

ABSTRACT
Paclitaxel is a widely used anti-cancer treatment that disrupts cell cycle progression by blocking cells in mitosis. The block at mitosis, with spindles assembled from short microtubules, is surprising given paclitaxel’s microtubule stabilizing activity and the need to depolymerize long interphase microtubules prior to spindle formation. Cells must antagonize paclitaxel’s microtubule stabilizing activity during a brief window of time at the transition from interphase to mitosis, allowing microtubule reorganization into a mitotic spindle, although the mechanism underlying microtubule depolymerization in the presence of paclitaxel has not been examined. Here we test the hypothesis that microtubule severing and/or depolymerizing proteins active at mitotic entry are necessary to clear the interphase array in paclitaxel-treated cells and allow subsequent formation of mitotic spindles formed of short microtubules. A549 and LLC-PK1 cells treated with 30nM paclitaxel approximately 4 h prior to mitotic entry successfully progress through the G2/M transition by clearing the interphase microtubule array from the cell interior outward to the cell periphery, a spatial pattern of reorganization that differs from that of cells possessing dynamic microtubules. Depletion of kinesin-8s, KIF18A and/or KIF18B obstructed interphase microtubule clearing at mitotic entry in paclitaxel-treated cells, with KIF18B making the larger contribution. Of the severing proteins, depletion of spastin, but not katanin, reduced microtubule loss as cells entered mitosis in the presence of paclitaxel. These results support a model in which KIF18A, KIF18B, and spastin promote interphase microtubule array disassembly at mitotic entry and can overcome paclitaxel-induced microtubule stability specifically at the G2/M transition.
KEYWORDS: Prophase, microtubules, paclitaxel, kinesin, spastin, katanin
Introduction
Rapid cell proliferation is a hallmark of cancer,1 and this property has been targeted by drugs that block proliferation by stalling cells in mitosis through disruption of normal cycles of microtubule polymer assembly or disassembly from tubulin subunits.2 Microtubule-targeting drugs induce either polymer depolymerization (i.e. vinblastine, colchicine) or stability (i.e. paclitaxel, docetaxel) and have had success in clinical applications as cancer therapies.3–5 By inhibiting microtubule function during mitosis, microtubule-targeting drugs block successful completion of mitosis and ultimately induce cell death,6–8 although these drug treatments also bring on significant side effects, and drug resistance remains a challenge.5,9–11
Microtubules, polymers of α and ß tubulin subunits, form one of the intracellular cytoskeletal filaments that function outside of mitosis to maintain the spatial organization of organelles and facilitate intracellular vesicular trafficking throughout the cell. The architecture of the microtubule cytoskeleton is specific to cell shape and biological process, for example elongated or polarized cells,12 as well as the leading and trailing edges during cell migration.13 Microtubules adopt a highly dynamic structure during cell division, forming the mitotic spindle, which is critical to the segregation of genetic material between daughter cells. This change in organization from the G2 stage of the cell cycle to mitosis is characterized by the dissolution of the interphase polymer array, consisting of a few hundred long microtubules, releasing tubulin subunits for assembly of the mitotic spindle, comprised of up to several thousand short microtubules; this entire process occurs during a small window of time during the G2/M transition.14,15 A subset of short microtubules is also transported by dynein motors toward the nucleus in preparation for mitosis, but a large fraction of the microtubule cytoskeleton is first depolymerized to tubulin subunits prior to spindle assembly.15,16
Microtubule organization and turnover is governed by dynamic instability, a process where tubulin subunits assemble or disassemble from microtubule ends. Transitions between these assembly and disassembly states are termed catastrophe, the molecular changes that drive the abrupt switch from assembly to disassembly states, and rescue, the molecular events underlying the transition from disassembly back to an assembly state. Measurements of dynamic instability in living cells have demonstrated a slower turnover of interphase microtubules compared to those in mitotic cells, including the brief transition from G2 to M phases of the cell cycle, coinciding with nuclear envelope breakdown (NEBD).16,17 The changes in overall microtubule dynamics are regulated primarily via changes in catastrophe and rescue frequencies, rather than in the rates of polymerization or depolymerization.16–18 Simulation of the dynamic microtubule array using a Monte Carlo-based algorithm supported the hypothesis that changes in the transition frequencies of dynamic instability are sufficient to disassemble the interphase array within minutes using parameters measured at NEBD.17,19
The measured shift in dynamic instability parameters as cells enter mitosis presents an interesting question: how can an interphase microtubule array, stabilized by paclitaxel, disassemble to form a spindle of short microtubules? Paclitaxel and related compounds induce microtubule stability by inhibiting dynamic instability, reducing catastrophes, promoting rescues, and driving nearly every tubulin dimer into polymer form.20–22 Previous studies have shown that cells readily form mitotic spindles in the presence of the microtubule stabilizing agent paclitaxel, despite the significantly reduced microtubule dynamics during both interphase and mitosis.10,23–25 The reorganization of the microtubule array in the presence of paclitaxel demonstrates that cells must have a mechanism other than a shift in dynamic instability to disassemble the G2 microtubule array at the G2/M transition. The spatially distinct pattern of microtubule clearing from the central region of the cytosol observed as paclitaxel-treated cells enter mitosis supports the idea that an alternative mechanism exists to clear the long interphase microtubules in preparation for mitosis.24
Here we used a candidate protein approach to examine the relative contributions of microtubule destabilizing proteins in depolymerization of paclitaxel-stabilized microtubule arrays during the G2 to prophase transition. Candidate proteins within the classes of severing proteins and depolymerizing kinesins were selected based on previously demonstrated ability to depolymerize paclitaxel-stabilized microtubules in vitro, including the kinesin-8 family,26,27 KIF2,28–30 KIF11 (Eg5),31,32 and the severing proteins katanin and spastin.33–36 By depleting these proteins, we find that the kinesin-8 family of depolymerizing kinesins (KIF18A and KIF18B) and the severing protein spastin are required for timely microtubule array remodeling in the presence of paclitaxel, with KIF18B making the most significant contribution in our assays.
Results
Interphase microtubule dissolution occurs with normal timing in paclitaxel-treated cells, but proceeds through a unique clearing pattern
By dampening dynamic instability and stabilizing microtubules, paclitaxel presumably should cause a cell cycle block prior to prophase by inhibiting efficient interphase microtubule array dissolution and, thus, preventing spindle assembly. However, the well-documented presence of abnormal spindles, with short microtubules, in paclitaxel-treated cells indicates that the stabilized interphase microtubule array is successfully cleared, despite stifled dynamics.8,23,24 We first considered that this process was still governed by microtubule dynamics, but just significantly slowed by the dampened microtubule dynamics in paclitaxel. Using long term live cell imaging, we tracked cell populations synchronized at the G1/S border as they progressed through the cell cycle. Cells were treated with 30nM paclitaxel at 4 h post-release from synchronization. The dosage and duration of paclitaxel treatment was selected such that the estimated intracellular concentration of paclitaxel, which accumulates within cells over time, corresponds with clinical intratumoral concentrations (~10–20 μM) coinciding with the peak in mitotic events.8,22 Image series were collected in phase contrast and analyzed to measure the time to mitotic entry, which we defined as the observed loss of nuclear envelope integrity (Figure 1(a)). Treatment with 30nM paclitaxel did not alter the timing of mitotic entry in either human Hela or A549 cells but was sufficient to modestly delay mitotic entry in porcine (Sus scrofa) LLC-PK1 cells by approximately 1 h compared to DMSO vehicle control-treated populations (Figure 1(b)). Timely mitotic entry in the presence of paclitaxel demonstrates that induced microtubule stability still allows for efficient transition from interphase to prophase, supporting previous observations.10,24,25 As an additional measure of cell cycle progress, we measured changes in the basal surface area of cells because cell rounding over approximately 30 min prior to mitosis is tightly correlated with CDK1 activity.37 Consistent with the overall timing, cell rounding was primarily observed over a 30 min time window just before mitosis (Figure 1(c)), consistent with normal activation of CDK1.
Figure 1.
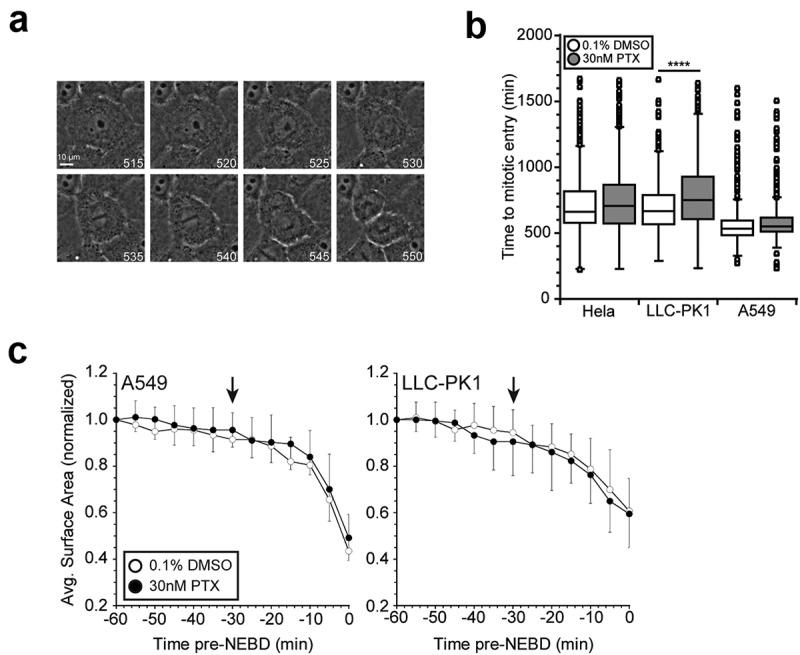
Paclitaxel treatment does not influence the timing of mitotic entry. (a) Representative image series of an LLC-PK1 cell entering mitosis, tracked by long term live cell phase contrast imaging. The time of image acquisition is indicated in minutes post-release from a double thymidine block. Mitotic entry was defined as the first frame where loss of nuclear envelope integrity was evident, as indicated by the white arrowhead. (b) Treatment with 30nM paclitaxel does not affect the timing of mitotic entry in Hela or A549 cells, but results in a modest delay of approximately 1 h in LLC-PK1 cells. Cells were synchronized at the G1/S border by double thymidine block. Drug treatments were added 4 h after the second release and cells were tracked by long term, live cell phase contrast imaging for 24 h. At least 100 cells were tracked for each experimental condition in each of 3 independent experiments. (c) Cell surface area in contact with the substrate showed cell rounding precedes mitotic entry (time 0) by approximately 30 min and is unaffected by paclitaxel treatment. Graphs represent the average of 10 cells for each condition. For clarity, standard deviations are shown in only one direction. PTX, paclitaxel. ****p < .0001.
Attempts to visualize the remodeling of the microtubule array at the interphase-prophase transition in a stable LLC-PK1 cell line expressing GFP-labeled α-tubulin by live cell imaging were largely unsuccessful. Despite high temporal resolution (image acquisition at 3 s intervals), several factors made use of this assay difficult and/or inefficient: (1) mitotic entry occurs over a short time window (~5 min) with a great degree of cell-to-cell asynchrony, even in cell populations synchronized by release from the G1/S border, and we currently lack adequate markers to predict impending mitotic entry in G2 cells; (2) quantification of tubulin in polymer was impeded by the background signal from soluble tubulin, especially as the microtubule array remodeled and polymer was disassembled; (3) interphase array disassembly and bipolar spindle formation do not occur discretely, such that overlap between the two processes obstructs accurate quantification.
To avoid the problems associated with live cell imaging and to allow measurement from a larger number of cells, we used an immunofluorescent assay in cells synchronized at the G1/S border and then fixed at the approximate mitotic peak to visualize and quantify the microtubule array. Cells were fixed following brief lysis to release soluble tubulin and this population-based approach enriched for the rare population of prophase cells, which were selected for analysis in comparison to neighboring G2 cells. Prophase cells were binned into two categories, early and late prophase, based on the extent of chromatin condensation. Early prophase cells were classified by modest chromatin condensation and a smooth contour around genetic material, indicating an intact nuclear envelope. Late prophase was characterized by tighter chromatin condensation and lack of a distinctly smooth outline of the presumptive nuclear envelope. Due to a lack of adequate markers that suited the required fixation method, however, we could not definitively confirm nuclear envelope integrity. Nevertheless, treatment with 30nM paclitaxel qualitatively demonstrated a unique pattern of microtubule array remodeling that differed from that of control populations (Figure 2(a)). In paclitaxel-treated cells, the interphase array clears in the center region of the cytosol, consistent with previous observations.24 The difference in array organization in the presence and absence of paclitaxel was especially apparent in late prophase.
Figure 2.
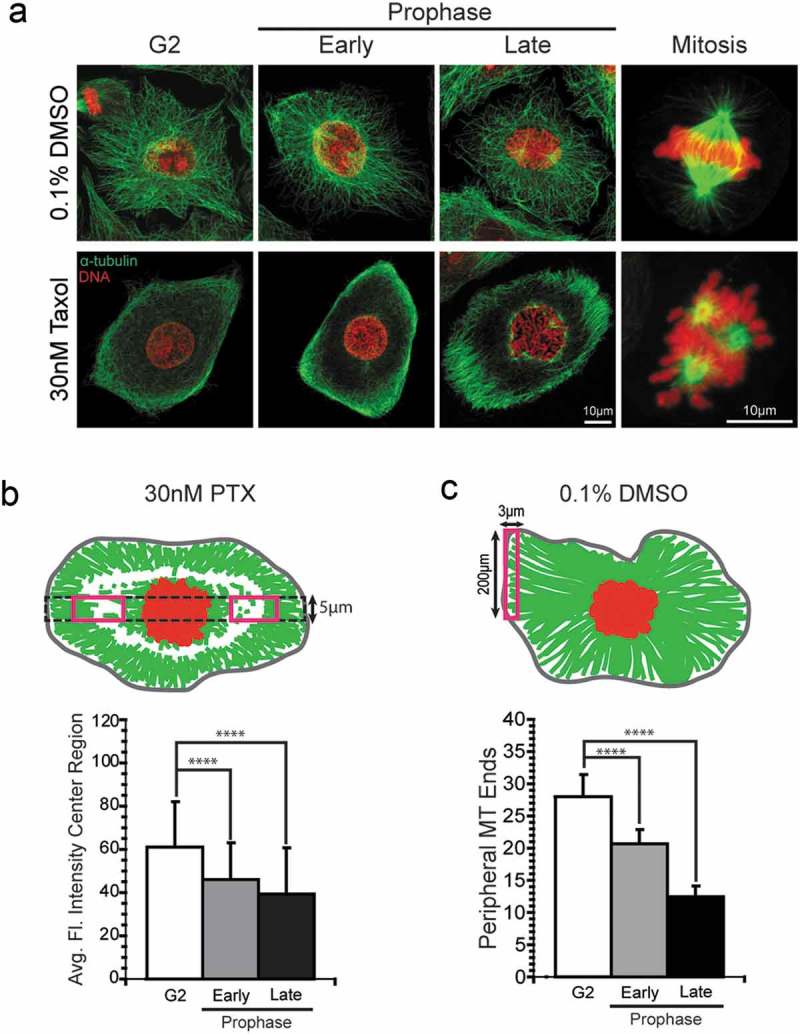
Assays to measure microtubule polymer loss at mitotic entry in A549 cells. (a) Representative images of A549 cells treated with 30nM paclitaxel or 0.1% DMSO (vehicle control) illustrate different patterns of microtubule array disassembly. A549 cells treated with 30nM paclitaxel or 0.1% DMSO were fixed at 8 h post-release from the second S-phase block. Cells at or approaching mitotic entry were characterized as G2, early prophase, or late prophase based on the extent of chromatin condensation. (b) Diagram (top) of area used to quantify microtubule polymer in 30nM paclitaxel-treated cells from the central regions on either side of the nucleus (red rectangles), measured along the long axis of the cell (dotted black rectangle). A plot of microtubule loss from this region in early and late prophase compared to neighboring G2 cells is shown (bottom). (c) Diagram (top) and graph (bottom) of assay used to quantify microtubule polymer loss from the peripheral region of A549 cells treated with DMSO. For B-C, at least 10 cells for each cell cycle stage of each experimental condition were measured in 3 independent experiments for a total of 30 cells per condition and stage. For paclitaxel-treated cells, both of the center cytoplasmic regions were used (red boxes in B, top panel), representing a total of 60 measurements per stage (plotted in B, lower panel). PTX, paclitaxel.****p < .0001.
In order to quantify the change in microtubule polymer in paclitaxel-treated cells, we developed an immunofluorescent assay measuring the average fluorescent intensity in central regions of the cytoplasm on either side of the nucleus (Figure 2(b); see Methods). This method of quantification was designed to specifically address the clearing pattern observed in paclitaxel-treated cells, but also to avoid the cytoplasmic region neighboring the nucleus, where concurrent interphase microtubule disassembly and bipolar spindle formation confounds the measurements. For this assay, we chose to proceed with A549 and LLC-PK1 cells because their flat morphologies were more amenable to visualizing the microtubule array compared to Hela cells. In the presence of paclitaxel, microtubule polymer significantly decreased in the center region of the cytoplasm as either A549 cells (Figure 2(b)) or LLC-PK cells (Supplemental Figure 1) progressed from G2 through late prophase.
Treatment of the two cell lines with the DMSO vehicle control yielded a different pattern of microtubule loss as cells approached mitotic entry (Figure 2(a)). In control-treated A549 cells, the central region did not show a large loss of microtubule polymer in early or late prophase (data not shown), which we attribute to cell rounding (see Figure 1(c)). As an alternative measure of microtubule loss in DMSO-treated cells, we measured the density of microtubule ends at the cell periphery by counting the number of ends located within 3 µm of the cell periphery (see Methods). As shown in Figure 2(c), DMSO control-treated A549 cells showed a 61 ± 13% decrease in microtubule ends in prophase cells compared to neighboring G2 cells. Similar results were obtained in LLC-PK1 cells, with a 50% decrease in microtubule ends in prophase cells compared to G2 cells (Supplemental Figure 1(d)). In general, our observations are consistent with the loss of microtubule polymer as cells transition from G2 to prophase reported previously.15 Overall, the pattern of microtubule array dissolution at mitotic entry clearly differs between controls (DMSO-treated) and paclitaxel-treated cells, consistent with a previous report.24
The central microtubule clearing, combined with the dampened microtubule dynamics in paclitaxel-treated cells,22 indicated that microtubule array remodeling likely does not proceed as a result of plus-end dynamics. We hypothesized that paclitaxel-stabilized microtubules are reorganized at the G2/M transition by microtubule depolymerizing kinesins and/or severing proteins. Candidate proteins were selected based on their previously demonstrated ability to depolymerize paclitaxel-stabilized microtubules in vitro and their contributions determined by examining microtubule clearing from the central region of the cytosol after either protein depletion by siRNA or chemical inhibition of protein activity.
KIF18A and KIF18B depletion obstructs clearing of paclitaxel-stabilized interphase arrays
Depolymerizing kinesins regulate microtubule dynamics by inhibiting the stability of microtubule plus- and/or minus-ends and evidence points to their functions during mitosis.38–40 We reasoned that specific activation of depolymerizing kinesins at mitotic entry could induce disassembly of paclitaxel-stabilized arrays to facilitate interphase array dissolution. Here we consider proteins within the kinesin-8, kinesin-5, and kinesin-13 families (see consensus nomenclature)41 as potential modulators the G2/M transition that may facilitate reorganization of stabilized microtubule arrays.
Kinesin-8 family members were depleted individually and/or in combination by siRNA knockdown in A549 and LLC-PK cell lines. The kinesin-8 family is expressed in a cell cycle-dependent manner and therefore siRNA transfections were performed during the second block of the double thymidine block/synchronization method to ensure that siRNA was present before kinesin-8 expression and so that we assessed the first mitotic entry following protein depletion to eliminate possible downstream consequences of a previous aberrant mitosis. KIF18B depletion was confirmed at 24 h after transfection by immunoblotting (Figure 3(a)), however, attempts to confirm KIF18A depletion by this method were inconclusive. We did not explore immunoblot issues further, but the inconclusive results could be due to cross-reactivity with Kif18B, or because control-transfected cells degrade Kif18A as they proceed through mitosis (reducing the Kif18A level detected in the cell population), while depleted cells are blocked in prometaphase and maintain any remaining Kif18A.42,43 To examine knockdown directly, we confirmed KIF18A depletion by immunofluorescence staining; mitotic cells depleted of KIF18A exhibited a loss of KIF18A staining adjacent to kinetochores (Figure 3(b)), consistent with a previous report.42 For DMSO-treated control A549 cells, depletion of KIF18A and/or KIF18B did not change the density of microtubule ends at the cell periphery in the cell cycle stages examined (G2, early, or late prophase cells; Figure 3(c,d)). These data indicate that KIF18A and/or KIF18B are not required for clearance of the G2 microtubule array in the absence of paclitaxel, conditions where changes in microtubule plus end dynamics should be sufficient to depolymerize the array. In contrast, depletion of KIF18A and KIF18B, either singly or in combination, reduced the loss of microtubule polymer in paclitaxel-treated cells at mitotic entry compared to cells transfected with a control siRNA (Figure 3(c,d)). There was no significant difference between the amount of polymer in G2 and early prophase in cells depleted of KIF18A, although microtubule levels were still reduced by late prophase. Depletion of KIF18B alone, or dual depletion of both KIF18A and KIF18B, eliminated the loss of microtubule polymer in paclitaxel-treated cells at early or late prophase (Figure 3(d)). In contrast, cells transfected with the control siRNA showed the same pattern of microtubule loss in early or late prophase as those treated with paclitaxel alone (compare Figures 2(b) and 3(e)).
Figure 3.

Depletion of KIF18A and KIF18B in A549 cells reduced disassembly of paclitaxel-stabilized interphase microtubules at mitotic entry. (a) KIF18B depletion by siRNA was confirmed by immunoblot. Gel samples were made following double thymidine synchronization approximately 24 h post-siRNA transfection. α-tubulin and GAPDH serve as loading controls. (b) Representative images of mitotic cells confirming KIF18A depletion approximately 24 h post-siRNA transfection. KIF18A-depleted cells lose staining (green) of KIF18A adjacent to kinetochores, as marked by CREST staining (red), during mitosis,42 shown at higher magnification in the boxed insets. DNA is shown in blue. (c) Representative images of cells in early or late prophase incubated in 30nM paclitaxel and depleted of KIF18A and/or KIF18B as indicated. Microtubules (green) and chromatin (red) are labeled. A549 cells were synchronized by double thymidine block and treated with 0.1% DMSO control (not shown) or 30nM paclitaxel approximately 4 h post-release. Cells were fixed approximately 8 h post-release and 24 h post-siRNA transfection. (d) For DMSO-treated cells, KIF18A and/or KIF18B depletion did not affect microtubule clearing from G2 through prophase. Shown are the number of microtubule ends at the cell periphery as described in Methods. (e) KIF18A and/or KIF18B depletion impedes interphase microtubule array clearing in the presence of paclitaxel. Microtubule polymer was quantified in the central region as described in Methods. In each of 3 independent experiments, at least 10 cells for each cell cycle stage were measured, including 2 measurements per cell in paclitaxel-treated cells (see diagram and legend in Figure 2). NT, Non-targeting; PTX, paclitaxel. *p < .05; **p < .01;***p < .001;****p < .0001.
Similar results were obtained in LLC-PK1 cells where the combination of KIF18A and KIF18B depletions abrogated the reduced microtubule polymer levels measured in the center region of the cytoplasm of paclitaxel-treated cells (Supplemental Figure 1). Depletion of KIF18A or KIF18B individually, or inhibition of KIF18A with a small molecule inhibitor (BTB1)44 did not change the pattern of microtubule loss, and therefore both KIF18A and KIF18B were required for microtubule loss at prophase in this cell line (Supplemental Figure 1). Consistent with A549 cells, KIF18A and KIF18B were not required for interphase microtubule loss in DMSO-treated control LLC-PK cells (Supplemental Figure 1).
Several other kinesin families able to depolymerize paclitaxel-stabilized microtubules were also considered as candidates for clearing stable microtubules during prophase. KIF11 (also known as kinesin-5 or Eg5)41 is a motor protein with an established role during mitosis as a microtubule depolymerase,31,32 However, inhibition of KIF11 by monastrol treatment in LLC-PK1 cells did not alter the timing of mitotic entry or microtubule array remodeling at the G2-prophase transition in the presence or absence of paclitaxel (data not shown).45,46 Other candidates included the kinesin-13 family of depolymerizing kinesins, consisting of KIF2A, KIF2B, and KIF2C/MCAK. Kinesin-13s are non-processive, reaching microtubule ends by diffusion where they function as microtubule depolymerases that use ATPase activity to directly depolymerize microtubules from both plus- and minus-ends.28–30 KIF2A and KIF2C/MCAK have established roles during mitosis, with evidence to suggest that their activity and localization are regulated by mitotic kinases.40,47 Thus, we assessed how depletion of these proteins affected G2 microtubule array disassembly in A549 cells treated with paclitaxel. While depletion of KIF2A and KIF2B was confirmed by immunoblotting at 48 h post-transfection (Figure 4(a)), our attempts to deplete KIF2C/MCAK were unsuccessful. Still, immunoblot analysis demonstrated that KIF2A and KIF2B depletion were enhanced by co-transfection with siRNA targeting KIF2C/MCAK. Therefore, we proceeded with triple transfection of KIF2A, KIF2B, and KIF2C/MCAK siRNAs and evaluated the effect on microtubule array remodeling at the G2/M transition. Following siRNA transfection in A549 cells, both in the presence and absence of paclitaxel, interphase array dissolution was not significantly different from control cells transfected with non-targeting siRNA (Figure 4(b,c)). These results illustrate that microtubule array remodeling in paclitaxel-treated cells proceeds despite the depletion of KIF2A and KIF2B, indicating that these depolymerizing kinesins do not contribute to the dissolution of stabilized microtubules at mitotic entry. It is possible, however, that KIF2C/MCAK may play a role in this process which remains uncharacterized due to unsuccessful attempts to deplete KIF2C in this study.
Figure 4.

Depletion of kinesin-13s in A549 cells did not affect interphase array clearing in the presence of paclitaxel. (a) Depletion of kinesin-13’s, KIF2A and KIF2B, by siRNA knockdown was confirmed at 48 h post-transfection by immunoblotting. Gel samples were made concomitant with double thymidine synchronization at 24 and 48 h post-siRNA transfection. Actin served as a loading control. Attempts to detect KIF2C/MCAK by immunoblot were unsuccessful; however, KIF2A and KIF2B depletion appeared to be enhanced following triple transfection of KIF18A, KIF18B, and KIF2C/MCAK siRNA. (b) For DMSO-treated control A549 cells, microtubules were lost at the cell periphery independent of depletion of KIF2A and KIF2B. (c) In paclitaxel-treated cells, microtubule density in the central region of the cytoplasm also showed a similar reduction in early and late prophase for either control siRNA transfected cells or cells transfected with siRNA targeting the kinesin 13’s. For each of 3 independent experiments, at least 10 cells for each cell cycle stage of each experimental condition were measured. NT, Non-targeting; PTX, paclitaxel. **p < .01; ***p < .001; ****p < .0001.
Spastin depletion reduces interphase array disassembly in paclitaxel-treated cells
The microtubule severing proteins constitute a class of molecular enzymes which cut microtubules into short fragments by an ATP-dependent mechanism, operating along microtubule lengths, and are able to sever microtubules stabilized by paclitaxel.33,34,48 Severing promotes microtubule disassembly and has been implicated in a variety of cellular functions, including well characterized roles in mitosis.48 During mitosis, the severing enzymes contribute to poleward microtubule flux that play a part in chromosome movement during metaphase and anaphase, presumably by removing stabilizing caps at microtubule plus- and minus-ends to provide a substrate suitable for depolymerizing kinesins.33,48–50 Here we tested whether severing by katanin or spastin was required to depolymerize paclitaxel-stabilized interphase microtubule arrays at mitotic entry. We did not examine fidgetin because we were unable to detect this protein in the cell lines studied.
We evaluated the contribution of katanin and spastin, singly and in combination, by siRNA depletion. Immunoblot analysis revealed that at least 48 h was required post-transfection to achieve detectable protein depletion; even still, spastin protein was depleted only ~50% after 48 h (Figure 5(a)). Depletion of either protein alone, or both proteins in combination, did not alter the loss of microtubules in DMSO-treated control cells (Figure 5(b)), indicating that these proteins are not normally required at mitotic entry. In contrast, depletion of spastin, but not katanin, in A549 cells hindered microtubule array disassembly in the presence of paclitaxel (Figure 5(c)). In spastin-depleted cells, we observed reduced microtubule polymer loss in early and late prophase cells in the presence of paclitaxel, with no statistical difference in the relative amount of microtubule polymer between G2 and early prophase cells. In late G2 cells, microtubule polymer was reduced compared to G2 levels, but the reduction was less than that observed in cells transfected with non-targeting siRNA. Thus, spastin depletion by only ~ 50%, was sufficient to cause a reduction in the extent of G2 microtubule array clearing at mitotic entry in paclitaxel-treated A549 cells. Katanin-depleted cells showed no defect in G2 microtubule array clearing in paclitaxel, showing a pattern of microtubule polymer loss through early and late prophase that matched well with the pattern in control siRNA-transfected cells.
Figure 5.
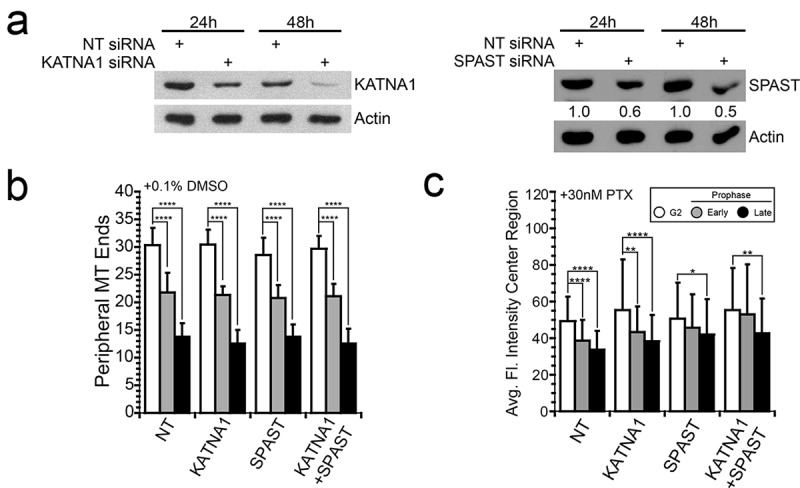
Depletion of spastin, but not katanin, hinders microtubule array disassembly in paclitaxel-treated A549 cells. (a) Depletion of katanin and spastin by siRNA knockdown was confirmed at 48 h post-transfection by immunoblotting. However, even after 48 h, spastin protein was only reduced by ~ 50%. A549 cells were transfected with siRNA targeting katanin, spastin, or non-targeting control; gel samples were made 24 or 48 h post-siRNA transfection. Actin served as a loading control. (b) In DMSO-treated control cells, the number of microtubule ends at the cell periphery was reduced in early (grey bars) and late prophase (black bars) compared to G2 (white bars); depletion of katanin and/or spastin did not alter the amount of microtubules lost under these conditions. (c) For cells treated with paclitaxel, interphase microtubule array disassembly was reduced by spastin depletion, but not by katanin depletion. This reduction was detectable in early prophase cells, where the amount of microtubule polymer was not significantly different from the G2 level. A549 cells were synchronized by double thymidine block and treated with 0.1% DMSO control or 30nM paclitaxel approximately 4 h after the second release. Cells were fixed approximately 8 h post-release and 48 h post-siRNA transfection. In each of 3 independent experiments, at least 10 cells for each cell cycle stage of each experimental condition were measured. NT, Non-targeting; KATNA1, katanin; SPAST, spastin; PTX, paclitaxel. *p < .05; **p < .01; ****p < .0001.
Parallel experiments depleting spastin from LLC-PK1 cells yielded a similar trend, but less robust abrogation of microtubule polymer loss in early or late prophase, and this abrogation was detectable in either DMSO-treated controls or paclitaxel-treated cells (Supplemental Figure 2). Collectively, these results indicate that spastin, but not katanin, plays a role in remodeling paclitaxel-stabilized microtubule arrays at mitotic entry.
Discussion
Despite its successful therapeutic use, paclitaxel’s mechanism of action has a paradox where its microtubule-stabilizing activity is briefly antagonized to allow microtubule array reorganization as cells approach mitotic entry.23,24,51 Paclitaxel-stabilized microtubule arrays reorganize in a spatially distinct pattern at mitotic entry (Figure 2; also Hornick et al.24). Here we identified roles for microtubule depolymerizing proteins, KIF18A, KIF18B and spastin, a microtubule severing protein, in remodeling of paclitaxel-stabilized microtubules arrays, specifically as cells transition from G2 to prophase. Depletion of KIF18A and KIF18B or spastin in either paclitaxel-treated A549 or LLC-PK1 cells abrogated interphase microtubule array clearing during prophase, with the effects of KIF18A and B depletion, or KIF18B depletion alone, showing the most robust phenotype. In all cases, aberrant spindles of short microtubules still formed indicating that other proteins likely contribute to clearing paclitaxel-stabilized interphase microtubule arrays. Because we were only able to deplete spastin by ~50%, we were unable to determine whether these three proteins are the sole proteins needed to clear the paclitaxel-stabilized microtubule array as cells transition from G2 to prophase. Additional proteins, possibly including residual spastin, or others able to depolymerize paclitaxel-stabilized microtubules, may contribute to G2 microtubule array depolymerization in paclitaxel. We assume that KIF18A, B, and spastin normally contribute to interphase array dissolution in the absence of paclitaxel as well, but that the change in microtubule dynamics at mitotic entry is the major driver of microtubule loss, thus masking the roles of KIF18A, B, and spastin unless dynamic turnover is blocked by paclitaxel. Alternatively, the change in microtubule structure caused by paclitaxel may render cellular microtubules susceptible to KIF18’s and/or spastin and these proteins may have a unique function in paclitaxel-treated cells to drive microtubule reorganization at mitotic entry.20,52
A remaining unanswered question is why KIF18A, KIF18B and/or spastin antagonize paclitaxel specifically in a brief window during the G2/prophase transition. Expression of both KIF18A and KIF18B is cell cycle-dependent, with expression rising in G2, and protein destruction after mitotic exit.53,54 KIF18A and KIF18B are imported into the nucleus after synthesis, where they reside until nuclear envelope breakdown.53,55 It is possible that the cell cycle-dependent expression and localization to the nucleus confines KIF18A- and KIF18B-dependent microtubule depolymerization to a finite time window beginning at nuclear envelope breakdown.38,53,55,56 In this regard, it is important to note that the nuclear envelope does not instantly break apart, but instead first becomes leaky as its structure is breaking down.57 In this model, KIF18A and KIF18B have access to the interphase microtubule array only during prophase and later stages of mitosis,38,53,55,56 where these proteins can antagonize paclitaxel to facilitate interphase microtubule depolymerization.
Possible mechanisms underlying spastin’s ability to antagonize paclitaxel specifically at mitotic entry are less clear, but several models are possible. First, the human spastin protein sequence is predicted to contain eight CDK1 phosphorylation sites, which could stimulate spastin’s severing activity as active CDK1 levels rise, but it is not yet known whether CDK1 phosphorylates spastin in cells, or whether phosphorylation stimulates spastin severing activity. Currently the only known modification that stimulates spastin’s microtubule severing activity is the polyglutamylation of tubulins within microtubules, a modification to the substrate rather than to the enzyme itself. Recent work has demonstrated that polyglutamylation induces microtubule severing by recruiting and honing katanin and spastin severing protein activity.58–60 Additionally, paclitaxel treatment increases tubulin polyglutamylation within microtubules and we hypothesized that the increased polyglutamylation could sensitize microtubules to severing by spastin.61 In a preliminary experiment, we measured the levels of tubulin polyglutamylation over time in synchronized A549 cells after release from a G1/S block. In the presence or absence of paclitaxel, polyglutamylation levels were high 6 hours after release from a G1/S block, 2 hours before the peak in mitotic cells. These preliminary results do not support a cell-cycle dependent rise in polyglutamylated tubulin in paclitaxel-treated cells that coincides with mitotic entry. It remains an open question how spastin-dependent severing of paclitaxel-stabilized microtubules is confined to a brief window of time at the G2/prophase transition.
In summary, the experiments presented here identified roles for microtubule destabilizing proteins, KIF18A, KIF18B, and spastin, in clearing the interphase microtubule array and allowing microtubule reorganization at mitotic entry. There has been a growing trend toward identifying microtubule associated proteins as targets for cancer therapy due to accumulating evidence that these candidates can affect cancer cell sensitivity to microtubule targeting agents by altering microtubule dynamics.45,46,62 Whether depletion or inhibition of the proteins addressed in this study could synergize with paclitaxel to control cell proliferation has not yet been addressed, but will be interesting to pursue in the future.
Materials and methods
Cell culture and drug treatments
Porcine (Sus scrofa; LLC-PK1) and human (A549) cells were obtained from the ATCC (Manassas, VA) and were grown in DMEM (Sigma; D5796) or Ham’s F-12K (Life Technologies; 21127022) medium, supplemented with 10% fetal bovine serum (FBS; Life Technologies; A3160401) and 1% antibiotic/antimycotic (Sigma; A5955). For preliminary imaging experiments, an LLC-PK1 cell line stably expressing GFP-tagged α-tubulin was used.17 Cells were synchronized through a double thymidine block by overnight incubation in 5mM thymidine (Sigma; T1895), an 8-h release in fresh medium after 5 washes in warm phosphate-buffered saline (PBS), followed by a 16-h incubation in 5mM thymidine. Cells were transferred to fresh medium following 5 washes with warmed PBS. Drug treatments, including 0.1% DMSO or 30nM paclitaxel (CalBiochem; 580555) were applied approximately 4 h following the second thymidine release. In the case of KIF18A or KIF11 (Eg5) inhibition, cells were treated with small molecule inhibitors, 50μM BTB1 (see ref. 44; R&D Systems; 5539) or 100μM monastrol (see refs. 45,46; R&D Systems; 1305), respectively, after synchronization.
siRNA and transient transfection
For RNA interference in synchronized cells, transfections were performed in serum free medium 1–2 days after plating using GeneSilencer (Genlantis; T500750) according to the manufacturer’s protocol, as described previously.63 The siRNA oligonucleotides were purchased from Dharmacon and include: Sus scrofa KIF18A 5ʹ-CAAGAUAAGUCAAGUGAUAUU-3ʹ; Sus scrofa KIF18B 5ʹ-CGAGGGAGCUGGAGAGUCAUU-3ʹ; Sus scrofa spastin 5ʹ-UGAUAUAGCUGGUCAAGAAUU-3ʹ; SMARTpool targeting human KIF18A (L-006894–00-0005); SMARTpool targeting human KIF18B (L-010460–01-0005); SMARTpool targeting human KIF2A (L-004959–00-0005); SMARTpool targeting human KIF2B (L-008345–00-0005); SMARTpool targeting human KIF2C (L-004955–00-0005); SMARTpool targeting human katanin (L-005157–02-0005); SMARTpool targeting human spastin (L-014070–00-0005). siGenome non-targeting siRNA (Dharmacon; D-001210–01-05) was used as a control siRNA sequence. In cases where protein depletion occurred within 24 h post-siRNA transfection (KIF18A and KIF18B knockdown in A549 and LLC-PK1 cells), siRNA was transfected during the first release of the double thymidine block protocol. For KIF2A, KIF2B, KIF2C/MCAK, katanin, and spastin, siRNA transfections occurred prior to the first thymidine block during the synchronization protocol in order to achieve protein depletion at 48 h.
Knockdowns were confirmed by immunoblotting (below), or observations of mitotic defects after fixation and immunofluorescence (below) to detect defects in chromosome alignment, spindle structure, or loss of KIF18A adjacent to kinetochores.
Immunoblotting
Soluble cell extracts were prepared as described previously and protein concentrations were measured by Bradford assay.63 Lysates were diluted in PAGE sample buffer; 10–20 µg total protein per lane was typically loaded and resolved in 10% polyacrylamide gels and transferred to PVDF membranes (Bio-Rad; 1704156) using the TurboTransfer apparatus (Bio-Rad, Hercules CA). Membranes were blocked with 5% nonfat milk in Tris-buffered saline with 0.1% Tween and then probed with primary antibodies: anti-α-tubulin B512 (1:10,000; Sigma; T5168), vinculin (1:1000; Cell Signaling; 13901), KIF2A (1:10,000; a generous gift from Dr. Duane Compton), KIF2B (1:2000; Novus Biologicals; NBP1-86002), actin (1:10,000; Sigma; A5060), KIF18A (1:2000; Bethyl Laboratories; A301-080A-T), KIF18B (1:5000; Bethyl Laboratories; A303-982A), GAPDH (1:1000; Abcam; ab9483), katanin (1:1000; Abcam; ab111881), or spastin (1:1000; Sigma; S7074); followed by horseradish peroxidase-linked secondary antibodies, anti-mouse IgG (1:10,000; Sigma; A2554) or anti-rabbit IgG (1:10,000; Bio-Rad; 170–6515). Immuno-reactive bands were developed using enhanced chemiluminescence (GE Amersham; RPN2232) and detected on X-ray film (Denville Scientific; E3012).
Indirect immunofluorescence and confocal microscopy
Cells were grown on glass coverslips and treated as described above. Prior to fixation, cells were rinsed with warmed PBS and briefly lysed in PEM (80 mM PIPES, 1 mM EGTA, 1 mM MgSO4) supplemented with 0.5% Triton X-100 for 15 s to remove soluble tubulin but preserve microtubules.64 Cells were then fixed with a mixture of 2% paraformaldehyde and 0.1% glutaraldehyde in PEM at 37°C for 10 min and then further permeabilized in −20°C methanol for 5 min. Alternatively, for scoring of mitotic phenotypes to confirm protein knockdowns, cells were fixed with −20°C methanol supplemented with 1mM EDTA for 10 min, and then rehydrated in PBS. Fixed cells were incubated with blocking reagent (10% FBS in PBS) for 30 min at 37°C, followed by a 45-min incubation with primary antibody at 37°C. Cells were then washed with PBS and incubated with secondary antibody and 1.5 µM propidium iodide (Sigma; P4864) or 2g/ml Hoechst 33342 (Life Technologies; H3570) for 45 min at 37°C. Primary antibodies used included anti-α-tubulin B512 (1:1000; Sigma; T5168), anti-KIF18A (1:100; Bethyl Laboratories; A301-080A-T), and anti-CREST (1:100; a generous gift from Dr. Bill Brinkley). Secondary antibodies used included goat anti-mouse IgG Alexa 488 (1:50; Life Technologies; A11029), goat anti-rabbit IgG Alexa 488 (1:50; Life Technologies; A11034), goat anti-rabbit IgG Alexa 568 (1:50; Life Technologies; A11036), or goat anti-human IgG Alexa 568 (1:50; Life Technologies; A21090). Coverslips were then washed with PBS and mounted on slides with Vectashield (Vector Labs; H-1000). Cells were imaged using a 63x/1.4 numerical aperture Plan-Apo objective on Zeiss LSM 880 confocal microscope (Zeiss USA, Thornwood, NY).
For quantitative measurement of microtubule polymer by immunofluorescence, maximum intensity projections were made from Z-stacks and then analyzed by 5 μm wide line scans that spanned the diameter of the cell using Zen Black software (Zen 2.1 SP2). The resulting line scan profiles in LLC-PK1 cells spanning from the nuclear boundary to the cell margin were divided into the following subcellular regions: interior (25%), center (50%), and periphery (25%). Due to greater cell rounding, line scan profiles in A549 cells were divided from nuclear boundary to cell margin into 3 subcellular regions: interior (37.5%), center (25%), and periphery (37.5%). Based on the pattern of microtubule clearing qualitatively observed in paclitaxel-treated cells, the average fluorescence intensity measured from the center region, as described above, was used for quantitative analyses. Microtubule loss in DMSO-treated control cells was quantified by the density of polymer at the cell margin. Microtubule ends within 3 µm of the cell periphery were counted within a 200 µm long line at the cell margin. For all measurements, at least 10 cells for each cell cycle stage and each experimental condition were measured in each of 3 independent experiments for a total of 30 cells for every stage and condition quantified. We assumed that all siRNA-transfected cells were depleted of their respective protein(s) and did not confirm knockdown for each individual cell.
Live-cell imaging
For long term cell fate tracking, cells were plated on glass bottom dishes (CellVis; D35201.5N) and imaged using a Biostation IM (Nikon Instruments, Melville NY), as described previously.65 Cells were imaged with phase-contrast optics using a 20x objective, and images were collected at 5-min intervals for at least 24 h. Cell fates were tracked from image series. Mitotic entry was marked either by the first image showing loss of the nuclear envelope or by significant cell rounding. The time from G1/S to mitosis was not changed by any of the siRNA depletions examined here. For assessment of cell rounding, the basal surface area of individual cells was measured from the collected image series using Fiji software (NIH, version 2.0.0-rc-69/1.52i) by tracing the cell periphery in the 12 frames prior to the visible loss of nuclear envelope integrity and were normalized to the initial basal surface area measured at 60 min prior to NEBD.37,66
Data analysis
Unless stated otherwise, all experiments were repeated in three independent experiments for each cell line and data from the three experiments pooled for analyses. Statistical analyses were performed using unpaired t-tests using either GraphPad (https://www.graphpad.com/quickcalcs/ttest1.cfm) or KaleidaGraph software (version 4.5; Synergy Software, Reading PA) and significance is noted on all graphs.
Funding Statement
This work was supported by the National Institutes of Health [GM117560].
Acknowledgments
This work was supported by the NIH under grant GM117560 to LC. JCL was partially supported by the Marjorie M. Nemes Fellowship awarded by the Department of Biological Sciences at Lehigh University.
Disclosure Statement
The authors report no conflict of interest.
Supplementary data
Supplemental data for this article can be accessed on the publisher’s website
References
- 1.Hanahan D, Weinberg RA.. 2011. Hallmarks of cancer: the next generation. Cell. 144:646–674. DOI: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 2.Steinmetz MO, Prota AE. 2018. Microtubule-targeting agents: strategies to hijack the cytoskeleton. Trends Cell Biol. 28:776–792. DOI: 10.1016/j.tcb.2018.05.001. [DOI] [PubMed] [Google Scholar]
- 3.Mukhtar E, Adhami M, Mukhtar H. 2014. Targeting microtubules by natural agents for cancer therapy. Mol Cancer Ther. 13:275–284. DOI: 10.1158/1535-7163.MCT-13-0791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Fanale D, Bronte G, Passiglia F, Calò V, Castiglia M, Di Piazza F, Barraco N, Cangemi A, Catarella MT, Insalaco L and Listì A.. Stabilizing versus destabilizing the microtubules: A double-edge sword for an effective cancer treatment option? Anal Cell Pathol. 2015;2015: 690916.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Yang CPH, Horwitz SB. Taxol®: the first microtubule stabilizing agent. Int J Mol Sci. 2017;18:1733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rohena CC, Peng J, Johnson TA, Crews P, Mooberry SL. Chemically diverse microtubule stabilizing agents initiate distinct mitotic defects and dysregulated expression of key mitotic kinases. Biochem Pharmacol. 2013;85:1104–1114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Weaver BA. How Taxol/paclitaxel kills cancer cells. Mol Biol Cell. 2014;25:2677–2681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zasadil L. M., Andersen KA, Yeum D, Rocque GB, Wilke LG, Tevaarwerk AJ, Raines RT, Burkard ME and Weaver BA. Cytotoxicity of paclitaxel in breast cancer is due to chromosome missegregation on multipolar spindles. Sci Transl Med. 2014;6:229ra43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Barbuti AM, Chen ZS. Paclitaxel through the ages of anticancer therapy: exploring its role in chemoresistance and radiation therapy. Cancers (Basel). 2015;7:2360–2371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gascoigne KE, Taylor SS. Cancer cells display profound intra- and interline variation following prolonged exposure to antimitotic drugs. Cancer Cell. 2008;14:111–122. DOI: 10.1016/j.ccr.2008.07.002. [DOI] [PubMed] [Google Scholar]
- 11.Kavallaris M. Microtubules and resistance to tubulin-binding agents. Nat Rev Cancer. 2010;10:194–204. DOI: 10.1038/nrc2803. [DOI] [PubMed] [Google Scholar]
- 12.Bartolini F, Gundersen GG. Generation of noncentrosomal microtubule arrays. J Cell Sci. 2006;119:4155–4163. DOI: 10.1242/jcs.02791. [DOI] [PubMed] [Google Scholar]
- 13.Salaycik KJ, Fagerstrom CJ, Murthy K, Tulu US, Wadsworth P. Quantification of microtubule nucleation, growth and dynamics in wound-edge cells. J Cell Sci. 2005;118:4113–4122. DOI: 10.1242/jcs.02391. [DOI] [PubMed] [Google Scholar]
- 14.Prosser SL, Pelletier L. Mitotic spindle assembly in animal cells: a fine balancing act. Nat Rev Mol Cell Biol. 2017;18:187–201. DOI: 10.1038/nrm.2016.162. [DOI] [PubMed] [Google Scholar]
- 15.Zhai Y, Kronebusch PJ, Simon PM, Borisy GG. Microtubule dynamics at the G2/M transition: abrupt breakdown of cytoplasmic microtubules at nuclear envelope breakdown and implications for spindle morphogenesis. J Cell Biol. 1996;135:201–214. DOI: 10.1083/jcb.135.1.201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rusan NM, Tulu US, Fagerstrom C, Wadsworth P. Reorganization of the microtubule array in prophase/prometaphase requires cytoplasmic dynein-dependent microtubule transport. J Cell Biol. 2002;158:997–1003. DOI: 10.1083/jcb.200206094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rusan NM, Fagerstrom CJ, Yvon A-MC, Wadsworth P. Cell cycle-dependent changes in microtubule dynamics in living cells expressing green fluorescent protein- Tubulin. Mol Biol Cell. 2001;12:971–980. DOI: 10.1091/mbc.12.4.971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Belmont LD, Hyman AA, Sawin KE, Mitchison TJ. Real-time visualization of cell cycle-dependent changes in microtubule dynamics in cytoplasmic extracts. Cell. 1990;62:579–589. DOI: 10.1016/0092-8674(90)90022-7. [DOI] [PubMed] [Google Scholar]
- 19.Cassimeris L, Leung JC, Odde DJ. Monte Carlo simulations of microtubule arrays: the critical roles of rescue transitions, the cell boundary, and tubulin concentration in shaping microtubule distributions. PLoS One. 2018;13:1–20. DOI: 10.1371/journal.pone.0197538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Castle BT, McCubbin S, Prahl LS, Bernens JN, Sept D, Odde DJ, Marshall W. Mechanisms of kinetic stabilization by the drugs paclitaxel and vinblastine. Mol Biol Cell. 2017;28:1238–1257. DOI: 10.1091/mbc.e16-08-0567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Schiff PB, Horwitz SB. Taxol stabilizes microtubules in mouse fibroblast cells. Proc Natl Acad Sci. 1980;77:1561–1565. DOI: 10.1073/pnas.77.3.1561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yvon A-M-M, Wadsworth P, Jordan MA. Taxol suppresses dynamics of individual microtubules in living human tumor cells. Mol Biol Cell. 1999;10:947–959. DOI: 10.1091/mbc.10.4.947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Amin-Hanjani S, Wadsworth P. Inhibition of Spindle Elongation by Taxol. Cell Motil Cytoskeleton. 1991;20:136–144. DOI: 10.1002/(ISSN)1097-0169. [DOI] [PubMed] [Google Scholar]
- 24.Hornick JE, Bader JR, Tribble EK, Trimble K, Breunig JS, Halpin ES, Vaughan KT, Hinchcliffe EH. Live-cell analysis of mitotic spindle formation in taxol-treated cells. Cell Motil Cytoskeleton. 2008;65:595–613. DOI: 10.1002/cm.v65:8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Shi J, Orth JD, Mitchison T. Cell type variation in responses to antimitotic drugs that target microtubules and kinesin-5. Cancer Res. 2008;68:3269–3276. DOI: 10.1158/0008-5472.CAN-07-6699. [DOI] [PubMed] [Google Scholar]
- 26.Gupta ML, Carvalho P, Roof DM, Pellman D. Plus end-specific depolymerase activity of Kip3, a kinesin-8 protein, explains its role in positioning the yeast mitotic spindle. Nat Cell Biol. 2006;8:913–923. DOI: 10.1038/ncb1457. [DOI] [PubMed] [Google Scholar]
- 27.Varga V, Helenius J, Tanaka K, Hyman AA, Tanaka TU, Howard J. Yeast kinesin-8 depolymerizes microtubules in a length-dependent manner. Nat Cell Biol. 2006;8:957–962. DOI: 10.1038/ncb1462. [DOI] [PubMed] [Google Scholar]
- 28.Akhmanova A, Steinmetz MO. Control of microtubule organization and dynamics: two ends in the limelight. Nat Rev Mol Cell Biol. 2015;16:711–726. DOI: 10.1038/nrm4084. [DOI] [PubMed] [Google Scholar]
- 29.Gardner MK, Zanic M, Howard J. Microtubule catastrophe and rescue. Curr Opin Cell Biol. 2013;25:1–9. DOI: 10.1016/j.ceb.2012.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wordeman L. Microtubule-depolymerizing kinesins. Curr Opin Cell Biol. 2005;17:82–88. DOI: 10.1016/j.ceb.2004.12.003. [DOI] [PubMed] [Google Scholar]
- 31.Myers KA, Baas PW. Kinesin-5 regulates the growth of the axon by acting as a brake on its microtubule array. J Cell Biol. 2007;178:1081–1091. DOI: 10.1083/jcb.200702074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gardner MK, Bouck DC, Paliulis LV, Meehl, JB, O'Toole ET, Haase J, Soubry A, Joglekar AP, Winey M, Salmon ED, et al. Chromosome congression by Kinesin-5 motor-mediated disassembly of longer kinetochore microtubules. Cell. 2008;135:894–906. DOI: 10.1016/j.cell.2008.09.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.McNally KP, Buster D, McNally FJ. Katanin-mediated microtubule severing can be regulated by multiple mechanisms. Cell Motil Cytoskeleton. 2002;53:337–349. DOI: 10.1002/(ISSN)1097-0169. [DOI] [PubMed] [Google Scholar]
- 34.Sudo H, Baas PW. Acetylation of microtubules influences their sensitivity to severing by katanin in neurons and fibroblasts. J Neurosci. 2010;30:7215–7226. DOI: 10.1523/JNEUROSCI.0048-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Roll-Mecak A, Vale RD. Structural basis of microtubule severing by the hereditary spastic paraplegia protein spastin. Nature. 2008;451:363–367. DOI: 10.1038/nature06482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Salinas S, Carazo-Salas RE, Proukakis C, Cooper JM, Weston AE, Schiavo G, Warner TT. Human spastin has multiple microtubule-related functions. J Neurochem. 2005;95:1411–1420. DOI: 10.1111/j.1471-4159.2005.03472.x. [DOI] [PubMed] [Google Scholar]
- 37.Gavet O, Pines J. Progressive activation of CyclinB1-Cdk1 coordinates entry to mitosis. Dev Cell. 2010;18:533–543. DOI: 10.1016/j.devcel.2010.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Gardner MK, Odde DJ, Bloom K. Kinesin-8 molecular motors: putting the brakes on chromosome oscillations. Trends Cell Biol. 2008;18:307–310. DOI: 10.1016/j.tcb.2008.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Manning AL, Ganem NJ, Bakhoum SF, Wagenbach M, Wordeman L, Compton DA, Salmon T. The Kinesin-13 Proteins Kif2a, Kif2b, and Kif2c/MCAK have distinct roles during mitosis in human cells. Mol Biol Cell. 2007;18:2970–2979. DOI: 10.1091/mbc.e07-02-0110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Walczak CE, Gayek S, Ohi R. Microtubule-Depolymerizing Kinesins. Annu Rev Cell Dev Biol. 2013;29:417–441. DOI: 10.1146/annurev-cellbio-101512-122345. [DOI] [PubMed] [Google Scholar]
- 41.Lawrence C. J., et al. A standardized kinesin nomenclature. J Cell Biol. 2004;167:19–22. DOI: 10.1083/jcb.200408113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Stumpff J, von Dassow G, Wagenbach M, Asbury C, Wordeman L. The Kinesin-8 Motor Kif18A suppresses kinetochore movements to control mitotic chromosome alignment. Dev Cell. 2008;14:252–262. DOI: 10.1016/j.devcel.2007.11.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Stumpff J, Wagenbach M, Franck A, Asbury CL, Wordeman L. Kif18A and chromokinesins confine centromere movements via microtubule growth suppression and spatial control of kinetochore tension. Dev Cell. 2012;22:1017–1029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Catarinella M, Grüner T, Strittmatter T, Marx A, Mayer TU. BTB-1 A small molecule inhibitor of the mitotic motor protein kif18A. Angew Chemie - Int Ed. 2009;48:9072–9076. DOI: 10.1002/anie.200904510. [DOI] [PubMed] [Google Scholar]
- 45.Bhat KMR, Setaluri V. Microtubule-associated proteins as targets in cancer chemotherapy. Clin Cancer Res. 2007;13:2849–2854. DOI: 10.1158/1078-0432.CCR-06-3040. [DOI] [PubMed] [Google Scholar]
- 46.Chandrasekaran G, Tátrai P, Gergely F. Hitting the brakes: targeting microtubule motors in cancer. Br J Cancer. 2015;113:693–698. DOI: 10.1038/bjc.2015.264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Tanenbaum ME, Medema RH, Akhmanova A. Regulation of localization and activity of the microtubule depolymerase MCAK. Bioarchitecture. 2011;1:80–87. DOI: 10.4161/bioa.1.1.14631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sharp DJ, Ross JL. Microtubule-severing enzymes at the cutting edge. J Cell Sci. 2012;125:2561–2569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Buster D, McNally K, McNally FJ. Katanin inhibition prevents the redistribution of gamma-tubulin at mitosis. J Cell Sci. 2002;115:1083–1092. [DOI] [PubMed] [Google Scholar]
- 50.Zhang D, Rogers GC, Buster DW, Sharp DJ. Three microtubule severing enzymes contribute to the ‘Pacman- flux’ machinery that moves chromosomes. J Cell Biol. 2007;177:231–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Rizk R. S., et al. MCAK and Paclitaxel have differential effects on spindle microtubule organization and dynamics. Mol Biol Cell. 2009;20:1639–1651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kellogg EH, Hejab NMA, Howes S, Northcote P, Miller JH, Diaz JF, Downing KH, Nogales E. Insights into the distinct mechanisms of action of taxane and non-taxane microtubule stabilizers from cryo-EM structures. J Mol Biol. 2017;429:633–646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Lee Y. M., Kim E, Park M, Moon E, Ahn SM, Kim W, Hwang KB, Kim YK, Choi W and Kim W. Cell cycle-regulated expression and subcellular localization of a kinesin-8 member human KIF18B. Gene. 2010;466:16–25. [DOI] [PubMed] [Google Scholar]
- 54.Mayr M. I., Hümmer S, Bormann J, Grüner T, Adio S, Woehlke G and Mayer TU. The human Kinesin Kif18A is a motile microtubule depolymerase essential for chromosome congression. Curr Biol. 2007;17:488–498. [DOI] [PubMed] [Google Scholar]
- 55.Messin LJ, Millar JBA. Role and regulation of kinesin-8 motors through the cell cycle. Syst Synth Biol. 2014;8:205–213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Stout J. R., Yount AL, Powers JA, LeBlanc C, Ems-McClung SC and Walczak CE. Kif18B interacts with EB1 and controls astral microtubule length during mitosis. Mol Biol Cell. 2011;22:3070–3080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Güttinger S, Laurell E, Kutay U. Orchestrating nuclear envelope disassembly and reassembly during mitosis. Nat Rev Mol Cell Biol. 2009;10:178–191. [DOI] [PubMed] [Google Scholar]
- 58.Lacroix B, van Dijk J, Gold ND, Guizetti J, Aldrian-Herrada G, Rogowski K, Gerlich DW and Janke C. Tubulin polyglutamylation stimulates spastin-mediated microtubule severing. J Cell Biol. 2010;189:945–954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Shin S. C., Im SK, Jang EH, Jin KS, Hur EM and Kim EE.. 2019. Structural and molecular basis for katanin-mediated severing of glutamylated microtubules. Cell Rep. DOI: 10.1016/j.celrep.2019.01.020. [DOI] [PubMed] [Google Scholar]
- 60.Valenstein ML, Roll-Mecak A. Graded control of microtubule severing by tubulin glutamylation. Cell. 2016;164:911–921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Audebert S., Desbruyeres E, Gruszczynski C, Koulakoff A, Gros F, Denoulet P and Eddé B.. Reversible polyglutamylation of alpha- and beta-tubulin and microtubule dynamics in mouse brain neurons. Mol Biol Cell. 2013;4:615–626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Rath O, Kozielski F. Kinesins and cancer. Nat Rev Cancer. 2012;12:527–539. [DOI] [PubMed] [Google Scholar]
- 63.Carney BK, Cassimeris L. Stathmin/oncoprotein 18, a microtubule regulatory protein, is required for survival of both normal and cancer cell lines lacking the tumor suppressor, p53. Cancer Biol Ther. 2010;9:699–709. [DOI] [PubMed] [Google Scholar]
- 64.Minotti AM, Barlow SB, Cabral F. Resistance to antimitotic drugs in Chinese hamster ovary cells correlates with changes in the level of polymerized tubulin. J Biol Chem. 1991;266:3987–3994. [PubMed] [Google Scholar]
- 65.Carney BK, Caruso Silva V, Cassimeris L. The microtubule cytoskeleton is required for a G2 cell cycle delay in cancer cells lacking stathmin and p53. Cytoskeleton. 2012;69:278–289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Schindelin J., Arganda-Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, Preibisch S, Rueden C, Saalfeld S, Schmid B and Tinevez JY.. Fiji: an open-source platform for biological-image analysis. Nat Methods. 2012;9:676–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.