

Introduction
Antimicrobial peptides were first described in the 1980’s, having been found in insect hemolymph 4, 5, mammalian neutrophil granules6 and frog skin secretions8. Subsequently, more than a thousand natural AMPs have been found in many tissues of many different species9, 10. Hundreds of synthetic AMPs have been also been created, often by mimicking natural sequences in combination with trial and error experimentation9,10, and sometimes by screening or computer aided design2, 3, 11–14. Since the beginning of the field, AMPs have been promoted as novel antibiotics that might improve human health and well-being. Yet, at the time of their initial discovery, there was little urgency to the translational applications of AMPs because it was not known, at the time, that drug resistant bacterial infections would grow over the next 30 years to become a global health crisis in morbidity and mortality15–17. We are now urgently and ever increasingly in need of novel antibiotic treatment options against drug-resistant bacteria. While some AMPs have been developed into potential topical drugs, and some are nearing or are in clinical trials18, AMPs have not succeeded, recently or in past decades19, to have any real impact on the systemic treatment options for drug resistant bacteria.
Thousands of AMPs are known that have good antibiotic activity in the culture tube, microwell plate, and petri dish. At least under standard laboratory conditions, many AMPs have potent, sterilizing activity at low μM concentrations against multiple species of bacteria20,21, often including both Gram positive and Gram negative species. AMP activity is observed at the same concentrations at which most conventional antibiotics are active under the laboratory conditions. AMPs often have equally potent activity against drug-susceptible, drug-resistant and multidrug resistant bacteria22–25 showing that the conventional mechanisms of drug resistance do not apply to AMPs. AMPs can also act against biofilms22, 26, and they can create antibacterial surfaces by covalent tethering or physical adsorption27. Importantly, AMPs may be less likely to induce resistance than conventional antibiotics28–30 although resistance to AMPs does occur31. Despite their potent, broad spectrum activity in the laboratory at low concentration, AMPs have not succeeded in the ultimate goal: the development of novel antibiotics that can be used systemically to prevent or treat drug resistant bacterial infections.
This dearth of systemically active AMPs has many causes, but may be due in part, to impediments to bioavailability and dosing such as host cell inhibition1, serum inhibition22, residual toxicity32, and proteolytic degradation7. Systemically active AMPs are almost certainly possible, as none of these impediments seems to be unsurmountable on its own. In fact, the possibility of a life-saving anti-infective, peptide drug has been proven by the record of the anti-HIV peptide enfuvirtide that has been approved for use in the US and Europe since 200333. Enfuvirtide is a linear 36-residue peptide drug with over $1 billion in net sales33–35. This peptide is typically administered subcutaneously in 90 mg doses and has extended the lives of many patients infected with HIV that had become resistant to other drugs. Enfuvirtide has a long half-life in the human body34 despite the lack of specific modifications to increase bioavailability or decrease proteolytic sensitivity. The peptide is an amphipathic α-helical peptide36 that likely binds to cells and serum proteins, and this may help it to remain intact and in circulation. Despite this, enfuvirtide has low toxicity and is able to effectively inhibit the fusion of HIV viruses with cell membranes in vivo, probably by interfering with the structure-function relationships of the GP41 fusion protein, from which enfuvirtide was obtained37.
If there are no insurmountable barriers to systemically active AMPs and we have thousands of known AMPs with activity in the laboratory, it is reasonable to ask: Why are there no systemically active AMP drugs? Why does there seem to be few in the development pipeline? In this chapter we hypothesize that the number of potential systemically active AMPs drugs in the drug development pipeline is small because i) the thousands of AMPs known have not evolved to be systemic AMPs or have not been discovered under the most relevant conditions, and because ii) rational engineering of AMP properties is not possible due to the fact that we do not have good sequence-function relationships for any of these impediments, including antibacterial activity. Below, we discuss an approach that may be especially well suited in this situation: Discovery of novel AMPs by iteratively screening small peptide libraries under experimental conditions that are increasingly relevant to physiological conditions. We have referred to this approach as “synthetic molecular evolution” 38–40. By these means we suggest approaching the most clinically relevant conditions in a stepwise manner and doing so as a first stage in the preclinical identification of peptide antibiotic drugs to feed a larger number of relevant candidates into the development pipeline. Below we detail some of the major impediments to systemic activity of AMPs, and then describe how screening for AMP activity can be done under conditions that much more closely mimic in vivo conditions to identify peptides without these impediments.
Impediments to systemic activity
Compared to conventional antibiotic drugs, AMPs have a fundamentally different mode of action on bacteria. Most conventional antibiotics inhibit a critical biochemical process by targeting one molecule (e.g. an enzyme or ribosome). The number of functional molecules decreases until the microbe loses so much of that essential function that it cannot replicate or survive. The rate or degree to which critical activity is lost depends on local drug concentration. AMPs on the other hand kill microbes in a cooperative, saturation-like process in which the peptides must massively accumulate on bacteria to levels that essentially saturate the cell in order to kill them through the effect of their interfacial activity41 on membrane integrity. Various measurements in the literature1, 42–45 have shown that the number of bound AMPs required to kill a bacterial cell is extremely high, from 107 to 108 peptides per cell. Since an E. coli cell can be expected to have perhaps 2×107 total lipids, this means that killing does not occur until there around one peptide bound for every bacterial lipid, which is more peptide than can possibly bind to the cytoplasmic membrane alone. This lethal amount of peptide is also equivalent to roughly one peptide per DNA base. If we assume that 108 peptides are evenly distributed in the volume of the cell, the local concentration of AMP is about 80 mM1. In reality, AMPs will specifically accumulate on anionic structures such as cell wall, LPS, cytoplasmic membrane, and DNA and could have local concentrations that approach molar. As a result, the mode of action of AMPs is a successive attack on the cell architecture. Membranes are permeabilized within minutes2, 3, followed by leakage of macromolecules, including DNA. Within 60 minutes of treatment of bacteria with AMPs the entire cell architecture is compromised and individual cells are sometimes not discernable. It is thus reasonable that resistance would be more difficult to evolve, given that mechanism of action of AMPs may involve the entire cell architecture. When resistance is observed, it is usually due to changes in LPS or cell wall components46, 47, making these structures less anionic so they do not accumulate large amounts of cationic AMPs.
Serum and Host Cell Inhibition
The need for accumulation of peptide means that systemic activity of AMPs faces different challenges than conventional drugs (Figure 1) and will exhibit threshold behavior. There may be only a narrow window between saturation/killing of bacteria and survivable accumulation of an AMP (Figure 2). Thus, any factor that competes for bacterial binding has the capacity to decrease accumulation on bacteria. Contrary to the commonly stated fact, external eukaryotic membranes are highly anionic overall, due to the large amount of anionic glycoconjugates on lipid and proteins. Thus, cationic AMPs bind to eukaryotic cells and tissues, at least moderately, and because host cells and tissue will always be orders of magnitude more abundant than pathogens, even weak competition can be problematic. Using a set of 12 natural and synthetic AMPs, we have shown that even a few minutes of preincubation of AMPs with human erythrocytes strongly reduced the activity of most of them1, see Figure 3, through some combination of host cell binding and proteolysis by the cytosolic proteases of RBCs7. This effect is not always observed, indicating that it is a surmountable impediment. For example, the insect peptide cecropin A was not affected in our study by human RBCs1. Similarly Stella and colleagues carefully examined the effect of RBCs on the activity of an AMP and found little inhibition43.
Figure 1.
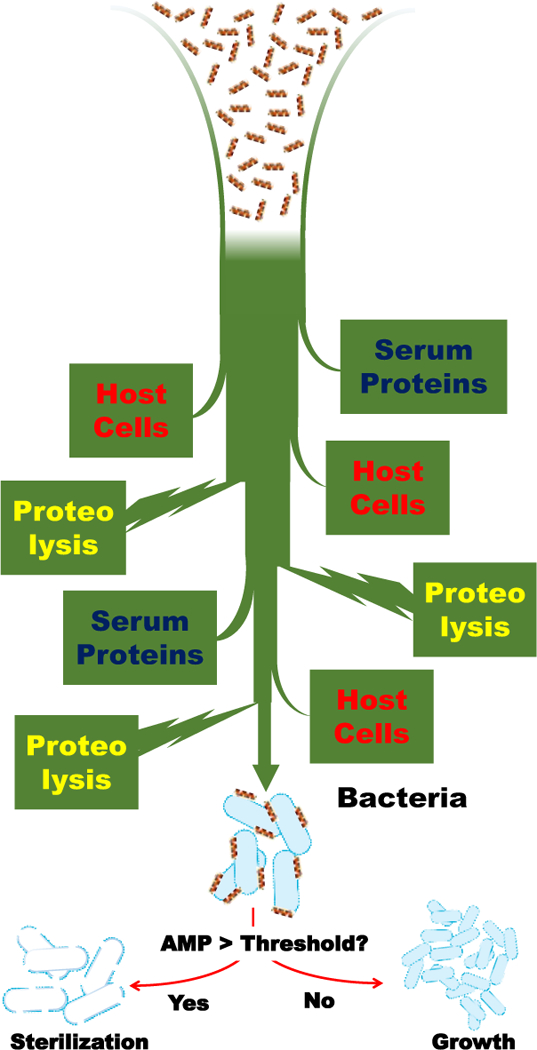
Some impediments to the bioavailablity and systemic activity of antimicrobial peptides. Antimicrobial peptides must accumulate significantly on bacteria to have bactericidal activity. In the body, interactions with host cells and tissue, interactions with serum proteins and proteolytic degradation can decrease accumulation, and decrease activity.
Figure 2.
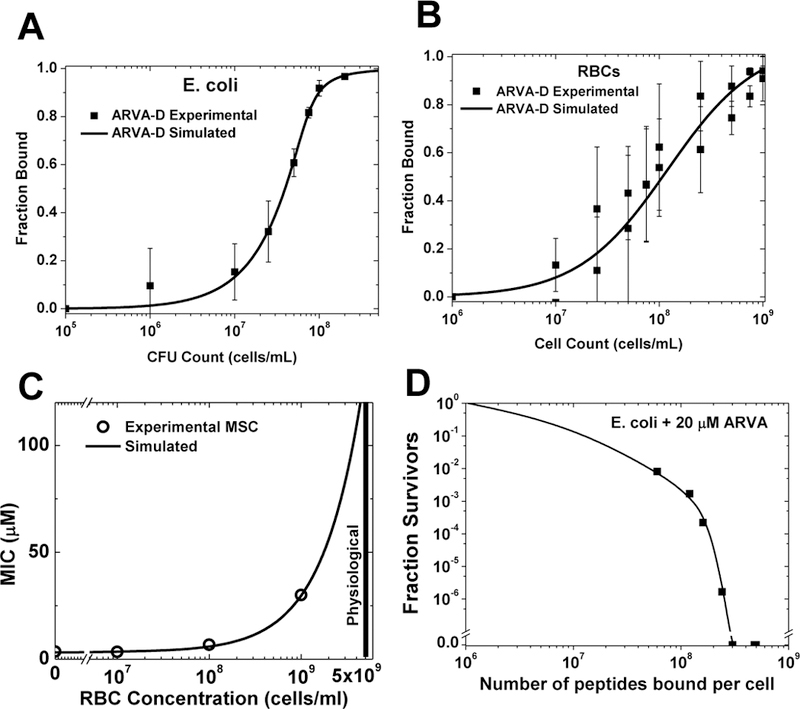
Saturation-dependent activity of an antimicrobial peptide and its inhibition by host cells. The protease resistant AMP D-ARVA (rrgwalrlvlay-NH2) was used in these experiments1–3. A: Measured binding of D-ARVA to E. coli cells. B: Measured binding of D-ARVA to human RBCs. C: Experimental measurements compared to simulation of a mixed experiment assuming simple competition between E. coli and RBC shows that the measured binding accounts for the loss of activity when host cells are present. D: Survival of different innocula of E. coli incubated with 20 μM D-ARVA in conjunction with the binding curve in panel A, enables comparison of peptide lethality and the number of peptides bound to each bacterial cell. More than 2×108 peptides bound per cell are required for sterility.
Figure 3.
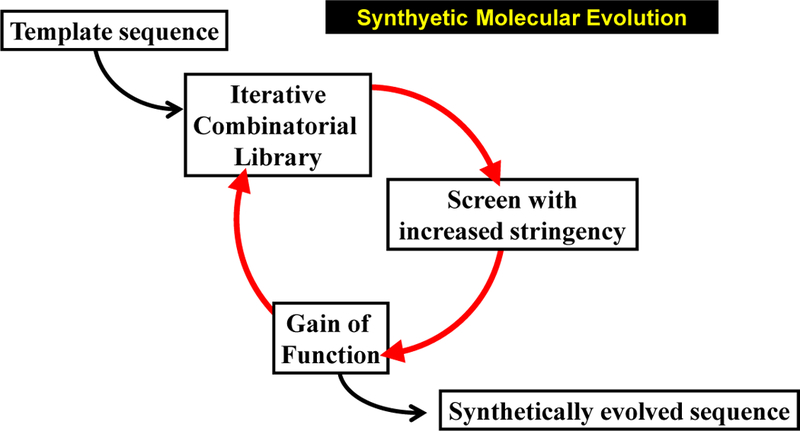
Synthetic molecular evolution of peptides. As we practice it, SME utilizes multiple small libraries (generations) which are iteratively screened for gain of function daughter sequences.
Serum proteins, especially serum albumin, can also bind cationic AMPs and are also highly concentrated in the body (35–50 mg/ml in blood), further potentially reducing the effective concentration of peptide available to bind to bacteria22. As we discuss in detail below, host cell and serum protein binding are rarely considered in the early stages of novel AMP discovery or design. If they are tested at all, it is determined how much these factors interfere with AMP activity only very late in the preclinical development pipeline. Here we are proposing that these factors be included during initial discovery of AMPs by screening. This will presumably give rise to a large number of relevant AMPs that can enter the pipeline, which will increase the probably of finding a few that can be developed into systemic drugs.
Toxicity Against Mammalian Cells
AMPs bind to anionic mammalian cells through electrostatic interactions1, 48,49, 50 and have interfacial activity41. As a result, many have at least some acute toxicity due to permeabilization of the plasma membrane. Toxicities vary significantly among known AMPs, yet those with relatively little toxicity still have relatively poor therapeutic indices compared to conventional antibiotics. Some AMPs may become less toxic in the presence of serum or when host cells are highly concentrated, although this effect is rarely tested.
In the abundant literature on AMPs, researchers often measure lysis of erythrocytes (hemolysis) as a surrogate for eukaryotic cell toxicity. This is an easy assay and its widespread use provides some uniformity in the AMP literature. However for the maximum sensitivity to toxicity, nucleated cells in culture may be a more sensitive and more informative model system. Nucleated cells can also respond to mechanisms of toxicity other than acute cytolysis. While some researchers have discussed how some AMPs may have useful selective activity against cancer cells48, 51, we argue that cancer cells would make an especially stringent test system for selecting against toxicity because they are especially sensitive to AMPs. In other words, a synthetically evolved AMP that has potent antibacterial activity under relevant conditions and no toxicity against mammalian cancer cells would seem to be an ideal candidate for development into a systemic drug. We discuss how this can be done in a screen below.
Proteolytic Degradation
Chemical stability of peptides, i.e. resistance to proteolysis, which does not come into play in laboratory assays, is also a critical consideration for systemic activity of peptides. Some peptides are degraded rapidly by serum exopeptidases, dipeptidases and other proteases7, 52, 53. Sensitivity to serum proteolysis is partially predictable based on sequence. In peptide drug development, serum stability competes with synthetic complexity (i.e. manufacturing cost). Shorter, linear, L-amino acid peptides are most economical to produce, but are susceptible to rapid proteolysis, while cyclic, crosslinked or chemically modified peptides are more costly to produce but are resistant. Proteolysis is a pervasive threat to AMPs. For example, we have shown that washed human RBCs contain a very high concentration of multiple proteases in their cytosol7. Incubation of a set of natural and synthetic linear AMPs with dilute RBCs leads to rapid degradation of peptide if there is even a small amount of hemolysis, which is almost always true. Cytosolic amino-and carboxy-exopeptidases removed amino acids one or two at a time from both termini7. For this reason, even standard hemolysis assays in the laboratory may be strongly affected by the proteolytic sensitivity of peptides.
It is likely that host cell binding and serum protein binding will decrease susceptibility to degradation, but as stated above they may also interfere with activity. Cyclization or crosslinking, as found in many natural AMPs will reduce proteolysis. For linear AMPs chemical modification of the termini52, 54, 55 non-natural terminal amino acids or selective substitution with D-amino acids can increase stability54. Perhaps the simplest approach is to replace all residues with D-aminoacids, as this will provide complete resistance to proteolysis while not changing activity1, 2, 42
Synthetic Molecular Evolution
To create an AMP that could have useful, systemic (in vivo) antibacterial activity, the factors describe above will need to be simultaneously optimized. Specifically it will be necessary to i) maximize selectivity for binding to bacteria over serum proteins and host cells; ii) maximize bactericidal activity of bound peptide; iii) minimize susceptibility to proteolytic degradation; iv) minimize residual cytotoxicity and v) maximize solubility under physiological conditions. Thus, the design of a systemically-active AMP is like a puzzle in which each of these coupled factors must be simultaneously minimized or maximized without negatively affecting the others. Yet, other than proteolytic susceptibility, the sequence-structure-function relationships for none of these factors are understood well enough to make useful predictions or to enable rational engineering. This is why most new AMPs described in the literature are either identified from natural sources, or are discovered in the laboratory by simple trial and error under standard conditions.
How can one simultaneously optimize these various factors when they are incompletely coupled and when the molecular mechanism are not understood in enough molecular detail to enable rational design? In this chapter, we discuss how this might be done using synthetic molecular evolution (SME). By this we mean iterative screening of rationally designed peptide libraries that are based on known AMPs and are designed using the known physical principles to allow rational variations in library members. SME is useful for the development of AMPs when screening is done under conditions that mimic the environment in which a systemically-active peptide must function. We call it “evolution” because it is most economical to screen iteratively such that each “generation” of gain-of-function AMP is selected from a library built around an active sequence from the previous generation, and each iterative screen further refines the selected sequences to have the properties that are sought. In this case we seek bactericidal activity at low μM concentration in the presence of concentrated host cells and serum, without toxicity.
Design and Synthesis of Combinatorial Peptide Libraries
Although there are many ways to synthesize and screen peptide libraries14, 56–63 we focus here on the approach we have taken recently to identify membrane active peptides with specific properties2, 3, 38–40, 63–67. We design small, iterative libraries of 10–30,000 members that are based on a template sequence with known activity. Using a library of this level of diversity means that we can design a library synthesis scheme that provides a relatively large amount of each library member to work with. This, in turn, enables us to use much more complex screens and it allows us to screen the same library member in multiple parallel assays, which is needed to screen for bactericidal activity against multiple microbes as well as toxicity against host cells.
Our approach to library synthesis and quality control is well described in many papers2, 3, 38, 61, 63–69. In short, a photocleavable linker is added to Tentagel-NH2 Megabeads, followed by library construction using the split and recombine approach58. Quality control is assured with HPLC, mass spec and sequencing performed on multiple individual beads. Each bead contains about 1 nmol of peptide which is released by UV light providing 100 μl of a 10 μM solution. This is sufficient peptide to perform multiple parallel assays on each library member. Screening of such small iterative libraries has long been routine in the laboratory2, 3, 38, 61, 63–69 and requires no special robotic instrumentation.
High Throughput Screening for Antibacterial Activity
As we envision the discovery of systemically active AMPs, screening must be done under conditions that most closely mimic the conditions experienced by a peptide antibiotic in vivo. At a minimum, there must be a high concentration of host cells and a high concentration of serum that has been heat inactivated to eliminate activated complement proteins. RBC also contain a very high concentration of proteases that are somewhat different that serum proteases7, so any screen with RBCs and/or serum will contain many realistic proteases. We have been experimenting with using human red blood cells (RBCs) as a host tissue analog. This is advantageous because it is easy to procure large amounts of fresh, concentrated human RBCs.
In Figure 5 we show the effect of preincubation of 1×109 RBC/ml on the antimicrobial activity of a set of natural and synthetic AMPs. In many cases, but not all, RBCs inhibit the antimicrobial activity when AMP are preincubated for a few minutes with RBCs. We subsequently showed that this inhibition is due to two things: RBC proteases, released by background autolysis or by a small amount of direct hemolysis, and direct interactions of AMPs with the cells that reduce the pool of available AMP by competition. Some peptides are more susceptible to the former and some are more susceptible to the latter1. Any AMP that will maintain antibacterial activity in vivo will need to be resistant to both proteolysis and direct host cell inhibition.
Figure 5.
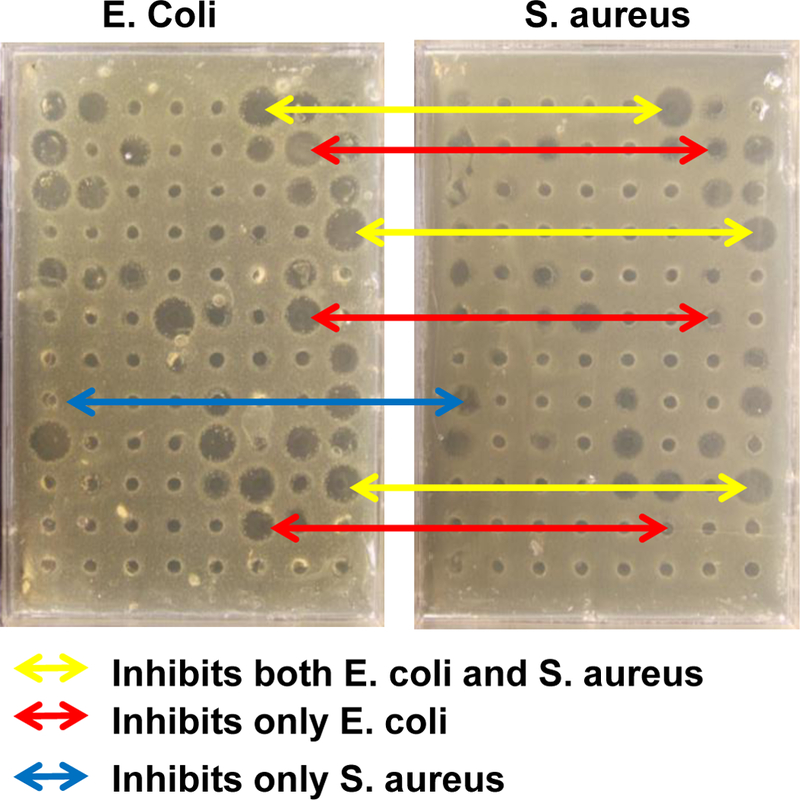
Example screening of members of a peptide library against two organisms using radial diffusion. The same set of peptide library members were screened again the Gram negative E. coli and the Gram positive S. aureus with radial diffusion. Zones of inhibition are observed. Some active library members inhibit both bacteria (yellow) while others inhibit only one of the two (red and blue).
Radial diffusion
There are a number of antimicrobial assays that can be used in a high throughput screen format. Here we will discuss the strengths and weaknesses of some of them in the context of SME. Radial diffusion is an assay in which a thin bacteria-seeded agar layer is overlaid with a sterile, nutrient rich agar enabling a lawn of bacteria to grow between the layers, except where growth is inhibited in a zone around a locally applied antibiotic. For peptides to be tested in the presence of host cells and serum, a small hole can be made in the lower agar layer and a small volume of peptide mixture can be introduced. After allowing some time for peptide diffusion into the agar, the nutrient overlay is added and the plate is allowed to grow overnight. The following day, the lawn of bacteria is visible and the zones of inhibition are readily observed and quantitated, as shown in Figure 5. Radial diffusion has the advantage that it is readily adapted to high throughput, and that it is quantitative. A larger zone of inhibition is related to better activity. However, the size of the zone of inhibition may also be strongly affected the ability of the antibiotic to diffuse in agar rather than by its inherent antibacterial activity. MIC-based quantitation by radial diffusion using serial dilution circumvents this problem, but cannot be accomplished in a screen. Further, radial diffusion reports on inhibition of bacterial growth, which does not necessarily indicate sterilization, a more desired property. In Figure 5 we show some examples of radial diffusion-based screens of a peptide library simultaneously against the Gram negative Escherichia coli and against the Gram positive Staphylococcus aureus. Some library members inhibit only one or the other species, while some inhibit both.
Incorporation of concentrated host cells and serum is easily accomplished in radial diffusion, as they can be mixed with peptide prior to introduction of the whole mixture into the well in the agar layer. In Figure 6 we show the effect of concentrated human RBCs on Radial diffusion against S. aureus. While powerful and quantitative, we have found that the most significant fault of radial diffusion as a screening method is that it does not necessarily select for peptides with sterilizing activity. To overcome this barrier, we also have developed screens based on broth sterilization, which we describe next.
Figure 6.
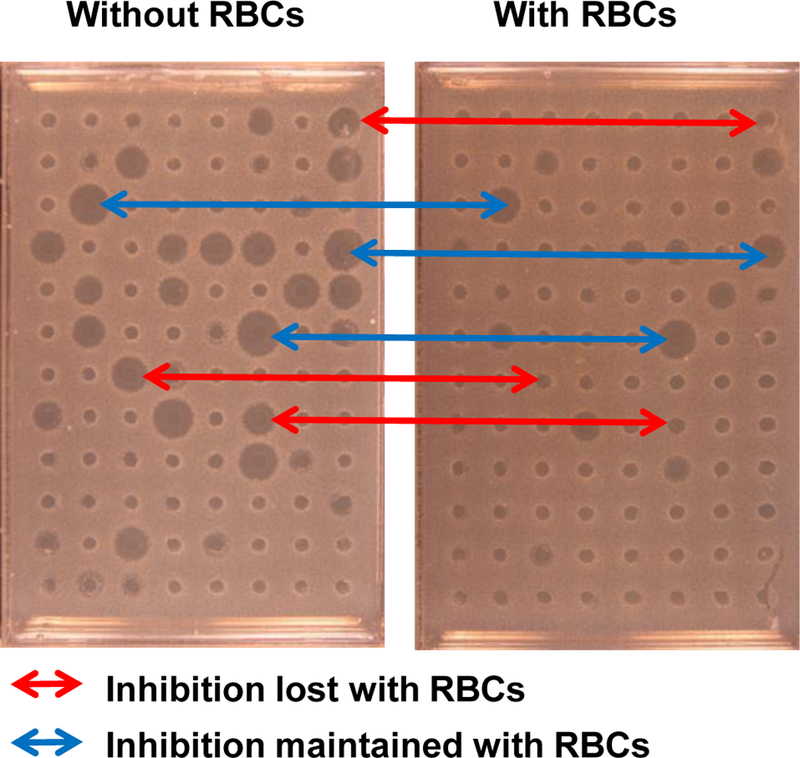
The effect of human RBCs on the activity of peptide library members against S. aureus using radial diffusion. Example screening of a peptide library S. aureus with radial diffusion. Both plates were screened with the same library members. The samples added to the right plate had been incubated with 1×109 human RBC/ml prior to use. Some active library members are inhibited by RBCs, while others are not.
Broth sterilization
Broth sterilization is an unambiguous, all-or-none assay for sterilization. Broth assays are done in liquid media inoculated with bacteria and treated with antibiotic. After overnight incubation, cultures are assayed for absence or presence of live bacteria. Survival of any bacteria generally means that they will grow to a high density after overnight incubation, while sterilized cultures will remain sterile. These two outcomes can easily be measured with optical density, and sterility can be verified by plating the sterile culture on nutrient agar and noting the presence or absence of colony forming units (CFU). Broth sterilization assays can be modified by the addition of concentrated host cells and heat inactivated serum to test for sterilization under physiologically relevant conditions. When concentrated host cells, such as concentrated human RBCs, are used the growth of bacteria cannot be measured directly by turbidity, so a secondary plate can inoculated and allowed to grow overnight. Alternately, aliquots can be spotted on nutrient agar and CFUs can be counted.
Broth dilution assays are not quantitative in a high throughput screen. They are binary tests in which library members will either be positive or negative for sterilization under the conditions of the screen. Typically screens can only be done under one condition for each library member tested. We propose testing antibiotic activity against multiple species simultaneously limiting the amount of each library member available. Thus it is critical to adjust the stringency such that a small number of leads are identified. The stringency of a broth sterilization assay can be modified by adjusting the inoculum size, antibiotic concentration, incubation time or other factors. A possible scheme for a broth dilution screen is shown in Figure 7, along with the results of screening members of an AMP library using broth dilution.
Figure 7.
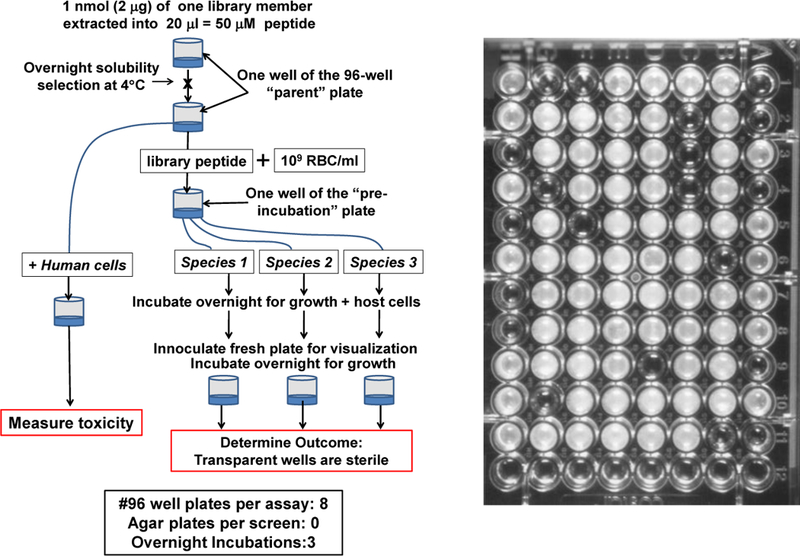
Peptide library screening using broth sterilization. Left: One possible scheme for screening a peptide library for solubility, for physiologically relevant broad spectrum bactericidal activity, and for lack of toxicity. Right: Example 96 well plate after screening a library for sterilizing activity against E. coli. Wells are either transparent or opaque. Transparent wells have no colony forming units on nutrient agar, confirming sterility.
Reduction of Colony Forming Units
CFU reduction is a hybrid assay that enables counting of live bacteria remaining in a solution after antibacterial treatment. It essentially reports on the same phenomenon as broth dilution, yet can be done with less labor and in less time. In this assay, which is readily adapted to high throughput, bacteria and antibiotic, with host cells and/or serum, are incubated together for an amount of time that enables killing, and then are spotted on a nutrient agar plate at high nominal CFU counts and grown overnight. In the absence of bactericidal activity, a dense mat of bacteria will grow. However, when only a small fraction of bacteria survive, or none at all, a countable number of colonies will grow, a quantitative result that can be used to rank order AMPs in a screen. CFU reduction assays can readily be modified by the addition of host cells and serum, as above. In Figure 8 we show a possible scheme for SME using CFU counts along with the results of a test screen of AMPs from a library. Note that positive, sterilizing sequences can readily be identified by the absence of CFUs, which amounts to more than 4 logs of CFU reduction.
Figure 8.
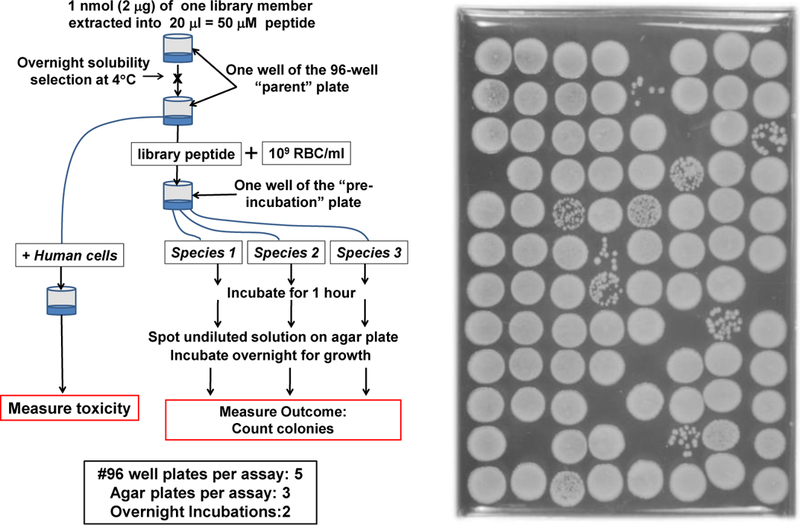
Peptide library screening using the reduction in colony forming units (CFU). Left: One possible scheme for screening a peptide library for solubility, for physiologically relevant broad spectrum bactericidal activity, and for lack of toxicity. Right: Example nutrient agar plate after spotting library members mixed with bacteria for 1 hour. Clear spots with no colonies have been sterilized.
Cytotoxicity and hemolysis
In a screen for antimicrobial assays under physiological conditions, toxicity must also be measured simultaneously. This can be accomplished in assays that are done in the presence of RBCs as host cells by also measuring hemolysis. However, hemolysis may not be sensitive enough to be useful, especially in the presence of concentrated serum which can be protective protective. Here we suggest that toxicity be measured in parallel using sensitive human cancer cell lines, such as HeLa cells such that peptides with very low toxicity can be identified during the screen.
ESKAPE Pathogens
While many bacteria can infect humans and harbor drug resistance, there are a small set that account for the majority of morbidity and mortality16, 70. These include Clostridium difficile, often associated with gastrointestinal infections, and the ESKAPE pathogens, whose acronym indicates Enterococcus faecalis, Staphylococcus aureus, Klebsiella pneumonia, Acinetobacter baumannii, Pseudomonas aeruginosa, and the Enterobacteriaceae, which includes Escherichia, Salmonella, Vibrio and Shigella species, among others. Since some AMPs have variable potencies against these different organisms, thus screening for the broadest activity must be done against multiple species simultaneously. We previously screened against two ESKAPE bacteria, E. coli and S. aureus, and a fungus, Cryptococcus neoformans, simultaneously and found very low overlap in activities. This enabled the identification of the rare peptides with broad spectrum activity2. We suggest screening in parallel against S. aureus two Gram negative ESKAPE pathogens.
Future Prospects
The physical chemistry-based action of AMPs on bacteria leads to broad-spectrum activity and difficulty in evolving resistance, which accounts for some of the appeal of AMPs as potential drugs. Yet, these same properties also drive nonspecific interactions with serum protein and host cells, which reduce the effectiveness of AMPs. In this chapter, we have presented the concept of synthetic molecular evolution as an tool to enable the discovery of AMPs, early in the development pipeline, that are less affected by host cell and serum protein binding. We remain hopeful that this new approach will finally enable AMP researchers to bridge the gap between the laboratory bench and the clinic.
Figure 4.
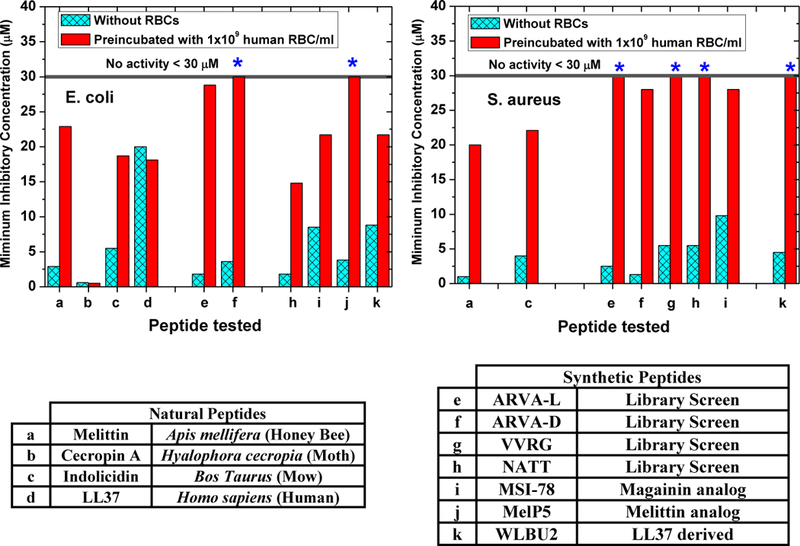
Human Red Blood cells inhibit antimicrobial peptides. As described elsewhere1 preincubation of natural and synthetic AMPs with 1×109 human RBC/ml (2% of physiological concentration) causes inhibition of most, but not all, of them. We have shown that such host cell inhibition is the result of direct RBC binding and also to proteolysis of the AMP by the cytosolic proteases found in human RBCs7.
Reference List
- 1.Starr CG, He J, & Wimley WC Host Cell Interactions Are a Significant Barrier to the Clinical Utility of Peptide Antibiotics. ACS Chem. Biol 11, 3391–3399 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Rathinakumar R & Wimley WC High-throughput discovery of broad-spectrum peptide antibiotics. FASEB J 24, 3232–3238 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Rathinakumar R, Walkenhorst WF, & Wimley WC Broad-spectrum antimicrobial peptides by rational combinatorial design and high-throughput screening: The importance of interfacial activity. J. Am. Chem. Soc 131, 7609–7617 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Steiner H, Hultmark D, Engstrom A, Bennich H, & Boman HG Sequence and specificity of two antibacterial proteins involved in insect immunity. Nature (London) 292, 246–248 (1981). [DOI] [PubMed] [Google Scholar]
- 5.Okada M & Natori S Purification and characterization of an antibacterial protein from haemolymph of Sarcophaga peregrina (flesh-fly) larvae. Biochem. J 211, 727–734 (1983). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zasloff M Magainins, a class of antimicrobial peptides from Xenopus skin: isolation, characterization of two active forms, and partial cDNA sequence of a precursor. Proc. Natl. Acad. Sci. U. S. A 84, 5449–5453 (1987). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Starr CG & Wimley WC Antimicrobial peptides are degraded by the cytosolic proteases of human erythrocytes. Biochim. Biophys Acta 1859, 2319–2326 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Patterson-Delafield J, Szklarek D, Martinez RJ, & Lehrer RI Microbicidal cationic proteins of rabbit alveolar macrophages: Amino acid composition and functional attributes. Infect. Immun 31, 723–731 (1981). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fjell CD, Hancock RE, & Cherkasov A AMPer: a database and an automated discovery tool for antimicrobial peptides. Bioinformatics 23, 1148–1155 (2007). [DOI] [PubMed] [Google Scholar]
- 10.Wang G, Li X, & Wang Z APD3: the antimicrobial peptide database as a tool for research and education. Nucleic Acids Res 44, D1087–D1093 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Moy TI et al. High-throughput screen for novel antimicrobials using a whole animal infection model. ACS Chem. Biol 4, 527–533 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kulagina NV, Shaffer KM, Ligler FS, & Taitt CR Antimicrobial peptides as new recognition molecules for screening challenging species. Sens. Actuators B Chem 121, 150–157 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kulagina NV, Shaffer KM, Anderson GP, Ligler FS, & Taitt CR Antimicrobial peptide-based array for Escherichia coli and Salmonella screening. Anal. Chim. Acta 575, 9–15 (2006). [DOI] [PubMed] [Google Scholar]
- 14.Hilpert K, Volkmer-Engert R, Walter T, & Hancock RE High-throughput generation of small antibacterial peptides with improved activity. Nat. Biotechnol 23, 1008–1012 (2005). [DOI] [PubMed] [Google Scholar]
- 15.Arias CA & Murray BE Antibiotic-resistant bugs in the 21st century--a clinical super-challenge. N. Engl. J. Med 360, 439–443 (2009). [DOI] [PubMed] [Google Scholar]
- 16.Boucher HW et al. Bad bugs, no drugs: no ESKAPE! An update from the Infectious Diseases Society of America. Clin. Infect. Dis 48, 1–12 (2009). [DOI] [PubMed] [Google Scholar]
- 17.Otto M MRSA virulence and spread. Cell Microbiol 14, 1513–1521 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Fox JL Antimicrobial peptides stage a comeback. Nat. Biotechnol 31, 379–382 (2013). [DOI] [PubMed] [Google Scholar]
- 19.Gordon YJ, Romanowski EG, & McDermott AM A review of antimicrobial peptides and their therapeutic potential as anti-infective drugs. Curr. Eye Res 30, 505–515 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Easton DM, Nijnik A, Mayer ML, & Hancock RE Potential of immunomodulatory host defense peptides as novel anti-infectives. Trends Biotechnol 27, 582–590 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hamill P, Brown K, Jenssen H, & Hancock RE Novel anti-infectives: is host defence the answer? Curr. Opin. Biotechnol 19, 628–636 (2008). [DOI] [PubMed] [Google Scholar]
- 22.de BA et al. The antimicrobial peptide SAAP-148 combats drug-resistant bacteria and biofilms. Sci. Transl. Med 10, (2018). [DOI] [PubMed] [Google Scholar]
- 23.Schlusselhuber M et al. In vitro effectiveness of the antimicrobial peptide eCATH1 against antibiotic-resistant bacterial pathogens of horses. FEMS Microbiol. Lett 350, 216–222 (2014). [DOI] [PubMed] [Google Scholar]
- 24.Mechkarska M et al. An analog of the host-defense peptide hymenochirin-1B with potent broad-spectrum activity against multidrug-resistant bacteria and immunomodulatory properties. Peptides 50, 153–159 (2013). [DOI] [PubMed] [Google Scholar]
- 25.Park SC et al. Synthetic diastereomeric-antimicrobial peptide: antibacterial activity against multiple drug resistant clinical isolates. Biopolymers 96, 130–136 (2011). [DOI] [PubMed] [Google Scholar]
- 26.Wolfmeier H, Pletzer D, Mansour SC, & Hancock REW New Perspectives in Biofilm Eradication. ACS Infect. Dis(2017). [DOI] [PubMed]
- 27.Kazemzadeh-Narbat M et al. Antimicrobial peptides on calcium phosphate-coated titanium for the prevention of implant-associated infections. Biomaterials 31, 9519–9526 (2010). [DOI] [PubMed] [Google Scholar]
- 28.Dobson AJ, Purves J, Kamysz W, & Rolff J Comparing selection on S. aureus between antimicrobial peptides and common antibiotics. PLoS. One 8, e76521 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Pollard JE et al. In vitro evaluation of the potential for resistance development to ceragenin CSA-13. J. Antimicrob. Chemother 67, 2665–2672 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Fedders H, Podschun R, & Leippe M The antimicrobial peptide Ci-MAM-A24 is highly active against multidrug-resistant and anaerobic bacteria pathogenic for humans. Int. J Antimicrob. Agents 36, 264–266 (2010). [DOI] [PubMed] [Google Scholar]
- 31.Perron GG, Zasloff M, & Bell G Experimental evolution of resistance to an antimicrobial peptide. Proc. Biol. Sci 273, 251–256 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yeaman MR & Yount NY Mechanisms of antimicrobial peptide action and resistance. Pharmacol. Rev 55, 27–55 (2003). [DOI] [PubMed] [Google Scholar]
- 33.Poveda E, Briz V, & Soriano V Enfuvirtide, the first fusion inhibitor to treat HIV infection. AIDS Rev 7, 139–147 (2005). [PubMed] [Google Scholar]
- 34.Joly V, Jidar K, Tatay M, & Yeni P Enfuvirtide: from basic investigations to current clinical use. Expert. Opin. Pharmacother 11, 2701–2713 (2010). [DOI] [PubMed] [Google Scholar]
- 35.LaBonte J, Lebbos J, & Kirkpatrick P Enfuvirtide. Nat. Rev Drug Discov 2, 345–346 (2003). [DOI] [PubMed] [Google Scholar]
- 36.Rapaport D, Ovadia M, & Shai Y A synthetic peptide corresponding to a conserved heptad repeat domain is a potent inhibitor of Sendai virus-cell fusion: An emerging similarity with functional domains of other viruses. EMBO J 14, 5524–5531 (1995). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Qureshi NM, Coy DH, Garry RF, & Henderson LA Characterization of a putative cellular receptor for HIV-1 transmembrane glycoprotein using synthetic peptides. AIDS 4, 553–558 (1990). [DOI] [PubMed] [Google Scholar]
- 38.Krauson AJ, He J, Hoffmann AR, Wimley AW, & Wimley WC Synthetic molecular evolution of pore-forming peptides by Iterative combinatorial library screening. ACS Chem. Biol 8, 823–831 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kauffman WB, Guha S, & Wimley WC Synthetic molecular evolution of hybrid cell penetrating peptides. Nat. Commun 9, 2568 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Li S et al. Potent Macromolecule-Sized Poration of Lipid Bilayers by the Macrolittins, A Synthetically Evolved Family of Pore-Forming Peptides. J Am. Chem. Soc 140, 6441–6447 (2018). [DOI] [PubMed] [Google Scholar]
- 41.Wimley WC Describing the mechanism of antimicrobial peptide action with the interfacial activity model. ACS Chem. Biol 5, 905–917 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Savini F, Bobone S, Roversi D, Mangoni M, & Stella L From liposomes to cells: Filling the gap between physicochemical and microbiological studies of the activity and selectivity of host-defense peptides. Peptide Sciencee 24041 (2018).
- 43.Savini F et al. Cell-Density Dependence of Host-Defense Peptide Activity and Selectivity in the Presence of Host Cells. ACS Chem. Biol 12, 52–56 (2017). [DOI] [PubMed] [Google Scholar]
- 44.Tran D et al. Homodimeric theta-defensins from rhesus macaque leukocytes: isolation, synthesis, antimicrobial activities, and bacterial binding properties of the cyclic peptides. J. Biol. Chem 277, 3079–3084 (2002). [DOI] [PubMed] [Google Scholar]
- 45.Steiner H, Andreu D, & Merrifield RB Binding and action of cecropin and cecropin analogues: antibacterial peptides from insects. Biochim. Biophys Acta 939, 260–266 (1988). [DOI] [PubMed] [Google Scholar]
- 46.Peschel A & Sahl HG The co-evolution of host cationic antimicrobial peptides and microbial resistance. Nat. Rev. Microbiol 4, 529–536 (2006). [DOI] [PubMed] [Google Scholar]
- 47.Peschel A How do bacteria resist human antimicrobial peptides? Trends Microbiol 10, 179–186 (2002). [DOI] [PubMed] [Google Scholar]
- 48.Riedl S, Zweytick D, & Lohner K Membrane-active host defense peptides--challenges and perspectives for the development of novel anticancer drugs. Chem. Phys. Lipids 164, 766–781 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Selsted ME, Brown DM, DeLange RJ, Harwig SSL, & Lehrer RI Primary structures of six antimicrobial peptides of rabbit peritoneal neutrophils. J. Biol. Chem 260, 4579–4584 (1985). [PubMed] [Google Scholar]
- 50.Agawa Y et al. Interaction with phospholipid bilayers, ion channel formation, and antimicrobial activity of basic amphipathic a-helical model peptides of various chain lengths. J. Biol. Chem 266, 20218–20222 (1991). [PubMed] [Google Scholar]
- 51.Riedl S et al. In search of a novel target -phosphatidylserine exposed by non-apoptotic tumor cells and metastases of malignancies with poor treatment efficacy. Biochim. Biophys. Acta 1808, 2638–2645 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Werle M & Bernkop-Schnurch A Strategies to improve plasma half life time of peptide and protein drugs. Amino. Acids 30, 351–367 (2006). [DOI] [PubMed] [Google Scholar]
- 53.Molhoek EM, van DA, Veldhuizen EJ, Haagsman HP, & Bikker FJ Improved proteolytic stability of chicken cathelicidin-2 derived peptides by D-amino acid substitutions and cyclization. Peptides 32, 875–880 (2011). [DOI] [PubMed] [Google Scholar]
- 54.Starr CG, Maderdrut JL, He J, Coy DH, & Wimley WC Pituitary adenylate cyclase-activating polypeptide is a potent broad-spectrum antimicrobial peptide: Structure-activity relationships. Peptides 104, 35–40 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Nguyen LT et al. Serum stabilities of short tryptophan-and arginine-rich antimicrobial peptide analogs. PLoS. One 5, (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Lam KS et al. A new type of synthetic peptide library for identifying ligand-binding activity. Nature (London) 354, 82–84 (1991). [DOI] [PubMed] [Google Scholar]
- 57.Dooley CT et al. An all D-amino acid opioid peptide with central analgesic activity from a combinatorial library. Science 266, 2019–2022 (1994). [DOI] [PubMed] [Google Scholar]
- 58.Chen CL, Strop P, Lebl M, & Lam KS One bead-one compound combinatorial peptide library: different types of screening. Methods Enzymol 267, 211–219 (1996). [DOI] [PubMed] [Google Scholar]
- 59.Frank R The SPOT-synthesis technique. Synthetic peptide arrays on membrane supports--principles and applications. J. Immunol. Methods 267, 13–26 (2002). [DOI] [PubMed] [Google Scholar]
- 60.Humet M et al. A positional scanning combinatorial library of peptoids as a source of biological active molecules: identification of antimicrobials. J. Comb. Chem 5, 597–605 (2003). [DOI] [PubMed] [Google Scholar]
- 61.Rathinakumar R & Wimley WC Biomolecular engineering by combinatorial design and high-throughput screening: small, soluble peptides that permeabilize membranes. J. Am. Chem. Soc 130, 9849–9858 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Deuss PJ, Arzumanov A, Williams DL, & Gait MJ Parallel synthesis and splicing redirection activity of cell-penetrating peptide conjugate libraries of a PNA cargo. Organic & biomolecular chemistry 11, 7621–7630 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Wiedman G, Kim SY, Zapata-Mercado E, Wimley WC, & Hristova K PH-Triggered, Macromolecule-Sized Poration of Lipid Bilayers by Synthetically Evolved Peptides. J. Am. Chem. Soc 139, 937–945 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Krauson AJ et al. Conformational Fine-Tuning of Pore-Forming Peptide Potency and Selectivity. J. Am. Chem. Soc 137, 16144–16152 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Krauson AJ, He J, & Wimley WC Gain-of-Function Analogues of the Pore-Forming Peptide Melittin Selected by Orthogonal High-Throughput Screening. J. Am. Chem. Soc 134, 12732–12741 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Marks JR, Placone J, Hristova K, & Wimley WC Spontaneous membrane-translocating peptides by orthogonal high-throughput screening. J. Am. Chem. Soc 133, 8995–9004 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Rausch JM, Marks JR, & Wimley WC Rational combinatorial design of pore-forming beta-sheet peptides. Proc. Natl. Acad. Sci. U. S. A 102, 10511–10515 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.He J et al. Direct Cytosolic Delivery of Polar Cargo to Cells by Spontaneous Membrane-translocating Peptides. J Biol. Chem 288, 29974–29986 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.He L, Hoffmann AR, Serrano C, Hristova K, & Wimley WC High-throughput selection of transmembrane sequences that enhance receptor tyrosine kinase activation. J. Mol. Biol 412, 43–54 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Centers For Disease Control. ANTIBIOTIC RESISTANCE THREATS in the United States, 2013 http://www.cdc.gov/drugresistance/pdf/ar-threats-2013-508.pdf 2014.