

Abstract
Targeted mutagenesis in mice demonstrates that the POU-domain gene Brn4/Pou3f4 plays a crucial role in the patterning of the mesenchymal compartment of the inner ear.Brn4 is expressed extensively throughout the condensing mesenchyme of the developing inner ear. Mutant animals displayed behavioral anomalies that resulted from functional deficits in both the auditory and vestibular systems, including vertical head bobbing, changes in gait, and hearing loss. Anatomical analyses of the temporal bone, which is derived in part from the otic mesenchyme, demonstrated several dysplastic features in the mutant animals, including enlargement of the internal auditory meatus. Many phenotypic features of the mutant animals resulted from the reduction or thinning of the bony compartment of the inner ear. Histological analyses demonstrated a hypoplasia of those regions of the cochlea derived from otic mesenchyme, including the spiral limbus, the scala tympani, and strial fibrocytes. Interestingly, we observed a reduction in the coiling of the cochlea, which suggests that Brn-4 plays a role in the epithelial–mesenchymal communication necessary for the cochlear anlage to develop correctly. Finally, the stapes demonstrated several malformations, including changes in the size and morphology of its footplate. Because the stapes anlage does not express theBrn4 gene, stapes malformations suggest that theBrn4 gene also plays a role in mesenchymal–mesenchymal signaling. On the basis of these data, we suggest that Brn-4 enhances the survival of mesodermal cells during the mesenchymal remodeling that forms the mature bony labyrinth and regulates inductive signaling mechanisms in the otic mesenchyme.
Keywords: inner ear development, Brn4/Pou3f4, POU-domain transcription factor, epithelial–mesenchymal interaction, stapes, targeted mutagenesis
Inner ear development requires a complex series of morphogenetic changes that result from the interaction of ectodermal epithelia and mesenchyme (Deol, 1966;Langman, 1982; Frenz and Van de Water, 1991). In vertebrates, the development of the inner ear is initiated by the invagination of somatic ectoderm that lies lateral to the hindbrain to form the otic vesicle (for review, see Lewis et al., 1985; Peck, 1994). From the simple spherical structure of the otic vesicle, a number of complex morphological changes occur as a result of reciprocal interactions between the ectodermally derived otic vesicle and the surrounding mesenchyme (Noden and Van de Water, 1986; Fekete, 1996). The molecular mechanisms that regulate this series of inductive interactions remain to be fully characterized. However, genetic and reverse genetic approaches are providing insights into the regulation of inner ear development (for review, see Noden and Van de Water, 1992; Steel and Brown, 1994; Smith, 1995).
The reverse genetic approach to the characterization of candidate regulatory genes is illustrated by the isolation and characterization of the POU-domain gene family, many members of which are expressed during inner ear development (for review, see Ryan and Rosenfeld, 1997). POU-domain genes encode transcription factors that regulate a number of developmental processes. Sequence-specific DNA binding of POU-domain genes is mediated by a bipartite motif, which consists of a POU homeodomain and POU-specific domain. The POU homeodomain is a 60 amino acid segment with similarity to the classic homeodomain transcription factors. The POU-specific domain is an additional ∼75 amino acid motif that cooperates with the POU homeodomain to enhance the binding affinity and specificity of DNA binding (for review, see Herr and Cleary, 1995; Ryan and Rosenfeld, 1997). These transcription factors display a broad domain of expression during embryogenesis (He et al., 1989; Ryan and Rosenfeld, 1997). Genetic analyses have demonstrated that these factors play crucial developmental roles in a number of organ systems, including the inner ear (de Kok et al., 1995b; Erkman et al., 1996; Xiang et al., 1996,1997).
For example, de Kok et al. (1995b) have shown that mutations in the human Brain-4 (Brn4) ortholog POU3F4 produce mixed conductive and sensorineural deafness associated with perilymphatic gusher during stapes surgery (DFN3; McKusick catalog #304400). High-resolution computed tomography has shown that these patients also displayed multiple defects of the bony labyrinth. These defects were classified as pseudo-Mondini stage II dysplasias and were characterized by several features, including (1) partial hypoplasia of the cochlea, (2) stapes fixation, and (3) a dilated internal auditory meatus associated with an abnormal communication with the base of the cochlear duct. The defects that occur in DFN3 patients have been hypothesized to result from abnormal otic capsule formation between 25 and 47 d of gestation (Piussan et al., 1995). This corresponds approximately to 10.0–13.5 d postcoitus (dpc) of mouse development (Theiler, 1989) and corresponds precisely with the period of development in which the mouse Brn4 gene is expressed in the developing otic capsule (Phippard et al., 1998). Further molecular analyses are necessary to determine the role of the Brn4gene during normal development.
To assess the embryological consequences of Brn4/Pou3f4mutations, we have generated a mutant mouse pedigree containing a null mutation of the Brn4 gene, using homologous recombination in embryonic stem (ES) cells. Previous analyses from our laboratory have demonstrated that the Brn4 gene is initially expressed in the otic capsule at 10.5 dpc, when the capsule initiates the mesenchymal condensation that gives rise to the temporal bone (Phippard et al., 1998). As the otic mesenchyme condenses around the otic vesicle, Brn4 is expressed throughout the otic capsule. Characterization of Brn4 null mouse mutants demonstrates that this gene plays a crucial role in the morphogenesis of the mesenchymal component of the developing inner ear.
MATERIALS AND METHODS
Gene targeting of the Brn4 gene. Genomic sequences for the Brn4/Pou3f4 gene were isolated from a λFIXII library derived from 129/SvJ genomic DNA (Stratagene, La Jolla, CA). The 5′ homologous sequences were derived from theXbaI (−3.2 kb) to the BamHI site that lies 14 bp 5′ of the initiator methionine codon. The lacZ gene from pCH110 was introduced into the knock-out locus such that it would be expressed from the endogenous Brn4 promoter. The PGK-neo gene was isolated from pTKAB′ (a gift of J. Bermingham), and the 3′ homologous sequences were derived from a 6.7 kb SpeI site that lies 3′ of the Brn4-coding sequences. The PGK-TK gene from pGEM7(TK)SalI was introduced into the targeting vector to enable counterselection (a gift of M. A. Rudnicki). As indicated in Figure 1, this gene-targeting vector removed the coding sequence of theBrn4 gene after integration. The targeting construct was linearized by restriction digestion with NotI, and 25–40 μg of linearized vector was transfected into 107R1 ES cells by electroporation according to published protocols (Joyner, 1993). Transfected colonies were isolated after selection with 280 μg/ml (active) G418 (Life Technologies, Gaithersburg, MD) and 1 μm gancyclovir (Cytovene). Genomic DNA from individual clones was isolated, digested with PstI, electrophoresed on a 0.7% agarose gel, transferred to Hybond N+(Amersham, Arlington Heights, IL), and hybridized with a random-labeled 450 bp PstI/XbaI fragment that lies just 5′ of the gene-targeting vector (see Fig. 1). Positive clones were expanded and analyzed by Southern blot analysis using the lacZ gene, theBrn4-coding sequences, and a probe that lies within theBrn4 gene 3′ of those sequences included in the targeting vector. A positive clone (2G-13) was injected into C57BL/6J blastocysts, according to published protocols (Joyner, 1993). The contribution of the ES cells to the resulting chimeric animals was determined by estimating the proportion of agouti coat color. Chimeric animals that were estimated to be >90% derived from ES cells were mated to C57BL/6J or CD-1 females. Because the Brn4 gene is X-linked, the null allele was transmitted to all of the female progeny that were derived from the ES cells. These heterozygous females were detected by Southern blot analyses, using the same protocol described above for the analysis of ES cell clones. Hemizygous null male animals were generated by mating of the heterozygous females to C57BL/6J males.
Fig. 1.

Targeted mutagenesis of the Brn4 locus.A, Diagram of the targeting vector (top line), the wild-type allele (middle line), and the mutated allele (bottom line). The entire intronless coding region (box labeled Brn4) and a small region of 3′-flanking sequences have been replaced by the lacZ reporter gene and the PGK-neo gene. The lacZ-coding sequences have been introduced into the Brn4 locus such that theBrn4 promoter drives the transcription of the reporter gene from the mutated allele. Homologous recombination was detected with the 5′ P/X probe that detects the conversion of a 4.6 kbPstI fragment to 8.0 kb. P,PstI; S, SalI;X, XbaI. B, Southern blot analysis of the genetic transmission of the X-linked locus. Analysis of progeny from the founder chimeric male animals indicated that the mutated allele is transmitted to only the female progeny in generation 2. Genomic DNA was digested with PstI and probed with the 5′ P/X probe. F, Females; M, males.
Embryonic tissue analyses. For analysis of lacZ expression, embryos were isolated from timed matings of hemizygous males that were null for the X-linked Brn4 allele. The morning that vaginal plugs were detected was designated 0.5 dpc. Verification of transgenic animals was performed by dot blot or PCR analyses of yolk sac DNA with lacZ-specific probes or oligonucleotides. PCR analyses to detect the lacZ gene were performed essentially as described (Hogan et al., 1994), using the sense strand oligonucleotide CGCCGAAATCCCGAATCTCTA and the antisense oligonucleotide TCACCGCCGTAAGCCGACCAC. PCR was run for 30 cycles with a temperature profile as follows: 94°C for 30 sec, 65°C for 40 sec, and 72°C for 45 sec.
For the whole-mount preparation, the embryos were fixed in PBS containing 2% paraformaldehyde, 0.2% glutaraldehyde, 0.02% NP-40, and 0.01% sodium deoxycholate on ice for a period of time that depended on gestational age; 8.5 and 9.5 dpc embryos were fixed for 1 hr, 10.5 and 11.5 dpc embryos were fixed for 1.5 hr, and 12.5 dpc and older embryos were fixed for 2 hr. After fixation, embryos were washed three times in PBS. Whole-mount preparations were then stained for 8 hr to overnight at 33°C in 1 mg/ml 5-bromo-4-chloro-3-indolyl-β-d-galactopyranoside (X-gal), 5 mm potassium ferricyanide, 5 mm potassium ferrocyanide, 1 mm MgCl2, 0.02% NP-40, and 0.01% sodium deoxycholate in PBS. Nontransgenic embryos control for background staining. After staining, the embryos were washed three times in PBS and post-fixed overnight in 4% paraformaldehyde in PBS at 4°C. Vibratome sections (150 μm) of whole-mount preparations were generated to examine the fine structure of the embryonic expression patterns. Embryos were photographed on a Leica MZ8 dissecting microscope (Nussloch, Germany) using Ektachrome 160T slide film. Images were then captured using an LS-1000 slide scanner (Nikon), and figures were constructed using Photoshop (Adobe Systems).
Adult tissue analyses. Temporal bone preparations were obtained by bisecting the head and dissecting away most soft tissue. Skulls were boiled in distilled water for 2–3 hr until the skull sutures were loosened enough to allow easy excision of the temporal bone. Temporal bones were then stained with 0.0015% alizarin red in 1% KOH for 48 hr (Peters, 1977).
Histological analysis of the cochlea began by perfusing the cochlea through the aorta or via the oval window. Mice (6–8 weeks of age) were anesthetized (Metofane) and sacrificed by cervical dislocation. The temporal bones were dissected from the rest of the calvarium, and the adherent tissues were removed. The bulla wall was punctured in the inferioposterior quadrant below the tympanic membrane and then further resected to expose the ossicles and oval window. In several cases, the ossicles were dissected free from the temporal bone, stained with alizarin red, as described above, and analyzed for malformations. Removal of an intact stapes required that the stapedial artery and stapedius muscle be severed with the tip of a 26 ga hypodermic needle. The head of the stapes was then disarticulated from the long arm (crus) of the incus. The round window was then punctured with the tip of the hypodermic needle, and the cochlea was perfused through the oval window with 4% paraformaldehyde in PBS. Fixation was continued overnight in 4% paraformaldehyde in PBS, followed by 4–5 d in Cal-EX II decalcifying solution (Fisher Scientific, Houston, TX). Decalcified temporal bones were imbedded in paraffin, sectioned (7 μm), and stained with hematoxylin and eosin.
To generate whole-mount cleared preparations of the cochlea, we perfused the temporal bones through the oval window and fixed the bones overnight in 4% paraformaldehyde in PBS, as described above. The temporal bones were bleached for 30 min in 6% hydrogen peroxide. A solution of 10% latex paint (Dutch Boy; white multipurpose primer sealer) in PBS was perfused into the cochlea through the oval window. Preparations were then dehydrated through a graded series of ethanols and cleared in methyl salicylate. Alternatively, the paint was applied after the temporal bones were cleared in methyl salicylate. In this case, a 10% solution of enamel paint (Duron; semigloss interior alkyd) in methyl salicylate was perfused into the cochlea through the oval window, such that the paint fills the perilymphatic space that normally surrounds the membranous labyrinth.
Preyer’s reflex measurements. Auditory function was assessed in mutant mice or their wild-type control siblings by ascertaining the pinna (Preyer’s) reflex threshold. Mice (6 weeks of age) were placed in a glass chamber and stimulated with a burst of high-frequency noise. Each burst was 50 msec in duration with a 5 msec rise and decay time. The burst consisted of a narrow-band noise whose half-power points were between 10.0 and 18.5 kHz. The signal on the high- and low-frequency side of these band limits declined at a rate of 48 dB/octave. The intensity of the noise burst was controlled by an attenuator. This band of noise overlapped with the optimal hearing range of the mouse (Fay, 1988). The noise bursts were delivered via a high-frequency tweeter and were initiated by the observer via a switch closure. The stimulus was calibrated at the bottom of the chamber using a 12.5 mm condenser microphone, and the sound pressure level (SPL) was expressed as decibels relative to 20 μPa. The occurrence of a Preyer’s reflex was determined by an observer who did not know the genotype of the mouse. A modified method of limits was used to measure the reflex threshold. Starting at a level of 90 dB SPL, a noise burst was presented. If a pinna or startle reflex was identified, the stimulus was attenuated 10 dB, and another burst was presented. This approach continued until no reflex could be identified. The stimulus level was then raised 5 dB, and a final trial was run. The threshold was defined as that SPL value halfway between the noise burst levels that could or could not elicit a reflex, and the error is given as an SD. If there was uncertainty in the threshold, the process was repeated. Care was taken to vary the interval between burst presentations (5–20 sec) to avoid habituation. This assessment of the reflex threshold provided us with an estimate of the animals’ hearing ability in the noise-band frequency range.
RESULTS
Gene targeting of the Brn4/Pou3f4 locus
The role of the Brn4/Pou3f4 gene during embryogenesis was investigated by generating a mutant mouse pedigree with a targeted deletion of this gene. As illustrated in Figure 1A, the entire coding sequence of the Brn4 gene was removed via homologous recombination in ES cells. Because the Brn4 gene is X-linked, a single targeted allele of the Brn4 gene was observed in the male R1 ES cell line (data not shown). Of 535 ES cell clones that were isolated after selection in G418 and gancyclovir, only one had undergone the appropriate targeting event. This ES cell clone was introduced into C57BL/6J blastocysts, and chimeras with high levels of contribution from the mutant ES cells were obtained. Germline transmission of the X-linked null allele resulted in the generation of heterozygous female animals (Fig. 1B). Hemizygous null male animals were then generated by mating the heterozygous female mice to C57BL/6J male animals (Fig. 1B).
Expression of a lacZ reporter gene introduced into the mutantBrn4 allele
The targeted insertional mutation was designed so that theBrn4-coding sequences were replaced by the lacZ reporter gene. This experimental approach allowed us to follow the expression pattern of the mutant allele in both heterozygous female and hemizygous null male animals. Our analyses indicated that the transcriptional regulation of the mutant allele corresponded precisely with the expression domain of the endogenous Brn4 gene. To illustrate the expression domain of the Brn4 gene, Figure2 depicts preparations of embryos containing a mutant Brn4 knock-out allele that have been stained for lacZ expression.
Fig. 2.
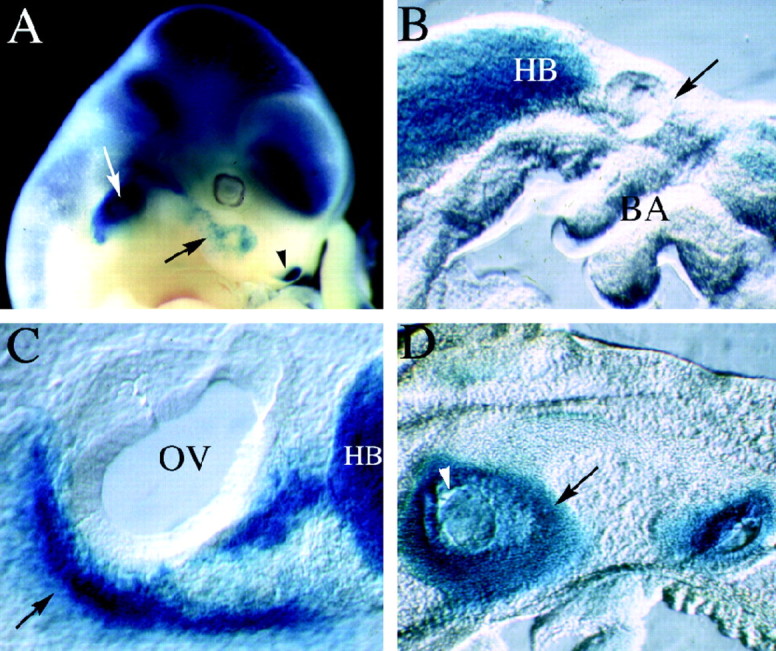
LacZ expression in mutant animals. Mutant embryos were stained with X-gal to visualize the expression of the lacZ gene, which was inserted into the Brn4 locus during the generation of the knock-out allele. Unless specified otherwise the embryos are oriented with rostral to the right and dorsal toward the top. A, The whole-mount preparation of a chimeric founder animal that corresponds to an 11.5 dpc embryo (chimeric embryos are developmentally delayed for ∼1 day because of experimental manipulation of the blastocysts). The expression of lacZ recapitulates precisely the pattern of endogenousBrn4 gene expression detected by hybridization histochemical analyses (Le Moine and Young, 1992; Mathis et al., 1992;Alvarez-Bolado et al., 1995; Phippard et al., 1998). Expression is found throughout most of the neuraxis and in a handful of mesodermally derived tissues in the head, including the otic capsule (white arrow), a small population of first branchial arch mesenchyme (black arrow), and the lateral nasal recess (black arrowhead). B, A parasagittal vibratome section (150 μm) through the otic vesicle of a 9.5 dpc embryo. Expression of lacZ is not detected in the mesenchyme surrounding the otic vesicle (black arrow), which lies dorsal to the branchial arches. However, expression is detected in the hindbrain of these embryos. C, A parasagittal vibratome section through the otic vesicle of a 10.5 dpc embryo. Expression of lacZ is detected in the condensing mesenchyme of the otic vesicle (black arrow), which lies ventral to the otic vesicle at this stage of embryogenesis. In this panel, the black arrow indicates the dorsal–ventral axis, with dorsal corresponding to the upper right-hand corner of the panel.D, Expression patterns of lacZ in a parasagittal section of a 14.5 dpc embryo. At this stage of development, lacZ expression is detected throughout the otic capsule but not in the otic epithelium (some regions of the otic epithelium appear blue, because they are covered with lacZ staining the otic capsule in these thick vibratome sections). BA, Branchial arches;HB, hindbrain; OV, otic vesicle.
At 9.5 dpc, the inner ear is just beginning to form as a hollow sphere of epithelial tissue, referred to as the otic vesicle (Fig.2B, black arrow). The Brn4 gene is not yet expressed in the developing inner ear but is broadly expressed throughout most of the neural tube (Fig.2A,B). Initial activation ofBrn4 gene expression in the inner ear is detected at 10.5 dpc in the condensing mesenchyme of the otic capsule (Fig.2C). Molecular signals from the otic vesicle induce surrounding mesenchyme to aggregate or condense to form the otic capsule (McPhee and Van de Water, 1986). The otic capsule represents the first stages in the formation of the temporal bone and will eventually give rise to the ossified and cartilaginous structures of the inner ear. These ossified and cartilaginous structures eventually encase the sensory and epithelial components of the inner ear, which are referred to as the membranous labyrinth. As the otic capsule develops, the mesenchyme surrounding the otic vesicle becomes progressively condensed and ultimately completely encapsulates the epithelial components of the developing inner ear. By 12.5 dpc, theBrn4 gene is expressed throughout the otic capsule, and this expression pattern is consistent with a role for Brn-4 in the regulation of inner ear formation.
Behavioral characteristics of the mutant mice
Animals with null mutations in the Brn4 locus demonstrated four behavioral phenotypes: (1) vertical head bobbing, (2) changes in gait, (3) reduced whisker mobility, and (4) hearing loss. Each of these phenotypes is consistent with malformations of structures that express the Brn4 gene, namely, the inner ear, the whisker follicles, and the neural tube (Phippard et al., 1998) (A. Heydemann and E. B. Crenshaw III, unpublished observations). The degree of vertical head bobbing is variable but was detected in the vast majority of the null mutants. The reduction in whisker mobility was observed as the mice probed their environment. In normal mice, the whiskers were moved in a sweeping motion when the animals explored their environment. In the mutant mice, the whiskers, which were reduced in size, did not demonstrate the typical sweeping motion and seldom moved.
As illustrated in Figure 3, we detected a hearing loss in the null Brn4 mutants, which was determined by an analysis of the auditory startle (Preyer’s) reflex. The mean SPL that elicited the threshold level of a Preyer’s reflex in wild-type littermates was 62 ± 3.1 dB SPL (n = 12). The mutant animals, however, elicited a reflex threshold at 71 ± 3.6 dB SPL (n = 12). A one-tailed Student’st test for independent groups demonstrated that these data were highly significant (p < 0.00001). The Preyer’s reflex estimate of hearing provides us with only a crude assessment of auditory sensitivity. Preyer’s reflex thresholds tend to be insensitive relative to other threshold estimates (e.g., operant conditioning techniques), and the broad-based stimulus used provides us with limited information on threshold loss at different frequencies. Nevertheless, the reflex assessments of hearing were completely reliable over numerous replications with the same animal, from animal to animal, and from observer to observer. From the observations summarized in Figure 3, we have concluded that hearing loss was fully penetrant in the mutant animals.
Fig. 3.

Preyer’s reflex in Brn4 knock-out (KO) mutants. The Brn4 null mutants demonstrate approximately a 10 dB SPL hearing loss in comparison with that in wild-type littermates, as assessed by Preyer’s reflex. Wild-type animals, 62 ± 3.1 dB SPL (n= 12). Brn4 null mutants, 71 ± 3.6 dB SPL (n = 12). Error bars represent SD. The Preyer’s reflex threshold is measured in decibels SPL relative to a standard of 20 μPa. One-tailed Student’s t test,p < 0.00001. If female animals are excluded from the comparison, then the wild-type male animals demonstrate a threshold of 62 ± 3.3 dB SPL (n = 9). Furthermore, there is no statistically significant difference in the threshold between male and female animals, whether they are mutant or wild type (data not shown).
Malformation of the stapes in the mutant mice
One of the major malformations contributing to hearing loss in human patients with mutations in the human Brn4 ortholog is stapes fixation. This is a condition resulting in reduced stapes movements in the oval window, which compromises sound conduction through the middle ear ossicular system. For this reason, we examined, in detail, the stapes of Brn4 null mice. Figure4 illustrates that the most profound malformations in the mutant stapes occur in the footplate region. The stapes normally conducts vibrational energy to the oval window and, subsequently, produces sound pressure waves in the cochlea that lead to hair cell transduction. The stapes footplate lies in the oval window, and the morphology and area of the footplate affect the efficiency with which sound energy is transmitted into the cochlear fluids. The normal footplate displays a convex surface (Fig.4A,C), whereas the surface of the mutant is flattened (Fig. 4B,D). When the sole of the normal stapes footplate is examined, it displays a rounded, but slightly eccentric, ovoid morphology (Fig.4E). The mutant footplate, however, typically displays a more polygonal morphology, with a marked narrowing on one end (Fig. 4F). Additionally, the stapes from the mutant animals are generally thinner than are those of the normal stapes (data not shown). The other ossicular bones, the incus and malleus, appeared normal in the mutant animals (data not shown).
Fig. 4.
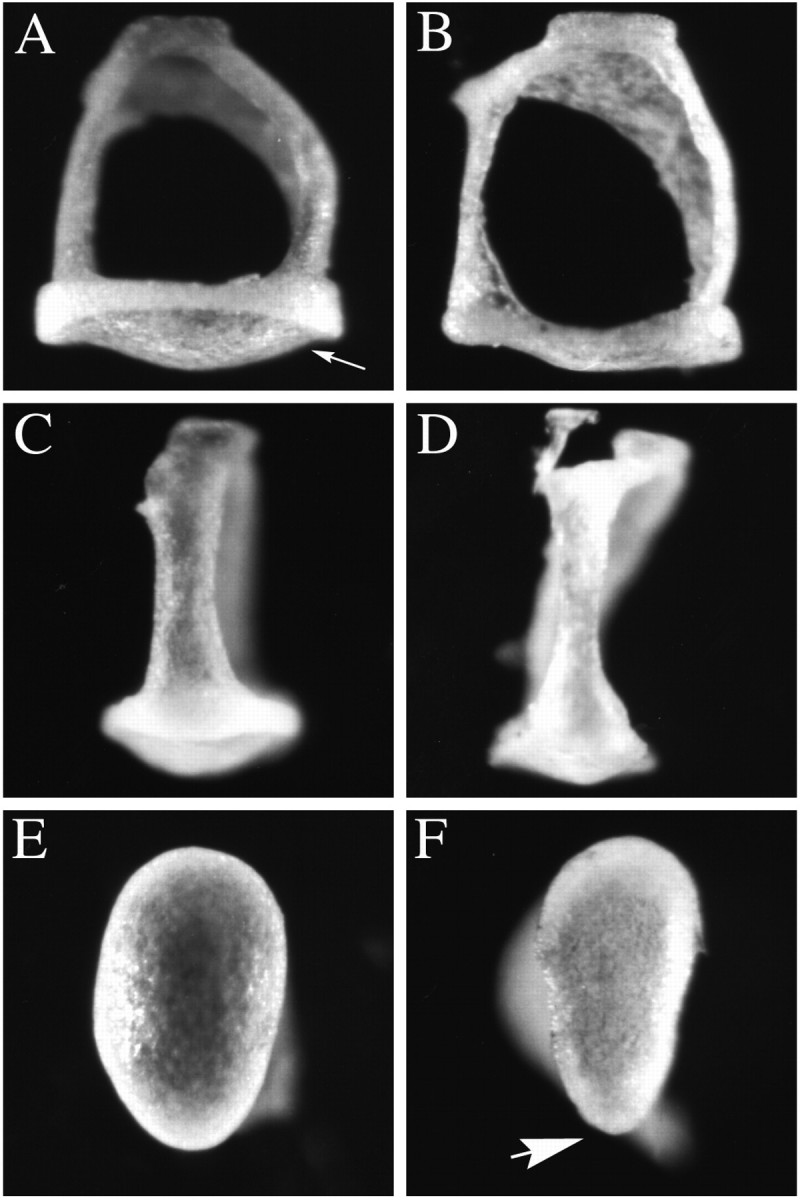
Malformations of the stapes observed inBrn4 null mice. The stapes of the mutant animals demonstrate several malformations, particularly in the stapedial footplate. A, C, E, Wild-type stapes are depicted. B, D,F, Mutant stapes are depicted. A,B, The footplate of the stapes (arrow inA) in mutant embryos (B) is flatter in comparison with that in wild-type stapes (A). C, D, A lateral view of the stapes demonstrates that the crus from which the stapedial ligament is attached is thinner in the mutant animals (D) than in the wild-type animals (C). E, F, Examination of the sole of the stapes footplate illustrates the slightly eccentric ovoid shape of the wild-type footplate (E). The mutant footplate adopts a more polygonal shape with an acutely angled tip on one end of the footplate (arrow in F). Stapes (n = 10) isolated from six mutant male animals were examined. Wild-type stapes (n = 15) were examined from eight male animals, including six wild-type littermates, two males from the inbred strain 129/SvJ, and two CBA/2J male animals.
Malformations of the cochlea in the mutant mice
Histological analyses demonstrated that adult cochleae from theBrn4 mutant mice had several dysplastic features (Fig.5). Midmodiolar sections demonstrated the overall hypoplasia of the cochlea (Fig.5A,B). This hypoplasia was most obvious in the basal turn and primarily resulted from a change in the scala tympani. The scala tympani of normal mice, which lies inferior to the organ of Corti, is ovoid in the basal turn of the cochlea (Fig.5C). However, in the mutant cochlea, the scala tympani adopts a more flattened elliptical shape with an apparent reduction in volume (Fig. 5D). Figure 5 depicts the typical reduction in size of the scala tympani and scala vestibuli in the mutant animals, but a few animals display a severe phenotype in which the scala tympani cross section is reduced by approximately one-half to one-third (data not shown). An additional component of the cochlear hypoplasia results from a reduction in the coiling, or number of turns, of the cochlea. This hypoplastic phenotype is demonstrated both in midmodiolar sections of the cochlea (Fig. 5) and in cleared temporal bone preparations (Fig.6). Midmodiolar sections of normal cochleae typically reveal cross sections through the organ of Corti at three different levels with an additional cross section near the apical end of this structure, the helicotrema (Fig. 5A,arrow). Midmodiolar sections of the mutant cochlea reveal a fewer number of cochlear turns (Fig. 5B). The reduction in cochlear coiling was most evident in cleared temporal bone preparations, which provide direct visualization of the cochlear morphology in whole mount (Fig. 6). The normal mouse cochlea consists of one and three-fourth turns, as shown in Figure 6A(Sher, 1971). Approximately 60% of the mutant cochleae show a reduction in the number of turns to one and one-half turns (Fig.6B). However, ∼1/4th of the cochlea demonstrated even fewer turns, with the least amount of coiling being a three-fourth turn (∼15% of the cochleae examined). Only 1/10th of the mutant cochleae examined demonstrated the normal one and three-fourth turns. Even when comparing the left and right bony labyrinths from the same animal, we observed variability in the number of turns (Fig.6B,C). Heterozygous female animals contained the normal one and three-fourth turns and were indistinguishable from wild-type animals.
Fig. 5.
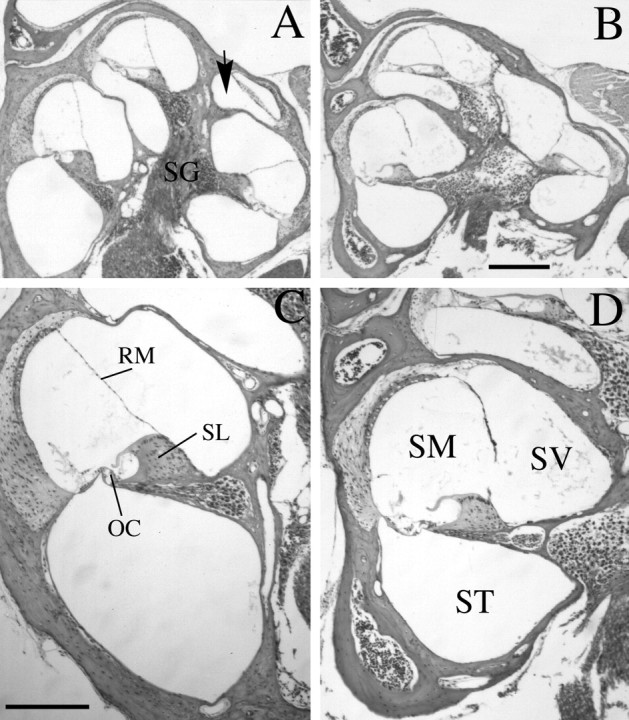
Brn4 knock-out mutants demonstrate cochlear dysplasias in adult mice. B, D, Midmodiolar sections from an adult (6-week-old) Brn4hemizygous null mutant are shown. A, C, Similar sections from a wild-type littermate are shown.A, B, The cochlea of the mutant mouse (B) demonstrates an overall hypoplastic structure compared with that of the wild-type animal (A). The arrow in A indicates the most apical turn of the normal cochlea, which is rarely detected in similar sections of the mutant animal. C, D, The scala tympani of the mutant mouse (D) is flattened and elliptical in comparison with that of the wild-type control mouse (C). Additionally, Reissner’s membrane displays the distended morphology seen in D in the mutant embryos, consistent with a hydrops condition in the mutant animals. In all cases examined, similar phenotypes were found in homozygous knock-out female animals (data not shown).OC, Organ of Corti; RM, Reissner’s membrane; SG, spiral ganglion; SL, spiral limbus; SM, scala media; ST, scala tympani; SV, scala vestibuli. Scale bars:A, B, 300 μm; C,D, 200 μm.
Fig. 6.

Cleared temporal bone preparations demonstrate cochlear hypoplasia. Temporal bone preparations were perfused through the oval window with white paint to visualize cochlear morphology. Cochleae are oriented such that the apical turn is at thetop of the photograph, as indicated by thearrow in B. A, A cochlea from a wild-type animal with one and three-fourth turns is shown.B, C, Mutant cochlea displayed a reduced number of turns, which could vary between less than one complete turn to one and one-half turns. Additionally, the amount of coiling can differ between left and right cochlea of the same mutant animal. For example, C demonstrates the right cochlea of a homozygous knock-out female animal, which has less than one coil. By contrast, B demonstrates the left cochlea of the same animal, which has one and one-half turns.
Histological analyses of the organ of Corti did not demonstrate gross abnormalities in the number or morphology of sensory hair cells (data not shown). However, a supporting structure for the organ of Corti, the spiral limbus, does show a reduction in size (Fig.7A,B). To quantitate this reduction, we have measured the height of the spiral limbus in the basal turn of the cochlea. The mean spiral limbus height in the wild-type animals was 91 μm with an SD of 11 μm (n = 10); in null male hemizygotes, the height was 75 ± 12 μm (n = 6). An additional phenotype in the cochlea of Brn4 mutant animals included the fibrocytes of the spiral ligament, which appear thinner and less adherent to one another than do those in the wild-type mouse (Fig.7C,D). This results in acellular spaces within the region where the fibrocytes normally underlie the stria vascularis. Reissner’s membrane often appears to be less tightly adherent to the fibrocytes in the mutant than in the wild-type mouse. These fibrocytes provide structural support for the stria vascularis, which regulates the ionic composition of the endolymph that bathes the organ of Corti. This malformation could compromise the function of the stria vascularis, leading to increased pressure or hydrops in the inner ear. In fact, all of the mutant animals examined (n = 12) appear to have hydrops, which is diagnosed by a distension of Reissner’s membrane (Fig. 5D).
Fig. 7.

Histological analysis of spiral limbus and strial fibrocyte dysplasias. B,D, Midmodiolar sections from an adult (6-week-old)Brn4 hemizygous null mutant are shown. A,C, Similar sections from a wild-type littermate are shown. A, B, This view demonstrates that the spiral limbus of mutant animals (B) is smaller than that detected in the wild-type animals (A). The height of the spiral limbus was calculated by drawing a baseline (height = 0) at the widest part of the spiral limbus from the tympanic lip of the internal spiral sulcus to the point where the spiral limbus meets the bony wall of the cochlea (see arrowheads in B). Measurements were made from photomicrographs, and the highest point attained by the interdental cells at the crest of the spiral limbus was measured in a plane perpendicular to the baseline. An additional aspect of the mutant phenotype seen in this figure includes acellular gaps that are often detected between the spiral ganglion cells of the mutants and the modiolus (arrow in B). In wild-type animals, such an acellular gap is rarely detected (B). C, D, This view demonstrates that the strial fibrocytes are not tightly adher-ent in the mutant (D) compared with the wild-type (C) mouse. This phenotype was fully penetrant in all 12 mutant animals examined. F, Fibrocytes; RM, Reissner’s membrane; SV, stria vascularis. Scale bars: A, B, 50 μm; C, D, 20 μm.
Additional features of the mutant phenotype sometimes include an incomplete fasciculation of the vestibuloacoustic nerve, as it enters the basal turn of the cochlea (data not shown). The apparent loss of cohesiveness of this nerve may be caused by the increased size of the internal auditory meatus, which conveys the nerve through the temporal bone.
Malformations of the temporal bone in Brn4null mutants
Because Brn4 is highly expressed throughout the anlage of the temporal bone, we examined the morphology of this bone in adult mutant animals (Fig. 8). The major external malformations that occur in the temporal bone result from enlargement of the internal auditory meatus. In rodents, this meatus consists of three foramina through which the auditory (VIII) nerve and facial (VII) nerve are conveyed through the temporal bone to the brainstem (Curthoys, 1981). The largest of these cavities contains the type I and II auditory nerve fibers, which originate at the auditory hair cells, have their cell bodies in the spiral ganglion, and have nerve fibers that project from the cochlea through the central axis or modiolus. The two smaller foramina convey the motor fibers of the facial (VII) nerve through the temporal bone and allow the vestibular component of the VIII nerve to communicate with the vestibular nuclei. All three foramina are enlarged in the mutant animals (Fig. 8).
Fig. 8.
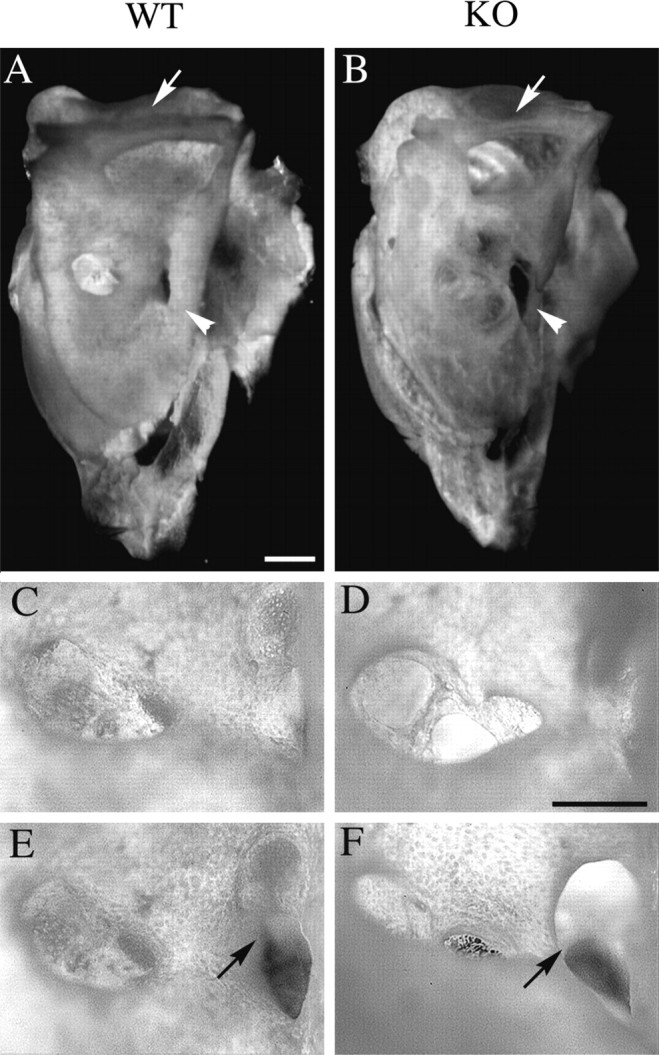
Malformations of the temporal bone inBrn4 null mice. Analysis of whole-mount preparations of adult temporal bone demonstrates dysplasia of the three foramina of the internal auditory meatus. A, C,E, A wild-type temporal bone is depicted.B, D, F, A mutant temporal bone is depicted. A, B, A medial view of the temporal bone demonstrates that the bony tissue appears thinner in the mutant (B) than in the wild type (A). The bone encompassing the superior semicircular canal is thinner in the mutant than in the wild-type animal (arrow), and the rostromedial ridge of the temporal bone is thinner in the mutant (arrowhead).C, D, The cochlear foramen of the internal auditory meatus (IAM) is enlarged in the mutant (D) animal when compared with that in the wild-type animal. E, F, The second and third foramen of the mutant IAM (F) appear to be fused (arrow) when compared with that in the wild type (E). Scale bars: A,B, 1 mm; C–F, 0.5 mm.
We have observed additional malformations of the temporal bone, which include (1) thinning of the lateral wall of the subarcuate fossa, which houses the lateral aspect of the cerebellum (data not shown), (2) thinning of the bone encompassing the superior semicircular canal (Fig.8A,B), and (3) reduction in the size of the rostromedial ridge of the temporal bone (Fig.8A,B). In each of these cases, the malformations appear to result from a reduction in the thickness of the bone. This observation is consistent with the hypothesis that theBrn4 gene may be necessary for the survival of mesenchymal cells during the extensive mesenchymal remodeling that occurs during development of the otic capsule (Phippard et al., 1998).
Malformations of the superior semicircular canals are observed inBrn4 null animals
Because the vertical head-bobbing phenotype in the mutant mice implies that the vestibular system is not functioning properly, we have examined the anatomy of the vestibular apparatus in greater detail. We consistently observed a constriction in the bony labyrinth encompassing the superior semicircular canals in the Brn4 null mice, which is ∼40% of the size of the wild-type superior semicircular canals (Fig. 9). Therefore, both the thickness of the bone encompassing the superior semicircular canal and the perilymphatic space encompassing the canal are reduced in size (Figs. 8A,B, 9). The orientation of the superior semicircular canal within the cranium is approximately parallel with the major body axis, and therefore, a dysfunctional superior semicircular canal would most easily explain the vertical nature of the head-bobbing phenotype. We have not observed malformations in the posterior semicircular canals in theBrn4 null mutants. However, the lateral semicircular canal shows a constriction or dysplastic phenotype in 4 of the 15 animals examined. Because the Brn4 gene is expressed throughout the otic capsule during development, it is not clear why any of the canals should be differentially sensitive to the loss of Brn4 gene function. In heterozygous female animals, the superior semicircular canals were identical in size to that in wild-type animals (data not shown). These data indicate that a constriction of the semicircular canal results in compromised vestibular function in the mutant animals. Furthermore, the correlation of the orientation plane of the superior semicircular and the vertical motion of the head-bobbing phenotype suggests that the dysplastic superior canal is the major dysfunctional component of the vestibular apparatus in the Brn4 null animals.
Fig. 9.

Constriction of the superior semicircular canal inBrn4 null mice. Temporal bone preparations were filled with latex paint and cleared in methyl salicylate to examine the structure of the bony labyrinth. A, Medial view of the left superior semicircular canal of a heterozygous female animal. The specimen is oriented with anterior to the right and dorsal toward the top of the photograph.B, The left superior semicircular canal of a homozygous female knock-out animal. The arrow indicates the typical constriction in the most dorsal extent of the superior semicircular canal. The narrowest constriction points of the mutant canals are ∼2.5 times smaller than are those of the wild-type mice. Scale bar, 300 μm.
DISCUSSION
Mice lacking the POU-domain transcription factor Brn-4 showed several malformations in the development of the inner ear. Many of these malformations are detected in inner ear structures derived from mesenchymal origins, which express the Brn4 gene. These malformations included structures, such as the scala tympani and the internal auditory meatus, that are derived from the otic capsule. Malformations in the mutant animals resulted in functional deficits in the adult mutant animals, including hearing loss and head bobbing. These mutant phenotypes suggest that Brn4 is crucial for the proper morphogenesis of both the auditory and vestibular systems of the inner ear.
The widespread distribution of defects in the inner ear correlated with the expression of Brn-4 throughout most of otic mesenchyme during development (Phippard et al., 1998). Brn4 gene expression was activated simultaneously with the initial mesenchymal condensation events that formed the otic capsule, suggesting the Brn4gene is induced by the same factors that regulate otic capsule formation. Later in embryogenesis, Brn4 gene expression is found throughout the otic capsule. Many of the defects found in the mutant mice are a result of reduced bone formation, including enlarged foramina of the internal auditory meatus and thinning of bone in the subarcuate fossa. These observations suggest that the Brn4gene is required for the proliferation or differentiation of temporal bone tissue. This conclusion is consistent with the role that has been demonstrated for another member of the POU-domain gene family,Pit1, which plays a crucial role in the regulation of proliferation and differentiation of the anterior pituitary gland (Li et al., 1990; Lin et al., 1992).
Not all of the malformations in the mutant animals were restricted exclusively to defects in otic mesenchyme. For example, the reduction in the number of turns of the cochlea suggests that dysplastic changes in the morphogenetic movements of the otic epithelia occur. Figure10A–C illustrates the morphogenetic movements of the otic epithelia that are required to generate cochlear coiling. Formation of the cochlea is initiated as a tubular outgrowth of the pars inferior of the otic epithelium. This outgrowth forms the cochlear duct that coils—within the surrounding otic mesenchyme—to give the overall “snail-shaped” appearance of the cochlea. In the Brn4 mutant mice, none of the cochleae that we examined had attained the correct number of turns, and some of the mutants demonstrated a severe reduction in cochlear coiling (Fig.6C). Because we have not detected the expression ofBrn4 in the otic epithelium, it seems likely that the coiling defect occurs because of phenotypic changes within the otic mesenchyme itself. These phenotypic changes could result from the disruption of cell signaling between the mesenchyme and epithelium. Alternatively, defective remodeling of the mesenchyme could impede the coiling of the cochlear duct by preventing the outgrowth of the tubular duct process. The absence of Brn-4 may be interfering with the signals required for epithelial–mesenchymal interaction such that the mesenchyme is not remodeled and the extension of the cochlear duct is physically blocked.
Fig. 10.

Schematic illustration of development of the cochlea. A–C, The formation of the coiled form of the cochlea during embryogenesis is depicted. Theseschematics illustrate the morphogenetic changes in the epithelial sac that is derived from the otic vesicle. The illustrated morphogenetic movements all occur within the surrounding condensed mesenchyme of the otic capsule, which expresses the Brn4gene. A, Formation of the cochlea is initiated by a tubular outgrowth of the pars inferior (ventral region) of the otic vesicle. B, Further extension of this tubular outgrowth gives rise to the cochlear duct. C, The cochlear duct coils progressively during development to give rise ultimately to one and three-fourth turns in the mouse, whereas the adjacent region of the otic just superior to the cochlea becomes the saccule (Sher, 1971;Morsli et al., 1998). The cochlear duct is the anlage of the middle fluid-filled sound-conducting chamber in the cochlea, referred to as the scala media. D–F, The mesenchymal remodeling of the otic capsule that gives rise to the two additional sound conduction compartments in the cochlea, the scala vestibuli and the scala tympani, is depicted. These panels depict a cross section through the cochlear duct. D, When the cochlear duct is initially formed, it is surrounded by the condensed mesenchyme of the otic capsule. E, The scala tympani and the scala vestibuli are formed by cavitation of the otic capsule. A time point during which these compartments are cavitating but are still filled with trabeculae of mesenchymal material is depicted inE. F, Illustration of the adult cochlea depicts the fully formed scala vestibuli and scala tympani. Both of these structures exhibit defects in the Brn4 mutant animals. This figure was schematized on the basis of figures found inLangman (1982) and modified to reflect mouse development more accurately.
Malformations in structures that result from mesenchymal remodeling of the otic capsule constitute the major class of abnormalities observed in Brn4 null animals. For example, remodeling of the otic mesenchyme surrounding the cochlear duct is necessary to generate two acellular fluid-filled chambers in the adult cochlea, the scala vestibuli and the scala tympani (Fig. 10D–F). These chambers, which are malformed in the mutant animals, are generated by cavitation or vacuolization of the otic mesenchyme (Langman, 1982). We suggest that the appropriate regulation of the Brn-4 factor within the otic mesenchyme is necessary for the mesenchymal remodeling that leads to the formation of the scala vestibuli and the scala tympani. This hypothesis is further supported by the observation that Brn-4 subcellular localization is regulated in regions of the otic capsule that undergo mesenchymal remodeling during embryogenesis (Phippard et al., 1998). In regions of the otic capsule that will form bone in the adult, the Brn-4 transcription factor retains a nuclear subcellular localization. However, in regions of the otic capsule that will be eliminated to form acellular caverns within the temporal bone, Brn-4 shifts from a nuclear to a perinuclear/cytoplasmic subcellular localization, suggesting that the ability of this transcription factor to function is downregulated in these regions. This shift in subcellular localization occurs in the foramen of the internal auditory meatus and the perilymphatic compartments of the cochlea. Most regions of the inner ear that demonstrated regulation of Brn-4 subcellular localization also demonstrate defects in Brn4 null mutants.
The defects demonstrated in the Brn4 null animals have been observed in human patients with mutations in the human Brn4ortholog POU3F4. These patients exhibited hypoplasia of the cochlea, an enlarged internal auditory meatus, and stapes fixation (de Kok et al., 1995a; Piussan et al., 1995). These pathologies suggest an evolutionary conservation of Brn-4 function in mammals. Interestingly, several differences can be noted between the mouse mutant and the human patients. First, the human patients demonstrate profound sensorineural deafness, arising from the loss of spiral ganglion cells in the cochlea. Although the mutant mice showed some hearing loss, it was mild compared with that of the human patients. To date, we have not observed any signs of sensorineural deafness in young animals. A more extensive analysis of sensorineural hearing loss in older animals is complicated by the fact that the genetic background (C57BL/6J) leads to age-onset sensorineural deafness. We are currently backcrossing theBrn4 mutation onto a different genetic background (CBA/J) that does not have age-onset hearing loss (for review, see Willott, 1996). The second difference between human and mouse mutants is found in the stapes. Stapes fixation clearly contributes to the conductive hearing loss of the patients. Although the stapes does exhibit malformations in the mutant embryos, we have yet to observe stapes fixation in dissected temporal bone preparations of transgenic mice. The differences with regard to the stapes may represent subtle differences in the size, morphology, and ontogeny of the human stapes when compared with that of the mouse.
Although defects were observed in the stapes of Brn4 null animals, Brn4 gene expression is not observed in the developing stapes (Phippard et al., 1998), which is formed from a mesenchymal condensation event that occurs adjacent to the otic capsule. Brn4 gene expression was observed, however, in those regions of the otic capsule that lie adjacent to the developing stapes from which the oval window develops. It is essential for the footplate of the stapes to sit in an appropriate manner in the oval window. Efficient sound conduction occurs when the footplate is firmly attached to the annular ligament and can move freely. Therefore, it seems likely that inductive signals from the otic capsule are required for the proper formation of the stapes and its positioning in the oval window. Further analyses of the mutant embryos may provide insight into the mesenchymal–mesenchymal signals necessary to coordinate the formation and orientation of the stapes and the oval window.
Interestingly, we did not see malformations of the inner ear in heterozygous female animals. Because of X chromosome inactivation, one would expect the inner ears of the heterozygous females to comprise a mosaic of cells with one-half expressing the functional Brn4allele and the other half expressing the knock-out allele. The anatomy of the temporal bones of heterozygous females was qualitatively unaffected. Also, all quantitative analyses examined, such as size of the superior semicircular canal and the number of cochlear turns, were identical to that of wild-type animals. One possible explanation is that cells that do not express the Brn4 gene are regulated by those neighboring cells that express a functional Brn4allele. Alternatively, regulatory mechanisms within the cells that contain the null Brn4 allele may have induced the expression of the Brn4 gene on the inactive X chromosome. Additional studies will be required to distinguish these hypotheses.
Despite the widespread expression of Brn-4 throughout the neural tube, we have not observed any defects in the development or function of the CNS (D. Phippard and E. B. Crenshaw III, unpublished observations). A likely explanation for this observation is that other POU-domain factors, including closely related members of the POU-domain subclass III, are also expressed widely in the neural tube, suggesting functional redundancy of these genes during neural tube ontogeny. All members of this subclass are broadly expressed throughout the neural tube during embryogenesis but show restricted domains of expression in late fetal and adult animals (He et al., 1989). As seen in theBrn4 mutants, the targeted disruption of the POU-domain subclass III members Brn2/Pou3f2 and Tst1/Pou3f3demonstrated rather restricted phenotypes in comparison with their broad domains of expression (Nakai et al., 1995; Schonemann et al., 1995; Bermingham et al., 1996). In fact, the defects in targeted disruptions of this subclass tended to be restricted to the regions of the CNS in which they showed expression patterns unique from that of other subclass members. For example, the targeted disruption of theTst1 gene demonstrated defects in glial cells and the LOTN region of the brainstem (Bermingham et al., 1996). Additionally, the defects tended to occur in cells in which the POU-domain gene was expressed in late fetal and adult stages. For example, the defects in the Brn2 mutants resulted in malformations of the hypothalamic nuclei that express theBrn2 gene in the adult (Nakai et al., 1995; Schonemann et al., 1995). In toto, these data strongly suggest that POU-domain factors are functionally redundant during neural tube development.
In conclusion, the absence of Brn-4 expression seems to affect three different properties of otic mesenchyme: (1) mesenchymal remodeling that is necessary to generate structures such as the scala tympani, (2) epithelial–mesenchymal interactions that are required to form the cochlear coils, and (3) mesenchymal–mesenchymal interactions that are necessary for the coordinated formation of the oval window and the stapes. The Brn4 gene is a useful marker gene for the formation of the otic mesenchyme, as well as a critical factor in pattern formation of the developing inner ear. Further analyses of the underlying processes that regulate the Brn4 gene as well as its role during inner ear development will provide insight into the molecular mechanisms of inner ear ontogeny.
Footnotes
This work was supported by National Institutes of Health Grant R01 NS-31674 and the March of Dimes Basil O’Connor Starter Scholar Award. We would like to thank Emily Howard, Tony Caggiano, and Jeff Neul with help generating the knock-out construct; Drs. Andras Nagy and J. Rossant for the R1 ES cells; Dr. John Bermingham for the pTKAB′ vector; and Dr. M. A. Rudnicki for the PGK-TK plasmid.
Correspondence should be addressed to Dr. E. Bryan Crenshaw III, Department of Neuroscience, University of Pennsylvania, Philadelphia, PA 19104-6074.
REFERENCES
- 1.Alvarez-Bolado G, Rosenfeld MG, Swanson LW. Model of forebrain regionalization based on spatiotemporal patterns of POU-III homeobox gene expression, birthdates, and morphological features. J Comp Neurol. 1995;355:237–295. doi: 10.1002/cne.903550207. [DOI] [PubMed] [Google Scholar]
- 2.Bermingham JR, Scherer SS, O’Connell S, Arroyo E, Kalla KA, Powell FL, Rosenfeld MG. Tst-1/Oct-6/SCIP regulates a unique step in peripheral myelination and is required for normal respiration. Genes Dev. 1996;10:1751–1762. doi: 10.1101/gad.10.14.1751. [DOI] [PubMed] [Google Scholar]
- 3.Curthoys IS. Scarpa’s ganglion in the rat and guinea pig. Acta Otolaryngol (Stockh) 1981;92:107–113. doi: 10.3109/00016488109133244. [DOI] [PubMed] [Google Scholar]
- 4.de Kok YJ, Merkx GF, van der Maarel SM, Huber I, Malcolm S, Ropers HH, Cremers FP. A duplication/paracentric inversion associated with familial X-linked deafness (DFN3) suggests the presence of a regulatory element more than 400 kb upstream of the POU3F4 gene. Hum Mol Genet. 1995a;4:2145–2150. doi: 10.1093/hmg/4.11.2145. [DOI] [PubMed] [Google Scholar]
- 5.de Kok YJ, van der Maarel SM, Bitner-Glindzicz M, Huber I, Monaco AP, Malcolm S, Pembrey ME, Ropers HH, Cremers FP. Association between X-linked mixed deafness and mutations in the POU domain gene POU3F4. Science. 1995b;267:685–688. doi: 10.1126/science.7839145. [DOI] [PubMed] [Google Scholar]
- 6.Deol MS. Influence of the neural tube on the differentiation of the inner ear in the mammalian embryo. Nature. 1966;209:219–220. doi: 10.1038/209219a0. [DOI] [PubMed] [Google Scholar]
- 7.Erkman L, McEvilly RJ, Luo L, Ryan AK, Hooshmand F, O’Connell SM, Keithley EM, Rapaport DH, Ryan AF, Rosenfeld MG. Role of transcription factors Brn-3.1 and Brn-3.2 in auditory and visual system development. Nature. 1996;381:603–606. doi: 10.1038/381603a0. [DOI] [PubMed] [Google Scholar]
- 8.Fay RR. Hearing in vertebrates: a psychophysics data book, pp 367–369. Hill-Fay; Winnetka, IL: 1988. [Google Scholar]
- 9.Fekete DM. Cell fate specification in the inner ear. Curr Opin Neurobiol. 1996;6:533–541. doi: 10.1016/s0959-4388(96)80061-4. [DOI] [PubMed] [Google Scholar]
- 10.Frenz DA, Van de Water TR. Epithelial control of periotic mesenchyme chondrogenesis. Dev Biol. 1991;144:38–46. doi: 10.1016/0012-1606(91)90476-j. [DOI] [PubMed] [Google Scholar]
- 11. He X, Treacy MN, Simmons DM, Ingraham HA, Swanson LW, Rosenfeld MG. Expression of a large family of POU-domain regulatory genes in mammalian brain development. Nature 340 1989. 35 41[Erratum (1989) 340:662]. [DOI] [PubMed] [Google Scholar]
- 12.Herr W, Cleary MA. The POU domain: versatility in transcriptional regulation by a flexible two-in-one DNA-binding domain. Genes Dev. 1995;9:1679–1693. doi: 10.1101/gad.9.14.1679. [DOI] [PubMed] [Google Scholar]
- 13.Hogan B, Beddington R, Costantini F, Lacy E. Manipulating the mouse embryo: a laboratory manual. Cold Spring Harbor Laboratory; Plainview, NY: 1994. [Google Scholar]
- 14.Joyner AL. Gene targeting. In: Rickwood D, Hames BD, editors. The practical approach series. Oxford UP; New York: 1993. p. 234. [Google Scholar]
- 15.Langman J. Medical embryology. Williams and Wilkins; Baltimore: 1982. [Google Scholar]
- 16.Le Moine C, Young WS. RHS2, a POU domain-containing gene, and its expression in developing and adult rat. Proc Natl Acad Sci USA. 1992;89:3285–3289. doi: 10.1073/pnas.89.8.3285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lewis ER, Leverenz EL, Bialek WS. The vertebrate inner ear. CRC; Boca Raton, FL: 1985. [Google Scholar]
- 18.Li S, Crenshaw EB, Rawson EJ, Simmons DM, Swanson LW, Rosenfeld MG. Dwarf locus mutants lacking three pituitary cell types result from mutations in the POU-domain gene pit-1. Nature. 1990;347:528–533. doi: 10.1038/347528a0. [DOI] [PubMed] [Google Scholar]
- 19.Lin C, Lin SC, Chang CP, Rosenfeld MG. Pit-1-dependent expression of the receptor for growth hormone releasing factor mediates pituitary cell growth [see comments]. Nature. 1992;360:765–768. doi: 10.1038/360765a0. [DOI] [PubMed] [Google Scholar]
- 20.Mathis JM, Simmons DM, He X, Swanson LW, Rosenfeld MG. Brain 4: a novel mammalian POU domain transcription factor exhibiting restricted brain specific expression. EMBO J. 1992;7:2551–2561. doi: 10.1002/j.1460-2075.1992.tb05320.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.McPhee JR, Van de Water TR. Epithelial-mesenchymal tissue interactions guiding otic capsule formation: the role of the otocyst. J Embryol Exp Morphol. 1986;97:1–24. [PubMed] [Google Scholar]
- 22.Morsli H, Choo D, Ryan A, Johnson R, Wu DK. Development of the mouse inner ear and origin of its sensory organs. J Neurosci. 1998;18:3327–3335. doi: 10.1523/JNEUROSCI.18-09-03327.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Nakai S, Kawano H, Yudate T, Nishi M, Kuno J, Nagata A, Jishage K, Hamada H, Fujii H, Kawamura K, Shiba K, Noda T. The POU domain transcription factor Brn-2 is required for the determination of specific neuronal lineages in the hypothalamus of the mouse. Genes Dev. 1995;9:3109–3121. doi: 10.1101/gad.9.24.3109. [DOI] [PubMed] [Google Scholar]
- 24.Noden DM, Van de Water TR. The developing ear: tissue origins and interactions. In: Ruben RW, editor. The biology of change in otolaryngology. Elsevier; Oxford, UK: 1986. pp. 15–46. [Google Scholar]
- 25.Noden DM, Van de Water TR. Genetic analyses of mammalian ear development [review]. Trends Neurosci. 1992;15:235–237. doi: 10.1016/0166-2236(92)90056-e. [DOI] [PubMed] [Google Scholar]
- 26.Peck JE. Development of hearing. Part II. Embryology. J Am Acad Audiol. 1994;5:359–365. [PubMed] [Google Scholar]
- 27.Peters PW. Double staining of fetal skeletons for cartilage and bone. In: Neubert D, editor. Methods in prenatal toxicology. Thieme; Stuttgart, Germany: 1977. pp. 153–154. [Google Scholar]
- 28.Phippard DJ, Heydemann A, Lechner M, Lu L, Lee D, Kyin T, Crenshaw EB., III Changes in the subcellular localization of the Brn4 gene product precede mesenchymal remodeling of the otic capsule. Hear Res. 1998;120:77–85. doi: 10.1016/s0378-5955(98)00059-8. [DOI] [PubMed] [Google Scholar]
- 29.Piussan C, Hanauer A, Dahl N, Mathieu M, Kolski C, Biancalana V, Heyberger S, Strunski V. X-linked progressive mixed deafness: a new microdeletion that involves a more proximal region in Xq21. Am J Hum Genet. 1995;56:224–230. [PMC free article] [PubMed] [Google Scholar]
- 30.Ryan AK, Rosenfeld MG. POU domain family values: flexibility, partnerships, and developmental codes. Genes Dev. 1997;11:1207–1225. doi: 10.1101/gad.11.10.1207. [DOI] [PubMed] [Google Scholar]
- 31.Schonemann MD, Ryan AK, McEvilly RJ, O’Connell SM, Arias CA, Kalla KA, Li P, Sawchenko PE, Rosenfeld MG. Development and survival of the endocrine hypothalamus and posterior pituitary gland requires the neuronal POU domain factor Brn-2. Genes Dev. 1995;9:3122–3135. doi: 10.1101/gad.9.24.3122. [DOI] [PubMed] [Google Scholar]
- 32.Sher AE. The embryonic and postnatal development of the inner ear of the mouse. Acta Otolaryngol Suppl (Stockh) 1971;285:1–77. [PubMed] [Google Scholar]
- 33.Smith SD. Overview of genetic auditory syndromes [review]. J Am Acad Audiol. 1995;6:1–14. [PubMed] [Google Scholar]
- 34.Steel KP, Brown SD. Genes and deafness [review]. Trends Genet. 1994;10:428–435. doi: 10.1016/0168-9525(94)90113-9. [DOI] [PubMed] [Google Scholar]
- 35.Theiler K. The house mouse: atlas of embryonic development, Second Edition. Springer; New York: 1989. [Google Scholar]
- 36.Willott JF. Anatomic and physiologic aging: a behavioral neuroscience perspective. J Am Acad Audiol. 1996;7:141–151. [PubMed] [Google Scholar]
- 37.Xiang M, Gan L, Zhou L, Klein WH, Nathans J. Targeted deletion of the mouse POU domain gene Brn-3a causes selective loss of neurons in the brainstem and trigeminal ganglion, uncoordinated limb movement, and impaired suckling. Proc Natl Acad Sci USA. 1996;93:11950–11955. doi: 10.1073/pnas.93.21.11950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Xiang M, Gan L, Li D, Chen ZY, Zhou L, O’Malley BW, Jr, Klein W, Nathans J. Essential role of POU-domain factor Brn-3c in auditory and vestibular hair cell development. Proc Natl Acad Sci USA. 1997;94:9445–9450. doi: 10.1073/pnas.94.17.9445. [DOI] [PMC free article] [PubMed] [Google Scholar]