholar]
- 39.Sabatini BL, Regehr WG. Timing of neurotransmission at fast synapses in the mammalian brain. Nature. 1996;384:170–172. doi: 10.1038/384170a0. [DOI] [PubMed] [Google Scholar]
- 40.Sakaguchi G, Orita S, Maeda M, Igarashi H, Takai Y. Molecular cloning of an isoform of Doc2 having two C2-like domains. Biochem Biophys Res Commun. 1995;217:1053–1061. doi: 10.1006/bbrc.1995.2876. [DOI] [PubMed] [Google Scholar]
- 41.Salin PA, Malenka RC, Nicoll RA. Cyclic AMP mediates a presynaptic form of LTP at cereballar parallel fiber synapses. Neuron. 1996;16:797–803. doi: 10.1016/s0896-6273(00)80099-9. [DOI] [PubMed] [Google Scholar]
- 42.Sambrook J, Fritsch EF, Maniatis T. Molecular cloning: a laboratory manual. Cold Spring Harbor Laboratory; Cold Spring Harbor, NY: 1989. [Google Scholar]
- 43.Shirataki H, Kaibuchi K, Sakoda T, Kishida S, Yamaguchi T, Wada K, Miyazaki M, Takai Y. Rabphilin-3A, a putative target protein for smg p25A/rab3A p25 small GTP-binding protein related to synaptotagmin. Mol Cell Biol. 1993;13:2061–2068. doi: 10.1128/mcb.13.4.2061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Shirataki H, Yamamoto T, Hagi S, Miura H, Oishi H, Jin-no Y, Senbonmatsu T, Takai Y. Rabphilin-3A is associated with synaptic vesicles through a vesicle protein in a manner independent of Rab3A. J Biol Chem. 1994;269:32717–32720. [PubMed] [Google Scholar]
- 45.Shisheva A, Südhof TC, Czech MP. Cloning, characterization, and expression of a novel GDP dissociation inhibitor isoform from skeletal muscle. Mol Cell Biol. 1994;14:3459–3468. doi: 10.1128/mcb.14.5.3459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Siegel SJ, Brose N, Janssen WG, Gasic GP, Jahn R, Heinemann SF, Morrison JH. Regional, cellular, and ultrastructural distribution of N-methyl-d-aspartate receptor subunit 1 in monkey hippocampus. Proc Natl Acad Sci USA. 1994;91:564–568. doi: 10.1073/pnas.91.2.564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Soriano P, Montgomery C, Geske R, Bradley A. Targeted disruption of the c-src proto-oncogene leads to osteopetrosis in mice. Cell. 1991;64:693–702. doi: 10.1016/0092-8674(91)90499-o. [DOI] [PubMed] [Google Scholar]
- 48.Stahl B, Chou JH, Li C, Südhof TC, Jahn R. Rab3 reversibly recruits rabphilin to synaptic vesicles by a mechanism analogous to raf recruitment by ras. EMBO J. 1996;15:1799–1809. [PMC free article] [PubMed] [Google Scholar]
- 49.Stevens CF, Wesseling JF. Activity-dependent modulation of the rate at which synaptic vesicles become available to undergo exocytosis. Neuron. 1998;21:415–424. doi: 10.1016/s0896-6273(00)80550-4. [DOI] [PubMed] [Google Scholar]
- 50.Südhof TC. The synaptic vesicle cycle: a cascade of protein-protein interactions. Nature. 1995;375:645–653. doi: 10.1038/375645a0. [DOI] [PubMed] [Google Scholar]
- 51.Südhof TC. Function of Rab3 GDP-GTP exchange. Neuron. 1997;18:519–522. doi: 10.1016/s0896-6273(00)80292-5. [DOI] [PubMed] [Google Scholar]
- 52.Sudhof TC, Rizo J. Synaptotagmins: C2−domain proteins that regulate membrane traffic. Neuron. 1996;17:379–388. doi: 10.1016/s0896-6273(00)80171-3. [DOI] [PubMed] [Google Scholar]
- 53.Telemenakis I, Benseler F, Stenius K, Südhof TC, Brose N. Rat homologues of yeast sec7p. Eur J Cell Biol. 1997;74:143–149. [PubMed] [Google Scholar]
- 54.Thomas KR, Capecchi MR. Site-directed mutagenesis by gene targeting in mouse embryo-derived stem cells. Cell. 1987;51:503–512. doi: 10.1016/0092-8674(87)90646-5. [DOI] [PubMed] [Google Scholar]
- 55.Towbin H, Staehelin T, Gordon J. Electrophoretic transfer of proteins from polyacrylamide gels to nitrocellulose sheets: procedure and some applications. Proc Natl Acad Sci USA. 1979;76:4350–4354. doi: 10.1073/pnas.76.9.4350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ullrich B, Südhof TC. Distribution of synaptic markers in the retina: implications for synaptic vesicle traffic in ribbon synapses. J Physiol (Paris) 1994;88:249–257. doi: 10.1016/0928-4257(94)90088-4. [DOI] [PubMed] [Google Scholar]
- 57.Verhage M, de Vries KJ, Roshol H, Burbach JP, Gispen WH, Südhof TC. DOC2 proteins in rat brain: complementary distributions and proposed functions as vesicular adapter proteins in early stages of secretion. Neuron. 1997;18:453–461. doi: 10.1016/s0896-6273(00)81245-3. [DOI] [PubMed] [Google Scholar]
- 58.Wang L-Y, Kaczmarek LK. High-frequency firing helps replenish the readily releasable pool of synaptic vesicles. Nature. 1998;394:384–388. doi: 10.1038/28645. [DOI] [PubMed] [Google Scholar]
- 59.Wang Y, Okamoto M, Schmitz F, Hofmann K, Südhof TC. RIM is a putative Rab3 effector in regulating synaptic-vesicle fusion. Nature. 1997;388:593–598. doi: 10.1038/41580. [DOI] [PubMed] [Google Scholar]
- 60.Zucker RS. Short-term synaptic plasticity. Annu Rev Neurosci. 1989;12:13–31. doi: 10.1146/annurev.ne.12.030189.000305. [DOI] [PubMed] [Google Scholar]