

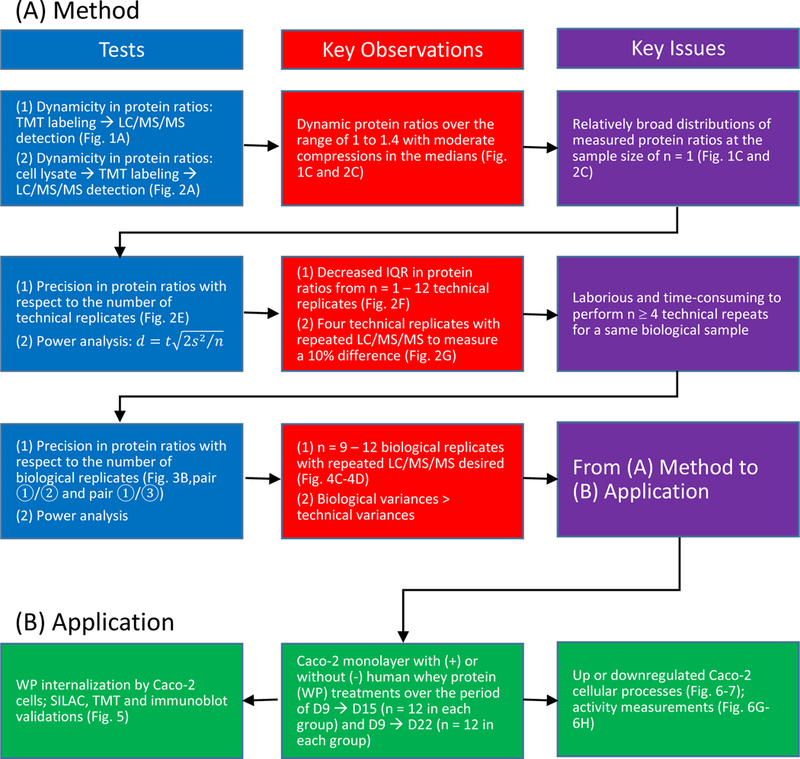
Abstract
Mass spectrometry (MS)-based proteomic approaches have largely facilitated our systemic understanding of cellular processes and biological functions. Cutoffs in protein expression fold changes (FC) are often arbitrarily determined in MS-based quantification with no demonstrable determination of small magnitude changes in protein expression. Therefore, many biological insights may remain veiled due to high FC cutoffs. Herein we employ the intestinal epithelial cell (IEC) line Caco-2 as a model system to demonstrate the dynamicity of tandem-mass-tag (TMT) labeling over a range of 5 – 40% changes in protein abundance, with the variance controls of ±5% FC for around 95% of TMT ratios when sampling nine to twelve biological replicates. We further applied this procedure to examine the temporal proteome of Caco-2 cells upon exposure to human whey proteins (WP). Pathway assessments predict subtle effects due to WP in moderating xenobiotic metabolism, promoting proliferation and various other cellular functions in differentiating enterocyte-like Caco-2 cells. This demonstration of a sensitive MS approach may open up new perspectives in the system-wide exploration of elusive or transient biological effects by facilitating scrutiny of narrow windows of proteome abundance changes. Furthermore, we anticipate this study will encourage more investigations of WP on infant gastrointestinal tract development.
Keywords: Caco-2, intestinal epithelial cells, milk proteins, quantitative proteomics, TMT
Graphical Abstract
INTRODUCTION
One important undertaking in MS-based proteomics is to generate comprehensive, in-depth protein reference maps covering biological systems.1–4 A vital addition for biological interrogation is to compare the regulation of protein expression in two or multiple states at a system-wide scale.5–8 Various MS-based strategies, including label-free (LF) and differential stable isotope labeling approaches, have been developed to quantitate relatively peptide levels and, by assembling peptide sequences, infer protein levels.9–12 The LF technology can be grouped approximately into two major categories, either comparing directly MS parent ion (MS1) currents from extracted ion chromatograms (XIC) or using MS/MS (MS2) spectral counts as a proxy of MS ion intensities. Stable isotope methods, on the other hand, label protein samples prior to MS analysis. Labeling can be achieved through the means of either metabolic incorporation such as stable isotope labeling by amino acids in cell culture (SILAC)13 or chemical derivatization such as commercially available mass differential tags for relative and absolute quantification (mTRAQ), isobaric tags for relative and absolute quantification (iTRAQ)14 or tandem mass tag (TMT).15,16
To obtain statistically significant information, quantitative approaches regularly involve the analysis of a sizable number of samples under each biological state, followed by a differential cutoff based on statistical significance p-value, abundance fold change (FC) or both, in order to distill real biological differences from technical artifacts and biological fluctuations. These cutoffs are often arbitrarily determined or may be inferred through means, such as power analysis, when relative standard deviations (r.s.d.) and sample sizes are known.17 Between the two LF procedures, a recent analysis showed the XIC-based approach outperforms the spectral counting method where the former is capable of separating a two-fold protein abundance change at the technical levels of false positive α = 0.05 (Type I error) and false negatives at β = 0.30 (Type II error), given triplicated LC/MS/MS analysis.8 The noble technical performance provides some basis for accurate quantitation of proteins in real biological systems using LF approaches. Nevertheless, LF quantification often depends highly on consistent sample processing and is expected to be lower in precision and sensitivity of detecting protein abundance changes when compared to labeling methods.18–21 For these reasons, stable isotope-based procedures are more widely considered the gold standard in MS-based quantitative proteomics.12
Extensive efforts have been made in ascertaining the technical merits of various stable isotope-labeling approaches with provisional exclusion of influences from biological fluctuations.22–25 Apart from the inherent technical quality of each method, additional factors such as the number of technical replicates and the number of repeated LC/MS/MS analysis for each technical replicate can affect the accuracy and precision of quantitation. In an example of SILAC quantitation,11 the technical r.s.d. of protein ratios was predicted to be 0.265 and 0.114 at n = 1 and 6 technical replicates, respectively.22 A more recent assessment of metabolic labeling approach suggested a r.s.d. of 0.059 in protein ratios when measuring four technical replicates, each with triplicated LC/MS/MS analysis.26 While every strategy has its own technical strengths and weaknesses, these comparisons focused primarily on relatively large protein fold changes (> 2×) with no demonstration of measuring smaller magnitude changes in protein abundances.
In various biological states, for instance with and without perturbation, the extents of biological implications that can be confidently distinguished are subjected to the minimum differences that can be reliably measured. Although it takes a different concept to MS1-based methods for quantification, MS2-based isobaric tagging approaches such as iTRAQ or TMT have been indicated to perform comparably to MS1-based metabolic labeling methods in both precision and accuracy at technical levels.23,27 On the other hand, multiplexed analysis can be conveniently implemented using isobaric labelling compared to metabolic labeling.12 To take the advantage of higher throughputs with isobaric tags, we set out to assess the performance of shotgun proteomics in measuring modest differences of protein abundance using, in this example, TMT isobaric tagging approach. We herein demonstrate a procedure of two-dimensional separation of protein tryptic digest from cell lysates. We chose electrostatic repulsion hydrophilic interaction chromatography (ERLIC)28 prefractionation coupled to second-dimension reversed-phase liquid chromatographic tandem mass spectrometric (RP-LC/MS/MS) analysis. Subsequently, we showed the dynamicity of tandem-mass-tag (TMT) labeling over a range of 5 – 40% change in abundance and described the control of ratio variances with respect to the number of proteomic measures. Combining these, we propose that the global quantitative analysis of cellular proteomes is feasible over a narrow range of abundance changes (Scheme 1A).
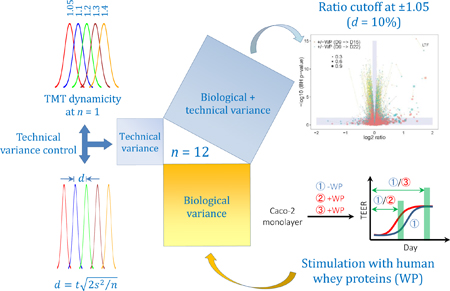
Scheme 1.
Overview of the study. (A) Demonstration of the feasibility in sensing subtle changes in protein abundances using shotgun proteomics. (B) Application of the procedure to examine the temporal proteome of Caco-2 monolayer with and without the stimulation of human WP.
To show the usefulness of this procedure, we examined the temporal proteome of enterocyte-like Caco-2 cells with and without the stimulation of human WP (Scheme 1B). In the absence of commercially available immortalized normal human intestinal epithelial cell (IEC) lines that are capable of differentiation in vitro, human colonic adenocarcinoma Caco-2 cell line has been widely adopted as a model for intestinal function as these cells differentiate spontaneously upon confluence, exhibiting the phenotype of polarized, small intestine-like enterocytes.29,30 The newborn gastrointestinal (GI) tract undergoes continual renewal, characterized by active proliferation and differentiation. Human milk proteins have been shown to promote the health and development of the neonatal GI tract in many ways.31–35 Still, our understanding of such benefits and their associated molecular basis remains superficial. It is largely unknown which intestinal epithelial processes can be modulated by the protein constituents in milk; even more obscure is the dynamics of these processes under the continuous influence of milk proteins. With our current demonstration in quantitative proteomics, we have been able to reveal various subtle effects of WP on the development of enterocyte-like Caco-2 cells. We anticipate this study will encourage more research investigating the effects of milk proteins on infant GI-tract development.
EXPERIMENTAL PROCEDURES
Reagents
Dulbecco’s Modified Eagle Medium (DMEM) with GlutaMAX™ (growth medium), DMEM without L-glutamine (standard experimental medium) and DMEM without L-glutamine, L-arginine and L-lysine (SILAC27,28 experimental medium), Dulbecco’s phosphate-buffered saline (DPBS), PBS-based enzyme-free cell dissociation buffer, heat-inactivated dialyzed fetal bovine serum (dFBS), L-glutamine and sodium pyruvate were from Life Technologies™ (Grand Island, NY). Penicillin-streptomycin solution, BD Falcon™ 24-well and 12-well cell culture inserts (0.4 μm pore size) and companion plates were from Corning Life Sciences (Tewksbury, MA). Pierce® RIPA buffer (25 mM Tris-HCl, 150 mM NaCl, 1% NP-40, 1% sodium deoxycholate, 0.1% SDS, pH 7.6), Halt™ protease inhibitor single-use cocktail, heat-inactivated HyClone™ FBS (FBS), SuperBlock™ and Microplate BCA™ (bicinchoninic acid) protein assay kit were from Thermo-Fisher Scientific (Rockford, IL). Complete Mini EDTA-free protease inhibitor cocktail was from Roche (Indianapolis, IN). VisionBlue™ fluorescence cell viability/proliferation assay kit was from BioVision (Milpitas, CA). Isotopically labeled 13C6,15N2-L-lysine (Lys8) and 13C6,15N4-L-arginine (Arg10) were from Cambridge Isotope Laboratories (Tewksbury, MA). L-proline was from MP Biomedicals (Solon, OH). Laemmli sample buffer, Criterion™ TGX™ (Tris-Glycine eXtended shelf life) precast gels, and Clarity™ Western ECL substrate were from Bio-Rad (Hercules, CA). PNGase F, S. cerevisiae, glutathione S-transferase (GST) assay kit, acetonitrile (ACN), acetic acid (HOAc), formic acid (FA) and all other chemicals were from Sigma-Aldrich (St. Louis, MO). Human breast milk was from Innovative Research, Inc. (Novi, MI) and the whey proteins (WP) extracted as previously described.36 Briefly, milk was thawed at 4 °C, then ultra-centrifuged at 4 °C (100,000 × g for 60 min) so that samples had a pellet of casein micelles on the bottom, a fat layer on the top, and delipidated whey supernatant in the middle. To obtain protein samples, the whey layer was filtered using a 10 kDa molecular-weight cut-off device (Millipore, Billerica, MA) and subjected to buffer-exchange with water.
Cell Culture for TMT Labeling
Human colorectal adenocarcinoma cell line Caco-2 (ATCC® HTB-37™) was from American Type Culture Collection (Manassas, VA) and routinely maintained in DMEM with GlutaMAX™ supplemented with 10% FBS, 100 IU/mL penicillin and 100 μg/mL streptomycin at 37°C with 5% CO2 humidified atmosphere. Cells between passage 26 and 30 were utilized for experiments. At least one passage prior to experimental plating, culture medium was changed to either DMEM supplemented with 10% dFBS, 3 mM L-glutamine, 1 mM sodium pyruvate, 100 units/mL penicillin and 100 μg/mL streptomycin for standard experiments or SILAC DMEM supplemented with Pro (400 mg/L), Arg10 (84 mg/L), Lys8 (146 mg/L), 10% dFBS, 3 mM L-glutamine, 1 mM sodium pyruvate, 100 units/mL penicillin and 100 μg/mL streptomycin for SILAC experiments. Cells transferred to the Arg10 and Lys8-containing SILAC DMEM underwent 5 or more doublings prior to experiment plating for incorporation of the isotopic amino acids. For experiments, cells at approximately 50% confluence were plated in either 24-well inserts at 100,000 cells/cm2/200 μL/700 μL or 12-well insets at 100,000 cells/cm2/600 μL/1500 μL and incubated at 37°C with 5% CO2 humidified atmosphere. After three days, and every 48 hours thereafter, the media was replaced (apical and basolateral) and incubation continued. At day 9, after allowing the replaced media to equilibrate for 2 hours at 37°C in 5% CO2 humidified atmosphere, apical cell surfaces were treated with (+) human WP (10%, v/v). This treatment was repeated after every media replacement throughout the duration of the experiment. For +/− (with or without) WP analysis, cells were harvested on D15 and D22, representing approximately partial or full differentiation to the enterocyte phenotype, respectively, and processed as described below. For time course studies without WP, cells were harvested on D9, 11, 13, 15, 17 and 21 and further processed.
Protein in-solution Tryptic Digestion and Peptide TMT Labeling
At the indicated times, cells were harvested using a PBS-based enzyme-free cell dissociation buffer according to the manufacturer’s instructions, lysed by resuspension in RIPA buffer containing a protease inhibitor cocktail, followed by a freeze-thaw cycle. Protein concentration for the cell lysates was determined by BCA. The cell lysates were then prepared for tandem mass tag (TMT) labeling using the Pierce® TMT six-plex isobaric mass tagging kit (Thermo-Fisher Scientific, Rockford, IL) according to the manufacturer’s instructions with slight modification. Briefly, cell lysates typically containing 60 μg proteins from each sample were dissolved in triethyl ammonium bicarbonate (TEAB), followed by reduction in tris(2-carboxyethyl)phosphine (TCEP) for 1 hour at 55 °C, and subsequent alkylation with iodoacetamide for 1 hour at room temperature. The alkylated proteins were then deglycosylated with PNGase F (1 U/50 μg protein) for 2 hours at 37°C, followed by acetone precipitation at −20 °C overnight. Following tryptic digestion overnight at 37°C, peptides were TMT labeled according to the manufacturer’s instructions. Each TMT 6-plex peptide mixture was pooled, lyophilized and resuspended in 200 μL of 90% ACN/0.1% HOAc for electrostatic repulsion-hydrophilic interaction chromatography (ERLIC) separation.
ERLIC Prefractionation of Tryptic Peptides
Approximately 360 μg tryptic peptides (6-plex with 60 μg from each sample) were injected onto a 4 mm i.d. × 10 mm WAX guard column (PolyWAX LP, 5 μm particle size, 1,000 Å pore size, PolyLC Inc., Columbia, MD) connected to a 2.1 mm i.d. × 200 mm WAX column (PolyWAX LP, 5 μm particle size, 300 Å pore size, PolyLC Inc.). ERLIC peptide separation was carried out via HPLC (U3000, Dionex, Sunnyvale, CA) at a flow rate of 200 μL/min. A gradient was started with 100% A (98% ACN, 0.1% HOAc) for 10 min and ramped to 28% B (30% ACN, 0.1% FA) over 55 min, to 85% B over 33 min, followed by a step gradient to 100% B and then held at 100% B for 10 min. UV absorption was monitored at 280 nm. Thirty fractions with retention times ranging from 20 – 90 min were collected at 2 to 3-min intervals. Each fraction was dried under reduced pressure, reconstituted in 20 μL of 0.1%FA, stored at −80 °C, and thawed at 4 °C when ready for nanocapillary liquid-chromatographic electrospray-ionization tandem mass-spectrometric (LC-ESI/MS/MS) analysis.
LC/MS/MS Analysis of Tryptic Peptides
LC-ESI/MS/MS analysis was conducted with a Q-Exactive mass spectrometer coupled to an EASY-nanoLC 1000 system (Thermo-Fisher Scientific). For each ERLIC fraction, 5 μL of sample was loaded onto a 75 μm i.d. × 2 cm Acclaim® PepMap 100 RP trap column (Thermo-Fisher Scientific). Peptide separations were carried out using an approximately 20-cm-long uncoated 75-μm i.d., 15-μm nanotip fused-silica column (New Objectives, Woburn, MA) packed in-house with 3-μm C18 particles (Bruker-Michrom, Auburn, CA). The separation was started with 98% mobile phase A (0.1% FA) and increased to 40% B (100% ACN, 0.1% FA) over 150 min, followed by a 10-min wash at 60% B, with a flow rate of 300 nL/min. Full-scan mass spectra were acquired by the Orbitrap mass analyzer in the mass-to-charge ratio (m/z) of 300 to 1650 and with a mass resolving power set to 70,000. Ten data-dependent high-energy collisional dissociations (HCD) were performed with a mass resolving power set to 17,500, a fixed first m/z 110, an isolation width of 4 m/z, and the normalized collision energy (NCE) setting of 30 with enabled stepped collision energy of 20% NCE. The maximum injection time was 100 ms for parent-ion analysis and 50 ms for product-ion analysis. Target ions already selected for MS/MS were dynamically excluded for 60 sec. An automatic gain control (AGC) target value of 3e6 ions was used for full MS scans and 1e5 ions for MS/MS scans. Only peptide ions with charge states of two or greater were selected for MS/MS interrogation.
Protein Identification and Quantification in TMT Experiments
MS/MS spectra with charges +2, +3 and +4 were analyzed using Mascot search engine (Matrix Science, London, UK; version 2.3.2). Mascot was set up to search against the human Uniprot/SwissProt database (20,319 entries; version 2011_08) assuming the digestion enzyme was trypsin with a maximum of 1 missed cleavage allowed. The searches were performed with a fragment ion mass tolerance of 0.02 Da and a parent ion tolerance of 10 ppm. Variable and fixed modifications were specified discretely for various TMT peptide searches (Table 1: (1a, 1b, 2a and 2b)). Peptide identities were accepted if they could be established at less than 1% probability of being a random match as specified in Mascot. The ratios of TMT reporter ions were determined with Mascot and used for peptide quantifications at a mass tolerance of 0.01 Da. The intensity values of peptides that can be assigned to a same protein were summed and the protein ratio were calculated from the summed values (weighted average). Dixon’s method was used in outlier removal when the number of peptide ratios is between 4 and 25 whereas Rosner’s method was used when the number of peptide ratios is greater than 25. Peptide identifications that can be assigned to more than one protein were removed from quantification. False-discovery rate (FDR) of peptides was estimated between 0.2 and 0.6% (Table S1A, Supporting Information; Supplemental Dataset 1, 2 and 3) by searching against a decoy database. The identities of proteins with two or more significant decoy peptide matches were considered false. Protein FDR for each data set containing 30 ERLIC fractions was estimated between 0.1 and 0.3% (Table S1A, Supporting Information) using the approximated formula . To estimate the protein FDR considering the data hierarchy of –WP (D9 → D21), +/−WP (D9 → D15), and +/−WP (D9 → D22), each of which contained repeated LC/MS/MS analysis (Figure 3C), the identities of false proteins and SwissProt proteins with two or more significant peptides were each pooled from the individual data sets and the ratio of was used an estimate of the overall protein FDR. The global protein FDR for the 10,179 quantified proteins (Figure 3D) was estimated to be less than 2%.
Table 1.
Selection of fixed and variable modifications in peptide characterization and quantitation.
| Experimental Procedures |
(1a) Protein Identification and Quantification in TMT Experiments |
(1b) Protein Identification and Quantification in TMT Experiments |
(2a) Estimation of TMT Ratio Compression |
(2b) Estimation of TMT Ratio Compression |
(3) Endocytosis Assay Using SILAC |
|---|---|---|---|---|---|
| Purposes or studies | Relative quantitation of 6-plex TMT peptides in studies of i) +/−WP, D9 -> D15 ii) +/−WP, D9 ->D22 iii) –WP, D9 -> D21 iv) WP endocytosis using TMT v) Caco-2 proteins with UPS-2 spiked | Labeling efficiency of TMT 6-plex (the number of peptides without TMT versus the number of peptides with TMT from (1a) | Relative quantitation of regular 6-plex TMT peptides under the presence of Lys-8, Arg-10-containing 6-plex TMT peptides | Relative quantitation of Lys-8, Arg-10-containing 6-plex TMT peptides under the presence of regular 6-plex TMT peptides | Endocytosis of WP by a Caco-2 monolayer |
| Types of peptide sequences for analysis | Regular tryptic peptides with TMT labels | Regular tryptic peptides without TMT labels | Regular tryptic peptides with TMT labels | Lys8, Arg10-containing tryptic peptides with TMT labels | Regular (L) versus Lys8, Arg10-containing (H) tryptic peptides |
| Fixed modifications | Carbamidomethyl (C), TMT6plex (N-term), TMT6plex (K) | Carbamidomethyl (C) | Carbamidomethyl (C), TMT6plex (N-term), TMT6plex (K) | Carbamidomethyl (C), Label:13C(6)15N(4) (R), TMT6plex (N-term), Label:13C(6)15N(2)+TMT6plex (K) | Carbamidomethyl (C) |
| Variable modifications | Acetyl (N-term), Deamidated (NQ), Oxidation (M) | Acetyl (N-term), Deamidated (NQ), Oxidation (M) | Acetyl (N-term), Deamidated (NQ), Oxidation (M) | Acetyl (N-term), Deamidated (NQ), Oxidation (M) | Acetyl (N-term), Deamidated (NQ), Oxidation (M), SILAC K+8 R+10 |
| Quantitation method in Mascot | TMT 6plex | N/A | TMT 6plex | TMT 6plex | L/H protein ratios from extracted ion chromatograms |
Figure 3.
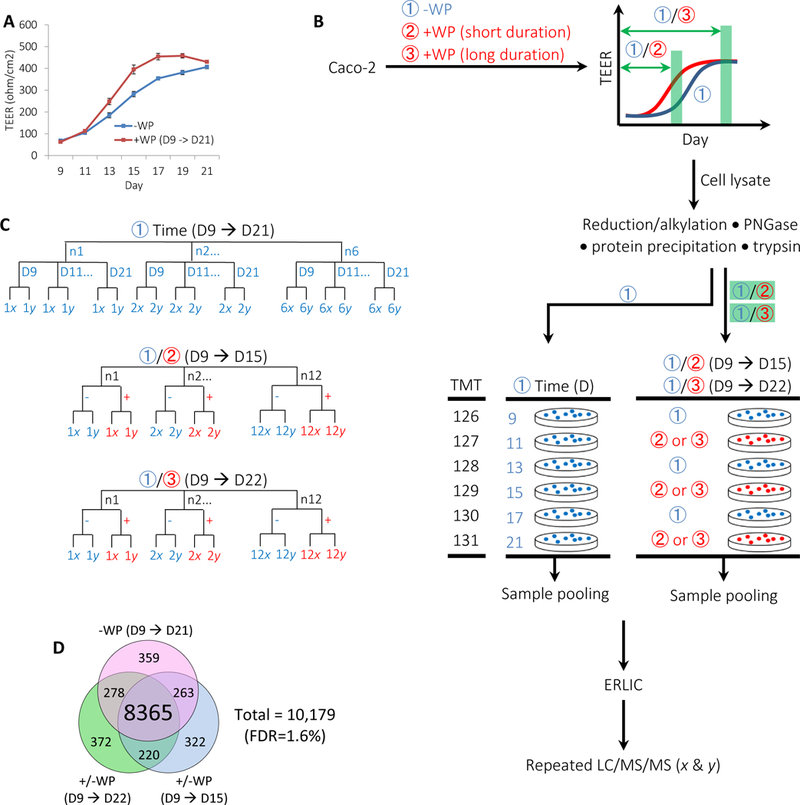
Strategy for comparing the proteome of Caco-2 monolayer with or without human whey protein (WP) treatment over time.
(A) Representative TEER trajectories of differentiating Caco-2 monolayer with (+) or without (−) successive application of WP beginning on D9. Days (D) are post seeding. Error bars represent s.e.m. (B) Schematic comparing Caco-2 proteome across six temporal stages and for two different +/−WP durations. To depict the temporal proteome of Caco-2 monolayer, cultured cells under –WP conditions (①) were harvested on D9, 11, 13, 15, 17 and 21, followed by TMT 6-plex-based proteomic procedure. To assess the effects of WP on the proteome of Caco-2 monolayer over time, cultured cells with successive +WP treatments beginning on D9 were harvested on D15 (②) or D22 (③), each grouped with respect –WP controls and followed by TMT 6-plex-based proteomic procedure. Combined results from the three studies of -WP (D9 → D21), +/−WP (D9 → D15) and +/−WP (D9 → D22) are used to depict the temporal proteome of Caco-2 monolayer with or without WP treatments. (C) Sample hierarchy including both biological replicates (n) and repeated LC/MS/MS analysis (x or y). N = 12 pairs for each of the +/−WP (D9 → D15) and the +/−WP (D9 → D22) analysis. N = 6 at each stage of D9, 11, 13, 15, 17 and 21 for the -WP (D9 → D21) analysis. Repeated LC/MS/MS were performed for each TMT sample set. (D) Venn diagram of proteins quantified from the three indicated studies. The number of proteins covered by each sample group is indicated.
Proteomic Data Analysis
Unless otherwise mentioned, normalization in protein ratios was applied in that the median ratios are log2 zero. Data analysis was performed with the free software environment for statistical computing and graphics, R (http://www.R-project.org). R package, plyr: Tools for Splitting, Applying and Combining Data, was used in data processing. R packages, ggplot2 and ggExtra, were used in data visualization. Gene ontology (GO) analysis was carried out using the Database for Annotation, Visualization and Integrated Discovery (DAVID).37,38 Pathway analysis was searched against Kyoto Encyclopedia of Genes and Genomes (KEGG)41 to assess differential protein expression under various conditions. Genomic suite (Partek Inc., St. Louis, MO, version 6.6) was used for principal components analysis (PCA).
Estimation of TMT Ratio Compression
Dried yeast cells (S. cerevisiae) were autolysed in DPBS (10%, w/v) containing Halt™ protease inhibitor at 30 °C for 2.5 hour, and subsequently pelleted by centrifugation at 12,000 ×g for 10 minutes at 4 °C. Normal L-lysine and L-arginine-containing Caco-2 lysates were prepared at the stoichiometric ratios of 1 : 1.05 : 1.1 : 1.2 : 1.3 : 1.4 (typically at 30 to 42 μg of protein), followed by the additions of equal-amount Lys8, Arg10-containing Caco-2 proteins, with or without the yeast proteins. Protein concentrations were determined using BCA. The aforementioned proteomic procedures were applied to obtain protein TMT ratios.
Endocytosis Assay Using TMT
Caco-2 cells were grown on cell culture inserts in standard experimental medium for 22 days as described in the section Cell Culture for TMT Labeling. For cells with WP treatments, 10% WP (v/v) was applied to the apical surfaces from D9 to D22 (13 days) or for 10 minutes on D22. Cells were then washed three times with cold PBS prior to harvesting with cell dissociation buffer. Four sets of samples, each comparing the condition of -WP (TMT channel 126 and 129), +WP (10 min, TMT channel 128 and 131) and +WP (13 days, TMT channel 127 and 130), were prepared as described in the sections Protein in-solution Tryptic Digestion and Peptide TMT Labeling and ERLIC Prefractionation of Tryptic Peptides, with the peptide fractions being stored at –80 °C until ready for LC-ESI/MS/MS analysis.
Endocytosis Assay Using SILAC
Caco-2 cells were routinely maintained and plated for experiments as described in the section Cell Culture for TMT Labeling, with the following specifications. Cells culture medium was changed to SILAC DMEM and the cells allowed to undergo 5 or more doublings prior to experiment plating in 12-well inserts at 100,000 cells/cm2/600 μL/1500 μL. Cell culture media was replaced as described; however, apical cell surface treatment with (+) human WP (10%, v/v) began on D22.
The cells were harvested on D24 as described in the section Protein in-solution Tryptic Digestion and Peptide TMT Labeling, with modifications to the reduction and alkylation of the proteins as follows. After determination of protein concentration by BCA, cell lysates were dissolved in 100 mM TEAB (final volume 200 uL), reduced in DTT (5 mM final concentration) for 45 min at 55°C, and subsequently alkylated with iodoacetamide (15 mM final concentration) for 45 min at room temperature protected from light. Approximately 360 μg tryptic peptides were prefractionated as described in the section ERLIC Prefractionation of Tryptic Peptides and the resultant peptide fractions stored at –80 °C until ready for LC-ESI/MS/MS analysis.
The SILAC K+8 R+10 method specified in Mascot was used in peptide searches (Table 1: (3)). The peptide identities were filtered using significance threshold of 0.01 and ion score cutoff of 0.01. The protein FDR was estimated to be 3%. Ratios of light L-lysine, L-arginine-containing proteins versus respective heavy Lys8, Arg10-containing proteins (L/H) were determined with Mascot Distiller (Matrix Science; version 2.4.3). Weighted averages of peptides specified in Distiller were chosen in calculating L/H protein ratios at a correlation threshold 0.7 without additional cutoffs using standard error threshold or fraction threshold.
Metabolic Cell Viability/Proliferation Assay
Caco-2 cells were maintained in standard experimental media and plated in 12-well inserts as described in the section Cell Culture for TMT Labeling. From D9 throughout the duration of the experiment, 10% WP (v/v) was applied onto the apical cell surfaces during medium changes. Cell viability/proliferation was assessed using the VisionBlue™ Fluorescence Cell Viability Assay Kit according to the manufacturer’s procedures with modifications. Briefly, on the days indicated, 60 μL of culture media was removed from the apical cell surfaces, replaced with an equal volume of the VisionBlue™ reagent and incubated for 2 hours at 37°C in 5% CO2 humidified atmosphere. Subsequently, three 100 μL aliquots of the apical surface media were transferred to a 96-well plate and the fluorescence (Ex=540 nm, Em=620 nm) determined using a Synergy™ 4 microplate reader (BioTek, Winooski, VT) with the subtraction of appropriate blanks. Aliquots of the basolateral surface media were also assessed to ensure cell confluence and insert membrane integrity. Cultures were washed twice with media post assay and the replacement media allowed to equilibrate for 2 hours prior to a subsequent application of WP.
Glutathione S-transferase (GST) Assay
Caco-2 cells grown on 12-well inserts in standard experimental media for the indicated time periods were washed 3 times with ice-cold DPBS and harvested by scraping into 60 μL cold NP-40 lysis buffer (Thermo-Fisher Scientific). Lysates were vortexed every 10 minutes for 30 minutes and then centrifuged at 16,000 ×g for 30 minutes at 4 °C. Supernatant were collected and protein concentrations determined using BCA. GST activity was determined in 96-well plates using a GST assay kit according to the manufacturer’s instructions. Briefly, 1.5 μg of proteins was reacted with 2-mM L-glutathione and 1-mM 1-chloro-2,4 dinitrobenzene (CDNB) in a total volume of 200 μL. Absorbance was measured at 340 nm kinetically every minute for 15 time points using a Synergy™ 4 microplate reader (BioTek).
Trans-Epithelial Electrical Resistance (TEER) Measurement
For cells grown on inserts, TEER was monitored prior to medium changes throughout the experiment duration using an STX2 electrovoltohmeter (World Precision Instruments, Sarasota, FL), according to the manufacturer’s instructions.
SDS-PAGE and WB Analysis
Caco-2 cells were maintained, plated in standard experimental media and treated with (+) human WP (10%, v/v) at the indicated time points as previously described in the section Cell Culture for TMT Labeling. Cells were harvested on the indicated days and prepared for SDS-PAGE, followed by analysis by immunoblot. Briefly, cells were washed with DPBS, harvested using enzyme-free cell dissociation buffer and centrifuged at 1200 ×g for 10 minutes at 4 °C. Cell pellets were lysed in RIPA buffer containing a protease inhibitor cocktail, with an additional freeze-thaw cycle. Lysate protein concentrations were determined using BCA. Samples were prepared in Laemmli buffer, subjected to SDS-PAGE, immunoblotted and analyzed as previously described.36 Antibodies against lactoferrin (LTF) and glyceraldehyde 3-phosphate dehydrogenase (GAPDH) were from Santa Cruz Biotechnology Inc. (Dallas, TX). GAPDH was used as loading control.
Statistical Analysis
Data from duplicated LC/MS/MS analysis were first averaged and protein abundance ratios were log2 transformed prior to statistical analysis. A 1-way ANOVA with step-up Benjamini-Hochberg (BH)42 correction was performed to assess the statistical significance in protein abundance changes between +/−WP conditions and between each two adjacent time points in -WP temporal studies. Student’s t-test was used to assess significance p-values in the analyses of protein endocytosis, VisionBlue™ assays and GST assays.
RESULTS
TMT-based 2-D LC/MS/MS Quantitation
We selected the human colorectal adenocarcinoma cell line Caco-2 as a testing ground to evaluate the hypothesis that higher resolution of FC measurements can be achieved in proteomic procedures. We investigated a strategy of two-dimensional (2-D) separation of TMT-labeled protein tryptic digest from Caco-2 cell lysates. Several prefractionation methods, such as ERLIC, concatenated ERLIC and concatenated high-pH reversed phase (Hp-RP) chromatography, have been recently reported to afford good orthogonality to second-dimension RP-LC/MS/MS.36,43–46 Here we use ERLIC for the tryptic peptide prefractionation, followed by online RP-LC/MS/MS analysis. It has been previously noted that glycopeptides bind strongly to ERLIC column materials,47 which could explain why heavily glycosylated proteins, such as milk collagens, were poorly characterized with similar ERLIC procedures.36 To potentially improve peptide recovery in ERLIC, proteins were enzymatically deglycosylated with PNGase prior to precipitation and tryptic digestion (EXPERIMENTAL PROCEDURES of Protein in-solution Tryptic Digestion and Peptide TMT Labeling).
TMT Dynamicity at Small Protein Stoichiometric Ratios
One known issue in TMT-based quantification is the underestimation of protein ratios biased towards unity, an observation that was ascribed primarily to the co-isolation and co-fragmentation of constitutively expressed contaminating ions together with target ions.19 It has been well documented that the extent of underestimation decreases from approximately 3.2-fold, 1.3-fold to less than 1.2-fold for theoretical ratio of 10 : 1, 2.5 : 1 and 2 : 1, respectively.21,48,49 Surprisingly, little attention has been paid to assessing such effects at smaller protein ratios. Thus, degrees of TMT ratio compression are not yet known for more subtle abundance differences.
To assess the extent of TMT ratio compression at low FC using our setup, we generated surrogate interference materials of 13C6,15N2-L-lysine (Lys8) and 13C6,15N4-L-arginine (Arg10)-labeled Caco-2 proteins via SILAC.13,26 Tryptic peptides were prepared for both regular and heavy isotope-labeled Caco-2 proteins and yeast proteins according to the proteomic procedure outlined in the EXPERIMENTAL PROCEDURES of Protein in-solution Tryptic Digestion and Peptide TMT Labeling. We then aliquoted regular peptides at stoichiometric (S.) ratios of 1.0 : 1.05 : 1.1 : 1.2 : 1.3 : 1.4, each mixed with equal-level heavy isotope-labeled peptides with or without yeast, followed by TMT labeling, ERLIC prefractionation and LC/MS/MS analysis (Figure 1A). When co-isolation and co-fragmentation occur between regular Caco-2 peptides and heavy Caco-2 peptides with or without yeast peptides, the regular peptides are expected to liberate TMT reporter ions of m/z 126 to 131 at theoretical ratios (Rtheo) of 1 to 1.4 whereas the heavy peptides are anticipated to provide reporter ions to the identical m/z channels at equal intensity (Rtheo = 1) The concurrent contribution of the reporter ions from regular Caco-2 peptides of Rtheo > 1 and interference materials of Rtheo = 1 provided a means to compress the ratios of regular Caco-2 peptides towards unity and to inflate those of heavy Caco-2 peptides above unity.45,46 The quotients of experimental median ratios to respective Rtheo were used to estimate the compression factors (FC) and the inflation factors (Fi) for regular and heavy Caco-2 proteins, respectively (Figure 1B). With the setup, we found that, in the presence of mimicking interference materials, the of regular Caco-2 protein are dynamic in accordance to their stoichiometric ratios with little systemic underestimation indicated by FC at the proximity of one (Figure 1C). We further observed that the increasing presence of regular Caco-2 proteins from stoichiometric ratios 1.0 to 1.4 enhanced accordingly the of equally loaded heavy Caco-2 proteins by factors Fi (Figure 1D).
Figure 1.

Analysis of TMT ratio compression (from labeling to LC/MS/MS detection).
(A) Schematic representation assessing TMT ratio compression. Regular Caco-2, heavy Caco-2 and yeast proteins were each prepared to obtain tryptic peptides. Regular Caco-2 peptides were then loaded at indicated stoichiometric (S.) ratios, whereas heavy Caco-2 peptides with or without yeast were loaded at equal levels, followed by TMT labeling, ERLIC prefractionation and repeated LC/MS/MS analyses. The dot frames indicate sample combinations for each 6-plex TMT channel. For simplicity, the schematic displays the interference containing both heavy Caco-2 and yeast materials. (B) Schematic representation showing the mutual interference in TMT ratios between regular and heavy Caco-2 peptides. The presence of heavy Caco-2 peptides at equal theoretical ratios (R theo) compresses the measurements of regular Caco-2 peptides from 1.05 – 1.4 Rtheo to respective experimental ratios for each TMT channel. Conversely, the presence of regular Caco-2 peptides at Rtheo inflates the ratios of heavy Caco-2 peptides from Rtheo to respective for each TMT channel.48,49 Comparisons of the experimental medians to respective Rtheo were used to calculate the compression factors (FC) and the inflation factors (Fi)for regular and heavy Caco-2 proteins, respectively. (C) Ratio distribution of regular Caco-2 proteins under HSILACi + YEASTi (C1) or HSILACi (C2) conditions. S. ratios of regular Caco-2 proteins are indicated. (D) Respective ratio distribution of equal-level heavy Caco-2 proteins under the presence of regular Caco-2 proteins at indicated S. ratios. The vertical lines in box plots indicate the median and the whisker caps indicate the lower 10th and upper 90th percentiles, respectively. HSILACi, heavy SILAC Caco-2 interference; YEASTi, yeast interference.
We then sampled regular Caco-2 cell lysates at protein stoichiometric ratios of 1.0 : 1.05 : 1.1 : 1.2 : 1.3 : 1.4 and combined each of the six regular Caco-2 protein samples at these ratios with equal-level surrogate interference materials containing heavy isotope-labeled Caco-2 proteins with or without yeast, followed by 6-plex TMT proteomic procedures (Figure 2A). In comparison to the prior analysis of TMT ratio compression where samples were aliquoted immediately before TMT labeling (Figure 1A), this protocol partitioned samples immediately after cell lysates collection. To account for variance in sample preparations, we first normalized the ratios of regular Caco-2 proteins to the median log2 zero of the heavy Caco-2 proteins. We found that the of regular Caco-2 protein were dynamic in accordance to their stoichiometric ratios (Figure 2B). Having learned that the increasing presence of regular Caco-2 proteins enhances the observed of equally loaded heavy Caco-2 proteins (Figure 1D), the mean log2 zero for heavy Caco-2 proteins should result in overestimated ratio compressions for the regular Caco-2 proteins. Alternatively, we referred to the of heavy Caco-2 proteins over the same ratio range (Figure 1D) and found smaller ratio compression for regular Caco-2 proteins (Figure 2C). We further observed moderate ratio divergence from high to low-intensity range (Figure 2D). The above observations suggest that, even at sample size of n = 1, the current TMT-based approach is dynamic over the narrow range of ratio 1 to 1.4 with moderate compressions in the medians of protein ratio.
Figure 2.
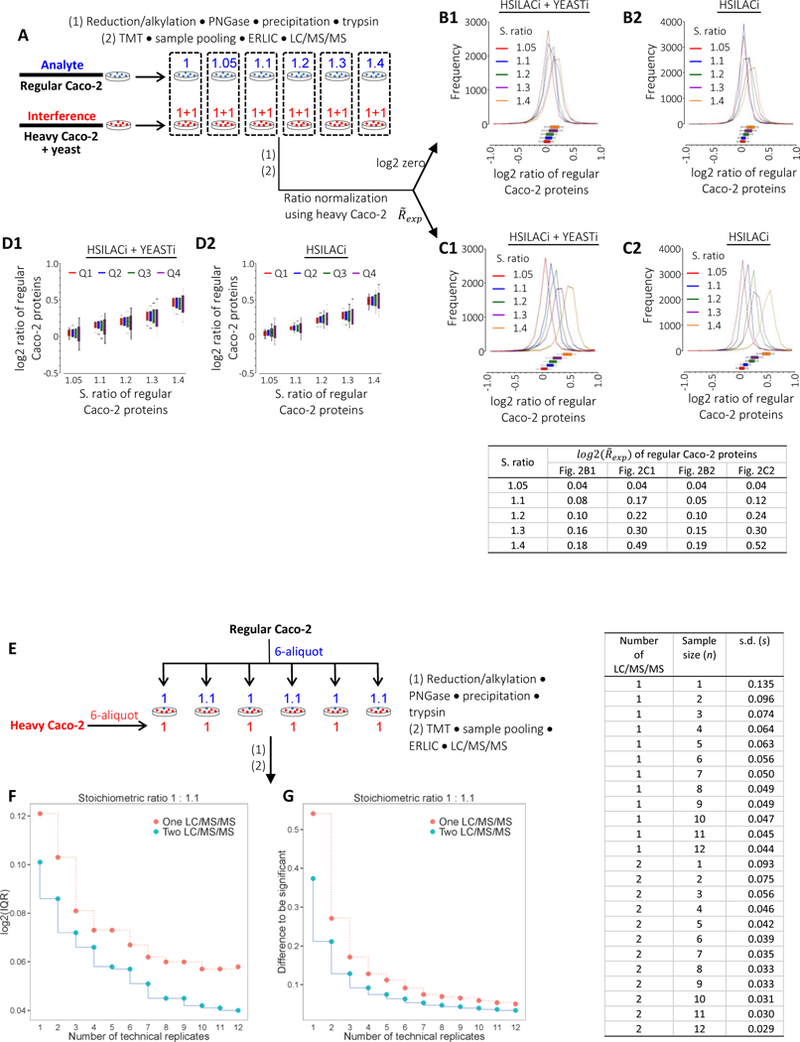
Analysis of TMT ratio compression (from cell lysis to LC/MS/MS detection).
(A) Schematic representation assessing TMT ratio compression. Regular Caco-2 proteins and interfering heavy Caco-2 proteins with or without yeast were mixed at indicated stoichiometric (S.) ratios immediately after cell lysis, followed by proteomic procedures and repeated LC/MS/MS analyses. The dot frames indicate sample combinations for each 6-plex TMT channel. For simplicity, the schematic displays the interference containing both heavy Caco-2 and yeast materials. (B) Ratio distribution of regular Caco-2 proteins under HSILACi + YEASTi (B1) or HSILACi (B2) conditions. S. ratios of regular Caco-2 proteins are indicated. Ratios of regular Caco-2 proteins were normalized to median log2 zero of the equally-loaded heavy Caco-2 proteins. (C) Ratios of regular Caco-2 protein in (B) were normalized to the experimental medians of heavy Caco-2 proteins (Figure 1D), instead of median log2 zero. (D) Ratio distribution of regular Caco-2 proteins with respect to relative protein abundance. Ratios of regular protein in (C) were divided into four quartiles from high to low emPAI. HSILACi, heavy SILAC Caco-2 interference; YEASTi, yeast interference. (E) Schematic representation assessing the precision of TMT ratio with respect to the number of technical replicate under independent sample preparation. Heavy Caco-2 proteins at equal levels were mixed with regular Caco-2 proteins at stoichiometric (S.) ratios of 1 to 1.1 immediately after cell lysis, followed by analogous proteomic procedures shown in Figure 2A. (F) Corresponding IQR of protein ratios at n = 1 – 12 technical replicates with single or duplicated LC/MS/MS. (G) Differences of protein ratios to be significant with respect to the number of technical replicates at significance levels α = 0.05 and β = 0.20.
Technical Variances in Sample Handling and LC/MS/MS Detection
While the above findings in TMT dynamicity provided some assurance in sensing small abundance changes at the levels of cellular proteome, the distribution of measured protein ratios at the sample size of n = 1 remains relatively broad, with a considerable portion of overlapping at various S. ratios. Presumably to some degree, random errors over sample preparation and LC/MS/MS detection contribute to the profile widening of biologically equivalent samples, which could be accommodated, in part, with increasing numbers of analysis. To assess such effects, we analyzed the extent of protein ratio variances with respect to the number of biologically equivalent Caco-2 cell lysates under the condition of independent sample preparation, using a protein S. ratio of 1.0 : 1.1 (Figure 2E). With single LC/MS/MS analysis, we found that the interquartile range (IQR, middle 50%) of protein ratios decreased from log2 0.12 at n = 1 to log2 0.08 at n = 3 and remained relatively constant at the vicinity of log2 0.06 from n = 7 to 12 (Figure 2F). Further improvements in ratio precision were found with duplicated LC/MS/MS analysis where the IQR of protein ratios decreased from log2 0.10 at n = 1 to log2 0.07 at n = 3 and from log2 0.05 at n = 7 to log2 0.04 at n = 10 – 12 (Figure 2F). The above observations suggest that ratio variances during sample handling and LC/MS/MS analysis are controllable via increasing the number of sampling events.
We next estimated the effects of technical sample sizes on the differences of protein ratios, d, being significant. For a two-sample t test (equivalent to one-way ANOVA), the value of test statistics is
| (1) |
and becomes
| (2) |
with equal variances, s2, and equal sample sizes, n, on each of the two groups. We performed power analysis with conventionally used probabilities of false positives at α = 0.05 (Type I error) and false negatives at β = 0.20 (Type II error). The quantile associated with a two-tailed normal distribution at α = 0.05 is 1.96 and the quantile associated with the power of a test at β = 0.20 is 0.84.17 These quantiles were added (t = 2.8) for sample size estimation and the formula for the difference can be calculated as
| (3) |
where Sn denotes s.d. at sample size of n. In the example of repeated LC/MS/MS analysis, the power analysis suggests a technical sample size of n = 4 to differentiate a 10% change in protein abundance at the significance level of α = 0.05 and β = 0.20 (Figure 2G).
Experimental Design Assessing the Effects of Biological Sample Size on Ratio Variance
The above findings in variance control provided additional assurance in sensing small abundance changes in the cellular proteome. Yet, it remains less than practical to perform many labor-intensive and time-consuming technical repeats for a same biological sample in MS-based proteomic analysis. Increasing numbers of biological sample being analyzed often lead to higher confidence levels between measured values and population values. Although less than ideal, one probable means to circumvent the constraint of human labor and instrument time is to allow the technical variances (for example in Figure 2F, at the levels of log2 IQR 0.10 at n = 1 or 0.07 at n = 3 under the conditions of repeated LC/MS/MS analysis) and handle them at the levels of biological replicate, which encompassed the natural variances of biological samples and the technical variances during both sample preparation and LC/MS/MS detection.
To assess the effects of biological sample size on the variance of protein ratios, we measured the temporal proteome of Caco-2 cells with and without the stimulation of human WP. The Caco-2 cell line is a well-established, in vitro model used to study enterocyte differentiation, secretion, digestion and immune response. These cells differentiate spontaneously upon confluence, typically at day (D) 5 post seeding in our lab, exhibiting the phenotype of polarized enterocytes (Figure S1A, Supporting Information). Our previous study of simulated infant stomach showed that a portion of human milk protein is retained or introduced during gastric digestion for release into the intestinal lumen.50 In the current study, we have identified conditions such that 10% WP (v/v) enhances the trans-epithelial electrical resistance (TEER) of differentiating Caco-2 cells (Figure 3A; Figure S1B, S1C and “Choice of Treatment Initiation Time”, Supporting Information). We then compared the Caco-2 proteomes with (+) or without (−) 10% WP (v/v) treatment for two different durations, which are post seeding D9 → D15 and D9 → D22. We further described the cellular proteome over D9 → D21 time course (TC) without WP treatment (Figure 3B). The combination of the two-stage +/−WP comparison and the temporal footprint of Caco-2 proteome may provide a model for comprehensive proteomic studies using a cell line.
A total of 10,179 proteins without the grouping of same-family identities were quantified, combining the results from the +/−WP pairs during both the D9 → D15 and the D9 → D22 periods and the -WP (D9 → D21) samples (Figure 3C, 3D and Supplemental Dataset 4–9), with 8,365 proteins being found in common. Recovery analysis of spiked UPS-2 protein standards suggests that the current proteomic procedure is capable of quantifying proteins over 6 orders of magnitude (Figure S1D and “Proteome Coverage”, Supporting Information) . These observations suggest that the proteomic procedure described herein is capable of protein quantification over a wide range of abundance.
The Effects of Biological Sample Size on Ratio Variance
We first evaluated quantification precision of the current setup by comparing FC variance (ΔFC) between repeated LC/MS/MS analysis of Caco-2 cells with and without the stimulation of WP. For each +/−WP sample pair (sample size n = 1), relatively small variances were found with IQR at the vicinity of log2 0.14 (Figure 4A). One caveat of TMT-like MS quantification is the heterogeneity in data precision, with greater variance existing for data from lower-abundance proteins.51,52 With the current method, we analyzed ΔFC with respect to an exponentially modified protein abundance index (emPAI) based protein relative intensity and found moderate divergence in ΔFC from the high to low-intensity range where a large majority of proteins having |ΔFC| < 0.1 between repeated analysis (Figure 4B). This suggests the general applicability of the proteomic procedure in global protein quantification.
Figure 4.

System-wide characterization of WP-induced abundance changes of Caco-2 proteins.
(A, B) Protein ratio variance between repeated LC/MS/MS analysis. (A) Profiles of protein ratio variance between repeated LC/MS/MS of 12 +/−WP sample pairs. (B) Scatter plot of protein ratio variance between repeated LC/MS/MS with respect to exponentially modified protein abundance index (emPAI) for an exemplary +/−WP sample pair. Proteins were ordered from high to low emPAI and assigned ranks where 1 corresponds to the highest emPAI. Color curves show the fraction of proteins with absolute FC variance (|ΔFC|) > 1.1 for each of the 12 sample pairs. FC, fold change. (C-D) Protein ratio variance with respect to the number of biological replicates. Ratios were from the average of two LC/MS/MS analysis. Ratio variances of +/−WP sample pair were compared between sample sizes of n+3 and n. Red, n = 3; blue, n = 6 and green, n = 9. s(n), sample size of n. (D) Scatter plots of protein ratio variance between s(n+3) and s(n) with respect to emPAI. Color curves show the fraction of proteins with |ΔFC| > 1.05. (E) Standard deviations of protein ratio at n = 9 biological replicates (“Std. Dev. (n=9)”, Supplemental Dataset 4 and 5) with respect to the number of significant peptide sequences for a linked protein; red dots and fitting line, BH p-value < 0.05; blue dots and fitting line, BH p-value ≥ 0.05; top marginal histogram, distribution of the number of unique peptide sequences; right marginal histogram, distribution of standard deviation. (F) Principal component analysis of the quantitative Caco-2 proteomes. Samples were grouped by ellipsoid according to treatment condition and duration. (G) Distribution of protein abundance changes induced by WP treatment over indicated periods. Proteins with positive or negative FCs are up- or down-regulated, respectively, under +WP conditions. Same proteins from the two different treatment durations are connected by solid lines with yellow ones highlighting significant abundance changes. The areas of dots are proportional to the standard deviations in protein abundance changes. The shade delimits −1.05 ≤ FC ≤ +1.05 or BH p-value ≤ 0.05. LTF, lactotransferrin. (H) Cross comparison of protein abundance change with respect to treatment duration. H1: mapping of 888 Caco-2 proteins (BH p < 0.05) from +/−WP (D9 → D15) dataset to +/−WP (D9 → D22) dataset. H2: mapping of 3,609 Caco-2 proteins (BH p < 0.05) from +/−WP (D9 → D22) dataset to +/−WP (D9 → D15) dataset. N = 12 for each group. To facilitate the visualization of small FCs, the color scale is limited to ±log2 1.0 with no distinction in colorimetric representation for greater magnitude of FC. In A and C: the vertical lines in box plots indicate the median and the whisker caps indicate the lower 10th and upper 90th percentiles, respectively.
To take into consideration the effects of sample size, we next assessed ratio variances with respect to the numbers of biological replicate. With duplicated LC/MS/MS, the IQR of ΔFCs decreased from the span of log2 0.049 between 3 and 6 pairs to log2 0.025 between 9 and 12 pairs for the +/−WP (D9 → D15) samples (Figure 4C1). By the same token, the IQR of ΔFCs decreased from the span of log2 0.044 to log2 0.017 for the +/−WP (D9 → D22) samples (Figure 4C2). We then assessed ΔFC with respect to emPAI-based relative protein abundance at different numbers of analysis. We found that, with repeated LC/MS/MS, around 95% of the ΔFCs fell within ±5% between 9 and 12 measures (Figure 4D). This observation suggests that small variances over the range of ±5% (d = 10%) are attainable for approximately 95% of TMT ratios when sampling nine to twelve biological replicates containing the natural variance of cells.
Notably, the earlier analysis of technical variances with repeated LC/MS/MS suggests a sample size of n = 4 to achieve the similar levels of significance at d = 10% (Figure 2G). The necessity of 9 or more biological replicates could be largely ascribed to the additional biological variances along with the technical variances. With approximately equal d = 10% between 4 technical replicates and 12 biological replicates, Equation 3 can be reapplied to form the following relationship
| (4) |
where ST,n and SB+T,n denote technical and technical-plus-biological s.d., respectively, at the sample size of n. Applying the earlier determinations in the technical s.d. (0.046 at n = 4 with two LC/MS/MS, Table related to Figure 2E–2G), the s.d. of protein ratios at n = 12 biological replicates can be estimated to be SB+T,9. = 0.087 We then compared the estimated to the experimentally determined s.d. of protein ratios at n = 12 (Supplemental Dataset 4 and 5). In line with the power analysis, half of the experimental s.d. are within 0.088 and 0.070 for +/−WP (D9 → D15) and +/−WP (D9 → D22) samples, respectively, with further reductions in value for proteins with significant abundance changes (Figure 4E1–4E2). Having learned the technical s.d. at n =12 (approximately ST,12 = 0.029 two LC/MS/MS, Table related to Figure 2E–2G) and , the s.d. contributed from the natural variances of biological specimens, SB,12, is approximately 0.082, which is noticeably greater than the technical s.d. under the same sample size with repeated LC/MS/MS.
A total of 1,492 and 1,296 proteins were quantified based on one significant peptide sequence for the +/−WP (D9 → D15) and the +/−WP (D9 → D22) samples, respectively (Figure 4E1–4E2, top marginal histograms). Among them, 837 and 701 proteins were quantitated based on one significant MS/MS match in the +/−WP (D9 → D15) and the +/−WP (D9 → D22) pairs, respectively (Supplemental Dataset 4 and 5). Greater variances in s.d. were generally observed for proteins with fewer significant peptide sequences being quantified (Figure 4E1–4E2). The observations are consistent with the conventional notion that fewer hits of tryptic peptide in protein quantification lead to lower accuracies in protein quantitation in bottom-up proteomics. PCA analysis showed increased separation of the fields from +WP (D9 → D15) to +WP (D9 → D22) to their respective -WP controls (Fig. 4F). Further comparison of the Caco-2 proteomes between the two treatment durations of D9 → D15 and D9 → D22 revealed broadly consistent changes of protein abundance with increasing magnitude of FC respective to longer treatment duration (as indicated by the connecting lines in Figure 4G). Between the two treatment periods, there are only two proteins whose expressions switch from significant upregulation to significant downregulation (|FC| > 1.05 and BH p-value < 0.05), or vice versa (indicated by the dark connecting lines in Figure 4G). We mapped the 1,201 proteins that were found to have significant abundance change (BH p-value < 0.05) over the treatment period of D9 → D15 to their respective identities over the period of D9 → D22. Heat map analysis showed that the patterns of protein regulation are broadly similar between the two treatment periods (Figure 4H1). The same is true when mapping the 2,836 proteins with significant abundance change (BH p-value < 0.05) in D9 → D22 to those in D9 → D15 (Figure 4H2). Having learned the consistency in WP-induced protein abundance changes, together with earlier demonstration of quantitation precision, we found it tempting to move the FC cutoff to ±1.05 for the +/−WP pairs over both treatment periods of D9 → D15 and D9 → D22 (vertical shade in Figure 4G).
Combining the above observations in the technical variances of sample preparation plus LC/MS/MS detection (Figure 2) and biological plus technical variances (Figure 4), we found it appealing to speculate that MS-based proteomics is capable of discriminating abundance changes beyond ±5% in complex sample types, such as cell lysates, when analyzing approximately 9 or more biological sample pairs with repeated LC/MS/MS analysis. This incentive would allow us to take advantage of quantitative proteomics for sensitive distinction of small abundance changes across different biological states.
Whey Proteins Internalization by Caco-2 Cells
To show the capability of the above proteomic procedure, we analyzed how WP modulates over time the expression of Caco-2 proteins in various biological processes and signaling pathways. We observed that lactotransferrin (LTF), a milk protein with putative bactericidal activities, was present in greater quantities upon WP treatment during both D9 → D15 and D9 → D22 periods (Figure 4G). Consistent with the low levels of LTF transcript and protein expression in human intestinal tissues (Human Protein Atlas53), we detected only trace amounts of LTF in untreated enterocyte-like or colonic adenocarcinoma-like Caco-2 cells. Although the higher levels of LTF under +WP condition are likely due to its uptake,54 the small FCs (< 4) from TMT measurements remain less than ideal given the negligible LTF production by Caco-2 cells. To gain a more detailed view of WP endocytosis by Caco-2 cells, we adapted a method of stable isotope labeling by amino acids in cell culture (SILAC)13,26 to differentiate between WP uptake and protein de novo synthesis (Figure 5A, left panel). Caco-2 cells, metabolically incorporated with 13C6,15N2-L-lysine and 13C6,15N4-L-arginine, were treated with WP and the ratio of light 12C,14N-containing peptides to their respective heavy 13C,15N-containing counterparts was measured. The SILAC quantification compares the light-to-heavy (L/H) parent-ion intensities of lysine/arginine-containing peptide pairs and does not have the same ratio compression issue found in the TMT approach. Analysis of LTF showed an L/H ratio average of 380 (Supplemental Dataset 10–11 and Figure 5A, right panel), representing an approximate 100-fold increase as opposed to the TMT ratios. Antimicrobial immunoglobulin A1 (IGHA1) is another human milk protein whose uptake by IEC is not yet known. Akin to LTF, the measured FC of IGHA1 increased from around 2.5 in TMT to an average of 1,261 in SILAC (Figure 5A, right panel). This suggests the protein’s primary source in Caco-2 cells is due to uptake from WP. The near exclusive presence of the light-forms of LTF and IGHA1 highlights the extended application of SILAC in assessing endocytosed proteins from a complex source, such as WP. Previous studies suggest that endocytosed cow’s milk IgG contributes to adaptive immunity.55 A systemic description of WP internalization by IEC would provide new perspectives in assessing dietary protein uptake. To assess the possibility that higher quantities of LTF present in +WP cells may be derived from non-specific protein binding, we evaluated the effects of WP treatment duration on the levels of LTF detected in Caco-2 cell lysates (Figure 5B, left panel). We found significantly higher LTF presence upon long-term WP exposure as opposed to lower LTF presence in -WP control and short-term +WP treatment (Figure 5B, right panel; Figure 5C). Analogous TMT observation can be made for IGHA1 (Figure 5B, right panel). Taken together, it can be postulated that protein endocytosis have contributed primarily to the LTF and IGHA1 found in the WP-treated Caco-2 cells.
Figure 5.

Analysis of WP endocytosis.
(A) 10% WP (light) was applied to Caco-2 cells with 13C6,15N2-L-lysine and 13C6,15N4-L-arginine-labeled proteins (heavy). Left panel: schematic of the SILAC procedure measuring light-to-heavy (L/H) protein ratios. Right panel: L/H protein ratios obtained from the SILAC setup. LTF, lactotransferrin; IGHA1, immunoglobulin heavy chain A1. (B) Caco-2 cells were cultured in standard medium for 22 days without WP (−) or with WP (+) over D9 → D22 period or on D22 for 10 minutes. Left panel, schematic of the TMT procedure assessing protein non-specific bindings. Right panel: corresponding TMT protein ratios of LTF and IGHA1 relative to -WP control. (C) Immunoblot analysis of LTF under -WP or +WP conditions over D9 → D15 or D9 → D22 periods. For samples with +WP treatment for 10 minutes, WP was applied on indicated end days. For samples with longer treatment durations, WP was applied from D9 to indicated end days.
Whey Proteins Regulate a Variety of Signaling Pathways in Differentiating Caco-2 Cells
Many additional insights can be envisioned with the advent in the ability to quantify smaller abundance differences in cellular proteomes, of which a few are outlined here. WP components have been thought to promote the maturation of GI tract in early infancy.32 Presumably, regulation of the signaling pathways for proliferation and differentiation in IECs are among the indicators of GI tract development. We first analyzed the -WP temporal Caco-2 proteomes in cell cycle, DNA replication and base/nucleotide excision repair pathways. We found that a great number of cell cycle proteins with significant abundance difference (SAD; BH p < 0.05 and |FC| < 1.05) were downregulated from D9 to D11, and subsequently constitutively expressed from D11 to the experimental end point (Figure 6A1). Examples include cyclin-dependent kinase 6 and 1 (CDK6, CDK1) which are key regulators of G1 progression and G2/M transition, respectively. This was also true for proteins involved in S-phase DNA replication and in the safeguarding processes of base/nucleotide excision repairs (Figure 6B1 and 6C1). These findings imply more resting cells after D11, consistent with the conventional notion that the proliferation of cultured cells transitions from a log phase to a stationary phase after confluence. Notwithstanding, we noted several proteins in these pathways were measured to be significantly upregulated from D9 to D11 (such as SMAD3 and CCND3 in Figure 6A1, POLD4 in Figure 6A2 and PARP4 in Figure 6C1). Given the relatively small sample size of n = 6 in the time course study, it may be plausible to move the FC cutoffs from ±1.05 to ±1.1 to improve the consistency in the distributions of protein regulation. The choice of a higher FC threshold to substantiate the regulation status of these signaling pathways is in line with the notion of reduced power in discriminating protein abundance changes when analyzing fewer samples. We further noted that, from D9 to D11 in the pathways of cell cycle and DNA replication, the significantly upregulated proteins were quantitated based on relatively small numbers of significant peptide sequence. An additional criteria with lower weights given to proteins with fewer numbers of quantified, significant peptide sequence may aid further the confidence levels in proteomic quantitation and in analysis such as enrichment analysis using gene sets (GSEA).39,40 The magnitude of protein abundance changes with WP stimulation is noticeably smaller compared to the temporal differences from D9 to D11 without WP (indicated by the widths of the vertical shades delimiting FC ±1.05 in Figure 6A–6F). CDK1, CDK6 and several other cell-cycle proteins were upregulated upon WP treatments from D9 to D15 (Figure 6A2). Between the two treatment periods, D9 → D15 and D9 → D22, the longer treatment time led to an increased magnitude of FC for a vast majority of the SAD members (indicated by the connecting lines in Figure 6A2). S-phase kinase-associated protein (SKP1 and SKP2) and 14–3-3 protein sigma (SFN) were indicated in mediating the ubiquitination of erroneously synthesized proteins in cell cycle progression, presumably for subsequent proteolysis.56 All three of these proteins were largely constitutively expressed with shorter WP treatment time from D9 to D15 and became downregulated with extended WP treatment time from D9 to D22 (Figure 6A2). Together with the observations of upregulated CDK1, CDK6 and several others under +WP conditions, it may be postulated that successive WP treatments confer a mechanism to promote cell cycle at no or little cost of increasing incidences in erroneous protein synthesis.
Figure 6.
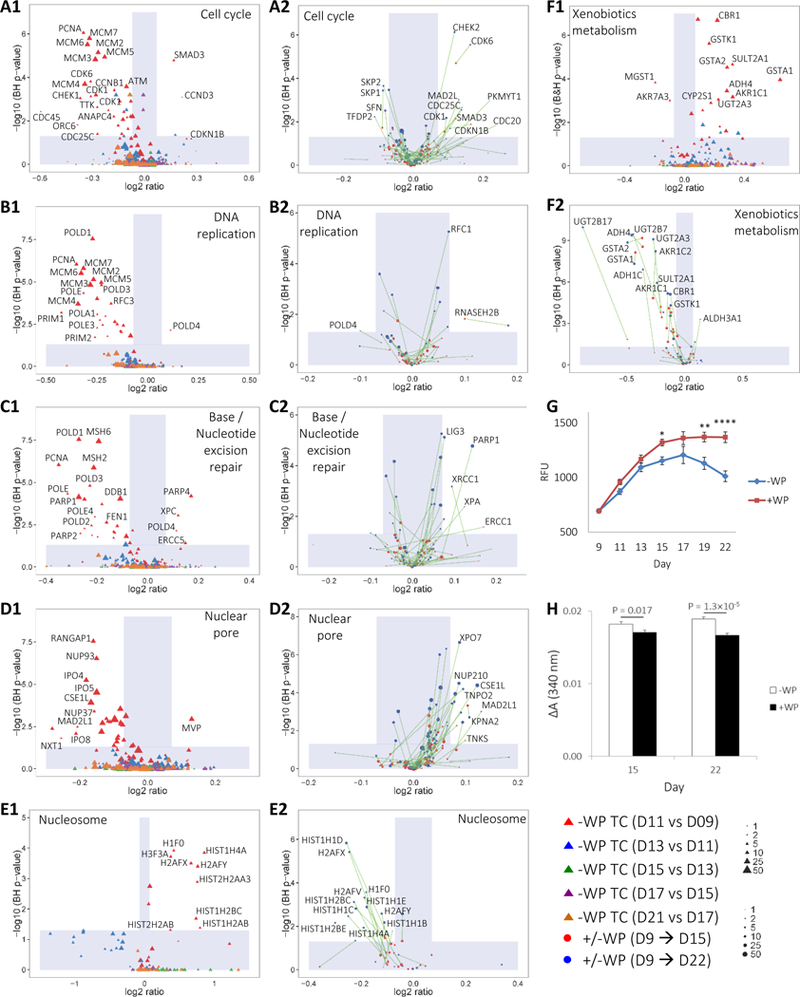
Regulation of Caco-2 pathways upon WP treatment.
(A1-F1) FC of Caco-2 proteins in KEGG pathways of (A1) cell cycle, (B1) DNA replication, (C1) base/nucleotide excision repair, (D1) nuclear pore, (E1) nucleasome and (F1) xenobiotics metabolism over TC without WP treatment. Proteins with positive or negative FCs are up or downregulated, respectively, in later days. The shade defines |FC| < 1.05 or Benjamini-Hochberg (BH) p-value < 0.05. (A2-F2) Parallel pathway analysis for +/−WP samples over D9 → D15 and D9 → D22 durations. Same proteins from the two different treatment durations are connected by solid lines. Proteins with positive or negative FCs are up- or down-regulated, respectively, under +WP conditions. The areas of dots are proportional to the number of significant peptide sequences that can be assigned to a protein. To facilitate the visualization of proteins with small number of significant sequences, the scale is limited from 1 to 50 with no distinction of greater values. The shade defines |FC| < 1.05 or BH p-value < 0.05. (G) Viability of +/−WP samples over D9 → D22 period. Values were normalized to D9 RFU. N = 12 – 36. * p < 0.05, ** p < 0.005, **** p < 0.00005. (H) GST activity of +/−WP samples over D9 → D15 and D9 → D22 periods. N = 12 – 18. Absorbance, 340 nm; ΔA, 0–6min.
Upon shorter WP treatment time of D9 → D15, pathway analysis showed largely confined protein expression between +/−WP pairs with nearly all |FC| < 1.05 in DNA replication and repair (Figure 6B2–6C2). Analogous to cell cycle, the longer treatment time of D9 → D22 tends to increase the magnitude of FC for both up and downregulated proteins in DNA replication and repair. Several pathway members of DNA repair were significantly upregulated upon extended WP stimulation from D9 to D22 (Figure 6C2). Examples include poly [ADP-ribose] polymerase 1 (PARP1), DNA repair protein XRCC1 (XRCC1) and complementing XP-A cells (XPA), which play an important role in defective DNA repair.56 The upregulation of these proteins implies increased needs in safeguarding error-prone DNA replication during cell proliferation. We performed a validating viability/proliferation assay and found significant enhancement in cell viability upon WP treatments compared to –WP control over both D9 → D15 and D9 → D22 periods (Figure 6G), supporting the proteomic prediction.
Additional indicators of cell proliferation or viability could include the regulation of proteins with cellular locations of nucleus organelles, such as nuclear pore, and nucleasome. We found that nearly all SAD members of nuclear pore were downregulated from D9 to D11, and largely constitutively expressed from D11 to the experimental end point under -WP conditions (Figure 6D1). Conversely, WP treatments upregulated all SAD members of nuclear pore with broadly progressive increases of FC from short to long treatment durations (Figure 6D2). Unexpectedly, SAD members of nucleasome (nucleasomal histones in this case) were upregulated temporally from D9 to D11; on the contrary, WP treatments downregulated all SAD nucleasomal histones (Figure 6E1–6E2). Nucleosomal histones play a primary role in wrapping chromatin. The downregulation of nucleosomal proteins upon WP stimulation, especially over the D9 → D22 period, infer a less compact chromatin, which is consistent with the notion of increasing DNA site exposure during replication. The above observations implies that WP continues to promote proliferation and various intracellular processes in differentiating Caco-2 cells.
One important feature conferred by living cells is their ability to detoxify xenobiotics.57 Previous analysis showed progressively increasing expression58 and activity59 of the detoxification enzyme, glutathione transferase (GST), during Caco-2 cell growth. This upregulation points to the likelihood of increasing cellular responses to environmental toxins over time. In this study, we found a nearly universal upregulation of SAD members involved in xenobiotics metabolism from D9 to D11 (Figure 6F1). Unexpectedly, WP was found to downregulate a vast majority of SAD protein expressions in this pathway over both D9 → D15 and D9 → D22 durations (Figure 6F2). Validating GST assays showed lower enzymatic activities in +WP samples for both treatment durations (Figure 6H). These findings indicate that WP provides a mechanism to mitigate cellular toxicity.
Whey Proteins Promote Functional Maturation of Differentiating Caco-2 Cells
A key aspect of developing enterocytes is the maturation of cellular function, including digestion, transportation, secretion, barrier function and immune response. We found that during both time intervals (D9 → D15 and D9 → D22) WP broadly upregulated the expression of ATP-binding cassette (ABC) transporters (Figure 7A), which play a primary role in the influx/efflux of small molecules across plasma membranes. While proteasomes often hydrolyze aberrant endogenous proteins, exogenous proteins with pathogenic origins are frequently proteolyzed through lysosomes. We found that WP stimulation has relatively little effects on the regulation of proteasomal proteins in Caco-2 cells (Figure 7B). In contrast, WP increasingly downregulated over time nearly all SAD members in lysosome pathway (Figure 7C). These findings indicate a defense property conferred by WP, in line with the protein downregulation in xenobiotics metabolism (Figure 6F2). Communication between basolateral surface adhesion receptors and extracellular matrix (ECM) proteins plays a pivotal role in cell differentiation, proliferation and migration. We analyzed proteins that are well-known to interact with or otherwise be secreted into the ECM. We noted that CD44 was increasingly upregulated upon longer WP treatment (Figure 7D). Its upregulation suggests that, apart from the apical interactions (i.e. LTF endocytosis), WP is capable of inducing far-reaching effects through basolateral interactions. This observation also raised the possibility that WP provides a means to aid important immune function such as lymphocyte activation through CD44. Fibronectin (FN1) and laminins (LAMB1, -B2, -B3 and -C2) are commonly destined for the ECM. This increased production of FN1 and LAMs upon WP treatment (Figure 7D) implies important WP function in developing the ECM underneath IECs. SAD members in fatty-acid (FA) degradation and metabolism were all downregulated upon WP treatment over the D9 → D15 period (Figure 7E and 7F). Interestingly, a number of the pathway members were differently regulated from constitutive expression (|FC| < 1.05) over the D9 → D15 period to upregulation over the D9 → D22 period between +WP and –WP. Similar observations can be made for peroxisome proteins and PPAR members which are known to play a role in FA oxidation and cell proliferation (Figure 7G and 7H), and glycolysis/gluconeogenesis and oxidative phosphorylation in energy metabolism (Figure 7I and 7J). The temporal diversity in the protein expressions highlights the intricate effects of WP on the FA and carbohydrate metabolism of differentiating Caco-2 cells. A comprehensive lipidomic and metabolomic analysis may distinguish the underlying mechanism of the protein regulations.
Figure 7.
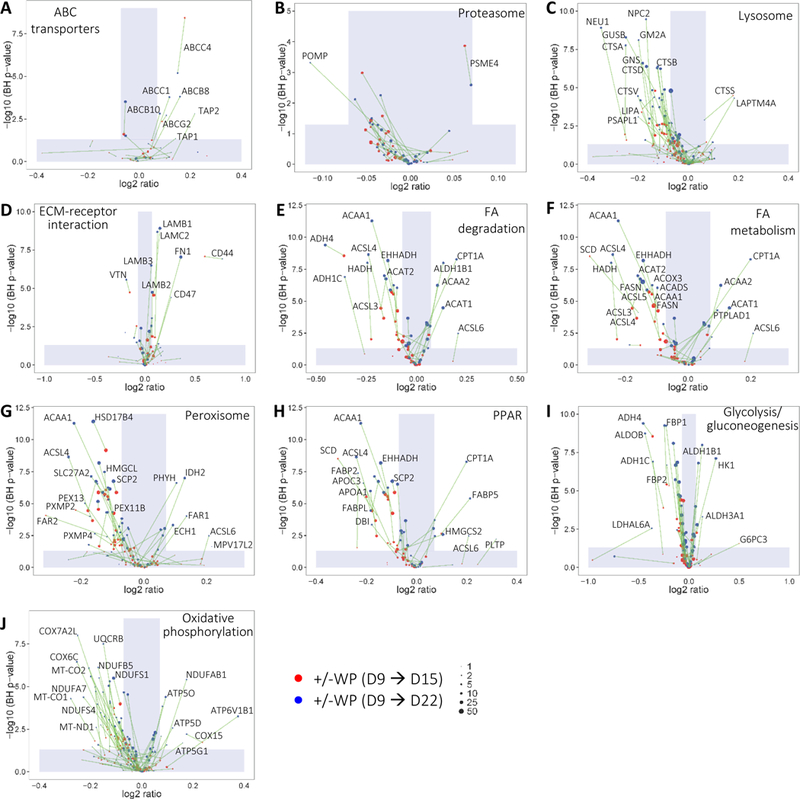
Abundance changes of Caco-2 protein in KEGG pathways depicting various apical, basolateral or paracellular processes.
Cells with (+) or without (–) WP treatments were compared over D9 → D15 and D9 → D22 periods. Same proteins from the two different treatment durations are connected by solid lines. Proteins with positive or negative FCs are up- or down-regulated, respectively, under +WP conditions. (A) ABC transporters. (B) Proteasome. (C) Lysosome. (D) ECM-receptor interaction. (E) Fatty acid degradation. (F) Fatty acid metabolism. (G) Peroxisome. (H) PPAR. (I) Glycolysis/gluconeogenesis. (J) Oxidative phosphorylation. The shade defines |FC| < 1.05 or BH p-value < 0.05. ABC, ATP-binding cassette; ECM, extracellular matrix.
Discussion
In-depth characterizations of cellular proteomes have recently been achieved with representative identification of 10,000 proteins in single human cell lines.60,61 However, their quantification has been achieved to a lesser extent7 and more scarce is the ability to measure small changes in protein abundance. Several issues remain in quantitative proteomics, including inadequate sensitivity in detecting smaller abundance changes and reduced precision in scrutinizing lower abundance proteins. In this report, we have described the feasibility of differentiating subtle changes in abundance with moderate decreases in precision for lower abundance proteins (Figures 1 and 4).
Despite numerous studies of human milk, our knowledge of its benefits remains far from perfect. The current proteomic analysis suggests a plethora of Caco-2 cellular processes that can be influenced by human whey proteins (Figures 6–7). This increased understanding can be largely attributed to the improved sensitivity described in this work. In addition to the various regulatory effects, the internalization of certain whey proteins, such as IgA, can be inferred (Figure 5). It has been speculated that milk IgA provides a means for mothers to confer immunity to their offspring.63 Our previous analysis showed that a portion of milk IgA can survive passage through the stomach.50 This additional line of evidence in protein internalization supports the hypothesis that milk IgA contributes to immunities in young infants.
Similar to many other in vitro models, the Caco-2 monolayer can only represent partially the intestinal epithelium in humans. For instances, the mucin barrier on the apical side of intestinal epithelium and interactions with immune cells underneath intestinal epithelium are essentially absent in this model. Moreover, the extent of the adenocarcinoma characteristics of Caco-2 cells during differentiation has not yet been fully determined at the molecular levels. Caution needs to be paid to avoid over-interpretation when extrapolating the results of in vitro work to the biology of living species. Animal and clinical studies are the important next steps to confirm or reject the observations from the in vitro simulations.
In summary, an MS-based quantitative proteomic procedure combining protein deglycosylation, TMT labeling and ERLIC prefractionation has been shown to allow finer distinction of protein abundance changes than were previously illustrated at a system-wide scale. This demonstration may encourage more investigations of proteomic procedures towards sensing small abundance changes in various biological entities. Using this proteomic procedure described in this work, we tested the progressive effects of human WP on enterocyte-like Caco-2 cell development over time. Broadly, we found that WP increasingly upregulated the expressions of Caco-2 proteins in various proliferation and differentiation processes, including cell cycle, basolateral interactions, immunity and small-molecule influx/efflux. Conversely, WP downregulated the expression of Caco-2 proteins involved in the pathway of lysosomal degradation and xenobiotics metabolism. Together with a validating SILAC study, a route for WP internalization by Caco-2 cells can be postulated. Similar arrangement may permit broader studies of this kind in the future, such as dietary protein uptake by IEC. Our findings indicate that WP exerts important function in many aspects of enterocyte development. This study improves our understanding of the multiple benefits conferred by human WP. Moreover, it showcases the MS-based proteomic application in ascertaining modest abundance changes across sample types, a central task in quantitative studies.
Supplementary Material
ACKNOWLEDGMENT
Q.Z. thanks A. J. Alpert at PolyLC for critical reading and discussion and anonymous reviewers for suggestions and critical comments. The authors thank J. Sanders for supporting the MS facility and R. Waworuntu for discussion in the early stage of research.
ABBREVIATIONS
- D
day
- emPAI
exponentially modified protein abundance index
- ERLIC
electrostatic repulsion hydrophilic interaction chromatography
- ESI
electrospray ionization
- FC
fold change
- FDR
false discovery rate
- GI tract
gastrointestinal tract
- GO
gene ontology
- HCD
high-energy collisional dissociation
- IEC
intestinal epithelial cell
- IQR
interquartile range
- KEGG
Kyoto Encyclopedia of Genes and Genomes
- LC-MS/MS
liquid-chromatographic tandem mass-spectrometry
- MW
molecular weight
- PCA
principal component analysis
- RP-LC/MS/MS
reversed-phase liquid chromatographic tandem mass spectrometry
- s.e.m.
standard error of the mean
- s.d.
standard deviation
- SILAC
stable isotope labeling by amino acids in cell culture
- TEER
trans-epithelial electrical resistance
- TMT
tandem mass tag
- UPS-2
universal protein standard 2
- WB
Western blot
- WP
whey protein (10%, v/v)
Footnotes
Conflict of Interest Statement
The authors contributed to this work during their employment of Mead Johnson Nutrition.
REFERENCES
- (1).Sharma K;D’Souza RC; Tyanova S;Schaab C;Wisniewski JR; Cox J;Mann M. Ultradeep human phosphoproteome reveals a distinct regulatory nature of tyr and ser/thr-based signaling. Cell reports 2014, 8, 1583–1594. [DOI] [PubMed] [Google Scholar]
- (2).Kim MS; Pinto SM; Getnet D;Nirujogi RS; Manda SS; Chaerkady R;Madugundu AK; Kelkar DS; Isserlin R;Jain S;Thomas JK; Muthusamy B;Leal-Rojas P;Kumar P;Sahasrabuddhe NA; Balakrishnan L;Advani J;George B;Renuse S;Selvan LD; Patil AH; Nanjappa V;Radhakrishnan A;Prasad S;Subbannayya T;Raju R;Kumar M;Sreenivasamurthy SK; Marimuthu A;Sathe GJ; Chavan S;Datta KK; Subbannayya Y;Sahu A;Yelamanchi SD; Jayaram S;Rajagopalan P;Sharma J;Murthy KR; Syed N;Goel R;Khan AA; Ahmad S;Dey G;Mudgal K;Chatterjee A;Huang TC; Zhong J;Wu X;Shaw PG; Freed D;Zahari MS; Mukherjee KK; Shankar S;Mahadevan A;Lam H;Mitchell CJ; Shankar SK; Satishchandra P;Schroeder JT; Sirdeshmukh R;Maitra A;Leach SD; Drake CG; Halushka MK; Prasad TS; Hruban RH; Kerr CL; Bader GD; Iacobuzio-Donahue CA; Gowda H;Pandey A. A draft map of the human proteome. Nature 2014, 509, 575–581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Wilhelm M;Schlegl J;Hahne H;Moghaddas Gholami A;Lieberenz M;Savitski MM; Ziegler E;Butzmann L;Gessulat S;Marx H;Mathieson T;Lemeer S;Schnatbaum K;Reimer U;Wenschuh H;Mollenhauer M;Slotta-Huspenina J;Boese JH; Bantscheff M;Gerstmair A;Faerber F;Kuster B. Mass-spectrometry-based draft of the human proteome. Nature 2014, 509, 582–587. [DOI] [PubMed] [Google Scholar]
- (4).Picotti P;Clement-Ziza M;Lam H;Campbell DS; Schmidt A;Deutsch EW; Rost H;Sun Z;Rinner O;Reiter L;Shen Q;Michaelson JJ; Frei A;Alberti S;Kusebauch U;Wollscheid B;Moritz RL; Beyer A;Aebersold R. A complete mass-spectrometric map of the yeast proteome applied to quantitative trait analysis. Nature 2013, 494, 266–270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).de Godoy LM; Olsen JV; Cox J;Nielsen ML; Hubner NC; Frohlich F;Walther TC; Mann M. Comprehensive mass-spectrometry-based proteome quantification of haploid versus diploid yeast. Nature 2008, 455, 1251–1254. [DOI] [PubMed] [Google Scholar]
- (6).Marguerat S;Schmidt A;Codlin S;Chen W;Aebersold R;Bahler J. Quantitative analysis of fission yeast transcriptomes and proteomes in proliferating and quiescent cells. Cell 2012, 151, 671–683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Weekes MP; Tomasec P;Huttlin EL; Fielding CA; Nusinow D;Stanton RJ; Wang EC; Aicheler R;Murrell I;Wilkinson GW; Lehner PJ; Gygi SP Quantitative temporal viromics: an approach to investigate host-pathogen interaction. Cell 2014, 157, 1460–1472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Cox J;Hein MY; Luber CA; Paron I;Nagaraj N;Mann M. Accurate proteome-wide label-free quantification by delayed normalization and maximal peptide ratio extraction, termed MaxLFQ. Mol. Cell. Proteomics 2014, 13, 2513–2526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Mueller LN; Brusniak MY; Mani DR; Aebersold R. An assessment of software solutions for the analysis of mass spectrometry based quantitative proteomics data. J. Proteome Res. 2008, 7, 51–61. [DOI] [PubMed] [Google Scholar]
- (10).Pan S;Aebersold R;Chen R;Rush J;Goodlett DR; McIntosh MW; Zhang J;Brentnall TA Mass spectrometry based targeted protein quantification: methods and applications. J. Proteome Res. 2009, 8, 787–797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Bantscheff M;Lemeer S;Savitski MM; Kuster B. Quantitative mass spectrometry in proteomics: critical review update from 2007 to the present. Anal. Bioanal. Chem. 2012, 404, 939–965. [DOI] [PubMed] [Google Scholar]
- (12).Rauniyar N;Yates JR 3rd Isobaric labeling-based relative quantification in shotgun proteomics. J Proteome Res 2014, 13, 5293–5309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Ong SE; Blagoev B;Kratchmarova I;Kristensen DB; Steen H;Pandey A;Mann M. Stable isotope labeling by amino acids in cell culture, SILAC, as a simple and accurate approach to expression proteomics. Mol. Cell. Proteomics 2002, 1, 376–386. [DOI] [PubMed] [Google Scholar]
- (14).Ross PL; Huang YN; Marchese JN; Williamson B;Parker K;Hattan S;Khainovski N;Pillai S;Dey S;Daniels S;Purkayastha S;Juhasz P;Martin S;Bartlet-Jones M;He F;Jacobson A;Pappin DJ Multiplexed protein quantitation in Saccharomyces cerevisiae using amine-reactive isobaric tagging reagents. Mol. Cell. Proteomics 2004, 3, 1154–1169. [DOI] [PubMed] [Google Scholar]
- (15).Thompson A;Schafer J;Kuhn K;Kienle S;Schwarz J;Schmidt G;Neumann T;Johnstone R;Mohammed AK; Hamon C. Tandem mass tags: a novel quantification strategy for comparative analysis of complex protein mixtures by MS/MS. Anal Chem 2003, 75, 1895–1904. [DOI] [PubMed] [Google Scholar]
- (16).Dayon L;Hainard A;Licker V;Turck N;Kuhn K;Hochstrasser DF; Burkhard PR; Sanchez JC Relative quantification of proteins in human cerebrospinal fluids by MS/MS using 6-plex isobaric tags. Anal. Chem. 2008, 80, 2921–2931. [DOI] [PubMed] [Google Scholar]
- (17).Crawley MJ In The R Book; Wiley: 2013, p 382–385. [Google Scholar]
- (18).Grossmann J;Roschitzki B;Panse C;Fortes C;Barkow-Oesterreicher S;Rutishauser D;Schlapbach R. Implementation and evaluation of relative and absolute quantification in shotgun proteomics with label-free methods. J Proteomics 2010, 73, 1740–1746. [DOI] [PubMed] [Google Scholar]
- (19).Karp NA; Huber W;Sadowski PG; Charles PD; Hester SV; Lilley KS Addressing accuracy and precision issues in iTRAQ quantitation. Mol. Cell. Proteomics 2010, 9, 1885–1897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Savitski MM; Mathieson T;Zinn N;Sweetman G;Doce C;Becher I;Pachl F;Kuster B;Bantscheff M. Measuring and managing ratio compression for accurate iTRAQ/TMT quantification. J. Proteome Res. 2013, 12, 3586–3598. [DOI] [PubMed] [Google Scholar]
- (21).Keshishian H;Burgess MW; Gillette MA; Mertins P;Clauser KR; Mani DR; Kuhn EW; Farrell LA; Gerszten RE; Carr SA Multiplexed, Quantitative Workflow for Sensitive Biomarker Discovery in Plasma Yields Novel Candidates for Early Myocardial Injury. Mol. Cell. Proteomics 2015, 14, 2375–2393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Zhang G;Fenyo D;Neubert TA Evaluation of the variation in sample preparation for comparative proteomics using stable isotope labeling by amino acids in cell culture. J Proteome Res 2009, 8, 1285–1292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Mertins P;Udeshi ND; Clauser KR; Mani DR; Patel J;Ong SE; Jaffe JD; Carr SA iTRAQ labeling is superior to mTRAQ for quantitative global proteomics and phosphoproteomics. Mol. Cell. Proteomics 2012, 11, M111 014423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Oppermann FS; Klammer M;Bobe C;Cox J;Schaab C;Tebbe A;Daub H. Comparison of SILAC and mTRAQ quantification for phosphoproteomics on a quadrupole orbitrap mass spectrometer. J Proteome Res 2013, 12, 4089–4100. [DOI] [PubMed] [Google Scholar]
- (25).Altelaar AF; Frese CK; Preisinger C;Hennrich ML; Schram AW; Timmers HT; Heck AJ; Mohammed S. Benchmarking stable isotope labeling based quantitative proteomics. J Proteomics 2013, 88, 14–26. [DOI] [PubMed] [Google Scholar]
- (26).Lau HT; Suh HW; Golkowski M;Ong SE Comparing SILAC- and Stable Isotope Dimethyl-Labeling Approaches for Quantitative Proteomics. J. Proteome Res. 2014, 13, 4164–4174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Li Z;Adams RM; Chourey K;Hurst GB; Hettich RL; Pan C. Systematic comparison of label-free, metabolic labeling, and isobaric chemical labeling for quantitative proteomics on LTQ Orbitrap Velos. J Proteome Res 2012, 11, 1582–1590. [DOI] [PubMed] [Google Scholar]
- (28).Alpert AJ Electrostatic repulsion hydrophilic interaction chromatography for isocratic separation of charged solutes and selective isolation of phosphopeptides. Anal. Chem. 2008, 80, 62–76. [DOI] [PubMed] [Google Scholar]
- (29).Simon-Assmann P;Turck N;Sidhoum-Jenny M;Gradwohl G;Kedinger M. In vitro models of intestinal epithelial cell differentiation. Cell biology and toxicology 2007, 23, 241–256. [DOI] [PubMed] [Google Scholar]
- (30).Chopra DP; Dombkowski AA; Stemmer PM; Parker GC Intestinal epithelial cells in vitro. Stem Cells Dev. 2010, 19, 131–142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Hamosh M. Bioactive factors in human milk. Pediatr. Clin. North Am. 2001, 48, 69–86. [DOI] [PubMed] [Google Scholar]
- (32).Goldman AS Modulation of the gastrointestinal tract of infants by human milk. Interfaces and interactions. An evolutionary perspective. J. Nutr. 2000, 130, 426S–431S. [DOI] [PubMed] [Google Scholar]
- (33).Karhumaa P;Leinonen J;Parkkila S;Kaunisto K;Tapanainen J;Rajaniemi H. The identification of secreted carbonic anhydrase VI as a constitutive glycoprotein of human and rat milk. Proc. Natl. Acad. Sci. U. S. A. 2001, 98, 11604–11608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Schack-Nielsen L;Michaelsen KF Advances in our understanding of the biology of human milk and its effects on the offspring. J. Nutr. 2007, 137, 503S–510S. [DOI] [PubMed] [Google Scholar]
- (35).Zivkovic AM; German JB; Lebrilla CB; Mills DA Human milk glycobiome and its impact on the infant gastrointestinal microbiota. Proc. Natl. Acad. Sci. U. S. A. 2011, 108 Suppl 1, 4653–4658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Zhang Q;Cundiff JK; Maria SD; McMahon RJ; Woo JG; Davidson BS; Morrow AL Quantitative Analysis of the Human Milk Whey Proteome Reveals Developing Milk and Mammary-Gland Functions across the First Year of Lactation. Proteomes 2013, 1, 128–158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Huang DW; Sherman BT; Lempicki RA Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 2009, 4, 44–57. [DOI] [PubMed] [Google Scholar]
- (38).Huang DW; Sherman BT; Lempicki RA Bioinformatics enrichment tools: paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res. 2009, 37, 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Subramanian A;Tamayo P;Mootha VK; Mukherjee S;Ebert BL; Gillette MA; Paulovich A;Pomeroy SL; Golub TR; Lander ES; Mesirov JP Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. U. S. A. 2005, 102, 15545-15550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Mootha VK; Lindgren CM; Eriksson KF; Subramanian A;Sihag S;Lehar J;Puigserver P;Carlsson E;Ridderstrale M;Laurila E;Houstis N;Daly MJ; Patterson N;Mesirov JP; Golub TR; Tamayo P;Spiegelman B;Lander ES; Hirschhorn JN; Altshuler D;Groop LC PGC-1alpha-responsive genes involved in oxidative phosphorylation are coordinately downregulated in human diabetes. Nat. Genet. 2003, 34, 267–273. [DOI] [PubMed] [Google Scholar]
- (41).Kanehisa M;Goto S. KEGG: kyoto encyclopedia of genes and genomes. Nucleic Acids Res 2000, 28, 27–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Benjamini Y;Hochberg Y. Controlling the False Discovery Rate: a Practical and Powerful Approach to Multiple Testing. J. R. Statist. Soc. B 1995, 57, 289–300. [Google Scholar]
- (43).Hao P;Guo T;Li X;Adav SS; Yang J;Wei M;Sze SK Novel application of electrostatic repulsion-hydrophilic interaction chromatography (ERLIC) in shotgun proteomics: comprehensive profiling of rat kidney proteome. J. Proteome Res. 2010, 9, 3520–3526. [DOI] [PubMed] [Google Scholar]
- (44).Wang Y;Yang F;Gritsenko MA; Clauss T;Liu T;Shen Y;Monroe ME; Lopez-Ferrer D;Reno T;Moore RJ; Klemke RL; Camp DG 2nd; Smith RD Reversed-phase chromatography with multiple fraction concatenation strategy for proteome profiling of human MCF10A cells. Proteomics 2011, 11, 2019–2026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).Hao P;Ren Y;Dutta B;Sze SK Comparative evaluation of electrostatic repulsion–hydrophilic interaction chromatography (ERLIC) and high-pH reversed phase (Hp-RP) chromatography in profiling of rat kidney proteome. J. Proteomics 2013, 82, 254–262. [DOI] [PubMed] [Google Scholar]
- (46).de Jong EP; Griffin TJ Online nanoscale ERLIC-MS outperforms RPLC-MS for shotgun proteomics in complex mixtures. J. Proteome Res. 2012, 11, 5059–5064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (47).Hao P;Guo T;Sze SK Simultaneous analysis of proteome, phospho- and glycoproteome of rat kidney tissue with electrostatic repulsion hydrophilic interaction chromatography. PLoS One 2011, 6, e16884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (48).Ting L;Rad R;Gygi SP; Haas W. MS3 eliminates ratio distortion in isobaric multiplexed quantitative proteomics. Nat. Methods 2011, 8, 937–940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (49).Wenger CD; Lee MV; Hebert AS; McAlister GC; Phanstiel DH; Westphall MS; Coon JJ Gas-phase purification enables accurate, multiplexed proteome quantification with isobaric tagging. Nat. Methods 2011, 8, 933–935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (50).Zhang Q;Cundiff JK; Maria SD; McMahon RJ; Wickham MSJ; Faulks RM; Tol EAF v. Differential Digestion of Human Milk Proteins in a Simulated Stomach Model. J. Proteome Res. 2014, 13, 1055–1064. [DOI] [PubMed] [Google Scholar]
- (51).Bantscheff M;Boesche M;Eberhard D;Matthieson T;Sweetman G;Kuster B. Robust and sensitive iTRAQ quantification on an LTQ Orbitrap mass spectrometer. Mol. Cell. Proteomics 2008, 7, 1702–1713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (52).Hultin-Rosenberg L;Forshed J;Branca RM; Lehtio J;Johansson HJ Defining, comparing, and improving iTRAQ quantification in mass spectrometry proteomics data. Mol. Cell. Proteomics 2013, 12, 2021–2031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (53).Taussig MJ; Stoevesandt O;Borrebaeck CA; Bradbury AR; Cahill D;Cambillau C;de Daruvar A;Dubel S;Eichler J;Frank R;Gibson TJ; Gloriam D;Gold L;Herberg FW; Hermjakob H;Hoheisel JD; Joos TO; Kallioniemi O;Koegl M;Konthur Z;Korn B;Kremmer E;Krobitsch S;Landegren U;van der Maarel S;McCafferty J;Muyldermans S;Nygren PA; Palcy S;Pluckthun A;Polic B;Przybylski M;Saviranta P;Sawyer A;Sherman DJ; Skerra A;Templin M;Ueffing M;Uhlen M. ProteomeBinders: planning a European resource of affinity reagents for analysis of the human proteome. Nat. Methods 2007, 4, 13–17. [DOI] [PubMed] [Google Scholar]
- (54).Jiang R;Lopez V;Kelleher SL; Lonnerdal B. Apo- and holo-lactoferrin are both internalized by lactoferrin receptor via clathrin-mediated endocytosis but differentially affect ERK-signaling and cell proliferation in Caco-2 cells. J. Cell. Physiol. 2011, 226, 3022–3031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (55).Rojas R;Apodaca G. Immunoglobulin transport across polarized epithelial cells. Nat. Rev. Mol. Cell Biol. 2002, 3, 944–955. [DOI] [PubMed] [Google Scholar]
- (56).UniProt: a hub for protein information. Nucleic Acids Res 2015, 43, D204–212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (57).Anzenbacher P;Anzenbacherova E. Cytochromes P450 and metabolism of xenobiotics. Cell. Mol. Life Sci. 2001, 58, 737–747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (58).Romero L;Andrews K;Ng L;O’Rourke K;Maslen A;Kirby G. Human GSTA1–1 reduces c-Jun N-terminal kinase signalling and apoptosis in Caco-2 cells. Biochem. J. 2006, 400, 135–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (59).Peters WH; Roelofs HM Time-dependent activity and expression of glutathione S-transferases in the human colon adenocarcinoma cell line Caco-2. Biochem. J. 1989, 264, 613–616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (60).Geiger T;Wehner A;Schaab C;Cox J;Mann M. Comparative proteomic analysis of eleven common cell lines reveals ubiquitous but varying expression of most proteins. Mol. Cell. Proteomics 2012, 11, M111 014050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (61).Nagaraj N;Wisniewski JR; Geiger T;Cox J;Kircher M;Kelso J;Paabo S;Mann M. Deep proteome and transcriptome mapping of a human cancer cell line. Mol. Syst. Biol. 2011, 7, 548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (62).Svensson M;Hakansson A;Mossberg AK; Linse S;Svanborg C. Conversion of alpha-lactalbumin to a protein inducing apoptosis. Proc. Natl. Acad. Sci. U. S. A. 2000, 97, 4221–4226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (63).Field CJ The immunological components of human milk and their effect on immune development in infants. J. Nutr. 2005, 135, 1–4. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.