

Abstract
Repeated cocaine exposure upregulates κ opioids and their receptors in the mesocorticolimbic system; the ensuing κ-mediated dysphoria appears to contribute to addiction and withdrawal. As a potential rehabilitation strategy to reverse cocaine-induced κ sensitization, the present study used tritiated dopamine release assays to examine the induction of κ-opioid tolerance in cultured mesencephalic neurons. Administration of the κ agonist U69,593 inhibited tetrodotoxin-sensitive, spontaneous (EC50 = 1.5 nm), and potassium-stimulated (EC50 = 10 nm) release. These effects were blocked by pertussis toxin and by the κ antagonist nor-binaltorphimine. The 2 d agonist exposure (1 μm) caused a shift in the U69,593 dose–response curve that was greater in the potassium-stimulated paradigm (140-fold) than in the spontaneous release assay (sixfold). These results were attributable to the attenuation of κ-receptor signaling mechanisms and to dependence. In the stimulated release assay, attenuation of κ signaling caused by 4 hr of U69,593 exposure recovered with a half-life of 1.1 hr, whereas attenuation after 144 hr of exposure recovered slowly (t½ = 20 hr). In the spontaneous release assay, attenuation of κ-opioid signaling occurred slowly (t½ = 22 hr), and resensitization after a 144 hr exposure was rapid (t½ < 1 hr). κ-Opioid dependence was observed after 144 hr of U69,593 exposure. Thus multiple mechanisms of adaptation to κ-opioid exposure occur in mesocorticolimbic neurons. These data support the idea that the administration of κ opioids might facilitate drug rehabilitation.
Keywords: κ-opioid receptor; tolerance; dependence; mesencephalon; dopaminergic neurons; primary neuronal culture; neurotransmitter release; U69,593
Drugs of abuse alter dopaminergic neurotransmission in the mesocorticolimbic system (MCL), which comprises the ventral tegmental neurons projecting to the striatum and cortex (White, 1996; Wise, 1996). Endogenous opioids contribute to the molecular adaptations that occur in response to drugs such as cocaine. For example, μ opioids facilitate dopamine release in the MCL (Di Chiara and Imperato, 1988; Koob and Le Moal, 1997), whereas κ opioids like dynorphin decrease dopamine levels in these circuits (Di Chiara and Imperato, 1988; Spanagel et al., 1990; Donzanti et al., 1992). Because κ opioids oppose the effects of cocaine in this system, they may be useful in treating cocaine addiction (Spanagel et al., 1992;Shippenberg et al., 1996). Indeed, κ opioids have been shown to attenuate the acute behavioral effects of cocaine in rodents and can prevent the development of behavioral sensitization to cocaine (Heidbreder et al., 1993, 1995). κ Opioids also decrease cocaine self-administration in rodents and primates (Glick et al., 1995; Negus et al., 1997).
Although the reinforcing properties of drugs like cocaine are important for their abuse, so too is the dysphoria associated with their withdrawal (Koob and Le Moal, 1997). Repeated cocaine exposure increases the expression of κ receptors (Unterwald et al., 1994;Staley et al., 1997) and dynorphin mRNA (Sivam, 1989; Smiley et al., 1990; Hurd and Herkenham, 1993; Spangler et al., 1993; Steiner and Gerfen, 1993; Cole et al., 1995; Carlezon et al., 1998) in the MCL. Although it is not known whether increased dynorphin release accompanies this response, conceivably, withdrawal dysphoria is attributable to these adaptations (Hurd and Herkenham, 1993; Hyman, 1996; Koob and Le Moal, 1997). Thus one therapeutic strategy to facilitate drug rehabilitation might be to reverse the cocaine-induced increase in κ opioids and their receptors via the induction of tolerance (Shippenberg and Rea, 1997). Such an approach depends, however, on how readily the MCL develops tolerance to κ opioids.
Tolerance, defined as diminished drug effect because of drug exposure, can occur during opioid therapy. Mechanistically, it involves (1) attenuated signaling via receptor and receptor-linked systems and (2) compensatory alterations in the basal activity of the neuron. The latter also produce opioid dependence (Nestler and Aghajanian, 1997). κ-Opioid tolerance has been shown to occur in intact animal systems as well as in hippocampal slices (Gmerek and Woods, 1986; Bhargava et al., 1989; Jin et al., 1997). However, regional differences in susceptibility to opioid tolerance exist within the brain (Sim et al., 1996; Paronis and Woods, 1997).
To address the question of whether κ-opioid tolerance occurs in the MCL, we have cultured midbrain neurons associated with the MCL and have studied κ-induced changes in tritiated dopamine release. This is a sensitive and specific measure of κ-opioid function in this system. We have found that the κ agonist U69,593 inhibits spontaneous and potassium (K+)-stimulated dopamine release. We also have found that U69,593 exposure induces tolerance and dependence in this defined model system of presynaptic brain reward circuitry.
MATERIALS AND METHODS
Primary culture. Primaria (Fisher Scientific, Pittsburgh, PA) 24-well plates were prepared with poly-d-lysine (PDL; 0.5 mg/ml; Sigma, St. Louis, MO) in water overnight at room temperature. The PDL was washed off, and the plates were treated with mouse laminin (2 mg/cm2; Becton Dickinson, Franklin Lakes, NJ) in PBS for 1–4 hr at room temperature. Laminin–PBS solution was removed before the addition of the cell suspension. Timed-pregnant Sprague Dawley (Harlan, Indianapolis, IN) rats at gestational day 15 were anesthetized and killed with carbon dioxide. Embryos were removed and the ventral mesencephalon was dissected out, using standard landmarks, and placed in L-15 medium on ice. Then the tissue was minced and incubated with 0.25% trypsin and 0.05% DNase I in PBS for 15 min at 37°C. The supernatant was removed and replaced with DMEM with 10% fetal calf serum, and the cells were dissociated by passage through a constricted-bore glass pipette. Dissociated cells were passed through a nylon filter and transferred to Neurobasal medium (Life Technologies, Gaithersburg, MD), B27 supplement [(Life Technologies), 0.5 mm glutamine, 100 U/ml penicillin, and 100 μg/ml streptomycin] and plated onto the prepared plates at 1.5 × 105 cells/cm2 in 500 ml of Neurobasal medium. At 3 d intervals the medium was refreshed by replacing one-half with fresh Neurobasal medium. Release assays were performed after 16–21 d in culture. No differences were noted in the characteristics of the cultures within this age range.
3H-dopamine release assays. To perform release assays, we removed the culture medium and washed the cultures three times in Krebs’–Ringer’s solution [KRS; containing (in mm) 119 NaCl, 2.5 KCl, 1.3 MgSO4, 2.5 CaCl2, 1 NaH2PO4H20, 26.2 NaHCO3, and 10 glucose]. Cultures were incubated with 50 nm 7,8-3H-dopamine (3H-DA; 43 Ci/mmol; Amersham, Arlington Heights, IL) in KRS for 15 min at 37°C in 5% CO2. Then the 3H-DA solution was removed; the cultures were washed three times with KRS and subsequently were placed in 330 μl of KRS. Next, KRS was removed for analysis and replaced with 330 μl of KRS at 30, 60, 66, 72, 78, 84, 90, and 96 min time points. Between KRS changes the cultures were kept in 5% CO2 at 37°C. After collection of the 96 min time point the cells were lysed with 0.4 m perchloric acid (PCA). All fractions, including the acid lysate, were analyzed by liquid scintillation in 4 ml of Biosafe II (Research Products International, Mt. Prospect, IL). For potassium (K+)-stimulated release conditions, 15 mm K+ in KRS (K+/KRS, NaCl-adjusted to 106.5 mm) was added after removal of the 78 min time point. The K+/KRS was removed at the 84 min point and replaced with KRS. To assay the effects of U69,593 [Research Biochemicals (RBI), Natick, MA], quinpirole (RBI), tetrodotoxin (a gift from Dr. C. Zorumski, Washington University Medical School, St. Louis, MO), nor-binaltorphimine (nor-BNI; RBI), or CNQX (RBI) on spontaneous and K+-stimulated release, we added these drugs in KRS after the removal of the 72 min fraction, and they were present in subsequent fractions until the addition of PCA. For PTX (RBI) treatment, 1 μg/ml of PTX in PBS was added to the culture medium, and incubations were performed for 22 hr at 37°C in 5% CO2. The tritium content of the 78 min fraction was used to quantitate drug effects on spontaneous release. To calculate K+-stimulated release for each condition, we subtracted the average tritium content of the 78, 90, and 96 min fractions from the content of the 84 min fraction. Although the amount of DA released in the presence of 15 mmK+ can influence the spontaneous release in the 90 and 96 min fractions, we find this has only a small effect (<10%) on the calculation of the amount of K+-stimulated release. Total 3H-DA uptake was calculated by summation of tritium content from all of the fractions collected, including the acid lysate. Except where noted, all data are expressed as mean ± SEM.
U69,593 tolerance studies. In experiments evaluating the kinetics of U69,593-induced attenuation of κ-receptor function, cultures were incubated with 1 μm U69,593 in Neurobasal medium. For multi-day exposures U69,593 was refreshed in the same manner as the media, at 3 d intervals. To evaluate recovery from U69,593-induced attenuation, we washed the cultures five times with Neurobasal media over a 1 hr period and then returned them to conditioned Neurobasal medium. For experiments in which the recovery time was 2 hr, U69,593 was removed along with the Neurobasal medium, and the drug was washed off during the KRS washes that are part of the usual 3H-DA loading protocol. Five washes over a period of 40 min were sufficient to reduce the U69,593 concentration to a point at which its effects on spontaneous release were undetectable. For experiments without recovery times, U69,593 at 1 μm was present in the KRS used for washing and 3H-DA loading and in all subsequent fractions. For evaluation of the effects of antagonism with nor-BNI (1 μm; RBI) on spontaneous and K+-stimulated release, nor-BNI in KRS was added after removal of the 66 or 72 min time points, and release was quantitated as shown in Figure 2. Unless otherwise noted, the U69,593 exposure time period is measured from the time of the addition of U69,593 until the time it is washed off or until the time of 15 mm K+ addition in the 3H-DA release assay.
Fig. 2.

Representative experiment showing that the κ agonist U69,593 inhibits spontaneous and K+-stimulated 3H-dopamine release from mesencephalic neurons. Mesencephalic neurons were maintained in culture for 16 d, and 3H-DA release assays were performed. Data are expressed as a percentage of total 3H-DA uptake. Shown are the fractions collected 72, 78, 84, 90, and 96 min after the completion of 3H-DA loading. In both conditions shown, 15 mm K+ in KRS was added for the 84 min time fraction. The 15 mm K+/KRS was replaced with regular KRS for the 90 min fraction. In the U69,593-treated condition, 1 μm U69,593 was present in all of the fractions after the 72 min time point. The 78 min fraction shows the U69,593 effect on spontaneous release. Error bars show the mean ± SEM for three determinations. Error bars are not shown for SEM <2%.
Reverse transcription/PCR. RNA was extracted from mesencephalic tissue with Trizol (Life Technologies) according to the manufacturer’s instructions, and reverse transcription was performed. Equal amounts of RNA were used for each reverse transcription reaction [after standardization with ribosomal RNA (O’Malley et al., 1990)] along with an opioid receptor primer common to all three subtypes (ATGCTGGTGAACATGTTGTAGTA). Then cDNA transcripts were amplified by PCR, using the reverse transcription primer in combination with one of three subtype-specific 5′ primers (μ, AACACCAGCGACTGCTCA; δ, GTGCGGAGCTGCAGTTTT; κ, AACAGCAGCTCTTGGTTC). Primers were selected to span an intron to minimize background signal caused by genomic templates. PCR products were fractionated by polyacrylamide gel electrophoresis and analyzed with Vistra Green (Amersham) and quantitative fluorimaging.
Nonlinear regression. Nonlinear regression and curve fitting were performed with Prism2 (GraphPad Software, San Diego, CA).
Statistical analyses. The significance of effects between control cultures and drug treatments was calculated with post hoc Student’s unpaired two-tailed t tests with statistical software (Prism2, GraphPad Software). The 95% confidence intervals (95% C.I.) for the EC50 values calculated for Figure 4 and the half-times calculated for Figures 5-7 are provided in Results. In all comparisons in which the 95% C.I. values were nonoverlapping, the data sets that were compared were also significantly different by two-way ANOVA (p < 0.001; Prism2, GraphPad Software).
Fig. 4.
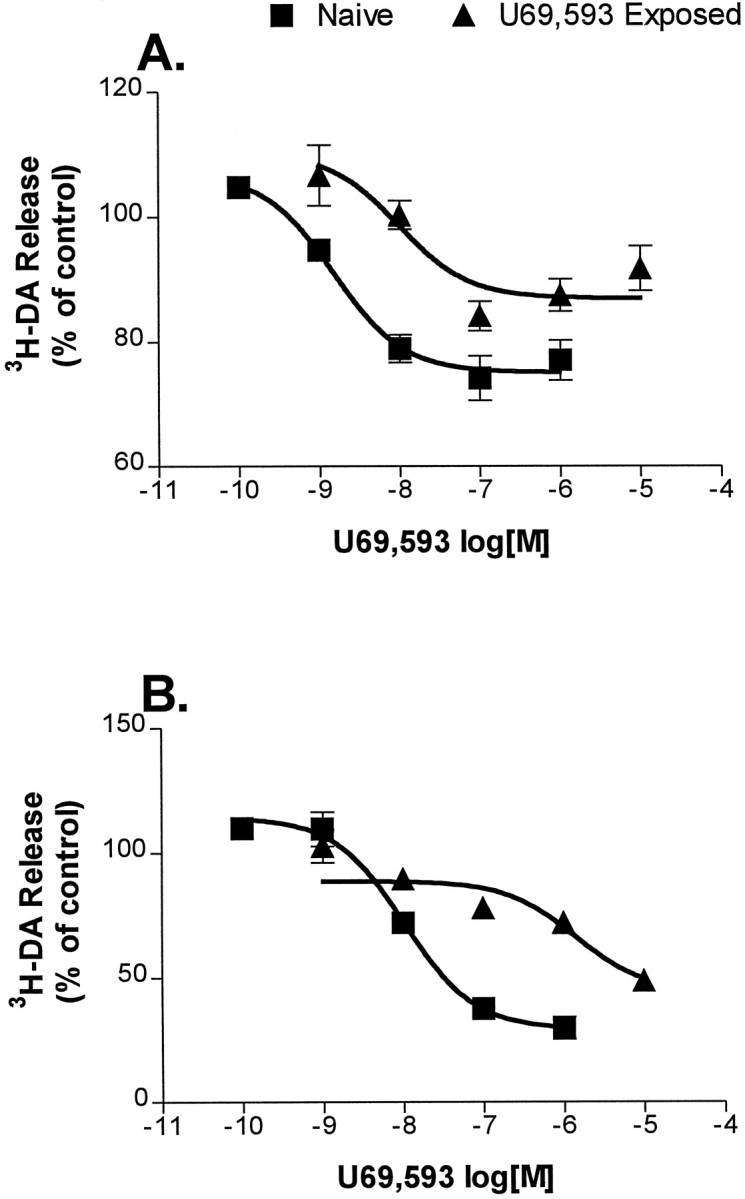
Cultures exposed to U69,593 for 48 hr show tolerance to U69,593. Cultures were incubated with or without 1 μm U69,593 for 48 hr. Subsequently, U69,593 was washed off and drug-free KRS was used for 3H-DA loading and for fractions up to 72 min thereafter. At that point, KRS containing the indicated concentrations of U69,593 was added to the cultures, and the U69,593 effect on spontaneous (A) and K+-stimulated (B) release was determined. Release is expressed as a percentage of release in U69,593-free, naïve cultures. The pointsrepresent the mean of 3–10 experiments ± SEM. Error bars are not shown for SEM <2%. Data are fit to a sigmoidal dose–response curve.
Fig. 5.
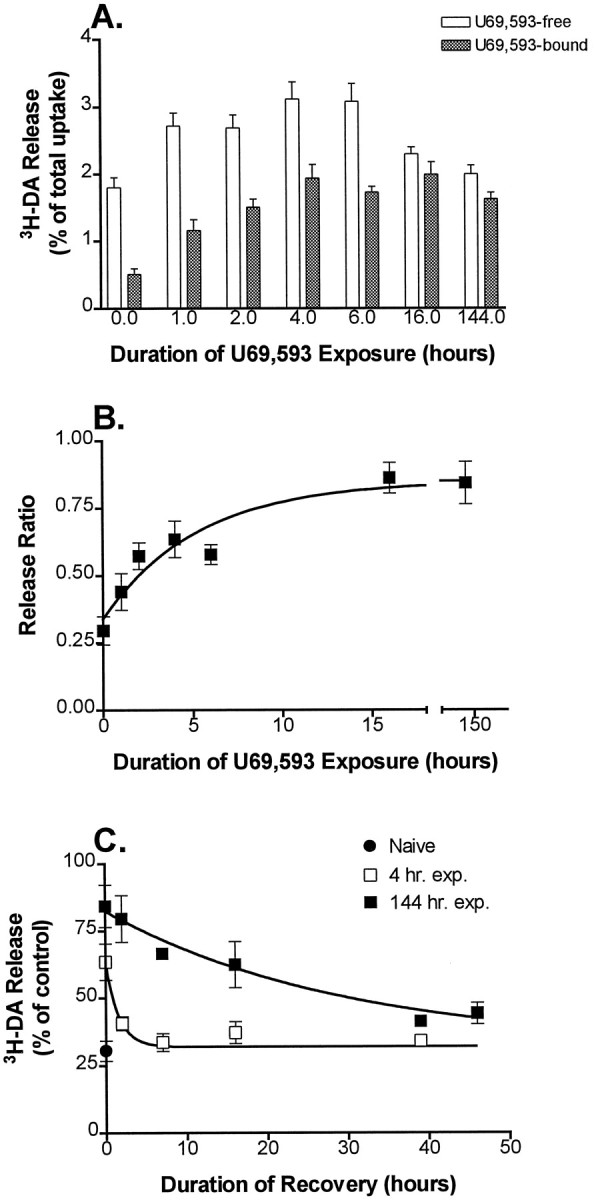
Onset and recovery kinetics of U69,593-mediated tolerance in the K+-stimulated 3H-DA release assay. A, B, Onset kinetics. Cultures were exposed to 1 μm U69,593 for 0, 1, 2, 4, 6, 16, or 144 hr before K+ stimulation. Release was determined in the presence of 1 μm U69,593 (filled bars) or after 1 μm nor-BNI was added to displace U69,593 from κ receptors (open bars). Thepoints at 0 hr of U69,593 exposure show the K+-stimulated release from naïve cultures in the absence (open bar) and presence (filled bar) of 1 μm U69,593.B, Data from A plotted versus time of U69,593 exposure. Points represent the ratio of release in the U69593-bound condition to the release after treatment with nor-BNI. Data are fit to a single exponential curve.Points represent the mean for 6–11 experiments; error bars show ± SEM. U69,593 exposure for 6 d slightly reduced (by ∼8%) the amount of 3H-DA taken up during the loading step of the release assay as compared with the naïve cultures, but this difference was not statistically significant (n = 6; p = 0.08).C, Recovery kinetics. Cultures were kept naïve (filled circle) or were exposed to 1 μm U69,593 for 4 hr (open squares) or 144 hr (filled squares). U69,593 was washed off, and the cultures were allowed to recover in KRS (2 hr time point) or in conditioned, drug-free medium (7, 16, 39, or 46 hr time points) before the assay of the U69,593 effect on stimulated release. The circle shows the effect of U69,593 on K+-stimulated release from naïve cultures. The squares show the effect of U69,593 after various time periods of recovery from the two treatment conditions. Results are expressed as a percentage of U69,593-free K+-stimulated release in control sister cultures that were pretreated, washed, and allowed to recover in an identical manner to the experimental cultures. Note that the values for release at 0 hr of recovery were carried over from the 4 and 144 hr time points of B. The curve represents the best fit of a single exponential. The points represent the mean for 3–14 experiments; error bars show ± SEM. Error bars are not shown for SEM <2%.
Fig. 6.

Onset and recovery kinetics of U69,593-mediated tolerance in the spontaneous 3H-DA release assay.A, Onset kinetics. Cultures were exposed to 1 μm U69,593 for various time periods, and spontaneous3H-DA release was determined in the presence of U69,593. Data are expressed as a percentage of 3H-DA release from naïve U69,593-free sister cultures. B, Recovery kinetics. Cultures were kept naïve or were U69,593-exposed for 144 hr. U69,593 effect on spontaneous release was determined at 0, 2, 6, 16, or 48 hr after the drug was washed off. Data are expressed as a percentage of U69,593-free spontaneous release in U69,593-exposed matched cultures. For the 0 hr recovery point the data are expressed as a percentage of release from naïve cultures. Thecurves represent the best fit of a single-phase exponential. The points are the mean ± SEM for 3–12 determinations. Error bars are not shown for SEM <2%.
Fig. 7.

Time course for U69,593-mediated dependence in the spontaneous 3H-DA release assay. Cultures were exposed to 1 μm U69,593 for various time periods; spontaneous3H-DA release was determined immediately after U69,593 was displaced by nor-BNI. Data are expressed as a percentage of release from naïve, matched cultures. The curverepresents the best fit of a single phase exponential. Thepoints are mean ± SEM for 3–12 determinations. Error bars are not shown for SEM <2%.
RESULTS
Characterization of the mesencephalic culture system
Of the total cells in the culture, ∼5% express tyrosine hydroxylase, as determined by immunofluorescent techniques (data not shown). RNA prepared from E15, E21, and adult mesencephalic tissue contains transcripts of μ- and κ-opioid receptors as determined by RT/PCR (Fig. 1A,C). In contrast, δ-opioid receptor message is detectable in adult preparations, but not in RNA preparations of embryonic tissue (Fig.1B).
Fig. 1.

μ- and κ-Receptor transcripts can be detected in embryonic and mesencephalic rat tissue. RNA was extracted from embryonic (E15, E21) and adult mesencephalic tissue, and reverse transcription was performed with a primer predicted to hybridize with μ-, δ-, and κ-receptor mRNA. PCR subsequently was performed with primers specific for either the μ, the δ, or the κ receptor. PCR primer specificity was confirmed with μ, δ, or κ cDNA templates in control reactions. PCR products were size-fractionated with PAGE and fluorescently stained. A, C, Primers specific for the μ receptor and primers specific for the κ-receptor-amplified segments that were the predicted size from E15, E21, or adult transcripts and from μ or κ cDNA sequences. C, Segments amplified by κ-specific primers contain anEcoRI restriction site as predicted. δ-Specific PCR primers (B) did not amplify a segment of predicted length from embryonic tissue.
To determine the functional relationship between κ-opioid receptors and dopaminergic neurons, we used a 3H-dopamine (3H-DA) release assay. Because dopamine transporters are localized exclusively on dopaminergic neurons in the midbrain (Kuhar et al., 1998), this approach allows for the selective analysis of dopaminergic function in the midst of a heterogeneous culture system. By analyzing multiple time points before and after K+-stimulated 3H-DA release, we could evaluate the effects of κ-specific ligands on spontaneous and stimulated release (Fig. 2). The data indicate that U69,593, a κ-specific agonist, inhibits both spontaneous and K+-stimulated 3H-DA release.
To determine the specificity of the U69,593-mediated effects on dopamine release, we examined several receptor or G-protein inhibitors in combination with U69,593 for their effects on spontaneous (Fig.3A) and K+-stimulated (Fig. 3B) release from the same set of cultures. The effect of tetrodotoxin indicates that ∼25% of the spontaneous release and all of the K+-stimulated release are action potential-dependent. U69,593 inhibited all of the action potential-dependent spontaneous release and ∼70% of the K+-stimulated release. The effects of U69,593 were blocked by a κ-specific antagonist nor-BNI (Portoghese et al., 1987) and by pertussis toxin. Quinpirole, a D2 dopamine receptor agonist, had an effect similar to U69,593 on spontaneous and K+-stimulated release. The individual effects of TTX, U69593, and quinpirole as compared with the no-drug controls were all statistically significant in both the spontaneous and K+-stimulated assay (p < 0.01; t test). Taken together, these results indicate that U69,593 is activating a κ-opioid receptor system in which Gi or Go couples the receptor to effector mechanisms. This system is capable of inhibiting the action potential-dependent release of 3H-DA. In this regard, the κ-receptor system is similar to the D2 autoreceptor system on dopaminergic neurons.
Fig. 3.
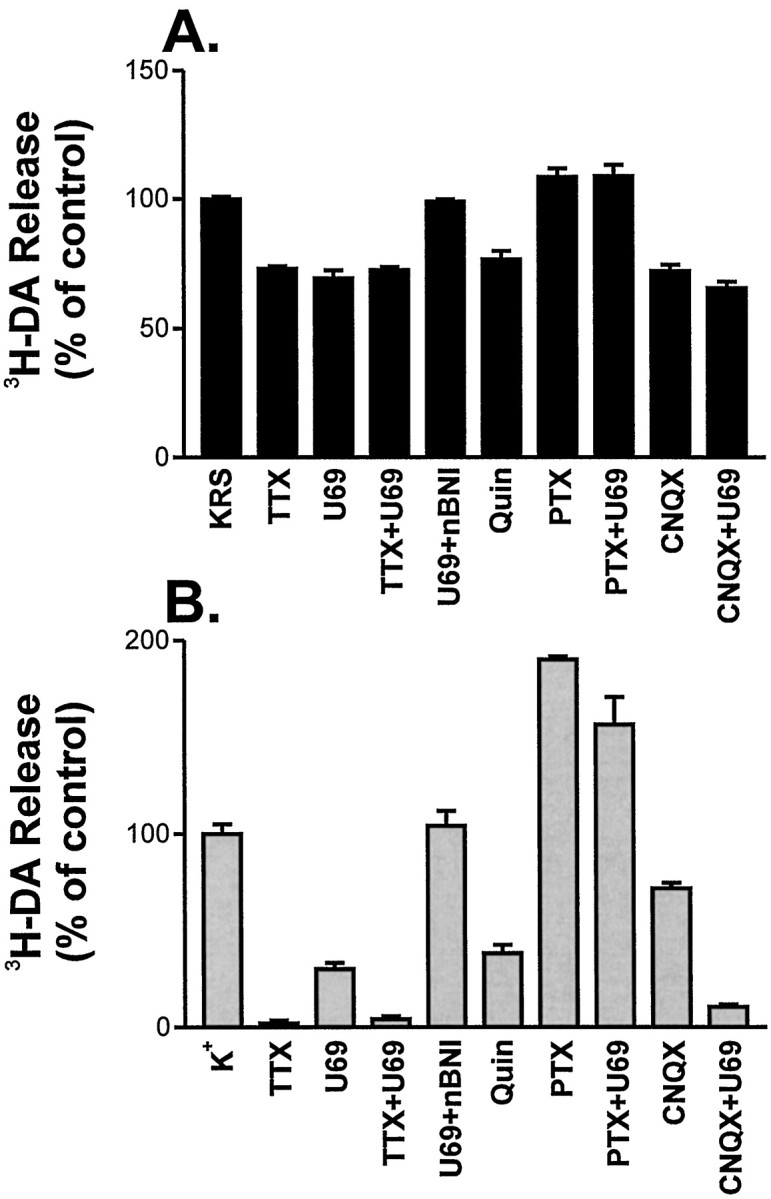
The activated κ-opioid receptor inhibits action potential-dependent spontaneous and K+-stimulated release of 3H-DA. The effects of various agents on spontaneous (A) and stimulated (B) release were quantitated as described in Materials and Methods. Results are expressed as a percentage of release from drug-free cultures (KRS; K+). For conditions containing TTX (1 μm), U69,593 (U69; 1 μm), quinpirole (Quin; 1 μm), nor-BNI (1 μm), and CNQX (10 μm), the drug was added in KRS after removal of the 72 min point. Pertussis toxin (1 μg/ml) treatment was for 22 hr before 3H-DA loading. Error bars are the mean of three to nine experiments ± SEM. Error bars are not shown for SEM <2%.
CNQX, an AMPA receptor antagonist, blocked 28% (p < 0.05; t test) of the K+-stimulated release and essentially all of the action potential-dependent spontaneous release (p < 0.01; t test). In the stimulated release assay CNQX and U69,593 showed additive effects; together they inhibited 89% of 3H-DA release (p < 0.01, t test, when compared with CNQX alone; p < 0.05, t test, when compared with U69593 alone). This indicates that the two drugs are working via distinct mechanisms to inhibit stimulated release. In the spontaneous release assay the two drugs were nonadditive, and CNQX occluded the effects of U69,593. This suggests that glutamate is the stimulus for action potential-dependent spontaneous release in this system.
Tolerance to U69,593
Concentration-dependent effects of U69,593 in naïve and U69,593-exposed cultures
Both spontaneous and K+-stimulated3H-DA release were inhibited by U69,593 in a dose-dependent manner (Fig. 4). The drug was a more potent inhibitor of spontaneous release (Fig. 4A; EC50 = 1.5 nm; 95% C.I. = 0.4–5 nm) than of K+-stimulated release (Fig.4B; EC50 = 10 nm; 95% C.I. = 6.4–17 nm). Cultures exposed to 1 μmU69,593 for 2 d showed decreased sensitivity to the effects of U69,593 on spontaneous (Fig. 4A; EC50 = 9 nm; 95% C.I. = 1.6–52 nm) and K+-stimulated (Fig.4B; EC50 = 1.4 μm; 95% C.I. = 0.4–49 μm) release. The EC50shift was larger in the K+-stimulated release assay (140-fold) than in the spontaneous release assay (sixfold). Because the protocol for this experiment involved washing off U69,593 from the U69,593-treated cultures ∼2 hr before the concentration dependence was determined, it is likely that some recovery from the drug treatment occurred during this 2 hr interval. These data demonstrate that prolonged exposure to U69,593 results in decreased receptor response, i.e., the development of tolerance.
Kinetics of U69,593-induced attenuation of its effect on K+-stimulated 3H-DA release
To measure the kinetics of tolerance onset, we exposed the cultures to 1 μm U69,593 (U69,593-exposed) for 0, 1, 2, 4, 6, 16, or 144 hr. After each period of U69,593 exposure the effect of U69,593 exposure on K+-stimulated release was measured in U69,593-bound cultures and in U69,593-free cultures. The latter was achieved by adding the κ-specific antagonist nor-BNI (1 μm) to drug-exposed cultures 12 min before K+ stimulation. Previous experiments indicated that 1 μm nor-BNI could displace U69,593 completely from its receptor within 6 min (data not shown). Moreover, no inverse agonist function or effects on spontaneous or stimulated 3H-DA release were seen after nor-BNI addition in naïve cultures. Thus its inclusion resulted in a U69,593-free condition after U69,593 exposure. As shown in Figure5A, the effects of U69,593 exposure on K+-stimulated release change with time in either the bound or free condition. When plotted as a ratio versus the duration of U69,593 exposure, the comparison of release in the U69,593-bound condition to the release in the U69,593-free condition shows the rate of attenuation of the capacity of the κ receptor to inhibit release (t½ = 3.7 hr; 95% C.I. = 2.4–7.5 hr, single phase exponential; Fig.5B).
Inhibition of release by U69,593 also resulted in increased basal activity or dependence. For example, in the cultures treated for 1, 2, 4, 6, and 16 hr with U69,593, the K+-stimulated release in the U69,593-free condition (i.e., nor-BNI-treated) was increased significantly (t test; p < 0.005) relative to the release from drug-free naïve cultures (Fig.5A). Thus U69,593 exposure induced κ dependence in this system.
To address the question of whether the agonist-induced attenuation of the κ-opioid receptor signaling system was the result of more than one mechanism, we compared the rate of recovery from a 4 hr U69,593 exposure to that from a 144 hr exposure. Thus after drug exposure U69,593 was washed off either with the KRS washes (2 hr recovery period) or with culture medium (7, 16, 39, or 46 hr recovery periods). The latter set of washes reduced total 3H-DA uptake in the release assay by 7–10%; otherwise, the total 3H-DA uptake was not affected by the duration of recovery. After a 4 hr exposure (Fig. 5C), attenuation of the κ-opioid receptor signaling system was ∼50% complete. After removal of the agonist, recovery to baseline sensitivity occurred rapidly (Fig. 5C;t½ = 1.1 hr; 95% C.I. = 0.6–4.0 hr). In contrast, the recovery rate after a 144 hr U69,593 exposure was slow (Fig. 5C; t½ = 20 hr; 95% C.I. = 13–52 hr). This large difference in the recovery rates indicated that the attenuation of the κ-receptor system attributable to 144 hr of U69,593 exposure was fundamentally different from the attenuation that occurred as a result of 4 hr of U69,593 exposure.
Kinetics of U69,593-induced attenuation of its effect on spontaneous 3H-DA release
Attenuation of the U69,593-mediated effects also is seen in the3H-DA spontaneous release paradigm. For example, at 1 hr of drug exposure 1 μm U69,593 retained its effect on spontaneous release (24 ± 2% inhibition; n = 12), but after 144 hr of U69,593 exposure the spontaneous release in the presence of U69,593 was not measurably different from the spontaneous release from naïve U69,593-free cultures. Evaluation of the U69,593 effect on spontaneous release as a function of U69,593 exposure time (Fig.6A) indicated that the rate of U69,593-induced attenuation in the spontaneous release assay occurred relatively slowly (t½ = 22 hr; 95% C.I. = 15–40 hr). In contrast, the recovery of spontaneous release was rapid (t½ ∼0.7 hr; Fig.6B).
U69,593-induced κ-opioid dependence also was observed in the spontaneous release assay. The addition of nor-BNI to U69,593-exposed cultures increased 3H-DA release relative to naïve cultures (Fig. 7). Evaluation of the magnitude of nor-BNI-evoked release in U69,593-exposed cultures as a function of U69,593 exposure time indicated that the effect was maximal at the earliest time that was measured (Fig. 7) and decayed to a plateau that was ∼120% of the release from naïve cultures (t½ = 41 hr; 95% C.I. = 19 to >100 hr). Even after reaching this plateau, the nor-BNI-induced release was significantly greater than the release from naïve sister cultures (t test; 144 or 220 hr exposed cultures vs naïve; p < 0.05). Similarly, when U69,593 was washed off before 3H-DA loading (Table1), spontaneous release from the 144 hr, U69,593-exposed cultures was significantly greater than the release from the 4 hr, U69,593-exposed cultures or from naïve cultures.
Table 1.
Spontaneous 3H-DA release in U69,593-exposed cultures after U69,593 washout
| Exposure time (hr) | 3H-DA release (n) |
|---|---|
| 0 | 24.4 ± 0.4 (6) |
| 4 | 25.8 ± 0.4 (6)* |
| 144 | 27.9 ± 0.4 (6)**# |
Cultures were kept naïve or were exposed to 1 μmU69,593 for 4 or 144 hr. Medium was removed, and the cultures were washed with U69,593-free KRS in preparation for 3H-DA load according to the standard release assay protocol. Spontaneous release was measured at 30 min post-load for each of the U69,593 exposure conditions. Release is expressed as a percentage of total uptake ±SEM. *p < 0.05 (vs 0 hr exposure); **p < 0.001 (vs 0 hr exposure);
p < 0.01 (vs 4 hr exposure).
DISCUSSION
Drugs of abuse produce long-lasting adaptations not only in dopaminergic pathways but also in interrelated systems. Repeated exposure to cocaine leads to the upregulation of κ opioids and their receptors. Although the effect of this response on dynorphin release and κ-opioid tone is unclear, this adaptation may be an important contributor to the dysphoric mood and aversive aspects of subsequent periods of abstinence. The induction of κ-opioid tolerance, particularly in the MCL, represents a potential clinical strategy to minimize the dysphoric aspects of abstinence and reduce the incentive for relapse into cocaine abuse. The present study directly assessed the degree to which κ tolerance can occur in cultures of MCL dopaminergic neurons. The major findings are that κ receptors inhibit both spontaneous and K+-stimulated dopamine release and that these processes exhibit different aspects of tolerance. κ-Opioid dependence is observed also. Thus these data support the idea that pharmacological administration of κ opioids may reverse cocaine-induced MCL adaptations.
Characterization of the cultured MCL system
The selective uptake of radioactive dopamine provides a sensitive assay by which both spontaneous and stimulated dopamine release can be measured (see Fig. 2). In the case of spontaneous release, endogenous glutamate appears to serve as the stimulus, whereas raising extracellular K+ levels to 15 mmgenerates a threefold increase in dopamine release (see Figs. 2, 3). As predicted from the RT/PCR studies (see Fig. 1), κ-opioid receptor function is apparent in MCL cultures (e.g., Fig. 2). Activation by the κ agonist U69,593 inhibits all of the action potential-triggered dopamine release in the spontaneous release paradigm (see Fig.3A) and up to 70% of the K+-stimulated release (see Fig. 3B). In either case the effects of U69,593 are blocked by nor-BNI as well as by pertussis toxin (see Fig. 3). Thus U69,593-activated κ receptors appear to be coupled to Gi- or Go-proteins in the modulation of dopamine release.
κ-Induced adaptations in MCL cultures: Tolerance
Prolonged U69,593 exposure results in decreased receptor response, or tolerance, in either the spontaneous or the stimulated release paradigm. The tolerance observed is attributable to decreased signaling via receptor and receptor-linked systems as well as to increased levels of basal neuronal activity. In the case of the stimulated release assay, the first process can be resolved into two mechanisms (see Fig.5). One mechanism develops early during U69,593 exposure and diminishes rapidly after removal of the drug. The other mechanism develops over a longer time period of exposure and is a more long-lasting adaptation. Conceivably, the first mechanism represents desensitization caused by receptor phosphorylation and arrestin binding, whereas the second represents receptor downregulation and/or an alteration in the activity level of another component of the signaling system such as a G-protein, an RGS protein, or an effector molecule (Loh et al., 1988; Freedman and Lefkowitz, 1996; Nestler and Aghajanian, 1997).
In contrast, the tolerant state is slower to develop in the spontaneous release assay, and recovery to full sensitivity is more rapid (see Fig.6). The difference in EC50 for U69,593 and the differences in the rates of tolerance onset and offset between the spontaneous and stimulated assays can be explained by the different degrees of κ-receptor activity and the corresponding levels of receptor occupancy required to counteract the two stimuli. For example, because 15 mm K+ is the greater of the two stimuli, the inhibition of its action requires a higher level of κ-receptor occupancy. Consequently, there are fewer “spare” receptors; thus the κ effect is more sensitive to processes inactivating receptors or receptor-linked systems. This inverse relationship between receptor reserve and EC50 in opioid systems has been well described in other systems (Cox and Chavkin, 1983; Chavkin and Goldstein, 1984).
Collectively, these data predict that κ-induced tolerance of MCL-mediated processes depends on the nature of the stimulus for dopamine release. For example, in the spontaneous release paradigm, half-maximal tolerance to the inhibitory effects of U69,593 was seen only after high concentrations of this drug were present in the culture system for 22 hr (see Fig. 6A). Sensitivity to the drug returned in approximately the same time period required to wash the drug off (see Fig. 6B). This suggests that dopamine release that is the result of spontaneous neuronal activity or that is attributable to weak stimuli will not show tolerance to κ opioids. Indeed, this has been observed (Donzanti et al., 1992). In contrast, dopamine release because of stronger stimuli such as the μ-opioid-induced disinhibition of dopaminergic neurons would be expected to develop tolerance rapidly to the effects of κ agonists. Thus selective tolerance to κ-opioid effects on MCL activity would occur. These observations are in agreement with recent primate data suggesting that selective tolerance occurs to κ-opioid effects on MCL-mediated behaviors (Negus et al., 1997).
κ-Induced adaptations in MCL cultures: Dependence
Agonist exposure also results in a compensatory increase in the basal activity of the dopaminergic neurons as measured by stimulated and spontaneous dopamine release (see Figs. 5A, 7). This effect is maximal at early time points and decays to a plateau that is above the level of release from naïve cultures (see Fig. 7). These data fit a model in which short-term U69,593 exposure induces an accumulation of dopamine within the neuron because of its capacity to reduce spontaneous dopamine release. Consequently, displacement of U69,593 with nor-BNI at early time points (<6 hr) triggers a large discharge of dopamine from the neuron because of the augmented intracellular reservoir (see Fig. 5A). In contrast, during prolonged exposure to U69,593, tolerance to this effect develops, and U69,593-induced intracellular accumulation of dopamine is minimal. In this model the residual 20% augmented dopamine release represents enhanced neuronal activity (see Fig. 7). Conceivably, this is the neuronal equivalent of κ-opioid dependence that has been observed in intact systems (Gmerek and Woods, 1986; Gmerek et al., 1987; Feng et al., 1997; Milanes et al., 1997; Mello and Negus, 1998). The augmented release seen after short U69,593 exposures is similar to the rebound effect seen in intact systems after single doses of κ agonists (Brent et al., 1993).
Role of κ tolerance in withdrawal syndromes
Both primates and rodents exhibit withdrawal behaviors after the termination of long-term κ-opioid exposure (Gmerek et al., 1987; Feng et al., 1997; Mello and Negus, 1998). For other opioids, characteristics of the withdrawal syndrome are the opposite of the immediate effects of acute administration of the opioid. Conceivably, then, withdrawal from κ-opioids might not be unpleasant because its acute administration produces dysphoria. Studies evaluating withdrawal from selective κ agonists have not been done in humans, although volunteers have been made tolerant to cyclazocine, a drug with some κ-agonist properties. The withdrawal syndrome in that study reportedly was mild (Martin et al., 1966). Moreover, in clinical trials testing the analgesic effect of enadoline, a κ-specific agonist, although the subjects reported unpleasant subjective effects of the drug, they did not withdraw from the study (Reece et al., 1994). Thus the pharmacological induction of κ tolerance may be possible.
In conclusion, in this paper we have demonstrated that κ tolerance and dependence can occur in midbrain dopaminergic neurons and, by implication, in the intact MCL system. As discussed above, exposure to drugs of abuse such as cocaine augments κ-opioid signaling in the MCL circuitry, and it has been suggested that this increase in κ-opioid tone contributes to the unpleasant and aversive quality of cocaine withdrawal and abstinence syndromes (Hurd and Herkenham, 1993;Shippenberg et al., 1996). Our results support the proposition that κ-opioid tone can be lowered by the pharmacological induction of κ-opioid tolerance and dependence in MCL circuitry. This type of intervention may help to ameliorate some of the symptoms of withdrawal and abstinence; as a result, it may be useful in the rehabilitation of drug-addicted individuals.
Footnotes
This work was supported by National Institutes of Health Grant DA08818. Technical assistance was provided by Mark Moffat and Steve Harmon. μ-, κ-, and δ-Opioid receptor cDNAs were generously provided by Huda Akil, University of Michigan Mental Health Research Unit.
Correspondence should be addressed to Dr. Friedrich C. Dalman, Department of Anesthesiology, Campus Box 8054, Washington University School of Medicine, 660 South Euclid Avenue, St. Louis, MO 63110.
REFERENCES
- 1.Bhargava HN, Gulati A, Ramarao P. Effect of chronic administration of U-50,488H on tolerance to its pharmacological actions and on multiple opioid receptors in rat brain regions and spinal cord. J Pharmacol Exp Ther. 1989;251:21–26. [PubMed] [Google Scholar]
- 2.Brent PJ, Chahl LA, Cantarell PA, Kavanagh C. The κ-opioid receptor agonist U50,488H induces acute physical dependence in guinea-pigs. Eur J Pharmacol. 1993;241:149–156. doi: 10.1016/0014-2999(93)90196-o. [DOI] [PubMed] [Google Scholar]
- 3.Carlezon JW, Thome J, Olson VG, Lane-Ladd SB, Brodkin ES, Hiroi N, Duman RS, Neve RL, Nestler EJ. Regulation of cocaine reward by CREB. Science. 1998;282:2272–2275. doi: 10.1126/science.282.5397.2272. [DOI] [PubMed] [Google Scholar]
- 4.Chavkin C, Goldstein A. Opioid receptor reserve in normal and morphine-tolerant guinea pig ileum myenteric plexus. Proc Natl Acad Sci USA. 1984;81:7253–7257. doi: 10.1073/pnas.81.22.7253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cole RL, Konradi C, Douglass J, Hyman SE. Neuronal adaptation to amphetamine and dopamine: molecular mechanisms of prodynorphin gene regulation in rat striatum. Neuron. 1995;14:813–823. doi: 10.1016/0896-6273(95)90225-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cox BM, Chavkin C. Comparison of dynorphin-selective κ receptors in mouse vas deferens and guinea pig ileum. Spare receptor fraction as a determinant of potency. Mol Pharmacol. 1983;23:36–43. [PubMed] [Google Scholar]
- 7.Di Chiara G, Imperato A. Drugs abused by humans preferentially increase synaptic dopamine concentrations in the mesolimbic system of freely moving rats. Proc Natl Acad Sci USA. 1988;85:5274–5278. doi: 10.1073/pnas.85.14.5274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Donzanti BA, Althaus JS, Payson MM, Von Voigtlander PF. κ-Agonist-induced reduction in dopamine release: site of action and tolerance. Res Commun Chem Pathol Pharmacol. 1992;78:193–210. [PubMed] [Google Scholar]
- 9.Feng Y, Rockhold RW, Ho IK. Nor-binaltorphimine precipitates withdrawal and excitatory amino acid release in the locus ceruleus of butorphanol–but not morphine-dependent rats. J Pharmacol Exp Ther. 1997;283:932–938. [PubMed] [Google Scholar]
- 10.Freedman NJ, Lefkowitz RJ. Desensitization of G-protein-coupled receptors. Recent Prog Horm Res. 1996;51:319–351. [PubMed] [Google Scholar]
- 11.Glick SD, Maisonneuve IM, Raucci J, Archer S. κ-Opioid inhibition of morphine and cocaine self-administration in rats. Brain Res. 1995;681:147–152. doi: 10.1016/0006-8993(95)00306-b. [DOI] [PubMed] [Google Scholar]
- 12.Gmerek DE, Woods JH. κ-Receptor-mediated opioid dependence in rhesus monkeys. Life Sci. 1986;39:987–992. doi: 10.1016/0024-3205(86)90287-0. [DOI] [PubMed] [Google Scholar]
- 13.Gmerek DE, Dykstra LA, Woods JH. κ Opioids in rhesus monkeys. III. Dependence associated with chronic administration. J Pharmacol Exp Ther. 1987;242:428–436. [PubMed] [Google Scholar]
- 14.Heidbreder CA, Goldberg SR, Shippenberg TS. The κ-opioid receptor agonist U-69593 attenuates cocaine-induced behavioral sensitization in the rat. Brain Res. 1993;616:335–338. doi: 10.1016/0006-8993(93)90228-f. [DOI] [PubMed] [Google Scholar]
- 15.Heidbreder CA, Babovic-Vuksanovic D, Shoaib M, Shippenberg TS. Development of behavioral sensitization to cocaine: influence of κ-opioid receptor agonists. J Pharmacol Exp Ther. 1995;275:150–163. [PubMed] [Google Scholar]
- 16.Hurd YL, Herkenham M. Molecular alterations in the neostriatum of human cocaine addicts. Synapse. 1993;13:357–369. doi: 10.1002/syn.890130408. [DOI] [PubMed] [Google Scholar]
- 17.Hyman SE. Addiction to cocaine and amphetamine. Neuron. 1996;16:901–904. doi: 10.1016/s0896-6273(00)80111-7. [DOI] [PubMed] [Google Scholar]
- 18.Jin W, Terman GW, Chavkin C. κ-Opioid receptor tolerance in the guinea pig hippocampus. J Pharmacol Exp Ther. 1997;281:123–128. [PubMed] [Google Scholar]
- 19.Koob GF, Le Moal M. Drug abuse: hedonic homeostatic dysregulation. Science. 1997;278:52–58. doi: 10.1126/science.278.5335.52. [DOI] [PubMed] [Google Scholar]
- 20.Kuhar MJ, Vaughan R, Uhl G, Cerruti C, Revay R, Freed C, Nirenburg M, Pickel V. Localization of dopamine transporter protein by microscopic histochemistry. Adv Pharmacol. 1998;42:168–170. doi: 10.1016/s1054-3589(08)60719-x. [DOI] [PubMed] [Google Scholar]
- 21.Loh HH, Tao PL, Smith AP. Role of receptor regulation in opioid tolerance mechanisms. Synapse. 1988;2:457–462. doi: 10.1002/syn.890020414. [DOI] [PubMed] [Google Scholar]
- 22.Martin WR, Gorodetzky CW, McClane TK. An experimental study in the treatment of narcotic addicts with cyclazocine. Clin Pharmacol Ther. 1966;7:455–465. doi: 10.1002/cpt196674455. [DOI] [PubMed] [Google Scholar]
- 23.Mello NK, Negus SS. Effects of κ-opioid agonists on cocaine-and food-maintained responding by rhesus monkeys. J Pharmacol Exp Ther. 1998;286:812–824. [PubMed] [Google Scholar]
- 24.Milanes MV, Rabadan JV, Laorden ML. Effects of chronic U-50,488H treatment on the isolated right atrium of the rat. Neuropeptides. 1997;31:511–515. doi: 10.1016/s0143-4179(97)90047-0. [DOI] [PubMed] [Google Scholar]
- 25.Negus SS, Mello NK, Portoghese PS, Lin CE. Effects of κ opioids on cocaine self-administration by rhesus monkeys. J Pharmacol Exp Ther. 1997;282:44–55. [PubMed] [Google Scholar]
- 26.Nestler EJ, Aghajanian GK. Molecular and cellular basis of addiction. Science. 1997;278:58–63. doi: 10.1126/science.278.5335.58. [DOI] [PubMed] [Google Scholar]
- 27.O’Malley KL, Mack KJ, Gandelman KY, Todd RD. Organization and expression of the rat D2A receptor gene: identification of alternative transcripts and a variant donor splice site. Biochemistry. 1990;29:1367–1371. doi: 10.1021/bi00458a003. [DOI] [PubMed] [Google Scholar]
- 28.Paronis CA, Woods JH. Ventilation in morphine-maintained rhesus monkeys. II. Tolerance to the antinociceptive but not the ventilatory effects of morphine. J Pharmacol Exp Ther. 1997;282:355–362. [PubMed] [Google Scholar]
- 29.Portoghese PS, Lipkowski AW, Takemori AE. Binaltorphimine and nor-binaltorphimine, potent and selective κ-opioid receptor antagonists. Life Sci. 1987;40:1287–1292. doi: 10.1016/0024-3205(87)90585-6. [DOI] [PubMed] [Google Scholar]
- 30.Reece PA, Sedman AJ, Rose S, Wright DS, Dawkins R, Rajagopalan R. Diuretic effects, pharmacokinetics, and safety of a new centrally acting κ-opioid agonist (CI-977) in humans. J Clin Pharmacol. 1994;34:1126–1132. doi: 10.1002/j.1552-4604.1994.tb01991.x. [DOI] [PubMed] [Google Scholar]
- 31.Shippenberg TS, Rea W. Sensitization to the behavioral effects of cocaine: modulation by dynorphin and κ-opioid receptor agonists. Pharmacol Biochem Behav. 1997;57:449–455. doi: 10.1016/s0091-3057(96)00450-9. [DOI] [PubMed] [Google Scholar]
- 32.Shippenberg TS, LeFevour A, Heidbreder C. κ-Opioid receptor agonists prevent sensitization to the conditioned rewarding effects of cocaine. J Pharmacol Exp Ther. 1996;276:545–554. [PubMed] [Google Scholar]
- 33.Sim LJ, Selley DE, Dworkin SI, Childers SR. Effects of chronic morphine administration on μ-opioid receptor-stimulated [35S]GTPγS autoradiography in rat brain. J Neurosci. 1996;16:2684–2692. doi: 10.1523/JNEUROSCI.16-08-02684.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sivam SP. Cocaine selectively increases striatonigral dynorphin levels by a dopaminergic mechanism. J Pharmacol Exp Ther. 1989;250:818–824. [PubMed] [Google Scholar]
- 35.Smiley PL, Johnson M, Bush L, Gibb JW, Hanson GR. Effects of cocaine on extrapyramidal and limbic dynorphin systems. J Pharmacol Exp Ther. 1990;253:938–943. [PubMed] [Google Scholar]
- 36.Spanagel R, Herz A, Shippenberg TS. The effects of opioid peptides on dopamine release in the nucleus accumbens: an in vivo microdialysis study. J Neurochem. 1990;55:1734–1740. doi: 10.1111/j.1471-4159.1990.tb04963.x. [DOI] [PubMed] [Google Scholar]
- 37.Spanagel R, Herz A, Shippenberg TS. Opposing tonically active endogenous opioid systems modulate the mesolimbic dopaminergic pathway. Proc Natl Acad Sci USA. 1992;89:2046–2050. doi: 10.1073/pnas.89.6.2046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Spangler R, Unterwald EM, Kreek MJ. “Binge” cocaine administration induces a sustained increase of prodynorphin mRNA in rat caudate-putamen. Brain Res Mol Brain Res. 1993;19:323–327. doi: 10.1016/0169-328x(93)90133-a. [DOI] [PubMed] [Google Scholar]
- 39.Staley JK, Rothman RB, Rice KC, Partilla J, Mash DC. κ2-Opioid receptors in limbic areas of the human brain are upregulated by cocaine in fatal overdose victims. J Neurosci. 1997;17:8225–8233. doi: 10.1523/JNEUROSCI.17-21-08225.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Steiner H, Gerfen CR. Cocaine-induced c-fos messenger RNA is inversely related to dynorphin expression in striatum. J Neurosci. 1993;13:5066–5081. doi: 10.1523/JNEUROSCI.13-12-05066.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Unterwald EM, Rubenfeld JM, Kreek MJ. Repeated cocaine administration upregulates κ and μ, but not δ, opioid receptors. NeuroReport. 1994;5:1613–1616. doi: 10.1097/00001756-199408150-00018. [DOI] [PubMed] [Google Scholar]
- 42.White FJ. Synaptic regulation of mesocorticolimbic dopamine neurons. Annu Rev Neurosci. 1996;19:405–436. doi: 10.1146/annurev.ne.19.030196.002201. [DOI] [PubMed] [Google Scholar]
- 43.Wise RA. Neurobiology of addiction. Curr Opin Neurobiol. 1996;6:243–251. doi: 10.1016/s0959-4388(96)80079-1. [DOI] [PubMed] [Google Scholar]