

Abstract
A number of studies have provided evidence that neuronal cell loss after stroke involves programmed cell death or apoptosis. In particular, recent biochemical and immunohistochemical studies have demonstrated the expression and activation of intracellular proteases, notably caspase-3, which act as both initiators and executors of the apoptotic process. To further elucidate the involvement of caspases in neuronal cell death induced by focal stroke we developed a panel of antibodies and investigated the spatial and temporal pattern of both caspase-8 and caspase-3 expression. Our efforts focused on caspase-8 because its “apical” position within the enzymatic cascade of caspases makes it a potentially important therapeutic target. Constitutive expression of procaspase-8 was detectable in most cortical neurons, and proteolytic processing yielding the active form of caspase-8 was found as early as 6 hr after focal stroke induced in rats by permanent middle cerebral artery occlusion. This active form of caspase-8 was predominantly seen in the large pyramidal neurons of lamina V. Active caspase-3 was evident only in neurons located within lamina II/III starting at 24 hr after injury and in microglia throughout the core infarct at all times examined. Terminal deoxynucleotidyl transferase-mediated biotinylated UTP nick end labeling, gel electrophoresis of DNA, and neuronal cell quantitation indicated that there was an early nonapoptotic loss of cortical neurons followed by a progressive elimination of neurons with features of apoptosis. These data indicate that the pattern of caspase expression occurring during delayed neuronal cell death after focal stroke will vary depending on the neuronal phenotype.
Keywords: stroke, ischemia, caspases, apoptosis, microglia, neuron
Ischemic insults affecting the CNS result in impaired cognitive abilities as well as compromised sensorimotor function. These deficits are generally attributed to a loss of neurons within the region of the ischemic insult. Several previous studies suggest that the loss of neurons via apoptosis might be important in this context (Linnik et al., 1993;MacManus et al., 1994; Li et al., 1995; Chen et al., 1998; Namura et al., 1998). Apoptosis is a form of programmed cell death (PCD) originally recognized by ultrastructural examination and characterized by degradation of nuclear chromatin, condensation of the cytoplasm and nucleus, and ultimately the fragmentation of the cell into “apoptotic bodies” (Kerr, 1969, 1971; Kerr et al., 1972). These distinct changes in cellular structure differentiate apoptosis from necrosis, an alternative form of cell death. The two processes of cell death, apoptosis and necrosis, are further distinguished by differing patterns of DNA degradation; the morphological changes of apoptosis have been shown to be associated with double-strand cleavage of nuclear DNA at the linker regions between nucleosomes (Wyllie, 1980; Afanas’ev et al., 1986). Genetic and biochemical studies have now demonstrated that a family of intracellular proteases participates in this coordinated disassembly of cellular structure.
Ellis and Horvitz (1986) reported the identification of two genes required for the elimination of a subset of cells via PCD in the nematode Caenorhabditis elegans during development. One of these genes, ced-3, was found to share extensive homology with human interleukin-1-converting enzyme (ICE) (Yuan et al., 1993), a novel cysteine protease required for the proteolytic activation of prointerleukin-1β into the active cytokine (Thornberry et al., 1992). ICE is now known to belong to a family of cysteine proteases, the caspases, which play a key role in both inflammation and PCD in mammals.
All identified members of the caspase family contain the conserved motif QAC(R/Q)G and typically reside within the cell as inactive proenzymes that are activated by proteolytic cleavage in response to a variety of cellular insults. Studies examining recombinant caspase activity (Faucheu et al., 1995; Fernandes-Alnemri et al., 1995, 1996;Tewari et al., 1995; Srinivasula et al., 1996), the sequential appearance of proteolytic activities cleaving fluorescent substrates (Enari et al., 1996), and affinity-labeling of active caspases (Takahashi et al., 1996, 1997; Yamin et al., 1996) support the hypothesis that multiple caspases are sequentially activated during apoptosis with caspase-8 often being the first caspase in the cascade.
Caspase-8 was identified via protein–protein interactions and has been shown to have protease activity toward most known caspases (Boldin et al., 1996; Fernandes-Alnemri et al., 1996; Muzio et al., 1996;Srinivasula et al., 1996). Unlike most other caspase family members, caspase-8 contains two death effector domain-like modules through which it interacts with FADD, an adaptor protein involved in recruiting caspase-8 for activation at either the CD95 death-inducing signaling complex (DISC) (Kischkel et al., 1995; Boldin et al., 1996;Muzio et al., 1996; Medema et al., 1997) or the tumor necrosis factor receptor-1 (Hsu et al., 1996). Activation of caspase-8 may also occur “downstream” of mitochondrial dysfunction and independent of signaling at the DISC (Fulda et al., 1997).
To investigate the possibility that caspase-8 activation might be involved in neuronal cell death after an ischemic insult we generated a panel of caspase antibodies and examined cellular expression and maturation of caspase-8 and caspase-3 after focal stroke. Our studies suggest that the death of cortical neurons involves multiple caspases and that the activation of caspase-3 in microglia may contribute to cell death in this cell population.
MATERIALS AND METHODS
Middle cerebral artery occlusion and tissue preparation. Adult male spontaneously hypertensive rats (SHR) (weight 250–350 gm) were obtained from Taconic Farms (Germantown, NY) and were maintained with food and water ad libitum at 23°C and 50% relative humidity for at least 7 d before surgery. Surgical procedures were as described previously (Barone et al., 1995). Briefly, rats were anesthetized with sodium pentobarbital (60 mg/kg, i.p.; Steris Laboratories, Phoenix, AZ) and placed in a stereotaxic head holder (David Kopf Instruments, Tujunga, CA), and body temperature was maintained at 37°C. Occlusion of the middle cerebral artery (MCA) was achieved at the level of the inferior cerebral vein. An incision was made between the orbit and the external auditory canal with dissection–retraction of the temporal muscle. The MCA was exposed through a 2–3 mm2 craniotomy made just rostral to the zygomatic–squamosal skull suture, directly over the artery. The dura over the MCA was opened, and the artery was positioned on the tip of a platinum–iridium wire (0.0045 inch diameter; Medwire, Mount Vernon, NY) mounted on a micromanipulator. The artery was pulled slightly away from the brain surface for simultaneous occlusion and transection by electrocoagulation (Force 2 Electrosurgical Generator; Valley Lab Inc.) without damaging the brain surface. In sham-operated rats, the dura mater was opened over the MCA, but the artery was not occluded. Blood pressure, blood gases, blood glucose concentrations, heart rate, and body temperature were monitored before and after surgery, and all parameters were within normal physiological range (Table 1). Rats were killed at 6 hr (n = 6), 24 hr (n = 6), 48 hr (n = 6), and 5 d (n = 3) after surgery; sham rats (n = 2) were killed 48 hr after surgery. Naive control rats (n = 2) were also examined. Rats were killed with 1 ml/kg sodium pentobarbital and perfused via the aorta with 50 mm Dulbecco’s PBS containing 2% paraformaldehyde for 15 min. The brain was then removed and post-fixed in PBS containing 2% paraformaldehyde for 4 d at 4°C. Brains were cryoprotected in 20% sucrose in PBS at 4°C, frozen in OCT (Tissue-Tek, Miles Inc., Elkhart, IN), and stored at −70°C until sectioned. Sections (12-μm-thick) were cut onto Fisher Scientific (Pittsburgh, PA) Superfrost Plus slides, dried on a warm plate at 37°C, and stored at −70°C until use.
Table 1.
Systemic parameters
| Occlusion (n = 5) | Sham (n = 3) | |||
|---|---|---|---|---|
| pre | post | pre | post | |
| MABP, mmHg | 136 ± 20 | 140 ± 15 | 137 ± 31 | 141 ± 27 |
| pH | 7.21 ± 0.02 | 7.24 ± 0.03 | 7.2 ± 0.02 | 7.23 ± 0.02 |
| PCO2, mmHg | 45 ± 10 | 42 ± 3.3 | 43 ± 6 | 43 ± 3 |
| PO2, mmHg | 54 ± 5 | 80 ± 9 | 58 ± 10 | 64 ± 9 |
| Rectal temp, °C | 37 ± 0.4 | 37 ± .3 | 37.5 ± 0.2 | 37 ± 0.4 |
| Heart rate | 321 ± 55 | 339 ± 27 | 342 ± 16 | 323 ± 20 |
| Glucose | 89 ± 20 | 103 ± 12 | 91 ± 7 | 80 ± 2 |
Values are mean ± SD; MABP, mean arterial blood pressure.
Terminal deoxynucleotidyl transferase-mediated biotinylated UTP nick end labeling. Terminal deoxynucleotidyl transferase-mediated biotinylated UTP nick end labeling (TUNEL) was performed according to the method of Gavrieli et al. (1992) with modifications of Davis et al. (1996). Slides were brought to room temperature and rehydrated in PBS. Endogenous peroxidase activity was quenched for 20 min in 3% H2O2 in methanol. Nuclei of tissue sections were stripped of proteins by incubation with 10 μg/ml proteinase K (Sigma, St. Louis, MO), 0.1 m Tris, pH 8, and 0.05 m EDTA at 37°C for 15 min. Reaction was stopped with wash in Tris for 5 min. A reaction mix of 400 pmol of biotinylated dATP (Life Technologies, San Diego, CA), 0.1 μl of CoCl2, and 25 U of terminal deoxynucleotidyl transferase (Promega, Madison, WI) was added to the sections, and slides were incubated at 37°C for 90 min. Slides were then washed in Tris for 5 min, incubated in ABC reagent for 60 min at room temperature, and washed in Tris three times for 5 min each. The sections were colorized with 0.02% DAB in 0.1 m Tris, pH 7.6, containing 0.02% H202 and then washed in Tris to end the reaction. Slides were washed in water, allowed to dry overnight, counterstained with hematoxylin, and coverslipped. Only dark-staining cells were counted as apoptotic phenotype.
DNA laddering. DNA was harvested from the infarcted area and the uninjured contralateral cortex of the rat brain 24 hr after permanent middle cerebral artery occlusion (pMCAO). The tissue was minced and then resuspended in digestion buffer (100 mmNaCl, 10 mm Tris, pH 8, 25 mm EDTA, 0.5% SDS, and 100 μg/ml proteinase K). Samples were digested for 16 hr at 50°C. After digestion, samples were treated with RNase (1 μg/ml) at 37°C for 1 hr, and then DNA was isolated by phenol/chloroform extraction. After precipitation, DNA was resuspended in Tris-EDTA, and then 10 μg of DNA was run on a 1% agarose gel containing ethidium bromide (FMC Bioproducts, Rockland, ME).
Antibody generation and Western blot analysis. The antibody used to detect procaspase-8 (SK441) was raised against full-length recombinant human caspase-8 purified from Escherichia coli. The antibody used to detect active caspase-8 (SK440) was raised against the p20/p10 fusion purified from E. coli. A neo-epitope peptide antibody (SK398) was generated to the C terminus of the p20 subunit of caspase-3 (GIETD). All antibodies are rabbit polyclonals. Western blot analysis was performed on rat brain, mouse liver, and Jurkat cell lysates spiked with 50 ng of recombinant purified human caspases to test for antibody specificity. Samples were run on a 16.5% SDS-PAGE gel under reducing conditions and electroblotted onto nitrocellulose. The blots were blocked with 5% dry milk in PBS plus 0.1% Tween 20. Antibodies were incubated with the blots in blocking buffer at 1:1000–1:10,000 for 1 hr and washed two times with PBS plus 0.1% Tween 20. Donkey anti-rabbit-HRP (1:5000; Amersham, Piscataway, NJ) was incubated in blocking buffer for 1 hr, washed, and visualized with ECL (Amersham).
Immunohistochemistry. For immunohistochemistry, slides were warmed to room temperature and placed under a vacuum for 7 min. Tissue was rehydrated in PBS, and endogenous peroxidase activity was quenched by immersion in 3% H2O2 in methanol for 10 min. A nonspecific blocking procedure making use of 1% bovine serum albumin was performed before application of primary antibodies. The antibody SK441 (procaspase-8) was used at a 1:4000 dilution. The antibody SK440 (active caspase-8) was used at a dilution of 1:2000, and the antibody SK398 (active caspase-3) was used at a dilution of 1:12,000. Cell-specific markers, OX-42 (microglia; 1:1000; Chemicon, Temecula, CA), GFAP (astrocytes; 1:2000; Chemicon), and NeuN (neurons; 1 μg/ml; Chemicon) were used to identify the phenotype of the caspase-expressing cells. All primary incubations were overnight at 4°C. A Vectastain ABC kit (Vector Laboratories, Burlingame, CA) was used to localize the primary antibody. A diaminobenzidine substrate was used for visualization of the catalyzed peroxidase-reaction product. After multiple rinses in PBS and a final rinse in distilled water, sections were counterstained with hematoxylin (Biomeda, Foster City, CA), dehydrated, cleared, and coverslipped. For double-labeling studies, fluorescent conjugated secondary antibodies (Molecular Probes, Eugene, OR) were used at a 1:400 dilution.
Adsorption studies. To test the specificity of the active caspase antibodies, we adsorbed the antibodies at their working dilution with either recombinant active human caspase-3 (p20/p10 fusion) or recombinant active human caspase-8 (p20/p10 fusion) overnight at 4°C and then used the primary antibody mixture to examine immunodetection of active caspase species in 24 hr brain tissue as per our usual immunohistochemical protocol.
Image analysis and quantitation. All images were collected and analyzed with an Olympus Optical (Tokyo, Japan) BX60 microscope equipped with the Spot digital camera (Diagnostic Instruments, Sterling Heights, MI) and Photoshop (Adobe Systems, San Jose, CA). For quantitative analysis of TUNEL and NeuN-positive cells, two fields from both lamina II/III and lamina V of the ischemic region in a single 12 μm section from each of three animals were randomly selected and quantitated. For the number of active caspase-8-positive neurons, three fields centered on lamina V and equally spaced along the dorsal to ventral extent of the core infarct in a single 12 μm section from each of three animals were quantitated. For the quantitation of active caspase-3-positive cells, four fields within the ischemic region in a single 12 μm section from each of three animals were randomly selected and quantitated. The total number of cells per field was assessed by counting the number of hematoxylin-counterstained nuclei in all of the above quantitated sections.
RESULTS
Time course of cortical apoptosis
We elected to use the terminal transferase enzyme and established TUNEL methodology to assess the time course of apoptosis within the core infarct of the ischemic rat brain. At 6 hr after pMCAO, there was no evidence of TUNEL-positive neurons in any of the cortical lamina (Fig.1A,B). By 24 hr after pMCAO, a moderate number of strongly labeled cells were found in lamina II/III (Fig. 1C) and lamina V (Fig.1D). Examination of the core infarct at 48 hr after pMCAO revealed robust positive staining of most cells situated within either lamina II/III (Fig. 1E) or lamina V (Fig.1F). This temporal sequence of developing presumptive apoptotic cell death was consistently observed in our experiments and is similar to that reported by Asahi et al. (1997) in a similar model of pMCAO.
Fig. 1.
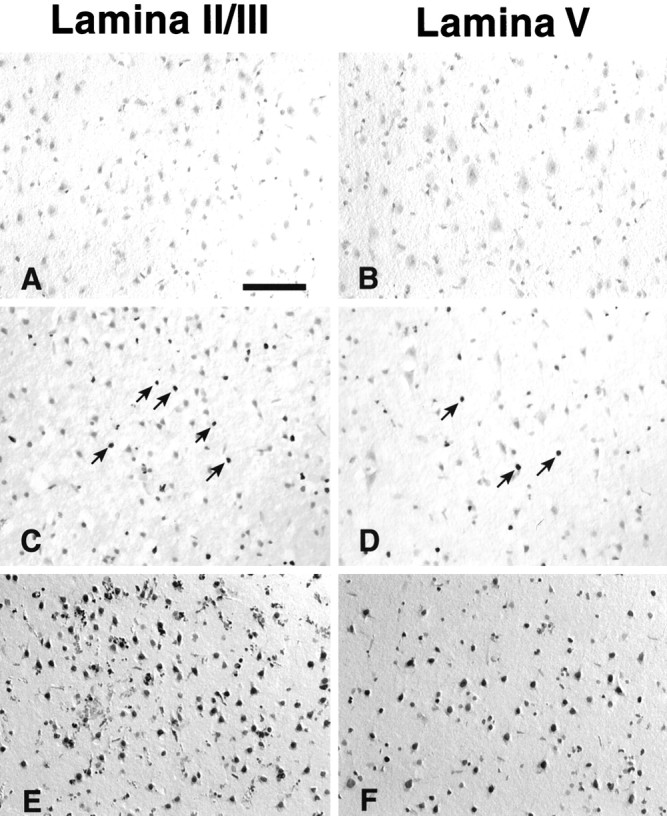
The time course of apoptosis within the core infarct as indicated by TUNEL. At 6 hr after injury there is no evidence of DNA damage within the core infarct (A,B). At 24 hr there is strong staining in a moderate number of cells distributed throughout lamina II/III (arrows, C) and lamina V (arrows, D). By 48 hr most of the cells within lamina II/III show robust staining (E), as do the majority of cells within lamina V (F). Scale bar: A–F, 250 μm.
Gel analysis of DNA
In addition to TUNEL analysis, we analyzed by gel electrophoresis the pattern of DNA cleavage. Using brain homogenates from 24 hr after injury, a time at which we could clearly identify apoptotic bodies by basic histological analysis (Fig.2A), we were able to demonstrate a laddered pattern of DNA in samples taken from the ipsilateral cortex (side of injury) but not from the uninjured contralateral cortex (Fig. 2B).
Fig. 2.

Gel analysis of DNA. Brain homogenates from 24 hr after injury were selected based on the TUNEL findings and basic histological analysis, which indicated the presence of apoptotic bodies at this time (A). A laddered pattern of DNA was observed in samples taken from the ipsilateral cortex (Ip; side of injury) but not from the uninjured contralateral cortex (Co) (B).St, Molecular weight standards.
Western blot analysis of caspases
Western blot analysis was performed to determine antibody specificity. Figure 3, left, shows that SK441, which recognizes an epitope(s) in the prodomain of caspase-8, detects full-length caspase-8, a 55 kDa band in alllanes (present in the Jurkat extract), as well as the processed prodomain (25 and 14 kDa bands) in the lane spiked with recombinant full-length caspase-8 (there is autoprocessing of the full-length recombinant caspase-8 resulting in a mixture of active and inactive enzyme). A single 55 kDa band is detected in normal rat brain. SK440 recognizes epitope(s) in the cleaved p20 subunit of caspase-8 (Fig. 3, middle). The epitope(s) appears to be unavailable for binding in the full-length caspase-8 because there is no recognition of either the endogenous caspase-8 in the Jurkat cell extracts or the recombinant full-length caspase-8 added to the sample (Fig. 3, middle). There is no cross reactivity of SK441 or SK440 to either recombinant human caspase-3 or caspase-4 (data not shown). The neo-epitope antibody generated to caspase-3, SK398, does not react with full-length caspase-3 but only recognizes the epitope after cleavage of the p10 subunit from the p20 subunit. Because of the sequence similarities between human caspase-3 and caspase-8, some crossreactivity was detected by Western blot using recombinant human caspase-3 and recombinant human caspase-8. Figure 3(right) shows SK398 reacts with the p20 subunit of human caspase-8 (18 kDa) and the prodomain plus p20 of human caspase-3 (21.5 kDa) and the p20 subunit of human caspase-3 (17 kDa). A single band corresponding to active caspase-3 is detected by SK398 in extracts of mouse liver after intravenous administration of anti-Fas. Crossreactivity between caspase-3 and caspase-8 when using SK398 is not a problem in the rodent because of sequence differences between human and rodent. The epitope for caspase-3 (CGIETD) is conserved in human, mouse, and rat (Mukasa et al., 1997; Ni et al., 1997), whereas the epitope from human caspase-8 (GIPVETD) is very different from rodent caspase-8 (FQGVPD) (GenBank accession number AJ000641). This explains the specificity of SK398 for active caspase-3 in the mouse liver extracts and rat brain.
Fig. 3.

Western blots demonstrating antibody specificity. SK441 (left) detects full-length endogenous caspase-8, a 55 kDa band, in all lanes of Jurkat extract and brain. In addition, SK441 recognizes the processed prodomain (there is autoprocessing of the full-length recombinant caspase-8, resulting in a mixture of active an inactive enzyme) as bands at 25 and 14 kDa in the lane spiked with recombinant full-length caspase-8. SK440 detects the cleaved p20 subunit of caspase-8 only (middle). SK398 (right) reacts with the p20 subunit of human caspase-8 (18 kDa) and the prodomain plus p20 of human caspase-3 (21.5 kDa) and the p20 subunit of human caspase-3 (17 kDa). A single band corresponding to active caspase-3 is detected in extracts of mouse liver after intravenous administration of anti-Fas. Mw, Molecular weight standards;8A, Jurkat extract spiked with recombinant active human caspase-8 (p20/p10 fusion); 8F, Jurkat extract spiked with full-length recombinant human caspase-8; Jk, Jurkat extract; Br, Rat brain; 3A, Jurkat extract spiked with recombinant human caspase-3 that has been cleaved to its active form; 3F, Jurkat extract spiked with full-length recombinant human caspase-3; Liver, mouse liver extract from animals that were not (−) injected with anti-Fas and animals that were injected with anti-Fas (+).
Multiple attempts to identify by Western analysis active forms of either caspase-8 or caspase-3 in rat brain samples taken from injured animals were all negative. Similarly, attempts to detect active caspases by enzymatic methods of analysis using suitable substrates all yielded negative results. Quantitative analysis of our in vivo findings suggests a plausible explanation for these negative results and will be presented later.
Expression of procaspase-8 in neurons
Initial studies examining caspase expression in uninjured rats were performed to assess procaspase-8 expression and to validate the specificity of antibodies recognizing active caspase forms. In both sham and normal rats, procaspase-8 was detected in neurons throughout all cortical lamina (Fig.4A). The pattern of intracellular staining for procaspase-8 was distinctly punctate and predominantly somatic, with only a small amount of immunoreactivity extending into proximal dendritic segments. In these uninjured animals, there was no observable positive immunostaining with either the active caspase-8 antibody (SK440) (Fig. 4B) or the active caspase-3 antibody (SK398) (data not shown).
Fig. 4.
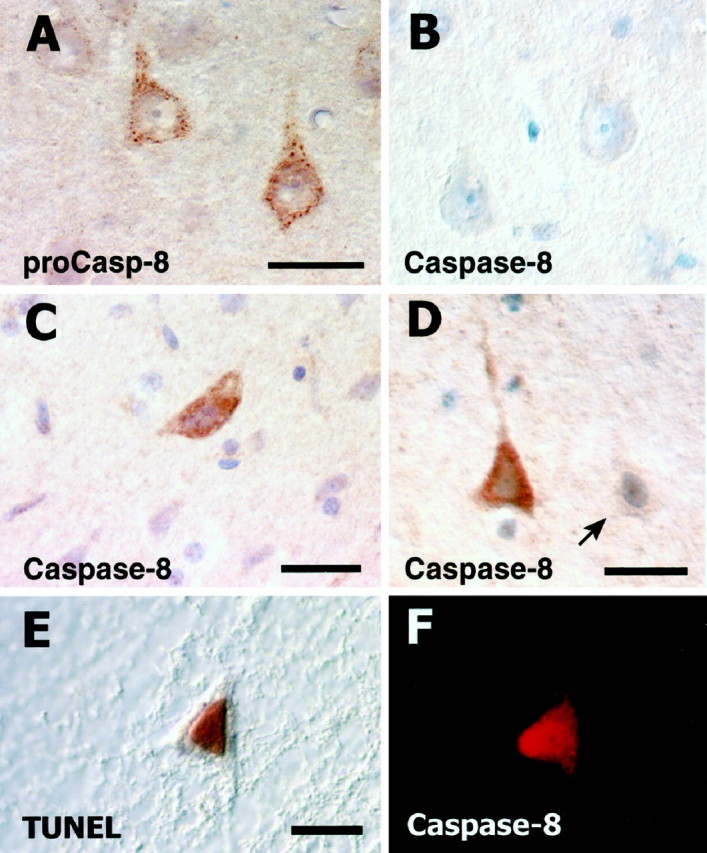
Expression of caspase-8 in cortical neurons of normal and ischemic rat brain. Punctate cytoplasmic staining for procaspase-8 is evident in two large pyramidal neurons of lamina V (A). The active caspase-8 antibody fails to demonstrate positive immunostaining in normal rat brain (B). At 6 hr after injury, positive immunoreactivity is readily detectable in the large pyramidal neurons of lamina V (C). Both positive and negative (arrow) immunostaining is still apparent in the large pyramidal neurons of lamina V at 48 hr (D). Colocalization of TUNEL reaction product (E) and positive immunofluorescence for active caspase-8 (F) could be found. Scale bars: (inA) A, B, 60 μm;C, D, 60 μm; (in E)E, F, 60 μm.
Cellular expression of active caspase-8 and caspase-3
Cell-specific antibodies and the antibodies SK440 (active caspase-8) and SK398 (active caspase-3) were used to assess (1) the temporal pattern of active caspase expression, (2) the phenotype of cells expressing active caspases, and (3) the intracellular localization of active caspase protein.
Positive immunoreactivity with SK440 (active caspase-8) was found within the core infarct as early as 6 hr in neurons scattered throughout the cortex but was most consistently observed in the large pyramidal neurons of lamina V (Fig. 4C). Positive immunoreactivity was also evident at 24 hr after pMCAO and as late as 48 hr (Fig. 4D) after injury. Unlike the discrete punctate staining seen for procaspase-8, the immunostaining for active caspase-8 was observed to be diffusely cytoplasmic. Neurons expressing active caspase-8 (Fig. 4F) demonstrated DNA damage as indicated by double labeling with TUNEL (Fig.4E).
A very different pattern of immunostaining was obtained with SK398, the active caspase-3 antibody. At 6 hr after pMCAO there was no positive neuronal staining with SK398. However, by 24 hr after pMCAO there was robust immunostaining of the small- to medium-sized pyramidal neurons of lamina II/III (Fig.5A,C); no staining of neurons was evident in the deeper cortical lamina (Fig.5B). The immunopositive neurons in lamina II/III often appeared in clusters or as a radial column of cells, and the intracellular localization of the detected active caspase was almost exclusively nuclear. A mixed nuclear–cytoplasmic compartmentalization was only rarely observed (Fig.5D). In addition to the positive immunostaining of neurons, there was also strong immunostaining of what appeared to be microglia based on morphological criteria; in double-labeling studies, colocalization was observed with the microglia marker OX42, confirming the presumed phenotypic classification (Fig. 5H). This staining could be found as early as 6 hr and in many instances revealed a cell with apparently fragmenting cellular processes (Fig.5E). Although many of these cells had typical microglial morphologies (Fig. 5F), others appeared atrophic with eccentrically located, pyknotic nuclei (Fig. 5G). No colocalization of active caspase-3 and GFAP, an astrocyte-specific marker, was observed.
Fig. 5.
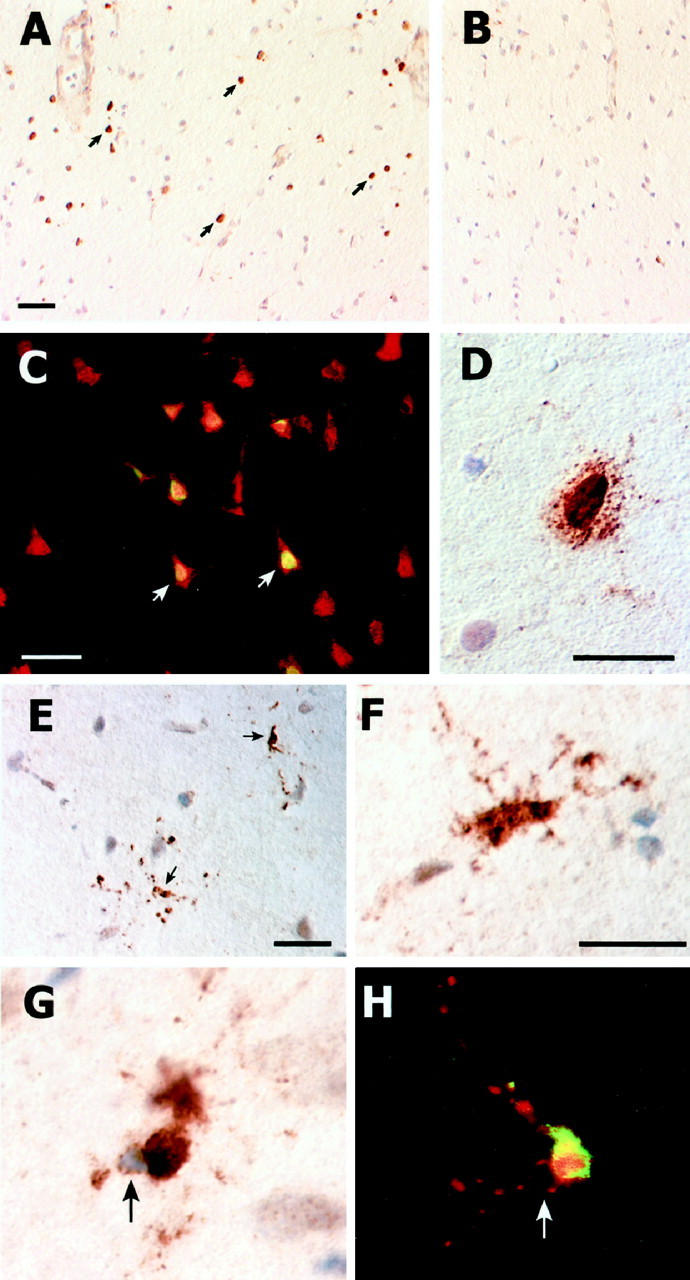
Detection of active caspase-3 in neurons and microglia located within the core infarct. All images are from 24 hr after injury. Positive immunostaining localized to the nucleus is seen in the small- and medium-sized pyramidal neurons of lamina II/III (A); several immunopositive neurons are indicated (arrows). No staining is evident in the deeper cortical lamina (B). Doubling labeling of active caspase-3-positive cells with NeuN confirms their neuronal phenotype (C). A mixed nuclear–cytoplasmic compartmentalization of immunostaining is only rarely observed (D). As early as 6 hr, small fragmenting cells with robust cytoplasmic immunostaining (arrows) could be seen (E). Immunopositive cells often had either a classical microglia morphology (F) or appeared atrophic (G) as evidenced by an abnormal cell shape and an eccentrically located pyknotic nuclei bounded by a thin rim of cytoplasm (arrow, G,H). Double labeling with the microglia-specific marker OX42, which is localized to the plasma membrane, confirms the microglial phenotype (H). Scale bars: (inA) A, B, 100 μm;C, E, 40 μm; D, 20 μm; (in F) F–H, 15 μm.
Adsorption control studies
To further evaluate the specificity of SK440 and SK398 we incubated samples of our working-dilution primary mixtures with both specific and irrelevant caspase peptides. Adsorption of SK440 with recombinant active human caspase-3 (irrelevant) did not affect immunoreactivity (Fig.6A), whereas adsorption with recombinant active human caspase-8 (relevant) completely abolished immunostaining (Fig. 6B). Adsorption of SK398 with recombinant active human caspase-8 (irrelevant) did not affect immunoreactivity (Fig. 6C), whereas adsorption with recombinant caspase-3 (relevant) abolished all immunostaining (Fig.6D).
Fig. 6.

The specificity of SB440 and SB398 was further evaluated by adsorption with caspase peptides. Adsorption of SB440 with recombinant human caspase-3 (irrelevant peptide control) did not have an affect on immunostaining (A), whereas adsorption with recombinant human caspase-8 completely abolished immunostaining (B). Adsorption of SB398 with recombinant human caspase-8 (irrelevant peptide control) did not have an affect on immunostaining (C), whereas adsorption with recombinant caspase-3 abolished all immunostaining (D).
Quantitative analyses
Using the neuron-specific marker NeuN and TUNEL we examined the temporal kinetics of neuronal cell loss and of cellular DNA damage. Counts of NeuN-positive cells (Fig.7A) revealed a biphasic loss of neurons in which an initial steady decline (0–24 hr) was followed by a plateau (24–48 hr) and subsequent loss (>48 hr). The decrease in neurons between 0 and 24 hr was found to be significant (p < 0.05) as was the difference between 48 hr and 5 d (p < 0.05) as measured by Tukey’s HSD test. Between 24 and 48 hr the decrease in neurons staining positively for NeuN was not significant. During the plateau phase, a rise in TUNEL-positive cells occurred (Fig. 7A), indicating the onset of DNA damage and presumptive apoptosis. The increase in TUNEL-positive cells was significantly greater at 48 hr compared with either 24 hr or 5 d (p < 0.05) as measured by Tukey’s HSD test.
Fig. 7.
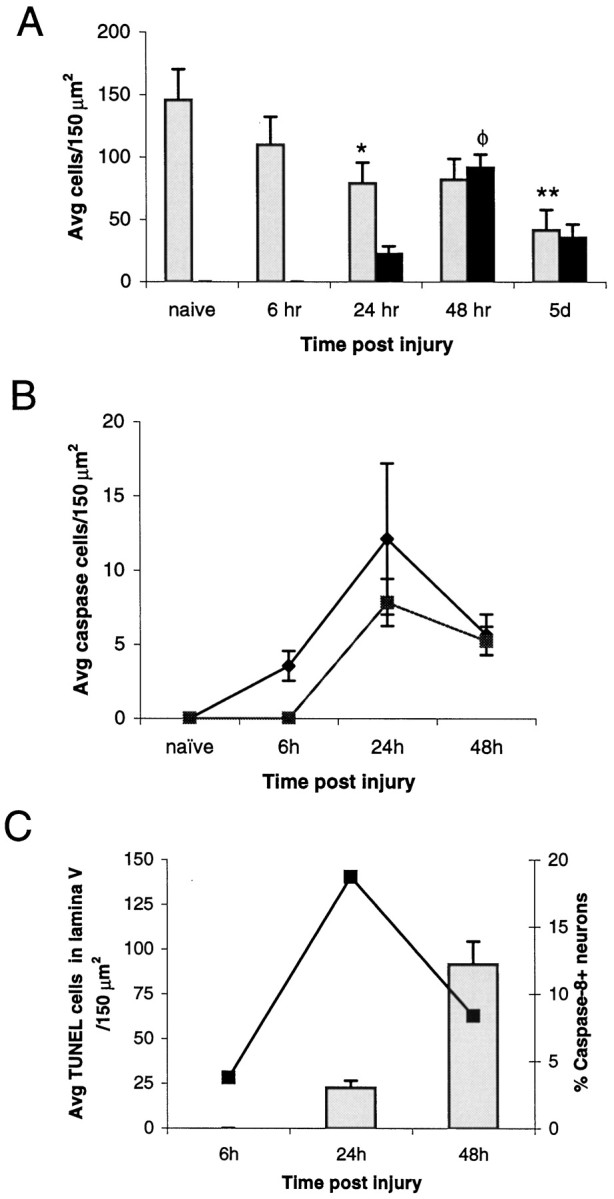
Quantitative analysis of cell death and active caspase expression. The quantitation of NeuN-positive cells (A, gray bars) indicated that there was a significant loss of neurons at 24 hr (*p < 0.05) compared with naïve, which was followed by a plateauing of neuronal cell loss (24–48 hr) and a subsequent significant loss of neurons detectable at 5 d (**p < 0.05) compared with 48 hr. The analysis of TUNEL (A,black bars) indicated a significant number of cells with DNA damage at 48 hr after injury (φp < 0.05) compared with naïve. The absence of TUNEL before 24 hr after injury indicates that the initial wave of neuronal loss was nonapoptotic. Temporal expression of active caspase-8 (B, ♦) preceded expression of caspase-3 (B, ▪). Peak expression of both caspases was observed at 24 hr, before the peak in TUNEL-positive cells (A). Analysis focusing on lamina V (C) indicated that the number of active caspase-8 (▪)-immunopositive neurons at 24 hr accounted for 19% of the total neuronal population of lamina V and that this peak of active caspase-8-expressing neurons preceded the rise of TUNEL (gray bars) within this lamina.
Quantitation of active caspase-expressing cells indicated that the maximum number of either active caspase-8- or active caspase-3-positive cells was found at 24 hr after injury (Fig. 7B). However, even at this time point, the number of immunoreactive cells accounted for a very small percentage of the total cell population; for active caspase-8-positive cells this amounted to 9% and for active caspase-3 this amounted to only 6%. Additional analysis indicated that the number of active caspase-8-positive neurons found in lamina V of the cortex at 24 hr accounted for 19% of the total neuronal population of lamina V. This peak in active caspase-8 expression directly preceded a sharp increase in the number of TUNEL-positive cells found in lamina V at 48 hr (Fig. 7C).
DISCUSSION
In the present study, we examined the expression of caspase-8 and caspase-3 to determine whether these enzymes play a role in the loss of cortical neurons subsequent to permanent focal stroke. A model of focal stroke was used that has consistent physiological parameters within normal range for the SHR rat (Table 1). Previous reports characterizing this occlusion model in SHR rats have demonstrated a significantly greater consistency in the infarct volume compared with that generated in either Wistar Kyoto or Sprague Dawley rats (Barone et al., 1992).
Positive immunostaining obtained with the procaspase-8-specific antibody SK441 demonstrated constitutive expression of procaspase-8 in the majority of cortical neurons. As early as 6 hr after focal stroke, a subpopulation of cortical neurons located predominantly in lamina V demonstrated proteolytic processing of caspase-8 as identified by the active caspase-8-specific antibody SK440. The number of active caspase-8-immunopositive neurons in lamina V reached a peak at 24 hr, when close to 20% of the total neuronal population in lamina V could be immunolabeled; this peak expression of active caspase-8 directly preceded the rise in TUNEL and presumptive apoptosis. In contrast, active caspase-3 expression in neurons was restricted to small clusters of cells distributed within lamina II/III and was first detectable at 24 hr after focal stroke. Besides this limited pattern of neuronal caspase-3 expression, microglia throughout the entire region of the infarct stained intensely for active caspase-3 beginning as early as 6 hr after stroke. The small percentage of cells expressing either active caspase-8 or active caspase-3 is probably the reason that neither active enzyme was detected by Western blot analysis or by enzymatic assay.
Our results indicate that there is an intracellular redistribution of caspase-8 associated with its activation. This change in intracellular distribution reflected a greater dispersal of caspase-8 throughout the cytoplasm because the proenzyme had a restricted localization that was of a discrete punctate nature. This punctate pattern of procaspase-8 immunostaining was suggestive of a lysosomal localization and bore a marked similarity to the pattern of neuronal staining obtained with antibodies to lysosomal enzymes reported previously (Cataldo et al., 1990, 1994). If caspase-8 activation involves signaling through CD95, whose expression has been shown to be upregulated in neurons after ischemic brain injury (Matsuyama et al., 1995), then the inactive form of caspase-8 may associate with the CD95/DISC within an endolysosomal compartment and become dissociated from this organelle on activation. A role for an acidic compartment in the induction of apoptosis by tumor necrosis factor-α (TNF) binding to the CD95-related protein TNFR-1 has been shown recently (Monney et al., 1998). Although this is only speculation, the diffuse cytoplasmic dispersal of active caspase-8 that we found would certainly facilitate its action on known caspase-8 substrates because most of these substrates are believed to reside within the cytoplasm of the cell.
In the current study, active caspase-3 was found to be localized within the nucleus of cortical neurons. This nuclear localization of active caspase-3 in neurons correlates with its well established role as an inactivator of the nuclear protein poly (ADP-ribose) polymerase (as well as other nuclear substrates) and is in agreement with previous studies demonstrating a nuclear localization of active caspase-3 in cells undergoing apoptosis (Martins et al., 1997a,b). Because procaspase-3 has been shown to reside within the cytoplasm of neurons (Namura et al., 1998), the observed nuclear localization would indicate an intracellular redistribution of caspase-3 on activation. The mixed nuclear–cytoplasmic localization of active caspase-3 in neurons that we occasionally observed is in agreement with this, as is the report of both nuclear and cytoplasmic localization of active caspase-3 in neurons after ischemia (Namura et al., 1998).
A distinct laminar pattern of active caspase expression in neurons was found: active caspase-8 expression was predominantly in lamina V, whereas active caspase-3 expression was restricted to lamina II/III. This pattern of active caspase expression suggests that the molecular mechanisms of cell death differ between the populations of neurons comprising the cerebral cortex and that selective inhibitors of specific caspases may ameliorate the loss of distinct neuronal classes. Indeed, the evidence presented here would indicate that an inhibitor of caspase-8 may prove effective in decreasing the loss of lamina V pyramidal neurons, a population of neurons functionally specialized for providing the efferent outflow of the cerebral cortex via long projection axons (i.e., corticospinal tract). Although future therapies for nervous system injury may involve cell grafts capable of restoring “associative” function via reformation of local synaptic connections, the ability of replacement cell populations to form axonal projections extending the entire rostral to caudal length of the adult CNS is questionable and would argue in favor of the development of therapeutics, whose mode of action is to preserve efferent neuronal populations such as the pyramidal neurons of lamina V.
The restricted pattern of active caspase-3 expression in neurons that we found was surprising because caspase-3 is considered to serve as an effector of the apoptotic process, and in previous reported work a widespread pattern of neuronal expression was demonstrated (Namura et al., 1998). This significant difference in findings may be related to interspecies variability or a difference in experimental procedure; in the Namura study the occlusion of the middle cerebral artery was transient (2 hr) followed by a period of reperfusion.
Reperfusion subsequent to ischemia has been postulated to adversely affect neuronal survival (reperfusion injury) (for review, seeHallenbeck and Dutka, 1990) and has been demonstrated to be associated with an earlier onset of neuronal apoptosis than permanent arterial occlusion (Li et al., 1995; Charriaut-Marlangue et al., 1996; Asahi et al., 1997; Namura et al., 1998). This difference in the temporal pattern of apoptosis observed between permanent and transient ischemia suggests that the dynamics and possibly the molecular mechanisms of neuronal apoptosis are affected by reperfusion. Curiously, the finding of nuclear caspase-3 immunoreactivity in only a few scattered neurons in lamina III and V recently reported by Chen et al. (1998) after transient (15 min) global ischemia in the rat is similar to our findings. Together, these results suggest that the pattern of active caspase-3 expression within the cerebral cortex after stroke is dependent on both the duration and extent of ischemia, as well as reperfusion.
In addition to the caspase-3 expression by neurons, microglial expression was also observed. Although the focus has been primarily on neuronal cell death after focal stroke, the glial cell population is also affected by ischemia. An examination of caspase expression by microglia after ischemia found upregulation of caspase-1 (also known as ICE or IL-1β-converting enzyme) in microglia of the gerbal hippocampus after global ischemia (Bhat et al., 1996). The authors suggest that expression of ICE mediates a component of the inflammatory response after injury by activating IL-1. Others have shown that microglia in this injury model undergo delayed apoptosis (Kato et al., 1996; Petito et al., 1998). Our findings of active caspase-3 expression in atrophic microglia in conjunction with the previous demonstration of caspase-1 expression suggest that microglia express multiple caspases before cell death after an ischemic insult.
The quantitative findings presented indicate that neuronal cell death within the core infarct after pMCAO occurs in two distinct phases. There is an early loss of neurons (∼46% of the entire population) within the first 24 hr that does not apparently involve DNA damage and is presumably a form of necrotic cell loss. These early and rapidly dying neurons were evenly distributed throughout the cortex and could not be discerned to possess any shared phenotypic trait other than an acute vulnerability to ischemic conditions, suggesting that mitigating loss within this population may best be accomplished by early restoration of blood flow. In contrast, during the subsequent 24 hr, many of the remaining neurons begin to undergo a process of cell death that featured DNA damage as evidenced by TUNEL and, in some neuronal populations, activation of either caspase-8 or caspase-3. Although other reports have demonstrated that TUNEL-positive neurons observed after cerebral ischemia fail to meet the ultrastructural criteria for apoptosis (Van Lookeren Campagne and Gill, 1996; Torres et al., 1997), the detection of active caspase species in neurons and a laddered pattern of DNA fragmentation shown here and previously (Namura et al., 1998; Chen et al., 1998; Linnik et al., 1993, 1995; Li et al., 1995) would indicate that neuronal cell death with characteristics of apoptosis does occur after focal stroke.
In conclusion, our results indicate that the death of cortical neurons after pMCAO involves at least two distinct forms of neuronal cell loss: an early necrotic loss and a delayed elimination of neurons that involves DNA damage and caspase activation. A role for caspase activation in microglial cell death is also suggested based on evidence of active caspase-3 expression in microglia, with morphological abnormalities indicative of cellular atrophy. Future studies to determine which other members of the caspase family are expressed after stroke are required for a greater understanding of the molecular mechanisms involved in neuronal cell death. This knowledge will in turn facilitate the development of therapeutics capable of ameliorating the incapacitating loss of function resulting from cerebral ischemia.
Footnotes
We thank Ray White, Paul Keller, and Kathy Gallagher for their excellent technical assistance. Animals were housed and cared for in accordance with the Guide for the Care and Use of Laboratory Animals [Department of Health, Education, and Welfare (Department of Health and Human Services) Publication No. (National Institutes of Health) 86–23, revised 1986, Office of Science and Health Reports, OPRR/National Institutes of Health, Bethesda, MD 20205]. The Institutional Animal Care and Use Committee of SmithKline Beecham Pharmaceuticals approved procedures using lab animals.
Drs. Velier and Ellison contributed equally to this work.
Correspondence should be addressed to Dr. Giora Z. Feuerstein’s present address: Cardiovascular Diseases Research, DuPont Pharmaceuticals, Route 141 and Henry Road, Building 400–3255, Wilmington, DE 19880-0400.
Dr. Ellison’s and Dr. Velier’s present address: Department of Neurological Surgery, University of California, 521 Parnassus Avenue, C224, San Francisco, CA 94143-0520.
REFERENCES
- 1.Afanas’ev VN, Korol BA, Mantsygin YA, Nelipovich PA, Pechatnikov VA, Umansky SR. Flow cytometry and biochemical analysis of DNA degradation characteristics of two types of cell death. FEBS Lett. 1986;194:347–350. doi: 10.1016/0014-5793(86)80115-6. [DOI] [PubMed] [Google Scholar]
- 2.Asahi M, Hoshimaru M, Uemura Y, Tokime T, Kojima M, Ohtsuka T, Matsuura N, Aoki T, Shibahara K, Kikuchi H. Expression of interleukin-1β converting enzyme gene family and bcl-2 gene family in the rat brain following permanent occlusion of the middle cerebral artery. J Cereb Blood Flow Metab. 1997;17:11–18. doi: 10.1097/00004647-199701000-00003. [DOI] [PubMed] [Google Scholar]
- 3.Barone FC, Price WJ, White RF, Willette RN, Feuerstein GZ. Genetic hypertension and increased susceptibility to cerebral ischemia. Neurosci Biobehav Rev. 1992;16:219–233. doi: 10.1016/s0149-7634(05)80182-4. [DOI] [PubMed] [Google Scholar]
- 4.Barone FC, Gillegass LM, Tzimas MN, Schmidt DB, Foley JJ, White RF, Price WJ, Feuerstein GZ, Clark RK, Griswold DE, Sarau HM. Time-related changes in myeloperoxidase activity and leukotriene by receptor binding reflect leukocyte influx in cerebral focal stroke. Mol Chem Neuropathol. 1995;24:13–30. doi: 10.1007/BF03160109. [DOI] [PubMed] [Google Scholar]
- 5.Bhat RV, DiRocco R, Marcy VR, Flood DG, Zhu Y, Dobrzanski P, Siman R, Scott R, Contreras PC, Miller M. Increased expression of IL-1β converting enzyme in hippocampus after ischemia: selective localization in microglia. J Neurosci. 1996;16:4146–4154. doi: 10.1523/JNEUROSCI.16-13-04146.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Boldin MP, Goncharov TM, Goltsev YY, Wallach D. Involvement of MACH, a novel MORT1/FADD-interacting protease, in FAS/APO-1 and TNF receptor induced cell death. Cell. 1996;85:803–815. doi: 10.1016/s0092-8674(00)81265-9. [DOI] [PubMed] [Google Scholar]
- 7.Cataldo AM, Thayer CY, Bird ED, Wheelock TR, Nixon RA. Lysosomal proteinase antigens are prominently localized within senile plaques of Alzheimer’s disease: evidence for a neuronal origin. Brain Res. 1990;513:181–192. doi: 10.1016/0006-8993(90)90456-l. [DOI] [PubMed] [Google Scholar]
- 8.Cataldo AM, Hamilton DJ, Nixon RA. Lysosomal abnormalities in degenerating neurons link neuronal compromise to senile plaque development in Alzheimer’s disease. Brain Res. 1994;640:68–80. doi: 10.1016/0006-8993(94)91858-9. [DOI] [PubMed] [Google Scholar]
- 9.Charriaut-Marlangue C, Margaill I, Represa A, Popovici RT, Pltokine M, Ben-Ari Y. Apoptosis and necrosis after reversible focal ischemia: an in situ DNA fragmentation analysis. J Cereb Blood Flow Metab. 1996;16:186–194. doi: 10.1097/00004647-199603000-00002. [DOI] [PubMed] [Google Scholar]
- 10.Chen J, Nagayama T, Jin K, Stetler RA, Zhu Rl, Graham SH, Simon RP. Induction of caspase-3-like protease may mediate delayed neuronal death in the hippocampus after transient cerebral ischemia. J Neurosci. 1998;18:4914–4928. doi: 10.1523/JNEUROSCI.18-13-04914.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Davis EC, Popper P, Gorski RA. The role of apoptosis in sexual differentiation of the rat sexually dimorphic nucleus of the preoptic area. Brain Res. 1996;734:10–18. [PubMed] [Google Scholar]
- 12.Ellis H, Horvitz H. Genetic control of programmed cell death in the nematode C. elegans. Cell. 1986;44:817–829. doi: 10.1016/0092-8674(86)90004-8. [DOI] [PubMed] [Google Scholar]
- 13.Enari M, Talanian RV, Wong WW, Nagata S. Sequential activation of ICE-like and CPP32-like proteases during Fas-mediated apoptosis. Nature. 1996;380:723–726. doi: 10.1038/380723a0. [DOI] [PubMed] [Google Scholar]
- 14.Faucheu C, Diu A, Chan AW, Blanchet AM, Miossec C, Herve F, Collard-Dutilleul V, Gu Y, Aldape RA, Lippke JA, Rocher C, Su M, Livingston DJ, Hercend T, Lalanne J-L. A novel human protease similar to the interleukin-1β converting enzyme induces apoptosis in transfected cells. EMBO J. 1995;14:1914–1922. doi: 10.1002/j.1460-2075.1995.tb07183.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fernandes-Alnemri T, Litwack G, Alnemri ES. Mch2, a new member of the apoptotic Ced- 3/Ice cysteine protease gene family. Cancer Res. 1995;55:2737–2742. [PubMed] [Google Scholar]
- 16.Fernandes-Alnemri T, Armstrong RC, Krebs J, Srinivasula SM, Wang L, Bullrich F, Fritz LC, Trapani JA, Tomaselli KJ, Litwack G, Alnemri ES. In vitro activation of CPP-32 and Mch3 by Mch4, a novel human apoptotic cysteine protease containing two FADD-like domains. Proc Natl Acad Sci USA. 1996;93:7464–7469. doi: 10.1073/pnas.93.15.7464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fulda S, Sieverts H, Friesen C, Herr I, Debatin K-M. The CD95 (APO-1/Fas) system mediates drug-induced apoptosis in neuroblastoma cells. Cancer Res. 1997;57:3823–3829. [PubMed] [Google Scholar]
- 18.Gavrieli Y, Sherman Y, Ben-Sasson SA. Identification of programmed cell death in situ via specific labeling of nuclear DNA fragmentation. J Cell Biol. 1992;119:493–501. doi: 10.1083/jcb.119.3.493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hallenback JM, Dutka AJ. Background review and current concepts of reperfusion injury. Arch Neurol. 1990;47:1245–1254. doi: 10.1001/archneur.1990.00530110107027. [DOI] [PubMed] [Google Scholar]
- 20.Hsu H, Shu HB, Pan MG, Goeddel DV. TRADD–TRAF2 and TRADD–FADD interactions define two distinct TNF receptor-1 signal transduction pathways. Cell. 1996;84:299–308. doi: 10.1016/s0092-8674(00)80984-8. [DOI] [PubMed] [Google Scholar]
- 21.Kato H, Kogure K, Liu X-H, Araki T, Itoyama Y. Progressive expression of immunomolecules on activated microglia and invading leukocytes following focal cerebral ischemia in the rat. Brain Res. 1996;734:203–212. [PubMed] [Google Scholar]
- 22.Kerr JFR. An electron-microscope study of liver cell necrosis due to heliotrine. J Pathol. 1969;97:557–562. doi: 10.1002/path.1710970314. [DOI] [PubMed] [Google Scholar]
- 23.Kerr JFR. Shrinkage necrosis: a distinct mode of cellular death. J Pathol. 1971;105:13–20. doi: 10.1002/path.1711050103. [DOI] [PubMed] [Google Scholar]
- 24.Kerr JFR, Wyllie AH, Currie AR. Apoptosis: a basic biological phenomenon with wide ranging implications in tissue kinetics. Br J Cancer. 1972;26:239–257. doi: 10.1038/bjc.1972.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kischkel FC, Hellbardt S, Behrmann I, Germer M, Pawlita M, Krammer PH, Peter ME. Cytotoxicity-dependent APO-1 (Fas/CD95)-associated proteins form a death-inducing signaling complex (DISC) with the receptor. EMBO J. 1995;14:5579–5588. doi: 10.1002/j.1460-2075.1995.tb00245.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Li Y, Chopp M, Jiang N, Yao F, Zaloga C. Temporal profile of in situ DNA fragmentation after transient middle cerebral artery occlusion in the rat. J Cereb Blood Flow Metab. 1995;15:389–397. doi: 10.1038/jcbfm.1995.49. [DOI] [PubMed] [Google Scholar]
- 27.Linnik MD, Zobrist RH, Hatfield MD. Evidence supporting a role for programmed cell death in focal cerebral ischemia in rats. Stroke. 1993;24:2002–2009. doi: 10.1161/01.str.24.12.2002. [DOI] [PubMed] [Google Scholar]
- 28.Linnik MD, Miller JA, Sprinkle-Cavallo J, Mason PJ, Thompson FY, Montgomery LR, Schroder KK. Apoptotic DNA fragmentation in the rat cerebral cortex induced by permanent middle cerebral artery occlusion. Mol Brain Res. 1995;32:116–124. doi: 10.1016/0169-328x(95)00069-5. [DOI] [PubMed] [Google Scholar]
- 29.MacManus JP, Hill IE, Huang Z-G, Rasquinha I, Xue D, Buchan AM. DNA damage consistent with apoptosis in transient focal cerebral ischemic neocortex. NeuroReport. 1994;5:493–496. doi: 10.1097/00001756-199401120-00031. [DOI] [PubMed] [Google Scholar]
- 30.Martins LM, Kottke T, Mesner PW, Basi GS, Sinha S, Frigon N, Tatar E, Tung JS, Bryant K, Takahashi A, Svingen PA, Madden BJ, McCormick DJ, Earnshaw WC, Kaufmann SH. Activation of multiple interleukin-1β converting enzyme homologues in cytosol and nuclei of HL-60 cells during etoposide-induced apoptosis. J Biol Chem. 1997a;272:7421–7430. doi: 10.1074/jbc.272.11.7421. [DOI] [PubMed] [Google Scholar]
- 31.Martins LM, Mesner PW, Kottke TJ, Basi GS, Sinha S, Tung JS, Svingen PA, Madden BJ, Takahashi A, McCormick DJ, Earnshaw WC, Kaufmann SH. Comparison of caspase activation and subcellular localization in HL-60 and K562 cells undergoing etoposide-induced apoptosis. Blood. 1997b;90:4283–4296. [PubMed] [Google Scholar]
- 32.Matsuyama T, Hata R, Yamamoto Y, Tagaya M, Akita H, Uno H, Wanaka A, Furuyama J, Sugita M. Localization of Fas antigen mRNA induced in postishemic murine forebrain by in situ hybridization. Mol Brain Res. 1995;34:166–172. doi: 10.1016/0169-328x(95)00162-l. [DOI] [PubMed] [Google Scholar]
- 33.Medema JP, Scaffidi C, Kischkel FC, Shevchenko A, Mann M, Krammer PH, Peter ME. FLICE is activated by association with the CD95 death-inducing signaling complex (DISC). EMBO J. 1997;16:2794–2804. doi: 10.1093/emboj/16.10.2794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Monney L, Olivier R, Otter I, Jansen B, Poirier GG, Borner C. Role of an acidic compartment in tumor necrosis factor α induced production of ceramide, activation of caspase-3 and apoptosis. Eur J Biochem. 1998;251:295–303. doi: 10.1046/j.1432-1327.1998.2510295.x. [DOI] [PubMed] [Google Scholar]
- 35.Mukasa T, Urase K, Momoi MY, Kimura I, Momoi T. Specific expression of CPP32 in sensory neurons of mouse embryos and activation of CPP32 in the apoptosis induced by NGF withdrawal. Biochem Biophys Res Commun. 1997;231:770–774. doi: 10.1006/bbrc.1996.6002. [DOI] [PubMed] [Google Scholar]
- 36.Muzio M, Chinnaiyan AM, Kischkel FC, O’Rourke K, Shevchenko A, Ni J, Scaffidi C, Bretz JD, Zhang J, Gentz R, Mann M, Krammer PH, Peter ME, Dixit VM. Flice, a novel FADD-homologous ICE/CED-3-like protease, is recruited to the CD95 (FAS/APO-1) death-inducing signaling complex. Cell. 1996;85:817–827. doi: 10.1016/s0092-8674(00)81266-0. [DOI] [PubMed] [Google Scholar]
- 37.Namura S, Zhu J, Fink K, Endres M, Srinivasan A, Tomaselli KJ, Yuan J, Moskowitz M. Activation and cleavage of caspase-3 in apoptosis induced by experimental cerebral ischemia. J Neurosci. 1998;18:3659–3668. doi: 10.1523/JNEUROSCI.18-10-03659.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ni B, Wu X, Du Y, Su Y, Hamilton-Byrd E, Rockey PK, Rosteck P, Poirier GG, Paul SM. Cloning and expression of a rat brain interleukin-1 β-converting enzyme (ICE)-related protease (IRP) and its possible role in apoptosis of cultured cerebellar granule neurons. J Neurosci. 1997;17:1561–1569. doi: 10.1523/JNEUROSCI.17-05-01561.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Petito CK, Olarte JP, Roberts B, Nowak TS, Pulsinelli W. Selective glial vulnerability following transient global ischemia in rat brain. J Neuropathol Exp Neurol. 1998;57:231–238. doi: 10.1097/00005072-199803000-00004. [DOI] [PubMed] [Google Scholar]
- 40.Srinivasula SM, Ahmad M, Fernandes-Alnemri T, Litwack G, Alnemri ES. Molecular ordering of the Fas-apoptotic pathway: the Fas/Apo-1 protease Mch5 is a CrmA-inhibitable protease that activates multiple Ced-3/ICE-like cysteine proteases. Proc Natl Acad Sci USA. 1996;93:14486–14491. doi: 10.1073/pnas.93.25.14486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Takahashi A, Alnemri ES, Lazebnik YA, Fernandes-Alnemri T, Litwack G, Moir RD, Goldman RD, Poirier GG, Kaufmann SH, Earnshaw WC. Cleavage of lamina A by Mch2a but not CPP32: multiple ICE-related proteases with distinct substrate recognition properties are active in apoptosis. Proc Natl Acad Sci USA. 1996;93:8395–8400. doi: 10.1073/pnas.93.16.8395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Takahashi A, Hirata H, Yonehara S, Imai Y, Lee KK, Moyer RW, Turner PC, Mesner PW, Okazaki T, Sawai H, Kishi S, Yamamoto K, Okuma M, Sasada M. Affinity labeling displays the stepwise activation of ICE-related proteases by Fas, staurosporine, and CrmA-sensitive caspase-8. Oncogene. 1997;14:2741–2752. doi: 10.1038/sj.onc.1201131. [DOI] [PubMed] [Google Scholar]
- 43.Tewari M, Quan LT, O’Rourke K, Desnoyers S, Zeng Z, Beidler, Poirier GG, Salvesen GS, Dixit VM. Yama/CPP32β, a mammalian homolog of CED-3, is a CrmA-inhibitable protease that cleaves the death substrate poly(ADP-ribose) polymerase. Cell. 1995;81:801–809. doi: 10.1016/0092-8674(95)90541-3. [DOI] [PubMed] [Google Scholar]
- 44.Thornberry N, Bull H, Calaycay J, Chapman KT, Howard AD, Kostura MJ, Miller DK, Molineaux SM, Weidner JR, Aunins J, Elliston KO, Ayala JM, Casano FJ, Chin J, Ding G, Egger LA, Guffney EP, Limjuco G, Palyha OC, Raju SM, Rolando AM, Salley JP, Yamin TT, Lee TD, Shively JE, MacCross M, Mumford RA, Schmidt JA, Tocci MJ. A novel heterodimeric cysteine protease is required for interleukin-1β processing in monocytes. Nature. 1992;356:768–774. doi: 10.1038/356768a0. [DOI] [PubMed] [Google Scholar]
- 45.Torres C, Munell F, Ferrer I, Reventos J, Macaya A. Identification of necrotic cell death by TUNEL assay in the hypoxic-ischemic neonatal rat brain. Neurosci Lett. 1997;230:1–4. doi: 10.1016/s0304-3940(97)00445-x. [DOI] [PubMed] [Google Scholar]
- 46.Van Lookeren Campagne M, Gill R. Ultrastructural morphological changes are not characteristic of apoptotic cell death following focal cerebral ischaemia in the rat. Neurosci Lett. 1996;213:111–114. doi: 10.1016/0304-3940(96)12839-1. [DOI] [PubMed] [Google Scholar]
- 47.Yamin TT, Ayala JM, Miller DK. Activation of the native 45-kDa precursor form of interleukin-1-converting enzyme. J Biol Chem. 1996;271:13273–13282. doi: 10.1074/jbc.271.22.13273. [DOI] [PubMed] [Google Scholar]
- 48.Yuan J, Shaham S, Ledoux S, Ellis H, Horvitz H. The C. elegans cell death gene ced-3 encodes a protein similar to mammalian interleukin-1β-converting enzyme. Cell. 1993;75:641–652. doi: 10.1016/0092-8674(93)90485-9. [DOI] [PubMed] [Google Scholar]
- 49.Wyllie AH. Glucocorticoid-induced thymocyte apoptosis is associated with endogenous endonuclease activation. Nature. 1980;284:555–556. doi: 10.1038/284555a0. [DOI] [PubMed] [Google Scholar]