

Abstract
α-Synuclein has been implicated in the pathophysiology of many neurodegenerative diseases, including Parkinson’s disease (PD) and Alzheimer’s disease. Mutations in α-synuclein cause some cases of familial PD (Polymeropoulos et al., 1997; Kruger et al., 1998). In addition, many neurodegenerative diseases show accumulation of α-synuclein in dystrophic neurites and in Lewy bodies (Spillantini et al., 1998). Here, we show that α-synuclein shares physical and functional homology with 14-3-3 proteins, which are a family of ubiquitous cytoplasmic chaperones. Regions of α-synuclein and 14-3-3 proteins share over 40% homology. In addition, α-synuclein binds to 14-3-3 proteins, as well as some proteins known to associate with 14-3-3, including protein kinase C, BAD, and extracellular regulated kinase, but not Raf-1. We also show that overexpression of α-synuclein inhibits protein kinase C activity. The association of α-synuclein with BAD and inhibition of protein kinase C suggests that increased expression of α-synuclein could be harmful. Consistent with this hypothesis, we observed that overexpression of wild-type α-synuclein is toxic, and overexpression of α-synuclein containing the A53T or A30P mutations exhibits even greater toxicity. The activity and binding profile of α-synuclein suggests that it might act as a protein chaperone and that accumulation of α-synuclein could contribute to cell death in neurodegenerative diseases.
Keywords: 14-3-3 proteins, Alzheimer’s disease, apoptosis, BAD, extracellular regulated kinase, Parkinson’s disease, protein kinase C, synuclein
α-Synuclein is part of a family of proteins consisting of α-, β-, and γ-synuclein (Jakes et al., 1994; Clayton and George, 1998). The biochemical properties of α-synuclein resemble that of a protein chaperone, capable of binding other proteins. It is a small 140 amino acid protein, which has an 11-mer repeat that recurs seven times (George et al., 1995). In solution, α-synuclein exists in a random open, rather than globular, conformation, consistent with a role as a chaperone (Weinreb et al., 1996). In vitro, the 11-mer repeat has been shown to promote binding to synthetic acidic phospholipid vesicles (Davidson et al., 1998). α-Synuclein is expressed in a wide variety of autosomal cells, as well as in neurons (Ueda et al., 1993). In neurons, α-synuclein is enriched in presynaptic terminals in which it is distributed between a soluble pool and a vesicle-bound pool of proteins. The only protein known to interact with α (and β)-synuclein is phospholipase D2, which is inhibited in vitro by α (and β)-synuclein with a KI of 10 nm (Jenco et al., 1998).
Recent research suggests that α-synuclein contributes to the pathophysiology of many neurodegenerative illnesses. In Parkinson’s disease (PD), α-synuclein accumulates in Lewy bodies. Dystrophic neurites in PD and amyotrophic lateral sclerosis show accumulations of α-synuclein, as do glia in multiple systems atrophy (Spillantini et al., 1998; Takeda et al., 1998; Tu et al., 1998). α-Synuclein also accumulates in neuritic plaques in Alzheimer’s disease. A number of studies suggest that α-synuclein can be toxic to some cells. Incubation of α-synuclein with the neuronal cells, SK-SY5Y, induces apoptosis (El-Agnaf et al., 1998a). Molecular genetic studies have identified two different point mutations in the α-synuclein gene, A53T and A30P, that appear to cause familial PD (Polymeropoulos et al., 1997; Kruger et al., 1998). The association between α-synuclein and disease suggests that perturbations of α-synuclein biology can be harmful to cells. The current knowledge of the biology of α-synuclein, however, is not sufficient to understand how α-synuclein might affect cell viability.
The 14-3-3 proteins constitute a family of protein chaperones that are particularly abundant in the brain, like α-synuclein. The 14-3-3 family of proteins consist of five different isoforms that share extensive sequence homology, both among the different isoforms and between similar isoforms in different species (Layfield et al., 1996;Broadie et al., 1997). 14-3-3 proteins bind to ligands at sites containing phospho-serine residues. Binding of 14-3-3 to phosphorylated Raf-1 stabilizes it in an active conformation (Tzivion et al., 1998). 14-3-3 binds to a phosphorylated epitope of protein kinase Cε (PKCε) and stabilizes PKCε in an inactive conformation that is unable to translocate to the membrane (Meller et al., 1996;Wheeler-Jones et al., 1996; Matto-Yelin et al., 1997). 14-3-3 also binds to phosphorylated BAD, which appears to stabilize maintenance of BAD in a cytoplasmic localization (Zha et al., 1996).
We now report that α-synuclein shares regions of homology with 14-3-3 proteins, binds to 14-3-3 proteins, binds to ligands of 14-3-3, and is toxic when overexpressed.
MATERIALS AND METHODS
Materials. α-Synuclein was cloned intoBamHI/NotI sites of pcDNA3. The FLAG sequence was inserted by PCR into the 5′ end of α-synuclein cDNA after the α-synuclein ATG start codon and confirmed by DNA sequencing. Antibodies used include: polyclonal anti-α-synuclein (SC1; 1:2000; against human α-synuclein; residues 116–131; sequence, MPVDPDNEAYEMPSEE), monoclonal anti-α-synuclein-1 (1:1000; Transduction Laboratories, Lexington, KY), monoclonal anti-FLAG (1:300; IBI/Kodak, New Haven, CT), polyclonal anti-BAD antibody (1:1000; Transduction Laboratories), monoclonal anti-PKC-III and polyclonal pan-PKC (1:1000; Upstate Biotechnology, Lake Placid, NY), polyclonal 14-3-3β, and monoclonal 14-3-3ε (1:1000; Transduction Laboratories).
Cell culture. Cells were grown in high glucose DMEM plus 10% FBS supplemented with 500 μg/ml G418, as needed. G418 was used for selection. Transfections used lipofectamine (Life Technologies, Gaithersburg, MD) with 2 μg/ml DNA and 6 μl/ml lipofectamine in Optimem.
Immunoblotting. Cells were harvested with SDS lysis solution (2% SDS, 10 mm Tris, pH 7.4, 2 mm β-glycerol phosphate, and 1 μm AEBSF). Protein was determined using the BCA assay (Pierce, Rockford, IL), and 30 μg/lane was run on 14% polyacrylamide gels and transferred to nitrocellulose (200 mA, 6 hr). The nitrocellulose was then incubated 1 hr in 5% milk–PBS, washed, incubated overnight in primary (1°) antibody, washed, incubated 3 hr in peroxidase-coupled secondary antibody, and developed with chemiluminescent reagent (DuPont, Billerica, MA).
Immunoprecipitations. Homogenates were extracted with 1% Triton X-100 in PBS plus protease and phosphatase inhibitors. Some immunoprecipitations of α-synuclein were also performed using an extraction buffer containing 1% Triton X-100–0.2% SDS plus protease and phosphatase inhibitors. The lysates were spun down (10,000 ×g, 15 min), and the supernatants were precleared with protein A Sepharose (Pharmacia, Piscataway, NJ). Agarose-coupled M2 anti-FLAG antibody (25 μl) or 1 μl of antibody was then added and incubated at 4°C overnight. For the precipitations with polyclonal SC1, pan-PKC, or 14-3-3 antibodies, the immunocomplexes were precipitated by incubation with protein A Sepharose at 4°C for 2 hr. After binding of the solid phase substrate, the samples were washed five times in TBS–1% Triton X-100 and immunoblotted.
PKC assay and cell fractionation. For the PKC assay, cells were incubated in serum-free conditions overnight, stimulated, washed, and scraped into 500 μl of 4°C extraction buffer [20 mm Tris, pH 7.4, 2 mm EDTA, 5 mmEGTA, 0.25 m sucrose, 5 mm β-mercaptoethanol, 0.1% Triton X-100, and 20 μg/ml protease inhibitors cocktail (Sigma, St. Louis, MO)]. The samples were then ultrasonicated and centrifuged at 100,000 × g at 4°C for 1 hr. PKC activity in the supernatant was then determined at 30°C for 10 min using 25 μg of protein and PKC assay buffer [50 mm Tris-Hcl, pH 7.4, 250 μg/ml BSA, 1 mm EGTA, 10 μm PKC substrate peptide (Ac-myelin basic protein4–14; Life Technologies), 100 μg/ml phosphatidylcholine/phosphatidylserine (80:20), 50 mm ATP, and 1 μCi γ-32P-ATP per tube]. For cell fractionation after harvesting in the PKC assay buffer without Triton X-100, the cell lysate was sonicated and centrifuged at 100,000 × g at 4°C for 1 hr.
Trypan blue staining. Transfections were performed as described above using the pGL3 luciferase control plasmid, a luciferase assay kit (Promega), and triplicate points. For the trypan blue viability, assay cells were trypsinized, spun down, and taken up in HBSS containing 0.4% trypan blue. The total number of cells and the number of blue-stained cells were then counted using a hemocytometer. Three different wells are used for each condition (triplicate plating), and four different fields were counted in each well.
DNA fragmentation. Cells from 100 mm dishes were harvested, and DNA was isolated by phenol chloroform extraction and analyzed on a 2% agarose gel using Syber Green for visualization.
RESULTS
α-Synuclein binds to 14-3-3
To explore whether α-synuclein might function as a protein chaperone, we searched the genetic database for homology between α-synuclein and other protein chaperones. Using the Multalin algorithm, we observed regions of sequence homology between members of the synuclein and 14-3-3 family of proteins (Fig.1) (Corpet, 1988; Dubois et al., 1997). This alignment program displays different levels of homology. Exact homologies are shown as white letters on a black background (Fig. 1), and homologies between amino acids with similar chemical properties, such as Ile and Val, are shown inblack letters on a gray background(Fig. 1). Two regions with 43 and 36% sequence homology were seen between amino acids 8 and 61 of α-synuclein and amino acids between 45 and 102 of 14-3-3 (Fig. 1A). This region contains domains of 14-3-3 that are thought to be phosphorylated by PKC and involved in dimerization of 14-3-3 (Dubois et al., 1997; Yaffe et al., 1997). In contrast, the C terminus of 14-3-3, which has been implicated in binding to phosphorylated Raf-1, shows no homology to α-synuclein (Ichimura et al., 1997).
Fig. 1.
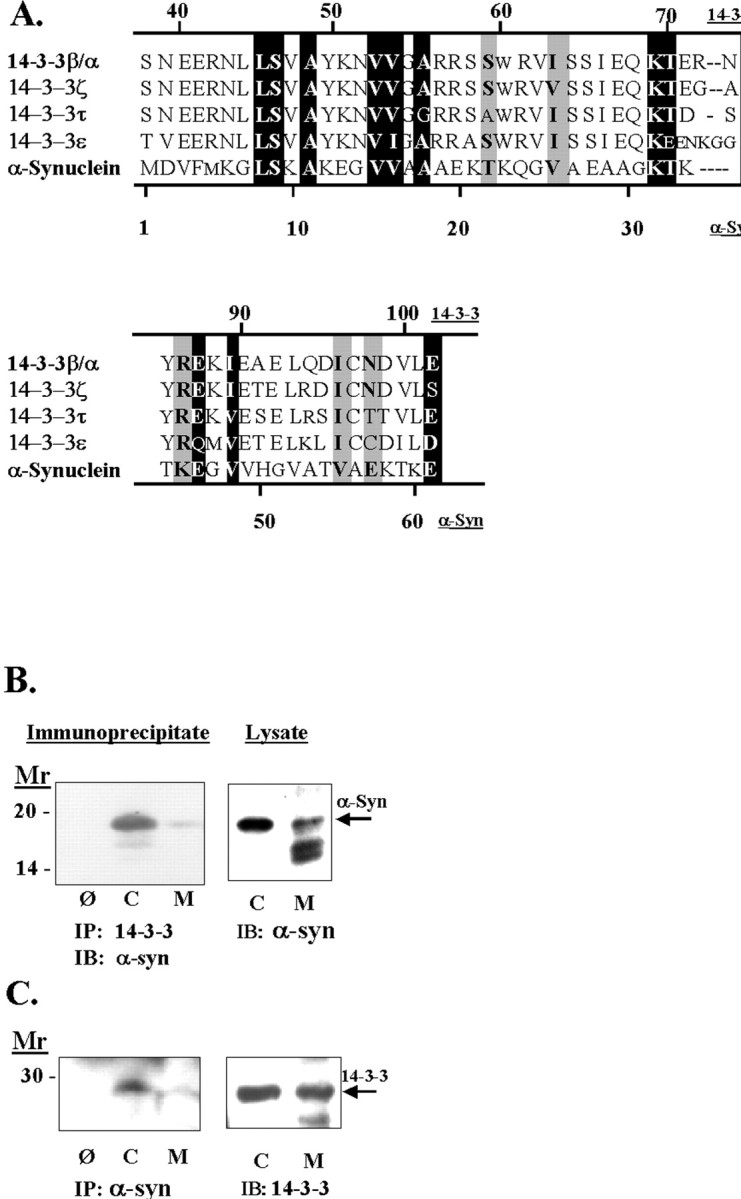
A, Alignment of α-synuclein and the 14-3-3 family of proteins. The alignment was performed using the Multalin program (http://www.toulouse.inra.fr/multalin.html). To observe homology, we excluded the first 30 amino acids of 14-3-3 proteins while performing the alignment algorithm. Exact matches are shown as white letters on a black background, and matches of proteins with similar properties are shown as black letters on a gray background. In addition, we have noted aligned serines and threonines in black because both amino acids can be phosphorylated by serine/threonine kinases. B, Association of α-synuclein with 14-3-3. Rat brain tissue was fractionated into cytoplasmic (C) and membrane (M) components, taken up in immunoprecipitation buffer, and treated as shown. Theleft shows an immunoblot of a 14-3-3β immunoprecipitate with anti-α-synuclein antibody, and theright shows an immunoblot of the lysates. The omit lane is labeled Ø and refers to immunoprecipitations done using protein A but no 1° antibody. C, Theleft shows an immunoblot of α-synuclein immunoprecipitates with anti-14-3-3ε antibody, and theright shows an immunoblot of the lysates. The omit lane is labeled Ø and refers to an immunoprecipitation done using protein A but no 1° antibody.
Because the homology between α-synuclein and 14-3-3 covers a region known to mediate dimerization of 14-3-3 proteins, we investigated whether α-synuclein associates with 14-3-3. Rat brain homogenates were fractionated into membrane and cytoplasmic components, and 14-3-3ε protein was immunoprecipitated. The resulting precipitates were immunoblotted using anti-α-synuclein antibody. As shown in Figure 1B, α-synuclein and 14-3-3β coimmunoprecipitated. Comparison of the exposure times for the immunoprecipitate and the lysate indicates that immunoprecipitation of 14-3-3 concentrated α-synuclein ∼40-fold because α-synuclein could be observed in the immunoprecipitates after an exposure of 0.5 sec but required a 20 sec exposure to be observed in the lysates (Fig.1B). α-Synuclein was most abundant in the 14-3-3ε immunoprecipitates obtained from the cytoplasmic samples (Fig.1B). The association between α-synuclein and 14-3-3 could also be observed by immunoprecipitating α-synuclein and probing the resulting immunoblots with anti-14-3-3ε antibody (Fig.1C). We used an antibody to the 14-3-3ε isoform because of the availability of a monoclonal mouse antibody (which can be distinguished from the polyclonal rabbit anti-synuclein antibody used for the immunoprecipitation). These data show that α-synuclein associates with 14-3-3.
α-Synuclein binds and inhibits PKC
To examine the significance of this association, we examined whether α-synuclein binds to other proteins that associate with 14-3-3 proteins. We first examined the binding to PKC. Rat brain tissue was homogenized in PKC assay buffer and stimulated for 30 min with 1 μm phorbol 12,13-myristate acetate (PMA). The homogenate was then separated into membrane and cytoplasmic components, the PKC was immunoprecipitated with pan-PKC antibody, and the precipitates were probed with monoclonal anti-α-synuclein-1 antibody. The presence of α-synuclein in the PKC immunoprecipitates was readily apparent, and immunoprecipitation of α-synuclein concentrated the PKC approximately fivefold based on exposure times (4 sec exposure for the immunoprecipitate and 20 sec exposure for the lysate) (Fig.2). Association between α-synuclein and PKC was also evident by immunoprecipitating α-synuclein and probing with antibodies specific to PKCα, β, γ, δ, or ε (Fig. 2). The pattern of association and amount of association with PKC was isoform-specific. Based on the intensity of the immunoprecipitations, α-synuclein appeared to give the following rank order of association with PKC isoforms: α = γ > ε > β = δ. Complexes of α-synuclein with PKCα were apparent in all samples, whereas complexes with PKCγ, δ, or ε were present only in the membrane fraction under basal or stimulated conditions, and complexes with PKCβ were apparent in the membrane fractions only after stimulation (data not shown).
Fig. 2.

Association of α-synuclein with PKC isoforms in rat brain tissue. Left panels show immunoblots of α-synuclein immunoprecipitates with isoform-specific PKC antibodies. Right panels show immunoblots of each PKC isoform in the membrane and cytoplasmic fractions. Before each immunoprecipitation, half of the brain homogenates were treated with 1 μm PMA plus 1 mm Ca2+ at 30°C for 30 min. The homogenates were then fractionated into membrane and cytoplasmic components taken up in immunoprecipitation buffer, and proteins were isolated as shown. Cytoplasmic, C; membrane, M; omit, Ø, which is an immunoprecipitation done using protein A but no 1° antibody.
The ability of α-synuclein to associate with PKC suggests that it might regulate PKC. To examine this issue, we generated lines of 293 human embryonic kidney (HEK) cells overexpressing wild-type or A53T α-synuclein, as well as a vector transfected control line (Fig.3A). First, we examined whether complexes of α-synuclein and PKC could be immunoprecipitated. Lysates were prepared from cells grown under basal conditions or conditions known to stimulate PKC. We then immunoprecipitated the α-synuclein PKC complex by precipitating with anti-α-synuclein antibody and probing with anti-PKCα (Fig. 3B) or by precipitating with anti pan-PKC antibody and probing with anti-α-synuclein (Fig. 3C). As with the brain tissue, we observed that α-synuclein in these cell lines also binds PKC. The association of α-synuclein with PKCα was most apparent after membrane translocation (Fig. 3B). Next, we determined whether overexpression of α-synuclein affected PKC activity. Control, wild-type, and A53T-α-synuclein 293 HEK cell lines were treated with 1 μm PMA for 30 min, and PKC activity in the lysates was analyzed in vitro. The cell lines overexpressing wild-type or A53T α-synuclein did not show any increases in PKC activity after PMA treatment, despite strong stimulation observed in the control cell lines and despite robust translocation of PKC observed in each line after PMA treatment (Fig. 3D,E). Similar results were observed in different monoclonal lines of 293 HEK cells stably transfected with wild-type or A53T α-synuclein (data not shown). These data show that α-synuclein binds to and inhibits PKC. Most of the PKC isoforms show strong association with membrane-bound α-synuclein (Figs. 2, 3B). This suggests that membrane-bound α-synuclein has a stronger affinity for PKC, which is consistent with observations that membrane-bound α-synuclein has a more stable secondary structure (Davidson et al., 1998). Inhibition of PKC by α-synuclein, however, does not appear to affect membrane translocation of PKC. 14-3-3 also binds to and inhibits PKC but does so in the cytoplasm.
Fig. 3.

A, An immunoblot of 293 HEK cell lines stably transfected with empty vector (Vec), wild-type (WT), or A53T α-synuclein using α-synuclein antibody SC1. Control 293 HEK cells do endogenously express low levels of α-synuclein (arrow), whereas the transfected cells show increased expression of the 19 kDa α-synuclein proteins. B, Immunoblot of PKCα in α-synuclein immunoprecipitates. 293 HEK cell lines stably transfected with empty vector (Vec), wild-type (WT), or A53T α-synuclein were grown under basal conditions or treated with 20 nm bradykinin for 30 min, and the lysates were immunoprecipitated using agarose-coupled anti-FLAG resin. Immunoblotting of the resulting samples with anti-PKC type III antibody showed coassociation of α-synuclein with PKCα only in the bradykinin-treated samples (top panel). Thebottom panel shows an immunoblot of PKCα in the corresponding total cell lysates. C, Immunoblot of α-synuclein in PKCα immunoprecipitates. Lane 1, Lysates were treated with 1 μm PMA for 30 min, and PKCα was immunoprecipitated using anti-pan-PKC antibody (lane 2) (this antibody recognizes PKCα, β, and γ; Upstate Biotechnology) and immunoblotted with anti-α-synuclein SC1 antibody. Lane 2 shows an immunoprecipitation with the anti-pan-PKC antibody omitted. Lane 3(Lys) shows a parallel anti-synuclein immunoblot of the lysate (30 μg of lysate). No reactivity was seen in absence of PMA stimulation. D, PKC activity does not increase in 293 HEK cell lines overexpressing wild-type or A53T α-synuclein after stimulation with PMA (1 μm, 30 min). *p < 0.001. E, Immunoblots of PKCα in fractionated cell lysates (cytoplasm to membrane) after treatment with PMA (1 μm, 30 min). All cell lines (vector, wild-type α-synuclein, and A53T α-synuclein) showed robust translocation of PKCα after PMA treatment. C, Cytoplasm; M, membrane.
α-Synuclein binds to BAD and extracellular regulated kinase
14-3-3 is also known to associate with BAD, which is a Bcl-2 homolog that regulates cell death (Yang et al., 1995). To examine whether α-synuclein also associates with BAD, we immunoprecipitated α-synuclein from rat cortex (Fig.4B). BAD was readily detected after immunoprecipitation of α-synuclein, and α-synuclein was readily detected after immunoprecipitation of BAD. The BAD was concentrated approximately ninefold in the α-synuclein immunoprecipitates (10 sec exposure for the immunoprecipitate vs 1.5 min exposure for the lysates) (Fig. 4A). We were also able to show an association between BAD and α-synuclein in cell lines. Coimmunoprecipitation of BAD and α-synuclein was apparent in lysates from 293 HEK cells transfected with α-synuclein (Fig.4B). We also observed coimmunoprecipitation of BAD and α-synuclein in HeLa cells stably transfected with wild-type or A53T α-synuclein (Fig. 4C). In these immunoprecipitations, we investigated the phosphorylation state of the immunoprecipitated BAD and found that the immunoprecipitated α-synuclein was positive for BAD but negative for phospho-BAD112 or phospho-BAD136 (Fig. 4C). Finally, we examined the regulation of α-synuclein binding to BAD and found a pattern opposite to that of PKCα. Stimulation of 293 HEK cells with either carbachol or bradykinin reduced α-synuclein–BAD complex formation (Fig. 4B). Similar results were observed in brain in which treatment of brain homogenates with 1 μmPMA decreased the association of BAD with α-synuclein (Fig.4A). Thus, binding of BAD and PKC to α-synuclein are inversely correlated. Binding of 14-3-3 and α-synuclein to BAD are also inversely correlated, because 14-3-3 has been shown to bind phospho-BAD, whereas α-synuclein binds dephospho-BAD (Zha et al., 1996).
Fig. 4.

A, Coimmunoprecipitation of α-synuclein and BAD from brain lysate. Homogenates from rat cortex were fractionated into membrane and cytoplasmic components and taken up in immunoprecipitation buffer. BAD protein was then immunoprecipitated, and precipitates were immunoblotted with monoclonal anti-α-synuclein. Cytoplasmic, C; membrane, M; omit,Ø, which is an immunoprecipitation done using protein A but no 1° antibody. B, Association of α-synuclein with BAD in 293 HEK cells and regulation by agents that stimulate PKC. 293 HEK cells (which express endogenous α-synuclein) were treated with carbachol (1 mm, 30 min) or bradykinin (20 nm, 30 min). α-Synuclein was then immunoprecipitated from total cellular lysates with anti-synuclein SC1 antibody, and the resulting immunoblots were probed with anti-BAD antibody.Ø represents an immunoprecipitation performed without 1° anti-α-synuclein antibody. Immunoblots of the lysates showed that equal amounts of protein were loaded in each lane (data not shown). C, Immunoblot of BAD (arrow,top) or phospho-BAD136(bottom, arrow points to absent band; 1:200; New England Biolabs, Beverly, MA) after immunoprecipitation of FLAG-tagged α-synuclein from HeLa cell lysates. α-Synuclein was immunoprecipitated with agarose-coupled anti-FLAG resin, and the immunoblots were probed with anti-BAD antibody (top) or anti-phospho-BAD136 antibody (bottom). No specific staining for phospho-BAD136 was observed. The bands in the phospho-BAD136 immunoblot were present in the control cell line that was not transfected with FLAG-tagged α-synuclein, and therefore these bands represent nonspecific binding. An immunoblot of the lysates showed equal expression of FLAG-tagged wild-type and A53T α-synuclein (data not shown).
We also examined binding of α-synuclein to Raf-1 and extracellular regulated kinase (ERK), which are key kinases in the ERK cascade. 14-3-3 has been shown to bind to phosphorylated Raf-1 protein. First, we immunoprecipitated α-synuclein from rat brain lysate and blotted with antibody to Raf-1. α-Synuclein did not bind to Raf-1 protein (5 sec exposure for the immunoprecipitate and lysate) (Fig.5). We investigated this issue further by examining whether α-synuclein might bind to another member of the ERK cascade. We next blotted the α-synuclein immunoprecipitate with anti-ERK 1 2 antibodies and observed that strong anti-ERK reactivity (Fig. 5). Based on exposure times, immunoprecipitation of α-synuclein concentrated ERK 20-fold (0.5 sec was used for exposure of the immunoprecipitate, and 10 sec was used for exposure of the lysate) (Fig. 5). This suggests that α-synuclein associates with ERK and might also play a role in regulating the ERK cascade. Because 14-3-3 is known to both Raf-1 and phospho-BAD, the inability of α-synuclein to bind Raf-1 or phospho-BAD suggests that the association of α-synuclein with its protein partners (PKC, BAD, and ERK) can occur independently of the association of α-synuclein with 14-3-3.
Fig. 5.

Association of α-synuclein with members of the ERK cascade. Left panels show immunoblots of α-synuclein immunoprecipitates with antibodies to either Raf-1 or ERK. No association was seen with Raf-1, whereas strong binding was seen with ERK. Right panels show immunoblots with the Raf-1 and ERK antibodies in the membrane and cytoplasmic fractions. Before each immunoprecipitation, half of the rat brain homogenates were treated with 1 μm PMA plus 1 mmCa2+ at 30°C for 30 min. The homogenates were then fractionated into membrane and cytoplasmic components and taken up in immunoprecipitation buffer, and proteins were isolated as shown. Cytoplasmic, C; membrane, M, and omit,Ø, which is an immunoprecipitation done using protein A but no 1° antibody.
Overexpression of α-synuclein is toxic
The correlation between mutations in α-synuclein and familial PD and the presence of α-synuclein pathology in multiple neurodegenerative diseases suggests that α-synuclein could be harmful to cells. Our observations that α-synuclein binds to proteins that are known to affect cell viability, such as BAD, suggests biochemical mechanisms that might underlie the toxicity of α-synuclein. Determining whether α-synuclein is indeed toxic is of fundamental importance to neuropathology. Expression of α-synuclein in cells has not yet been shown to be toxic to cells. The only study performed to date addressing this issue has shown that extracellular administration of aggregates of α-synuclein is toxic (El-Agnaf et al., 1998a). The conditions used in this study differ from physiological conditions because α-synuclein is an intracellular protein and not an extracellular protein. Because α-synuclein interacts with three proteins known to affect cell viability, BAD, PKC, and ERK, we sought to address directly whether increases in intracellular α-synuclein were toxic.
We first examined whether the regulation of α-synuclein levels was responsive to cell stress. To examine the regulation of α-synuclein, we measured the amount of α-synuclein that was endogenously expressed in 293 HEK cells under basal conditions and after serum deprivation for 24 or 48 hr. We observed that serum deprivation increased expression of α-synuclein after 24 hr (Fig.6A). The level of α-synuclein remained elevated at the 48 hr time point as well. Thus, cell stress can increase α-synuclein expression.
Fig. 6.

Toxicity of α-synuclein. A, α-Synuclein expression increased during incubation of 293 HEK cells in serum-free medium. The left shows an immunoblot of α-synuclein with SC1 antibody, and the right shows the same immunoblot reprobed with anti-actin antibody (Sigma).B, Transfection of 293 HEK cells with an antisense α-synuclein construct (AS; 2 μg) reduced the amount of endogenous α-synuclein expressed in the cells (left). The control lane(Ctrl) shows lysates from cells transfected with an empty pcDNA3 plasmid under the same conditions at the same time. The immunoblot was then reprobed with anti-actin antibody to show that equal amounts of protein were loaded in each lane(right). C, Transient transfection of 293 HEK (left) or SK-N-SH cells (middle) with a pGL3 luciferase plasmid and vector, wild-type, A53T, or A30P α-synuclein constructs induces a dose-dependent decrease in luciferase activity. In contrast, transfection with antisense α-synuclein increased luciferase expression (right). For the antisense experiments in the right, cells were transfected and then serum-deprived for 24 hr, after which luciferase activity was measured. Cells transfected with antisense α-synuclein showed less toxicity than cells transfected with vector. *p < 0.05; **p < 0.01;n = 4 for each point. D, Similar experiments showed a dose-dependent increase in toxicity as shown by trypan blue staining after serum deprivation for 0, 24, or 48 hr. Parallel experiments with a β-galactosidase vector showed a 40% transfection rate in 293 HEK cells. Based on 20% of the cells being positive for trypan blue after transfection, we estimate that transfection of 1 μg of A53T α-synuclein induced ∼50% cell death in 293 HEK cells. +p < 0.05; *p < 0.01; **p < 0.001.E, Effects of α-synuclein on DNA fragmentation. No fragmentation was seen under basal growth conditions in the control cell line (Vec, lane 1), wild-type α-synuclein overexpresser (WT, lane 2), or A53T α-synuclein expresser (A53T, lane 3). After 24 hr incubation in serum-free medium, oligomeric DNA fragmentation was strong in the control cell line (lane 4), moderate in the wild-type α-synuclein cell line (lane 5), and absent in the A53T α-synuclein cell line (lane 6). A slight increase in highly degraded DNA is apparent in lane 6 above the dye front, suggesting increased necrotic DNA. F, Wild-type α-synuclein increases BAD toxicity, but mutant α-synuclein (A53T and A30P) does not increase BAD toxicity. 293 HEK cells were cotransfected with the constitutively active 1 μg of pGL3 luciferase plasmid with or without 100 ng of BAD, and with or without 500 ng of α-synuclein (wild-type, A53T, or A30P). A β-galactosidase vector was used as a ballast to maintain the DNA amount at 2 μg; this vector does not affect luciferase activity. *p < 0.0001; n = 4; comparing samples with or without BAD. The A53T and A30P transfections alone were also significantly different from vector at p < 0.0001;n = 4.
To examine whether increased expression of α-synuclein was harmful to cells, we analyzed the expression of a constitutively active luciferase reporter vector after transient transfection of α-synuclein or control constructs. Apoptosis or necrosis reduces the amount of luciferase expressed because protein synthesis decreases during cell death. 293 HEK cells or SK-N-SH cells, a human neuroblastoma cell line, were transiently transfected with constructs containing vector, wild-type, A53T, or A30P α-synuclein, as well as with a pGL3 plasmid that constitutively expresses luciferase. Transfection of wild-type, A53T, and A30P α-synuclein induced dose-dependent increases in toxicity in both the 293 HEK cells and the SK-N-SH cells (Fig.6C). The A53T α-synuclein construct was more toxic than the wild-type construct in both cell lines at each dose tested. The A30P α-synuclein was more toxic than wild type up to 500 ng of DNA, after which the toxicity reached a plateau.
In separate experiments, we observed that transfection with antisense α-synuclein was cytoprotective (Fig.6B,C). 293 HEK cells were transfected with the constitutively active pGL3 luciferase vector and either antisense α-synuclein or empty pcDNA3. Immunoblots of the cells with anti-α-synuclein antibody showed that the antisense construct reduced the expression of endogenous α-synuclein (Fig.6B). α-Synuclein reactivity was not completely eliminated because, in transient transfections, some of the cells do not receive the antisense construct. The cells were then serum-deprived for 24 hr, and luciferase activity was measured. Serum deprivation caused a 25% decrease in luciferase activity in control cells that had been transfected with pGL3 and pcDNA3 (Fig. 6C, right panel). In contrast, cells transfected with pGL3 and antisense α-synuclein showed a 26% increase in luciferase activity after serum deprivation (Fig. 6C, right panel). The ability of the antisense α-synuclein construct to protect against toxicity suggests that α-synuclein participates in cell death processes and that reducing α-synuclein interferes with cell death processes.
We confirmed the link between α-synuclein and toxicity using trypan blue staining. 293 HEK cells were transiently transfected with empty vector, wild-type, or A53T α-synuclein and subsequently serum-starved for 0, 24, or 48 hr. The percentage of dead cells, as determined using trypan blue staining, was much greater in cells transfected with A53T α-synuclein than either the wild-type α-synuclein or the vector (Fig. 6D). Because the trypan blue assay is better at detecting necrosis than apoptosis, the increased cell death induced by A53T α-synuclein suggests that A53T α-synuclein induces more necrosis than apoptosis.
To analyze the mechanism of cell death further, we examined DNA fragmentation in the 293 HEK cell lines stably transfected with vector, wild-type, or A53T α-synuclein. 293 HEK cells were subjected to serum withdrawal for 12, 18, 24, 36, or 48 hr to induce apoptosis, and the DNA was analyzed by agarose gel electrophoresis. Maximal amount of DNA fragmentation was observed at 24 hr. The same experiment was then repeated using 293 HEK cells expressing vector, wild-type, or A53T α-synuclein. After 24 hr of serum deprivation, the fragmented DNA ran as a ladder in the serum-deprived control cell line, which is characteristic of apoptosis, but was less apparent and more smeared in the cell line expressing wild-type α-synuclein (Fig.6E). In the A53T α-synuclein cell line, the DNA was highly degraded and appeared mainly as smear at the dye front (Fig.6E). This suggests that overexpression of wild-type α-synuclein allows some apoptosis, but expression of A53T α-synuclein potentiates necrosis.
Finally, as an initial step in examining whether interaction with BAD contributes to the mechanism of cell death induced by overexpressed wild-type or mutant α-synuclein, we investigated whether overexpression of α-synuclein affects the toxicity of BAD. 293 HEK cells were cotransfected with 1 μg of the constitutively active pGL3 luciferase vector, with 500 ng of vector, wild-type, A53T, or A30P α-synuclein, and 100 ng of BAD. The amount of luciferase reactivity quantitates cell viability. The total amount of plasmid DNA used for transfections was kept constant by use of a β-galactosidase ballast plasmid, which we have shown does not affect luciferase activity. Transfection with wild-type α-synuclein added to the toxicity of BAD (Fig. 6F). Transfection with BAD alone produced 56% (p < 0.0001; n = 4) toxicity, whereas cotransfection with BAD plus wild-type α-synuclein produced 65% (p < 0.0001; n = 4) toxicity. In contrast, cotransfection of mutant α-synuclein (A53T or A30P) with BAD did not show any increase in toxicity over transfection with BAD alone (Fig. 6F). Transfection with BAD alone produced 56% toxicity, whereas transfection with BAD plus A53T or A30P α-synuclein produced only 49 and 51% (p < 0.0001; n = 4) toxicity. The inability of mutant α-synuclein constructs to add to toxicity when cotransfected with BAD suggests that A53T and A30P mutant α-synucleins cause toxicity through a mechanism utilizing BAD, although further experiments will need to be done to prove this point.
DISCUSSION
Our data show that α-synuclein binds to the protein chaperone 14-3-3 and shares a small region of homology with 14-3-3. By examining proteins known to associate with 14-3-3, we were able to identify three groups of proteins that also associate with α-synuclein. The proteins that bind both 14-3-3 and α-synuclein include five different isoforms of PKC, ERK, and BAD (Meller et al., 1996; Broadie et al., 1997). The ability of α-synuclein to concentrate ligands during immunoprecipitation appears to follow a rank order association of 14-3-3 > ERK > BAD > PKC isoforms. Based on this rank order, the association between α-synuclein and 14-3-3, ERK, or BAD might be stronger than that for PKC. However, the ability of overexpressed α-synuclein to inhibit PKC activity indicates that there is a significant functional association between the two proteins and suggests that the limited ability of α-synuclein to concentrate PKC during immunoprecipitations reflects technical rather than functional issues.
α-Synuclein shares a region of homology with 14-3-3 proteins, binds to many of the same proteins as 14-3-3, and modifies the activity of these proteins. Based on this, we propose that α-synuclein functions like 14-3-3 proteins and could be considered as part of a 14-3-3 superfamily. Interestingly, the same amino acids that are homologous between α-synuclein and 14-3-3 are also present in β- and γ-synuclein, which suggests that these two proteins might also be part of this superfamily. In addition, β- and γ-synuclein might also bind 14-3-3, PKC, BAD, and ERK, like α-synuclein. The 14-3-3 family of proteins are thought to be protein chaperones that bind to kinases and stabilize their activity (Tzivion et al., 1998). Examination of the mechanism of action of 14-3-3 suggests potential models for how α-synuclein might act. In the case of PKC, 14-3-3 binds to a phosphorylated epitope on PKC and holds the protein in an inactive conformation that prevents it from translocating to the membrane, despite the presence of diacylglycerol and calcium (Aitken et al., 1995; Meller et al., 1996; Reurther and Pendergast, 1996;Matto-Yelin et al., 1997). For Raf-1, this interaction is understood in even more detail (Muslin et al., 1996; Yaffe et al., 1997). After activation of Raf-1 by Ras, dimeric 14-3-3 binds to Raf-1 and stabilizes it in a conformation that remains active even after Ras has dissociated (Tzivion et al., 1998). Thus, 14-3-3 prolongs the duration of Raf-1 activation. For both PKC and Raf-1, the essential function of 14-3-3 is to stabilize the protein in a particular conformation, either active, as with Raf-1, or inactive, as with PKC. Based on the functional and physical homology to 14-3-3, we hypothesize that α-synuclein has a similar chaperone function. Previous reports have suggested that α-synuclein is a chaperone because of its open, unstructured character in solution (Weinreb et al., 1996). Our data now provide biochemical support for this hypothesis.
14-3-3 might therefore be a useful model for studying α-synuclein. The specific binding patterns of 14-3-3 and α-synuclein, however, appear to differ. Raf-1, which is a protein known to bind to 14-3-3, does not bind to α-synuclein (Tzivion et al., 1998). In addition, whereas 14-3-3 binds to phosphorylated BAD, α-synuclein binds to dephospho-BAD (Zha et al., 1996). If α-synuclein binds to proteins as part of a complex with 14-3-3, we would expect α-synuclein to associate with all proteins that associate with 14-3-3, but this is not what we observe. The lack of binding to Raf-1 and the binding to dephospho-BAD suggest that α-synuclein binds to proteins independently of 14-3-3 rather than as part of a larger 14-3-3–α-synuclein complex. Raf-1 has been shown to bind to the C terminus of 14-3-3, which is a region of the protein that shares no homology with α-synuclein (Tzivion et al., 1998). Thus, the inability of α-synuclein to bind to Raf-1 is consistent with the lack of homology between α-synuclein and the C-terminal domain of 14-3-3.
Based on protein binding patterns of α-synuclein, we were also able to identify functional consequences of increased expression of α-synuclein. Overexpression of α-synuclein inhibits PKC activity. Interestingly, despite inhibiting PKC activity, α-synuclein does not inhibit PKC membrane translocation. This suggests that α-synuclein acts by blocking the catalytic site of PKC but does not prevent the conformational change associated with membrane translocation. Multiple studies have documented the dissociation between PKC translocation and PKC activation (Lu et al., 1994; Mochly-Rosen and Kauvlar, 1998). The interaction of α-synuclein with PKC provides a mechanism through which the a dissociation of PKC translocation and activation could occur.
Another functional consequence of α-synuclein expression that we investigated was toxicity. The ability of α-synuclein to inhibit PKC and to bind to BAD suggested to us that overexpression of α-synuclein might be toxic. Consistent with this hypothesis, we observed that α-synuclein levels increase during conditions promoting cell stress, overexpression of α-synuclein induces toxicity, apparently by increasing necrosis, and reduced expression of α-synuclein protects against toxicity. Because α-synuclein appears to associate with multiple proteins, the mechanism of α-synuclein toxicity may be multifactorial. Our preliminary data, however, suggest that the added toxicity associated with overexpression of A53T and A30P α-synuclein is caused by the actions of BAD. Although the mutant α-synuclein constructs A53T and A30P are toxic when transfected alone, neither mutant form of α-synuclein induces significant toxicity when cotransfected with BAD. The inability of mutant α-synuclein to increase toxicity in cells that have been cotransfected with BAD is consistent with a hypothesis that the two proteins (mutant α-synuclein and BAD) use the same mechanism of toxicity and that the additional toxicity seen with both mutant α-synucleins is caused by activation of BAD. Proof of this point, however, awaits a demonstration that inhibiting BAD activity prevents the added toxicity associated with the mutant α-synucleins.
Unlike mutant α-synucleins, wild-type α-synuclein causes similar toxicity in control or BAD transfected cells. This suggests that wild-type α-synuclein does not activate BAD. The binding of wild-type α-synuclein to BAD contrasts with its apparent inability to affect BAD toxicity. This discrepancy between binding and activity is also seen with 14-3-3 protein, which binds to BAD but has not been shown to affect BAD function (Zha et al., 1996). The inability to detect regulation of BAD can occur because protein chaperones, such as 14-3-3, sometimes regulate the trafficking of their ligands rather than directly affecting their activity. In such cases, measurements of activity fail to detect regulation by the chaperone, despite a clear interaction between the chaperone and its ligand (Zha et al., 1996). Because of the physical and functional homology between α-synuclein and 14-3-3, we believe that the inability to detect regulation of BAD by wild-type α-synuclein might result from its function as a chaperone.
The unstructured character of α-synuclein may contribute to its tendency to aggregate in neurodegenerative diseases. Several studies have noted that α-synuclein has a tendency to aggregate in solution and that the A53T α-synuclein, but not the A30P α-synuclein, has an increased tendency to aggregate in vitro (Conway et al., 1998; Hashimoto et al., 1998; Paik et al., 1998). The A30P α-synuclein mutant might have a reduced tendency to associate with membranes, which could also increase its tendency to aggregate (El-Agnaf et al., 1998b; Jensen et al., 1998). The propensity of α-synuclein to aggregate may underlie its tendency to accumulate as focal accumulations in dystrophic neurites in neurodegenerative diseases such as PD, Alzheimer’s disease, multiple systems atrophy, or amyotrophic lateral sclerosis (Spillantini et al., 1998; Takeda et al., 1998).
The significance of accumulations of α-synuclein is unknown, but our study suggests that α-synuclein can be toxic. Our findings are supported by a recent study showing that α-synuclein is toxic when incubated with cells (El-Agnaf et al., 1998a). The ability of α-synuclein to cause toxicity suggests that accumulation of α-synuclein might contribute to the synaptic loss and cell death that underlies neurodegenerative diseases. The mechanism of toxicity is unknown. In the case of other protein aggregates, such as Aβ, the aggregated protein induces production of free radicals by binding to proteins that stimulate free radical production or by binding metals and stimulating free radical production through Fenton reactions (Behl et al., 1994; Yan et al., 1996; Atwood et al., 1998). Free radical production is also thought to be important in PD because the neurons of the substantia nigra contain dopamine and iron, both of which generate free radicals. In addition, the neuroprotective transcription factor NF-κB has been shown to be activated in the substantia nigra in PD (Hunot et al., 1997). In our studies we observed that α-synuclein binds to the neurotoxic protein BAD, and the cell death induced by A53T and the A30P α-synuclein in cell lines appears to involve BAD. The linkage between α-synuclein and BAD raises the possibility that BAD contributes to neurodegeneration in familial PD. The putative contribution of BAD to neurodegeneration in the CNS needs to be investigated.
Footnotes
This research was supported in part by grants from the Neuroscience Research and Education Fund and the National Parkinson Foundation. We thank Michael Comb (New England Biolabs) for providing anti-phospho-BAD antibodies and Yahong Zhang for her technical assistance.
N. Ostrerova and L. Petrucelli contributed equally to this work.
Correspondence should be addressed to Dr. Benjamin Wolozin, Department of Pharmacology, Loyola University Medical Center, Building 102, Room 4644, 2160 South 1st Avenue, Maywood, IL 60154.
REFERENCES
- 1.Aitken A, Howell S, Jones D, Madrazo J, Martin H, Patel Y, Robinson K. Post-translationally modified 14-3-3 isoforms and inhibition of protein kinase C. Mol Cell Biochem. 1995;149:41–49. doi: 10.1007/BF01076562. [DOI] [PubMed] [Google Scholar]
- 2.Atwood C, Moir R, Huang X, Scarpa R, Bacarra N, Romano D, Hartshorn M, Tanzi R, Bush A. Dramatic aggregation of Alzheimer abeta by Cu(II) is induced by conditions representing physiological acidosis. J Biol Chem. 1998;273:12817–12826. doi: 10.1074/jbc.273.21.12817. [DOI] [PubMed] [Google Scholar]
- 3.Behl C, Davis J, Klier F, Schubert D. Amyloid β peptide induces necrosis rather than apoptosis. Brain Res. 1994;645:253–264. doi: 10.1016/0006-8993(94)91659-4. [DOI] [PubMed] [Google Scholar]
- 4.Broadie K, Rushton E, Skoulakis E, Davis R. Leonardo, a Drosophila 14-3-3 protein involved in learning, regulates presynaptic function. Neuron. 1997;19:391–402. doi: 10.1016/s0896-6273(00)80948-4. [DOI] [PubMed] [Google Scholar]
- 5.Clayton D, George J. The synucleins: a family of proteins involved in synaptic function, plasticity, neurodegeneration and disease. Trends Neurosci. 1998;21:249–254. doi: 10.1016/s0166-2236(97)01213-7. [DOI] [PubMed] [Google Scholar]
- 6.Conway K, Harper J, Lansbury P. Accelerated in vitro fibril formation by a mutant α-synuclein linked to early-onset Parkinson’s disease. Nature Med. 1998;4:1318–1320. doi: 10.1038/3311. [DOI] [PubMed] [Google Scholar]
- 7.Corpet F. Multiple sequence alignment with hierarchical clustering. Nucleic Acids Res. 1988;16:10881–10890. doi: 10.1093/nar/16.22.10881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Davidson W, Jonas A, Clayton D, George J. Stabilization of α-synuclein secondary structure on binding to synthetic membranes. J Biol Chem. 1998;273:9443–9449. doi: 10.1074/jbc.273.16.9443. [DOI] [PubMed] [Google Scholar]
- 9.Dubois T, Howell S, Amess B, Kerai P, Learmonth M, Madrazo J, Chaudhri M, Rittinger K, Scarabel M, Soneji Y, Aitken A. Structure and sites of phosphorylation of 14-3-3 protein: role in coordinating signal transduction pathways. J Protein Chem. 1997;16:513–522. doi: 10.1023/a:1026321813463. [DOI] [PubMed] [Google Scholar]
- 10.El-Agnaf O, Jakes R, Curran M, Middleton D, Ingenito R, Bianchi E, Pessi A, Neill D, Wallace A. Aggregates from mutant and wild-type α-synuclein proteins and NAC peptide induce apoptotic cell death in human neuroblastoma cells by formation of β-sheet and amyloid-like filaments. FEBS Lett. 1998a;440:71–75. doi: 10.1016/s0014-5793(98)01418-5. [DOI] [PubMed] [Google Scholar]
- 11.El-Agnaf O, Jakes R, Curran M, Wallace A. Effects of the mutations Ala30 to Pro and Ala53 to Thr on the physical and morphological properties of α-synuclein protein implicated in Parkinson’s disease. FEBS Lett. 1998b;440:67–70. doi: 10.1016/s0014-5793(98)01419-7. [DOI] [PubMed] [Google Scholar]
- 12.George JM, Jin H, Woods WS, Clayton DF. Characterization of a novel protein regulated during the critical period for song learning in the zebra finch. Neuron. 1995;15:361–372. doi: 10.1016/0896-6273(95)90040-3. [DOI] [PubMed] [Google Scholar]
- 13.Hashimoto M, Hsu L, Sisk A, Xia Y, Takeda A, Sundsmo M, Masliah E. Human recombinant NACP/α-synuclein is aggregated and fibrillated in vitro: relevance for Lewy body disease. Brain Res. 1998;799:301–306. doi: 10.1016/s0006-8993(98)00514-9. [DOI] [PubMed] [Google Scholar]
- 14.Hunot S, Brugg B, Ricard D, Michel P, Muriel M, Ruberg M, Faucheux B, Agid Y, Hirsch E. Nuclear translocation of NF-κB is increased in dopaminergic neurons of patients with Parkinson’s disease. Proc Natl Acad Sci USA. 1997;94:7531–7536. doi: 10.1073/pnas.94.14.7531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ichimura T, Ito M, Itagaki C, Takahashi M, Horigome T, Omata S, Ohno S, Isobe T. The 14-3-3 protein binds its target proteins with a common site located towards the C-terminus. FEBS Lett. 1997;413:273–276. doi: 10.1016/s0014-5793(97)00910-1. [DOI] [PubMed] [Google Scholar]
- 16.Jakes R, Spillantini M, Goedert M. Identification of two distinct synucleins from human brain. FEBS Lett. 1994;345:27–32. doi: 10.1016/0014-5793(94)00395-5. [DOI] [PubMed] [Google Scholar]
- 17.Jenco J, Rawlingson A, Daniels B, Morris A. Regulation of phospholipase D2: selective inhibition of mammalian phospholipase D isoenzymes by α and β synucleins. Biochemistry. 1998;37:4901–4909. doi: 10.1021/bi972776r. [DOI] [PubMed] [Google Scholar]
- 18.Jensen P, Nielsen M, Jakes R, Dotti C, Goedert M. Binding of α-synuclein to brain vesicles is abolished by familial Parkinson’s disease mutation. J Biol Chem. 1998;273:26292–26294. doi: 10.1074/jbc.273.41.26292. [DOI] [PubMed] [Google Scholar]
- 19.Kruger R, Kuhn W, Muller T, Woitalla D, Graeber M, Kosel S, Przuntek H, Epplen J, Schols L, Riess O. Ala30Pro mutation in the gene encoding α-synuclein in Parkinson’s disease. Nat Genet. 1998;18:106–108. doi: 10.1038/ng0298-106. [DOI] [PubMed] [Google Scholar]
- 20.Layfield R, Fergusson J, Aitken A, Lowe J, Landon M, Mayer R. Neurofibrillary tangles of Alzheimer’s disease brains contain 14-3-3 proteins. Neurosci Lett. 1996;209:57–60. doi: 10.1016/0304-3940(96)12598-2. [DOI] [PubMed] [Google Scholar]
- 21.Lu Y, Tremblay R, Jouishomme H, Chakravarthy B, Durkin JP. Evidence that the activation of an inactive pool of membrane-associated protein kinase C is linked to the IL-2-dependent survival of T lymphocytes. J Immunol. 1994;153:1495–1504. [PubMed] [Google Scholar]
- 22.Matto-Yelin M, Aitken A, Ravid S. 14-3-3 inhibits the dictyostelium mysoin II heavy-chain-specific protein kinase C activity by a direct interaction: identification of the 14-3-3 binding domain. Mol Biol Cell. 1997;8:1889–1899. doi: 10.1091/mbc.8.10.1889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Meller N, Liu YC, Collins TL, Bonnefoy-Berard N, Baier G, Isakov N, Altman A. Direct interaction between protein kinase C theta (PKC theta) and 14-3-3 tau in T cells: 14-3-3 overexpression results in inhibition of PKC theta translocation and function. Mol Cell Biol. 1996;16:5782–5791. doi: 10.1128/mcb.16.10.5782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mochly-Rosen D, Kauvlar L. Modulating protein kinase C signal transduction. Adv Pharmacol. 1998;44:91–145. doi: 10.1016/s1054-3589(08)60126-x. [DOI] [PubMed] [Google Scholar]
- 25.Muslin A, Tanner J, Allen P, Shaw A. Interaction of 14-3-3 with signaling proteins is mediated by the recognition of phosphoserine. Cell. 1996;84:889–897. doi: 10.1016/s0092-8674(00)81067-3. [DOI] [PubMed] [Google Scholar]
- 26.Paik SR, Lee JH, Kim DH, Chang CS, Kim YS. Self-oligomerization of NACP, the precursor protein of the non-amyloid β/A4 protein (A β) component of Alzheimer’s disease amyloid, observed in the presence of a C-terminal A β fragment (residues 25–35). FEBS Lett. 1998;421:73–76. doi: 10.1016/s0014-5793(97)01537-8. [DOI] [PubMed] [Google Scholar]
- 27.Polymeropoulos M, Higgins J, Golbe L, Johnson W, Ide S, Di Iorio G, Sanges G, Stenroos E, Pho L, Schaffer A, Lazzarini A, Nussbaum R, Duvoisin R (1997) Mapping of a gene for Parkinson’s disease to chromosome 4q21–q23. Science 1197–1199. [DOI] [PubMed]
- 28.Reurther G, Pendergast M. The roles of 14-3-3 proteins in signal transduction. Vitam Horm. 1996;52:149–175. doi: 10.1016/s0083-6729(08)60410-0. [DOI] [PubMed] [Google Scholar]
- 29.Spillantini MG, Crowther RA, Jakes R, Hasegawa M, Goedert M. α-Synuclein in filamentous inclusions of Lewy bodies from Parkinson’s disease and dementia with Lewy bodies. Proc Natl Acad Sci USA. 1998;95:6469–6473. doi: 10.1073/pnas.95.11.6469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Takeda A, Mallory M, Sundsmo M, Honer W, Hansen L, Masliah E. Abnormal accumulation of NACP/-synuclein in neurodegenerative disorders. Am J Pathol. 1998;152:367–372. [PMC free article] [PubMed] [Google Scholar]
- 31.Tu P, Galvin J, Baba M, Giasson B, Tomita T, Leight S, Nakajo S, Iwatsubo T, Trojanowski J, Lee V. Glial cytoplasmic inclusions in white matter oligodendrocytes of multiple system atrophy brains contain insoluble α-synuclein. Ann Neurol. 1998;44:415–422. doi: 10.1002/ana.410440324. [DOI] [PubMed] [Google Scholar]
- 32.Tzivion G, Luo Z, Avruch J. A dimeric 14-3-3 protein is an essential cofactor for Raf kinase activity. Nature. 1998;394:88–92. doi: 10.1038/27938. [DOI] [PubMed] [Google Scholar]
- 33.Ueda K, Fukushima H, Masliah E, Xia Y, Iwai A, Yoshimoto M, Otero D, Kondo J, Ihara Y, Saitoh T. Molecular cloning of cDNA encoding an unrecognized component of amyloid in Alzheimer’s disease. Proc Natl Acad Sci USA. 1993;90:11282–11286. doi: 10.1073/pnas.90.23.11282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Weinreb P, Zhen W, Poon A, Conway K, Lansbury P. NACP, a protein implicated in Alzheimer’s disease and learning, is natively unfolded. Biochemistry. 1996;35:13709–13715. doi: 10.1021/bi961799n. [DOI] [PubMed] [Google Scholar]
- 35.Wheeler-Jones C, Learmonth M, Martin H, Aitken A. Identification of 14-3-3 proteins in human platelets: effects of synthetic peptides on protein kinase C activation. Biochem J. 1996;315:41–47. doi: 10.1042/bj3150041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Yaffe M, Rittinger K, Volinia S, Caron P, Aitken A, Leffers H, Gamblin S, Smerdon S, Cantley L. The structural basis for 14-3-3:phosphopeptide binding specificity. Cell. 1997;91:961–971. doi: 10.1016/s0092-8674(00)80487-0. [DOI] [PubMed] [Google Scholar]
- 37.Yan S, Chen X, Fu J, Chen M, Zhu H, Roher A, Slattery T, Zhao L, Nagashima M, Morser J, Migheli A, Nawroth P, Stern D, Schmidt A. RAGE and amyloid-β peptide neurotoxicity in Alzheimer’s disease. Nature. 1996;382:685–691. doi: 10.1038/382685a0. [DOI] [PubMed] [Google Scholar]
- 38.Yang E, Zha J, Jockel J, Boise LH, Thompson CB, Korsmeyer SJ. Bad, a heterodimeric partner for Bcl-XL and Bcl-2, displaces Bax and promotes cell death. Cell. 1995;80:285–291. doi: 10.1016/0092-8674(95)90411-5. [DOI] [PubMed] [Google Scholar]
- 39.Zha J, Harada H, Yang E, Jockel J, Korsmeyer SJ. Serine phosphorylation of death agonist BAD in response to survival factor results in binding to 14-3-3 not BCL-X(L). Cell. 1996;87:619–628. doi: 10.1016/s0092-8674(00)81382-3. [DOI] [PubMed] [Google Scholar]