

Abstract
The ClpXP machinery is a two component protease complex performing targeted protein degradation in bacteria and mitochondria. The complex consists of the AAA+ chaperone ClpX and the peptidase ClpP. The hexameric ClpX utilizes the energy of ATP binding and hydrolysis to engage, unfold and translocate substrates into the catalytic chamber of tetradecameric ClpP where they are degraded. Formation of the complex involves a symmetry mismatch, since hexameric AAA+ rings bind axially to the opposing stacked heptameric rings of the tetradecameric ClpP. Here we present the cryo-EM structure of ClpXP from Listeria monocytogenes. We unravel the heptamer-hexamer binding interface and provide novel insights into the ClpX-ClpP crosstalk and activation mechanism. The comparison with available crystal structures of ClpP and ClpX in different states allows us to understand important aspects of ClpXP’s complex mode of action and provides a structural framework for future pharmacological applications.
Introduction
Caseinolytic protease P (ClpP) represents a major proteolytic protein in prokaryotes and in organelles of eukaryotes which is involved in protein homeostasis, bacterial pathogenesis as well as cancer progression1–3. ClpP is highly conserved, essential for virulence and regulation of stress responses in several pathogenic bacteria and therefore considered as a promising therapeutic target for novel antibiotics4. ClpP associates with diverse ATP-dependent AAA+ chaperones such as ClpX, ClpC and ClpA to form a complex for the recognition, unfolding and digestion of substrate proteins5. To date, a large fraction of research has been dedicated to functionally exploit ClpP and its cognate chaperones, foremost ClpX, in terms of their enzymatic activity, individual structures and conformational control.
Previous low resolution electron microscopy (EM) studies of ClpXP and ClpAP from Escherichia coli revealed that up to two hexameric ClpX chaperones bind to a ClpP tetradecameric barrel6,7. The barrel consists of two stacked heptameric rings, forming a degradation chamber with 14 proteolytic sites8. Each ClpX subunit consists of an N-terminal zinc binding domain (ZBD) and a C-terminal AAA+ domain. The ZBDs at the periphery of ClpX are responsible for recognition and engagement of several substrates9. ClpX hydrolyzes ATP to unfold the target substrates and translocate the unfolded polypeptides through a central pore into the proteolytic chamber of the ClpP barrel (for review see10).
Early on, the hexamer-heptamer ClpX-ClpP interface fascinated researchers and several studies characterizing the role of putative interaction motifs have led to models explaining the symmetry mismatch and functional interaction between the two proteins7,11–13. Sequence alignments and mutational studies of AAA+ chaperones identified loops in ClpX, that interact with the hydrophobic clefts on the periphery of ClpP. They contain the highly conserved (I/L/V)-G-(F/L) motif and are essential for complex formation14.
More recently, cyclic acyldespipeptides (ADEPs), a novel class of anti-bacterial compounds, have been identified to bind to the same peripheral hydrophobic clefts on ClpP and to induce the opening of the axial pores of ClpP4,15–17. They stabilize ClpP in an “open” activated state in the absence of the chaperone, leading to unregulated proteolysis of substrates and finally to cell death18. This suggests that the protruding loops in ClpX that contain the (I/L/V)-G-(F/L) motif, also called IGF loops, are sufficient to activate ClpP. It has also been speculated that this activation involves the opening of the axial pore to allow translocation of the substrate into the proteolytic chamber of ClpP. However, due to the lack of high-resolution structures, a detailed understanding of the interaction between ClpX and ClpP is missing.
Contacts between the pore-2 loops of ClpX and the N-termini of ClpP represent a second set of well-characterized interactions between ClpX and ClpP, which are, however, more dynamic and dependent on the nucleotide state of ClpX13. A crucial function of the ClpP N-termini is to gate the entrance of the proteolytic chamber11. Despite these detailed biochemical insights, a high-resolution structure of the whole proteolytic complex is lacking, thereby limiting our understanding of this important protein degradation machinery. Here we present a 4 Å cryo-EM structure of ClpXP1/2 from Listeria monocytogenes.
Results
Cryo-EM structure of ClpXP1/2
In order to obtain a ClpXP complex that is suitable for structural studies, we used the ClpP1/2 from L. monocytogenes. In contrast to other bacteria, L. monocytogenes encodes two ClpP isoforms, LmClpP1 and LmClpP2, which can assemble into heterooligomers composed of two homoheptameric rings. Recent studies have revealed that ClpP1/2 has a higher affinity to ClpX in comparison to the more conserved ClpP2 homocomplex19,20, suggesting a superior stability of the heterooligomer. As ClpP1/2 might cleave ClpX to a small extent during sample preparation, we mutated one residue of the catalytic triad (S98A) in both ClpP isoforms. Furthermore, we mutated the nucleotide binding site of ClpX (E183Q) to allow ATP binding, but to prevent hydrolysis, which results in a tighter binding to ClpP21,22.
We formed a complex of ClpX and ClpP1/2 and obtained a large fraction of ClpXP1/2 dimers (ClpP1/2-ClpX-ClpX-ClpP1/2) that were in equilibrium with ClpXP1/2 monomers (Supplementary Fig. 1a-c). It has been demonstrated before that two ClpX or ClpA hexamers can bind to one ClpP barrel from both sites, resulting in a ClpX-ClpP-ClpX or ClpA-ClpP-ClpA complex6,7,23. However, ClpXP1/2 dimers (Supplementary Figure 1a-d) have, to our knowledge, not been described so far. We therefore concentrated our structural analysis first on these intriguing dimers and determined their structure by cryo-EM and single particle analysis using crYOLO24 and SPHIRE25 (Figure 1a-b, Supplementary Fig. 1e-g, Table 1). Although the intrinsic flexibility of the complexes did not allow the determination of a high-resolution structure (Supplementary Video 1, Supplementary Figure 1e-g), the fitting of the crystal structure of ClpX into the cryo-EM density suggests that the flexible N-terminal zinc binding domains (ZBDs) of ClpX mediate the interaction between two ClpX hexamers (Figure 1c). While ZBD-deleted ClpX still associated with ClpP to a small extent, ClpX dimerization was completely abolished supporting our structural data (Supplementary Fig. 1a).
Figure 1. LmClpXP1/2 forms flexible dimers via the ZBDs.

a) Typical low-dose cryo-EM micrograph of the ClpXP1/2 dimer from L. monocytogenes. Some particles are highlighted with ovals. Scale bar, 100nm b) Typical reference-free 2D class averages. Arrows indicate additional densities corresponding to ZBDs at the interface between two ClpX hexamers. Scale bar, 20nm c) Ribbon Model of ClpP1 (yellow), ClpP2 (green) and ClpX (orange) superimposed with the cryo-EM density map of the ClpXP1/2 dimer (white and transparent). The upper inset shows the complex shown as slice at the position of the axial pore entry of the upper ClpXP1/2 complex. ClpX and ClpX-ZBD densities are colored magenta and gray transparent, respectively. The arrow indicates the spiral arrangement of the ZBD domains. The lower inset shows four copies of ZBD-dimers (PDB: 1OVX) placed into the cryo-EM density at the interface between the ClpX hexamers. The low resolution density did not allow automated rigid-body fitting, therefore the dimers were placed manually and interconnected as proposed in 26. d) Cartoon depicting ClpXP1/2 dimerization via the ZBD domains of two opposing ClpX hexamers. Arrows indicate the flexibility of the complex.
Table 1. Cryo-EM data collection, refinement and validation statistics.
| LmClpX1/2 dimer | LmClpXP1/2 EMD-10162, (PDB-6SFX, 6SFW) |
||
|---|---|---|---|
| Data collection and processing | |||
| Magnification | x112,807 | X112,807 | |
| Voltage (kV) | 300 | 300 | |
| Electron exposure (e–/Å2) | 114 | 114 | |
| Defocus range (μm) | 0.5 - -3.0 | -0.5 - -3.0 | |
| Pixel size (Å) | 1.1 | 1.1 | |
| Symmetry imposed | C1 | C1 | |
| Initial particle images (no.) | 273,300 | 613,322 | |
| Final particle images (no.) | 143,901 | 613,322 | |
| Map resolution (Å) | 13 | 4 | |
| FSC threshold | 0.143 | 0.143 | |
| Map resolution range (Å) | - | 3.2-10 | |
| Refinement | 6SFX 6SFW | ||
| Initial model used (PDB code) | - | 4RYF | - |
| Model resolution (Å) | - | 2.8, | - |
| FSC threshold | - | - | - |
| Model resolution range (Å) | - | - | - |
| Map sharpening B factor (Å2) | - | -214 | -240 |
| Model composition | |||
| Nonhydrogen atoms | - | 20196 | 15225 |
| Protein residues | - | 2602 | 1955 |
| Ligands | - | - | |
| B factors (Å2) | 100.8 | 187.87 | |
| Protein | - | - | |
| Ligand | - | - | |
| R.m.s. deviations | |||
| Bond lengths (Å) | 0.012 | 0.018 | |
| Bond angles (°) | 1.233 | 1.978 | |
| Validation | |||
| MolProbity score | - | 2.34 | 2.30 |
| Clashscore | - | 22.88 | 22.43 |
| Poor rotamers (%) | - | 0.18 | 0.2 |
| Ramachandran plot | |||
| Favored (%) | - | 92.04 | 92.9 |
| Allowed (%) | - | 7.65 | 6.4 |
| Disallowed (%) | - | 0.31 | 0.7 |
The ZBDs are involved in substrate binding and cofactor recognition and were shown to dimerize when expressed as single domain26,27. Based on these results it has been previously proposed that the ZBDs of neighboring subunits within a single ClpX hexamer dimerize resulting in a trimer-of-dimer model26. In this model the ZBD dimers interact with the adjacent dimers, creating a ring structure that is aligned with the central channel of ClpX. The structure of the ClpXP1/2 dimer, however, reveals that the ZBDs do not form rings, but arrange in a flexible half-cone spiral with the first and last ZBD dimer positioned directly above or at the rim of the axial pore entry of the upper and lower ClpX hexamer, respectively (Figure 1c, Supplementary Figure 1e). The ZBDs are apparently interacting with the ZBDs from oppositely positioned subunits leading to the cross-linking of the two opposing ClpX hexamers (Figure 1c, d). In total, four ZBD dimers fit into the cryo-EM density (Figure 1c). Because of the limited resolution in this region, however, we cannot determine if the cross-bridges are mediated by single ZBDs that dimerize with ZBDs of the other ClpX or by ZBD dimers that interact with dimers of the other ClpX. Based on these results and the fact that the ZBDs are flexible and not resolved in the crystal structure of ClpX28, we propose that ZBD dimers form stable structures only at the interface between two oppositely positioned ClpX hexamers (Figure 1d).
To obtain a cryo-EM structure at higher resolution, we focused the structural analysis on one ClpXP1/2 subunit in the dimer and solved its structure using the same dataset (Figure 2a-d, Supplementary Fig. 2, Table 1). The final cryo-EM reconstruction has an average resolution of 3.6 - 4 Å for ClpP1/2 and 6 - 7 Å for ClpX (Supplementary Fig. 2e-g). The overall lower resolution of ClpX indicates that the chaperone is intrinsically more flexible and heterogeneous than the ClpP barrel in the ClpXP1/2 complex. To build a complete atomic model of ClpXP1/2, we fitted a homology model of ClpX and the available crystal structure of ClpP1/2 (PDB-ID 4RYF) into the cryo-EM density and refined the model using Molecular Dynamics Flexible Fitting (MDFF)29.
Figure 2. Cryo-EM structure of the ClpXP1/2 protein degradation machinery.
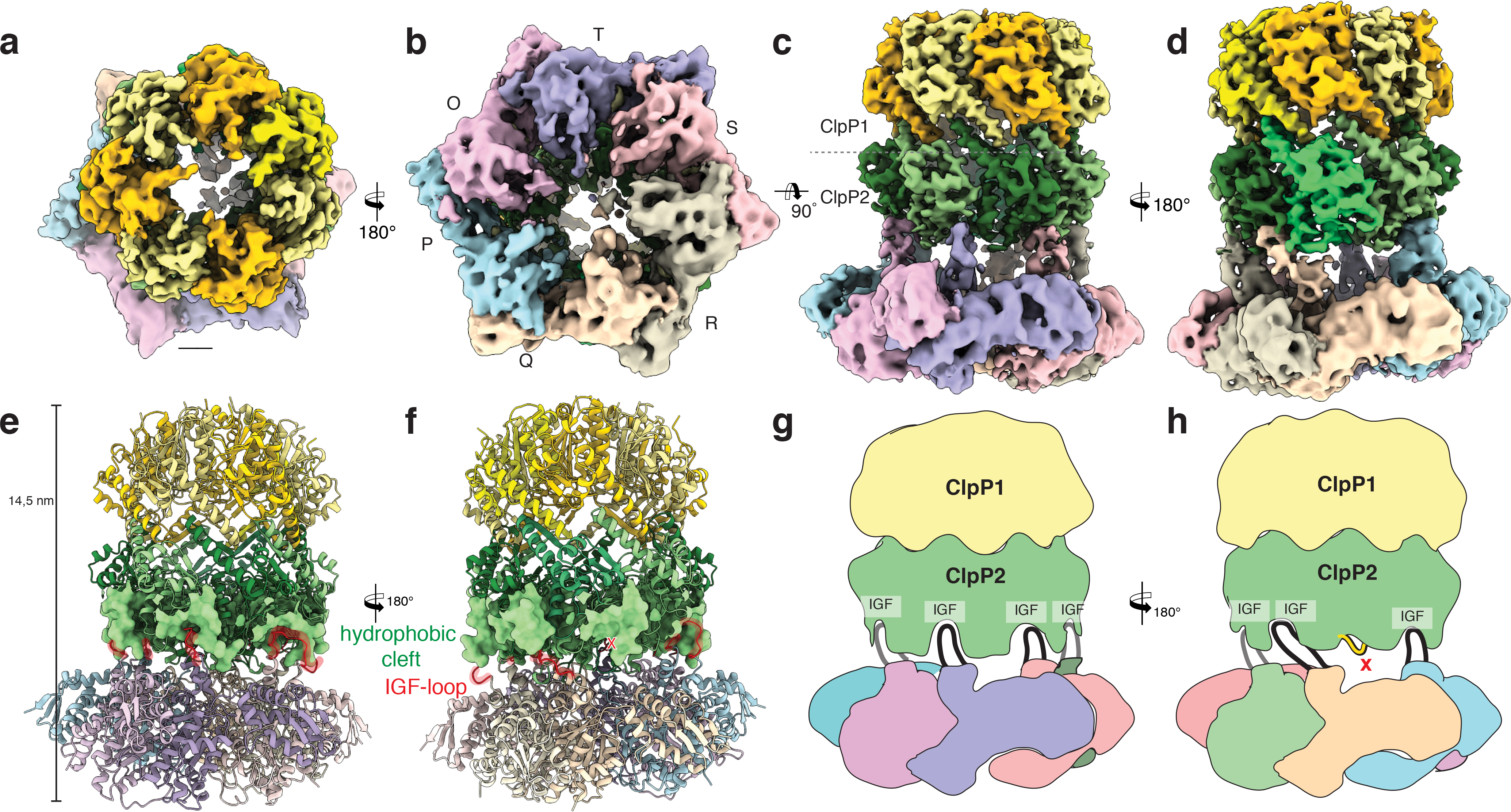
a-d) Cryo-EM density of ClpXP1/2 shown from the top (a), bottom (b) and side (c,d). ClpP1 and ClpP2 subunits are colored in khaki, orange and dark, light green, respectively. ClpP2 subunit J is highlighted in mint green. Note that this is the only ClpP2 subunit not interacting with ClpX via an IGF-loop. Each subunit of ClpX is assigned a different color. This color code is maintained throughout the manuscript. e-f) Molecular model of ClpXP. The hydrophobic pockets of ClpP2, each spanning two ClpP2 subunits, are shown as surface. The IGF interaction loops are highlighted in red. g-h) Cartoon depicting how the ClpX hexamer interacts with the ClpP2 heptamer via the six IGF-loops. Note the extended conformation of IGF-loop of ClpX subunit Q.
The structure of ClpXP1/2 reveals that ClpP1 forms the upper homoheptamer of the ClpP barrel, whereas ClpP2 sits below and interacts with ClpX (Figure 2c-h). Our cryo-EM structure is consistent with previous binding studies on Listeria monocytogenes and Mycobacterium tuberculosis ClpP proteases, showing ClpX-ClpP1/2 interactions exclusively via the ClpP2 ring surface30–32.
Interestingly, the ClpX hexamer is not centrally aligned, but slightly tilted by ~11° towards ClpP2. The structure of ClpP1/2 is almost identical to the available crystal structure of apo-ClpP1/2 (PDB-ID 4RYF), indicating that the binding of ClpX does not induce large conformational changes in ClpP1/2. In contrast, interaction with ClpP1/2 has an effect on the overall conformation of ClpX. Whereas the crystal structure of E. coli ClpX shows the ATPase domains in a “dimer-of-trimers” arrangement33, our structure shows that upon ClpP1/2 binding, these domains become more regularly arranged and are related by pseudo-six-fold symmetry. Unlike recent substrate bound AAA+ structures that show a “spiral-staircase” arrangement with one “seam” subunit moderately displaced from the pore34–36, all neighboring AAA+ domains of ClpX pack closely with each other. The resolution at the nucleotide pocket is not high enough to visualize nucleotides, but the structure reveals that all six ClpX protomers are in the “loadable” conformation (Supplementary Fig. 3). This is in contrast to ClpX with the E183Q mutation in its apo-state28,33. There, two subunits are in the “loadable” (L) and four are in the “unloadable” (U) conformation (Supplementary Fig. 3). In the L state, the arrangement of the small and large AAA+ domains results in an open binding cleft, to which the nucleotide can bind. In the U state, this site is blocked. A dynamic interconversion between L and U conformations is required to couple ATP hydrolysis by ClpX to mechanical work.
However, the arrangement is not a direct consequence of the bound nucleotide or the presence of specific mutations28. To further examine the interaction between ClpP1/2 and ClpX we used hydrogen-deuterium exchange with mass spectrometry (HDX-MS) to monitor the accessibility of residues at the interface. In line with our structural observations, complex formation between ClpP1/2 and ClpX only changes the accessibility of residues of ClpX and ClpP2, but not of ClpP1 (Supplementary Fig. 4). This not only corroborates that ClpX solely interacts with the ClpP2 isoform, but also indicates that ClpX binding does not induce major allosteric conformational changes in the ClpP1 heptamer.
Symmetry mismatch of IGF-loop interaction
The most interesting part of the structure is the interface between ClpP2 and ClpX, which involves a C6/C7 symmetry mismatch. As predicted by biochemical studies8,12,14, it is mediated mainly by the flexible IGF loops of ClpX interacting with hydrophobic grooves in ClpP2 (Figure 2c-d, Supplementary Figure 5a). The tilted arrangement of ClpX results in part of the loops interacting stronger with ClpP2 than others (Figure 3a).
Figure 3. Symmetry mismatch between ClpP1/2 and ClpX.

a) Molecular model of ClpXP1/2. The symmetry axes of the ClpP1/2 and ClpX are shown in green and orange, respectively. b-c) The ClpP2 heptamer (b) and the ClpX hexamer (c) are shown from the bottom and the top, respectively, perpendicular to the plane of the ClpP2-ClpX interface. The positions of the IGF-loops and the hydrophobic grooves are highlighted in yellow and connected by dashed lines. d) Cut-away view of the ClpP density. Secondary structure elements directly prior (residues 170-189) and after the pore-2-loops (residues 202-220) of ClpX are shown in ribbon representation. The pore-2-loops are not resolved in the cryo-EM density and not shown here. In order to indicate the arrangement and positioning of the pore-2-loops, as well as the position of the upper opening of the ClpX channel relative to the ClpP2 pore, a plane was calculated using the Cα atoms of Gly202 as anchor points and depicted here in orange. The plane is tilted and shifted relative to the ClpP channel axis, suggesting a spiral staircase-like arrangement of the pore-2-loops. The dashed line with the arrowhead indicates the pathway of substrate translocation from ClpX towards the ClpP proteolytic chamber. The inset shows the skin surface of the ClpXP pore. e) Molecular surface of ClpP2 shown from the bottom. Rosetta models of the pore-2-loops of ClpX are shown as ribbons. The black star indicates the positioning of the ClpX channel opening relative to the ClpP channel opening (yellow star). f) Schematic model of the ClpX-ClpP2-binding mechanism. Left images depict axial views of the ClpP2 heptamer (green) and the ClpX hexamer prior assembly of the ClpXP protease. The main interaction elements, the ClpX IGF-loops and ClpP2 hydrophobic grooves are highlighted. The remaining “free” ClpP2 hydrophobic groove stays shielded by the respective C-terminus (arrow).
The large domains of the respective ClpX subunit from which the loops protrude are positioned directly below the deep hydrophobic grooves of ClpP2 which are formed at the interface of two subunits. This arrangement allows a direct interaction of the IGF-loops with the opposing grooves. The hydrophobic grooves of ClpP are arranged in a circular manner with seven-fold symmetry and the positions of the ClpX IGF-loops in the complex, perfectly match this arrangement. Interestingly, both rings display similar diameters (Figure 3b-c), except that the IGF-ring remains open at the position of the seventh, free hydrophobic cleft.
Five of the six IGF loops (subunits O, P, R, S, T) display an overall similar arrangement. Due to the symmetry mismatch the large domain of the sixth subunit (subunit Q), is positioned in-between two hydrophobic grooves. The respective IGF-loop, however, still interacts with one of the opposing grooves by adopting an “extended” conformation (Figure 2c-h). The other groove stays empty. Although the distance between the IGF-loop and the “left” or “right” ClpP hydrophobic groove are similar, we only obtained a high-resolution structure with the IGF-loop binding exclusively to the left binding pocket.
To support our structural findings, we performed HDX-MS measurements and mutational studies. Upon complex formation deuterium uptake of the IGF-loop is strongly reduced (Figure 4, Supplementary Fig. 4) and mutations in the IGF loops of ClpX and the hydrophobic grooves of ClpP2 result in impaired complex formation (Supplementary Fig. 6). This is in line with our ClpXP1/2 structure that demonstrates that the interaction between the IGF loops with the hydrophobic grooves is crucial for complex formation and function.
Figure 4. HDX-MS analysis of ClpXP1/2 complex formation.

a) Difference in relative deuterium uptake after 10 s exposure is mapped on the structure of ClpXP1/2 (left), ClpP2 monomer (right top) and ClpX monomer (right bottom). Increased deuterium uptake upon complex formation is shown in red, decreased deuterium uptake is depicted in blue. Dark gray represents no coverage. The MS data are available online as source data. b) HDX kinetics of exemplary peptides in the N-terminus of ClpP2 (top) and in the IGF-loop of ClpX (bottom). Solid lines and filled circles represent the ClpXP1/2 complex, dashed lines and empty circles represent ClpP1/2 or ClpX. Two independent replicates are shown, lines denote the mean.
Taken together, tilting of the ClpX ring and stretching of one of the IGF-loops is sufficient for the hexameric ClpX to adapt to the seven-fold symmetry of the heptameric ClpP, leaving out one of the binding pockets (Figure 2g-h). Due to multivalence, this results in strong, but at the same time flexible binding, which is likely necessary to accommodate the different conformations of ClpX protomers during ATP hydrolysis and substrate processing12,21,33.
N-termini of ClpP2 and pore-2 loops of ClpX regulate the entry portal
ClpX is not only tilted, but also laterally shifted respective to ClpP2 (Figure 3a, d, e). Such an arrangement has also been described for other complexes that display a symmetry mismatch37–39. In the case of ClpXP1/2, this results in a misalignment of the central channels of ClpP and ClpX, creating in a twisted translocation channel with a constriction site at the interface between ClpP2 and ClpX (Figure 3d). At this position, the N-terminal loops of ClpP2 and pore-2 loops of ClpX interact with each other. These interactions are expected to be even more dynamic than the flexible contacts mediated by the IGF loops, and coupled to ATP-hydrolysis12,14,40. Indeed, the densities corresponding to the N-terminal loops of ClpP2 and pore-2 loops of ClpX are very weak indicating a higher degree of flexibility in this region of the complex (Supplementary Figures 7,8).
Different conformations of the ClpP N-terminal loops have been previously identified in crystal structures of apo and ADEP-bound ClpPs11,41,42. In the E. coli apo ClpP structure, the N-termini on the apical side of the ClpP barrel are in the “down” conformation, opening one axial pore of the barrel. On the basal side six of the N-termini are in the “up” conformation, with the loops moving out of the axial pore, thereby covering and closing it. It was speculated that the six ClpP N-termini in the “down” conformation would open to match the six-fold symmetry of ClpX and the seventh non-interacting N-terminus would stay in the “down” conformation upon binding to the chaperone. However, in the ADEP-bound structure of E. coli ClpP all loops point upwards while they are not resolved in a B. subtilis ADEP-bound ClpP structure having made general conclusions difficult so far41,42.
In our cryo-EM structure, residues 6 to 17 are not resolved, but the rest of the density reveals that all seven N-termini of ClpP2 (the apical side of the barrel facing the chaperone) adopt the “up”-conformation resolving the controversy about their positioning and the accessibility of the pore (Supplementary Fig. 7). The cryo-EM structure demonstrates that the interaction site between the ClpP2 N-termini and the ClpX pore-2 loops is not shielded and freely solvent accessible. In addition, the N-termini undergo a conformational change upon complex formation and adopt the “up” conformation, by which the protein backbone likely gets more solvent exposed and/or flexible. In line with this, deuteration of the ClpP2 N-terminus increased after complex formation (Figure 4, Supplementary Fig. 4). This observation is also supported by reported synchrotron hydroxyl radical footprinting data showing that ClpA binding enhanced the modification rate of an N-terminal peptide of ClpP, pointing towards a higher solvent accessibility43.
The C-terminus of ClpP2 shields the hydrophobic groove prior to ClpX binding
The C-termini of the ClpP2 show two conformations in our structure: a compact conformation that blocks the hydrophobic groove when it does not accommodate an IGF loop, and an extended conformation enlarging the groove when occupied by an IGF loop (Figure 5a). Since the residues of the C-terminus are not conserved (Supplementary Figure 9) and the conformational change is not transmitted to the rest of the protein, an allosteric regulation is rather unlikely. The C-termini probably shield the hydrophobic grooves, when ClpX is not bound and thereby prevent the interaction with other hydrophobic molecules and increase the stability of the protein in a hydrophilic environment.
Figure 5. Role of the ClpP2 C-terminus in ClpXP1/2 binding.

a) Molecular model and cryo-EM density of IGF-loop bound (upper image) and not bound to hydrophobic pockets of ClpP2 (lower image). The insets show the respective IGF-loops in ribbon representation. Arrows indicate the C-terminus of ClpP2. b) Peptidase activity of ClpP1/2 with C-terminally truncated ClpP2 (714 nM (ClpP1/2)14, 100 μM Ac-Ala-hArg-2-Aoc-ACC). c) Protease activity of ClpXP1/2 with C-terminally truncated ClpP2 (0.2 μM (ClpP1/2)14, 0.4 μM ClpX6, 0.8 μM GFP-SsrA). Data are normalized to the wild type as 100% (n = 6, data were recorded in triplicate and two independent experiments were performed, black lines denote means). Source data for graphs in b-c are available online.
To probe this, we deleted the last three to six amino acids of ClpP2. ClpP1/2ΔC-6 precipitated during purification, suggesting that a certain length of the C-terminus is important to protect the hydrophobic groove and facilitate protein stability. ClpP2 mutants bearing three to five amino acid deletions were however soluble and exhibited a similar peptidolytic activity as the wild type complex (Figure 5b). Interestingly, in protease assays requiring the binding of ClpX, the activity increased with a growing number of amino acid deletions in comparison to the wild type complex (Figure 5c). We interpret this result such that when the C-termini are shorter more complexes are formed because ClpX can easier access the hydrophobic grooves via the IGF-loops. Indeed, in line with this finding the C-termini of most ClpPs which were shown to interact with ClpX are shorter in length (Supplementary Figure 9).
ClpP activation mechanism by ClpX
Previous crystal structures of ClpP in its apo-form, i.e. without ClpX or compound bound, revealed three different conformational states of the protein: “compressed”, “compact” and “extended”44–48 (Figure 6). The catalytic triad of the peptidase is only intact in the extended state, suggesting that this is the only active state. ADEPs, that bind to the same site on ClpP as the IGF loops, can induce the transition from the compressed to the extended conformation15. In addition, a ~90° rotation of Tyr63 in the hydrophobic pocket results in the widening of the axial pore by 10 – 15 Å. A mutation of this residue to alanine has the same effect49. This “open” extended conformation of ClpP deregulates the protein. Instead of only processing short peptides of five to six residues, it is now capable to degrade large unfolded polypeptides that otherwise could not be processed in the absence of the chaperone (Figure 7)42,43,50. It has been speculated that the mechanism of ClpP activation by ClpX would imply similar conformational changes18,49.
Figure 6. Comparison of ClpX-bound ClpP1/2 with available structures of active and inactive ClpP.

a) Side view of the structure of ClpX-bound LmClpP1/2 (gold) and the crystal structures of LmClpP1/2 in the extended active state (PDB4RYF) (purple), Bacillus subtilis ClpP (BsClpP) in complex with ADEP2 in the extended open active state (PDB 3KTK) (gray), Staphylococcus aureus ClpP (SaClpP) in the extended active state (PDB 3V5E) (cyan), SaClpP in the compact inactive state (PDB 4EMM) (red) and SaClpP in the compressed inactive (PDB 3QWD) (purple) conformation are shown in ribbon representation. b) Structural superposition of ClpX-bound and unbound (PDB 4RYF) LmClpP1/2. The low R.M.S.D suggests that binding of ClpX to ClpP1/2 does not induce large conformational changes to ClpP1/2. c) Structural superposition of ClpX bound ClpP1/2 heterocomplex and ADEP2-bound ClpP homocomplex (PDB 3KTK) shown in top- and bottom view. Black arrows indicate the characteristic opening of the ClpP pore upon ADEP binding. d) Superposition of the catalytic residues S98 (S98A), H123 and D172 (N172) in ClpX-bound LmClpP1-S98A/P2-S98A, LmClpP1/2 (extended active state) (PDB 4RYF), SaClpP (compact inactive state) (PDB 4EMM). Note that despite the S98A mutation, the catalytic residues of ClpX-bound LmClpP1/P2 adopt the active conformation. e) Opposing subunits of ClpX-bound ClpP1 and ClpP2 rings interact via an antiparallel β-sheet.
Figure 7. ClpX binds to ClpP in a similar manner like ADEP, but does not induce ClpP pore widening.
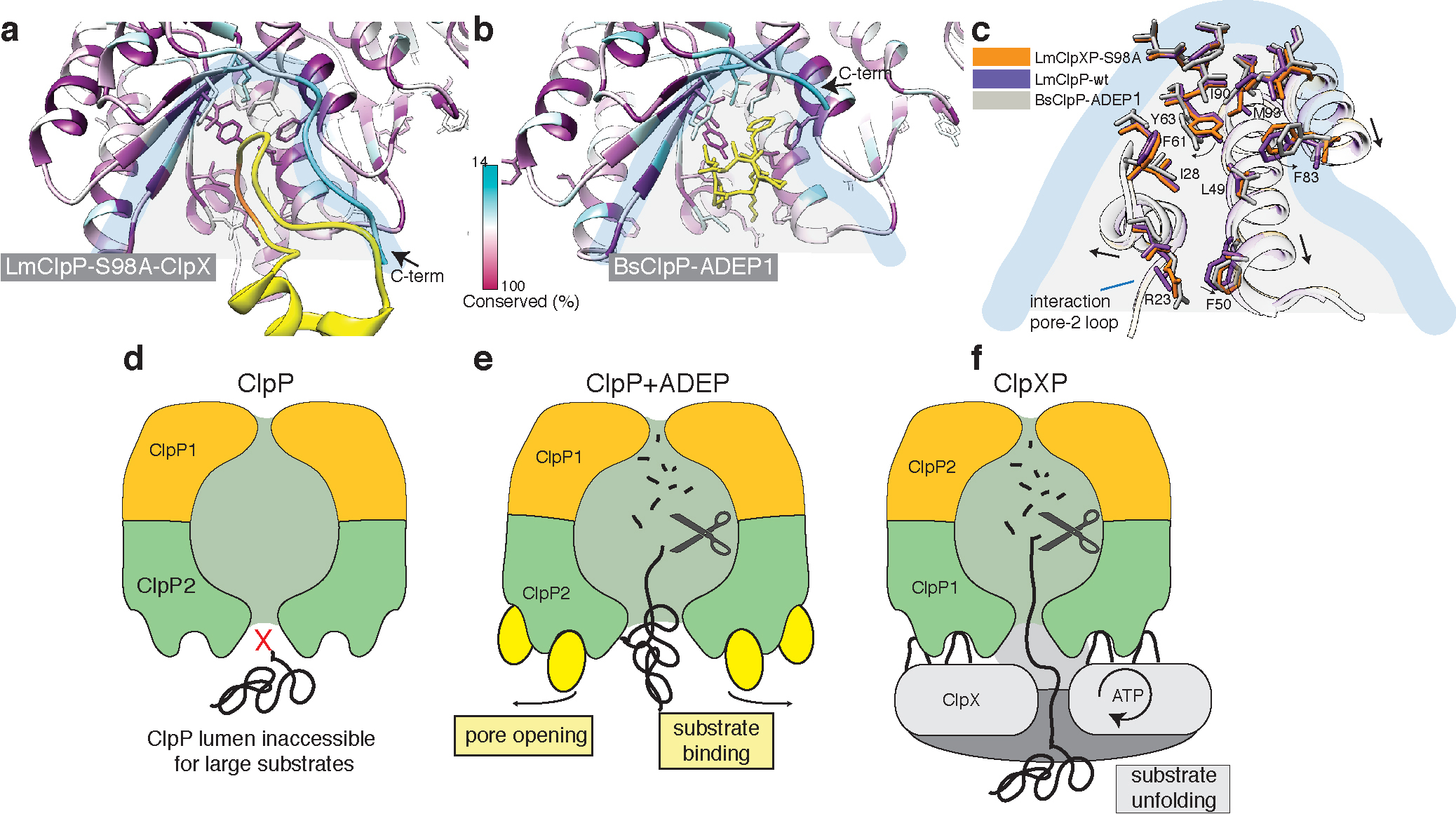
a) Local molecular interactions at one of the seven binding pockets between ClpP and the IGF-loop of ClpX. Residues of ClpP are colored by sequence conservation. The IGF-loop is shown in yellow with the IGF residues highlighted in orange. b) Interface of ADEP2 (yellow) with BsClpP (PDB 3KTI) (colored by conservation). c) Structural superposition of the binding pockets of ClpX-bound LmClpP2-S98A, ADEP1-bound BsClpP (PDB 3KTI) and “free” LmClpP2 (PDB 4RYF). Arrows indicate changes upon ADEP binding. d-f) Regulation of ClpP by ClpX and ADEP. The central pore of the ClpP protease is closed and entry of folded proteins into the proteolytic chamber is not allowed (d). ADEP binding to the binding pockets of ClpP induces pore opening. The proteolytic chamber is now accessible for unfolded proteins, leading to unregulated protein degradation and cell death. (e) ClpX binds in the same hydrophobic pockets on ClpP but does not induce pore opening. ClpP and ClpX form a continuous pore instead, with ClpX unfolding target proteins and forwarding them to the proteolytic chamber of ClpP for degradation in a regulated manner (f).
Our ClpXP1/2 structure demonstrates that this is not the case. ClpP is in the active extended conformation which is very similar to its conformation in the apo-state (Figure 6a, b). Despite the S98A mutation, the catalytic triad is aligned and in its active conformation (Figure 6d, Supplementary Fig. 5b). The ClpP1-P2 heptamers are interconnected via typical interactions of antiparallel β9 strands, characteristic for the “extended” active conformation45 (Figure 6e). Importantly, the axial pore of ClpP is not widened, when compared to the crystal structure of B subtilis ADEP-bound ClpP (Figure 6c, Supplementary Video 2). A comparison of the interface between the IGF-loop and ADEP with the hydrophobic ClpP pocket reveals that both interact with the same non-polar residues including Ile28, Leu49, Tyr63, Phe83, Ile90, Leu115 (Figure 7a-c). However, binding of ClpX does not induce the rotation of Tyr63 (Figure 7c), which is key to opening the pore. Thus, despite the fact that ADEPs and ClpX share the same binding sites, ClpX does not induce the conformational changes resulting in the opening of ClpP. Instead, binding does not induce any major conformational changes and the diameter of the ClpP channel is sufficient to accommodate the unfolded peptides that are threaded into the ClpP pore by the chaperone to be processed sequentially within the chamber of the peptidase (Figure 7f).
Discussion
ClpXP plays a significant role in the production and regulation of bacterial virulence factors during host infection and is therefore considered as a promising target for antimicrobial therapy51,52. On the other hand, targeting of the mitochondrial homologues is considered as a novel approach to halt tumor cell proliferation and metastatic competence53. Despite the important role of ClpXP in protein degradation, biology and medicine in general, structural knowledge of the dynamic two-component proteolytic machinery has lagged behind. The flexible and dynamic interaction between ClpX and ClpP via long flexible IGF- and pore-2 loops, involving a symmetry mismatch, together with the asymmetry of the ClpX ATPase make this complex a difficult specimen for structural analysis and probably explain why a high-resolution structure of the complex has been missing so far.
In contrast to previous works, here we utilized the ClpP1/2 heterocomplex from L. monocytogenes, showing a higher affinity to ClpX than the homocomplex. We mutated the proteolytic site and nucleotide binding site of ClpP1/2 and ClpX, respectively, and cross-linked the sample, in order to obtain a ClpXP1/2 complex with superior stability for cryo-EM studies. We believe that this was key to determine the ClpXP1/2 structure at an average resolution of 4 Å. The resolution for ClpX, however, is lower and therefore does not allow modeling of side chains.
An interesting finding of the current study is the structural visualization of the interface between the hexameric ClpX ATPase and the heptameric ClpP protease, which involves a symmetry mismatch. The structural plasticity, which is necessary for the interaction of the symmetrically different proteins is provided by the flexibility of the IGF-loops. The binding of ClpP to ClpX does not induce major conformational changes of ClpX and delocalization of distinct AAA+ subunits. The flexibility of ClpP-ClpX interface might be crucial to accommodate different conformations of the ATPase during hydrolysis and proteolysis, and might even allow rotational movement of the ATPase during the repeating cycles of substrate unfolding and translocation. However, further studies are necessary in order to support this scenario.
ClpX is tilted and slightly shifted relative to ClpP2 and the symmetry-axes of the protease and the ATPase are therefore not aligned. Thus, upon complex formation, the translocation pathway for unfolded peptides is not straight, but twisted. A similar arrangement involving a symmetry mismatch and formation of a twisted peptide translocation channel has been recently described for the PAN-proteasome39 and the bacterial ABC toxin complex37. The binding of proteasomal ATPases to the 20S core particle also involves a six-seven symmetry mismatch. However, in this case, the interface is more rigid, since the ATPases bind with their hydrophobic C-termini tightly into pockets at the surface of the 20S core particle (“key-in-lock” mechanism)54,55. Noteworthy, whereas most of ATPases induce pore opening to allow substrate entry into the proteasomal core, several eukaryotic ATPases (Rpt2, Rpt3 and Rpt5) stably bind to the same pockets of the core particle, but similar to ClpX, do not trigger gate-opening54,56.
Surprisingly, although ClpX interacts via the IGF-loops with the same site on ClpP as the potential antibiotic ADEP17, it does not induce the opening of the ClpP1/2 pore, as previously suggested. Thus, the underlying mechanisms of ClpP activation by ClpX and ADEP are distinct.
Our structure further reveals, that the extended C-terminus of L. monocytogenes ClpP1/2 shields the IGF-binding sites prior to ClpX binding. The length of the C-terminus is apparently crucial to fine-tune the binding affinity to ClpX, among the different species, which might be important for the future design of ClpP-based antibiotics.
The pore-2 loops, that control the peptidase gate and thread the substrate into the ClpP1/2 chamber, are disordered in our structure, underlining the dynamic nature of these interactions. However, the overall arrangement of adjacent structural elements suggest that the pore-2 loops are arranged in a spiral-staircase-like manner, similarly to other AAA+ complexes38,57.
Interestingly, the ClpXP1/2 complex from L. monocytogenes dimerizes. Only ClpP2 binds to ClpX and two opposing ClpX hexamers dimerize head-to-head through the ZBDs. In contrast, the E. coli ClpP homocomplex is doubly-capped by ClpX23. It is unclear whether the dimerization of the ClpXP1/2 complexes is biologically relevant. The termini of this arrangement of up to four ZBD dimers linking the ClpX hexamers, point directly to their distal pore entries. It is therefore tempting to speculate that this interaction might play a role in substrate binding and even help guiding it into the ClpX pores. Another explanation might be that, at the high concentrations used for EM, two copies of ClpX might recognize each other as substrate. This scenario is however unlikely, because most of ClpX stays intact after incubation of WT ClpX with WT ClpP1/2.
In summary, the cryo-EM structure of ClpXP1/2 provides the necessary basic insights into ClpXP architecture, essential to understand the molecular mode of action of this dynamic and highly flexible protein degradation machinery. Our results set the stage for future investigations into conformational changes underlying ClpXP ATP hydrolysis and substrate translocation during protein degradation.
Methods
Cloning
The cloning of pETDuet-1_ClpP1/2 and pET300_ClpX were described previously32. ClpX and ClpP1/2 point mutants, ClpP1/2ΔC-3, ClpP1/2ΔC-4 and ClpP1/2ΔC-5 were generated using the QuikChange™ technology. For ClpP1/2ΔC-4 and ClpP1/2ΔC-5, the pETDuet-1_ClpP1/2ΔC-3 plasmid was used as a template. ClpP1/2ΔC-6 and ClpXΔZBD(E183Q) were obtained with primers containing non-overlapping sequences 58. All primers are listed in Supplementary Table 1
Protein overexpression and purification
ClpP1/2 and its mutants’ variants were overexpressed and purified as follows. The proteins were overexpressed in E. coli BL21(DE3) bearing a pETDuet-1 vector with C-terminally Strep-II-tagged ClpP1 and C-terminally His6-tagged ClpP232. The bacteria were grown in LB medium until OD600 0.6 at 37 °C. Following induction with 1 mM isopropyl-β-D-thiogalactoside (IPTG), the bacteria were incubated at 37 °C for 6 h. After harvest, the cells were sonicated on ice in lysis buffer (20 mM MOPS, 300 mM KCl, 1% CHAPS, 10% glycerol, pH 7.5) and then kept at room temperature during the rest of the purification. The proteins from the cleared cell lysate were captured in a HisTrap HP 5 ml column (GE Healthcare) in His buffers (20 mM MOPS, 300 mM KCl, 10% glycerol, pH 7.5; +40 mM imidazole for washing) using an ÄKTA Purifier 10 system (GE Healthcare). The proteins were eluted by a 15 mL gradient from 40 mM to 300 mM imidazole, and the second elution peak was collected. A subsequent chromatography step was carried out on a StrepTrap HP 5 ml column (GE Healthcare) in Strep buffers (20 mM MOPS, 300 mM KCl, 10% glycerol, pH 7.5; +2.5 mM desthiobiotin for elution). A final gel filtration was performed on a Superdex200 pg 16/60 column (GE Healthcare) in ClpP SEC buffer (20 mM MOPS, 300 mM KCl, 15% glycerol, pH 7.0). In the case of the cystein-containing mutants, 1 mM TCEP was added to all buffers.
ClpX(E183Q) and ClpXΔZBD(E183Q) were overexpressed in E. coli BL21(DE3). An expression construct equipped with an N-terminal His6-tag and a TEV cleavage site in pET300 vector was used32. The bacteria were grown in LB medium to OD600 0.6 at 37 °C. After induction with 0.5 mM IPTG, the cells were incubated overnight at 25 °C. After harvest, the cells were resuspended in ClpX lysis buffer (25 mM HEPES, 200 mM KCl, 1 mM DTT, 0.5 mM ATP, 5 mM MgCl2, 10 mM imidazole, 5% glycerol, pH 7.6) and lysed by ultrasonication. The cleared cell lysate was loaded on a 5 mL HisTrap HP column (GE Healthcare). The column was washed with ClpX wash buffer (25 mM HEPES, 200 mM KCl, 1 mM DTT, 5% glycerol, 40 mM imidazole, pH 7.6). The protein was eluted with ClpX elution buffer (25 mM HEPES, 200 mM KCl, 1 mM DTT, 5% glycerol, 300 mM imidazole, pH 7.6). The protein fractions were pooled, 1 mM EDTA and TEV protease [1.25 mg for ClpX(E183Q) and 3.75 mg for ClpXΔZBD(E183Q)] were added and the reaction mixture was incubated at 10 °C overnight. Complete TEV cleavage was verified by intact-protein mass-spectrometry. The protein solution was loaded on a Superdex200 pg 16/60 column (GE Healthcare) and eluted in ClpX SEC buffer (25 mM HEPES, 200 mM KCl, 1 mM DTT, 0.5 mM ATP, 5 mM MgCl2, 5% glycerol, pH 7.6). ClpX(WT), ClpX(V264C), ClpX(I265C), ClpX(G266C) and ClpX(F267C) were overexpressed and purified similarly with the following modifications: the buffers contained 1 mM TCEP instead of DTT, and the ClpX wash buffer and ClpX elution buffer contained additionally 0.5 mM ATP and 5 mM MgCl2. The TEV digestion step was omitted.
N-terminally Strep-II-tagged eGFP with a C-terminal SsrA tag (AGKEKQNLAFAA) was overexpressed in E. coli SG1146a (ΔclpP) using pET55-Dest expression vector and purified by affinity chromatography and gel filtration as described previously 15,32.
Creatine kinase (product no. 10 127 566 001), lactate dehydrogenase (product no. 10 128 155 001) and pyruvate kinase (product no. 10 127 876 001) were purchased from Roche.
Isolation of the ClpXP complex
4.4 nmol (ClpP1/2)14 and 3.3 nmol ClpX6 were incubated for 10 min at 37 °C in PZA buffer (25 mM HEPES, 200 mM KCl, 5 mM MgCl2, 1 mM DTT, 0.5 mM ATP, 15% glycerol, pH 7.6). The samples were loaded onto a Superose 6 increase 10/300 column (GE Healthcare) connected to an ÄKTA Purifier 10 system (GE Healthcare) and eluted at 0.2 mL/min flow rate. Samples were taken at 12 mL retention volume for EM and HDX-MS measurements. For cryoEM, the sample was diluted 1:3 with glycerol-free PZA buffer and 0.1% glutaraldehyde was added. The reaction was quenched after 30 s with 2 eq. Tris-HCl. For SDS-PAGE, 4.4 μg protein was loaded on a gel and stained with Coomassie blue after separation.
Electron microscopy
Sample quality was examined by negative stain EM. Sample from the respective fraction was further diluted to a concentration of 0.01-0.03 mg ml-1 and negative stain EM was performed as described previously 59. Images were recorded with a JEOL JEM-1400 equipped with a 4K CMOS detector F416 (TVIPS) at a pixel size of 1.84 Å. For cryoEM, 4 μl of cross-linked ClpXP1/2 dimers at a concentration of 0.045 mg ml-1 were applied to a glow-discharged quantifoil 2/1 Cu grid with an additional 2nm thin carbon layer and after an incubation time of 45 sec, rapidly plunge-frozen using a CryoPlunge3 (Cp3, Gatan) at 90% humidity. To improve ice quality and thickness distribution, 0.01% Tween-20 was added shortly prior plunging. The quality of the grids was screened with a JEOL JEM 1400 and a FEI Tecnai Spirit, both equipped with a LaB6 cathode and a 4K CMOS detector F416 (TVIPS). A cryoEM dataset was acquired on a FEI Titan KRIOS at 300 kV equipped with spherical aberration corrector and a Falcon III direct detector (linear mode) at a x112,807 magnification (x59,000 nominal magnification), corresponding to a pixel size of 1.1 Å. Each exposure was recorded with a total dose of ~114 electrons/Å2 and a total exposure time of 2 sec (frame rate of 50 msec). A total of 3200 micrographs were collected using the EPU software (FEI).
Image processing and reconstruction
The frames were aligned, averaged and dose-weighted using unblur and sum_movie60. Unweighted full-dose images were further used to estimate the CTF parameters using CTER 61 (SPHIRE)25. Dose weighted full-dose images were used for all other steps of image processing. ClpXP1/2 dimers were picked automatically using EMAN2’s 62 neuralnet e2boxer. Further data processing was performed using the software package SPHIRE25. After inspection of micrographs using the CTF-assessment-GUI, 273,300 single particles were selected for further processing. The particle stack was subjected to 2D-clustering using ISAC2 (SPHIRE), resulting in a “clean” stack of 143,901 single particles producing stable and reproducible 2D-class averages. The 2D class-averages were used to calculate a 3D volume, using VIPER. After masking, this volume was used as the reference for a 3D refinement using Meridien (SPHIRE), which resulted in a 13 Å density map, as estimated by the “gold-standard” FSC. In agreement to the 2D clustering results (Supplementary Video 1), further 3D clustering using Sort3D (SPHIRE) confirmed that the ClpXP1/2 dimer is a continuously flexible structure (Supplementary Figure 1g). Independent refinement of the resulting subsets did not, however, further improve the resolution of the volume.
We then manually picked the ClpXP1/2 monomers within each ClpXP1/2-dimer for 10 representative micrographs of the dataset and used these data to train crYOLO63, which then automatically selected 613,322 single particles. After 2D and 3D clustering, a final “clean” stack of 383.927 particles was used for further refinement. During the first rounds of the refinement, we applied local symmetrization of the reference after each refinement round, as previously described 64,65 i.e. after each refinement round the density of ClpP was symmetrized using D7 symmetry, whereas the density of ClpX was scaled in order to put an additional weight on this region during the asymmetric refinement. Finally, both densities (ClpX and ClpP) were combined and the resulting volume was used as a reference for the subsequent refinement iteration. This procedure was performed during the initial rounds in order to obtain global projection parameters. The user function was not applied during the local refinements. This resulted in a density map with an average resolution of 4 Å, where the resolution of the density decreases towards ClpX (Supplementary Figure 2). The average resolution was calculated between two independently refined “half maps” at the 0.143 FSC criterion. The estimated accuracy of rotation and translation search during the last refinement round was estimated to 1.78° and 1.02 pixels, respectively. Local resolution was computed using the “Local Resolution” tool in SPHIRE. 3D clustering into four groups was performed using the RSORT3D tool of SPHIRE. However, according to the ANOVA analysis, the resulting volumes were not reproducible and were therefore not considered for further analysis. 3D Refinement and Clustering focusing on the density of ClpX, after removing the ClpP signal from the dataset, did also not result into further improvement of the ClpX density. The density of ClpP was auto-sharpened locally using phenix.auto_sharpen 66 and filtered to its average resolution of 3.9 Å. The ClpX desnity was filtered to an average resolution of 6.5 Å and sharpened with an ad-hoc b-factor of -240 Å2. Angular distribution plots were computed using SPHIRE. Sharpened 2D class averages were computed with 3500 members per group.
Atomic modelling
We built a homology model of ClpX with SWISS-MODEL 67 using ADP-bound E. coli ClpX (PDB-ID 3HWS, Chain A) and ATPγS-bound E. coli ClpX (PDB-ID 4I81, Chain B). We then used UCSF Chimera 68 to fit the structures of ClpX’s homology model and ClpP1/2 (PDBID 4RYF 32 into the cryo-EM density. We used the RosettaES protocol 69 to build the missing residues 9-16 for each ClpP2 subunit. Residues 1-2 were manually built in Coot70.
With the complete model, we performed several iterative runs of molecular dynamics flexible fitting (MDFF) 71and manual adjustment with Coot, paying particular attention to the fitting of the IGF loops. In the initial run, we applied 6-fold symmetry to ClpX, allowing regions poorly supported by the density to settle into reasonable conformations. This restraint was later removed. For the final iterations, we also included a step of real-space refinement in Phenix72, to decrease the number of Ramachandran outliers and to fit the atomic B-factors.
The necessary files for the MDFF runs were set up with VMD 73 and all simulations were performed in NAMD74, using the CHARMM 36m force field 75 with the implicit solvation model implemented in NAMD.
For the proper modeling of the structure with MDFF, we included all missing regions of the structures, even if their density does not allow full atomic modeling. After refinement, we removed all those from the final model. The quality of this model was assessed in Phenix, using the Molprobity 76 and EMRinger scores 77 as well as the overall geometry of the structure.
Sequence conservation was analyzed using the ConSurfserver78. Analysis of the channel pathway was performed with ChExVis79. Electron density maps and models were visualized using Chimera 69 and Chimera X80.
Peptidase assay
In this assay, the degradation of a fluorogenic tripeptide was measured, for which ClpX was not required. 99 μL 1 μM ClpP1/2 was incubated in PZ buffer (25 mM HEPES, 200 mM KCl, 5 mM MgCl2, 1 mM DTT, 10% glycerol, pH 7.6) in flat bottom black 96-well plates for 15 min at 30 °C. 1 μL acetylalanyl-homoarginyl-2-aminooctanoyl-7-amino-4-carbamoylmethylcoumarin (Ac-Ala-hArg-2-Aoc-ACC) substrate (10 mM stock in DMSO) was added and the fluorescence was measured (380 nm, 430 nm) with an infinite M200Pro plate reader (Tecan) at 30 °C. Data were recorded in triplicate and two independent experiments were performed. Peptidase activity was determined by linear regression using Microsoft Excel and plots were made with GraphPad Prism 6.
Protease assay
Protease assays were carried out in flat bottom white 96-well plates in a final volume of 60 μL. (ClpP1/2)14 (0.2 μM), ClpX6 (0.4 μM) and ATP regeneration mix (4 mM ATP, 16 mM creatine phosphate, 20 U/mL creatine kinase) were pre-incubated for 15 min at 30 °C in PZ buffer. 0.8 μM eGFP-SsrA substrate was added and fluorescence was measured (485 nm, 535 nm) at 30 °C. Data were recorded in triplicate and at least two independent experiments were performed. Protease activity was determined by linear regression using Microsoft Excel and plots were made with GraphPad Prism 6.
ATPase assay
90 μL 2 μM ClpX in ATPase buffer (100 mM HEPES, 200 mM KCl, 20mM MgCl2, 1 mM DTT, 1 mM NADH, 2 mM phosphoenolpyruvate, 50 U/mL lactate dehydrogenase, 50 U/mL pyruvate kinase, 5% glycerol, pH 7.5) was added to a flat bottom transparent 96-well plate and incubated for 15 min at 37 °C. The reaction was started by the addition of 10 μL 200 mM ATP in 100 mM HEPES, pH 7.5. Absorption at 340 nm was measured at 37 °C. Two independent experiments with three replicates each were carried out. ATPase activity was determined by linear regression using Microsoft Excel after subtraction of the background signal (measurement without ClpX), and the plot was made with GraphPad Prism 6.
Hydrogen/deuterium exchange mass-spectrometry (HDX-MS)
HDX-MS experiments were performed using an ACQUITY UPLC M-class system equipped with automated HDX technology (Waters). HDX kinetics were determined by taking data points at 0, 10, 60, 600, 1800 and 7200 s at 20 °C. At each data point of the kinetic, 3 µL of a solution of 30 µM „free” ClpP1/2 and „free” ClpX were analyzed and compared to the (ClpXP1/2)2 complex (1.4 µM). The respective protein solutions were diluted automatically 1:20 into 99.9% D2O-containing buffer (25 mM HEPES, 200 mM KCl, 5 mM MgCl2, 0.5 mM ATP, 1 mM TCEP, 5% glycerol, pH 7.6). As reference, all samples were analyzed in H2O –containing buffers. The reaction mixture was quenched by the addition of 1:1 200 mM KH2PO4, 200 mM Na2HPO4, pH 2.3 (titrated with HCl) at 1 °C and 50 µL of the resulting sample were subjected to on-column peptic digest on a Waters Enzymate BEH pepsin column 2.1 × 30 mm at 20 °C. Peptides were separated by reverse phase chromatography at 0 °C in trapping mode using a Waters Acquity UPLC C18 1.7 µm Vangard 2.1 × 5 mm pre-column and a Waters Aquity UPLC BEH C18 1.7 µm 1 × 100 mm separation column. For separation, a gradient increasing the acetonitrile concentration stepwise from 5 to 35% in 6 min, from 35 to 40% in 1 min and from 40 to 95% in 1 min was applied and the eluted peptides were analyzed using an in-line Synapt G2-S QTOF HDMS mass spectrometer (Waters). UPLC was performed in protonated solvents (0.1% formic acid), allowing deuterium to be replaced with hydrogen from side chains and amino/carboxyl termini that exchange much faster than backbone amide linkages81. All experiments were performed in duplicate. Deuterium levels were not corrected for back exchange and are therefore reported as relative deuterium levels82. The use of an automated system, i.e. handling all samples at identical conditions, negotiates the need for back exchange correction. MS data were collected over an m/z range of 100-2000, and are available online as source data [AU: correct?]. Mass accuracy was ensured by calibration with Glu-fibrino peptide B (Waters) and peptides were identified by triplicates MSE ramping the collision energy from 20-50 V. MS data were analyzed with the PLGS 3.0.3 and DynamX 3.0 software packages and all spectra were checked manually. For each peptide, relative uptake values were determined as follows: relative uptake [%] = deuterium uptake × 100 / maximal uptake. For each amino acid, the average of the relative uptake of all peptides covering the amino acid was calculated. The difference of the relative deuterium uptake between the “free” and “complex” states was calculated for each amino acid. Data were analyzed and visualized using custom MATLAB and python scripts, UCSF Chimera 1.12 and OriginPro 2016.
Supplementary Material
Acknowledgements
We thank O. Hofnagel for assistance in electron microscopy and Dr. M. Lakemeyer for building ClpX homology models. We are grateful to Dr. M. Haslbeck, G. M. Feind and F. Rührnößl for HDX-MS measurements. This work was supported by the Max Planck Society (to S.R.), the European Research Council (FP7/2007-2013) (grant no. 615984) (to S.R.) and the Deutsche Forschungsgemeinschaft (SFB1035) (to S.A.S).
Footnotes
Author Contributions S.A.S. and S.R. designed the study. C.G. screened and optimized samples, prepared cryo-EM grids, processed, and analyzed cryo-EM data. D.B. cloned, overexpressed and purified proteins, optimized sample preparation, conducted activity assays and gel filtration measurements, analyzed HDX-MS data. C.G. and F.M. built atomic models. C.G. and D.B. prepared figures, C.G., D.B., S.A.S., S.R. wrote the manuscript. All authors discussed the results.
Competing Interests The authors declare no competing interests.
Reporting summary:
Further information on experimental design is available in the Nature Research Reporting Summary linked to this article.
Data availability:
The cryoEM map of LmClpXP1/2 has been deposited at the EMDB with the accession code EMD-10162. The corresponding molecular models of LmClpX and LmClpP1/2 have been deposited at the wwPDB with accession codes PDB 6SFW and PDB 6SFX, respectively. Source data for Figure 4, Figure 5b-c, Supplementary Figure 1a and Supplementary Figure 6 are available online. All data used in this study are available from the corresponding authors upon reasonable request.
References
- 1.Bhandari V, et al. The Role of ClpP Protease in Bacterial Pathogenesis and Human Diseases. ACS Chem Biol. 2018;13:1413–1425. doi: 10.1021/acschembio.8b00124. [DOI] [PubMed] [Google Scholar]
- 2.Gaillot O, Pellegrini E, Bregenholt S, Nair S, Berche P. The ClpP serine protease is essential for the intracellular parasitism and virulence of Listeria monocytogenes. Mol Microbiol. 2000;35:1286–1294. doi: 10.1046/j.1365-2958.2000.01773.x. [DOI] [PubMed] [Google Scholar]
- 3.Yu AYH, Houry WA. ClpP: a distinctive family of cylindrical energy-dependent serine proteases. FEBS Lett. 2007;581:3749–3757. doi: 10.1016/j.febslet.2007.04.076. [DOI] [PubMed] [Google Scholar]
- 4.Brötz-Oesterhelt H, et al. Dysregulation of bacterial proteolytic machinery by a new class of antibiotics. Nat Med. 2005;11:1082–1087. doi: 10.1038/nm1306. [DOI] [PubMed] [Google Scholar]
- 5.Sauer RT, Baker TA. AAA+ proteases: ATP-fueled machines of protein destruction. Annu Rev Biochem. 2011;80:587–612. doi: 10.1146/annurev-biochem-060408-172623. [DOI] [PubMed] [Google Scholar]
- 6.Beuron F, et al. At sixes and sevens: characterization of the symmetry mismatch of the ClpAP chaperone-assisted protease. Journal of Structural Biology. 1998;123:248–259. doi: 10.1006/jsbi.1998.4039. [DOI] [PubMed] [Google Scholar]
- 7.Grimaud R, Kessel M, Beuron F, Steven AC, Maurizi MR. Enzymatic and structural similarities between the Escherichia coli ATP-dependent proteases, ClpXP and ClpAP. Journal of Biological Chemistry. 1998;273:12476–12481. doi: 10.1074/jbc.273.20.12476. [DOI] [PubMed] [Google Scholar]
- 8.Wang J, Hartling JA, Flanagan JM. The structure of ClpP at 2.3 A resolution suggests a model for ATP-dependent proteolysis. Cell. 1997;91:447–456. doi: 10.1016/s0092-8674(00)80431-6. [DOI] [PubMed] [Google Scholar]
- 9.Banecki B, Wawrzynow A, Puzewicz J, Georgopoulos C, Zylicz M. Structure-function analysis of the zinc-binding region of the Clpx molecular chaperone. Journal of Biological Chemistry. 2001;276:18843–18848. doi: 10.1074/jbc.M007507200. [DOI] [PubMed] [Google Scholar]
- 10.Baker TA, Sauer RT. ClpXP, an ATP-powered unfolding and protein-degradation machine. Biochim Biophys Acta. 2012;1823:15–28. doi: 10.1016/j.bbamcr.2011.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bewley MC, Graziano V, Griffin K, Flanagan JM. The asymmetry in the mature amino-terminus of ClpP facilitates a local symmetry match in ClpAP and ClpXP complexes. Journal of Structural Biology. 2006;153:113–128. doi: 10.1016/j.jsb.2005.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Joshi SA, Hersch GL, Baker TA, Sauer RT. Communication between ClpX and ClpP during substrate processing and degradation. Nat Struct Mol Biol. 2004;11:404–411. doi: 10.1038/nsmb752. [DOI] [PubMed] [Google Scholar]
- 13.Martin A, Baker TA, Sauer RT. Distinct static and dynamic interactions control ATPase-peptidase communication in a AAA+ protease. Mol Cell. 2007;27:41–52. doi: 10.1016/j.molcel.2007.05.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kim YI, et al. Molecular determinants of complex formation between Clp/Hsp100 ATPases and the ClpP peptidase. Nat Struct Biol. 2001;8:230–233. doi: 10.1038/84967. [DOI] [PubMed] [Google Scholar]
- 15.Gersch M, et al. AAA+ chaperones and acyldepsipeptides activate the ClpP protease via conformational control. Nat Commun. 2015;6:6320. doi: 10.1038/ncomms7320. [DOI] [PubMed] [Google Scholar]
- 16.Kirstein J, et al. The antibiotic ADEP reprogrammes ClpP, switching it from a regulated to an uncontrolled protease. EMBO Mol Med. 2009;1:37–49. doi: 10.1002/emmm.200900002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Schmitz KR, Carney DW, Sello JK, Sauer RT. Crystal structure of Mycobacterium tuberculosis ClpP1P2 suggests a model for peptidase activation by AAA+ partner binding and substrate delivery. Proc Natl Acad Sci USA. 2014;111:E4587–95. doi: 10.1073/pnas.1417120111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Alexopoulos J, et al. Structural determinants stabilizing the axial channel of ClpP for substrate translocation. Mol Microbiol. 2013;90:167–180. doi: 10.1111/mmi.12356. [DOI] [PubMed] [Google Scholar]
- 19.Zeiler E, et al. Vibralactone as a tool to study the activity and structure of the ClpP1P2 complex from Listeria monocytogenes. Angew Chem Int Ed Engl. 2011;50:11001–11004. doi: 10.1002/anie.201104391. [DOI] [PubMed] [Google Scholar]
- 20.Balogh D, et al. Insights into ClpXP proteolysis: heterooligomerization and partial deactivation enhance chaperone affinity and substrate turnover in Listeria monocytogenes. Chem Sci. 2017;8:1592–1600. doi: 10.1039/c6sc03438a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Amor AJ, Schmitz KR, Sello JK, Baker TA, Sauer RT. Highly Dynamic Interactions Maintain Kinetic Stability of the ClpXP Protease During the ATP-Fueled Mechanical Cycle. ACS Chem Biol. 2016;11:1552–1560. doi: 10.1021/acschembio.6b00083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hersch GL, Burton RE, Bolon DN, Baker TA, Sauer RT. Asymmetric interactions of ATP with the AAA+ ClpX6 unfoldase: allosteric control of a protein machine. Cell. 2005;121:1017–1027. doi: 10.1016/j.cell.2005.05.024. [DOI] [PubMed] [Google Scholar]
- 23.Ortega J, Singh SK, Ishikawa T, Maurizi MR, Steven AC. Visualization of substrate binding and translocation by the ATP-dependent protease, ClpXP. Mol Cell. 2000;6:1515–1521. doi: 10.1016/s1097-2765(00)00148-9. [DOI] [PubMed] [Google Scholar]
- 24.Wagner T, et al. SPHIRE-crYOLO is a fast and accurate fully automated particle picker for cryo-EM. Communications Biology. 2019;2:218. doi: 10.1038/s42003-019-0437-z. 2019 2:1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Moriya T, et al. High-resolution Single Particle Analysis from Electron Cryo-microscopy Images Using SPHIRE. J Vis Exp. 2017 doi: 10.3791/55448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Donaldson LW, Wojtyra U, Houry WA. Solution structure of the dimeric zinc binding domain of the chaperone ClpX. Journal of Biological Chemistry. 2003;278:48991–48996. doi: 10.1074/jbc.M307826200. [DOI] [PubMed] [Google Scholar]
- 27.Wojtyra UA, Thibault G, Tuite A, Houry WA. The N-terminal zinc binding domain of ClpX is a dimerization domain that modulates the chaperone function. Journal of Biological Chemistry. 2003;278:48981–48990. doi: 10.1074/jbc.M307825200. [DOI] [PubMed] [Google Scholar]
- 28.Stinson BM, et al. Nucleotide binding and conformational switching in the hexameric ring of a AAA+ machine. Cell. 2013;153:628–639. doi: 10.1016/j.cell.2013.03.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Trabuco LG, Villa E, Mitra K, Frank J, Schulten K. Flexible fitting of atomic structures into electron microscopy maps using molecular dynamics. Structure/Folding and Design. 2008;16:673–683. doi: 10.1016/j.str.2008.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Fux A, Korotkov VS, Schneider M, Antes I, Sieber SA. Chemical Cross-Linking Enables Drafting ClpXP Proximity Maps and Taking Snapshots of In Situ Interaction Networks. Cell Chem Biol. 2018 doi: 10.1016/j.chembiol.2018.10.007. [DOI] [PubMed] [Google Scholar]
- 31.Leodolter J, Warweg J, Weber-Ban E. The Mycobacterium tuberculosis ClpP1P2 Protease Interacts Asymmetrically with Its ATPase Partners ClpX and ClpC1. PLOS ONE. 2015;10:e0125345. doi: 10.1371/journal.pone.0125345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Dahmen M, Vielberg M-T, Groll M, Sieber SA. Structure and mechanism of the caseinolytic protease ClpP1/2 heterocomplex from Listeria monocytogenes. Angew Chem Int Ed Engl. 2015;54:3598–3602. doi: 10.1002/anie.201409325. [DOI] [PubMed] [Google Scholar]
- 33.Glynn SE, Martin A, Nager AR, Baker TA, Sauer RT. Structures of asymmetric ClpX hexamers reveal nucleotide-dependent motions in a AAA+ protein-unfolding machine. Cell. 2009;139:744–756. doi: 10.1016/j.cell.2009.09.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gates SN, et al. Ratchet-like polypeptide translocation mechanism of the AAA+ disaggregase Hsp104. Science. 2017;357:273–279. doi: 10.1126/science.aan1052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lo Y-H, et al. Cryo-EM structure of the essential ribosome assembly AAA-ATPase Rix7. Nat Commun. 2019;10:513. doi: 10.1038/s41467-019-08373-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ripstein ZA, Huang R, Augustyniak R, Kay LE, Rubinstein JL. Structure of a AAA+ unfoldase in the process of unfolding substrate. Elife. 2017;6:43. doi: 10.7554/eLife.25754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gatsogiannis C, et al. Tc toxin activation requires unfolding and refolding of a β-propeller. Nature. 2018;9:1. doi: 10.1038/s41586-018-0556-6. [DOI] [PubMed] [Google Scholar]
- 38.de la Peña AH, Goodall EA, Gates SN, Lander GC, Martin A. Substrate-engaged 26S proteasome structures reveal mechanisms for ATP-hydrolysis-driven translocation. Science. 2018;362 doi: 10.1126/science.aav0725. eaav0725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Majumder P, et al. Cryo-EM structures of the archaeal PAN-proteasome reveal an around-the-ring ATPase cycle. Proc Natl Acad Sci USA. 2019;116:534–539. doi: 10.1073/pnas.1817752116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Martin A, Baker TA, Sauer RT. Diverse pore loops of the AAA+ ClpX machine mediate unassisted and adaptor-dependent recognition of ssrA-tagged substrates. Mol Cell. 2008;29:441–450. doi: 10.1016/j.molcel.2008.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lee B-G, et al. Structures of ClpP in complex with acyldepsipeptide antibiotics reveal its activation mechanism. Nature Publishing Group. 2010;17:471–478. doi: 10.1038/nsmb.1787. [DOI] [PubMed] [Google Scholar]
- 42.Li DHS, et al. Acyldepsipeptide antibiotics induce the formation of a structured axial channel in ClpP: A model for the ClpX/ClpA-bound state of ClpP. Chem Biol. 2010;17:959–969. doi: 10.1016/j.chembiol.2010.07.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Jennings LD, Bohon J, Chance MR, Licht S. The ClpP N-terminus coordinates substrate access with protease active site reactivity. Biochemistry. 2008;47:11031–11040. doi: 10.1021/bi8010169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Geiger SR, Böttcher T, Sieber SA, Cramer P. A conformational switch underlies ClpP protease function. Angew Chem Int Ed Engl. 2011;50:5749–5752. doi: 10.1002/anie.201100666. [DOI] [PubMed] [Google Scholar]
- 45.Gersch M, List A, Groll M, Sieber SA. Insights into structural network responsible for oligomerization and activity of bacterial virulence regulator caseinolytic protease P (ClpP) protein. J Biol Chem. 2012;287:9484–9494. doi: 10.1074/jbc.M111.336222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kimber MS, et al. Structural and theoretical studies indicate that the cylindrical protease ClpP samples extended and compact conformations. Structure. 2010;18:798–808. doi: 10.1016/j.str.2010.04.008. [DOI] [PubMed] [Google Scholar]
- 47.Ye F, et al. Helix unfolding/refolding characterizes the functional dynamics of Staphylococcus aureus Clp protease. J Biol Chem. 2013;288:17643–17653. doi: 10.1074/jbc.M113.452714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zhang J, et al. Structural switching of Staphylococcus aureus Clp protease: a key to understanding protease dynamics. J Biol Chem. 2011;286:37590–37601. doi: 10.1074/jbc.M111.277848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ni T, et al. Characterization of Gain-of-Function Mutant Provides New Insights into ClpP Structure. ACS Chem Biol. 2016;11:1964–1972. doi: 10.1021/acschembio.6b00390. [DOI] [PubMed] [Google Scholar]
- 50.Stahl M, Sieber SA. An amino acid domino effect orchestrates ClpP's conformational states. Curr Opin Chem Biol. 2017;40:102–110. doi: 10.1016/j.cbpa.2017.08.007. [DOI] [PubMed] [Google Scholar]
- 51.Böttcher T, Sieber SA. Beta-lactones as privileged structures for the active-site labeling of versatile bacterial enzyme classes. Angew Chem Int Ed Engl. 2008;47:4600–4603. doi: 10.1002/anie.200705768. [DOI] [PubMed] [Google Scholar]
- 52.Frees D, et al. Clp ATPases are required for stress tolerance, intracellular replication and biofilm formation in Staphylococcus aureus. Mol Microbiol. 2004;54:1445–1462. doi: 10.1111/j.1365-2958.2004.04368.x. [DOI] [PubMed] [Google Scholar]
- 53.Cole A, et al. Inhibition of the Mitochondrial Protease ClpP as a Therapeutic Strategy for Human Acute Myeloid Leukemia. Cancer Cell. 2015;27:864–876. doi: 10.1016/j.ccell.2015.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Eisele MR, et al. Expanded Coverage of the 26S Proteasome Conformational Landscape Reveals Mechanisms of Peptidase Gating. Cell Rep. 2018;24:1301–1315.e5. doi: 10.1016/j.celrep.2018.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Smith DM, et al. Docking of the proteasomal ATPases‘ carboxyl termini in the 20S proteasome’s alpha ring opens the gate for substrate entry. Mol Cell. 2007;27:731–744. doi: 10.1016/j.molcel.2007.06.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Wehmer M, et al. Structural insights into the functional cycle of the ATPase module of the 26S proteasome. Proc Natl Acad Sci USA. 2017;114:1305–1310. doi: 10.1073/pnas.1621129114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Deville C, Franke K, Mogk A, Bukau B, Saibil HR. Two-Step Activation Mechanism of the ClpB Disaggregase for Sequential Substrate Threading by the Main ATPase Motor. Cell Rep. 2019;27:3433–3446.e4. doi: 10.1016/j.celrep.2019.05.075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Liu H, Naismith JH. An efficient one-step site-directed deletion, insertion, single and multiple-site plasmid mutagenesis protocol. BMC Biotechnol. 2008;8:91. doi: 10.1186/1472-6750-8-91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Gatsogiannis C, et al. Membrane insertion of a Tc toxin in near-atomic detail. Nature Publishing Group. 2016 doi: 10.1038/nsmb.3281. [DOI] [PubMed] [Google Scholar]
- 60.Grant T, Grigorieff N. Measuring the optimal exposure for single particle cryo-EM using a 2.6 Å reconstruction of rotavirus VP6. Elife. 2015;4:e06980. doi: 10.7554/eLife.06980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Penczek PA, et al. CTER-rapid estimation of CTF parameters with error assessment. Ultramicroscopy. 2014;140:9–19. doi: 10.1016/j.ultramic.2014.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Tang G, et al. EMAN2: An extensible image processing suite for electron microscopy. Journal of Structural Biology. 2007;157:38–46. doi: 10.1016/j.jsb.2006.05.009. [DOI] [PubMed] [Google Scholar]
- 63.Wagner T, et al. SPHIRE-crYOLO: A fast and well-centering automated particle picker for cryo-EM. bioRxiv. 2018 doi: 10.1101/356584. 356584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Tanaka Y, et al. Cryo-EM reveals the asymmetric assembly of squid hemocyanin. IUCrJ. 2019;6:426–437. doi: 10.1107/S205225251900321X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Gatsogiannis C, et al. Tc toxin activation requires unfolding and refolding of a β-propeller. Nature. 2018:1–5. doi: 10.1038/s41586-018-0556-6. [DOI] [PubMed] [Google Scholar]
- 66.Terwilliger TC, Sobolev OV, Afonine PV, Adams PD. Automated map sharpening by maximization of detail and connectivity. Acta Crystallogr D Struct Biol. 2018;74:545–559. doi: 10.1107/S2059798318004655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Biasini M, et al. SWISS-MODEL: modelling protein tertiary and quaternary structure using evolutionary information. Nucleic Acids Res. 2014;42:W252–8. doi: 10.1093/nar/gku340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Pettersen EF, et al. UCSF Chimera?A visualization system for exploratory research and analysis. J Comput Chem. 2004;25:1605–1612. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- 69.Frenz B, Walls AC, Egelman EH, Veesler D, DiMaio F. RosettaES: a sampling strategy enabling automated interpretation of difficult cryo-EM maps. Nature Methods. 2017;14:797–800. doi: 10.1038/nmeth.4340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Emsley P, Lohkamp B, Scott WG, Cowtan K. Features and development of Coot. Acta Crystallogr D Biol Crystallogr. 2010;66:486–501. doi: 10.1107/S0907444910007493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Singharoy A, et al. Molecular dynamics-based refinement and validation for sub-5 Å cryo-electron microscopy maps. Elife. 2016;5:213. doi: 10.7554/eLife.16105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Adams PD, et al. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr D Biol Crystallogr. 2010;66:213–221. doi: 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Humphrey W, Dalke A, Schulten K. VMD: visual molecular dynamics. J Mol Graph. 1996;14:33–8–27–8. doi: 10.1016/0263-7855(96)00018-5. [DOI] [PubMed] [Google Scholar]
- 74.Phillips JC, et al. Scalable molecular dynamics with NAMD. J Comput Chem. 2005;26:1781–1802. doi: 10.1002/jcc.20289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Huang J, et al. CHARMM36m: an improved force field for folded and intrinsically disordered proteins. Nature Methods. 2017;14:71–73. doi: 10.1038/nmeth.4067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Chen VB, et al. MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr D Biol Crystallogr. 2010;66:12–21. doi: 10.1107/S0907444909042073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Barad BA, et al. EMRinger: side chain-directed model and map validation for 3D cryo-electron microscopy. Nature Methods. 2015;12:943–946. doi: 10.1038/nmeth.3541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Ashkenazy H, Erez E, Martz E, Pupko T, Ben-Tal N. ConSurf 2010: calculating evolutionary conservation in sequence and structure of proteins and nucleic acids. Nucleic Acids Res. 2010;38:W529–33. doi: 10.1093/nar/gkq399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Masood TB, Sandhya S, Chandra N, Natarajan V. CHEXVIS: a tool for molecular channel extraction and visualization. BMC Bioinformatics. 2015;16:119. doi: 10.1186/s12859-015-0545-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Goddard TD, et al. UCSF ChimeraX: Meeting modern challenges in visualization and analysis. Protein Sci. 2018;27:14–25. doi: 10.1002/pro.3235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Englander SW, Kallenbach NR. Hydrogen exchange and structural dynamics of proteins and nucleic acids. Q Rev Biophys. 1983;16:521–655. doi: 10.1017/s0033583500005217. [DOI] [PubMed] [Google Scholar]
- 82.Wales TE, Engen JR. Hydrogen exchange mass spectrometry for the analysis of protein dynamics. Mass Spectrom Rev. 2006;25:158–170. doi: 10.1002/mas.20064. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.